

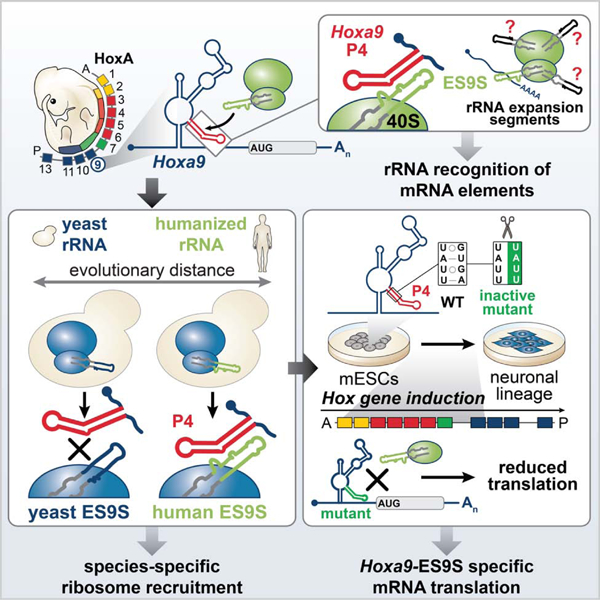
SUMMARY
Ribosomes have been suggested to directly control gene regulation, however regulatory roles for ribosomal RNA (rRNA) remain largely unexplored. Expansion segments (ESs) consist of multitudes of tentacle-like rRNA structures extending from the core ribosome in eukaryotes. ESs are remarkably variable in sequence and size across eukaryotic evolution with largely unknown functions. In characterizing ribosome binding to a regulatory element within a Homeobox (Hox) 5’ UTR, we unexpectedly identify a modular stem-loop within this element that binds to a single ES, ES9S. Engineering chimeric, “humanized” yeast ribosomes for ES9S reveals that an evolutionary change in the sequence of ES9S endows species-specific binding of Hoxa9 mRNA to the ribosome. Genome editing to site-specifically disrupt the Hoxa9-ES9S interaction demonstrates the functional importance for such selective mRNA-rRNA binding in translation control. Together, these studies unravel unexpected gene regulation directly mediated by rRNA and how ribosome evolution drives translation of critical developmental regulators.
Keywords: RNA structure, mRNA translation, ribosome, internal initiation, internal ribosome entry site, stem-loop, Hox cluster, RNA-protein interaction, yeast, ribosomal RNA, ribosome engineering, expansion segment, ES9S
Graphical Abstract
eTOC Blurb
Leppek and colleagues describe a novel layer of gene regulation mediated directly by ribosomal RNA (rRNA). They demonstrate that extended regions of rRNA, called expansion segments, which vary in sequence and size across evolution, selectively interact with an RNA stem-loop in the Hoxa9 5’UTR to guide species- and gene-specific translation.
INTRODUCTION
A central question in biology is how the genome is differentially expressed to enable the development of complex organisms. Recently, the ribosome has emerged as a direct, regulatory participant in control of gene expression (Genuth and Barna, 2018; Jackson et al., 2010; Kondrashov et al., 2011; Shi et al., 2017; Simsek et al., 2017; Sonenberg and Hinnebusch, 2009), during embryonic development (Kondrashov et al., 2011). However, beyond its fundamental role in the ribosomal peptidyl center, a functional contribution of ribosomal RNA (rRNA) to mRNA translation has been largely unexplored. The ribosome has increased in mass across eukaryotic evolution due in part to the insertions of sequence blocks called expansion segments (ESs), that “expand” eukaryotic rRNA (Gerbi, 1996). ESs are located in rRNA regions of lower primary sequence conservation, which implies that they are tolerated because they do not interfere with essential rRNA function. They vary in length and sequence both within and among different species, including different tissue types (Kuo et al., 1996; Leffers and Andersen, 1993; Parks et al., 2018). The longest ESs resemble tentacle-like, highly flexible extensions (Anger et al., 2013; Armache et al., 2010; Gerbi, 1996). For several decades, it has remained poorly understood whether ESs have a function in translation control and if there is a role for their dramatic variability across species. Thereby, we lack understanding of a critical facet in the evolution of an ancient molecular machinery and its biological impact on gene regulation and organismal development.
This study investigates how the ribosome is recruited to a structured 5’ UTR regulatory RNA element in a Homeobox (Hox) mRNA for ribosome-directed regulation of gene expression. As master regulators of metazoan body plan formation, the Hox clusters of transcription factors are one of the most spatiotemporally controlled transcripts already under broad regulation (Mallo and Alonso, 2013). Adding to the array of regulatory mechanisms, our lab has previously shown that a subset of Hox transcripts contain structured RNA internal ribosome entry sites (IRES)-like elements (Xue et al., 2015). Such regulatory elements (Leppek et al., 2017; Plank and Kieft, 2012) are critical for gene expression of several Hoxa mRNAs and anterior-posterior patterning of the axial skeleton (Kondrashov et al., 2011; Xue et al., 2015). A cap-proximal translation inhibitory element (TIE), a potent repressor of cap-dependent translation, within these 5’ UTRs, allows these Hox genes to be translated by the downstream IRES-like element as a means to more specifically control their spatiotemporal translation in development. As a paradigm example, we investigate the Hoxa9 IRES-like element, the first to have been selectively knocked-out in mice, leading to a homeotic transformation and diminished HOXA9 protein expression in developing somites and neural tube (Xue et al., 2015). Here, integrative mechanistic studies using different model systems ranging from mouse embryos, engineered yeast, to stem cell differentiation systems, as well as genome editing and structural biology, have revealed an unexpected function of ESs in gene regulation through mRNA-specific binding. Structural analysis of the Hoxa9 5’ UTR IRES-like element bound to the human ribosome by cryo-EM, reveals an interaction mediated by a single ES, ES9S, on the 40S small ribosomal subunit, particularly via a short RNA stem-loop. Evolutionarily distant yeast ribosomes, which possess a different ES9S sequence compared to mammalian ribosomes, cannot bind to this Hox 5’ UTR element. To functionally test the importance of ES sequences for species-specific mRNA-binding, we engineered chimeric ribosomes by “humanizing” yeast 18S rRNA exclusively in the distal part of ES9S, which is divergent between the two species. Such humanized ribosomes are sufficient to reconstitute Hoxa9 mRNA binding which highlights the ES specificity of this mRNA-rRNA interaction. Moreover, we interfered with the Hoxa9-ES9S interaction by selectively mutating the functional Hoxa9 5’ UTR binding site to ES9S in neural stem cells. These experiments revealed the critical importance of such mRNA-rRNA binding for accurate translational control in a physiological Hox gene expression system. Together, these findings suggest that the tentacle-like rRNA expansions of the ribosome may shape evolutionary diversity and endow greater modularity to this ancient molecular machine to guide gene and species-specific mRNA translation.
RESULTS
A short stem-loop in the Hoxa9 5’ UTR is sufficient to recruit the ribosome.
This study investigates how the ribosome is recruited to a structured 5’ UTR regulatory RNA element in the Hoxa9 mRNA to further understand ribosome-regulated translation (Figure 1A). The highly conserved mouse Hoxa9 IRES-like element expressed in mouse embryos (Figure S1A) folds into a 180 nucleotide (nt) long RNA secondary structure (termed a9 IRES180) (Figure S1B), that includes four pairing (P) elements P1–P4 (Cheng et al., 2015; Xue et al., 2015) (Figure 1A–C; S1B) within the 1.2 kb 5’UTR. We set out to identify the minimal RNA element within the a9 IRES180 required for translation initiation. First, our data show that the a9 IRES180 element is sufficient for IRES-like activity but is dependent on the distance from the start codon, possibly for correct ribosome placement or scanning. In particular, employing mouse C3H/10T1/2 embryonic mesenchymal cells that normally express Hox genes and support Hox IRES-like activity (Xue et al., 2015), either the 130 nt native spacer sequence downstream of a9 IRES180 (Figure 1B, D, E) or the unrelated actin 5’ UTR of similar length (100 nt) acting as a spacer (a9 IRES180-actin) promotes translation initiation compared to a9 IRES180 alone and actin-a9 IRES180 (Figure 1D, E). This enabled us to individually test the two highest conserved stem-loops of the a9 IRES180: P3 and P4. Both have previously been shown to contribute, and predominantly P4, to the overall IRES-like activity of the Hoxa9 5’ UTR (Xue et al., 2015), but it remained unclear if they are individually sufficient to confer IRES-like activity. Surprisingly, the P4 stem-loop of only 35 nts fused to actin is sufficient for IRES-like activity (Figure 1F). P4 activity also decreased by further shortening the spacer sequence (Figure S1 H, I). This is in contrast to P3 and P4 alone, or the P3-actin fusion. Next, extensive structural mutagenesis of the P4 stem-loop (Figure 1C, S1C–G) identified the smallest inactive P4 mutant, M5, that introduces only a 4-nt sequence mutation in the 3’ arm of P4 which diminishes IRES-like activity (Figure 1F, G, S1E). The activity of P4 fused to the 130 nt native or 100 nt actin(inv) spacer is abrogated by the P4(M5) mutation (Figure 1G). While a GUG codon is present in the P4(M5), it is out of frame with the main AUG, which excludes usage of the GUG as a start site in these reporters (Figure 1C, S1F, G). To more physiologically mirror the topology of the endogenous Hoxa9 5’ UTR, we designed a monocistronic “mini-UTR” reporter mRNA. It only contains the a9 TIE at the 5’ cap, which suppresses cap-dependent translation, immediately followed by the full-length a9 IRES-like element (Figure 1H). The P4-native-containing mini-UTR reporter mRNA specifically promoted translation initiation to the same extent as the a9 IRES180 and is abrogated by the M5 mutation. The hepatitis C virus (HCV) IRES that serves as an IRES control only mediates moderate IRES activity in this context. These data suggest that the P4 stem-loop is the critical, minimal active RNA element in the a9 IRES180 element.
Figure 1. A short stem-loop in the Hoxa9 5’ UTR is sufficient for recruitment of the ribosome.
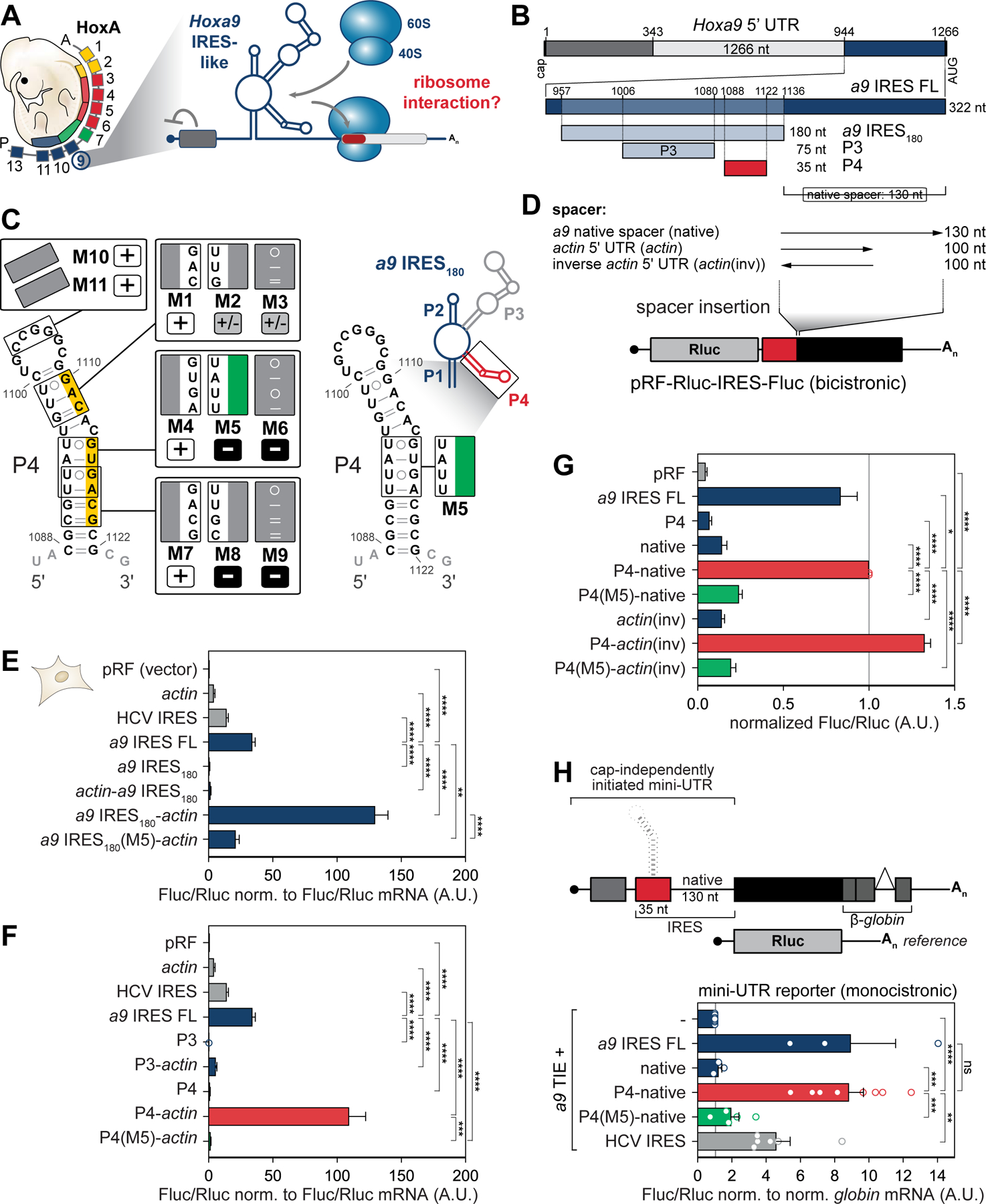
(A) Model of functional RNA elements in the Hoxa9 5’ UTR that regulate the translation of subsets of Hoxa mRNAs in the embryo (Xue et al., 2015). TIE, translation inhibitory element; IRES, internal ribosome entry site.
(B) Schematic of the topology of regulatory elements in the mouse Hoxa9 5’ UTR. The 180 nucleotides (nt)-long Hoxa9 IRES-like RNA element (a9 IRES180) harbors the P3 and P4 stem-loops and resides 130 nt upstream of the AUG (native spacer).
(C) Secondary structure model of a9 IRES180, a zoomed-in view of the P4 stem-loop (red), and substitution mutations mapped onto the P4 structure. Nts mutated in P4(M5) (green). Numbers refer to nt positions in the Hoxa9 5’ UTR. Active P4 mutants (normalized Fluc/Rluc < 0.5 A.U.) are labeled “+”, moderately active mutants (Fluc/Rluc < 0.5, > 1.0 A.U.) are labeled “+/–”, and inactive mutants (Fluc/Rluc > 0.5 A.U.) are labeled “–”. Yellow: Sequence critical for IRES-like activity. See also Figure S1.
(D) Spacer sequence requirement for a9 IRES-like element activity is tested by inserting spacers of different lengths downstream of an IRES-like element in a bicistronic reporter mRNA plasmid (pRF). Rluc, renilla luciferase; Fluc, firefly luciferase.
(E) Bicistronic reporter genes were transiently transfected into mouse C3H/10T1/2 cells and expressed from plasmids. Cells from the same transfection were split in half for protein and mRNA analysis. Relative luciferase activity is expressed as a Fluc(IRES)/Rluc(cap-initiation) ratio normalized to respective Fluc/Rluc mRNA levels. Average IRES-like activity ± standard error of the mean (SEM), n = 4–15. pRF and actin 5’ UTR serve as negative controls, HCV IRES as an IRES control. a9 IRES FL: FL, full-length; pRF (vector), no insert in the intergenic region; A.U., arbitrary units.
(F) Bicistronic reporter mRNAs were transiently expressed as described in (E). Average IRES-like activity normalized to respective Fluc/Rluc mRNA levels ± SEM, n = 4–15.
(G) Bicistronic reporter mRNAs were transiently expressed as described in (E). actin(inv) serves as a spacer sequence control. Average Fluc/Rluc IRES-like activity ± SEM, n = 3–8.
(H) Schematic of monocistronic “mini UTR” Fluc and control Rluc reporter mRNAs. IRES-like elements and spacer-derivatives were introduced into the Fluc 5’ UTR, and Fluc/Rluc luciferase activity was measured in transiently plasmid-transfected C3H/10T1/2 cells. A co-expressed Rluc reporter served as reference. Average Fluc/Rluc activity is normalized to respective globin/NupL1 mRNA levels ± SEM, n = 3–7; -, TIE alone; ns, not significant.
The Hoxa9 P4 stem-loop is a modular translation enhancer.
Further confirmation of P4’s activity to promote internal translation initiation was provided using A-capped Nanoluc (Nluc) reporter mRNAs (Figure 2A, B). RNA transfection of A-capped (ApppG) mRNA reporters that are exclusively initiated in a cap-independent manner are an established tool to assess cellular IRES-like activity (Hundsdoerfer et al., 2005). The increased internal initiation of such mRNA reporters by P4-actin(inv) was specifically reduced by introducing M5. In addition, when this P4-actin(inv) reporter mRNA is m7G-capped, as are most endogenous mRNAs (Figure 2C, D), a 1.8-fold increase in translation was observed compared to actin(inv) alone (Figure 2D). This finding highlights the modularity of the P4 stem-loop and suggests that it functions as a translation enhancer (TE) in multiple contexts. Therefore, P4 may serve as a modular TE in any 5’ UTR if placed upstream of a spacer rendering it a very attractive RNA element that promotes translation initiation beyond its importance for Hoxa9 mRNA expression. We therefore from here on in refer to P4 as a 5’ UTR TE. To further characterize P4’s modular TE activity, we interfered with cap-mediated initiation by treating cells with the small molecule inhibitor 4EGI-1 when the P4-actin(inv) reporter mRNA is m7G-capped (McMahon et al., 2011; Moerke et al., 2007). This molecule binds to the cap-binding protein eIF4E to displace the translation initiation factor eIF4G and inhibits its association with 4E, which blocks 40S ribosome recruitment and cap-dependent translation (Figure 2E, S1J, K). Transfection of a m7G-capped P4-actin(inv) reporter mRNA into 4EGI-1-treated cells compared to untreated/DMSO-treated control cells revealed a relative increase in translation initiation under conditions of strong cap-dependent translation inhibition (Figure 2F, S1J, K). These data indicate that P4 acts as a cap-independent TE.
Figure 2. The P4 stem-loop is the minimal TE element in the Hoxa9 5’ UTR.
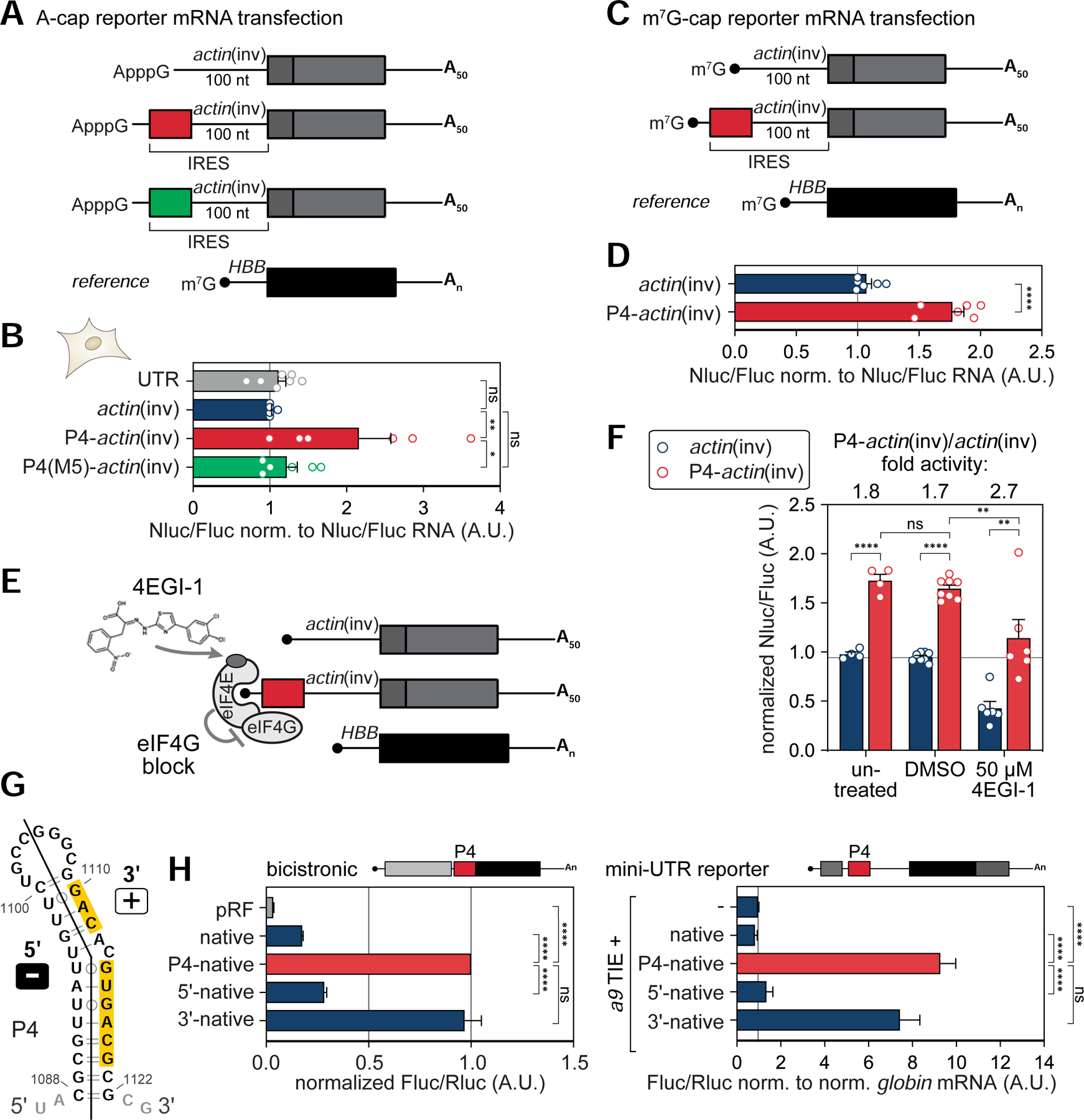
(A) In vitro transcribed, A-capped, and polyadenylated (A50-tail) HA-Nanoluc (Nluc) reporter mRNAs containing 5’ UTR elements. m7G-capped and polyadenylated HBB-Fluc mRNA served as reference.
(B) A-capped reporter mRNAs shown in (A) were directly transfected into mouse C3H/10T1/2 cells. IRES-like activity of P4-actin(inv) derivatives were compared to a 46 nt-long scrambled control (UTR). m7G-capped HBB-Fluc mRNA was co-transfected as reference. Cells were transiently transfected with RNA for 6 hours, cells from the same transfection were split in half for protein and mRNA analysis, and average Nluc/Fluc luciferase activity was normalized to respective Nluc/Fluc mRNA levels ± SEM, n = 6–9; actin(inv) alone was set to 1.
(C) In vitro transcribed, m7G-capped, and polyadenylated (A50-tail) HA-Nanoluc (Nluc) reporter mRNAs as in (A) that contain a conventional m7G cap.
(D) m7G-capped reporter mRNAs shown in (C) were directly transfected into human HEK293T cells as in (B). Average Nluc/Fluc luciferase activity was normalized to respective Nluc/Fluc mRNA levels ± SEM, n = 6.
(E) Schematic of the small molecule inhibitor 4EGI-1 that binds to eIF4E and blocks eIF4G association, thus uncouples cap-dependent initiation from P4 translation enhancer function. See also Figure S1.
(F) Luciferase activity analysis in mouse C3H/10T1/2 cells was carried out as in (D). Cells were treated for 3 h, transiently transfected with RNA for 6 hours in presence of drug or carrier, and harvested for luciferase analysis. Average Nluc/Fluc luciferase activity ± SEM, n = 4–8; actin(inv), untreated was set to 1.
(G) Secondary structure model of the 5’ and 3’ arms of the P4 stem-loop mapped onto the structure. Yellow: Sequence critical for P4 activity.
(H) P4 derivatives fused to the native spacer were tested for IRES-like activity in bicistronic (left, average Fluc/Rluc IRES-like activity ± SEM, n = 4–6) or monocistronic mini-UTR reporter mRNAs (right, average Fluc/Rluc activity normalized to respective globin/NupL1 mRNA levels ± SEM, n = 3–8). Reporter mRNAs were transiently expressed from plasmids as described in Figure 1E, H. The inactive 5’ arm (Fluc/Rluc > 0.5 A.U.) is labeled “–”, and the active 3’ arm (normalized Fluc/Rluc < 0.5 A.U.) is labeled “+” in (G).
Given that mutations in the basal stem of P4 strongly affect its activity (Figure 1C, S1C–G), the functional contribution of the P4 5’ and 3’ stem portions individually were next examined (Figure 2G, H). Particularly, mutations affecting the 5’ arm and the loop of P4 (M1, M4, M7, M10, and M11) do not affect its activity, whereas 3’ arm mutants (M5, M6, M8, and M9) are inactive (Figure 1C, S1F, G). These data suggest that a sequence motif in the P4 3’ arm is crucial for P4 TE activity. Indeed, only the 3’ arm has equal activity as the full P4 (Figure 2G, H). Together, these data suggest that unexpectedly the 35 nt-long P4 stem-loop of the a9 IRES180 element by itself, and particularly the 18 nt sequence of the 3’ P4 stem-loop, is important for its activity as a TE, which is sufficient to mediate internal translation initiation.
The Hoxa9 IRES-like element binds to the 40S ribosome.
The next question was whether and how the Hoxa9 5’ UTR, and P4 in particular, interacts with the ribosome. Ribonucleoprotein (RNP) affinity purification through streptavidin (SA)-binding 4xS1m aptamers was employed that we had previously established (Leppek and Stoecklin, 2014; Leppek et al., 2013) (Figure 3A, S2). RNPs were formed in vitro by incubating cell lysates with RNA-coupled beads, releasing them by RNase A, and enrichment of components of the translation machinery were monitored by western blot (WB) analysis. Among different regions of the Hoxa9 5’ UTR tested, combined with C3H/10T1/2 cell lysate, compared to the P3 that shows overall weak direct ribosome binding, P4 most highly enriched for the 40S component RPS6/eS6 and to a lesser extent for RPL10A/uL1 (60S) (Figure 3B, S2A). Beyond this qualitative assessment by WB analysis that is limited in its sensitivity for highly abundant proteins such as RPs as probed for by individual antibodies, the samples were subjected to quantitative mass spectrometry (MS) for P3 and P4 in comparison to the 4xS1m control to assess the relative enrichment of whole ribosomal subunits in the pulldown (Thompson et al., 2003) (Figure 3C; Table S4). P3, that by itself cannot mediate IRES-like activity (Figure 1F), does indeed not enrich for 40S nor 60S components (Figure 3D, S2B). In contrast, P4 preferentially enriches for almost all detected 40S proteins (29/30) and significantly less and only partially for 60S components. To control for the specificity of the observed 40S binding preference due to the overall RNA-binding preference of ribosomal subunits, compared to P4, P4(M5) and the inverse P4 sequence show reduced and abrogated binding to a representative 40S RP (Figure 3E, S2C). Similarly, the full a9 IRES180 structure can bind to this 40S component, which is specifically abolished by its inverse sequence as a negative control.
Figure 3. The Hoxa9 IRES-like element binds to the 40S ribosomal subunit via P4.
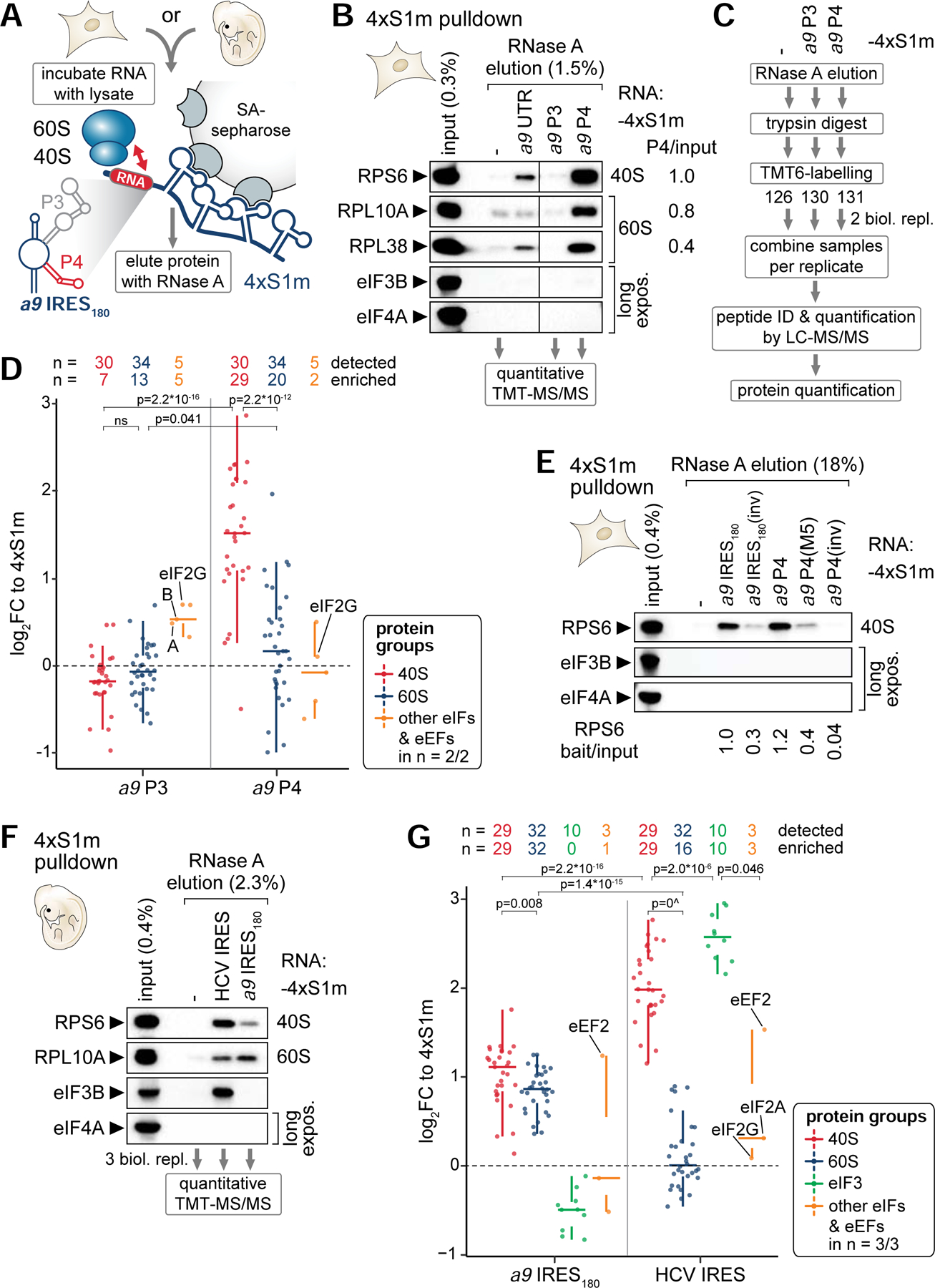
(A) Schematic of the 4xS1m pulldown to probe interactions of in vitro transcribed 4xS1m-aptamer fusion RNA with lysate components from C3H/10T1/2 cells or mouse embryos to form ribonucleoproteins (RNPs) in vitro. SA, streptavidin.
(B) 4xS1m pulldown is performed by combining mouse Hoxa9 mRNA elements with C3H/10T1/2 cell lysates. The aptamer alone (−) served as negative control. RPs of the 40S and 60S subunit and eIFs were monitored by western blot (WB) analysis. The fraction loaded of input and elution samples is expressed as percentage of the original lysate volume. Representative of n = 2 is shown. RNase A elutions of the aptamer control, P3 and P4 were subjected to mass spectrometry (MS) analysis. Differential enrichment of RPs compared to input with P4 was normalized to RPS6 set to 1. UTR, full-length Hoxa9 5’ UTR.
(C) Workflow for identifying and quantifying proteins in RNase A elutions by quantitative MS using tandem mass tag (TMT) peptide labelling. Proteins were trypsin-digested into peptides, labeled with a distinct TMT, mixed equally per replicate, and subjected to liquid chromatography (LC)-tandem MS (MS/MS) analysis for multiplex quantification.
(D) Analysis of TMT-MS/MS data displayed as log2 fold change (FC) relative to the aptamer control (4xS1m) for a9 P3 and P4 shows the relative abundance of proteins detected and enriched in respective protein groups compared to their levels in the control. Samples correspond to the pulldown in (B) from C3H/10T1/2 cells. Only proteins detected in 2/2 biological replicates are shown. See also Table S4.
(E) 4xS1m-pulldown as described in (B) comparing the a9 IRES180 and a9 P4 to control constructs. Lysates of C3H/10T1/2 cells were used as input. Representative of n = 3 is shown. Differential enrichment of RPS6 compared to input with RNA baits was normalized to a9 IRES180 set to 1.
(F) 4xS1m-pulldown as described in (B) comparing the a9 IRES180 to an unrelated viral IRES, HCV. Lysates of FVB stage E11.5 mouse embryos were used as input. Representative of n = 3 is shown. RNase A elutions were subjected to TMT-MS/MS analysis.
(G) Analysis of TMT-MS/MS data displayed as log2 fold change (FC) relative to 4xS1m for a9 IRES180 and HCV IRES as in (D). Samples correspond to the pulldown in (F) from FVB stage E11.5 mouse embryos. Only proteins detected in 3/3 biological replicates are shown. See also Table S5.
To recapitulate the cellular environment in which Hox genes are expressed, the ribosome interaction of the a9 IRES180 was further confirmed by employing E11.5 mouse embryo lysates (Figure 3F, S2D). Interestingly, MS-quantification revealed that the a9 IRES180 binds to both full 40S and 60S subunits, with a preference for the 40S (Figure 3G, S2E, F; Table S5). This is in contrast to the HCV IRES that strongly enriches for only the 40S, and the 13-subunit eIF3 complex (10/10 subunits enriched, Figure 3G, S2G). eIF3B binding was confirmed for the HCV IRES, but not the Hoxa9 IRES-like element (Figure 3F). However, in the full a9 IRES180, additional regions or more extensive tertiary RNA structure may contribute or be needed to recruit the 60S, consistent with the previous observation that a large subunit protein, RPL38/eL38, is functionally important for IRES-like activity (Kondrashov et al., 2011; Xue et al., 2015). Together, these data suggest that P4 alone without additional binding factors serves as the minimal RNA element sufficient to recruit the 40S to the Hoxa9 5’ UTR. However, it is part of a more complex RNA sequence integrated in an unusually long 5’ UTR, wherein future studies are required to fully understand how, in particular, a specific RP can promote translation initiation from this 5’ UTR.
The Hoxa9 IRES-like element and P4 bind the ribosome via 18S rRNA ES9S.
We next obtained the cryo-EM structure of the Hoxa9 IRES-like RNA bound to the ribosome for which 40S and 80S subunits from human cells were used. A 3.9 Å cryo-EM reconstruction of the full-length Hoxa9 IRES-like element in complex with the human 40S was obtained (Figure 4A, S3A–C). At the top of the head of the 40S, an extra density is visible (orange) corresponding in size and shape to an RNA helix that is part of the Hoxa9 IRES-like element. A similar extra density (orange) is also seen in context of the 80S ribosome at 4.40 Å (Figure 4B, S3D–F). Strikingly, cryo-EM analysis of the 40S bound to the P4 stem-loop RNA alone revealed a similar interaction (orange) at the 40S head at 4.1 Å (Figure 4C, S4) as with the full-length RNA, which is absent in a reconstruction of the 40S alone (Figure S4B). Unexpectedly, closer examination of the interaction site of the RNA revealed a single ES, ES9S, in the 18S rRNA as the direct binding site for both the full-length Hoxa9 IRES-like element (Figure 4A, B) and P4 alone (Figure 4C). Importantly, while human ribosomes were used for cryo-EM analyses, the ES9S sequence is identical in human and mouse 18S rRNA. The resolution of the 40S head allows to fit an atomic model of the corresponding portion of the human ribosome (Natchiar et al., 2017) which only required minor adjustments to the tip of ES9S. With this higher resolution reconstruction of the P4–40S complex (Figure 4D, E), the extra density has clear features and the shape of an RNA helix that corresponds in size to the P4 stem-loop helix. However, the lower local resolution of these reconstructions at the interaction site, probably due to the flexibility of ES9S and P4, does not allow for a more detailed interpretation of the mode of RNA-RNA interaction at present. Based on loop mutations in P4 (M10 and M11; Figure 1C, S1) not affecting P4 activity, we assume this interaction is less likely to represent a loop-loop interaction. Nevertheless, these data clearly reveal ES9S as the binding site on the ribosome. Together, these findings unexpectedly reveal mRNA-rRNA contacts between a very specific rRNA ES and the Hoxa9 5’ UTR, which may serve as the entry point for 40S ribosome recruitment required for translation initiation.
Figure 4. Cryo-EM reveals that the Hoxa9 IRES-like and P4 RNA bind to the ribosome via ES9S.
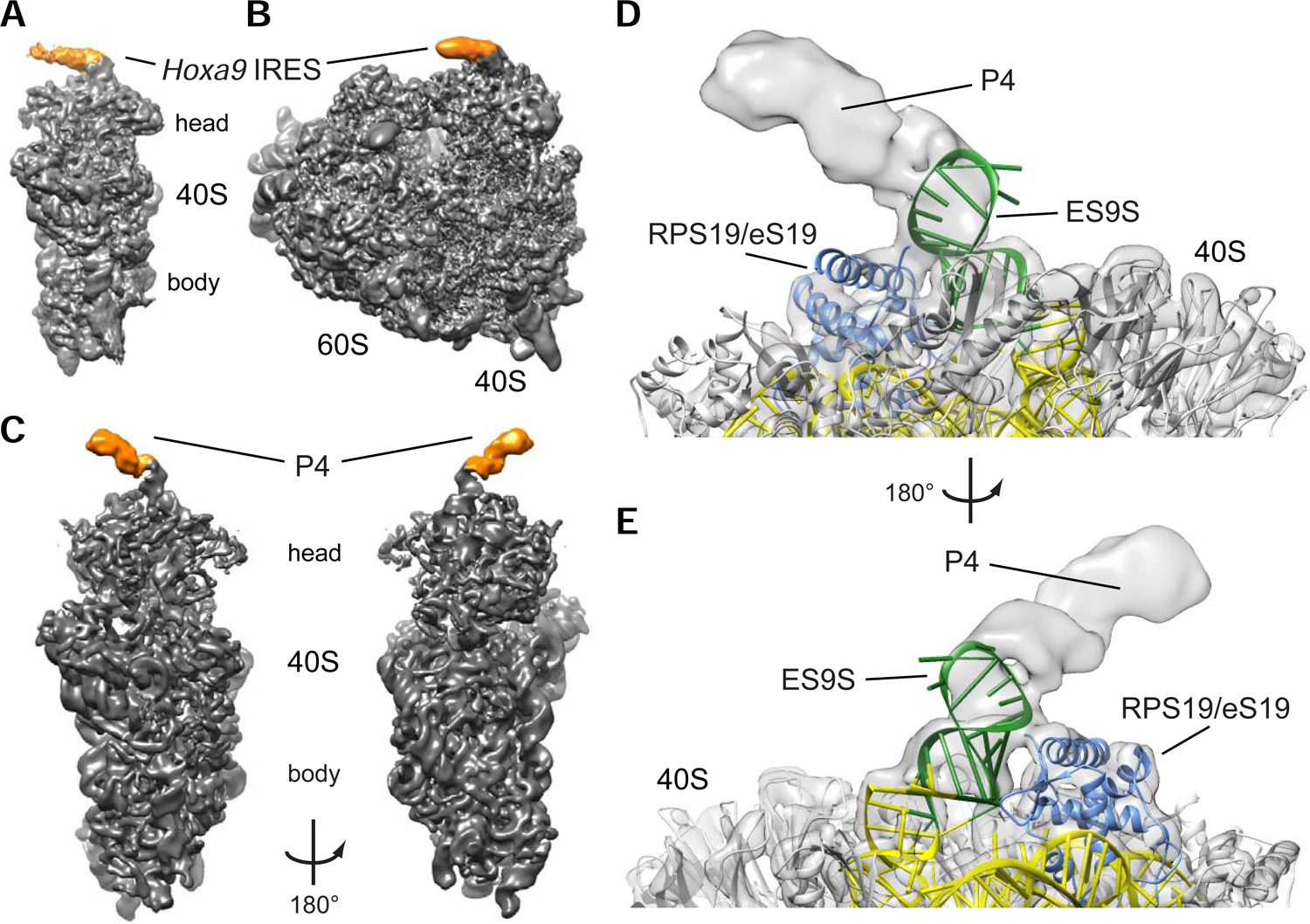
(A) Reconstruction of the human 40S ribosomal subunit with the mouse Hoxa9 IRES-like element (a9 IRES FL) at 3.9 Å resolution. Additional density for a9 IRES FL is indicated in orange.
(B) Reconstruction of the human 80S ribosome at 4.40 Å resolution with the additional density for the mouse a9 IRES FL indicated in orange.
(C) The a9 P4 stem-loop binds to the head of the small 40S ribosomal subunit. Reconstruction of the P4 stem-loop (orange) bound to human 40S ribosomal subunit (grey) at 4.1 Å for the 40S head and 3.1 Å resolution for the 40S body. The density is low-pass filtered to 7 Å to show RNA helical features of P4 (orange).
(D) Cryo-EM analysis of the mouse a9 P4 stem-loop in complex with the human ribosome. Reconstruction of the 40S ribosomal subunit head in complex with P4 at 4.1 Å resolution. P4 binds to ES9S (green) of the 18S rRNA (yellow) near ribosomal protein RPS19/eS19 (blue). The tip of ES9S was modelled onto pdb 5a2q to better visualize the bound P4 element.
(E) 180° rotation of the reconstruction in (D).
Engineering of humanized ribosomes in yeast exclusively harboring human ES9S.
We next investigated whether the ES9S in 18S rRNA is functionally required for the ribosome-Hoxa9 5’ UTR contact. Due to thousands of repeats of ribosomal DNA (rDNA) loci distributed across multiple chromosomes in metazoans (Romanova et al., 2006), it is not presently possible to genetically manipulate specific rRNA regions in mammalian cells. In the yeast Saccharomyces cerevisiae (S. cerevisiae) (Armache et al., 2010) and the human (Natchiar et al., 2017) (H. sapiens) 18S rRNA secondary structure, the basal stem of helix h39 adjacent to ES9S is highly conserved while the distal portion of ES9S is variable in length, structure and sequence (Figure 5A, boxed region). This ES9S divergence is also apparent in comparison to other species across evolution (Figure 5B). The distal human ES9S structure was revised based on our cryo-EM data (Figure 4). We next harnessed the interspecies variability of ES9S to test the specificity of P4 TE binding to the ribosome via this ES and the functional importance of the evolutionary change in ES9S sequence. To accomplish this, we turned to yeast as a model system, which only has a single rDNA locus, containing hundreds of tandem-repeated rDNA copies. This allows for deletion of the entire rDNA locus and complementation with edited rDNA from an exogenous plasmid (Nemoto et al., 2010; Wai et al., 2000). We thereby engineered “humanized” (termed hES9S) hybrid ribosomes (Figure 5C), for which hES9S was introduced scarlessly into the conserved h39 stem of yeast 18S rRNA (boxed region in Figure 5A). To distinguish humanized rRNA-containing ribosomes from possibly remaining untagged wildtype (WT) ribosomes, unique sequence tags for RT-qPCR were introduced into both 18S and 25S rRNA (Figure 5D, S5A–C). As rRNA is sensitive to manipulation and exchanging large regions can lead to ribosome biogenesis defects (Jeeninga et al., 1997; Ramesh and Woolford, 2016; Sweeney et al., 1994), it was initially confirmed that yeast cells induced to exclusively contain hES9S-ribosomes are viable and only show a slight growth defect in comparison to WT rRNA-containing cells (Figure S5A, D). This enabled the successful isolation of yeast strains after rDNA plasmid shuffling (Figure 5E, S5B) (Nemoto et al., 2010) that solely contain tagged hES9S 18S rRNA-ribosomes, as well as control strains harboring tagged WT 18S rRNA-ribosomes (Figure S5B–D) that could then be used as a tool to study species-specific mRNA-ES interactions.
Figure 5. Engineering of chimeric hES9S-humanized ribosomes in yeast.
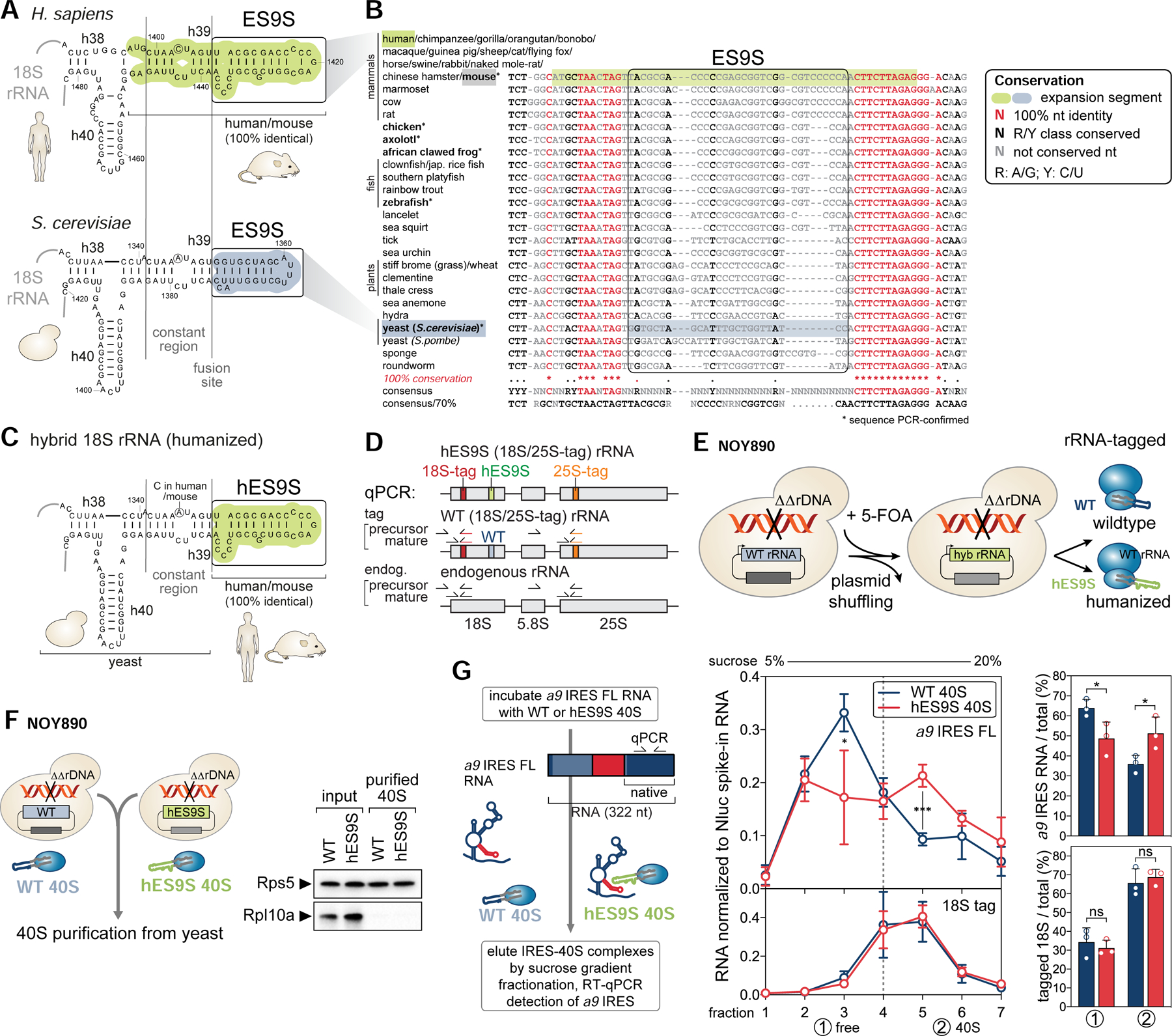
(A) Secondary structure models of the human (green) and yeast (blue) 18S rRNA region containing ES9S. The constant region (h39) and ES9S-fusion site selected for chimeric 18S rRNA engineering are indicated.
(B) A 40-way multiple sequence alignment (MSA) and conservation analysis of the highly conserved 18S rRNA region in which the more variable ES9S is embedded. Nts are color coded according to conservation. For six species (bold, asterisk) annotated 18S rRNA sequences were confirmed by RT-PCR spanning the ES9S region. R, purine; Y, pyrimidine.
(C) Structure model of the engineered yeast 18S rRNA after exchange of the yeast to human ES9S (hES9S, green).
(D) The rRNA cassette that encodes 18S, 5.8S and 25S rRNA indicating the position of unique sequence tags in 18S (red) and 25S (orange) rRNA used for RT-qPCR to detect precursor and mature forms of endogenous and tagged yeast rRNA.
(E) The plasmid shuffling approach to generate yeast strains that contain a homozygous knock-out of the rDNA locus (NOY890) and exclusively express plasmid-encoded tagged chimeric 18S rRNA as in (C). See also Figure S5.
(F) 40S subunits of WT and hES9S yeast strains (NOY890) were purified by sequential sucrose gradient sedimentation (see also Figure S6). The purity of the isolated 40S was confirmed by WB analysis of RPs compared to the input lysate. RPL10A/uL1 is yeast Rpl1 (referred to as Rpl10a).
(G) In vitro binding assays using purified WT and hES9S 40S subunits to test direct binding to the a9 IRES FL RNA. IRES-40S complexes were eluted by 5–20% sucrose gradient fractionation. Co-sedimentation of 40S (18S rRNA tag) and bound RNA (a9 IRES FL) was detected by RT-qPCR, normalized to a Nluc spike-in RNA (average ± SD, n = 3). IRES-40S co-sedimentation was assessed by integrating the gradient distribution and expressed for the free (1) and 40S (2) fractions as the percentage relative to the total (average ± SD, n = 3).
Humanized yeast ribosomes reconstitute binding of the Hoxa9 IRES-like RNA and P4 TE to hES9S.
Initially, we asked whether humanized hES9S 40S subunits can directly bind in vitro to the Hoxa9 IRES-like RNA element compared to WT 40S. For that, 40S subunits were purified from WT and hES9S yeast strains by high salt/puromycin treatment and sequential gradient fractionation (Figure S6A), and their purity and integrity were confirmed (Figure 5F, S6B). The a9 IRES FL RNA was then incubated with WT or hES9S 40S and RNP complexes were separated on a sucrose gradient (Figure 5G). The ability of the a9 IRES FL RNA to co-migrate with the 40S was assessed using the 18S tag as a reference (Figure 5G). The a9 IRES-like RNA co-sediments with hES9S 40S in high molecular weight fractions in contrast to the WT 40S, in which the majority of RNA is detected in the unbound, free fractions. These data support ES9S as the direct interaction site for the a9 IRES-like RNA on the 40S subunit.
Next, the importance of ES9S-Hoxa9 IRES-like interaction was investigated in complex yeast lysates. In vitro 4xS1m pulldowns (Figure 6A, Figure S6) (Leppek and Stoecklin, 2014) with lysates of tagged WT or humanized yeast strains revealed no binding of P4 and a9 IRES180 to WT yeast ribosomes, but a 36- and 11-fold enrichment of binding, respectively, to 18S rRNA from humanized ribosomes (Figure 6B, Figure S6C, D). Consistently, a 40S RP, RPS5/uS7, was found enriched with both P4 TE and a9 IRES180 RNAs by WB analysis. The specificity of these interactions was corroborated by no enrichment of tagged 25S rRNA, nor other classes of endogenous yeast RNAs, such as act1 mRNA or UsnRNA1. The viral HCV IRES served as a negative control, as it is known to bind to the 40S through a distinct, ES9S-independent mechanism (Malygin et al., 2013; Matsuda and Mauro, 2014).
Figure 6. Chimeric hES9S-humanized yeast ribosomes reconstitute Hoxa9- and P4-ES9S binding.
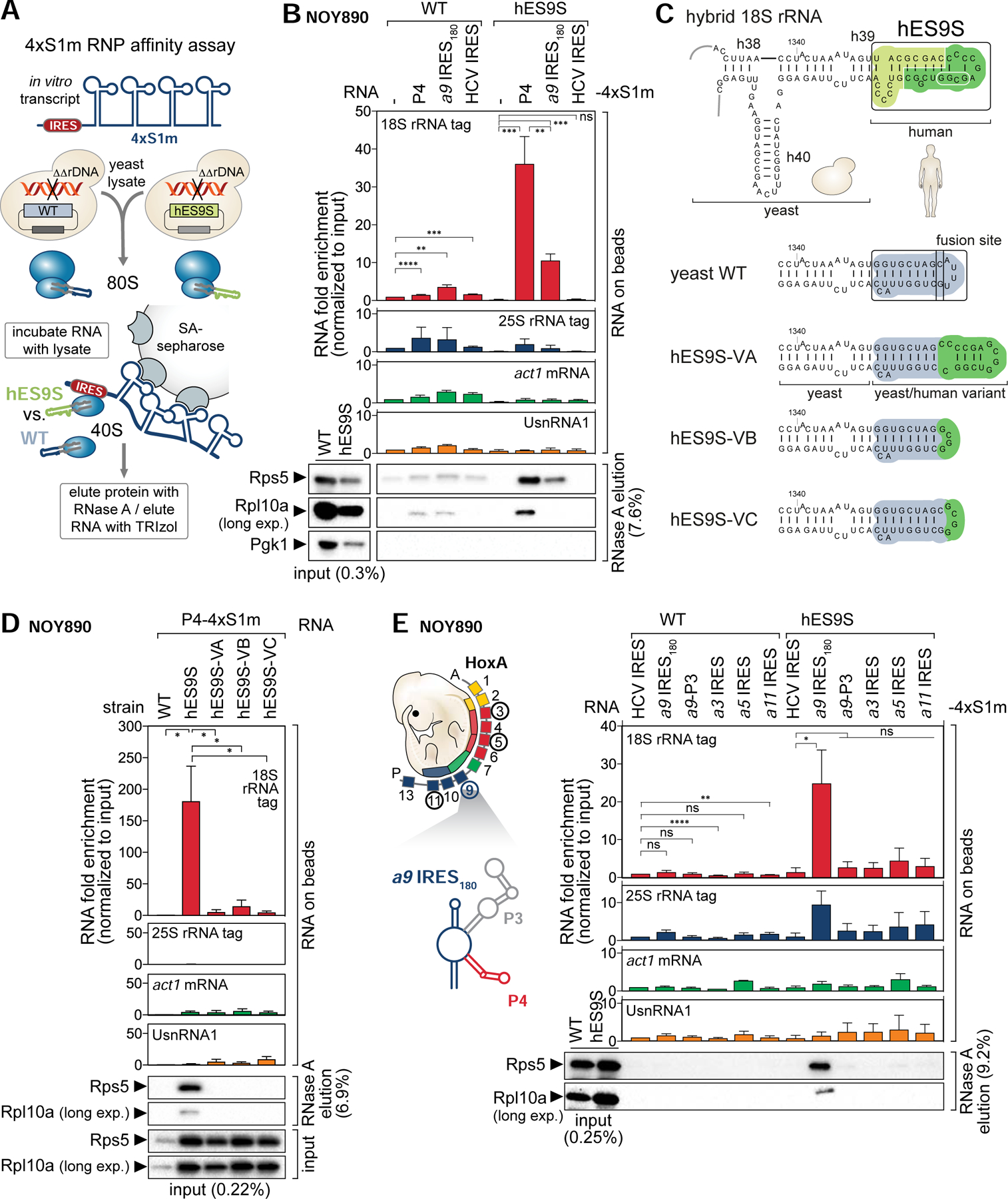
(A) Schematic of the 4xS1m pulldown to probe interactions of IRES-4xS1m RNA with WT and hES9S ribosomes from yeast (NOY890) lysates. Ribosome enrichment is monitored by RT-qPCR for tagged rRNA and other RNA classes normalized to the input and WB analysis for RPs.
(B) rRNA bound to 4xS1m-fused RNA is quantified with primers specific for 18S and 25S rRNA tags (RNA on beads). 4xS1m aptamer alone (−) and the HCV IRES serve as a negative and IRES control, respectively. The 4xS1m aptamer/WT sample was used to normalize for fold enrichment of detected RNA (set to 1). Yeast actin (act1) and yeast UsnRNA1 serve as negative controls for an mRNA and a non-coding RNA, respectively. Ribosome enrichment was assessed by WB analysis of same volumes of protein released from beads by RNase A. The fraction loaded of input and elution samples is expressed as percentage of the original lysate volume. Cytoplasmic enzyme PGK1 serves as a negative control. Average RNA fold enrichment, standard deviation (SD), n = 3; long exp., long exposure. See also Figure S6.
(C) Structure model of the engineered (hES9S, green) yeast 18S rRNA with annotations of the tested sequences (dark green; white circle) for hES9S-variants VA-C which contain partial hES9S sequences. Structure models of hES9S variants were predicted using Vienna RNAfold which predicted the assumed correct RNA folds for the yeast and human WT ES9S.
(D) Same analysis was performed as in (B), comparing hES9S variants VA-C for their ability to bind to P4. The full hES9S serves as a positive control. The P4–4xS1m/WT sample was used to normalize for fold enrichment (set to 1). Average RNA fold enrichment, SEM, n = 3.
(E) Same analysis was performed as in (B), comparing IRES-like elements of the Hoxa cluster, a9 IRES180, a3, a5, a11, and a9 P3. The HCV IRES serves as a negative control. The structure model of a9 IRES180 with P3 and P4, and the HoxA gene cluster chromosomal arrangement is given. The HCV-4xS1m/WT sample was used to normalize for fold enrichment (set to 1). Average RNA fold enrichment, SD, n = 3.
To confirm the specificity of the hES9S-P4 TE interaction, ES9S variants (V) were generated, which instead of the full hES9S, contain only half of the distal ES9S sequence (variant A (VA)) transplanted onto the yeast 18S rRNA or replaced only the yeast ES9S distal loop (VB and VC) (Figure 6C, S5E). All hES9S variant-containing yeast cells are viable and if at all only show a slight growth defect in comparison to WT cells (Figure S5F, G), which allowed variant strain isolation (Figure S5G). In the 4xS1m pulldown, neither of the variants VA-C were able to rescue P4 TE binding compared to the full hES9S, that efficiently enriched for P4 TE RNA (Figure 6D, S6E). Thus, the full hES9S is required for P4 TE interaction which implies that either the full hES9S is required to fold correctly in context of the h39 stem or its full sequence is needed to recognize the P4 stem-loop. Further, our data did not reveal the same binding to humanized ribosomes for additional IRES-like elements from other genes in the HoxA cluster (Xue et al., 2015), nor P3 (Figure 6E, Figure S6F). Consistently, a P4-like sequence or the 18 nt P4 3’ motif is only present in the Hoxa9 5’ UTR but no other Hoxa IRES-like element (Figure S6G, H). Together, these findings highlight the intricate specificity for hES9S to interact with a selective transcript.
The P4-ES9S interaction is important for endogenous Hoxa9 mRNA translation.
A series of experiments aimed at corroborating the physiological significance of the P4-ES9S interaction were next performed (Figure 7A). Importantly, given that the 4 nts mutated in P4(M5) are critical for 40S binding, compared to P4, P4(M5) markedly shows 4–5-fold reduced binding to hES9S 40S ribosomes from yeast lysates compared to WT (Figure 7B, S6I). This revealed the importance of these 4 nts in the P4-ES9S binding event. Our collective mRNA-rRNA interaction data (Figure 6; 7B) paved the way for performing CRISPR/Cas9 genome editing in mouse embryonic stem cells (mESCs) to site-specifically introduce the 4 nt mutation of M5 into P4 in the endogenous 1.2 kb long 5’ UTR of Hoxa9, as a means of selectively disrupting the P4-ES9S interaction in primary mammalian cells (Figure 7C, S7). Homozygous clones were selected after templated genome editing that scarlessly contain the 4 nt M5 mutation (Figure S7F).
Figure 7. The P4-ES9S interaction is important for endogenous Hoxa9 mRNA translation.
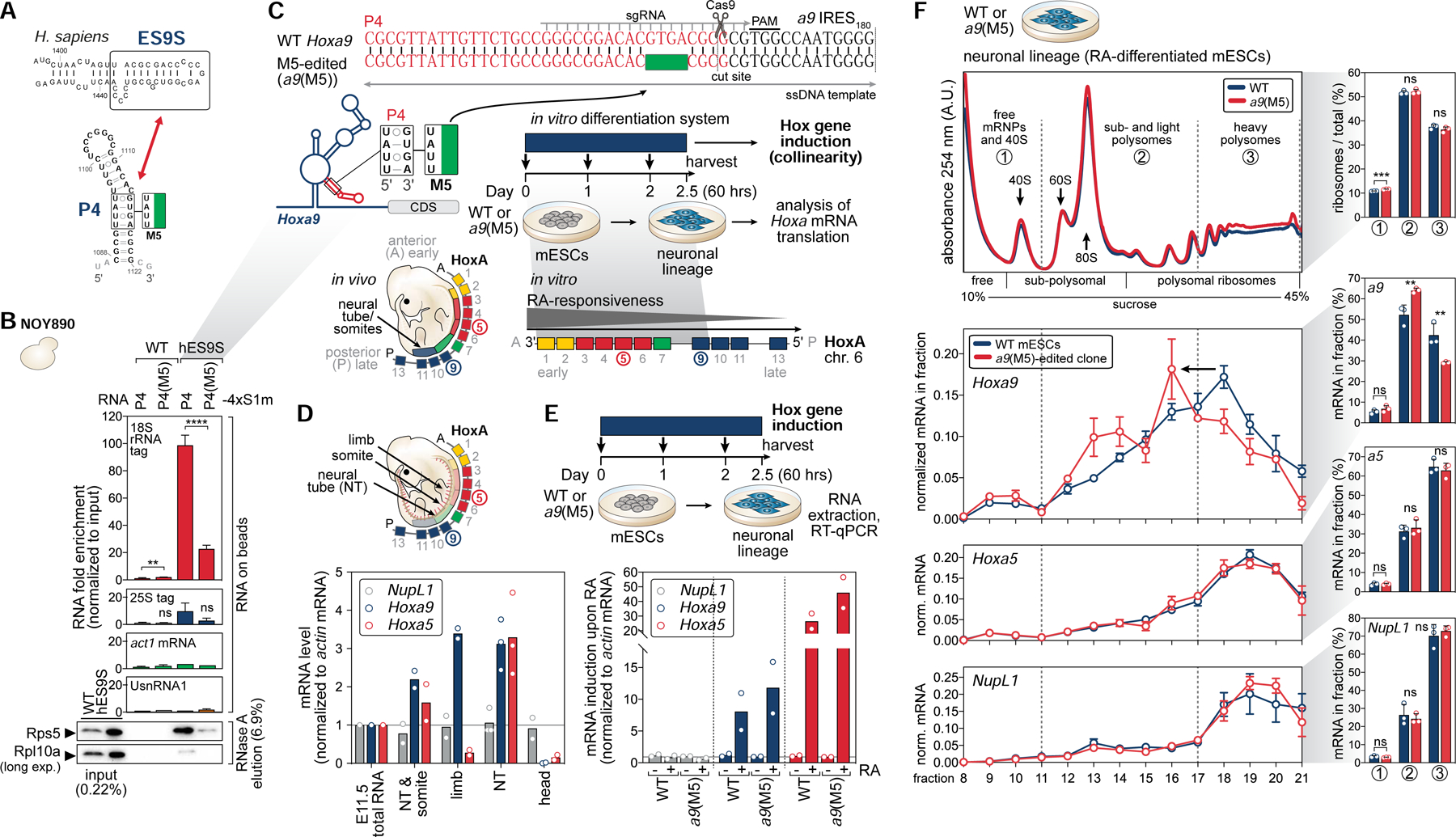
(A) Schematic of the secondary structures of human ES9S, which is identical to mouse, and the P4 stem-loop indicating the inactive M5 mutation used to test the functional relevance of their interaction for Hoxa9 mRNA translation.
(B) 4xS1m pulldown analysis was performed as in Figure 6B, comparing a9 P4 with P4(M5). The P4–4xS1m/WT sample was used to normalize for fold enrichment (set to 1). Average RNA fold enrichment, SD, n = 4.
(C) Schematic of targeted CRISPR/Cas9-editing of the 4 nt-mutation (TATT) of P4(M5) into the genomic Hoxa9 locus of mESCs. In vitro differentiation of mESCs by retinoic acid (RA)-treatment into the neuronal lineage (neural stem cells) induces colinear Hox gene expression. Genome-edited clones and WT cells were subjected to endogenous Hoxa mRNA translation analysis. See also Figure S7.
(D) The expression levels of NupL1, Hoxa9, and Hoxa5 mRNAs relative to actin mRNA in whole stage E11.5 FVB mouse embryos (E11.5 total RNA) compared to embryonic tissues of the same stage. DNase-treated total RNA from a whole embryo (E11.5 total RNA) was set to 1, n = 2–3; NT, neural tube.
(E) Schematic of Hox gene induction in WT and a9(M5)-edited mESCs (clone D6) upon 60 hours (2.5 days) of 33 nM RA-treatment or DMSO (control). mRNA induction of Hoxa9, Hoxa5, and NupL1 (control), normalized to actin mRNA in RA (+) and DMSO (−)-treated mESCs. Respective DMSO/WT or DMSO/a9(M5) samples were set to 1 to indicate mRNA induction, n = 2.
(F) Sucrose gradient fractionation analysis of lysates derived from RA-treated WT and a9(M5)-edited cells on 10–45% sucrose gradients. RNA extraction of individual fractions, RT-qPCR specific for Hoxa9, as well as Hoxa5 and NupL1 mRNAs as controls, and normalization to a Fluc-Rluc spike-in RNA, reflects the normalized distribution of the endogenous mRNAs in fractions to determine their translation efficiencies (average ± SEM, n = 3). Gradient distribution was quantified by integrating free and 40S (1), sub- and light (2), heavy polysomes (3) which was expressed as the percentage relative to the total (average ± SD, n = 3).
Clustered Hox genes are unique in that there is a direct relationship between their chromosomal organization, expression, and function in time and space during development (termed collinearity), such that the 3′ genes are sequentially activated before 5′ members (Kmita and Duboule, 2003). Sequential expression of Hox genes is established during embryogenesis through combinatorial inputs from multiple signaling pathways, including transcription-inducing signals such as 9-cis retinoic acid (RA) (Conlon and Rossant, 1992; Nolte et al., 2019) that affects RA response elements present in Hox clusters (Ghyselinck and Duester, 2019). mESCs do not usually express Hox transcripts, but can be differentiated into the neuronal lineage (neural stem cells) upon RA-treatment which induces highly temporal collinear Hox gene expression (Papalopulu et al., 1991; Simeone et al., 1990).
RA concentrations in developing mouse embryos normally range from 16 to 35 nM (Horton and Maden, 1995; Sheikh et al., 2014). We found that Hoxa9 mRNA expression is highest after 60 hours of 33 nM RA-treatment of mESCs, which is consistent with reported oscillating Hoxa9 mRNA expression and mimics sequential embryonic Hox gene activation (De Kumar et al., 2015) (Figure 7C, D; S7A–C). Additionally, the a9 mRNA expressed upon RA induction was confirmed to contain the same 5’ UTR IRES-like element as present in the embryo (Figure S7D, E). Hox gene induction upon 60 hours of RA-treatment was comparable in M5-edited and WT cells as seen for Hoxa9 and Hoxa5 mRNAs located on the same chromosomal locus (Figure 7E). Then, induced WT and edited cells were subjected to sucrose gradient fractionation analysis to quantify translation efficiency (Figure 7C, F). This analysis showed a clear, significant shift of translated Hoxa9 mRNA from the well translated pool of mRNAs (heavy polysomes) in WT cells to less translated mRNAs (light polysomes) in the M5-edited cells (Figure 7F). Importantly, the translation of Hoxa5 mRNA in the same cluster and a control mRNA, NupL1, were unaffected in edited cells. Together, using gene editing of M5 into the Hoxa9 genomic locus and neural stem cell differentiation, these findings underscore the physiological importance of the specific P4-ES9S interaction for ultimate Hoxa9 mRNA translation.
DISCUSSION
Our study unravels that the ribosome, a highly conserved fossil of the RNA world, has evolved to accommodate species-specific adaptations in the form of a previously unknown functional role for ESs in selective mRNA interactions. This sheds light on the unexpected regulatory potential of rRNA as a trans-acting player in establishing specific and direct mRNA contacts, distinct from its essential function in peptide-bond formation (Dahlberg, 1989), and from established interactions with proteins, such as eIFs and ribosome-associated proteins (RAPs) (Chen et al., 2014; Fujii et al., 2018; Shao et al., 2015; Simsek et al., 2017). ESs have been notoriously hard to visualize on ribosome structures as they are very flexible and dynamic (Armache et al., 2010). Hoxa9 IRES-like RNA binding may have stabilized the ES9S structure, which has facilitated the resolution of this interaction and additionally improves the so far only modelled ES9S structure (Figure 5A).
Ribosomal subunits have been proposed to have regulatory roles in translation themselves via differential mRNA binding (Mauro and Edelman, 2002), but clear evidence was sparse. To date, mRNA-rRNA interactions have been mainly implicated for translational control in viruses or bacteria, such as the classic example of base pairing between the Shine-Dalgarno sequence and the 3’ end of prokaryotic 16S rRNA to identify translation start sites (Shine and Dalgarno, 1974; Steitz and Jakes, 1975). For eukaryotic mRNA-rRNA interactions, base pairing sequence complementarity with the 18S rRNA has been suggested for a few individual mRNA 5’ UTRs or solely predicted genome-wide (Dresios et al., 2006; Pánek et al., 2013; Panopoulos and Mauro, 2008; Parker et al., 2018; Tranque et al., 1998). Particularly, a purine-rich sequence in the histone H4 mRNA coding region has been found to base pair with 18S helix h16 to tether the 40S to the start codon (Martin et al., 2016). However, ES9S represents the first confirmed binding site for such mRNA-rRNA interactions within ESs.
ESs display a wide variability in sequence, structure, and length between species (Figure 5B) (Kuo et al., 1996; Leffers and Andersen, 1993; Parks et al., 2018). Direct ES-mRNA-binding may represent an important determinant for tissue-specific translation regulation as rRNA variants may be differentially expressed between tissues in the same organism (Parks et al., 2018; Tseng et al., 2008), or in organismal development (Teixeira and Lehmann, 2018). For example, one of the largest estimated differences in rRNA sequences between mouse tissues are due to ES variation (Parks et al., 2018; Tseng et al., 2008). While the potential tissue-specific variability in ES9S remains to be addressed, ten SNPs in ES9S were found across human populations. Species- and organ-selective rRNA variants could thereby reflect an evolutionary adaptation of the ribosome itself to the transcriptome expressed in a species. With respect to the Hox gene cluster, regulatory mechanisms that ensure their remarkable spaciotemporal collinear expression have been a fascination for decades (Kmita and Duboule, 2003; Krumlauf, 1994). Hox genes are among the most tightly regulated transcripts, essential for metazoan body plan formation. The evolution of the ribosome to facilitate the translation of a Hox mRNA reflects an additional, important layer of control that may have co-adapted species-specific changes to Hox gene expression underlying embryonic development. Beyond that, we extended these findings and identified hundreds of mouse embryonic mRNAs translationally regulated in an ES9S-depedent manner by defining the mRNA interactome of ES9S-humanized ribosomes genome-wide (Leppek et al., 2020). Moreover, the modular activity of the Hoxa9 P4 stem-loop to enhance translation initiation from any 5’ UTR by 40S-binding, and particularly via the 3’ 18-nt motif (Figure 2) reflects, to our knowledge, the shortest sequence harboring ribosome-binding activity in itself. Thereby, this may be a very attractive short RNA element beneficial for recently emerging RNA-based therapeutics, e.g. mRNA vaccines (Pardi et al., 2018). Indeed, in efforts to develop optimized mRNA vaccines for SARS-CoV-2 we find that the P4 stem-loop performs exceptionally well to enhance translation of mRNA therapeutics (Leppek et al., in preparation). Thereby, the direct interplay between a select mRNA and an rRNA ES identified in this study provides a so far unrecognized mechanism by which specific features of eukaryotic 5’ UTRs serve to recruit the ribosome and modulate gene regulation at the post-transcriptional level.
LIMITATIONS
While we have been able to detect a pronounced change in Hoxa9 translation efficiency upon interruption of the P4-ES9S interaction, ultimately, we could not find a suitable antibody to further examine Hoxa9 protein expression by WB. HOX proteins are highly conserved, and similar in sequence and structure due to their common Homeodomain domain. The lack of specific tools such as antibodies to specifically detect and isolate them - the “Hox specificity paradox” (Luo et al., 2019) – has long been known to be a technical bottleneck in the field. The diversity of translational regulation even among Hoxa mRNAs is apparent in the different translation profiles of Hoxa5 and Hoxa9 mRNAs (Figure 7F), which, in absence of any specific antibodies for these mouse HOX proteins, is currently the best method to probe their translation.
Our studies reveal that the a9 P4 stem-loop may serve as the default, underlying regulatory element to recruit the 40S via 18S rRNA-ES9S to the Hoxa9 5’ UTR. However, it is part of a more complex unusually long 5’ UTR, wherein the possible dynamic interplay and hierarchy of RNA sequences or structures remains unknown. While the TIE-IRES topology is not exclusive to the Hoxa9 5’ UTR in the HoxA gene cluster (Xue et al., 2015), the P4 stem-loop is. Additionally, multiple Hoxa IRES-like elements including Hoxa9, selectively require RPL38/eL38 on the 60S for their activity (Kondrashov et al., 2011; Xue et al., 2015). Our cryo-EM IRES-ribosome complexes reconstituted from purified components may thus represent the minimal interactions for recruitment of the 40S, and further structural work should address the native complex in developing mouse tissues to reveal the full interplay of interactions required for regulation of Hox mRNA translation.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Maria Barna (mbarna@stanford.edu).
Materials Availability
All plasmids, yeast strains, and cell lines generated in this study are available upon request and will be fulfilled by the Lead Contact, Maria Barna (mbarna@stanford.edu).
Data and Code Availability
Mass Spectrometry data are available in Table S4 and S5 and have been uploaded to the ProteomeXchange database via the PRIDE repository; data are available via ProteomeXchange with the identifier PXD021678 (PRIDE database: PXD021678). Cryo-EM map data have been deposited in the Electron Microscopy Data Bank (EMDB) under accession numbers EMD 11562 - EMD 11568: mouse Hoxa9 IRES-like element bound to the human 80S ribosome EMD-11562; mouse Hoxa9 IRES-like element bound to the human 40S ribosomal subunit head - IRES binding site EMD-11563; mouse Hoxa9 IRES-like element bound to the human 40S ribosomal subunit - 40S head EMD-11564; mouse Hoxa9 IRES-like element bound to the human 40S ribosomal subunit - 40S body EMD-11565; Hoxa9 IRES P4 stem-loop bound to the 40S ribosomal subunit - 40S head EMD-11566; Hoxa9 IRES P4 stem-loop bound to the 40S ribosomal subunit - 40S body EMD-11567; human 40S ribosomal subunit (control) EMD-11568. Original/source data for figures in the paper is available at Mendeley Data: http://dx.doi.org/10.17632/45zmm935g2.1.
EXPERIMETNAL MODEL AND SUBJECT DETAILS
Cell Culture and Transfection or Treatment
C3H/10T1/2 (ATCC: CCL-226) cells or HEK393T (ATCC: CRL-3216) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco, 11965–118) containing 2 mM L-glutamine, supplemented with 10% fetal calf serum (EMD Millipore, TMS-013-B), 100 U/ml penicillin and 0.1 mg/ml streptomycin (EmbryoMax ES Cell Qualified Penicillin-Streptomycin Solution 100X; EMD Millipore, TMS-AB2-C or Gibco, 15140–122) at 37°C in 5% CO2−- buffered incubators. ~0.6 X 106 C3H/10T1/2 cells were seeded per well in 12-well dishes and transfected the following day with 0.8–1.6 μg of plasmid using 4 μL Lipofectamine 2000 (Invitrogen, 11668–019) and Opti-MEM (Gibco, 11058–021) according to the manufacturer’s instructions in serum-free and antibiotic-free DMEM. For transfection with monocistronic Fluc constructs (pGL3 or pGL3-FLB), 12 ng of a Rluc-control plasmid (pRL) was co-transfected per well. The medium was changed to regular DMEM 4–6 hours after transfection and cells were collected 24 hours post-transfection. For transfection of A-capped RNA, 1 μg A-capped Nluc mRNA and 100 ng m7G-capped HBB-Fluc control mRNA, both polyadenylated, were reverse transfected per 0.12 × 106 cells in a 12-well dish using 4 μL Lipofectamine 2000 (Invitrogen, 11668–019) and Opti-MEM (Gibco, 11058–021). Cells were harvested 6 hours post-transfection and samples were split in half for protein and mRNA analysis. For transfection of m7G-capped RNA into HEK393T or C3H/10T1/2, 200 ng m7G-capped Nluc mRNA and 100 ng m7G-capped HBB-Fluc control mRNA, both polyadenylated, were transfected per 0.04 × 106 cells in a 24-well dish seeded the day before, using 2 μL Lipofectamine MessengerMAX (Invitrogen, LMRNA001) and Opti-MEM (Gibco, 11058–021) in antibiotics-free medium and 5% FCS. Media was changed to regular media after 3h, cells were harvested 6 hours post-transfection, and samples were split in half for protein and mRNA analysis. For drug treatment, cells were pre-treated for 3 h with 50 μM 4EGI-1 (eIF4E/eIF4G interaction inhibitor, Sigma, 324517) in DMSO (Sigma, D2650), with DMSO only or left untreated. Then, cells were transiently transfected with RNA for 6 hours in presence of drug or carrier, media was changed to regular media with drug or carrier, and cells were harvested for luciferase analysis. No effect on cell viability or morphology was observed after total 9 h of treatment.
Low-passage E14 mouse embryonic stem cells (mESCs) were cultured on 0.1% gelatin-coated dishes in 5% CO2-buffered incubators at 37°C using media comprised of Knockout-DMEM (Life Technologies, 10829018) with 15% Embryomax FBS (EMD Milipore, ES-009-B), 2mM non-essential amino acids (EMD Milipore, TMS-001-C), 2 mM L-Glutamine (EMD Milipore, TMS-002-C), 0.055 mM 2-mercaptoethanol (GIBCO, 21985023), 103 U/ml LIF (EMD Millipore, ESG1107), and 1x ES-grade penicillin/streptomycin (EMD Millipore, TMS-AB2-C) in a 37°C, 5% CO2 incubator. Cells were split every other day to culture about 5 × 106 cells/10 cm dish and were used up to passage 35. For Hox gene induction, 0.17 × 106 mES cells were seeded onto 12-well dishes pre-coated with 0.1% gelatin. After 4 hours, cells were treated with 33 nM retinoic acid (RA, Sigma, R2625) in DMSO (Sigma, D2650) or with DMSO alone as a negative control. An RA concentration of 33 nM more closely mimicked physiological oscillation of Hox gene induction than commonly used 10 μM concentration, which agrees with previous work (De Kumar et al., 2015). Fresh media with RA was provided every 12 hours and cells were harvested after 60 hours, or the latest 72 hours for time course experiments, and subjected to RNA extraction and RT-qPCR.
Mice
Mice were housed under a 12 h light/dark cycle with free access to food and water. FVB/NJ (Stock# 001800) mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and used as wildtype. Pregnant FVB females, 3–8 months of age, were euthanized at E11.5, the uterus was dissected and embryos were taken out and placed into 1x PBS (Gibco, 14190–250). Embryos were individually collected in either TRIzol (Invitrogen, 15596) and lysed by pipetting for total RNA isolation or collected in 2 ml safe-lock tubes (Eppendorf) in 1x PBS, supernatant was removed and embryos were snap frozen in liquid nitrogen. For lysates, embryo pellets were homogenized by cryo-milling after addition of a 2.5 or 5 mm steel bead using a tissue lyser (QIAgen TissueLyser II) at 25 Hz for 15 seconds 3–6 times, and the powder was either processed directly or snap frozen in liquid nitrogen and stored at −80°C. All animal work was performed in accordance with protocols approved by Stanford University’s Administrative Panel on Laboratory Animal Care.
Yeast Strains and Transformation
Yeast plasmids and strains (Saccharomyces cerevisiae) used in this paper are listed in Table S1 and Table S2, respectively. Yeast strains were grown in YPD medium (10 g/L yeast extract, 20 g/L peptone, and 20 g/L glucose), YPAD medium (10 g/L yeast extract, 20 g/L peptone, 40 mg/L adenine sulfate, and 20 g/L glucose), or Synthetic Dextrose (SD) medium (6.7 g/L yeast nitrogen base, 20 g/L glucose, 1.6 g/L amino acids drop out mix (Complete Supplement Mixture, CSM, Sunrise Science Products)). All yeast strains were cultured at 30°C, unless specified otherwise. Cells were harvested in mid-log phase growth (OD600 = ~0.8). Plasmid transformation of yeast cells was performed using mid-log phase cells grown in YPD, YPAD, or SD medium and standard lithium acetate-mediated transformation of 1 μg DNA and selection of transformants on SD plates of appropriate amino acids drop-out for 2–3 days at 30°C was performed.
For viability assays, we used a RNA polymerase I (pol I) temperature sensitive yeast strain (NOY401) to suppress endogenous rRNA expression, complementing it with exogenous rRNA expression from a plasmid driven by RNA polymerase II through the GAL7 promoter (pNOY102, uracil (URA3) auxotrophic marker gene) (Nogi et al., 1991). In this system, the endogenous rRNA can be compensated by rRNA derived from a plasmid (pNOY102), or pNOY102-derivatives encoding tagged hES9S-containing rRNA, at restrictive temperature (37°C) on galactose plates compared to growth at permissive temperature (25°C). Growth on galactose of these cells at 37°C inactivates their pol I and induces the GAL7 promoter. Control cells were grown on -URA3/glucose plates at both temperatures. For the spot assay, fresh overnight liquid cultures in SD-URA3/raffinose were grown at 25°C and day cultures in SD-URA3/galactose were adjusted to a concentration of OD600 = 1, and 10-fold serial dilutions were spotted on fresh SD-URA3/galactose or SD-URA3/glucose plates. The plates were then incubated at 37°C or 25°C for 3–6 days and documented by scanning. To monitor rRNA processing, 5’ end processing of endogenous and tagged 18S and 25S rRNA were analyzed by RT-qPCR using pre-mature rRNA-specific or total rRNA primers (Fujii et al., 2009). Total RNA was extracted according to the manufacturer’s instructions (MasterPure Yeast RNA Purification Kit, Epicentre, MPY03100) from cells grown in SD-URA3/galactose before plating.
The rDNA mutant strains were produced from the genomic rDNA deletion strain (KAY488 (NOY890)) (Nemoto et al., 2010), complemented rDNA with an exogenous plasmid, pRDN-hyg (RDNAhyg URA3) (Nemoto et al., 2010; Wai et al., 2000), which was exchanged by plasmid shuffling to pNOY373 (RDNA LEU2) or derivatives containing human ES9S and 18S and 25S rRNA tags. To remove the pRDN-hyg plasmid, strains were negatively selected against the URA3 marker gene using 1 mg/mL of 5-Fluoroorotic Acid (5-FOA) (Fisher Scientific, F10501–5.0) in SD-plates, which is processed to a toxic product by the Ura3 enzyme. Successful plasmid shuffling was confirmed by total RNA extraction and RT-qPCR for rRNA tags, as well as by plasmid miniprep and RT-PCR specific for the ES9S region and the 18S rRNA tag.
METHOD DETAILS
Plasmid Construction
The following plasmids have been described previously: pFLB (pcDNA3-Fluc-β-globin, p2524) (Ozgur et al., 2010) containing the Firefly luciferase (FL) and rabbit β-globin (B) reporter genes, as well as pSP73 (p2008) and pSP73–4xS1m (p2880) (Leppek and Stoecklin, 2014) were kindly provided by Georg Stoecklin; pRF and pRF-HCV(Yoon et al., 2006) were kindly provided by Davide Ruggero (UCSF); pRL (Promega) encoding the Renilla luciferase reporter gene and pGL3 (Promega) encoding the Firefly luciferase reporter gene driven by the SV40 promoter; pRF derivatives containing full-length Hox 5’ UTRs and IRES-like elements (Xue et al., 2015).
Plasmid pSP73–4xS1m(MCS) was generated by PCR-amplifying 4xS1m from pSP73–4xS1m (p2880) using an EcoRV-flanked forward and EcoRI-flanked reverse primer using AccuPrime Pfx DNA Polymerase (Thermo, Invitrogen, 12344024) or KOD Xtreme Hot Start DNA Polymerase (EMD Millipore, 71975). The amplicon was then digested with BglII, gel-extracted (QIAquick Gel Extraction Kit (QIAgen, 28706) or Monarch Gel Extraction Kit (NEB, T1020S)), and inserted into the EcoRV/EcoRI-sites of pSP73 (Promega). This plasmid enables more convenient directed cloning of RNA elements as digested PCR amplicons or phosphorylated, annealed oligonucleotides into the BglII/EcoRV-sites 5’ of the 4xS1m aptamer. pSP73–4xS1m(MCS) and derivatives can then be linearized at the EcoRI site for run-off in vitro transcription.
All bicistronic Rluc-IRES-Fluc constructs were generated by amplification of plasmid templates or E11.5 mouse cDNA and inserts were cloned into the EcoRI/NcoI-sites of the bicistronic pRF vector (Yoon et al., 2006). For native spacer constructs, spacers were fused to P4, P3 or a9 IRES180 amplicons, or their M5-derivatives, by overlap PCR and inserted into the EcoRI/NcoI-sites of pRF. Actin and actin(inverse) spacers were cloned as EcoRI/NcoI-flanked inserts in EcoRI-IRES-NcoI/EcoRI-spacer-NcoI-Fluc topology into pRF. For insert cloning either conventional restriction site-directed cloning or Gibson assembly using the NEBuilder HiFi DNA Assembly Master Mix (NEB, E2621S) was used. For monocistronic pGL3 reporter constructs, inserts were cloned into the HindIII/NcoI-sites of pGL3 (Promega). The “mini UTR” reporter mRNA was constructed by inserting the a9 TIE followed by P4 and the native spacer into the 5’ UTR of the Fluc reporter mRNA. For pGL3-FLB, which encodes a FLuc/β-globin fusion construct under the control of a SV40 promoter, retaining the stop codon of the Fluc ORF, β-globin was amplified from pcDNA3-FLB, a kind gift of Georg Stoecklin (Heidelberg University, Germany), with XbaI/XbaI-flanked primers. First, an EcoRV site was introduced as a phosphorylated annealed oligo in between the HindIII and AUG-containing NcoI-site of pGL3 for pGL3-(EcoRV). Consecutively, β-globin was cloned as a XbaI-XbaI fragment into the XbaI/XbaI sites of pGL3-(EcoRV) to generate pGL3-FLB. pGL3-FLB-TIE-FL was generated by amplifying the full-length a9 TIE from a plasmid template and insertion into the HindIII/EcoRV-sites of pGL3-FLB by Gibson assembly. IRES inserts were amplified from plasmids and fused to the TIE by overlap PCR before insertion into the HindIII/EcoRV-sites of pGL3-FLB. For RNA transfection of luciferase mRNAs, we inserted P4-actin(inv), P4(M5)-actin(inv) or control sequences (a 46 nt scrambled UTR (UTR) or actin(inv)) into the 5’ UTR of plasmid pcDNA3.1–5’UTR-3xHA-Nluc (Osuna et al., 2017), a kind gift of Conor J. Howard (UCSF, San Francisco, CA, USA). It encodes HA-tagged Nanoluc followed by a 50 nt-poly(A) tail, which allows insertion of 5’ UTR sequences between a T7 promoter and 3xHA-Nluc.
Into the yeast plasmid derivatives of pNOY102 or pNOY373, we inserted rRNA tag sequences, a 16-nt tag into 18S rRNA (Beltrame et al., 1994) and a 24-nt tag into 25S rRNA (Musters et al., 1989), for RT-PCR and RT-qPCR analysis. For pNOY373–18S/25S-tag, the 18S tag was inserted into pNOY373–25S-tag by amplifying a NdeI/SacII fragment from pNOY102–18S/25S-tag and cloning it into the NdeI/SacII-sites of pNOY373–25S-tag. In a second step, the yeast ES9S was exchanged for the human ES9S or ES9S variants in pNOY102–18S/25S-tag and pNOY373–18S/25S-tag, which were generated by overlap extension PCR and were subsequently introduced into the SacII-XhoI-sites of pNOY102–18S/25S-tag and SacII-MluI-sites of pNOY373–18S/25S-tag, respectively. rDNA plasmids for plasmid shuffling (pNOY373) thus encode either tagged WT or hES9S 18S rRNA. A list of all plasmids and primer sequences used are provided in Table S1 and Table S3, respectively. All oligonucleotides were purchased from IDT. Mutations, cloning boundaries and coding sequences in all plasmids were verified by DNA sequencing (QuintaraBio).
In vitro transcription of reporter mRNAs
For mRNA transfection of C3H/10T1/2 cells, A-capped or m7G-capped and polyadenylated mRNAs for RNA transfection were generated by in vitro transcription. For A-capped mRNAs, DNA templates were generated by PCR by amplification from pcDNA3.1–5’UTR-3xHA-Nluc plasmids that were flanked by the T7-promoter 5’ and poly(A)50 3’ and gel extracted. A-capped RNAs were in vitro transcribed using the MEGAscript T7 kit (Ambion, AM1333). A 60 μl transcription reaction contained 5 μg linear DNA template, 4 mM of each NTP (Ambion), 6 μL/ 600 U MEGAscript T7 RNA polymerase (Ambion) and 1x T7 MEGAscript Transcription Buffer (Ambion). Importantly, for A-capped RNA, G was substituted with G(5’)ppp(5’)A RNA Cap Structure Analog (NEB, S1406L) in a 1:5 ratio to yield at least 80% A-capped RNA. After 2 hours incubation at 37°C, 3 μL GTP was added to the reaction. After a total incubation for 4 hours at 37°C, the DNA was digested by addition of 3 μL/6 U Turbo DNase (Ambion, AM2238) for 15 min at 37°C. Synthesized RNA was purified by gel filtration using pre-packed G-50 Mini Quick Spin Sephadex RNA columns (Roche, 11814427001) according to the manufacturer’s instructions, and extracted using acid Phenol:Chloroform (Ambion, AM9722). m7G-capped and polyadenylated Nluc reporter mRNAs and HBB-Fluc control mRNA was generated by PCR amplification from pcDNA3.1–5’UTR-3xHA-Nluc plasmids and of HBB-Fluc from pGL3-HBB and in vitro transcription using the MEGAscript T7 kit (Ambion, AM1333) according to the manufacturer’s instructions. RNA was purified using RNA PureLink columns (Thermo Scientific, Ambion, 12183018) and sequentially m7G-capped and polyadenylated using the ScriptCap m7G Capping System (CellScript, C-SCCE0625) and A-Plus Poly(A) Polymerase Tailing Kit (CellScript, C-PAP5104H), respectively, according to the manufacturer’s instructions. RNA was again purified using RNA PureLink columns. RNA concentration and quality were determined by Nanodrop and 4% urea-PAGE or 1% formaldehyde agarose gel, respectively.
Luciferase Activity Assay after plasmid or mRNA transfection
Transiently transfected C3H/10T1/2 cells in 12-well plates were washed twice with 1x PBS (Gibco, 14190–250) and collected by trypsinization 24 hours post-transfection for luciferase activity assays. Half the cells were used for assaying luciferase activity using the Dual-Luciferase Reporter Assay System (Promega, E1980) to measure Firefly (Fluc) and Renilla (Rluc) luciferase activities, the other half was collected in TRIzol (Invitrogen, 15596) for total RNA extraction and normalization to mRNA levels by RT-qPCR (see RT-qPCR section). For luciferase assays, cells were lysed in 60 μl of 1x passive lysis buffer of the Dual-Luciferase Reporter Assay System (Promega, E1980) and directly assayed or frozen at −20°C. After thawing, cell debris and nuclei were removed by centrifugation for 1 min at 13,000 rpm. 20 μl of supernatant was assayed for luciferase activity in technical replicates by mixing with 50 μl of Dual-Luciferase Reporter Assay System substrates. Fluc and Rluc activities were measured on a GloMax-Multi (Promega) plate reader. Luciferase reporter activity is expressed as a ratio between Fluc and Rluc which was normalized to the ratio of Fluc to Rluc mRNA levels for bicistronic pRF constructs to verify the integrity of the bicistronic mRNA construct. For monocistronic Fluc-β-globin-fusion constructs, Fluc/Rluc luciferase activity was normalized to β-globin, β-actin or NupL1 mRNA levels to quantify variation in mRNA expression.
For transfection of C3H/10T1/2 or HEK293T cells with A-capped or m7G-capped reporter mRNAs, cells in 12-well plates were harvested 6 hours post-transfection and split in half for luciferase measurement as well as RNA extraction for RT-qPCR. Co-transfection of a m7G-capped HBB-Fluc mRNA served as internal control. Luciferase activity was assayed using the Nano-Glo Dual-Luciferase Reporter Assay System (Promega, N1610) to measure Firefly (Fluc) and Nanoluc (Nluc) luciferase activities. Relative Nluc/Fluc luciferase reporter activity was normalized to respective Fluc/Nluc mRNA levels. Each experiment was performed a minimum of three independent times. Statistical analysis was performed using unpaired two-tailed Student’s t-test.
Quantitative RT-PCR (RT-qPCR) Analysis
Cells transfected with pGL3, pGL3-FLB-stop or pRF constructs were collected in 500 μL TRIzol (Invitrogen, 15596). Total RNA was isolated from the aqueous phase using RNA PureLink columns (Thermo Scientific, Ambion, 12183018) and treated with TURBO DNase (Ambion, AM2238) twice, followed by a second RNA PureLink column purification to remove plasmid DNA. DNase treatment and a second column purification were omitted for pGL3-FLB-stop constructs. For reverse transcription-quantitative PCR (RT-qPCR) analysis, cDNA was synthesized from 100–200 ng of total RNA using iScript Supermix (Bio-Rad, 1708840) containing random hexamer primers, according to the manufacturer’s instructions. PCR reactions were assembled in 384-well plates using 2.5 μL of a 1:4–1:5 dilution of a cDNA reaction, 300 nM of target-specific primer mix and the SsoAdvanced SYBR Green supermix (Bio-Rad, 1725270) in a final volume of 10 μl per well. SYBR green detection qPCR was performed on a CFX384 machine (Bio-Rad). Data was analyzed and converted to relative RNA quantity manually or using CFX manager (Bio-Rad). Gene-specific qPCR primer sequences used for detection of mRNAs and rRNAs are given in Table S3.
In vitro RNP affinity purification via 4xS1m-aptamers
The 4xS1m-pulldown of RNP complexes was performed similar to as previously reported (Leppek and Stoecklin, 2014). RNAs were synthesized by in vitro transcription: RNA elements were fused to 4xS1m aptamers by cloning IRES amplicons into the BglII/EcoRV sites of pSP73–4xS1m(MCS). 4xS1m alone served as negative control RNA. Since amplification of the highly structured 4xS1m tag by PCR is problematic, linearized pSP73 plasmids served as DNA templates. Up to 20 μg template plasmid was linearized at the EcoRI-site downstream of the 4xS1m sequence in a 50 μL reaction for 6 hours or overnight, purified with the QIAquick PCR Purification Kit (QIAgen) and used as DNA templates for run-off in vitro transcription using MEGAscript SP6 kit (Ambion, AM1330). A 40 μl transcription reaction contained 8 μg linear DNA template, 4 mM of each NTP (Ambion), 4 μL/ 400 U MEGAscript SP6 RNA polymerase (Ambion) and 1x SP6 MEGAscript Transcription Buffer (Ambion). After incubation for 4–6 hours at 37°C, the DNA was digested by addition of 2 μL/4 U Turbo DNase (Ambion, AM2238) for 15 min at 37°C. Synthesized RNA was purified by gel filtration using pre-packed G-50 Mini Quick Spin Sephadex RNA columns (Roche, 11814427001) according to the manufacturer’s instructions, and RNA concentration and quality was determined by Nanodrop and 4% urea-PAGE, respectively. One reaction typically yielded 50–200 μg of RNA.
For all steps in the pulldown experiments, 1.5 mL DNA/RNA LoBind tubes (Eppendorf) were used to reduce unspecific binding. Per sample, 100 μl 50% slurry of Streptavidin Sepharose High Performance (GE Healthcare) beads were washed three times with 0.5–1 ml of SA-RNP lysis buffer (20 mM Tris-HCl (pH 7.5, Ambion, AM9850G, and Ambion, AM9855G), 150 mM NaCl (Ambion, AM9759), 1.5 mM MgCl2 (Ambion, AM9530G), 2 mM DTT (Ambion, 10197777001), and 1 tablet/10 ml Mini Complete Protease Inhibitors, EDTA-free (Sigma-Aldrich, Roche, 11836170001) in nuclease-free water (Thermo Fisher, Invitrogen, 10977023). At each step, beads were gently pelleted at 500 rpm (~20 x g) for 1 min at 4°C. ~30 μg of the in vitro transcribed 4xS1m or IRES-4xS1m RNAs per sample for pulldown from mouse or embryo powder for protein analysis or 2.5–7.5 μg of the in vitro transcribed RNAs per sample for pulldown of ribosomes from yeast was renatured in 50 μl SA-RNP lysis buffer by heating at 56°C for 5 min, 10 min at 37°C, and incubation at room temperature for several minutes to refold RNA structures. The RNA was added to the 100 μl SA Sepharose slurry together with 1 μl RNasin Plus RNase inhibitor (40 U/μL, Promega, N261A). 10 μl of the supernatant was saved for extraction of input RNA using TRIzol (Invitrogen, 15596), 2.5 μl of the supernatant (input RNA) was saved for urea-PAGE analysis, and 20 μL for an input protein sample. The mixture was incubated at 4°C for 2–3 hours under rotation to permit binding of the RNA to the column. Then, beads were sedimented and 2.5 μl of the supernatant (unbound RNA) was saved for urea-PAGE analysis, while the remaining supernatant was discarded. Input and unbound RNA samples were compared side by side by 4% polyacrylamide (Ambion)/0.5x TBE (Sigma)/urea (Sigma) gel electrophoresis and SYBR Gold (10,000x, Thermo Fisher, Invitrogen, S11494) staining in 0.5x TBE to assess the efficiency of RNA coupling.
For WB analysis of RNA-associated proteins from cultured cells and mouse embryos, the following harvest and lysis was performed. Cellular extracts were prepared from 20 confluent 15-cm dishes of untransfected C3H/10T1/2 cells. A total of 1.5 g cells was collected, washed once in PBS, divided into ~300 mg portions and aliquoted in 2 ml safe-lock tubes (Eppendorf). Lysates of FVB stage E11.5 mouse embryos were used as input to recapitulate the cellular environment of Hox gene expression. FVB E11.5 mouse embryos were harvested in 1xPBS as described in the mouse section and individually added to 2 ml safe-lock tubes. For analysis of RNA-associated proteins and RNA from yeast cells, mid-log phase cells from a 1 L SD-LEU medium culture was harvested as described in the yeast section, washed once with water, and the cell pellet was split into 16 equal aliquots into 2 ml safe-lock tubes. The cell, embryo, or yeast pellets were then snap frozen in liquid nitrogen, homogenized by cryomilling after addition of a 2.5 mm steel bead using a tissue lyser (QIAgen TissueLyser II) at 25 Hz for 30 seconds 3–6 times, or until the tissue was powderized, and the powder was either processed directly or stored at −80°C. For the embryo experiments, two embryos per samples were used. The frozen homogenate of one aliquot (~300 mg) was solubilized by the addition of 100 μl ice-cold RNP lysis buffer per sample and allowed to thaw for 5 min at room temperature or until thawed. Cell debris was removed by centrifugation for 5 min at 17.000 x g at 4°C, resulting in a supernatant of ~500 μl. Yeast samples were centrifuged again for 10 min at 17.000 x g at 4°C to remove remaining cell debris. The protein concentration in the extract was determined by Nanodrop to be ~25–70 mg/ml.
Next, the extract (~500 μl) was pre-cleared by addition of 25 μl of a 50% slurry of Avidin Agarose (Thermo Pierce) beads, 100 μl of a 50% slurry of SA Sepharose beads, and 5 μL RNasin (Promega), and tumbling for 2 hours at 4°C. Beads were collected and discarded, and the pre-cleared lysate was supplemented with 2 μl of RNasin Plus (Promega), added onto the freshly prepared, RNA-coupled SA Sepharose matrix, and incubated at 4°C for 2–3 hours under rotation to form RNP complexes. Beads were rinsed once and washed 3 times for 2–5 min with 1 ml SA-RNP wash buffer (20 mM Tris-HCl (pH 7.5), 300 mM NaCl, 5 mM MgCl2, 2 mM DTT, and 1 tablet/50 ml Complete Protease Inhibitors, EDTA-free (Roche) in nuclease-free water).
For WB analysis of proteins from cultured cells and embryos, elution was performed as follows. After the last wash of 6 washes, beads were transferred to a fresh tube and RNA-bound proteins were eluted by addition of 5 μg RNase A (Invitrogen, AM2271, 1μg/μL) in 100 μl Low Salt Buffer (20 mM Tris-HCl (pH 7.5), 30 mM NaCl, 5 mM MgCl2, 2 mM DTT, 1 tablet/10 ml Mini Complete Protease Inhibitors, EDTA-free (Roche)) and rotation for 15 min at 4°C. The RNase A eluate was recovered and 10 μl of the eluate was analyzed by SDS-PAGE and WB.
For RT-qPCR analysis of RNA and WB analysis of proteins from yeast cells, elution was performed as follows. After formation of ribosome-RNA ribonucleoproteins (RNPs) in vitro, beads are split in half: total RNA is eluted with TRIzol, and protein is eluted with RNase A. After the last wash, beads were transferred to a fresh tube and resuspended in 500 μL SA-RNP lysis buffer. 250 μL were saved and used for TRIzol extraction of bound RNA according to the manufacturer’s instructions. 15 μg GlycoBlue (Ambion, LSAM9516) was added to the RNA prior to precipitation. RNA-bound proteins were eluted from the rest 250 μL of beads by addition of 2 μg RNase A (Invitrogen, AM2271, 1μg/μL) in 30 μl Low Salt Buffer and rotation for 20 min at 4°C. The RNase A eluate was recovered, supplemented with SDS sample buffer and 8 μl of the eluate was analyzed by SDS-PAGE and WB. After RNase A elution, the beads were extracted with 30 μl 2x SDS sample buffer, 10 μl of which were analyzed by SDS-PAGE and WB. The fraction loaded of input and elution samples is expressed as percentage of the original lysate volume. For qualitative assessment of binding and elution efficiencies, an RNA fraction at each step was analysed by 4% polyacrylamide/0.5x TBE/urea gel electrophoresis and SYBR Gold staining. For qPCR analysis following RNA-IP, a fixed volume of 1:100 diluted RNA extracted from IP and input samples was used for RT. To indicate specific enrichment of RNA, fold enrichment of RNAs was determined by RT-qPCR using same volumes of eluted RNA and normalizing Ct values of each sample to their respective RNA input (WT or hES9S). Each sample was normalized to the 18S-tag Ct values for that respective sample to control for ribosome-IP efficiency.
Relative Protein Quantification by Tandem Mass Tag (TMT) Labeling
TMT labelling has been performed similar to in (Shi et al., 2017). In brief, proteins in RNase A elutions after 4xS1m pulldown were denatured with 5 mM DTT and 2 M urea for 1 hour at 65°C, then alkylated with 15 mM iodoacetamide (Sigma) for 30 min in the dark at room temperature. Proteins were digested with sequencing grade modified trypsin (Promega, V5111) with a ratio of 1:50 w/w (trypsin:protein) at 37°C water bath for 16 hours. Digested peptides were desalted using the OMIX C18 pipette tips column (Agilent, A57003100) following the manufacturer’s manual. Each sample was labeled with a distinct TMT label (TMTsixplex, Thermo Scientific, 90066) following the manufacturer’s manual, mixed equally and desalted again through the OMIX C18 pipette tips column. The solution was then dried with a Speed Vac. Peptides were resuspended with 10 μL 0.1% formic acid and subjected to ultra performance liquid chromatography (UPLC)-tandem mass spectrometery (MS/MS) analysis using an ACQUITY UPLC M-class system (Waters) coupled online to an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific). More precisely, for each TMT sample, 3 μl was loaded on a Self-Pack PicoFrit column (New Objective) with a 360 μm outer diameter, 75 μm inner diameter, and a tip size of 15 μm packed to approximately 22 cm with HALO Peptide ES-C18 Bulk Packing 2.7 μm beads (MAC-MOD Analytical). UPLC solvent A was 0.1% formic acid, and solvent B was 0.1% formic acid/100% acetonitrile. Peptides were loaded for 30 minutes at 1% B at a flow rate of 0.3 μl/min. Peptides were eluted at the same flow rate using a linear gradient from 5% B to 40% B for 180 minutes, followed by a linear ramp to 100% B for 10 minutes, followed by constant flow at 100% B for 10 minutes. Then, there was a ramp down to 1% B over 1 minute, followed by constant flow at 1% B for 9 minutes. The Orbitrap Elite (Thermo Fisher Scientific) was operated in the data-dependent mode using Xcalibur v3.0 (Thermo Fisher Scientific) to acquire higher energy collisional dissociation (HCD) MS/MS scans (R = 15,000) after each MS1 scan (R = 60,000) on the top 15 most abundant ions in the Orbitrap. HCD parameters were set to an isolation width of 1.6 m/z, normalized collision energy of 40%, and an activation time of 0.1 milliseconds. Dynamic exclusion parameters were set at repeat count 1 with a 30 second repeat duration, exclusion list size of 500, exclusion duration of 60 seconds, and a +/− 10 ppm exclusion mass width. Charge state rejection was enabled for charge states that were unassigned or 1.
The results were analyzed using the Proteome Discoverer 1.4 (Thermo Scientific) employing the Mascot search engine (Perkins et al., 1999), searching against Mus musculus SwissProt database with the following parameters: 1 maximum missed trypsin enzyme cleavage site, precursor mass tolerance of 10 ppm, and fragment mass tolerance of 0.8 Da, with deamidation (NQ) and oxidation (M) dynamic modifications, and TMT6plex (N-term and K) and carbamidomethyl (C) static modifications. Identified peptides were subsequently filtered for 1% FDR. Relative abundance of each protein was calculated by their levels in the individual samples compared to the 4xS1m control.
Protein abundance ratios were then log2 transformed, averaged between replicates, and median normalized, to represent detected and enriched proteins. Subsequently, proteins were categorized into curated gene sets corresponding to core components of the translational process: ribosomal subunits and translation factors. Statistical significance between distributions of log2 ratios of each gene set was calculated with the Brunner-Munzel test. Standard linear regression was performed between samples, and on certain gene sets, to determine the Pearson correlation coefficient.
Cryo-EM Sample Preparation, Data Acquisition and Analysis
Cryo-EM sample preparation, data acquisition and analysis using human 40S and 80S ribosomal subunits purified from HEK293 cells and in vitro transcribed RNAs was prepared as described in (Quade et al., 2015) with the following parameters.
Cryo-EM: Purification of 40S ribosomal subunits
Human 40S ribosomal subunits were purified similar to previously described protocols to isolate human 80S ribosomes (Quade et al., 2015). The whole purification was carried out at 4°C. For lysis, HEK293–6E cells were stirred for 30 min in a buffer containing 50 mM HEPES/KOH pH 7.6, 300 mM NaCl, 6 mM Mg-acetate, 0.5 % NP-40, 5 μM E-64, 20 μM Leupeptin, 20 μm Bestatin, 5 μM Pepstatin A, 1 mM PMSF and 2 mM DTT. Cell debris was removed from the lysate by centrifugation for 20 min at 45,000 ×g in an SS-34 rotor (Thermo Fisher Scientific, Waltham). The ribosomes were pelleted by centrifugation for 20 hours at 257,000 ×g in a Type 70 Ti rotor (Beckmann Coulter, Indianapolis) through a sucrose cushion containing 50 mM HEPES/KOH pH 7.6, 50 mM KCl, 6 mM Mg-acetate, 60% (w/v) sucrose and 2 mM DTT. The pelleted ribosomes were resuspended in a buffer containing 50 mM HEPES/KOH pH 7.6, 150 mM KCl, 6 mM Mg-acetate and 2 mM DTT by shaking for 1 hour and subsequently centrifuged for 20 min at 20,000 ×g in a table top centrifuge (Eppendorf AG, Hamburg). The ribosomal subunits were separated by loading the supernatant onto a 12% (w/v) to 48% (w/v) sucrose gradient in 50 mM HEPES/KOH pH 7.6, 500 mM KCl, 6 mM Mg-acetate and 2 mM DTT and centrifugation for 18.5 hours at 78,000 ×g in a SW 32 Ti rotor (Beckmann Coulter, Indianapolis). Light scattering was used to identify the bands of the ribosomal subunits and the 40S and 60S ribosomes were harvested from the gradient with a syringe. After that, they were concentrated and buffer exchanged into 80S buffer (20 mM HEPES pH 7.6, 100 mM KCl, 5 mM MgCl2) using 100,000 kDa MWCO tabletop-centrifuge concentrators (Sartorius).
Cryo-EM: Preparation of IRES RNA
Hoxa9 IRES was produced by in vitro transcription of a linearized plasmid containing the Hoxa9 IRES sequence, followed by LiCl precipitation and resuspension in water. The IRES was then diluted to 1 mg/mL in folding buffer (20 mM HEPES pH 7.6, 100 mM K-acetate, 2.5 mM MgCl2, 0.25 mM spermidine) and folded by two times heating to 95°C for one minute and cooling on ice.
Cryo-EM: Reconstitution of the Hoxa9 IRES-80S complex
The ribosomal subunits were buffer exchanged into 20 mM HEPES/KOH pH 7.6, 100 mM KCl, 5 mM MgCl2, 2 mM DTT by a centrifugation filter with 100 kDa molecular mass cut-off. For complex formation, human 40S ribosomal subunit were incubated at 100 nM concentration with 1 μM Hoxa9 IRES for 5 min at 37°C, 60S ribosomal subunits were added at 100 nM concentration and incubated for 5 min at 37°C before storing the sample on ice.
Cryo-EM: Sample preparation and cryo-EM image acquisition of the Hoxa9 IRES-80S ribosome complex
5 μl of the Hoxa9 IRES-80S complex were applied for 30 sec to glow discharged R2/2 holey carbon grids (Quantifoil Micro Tools, Großlöbichau) which was coated with a thin film of carbon using a BAE 120 thin-film coating system (Balzers, Pfäffikon). Excess liquid was blotted away for 12 sec and the grid were frozen in liquid ethane/propane (1:2) using a Vitrobot Mk IV (FEI Company, Hillsboro) at 4°C and 100% relative humidity. The grids were imaged in a Titan Krios cryo-electron microscope (FEI Company, Hillsboro) at 300 kV and a magnification of 100 720 × with a Falcon II direct electron detector (FEI Company, Hillsboro). Micrographs were recorded using dose fractionation with 37 frames and a total dose of 40 e− /A2 with the EPU software for automated data collection. Defocus values of the micrographs in the final dataset range from −700 nm to −3800 nm.
Cryo-EM: Image processing of the Hoxa9 IRES-80S ribosome complex sample
After initial screening of the micrographs for ice quality, the contrast transfer function (CTF) was determined using CTFFIND (Mindell and Grigorieff, 2003) and 4307 micrographs were retained with Thon rings extending to higher resolution in the power spectra. Initial evaluation of the micrographs revealed the presence of 40S ribosomal subunits and 80S ribosomal particle. At total set of 687’603 single particle images of the 40S ribosomal subunit were selected with Batchboxer (Ludtke et al., 1999) using projections of a mammalian 40S ribosomal subunit as a reference (Quade et al., 2015). RELION (Scheres, 2012) was used for further image processing (see Figure S3A–C), an initial round of two-dimensional classification was performed with 100 classes at a pixel size of 5.6 Å on the object scale (80-pixel frames). Classes containing 40S ribosomal subunits, 131’970 particles, were selected and subjected to three-dimensional classification with 10 classes using a low-pass filtered reconstruction of the 40S ribosome as an initial reference. 6 classes showing a well-defined 40S density were selected, 92118 particles, and used for an initial 3D refinement with unbinned images with a pixel size of 1.39 Å on the object scale. The reconstruction was further refined using the Multi-Body refinement in RELION 3 (Nakane et al., 2018) with 40S ribosomal subunit head and body as separate bodies. The reconstruction revealed a smeared out density on top of the 40S subunit head. To classify for different conformation in this region a spherical mask was used in a 3D classification with 10 classes. A single class, 19’342 particles, displayed a well-defined rod-shaped density on top of the 40S head. This subset of images was taken to calculate the final maps using the 3D refinement parameters from the Multi-Body refinement resulting in a reconstruction at 3.9 Å resolution. The structure of the high resolution reconstruction of the human ribosome (Natchiar et al., 2017) (pdb 6ek0) was used split into head and body to interpret the reconstruction. The tip of RNA expansion segment ES9S was manually readjusted in COOT (Emsley et al., 2010). For figure generation the reconstruction was filtered to the local resolution using RELION 3.
The reconstruction of a9 IRES FL-IRES in complex with the 80S ribosome was obtained similarly to the reconstruction of 40S ribosomal subunit complex using the structure of a mammalian 80S ribosome (Quade et al., 2015) as a reference for particle picking and reconstruction (Figure S3D–F): Particle were picked with RELION using a low pass filtered 80S ribosome as a reference from the set of processed micrographs described above for the 40S subunit complex processing. Particles were extracted with a box of 360 × 360 pixel and 4 times binned for initial processing to a pixel size of 5.6 Å on the object scale. After a 2D classification with 100 classes, 134469 single particle images were subjected to a 3D classification with 8 classes. Particles from 3D classes with well-defined density for both subunits (87583 particles) were re-extracted at a pixel size of 2.8 Å and refined. A mask was placed at the head of the 40S subunits for 3D classification without alignment using 10 classes. One class (9305 particles) showed an elongated, well defined density at the 40S subunit head. Particles from this class were re-extracted at an unbinned pixel size of 1.39 Å. This final set of 9305 particles was subjected to a local refinement with a mask for the 40S subunit to reconstruct the a9 IRES FL-IRES in complex with the 80S ribosome at 4.4 Å resolution. For figure generation the reconstruction was filtered to the local resolution using RELION 3.
Cryo-EM of the Hoxa9 IRES P4–40S complex: Preparation of Hoxa9 IRES P4 RNA
Hoxa9 IRES P4 RNA was purchased from Microsynth. The synthesized RNA was dissolved as 100 μM in folding buffer (20 mM HEPES pH 7.6, 100 mM K-acetate, 2.5 mM MgCl2, 0.25 mM spermidine) and folded by heating to 95°C for one minute and cooling on ice.
CryoEM: Reconstruction of Hoxa9 IRES P4–40S complex and 40S control
Human 40S ribosomal subunits were buffer exchanged into 20 mM HEPES/KOH pH 7.6, 100 mM KCl, 5 mM MgCl2, 2 mM DTT using 100 kDa MWCO tabletop-centrifuge concentrators (Amicon®Ultra, Merck). For reconstruction of the Hoxa9 IRES P4–40S complex, human 40S ribosomal subunits were incubated at a concentration of 120 nM with 1 μM Hoxa9 IRES P4 RNA for 5 min at 3°C. Concurrently, 120 nM 40S ribosomal subunits without Hoxa9 IRES P4 RNA were used as a control.
CryoEM: Sample preparation and data acquisition for Hoxa9 IRES P4–40S complex and 40S control
The grids were prepared as described above for the Hoxa9 IRES-80S complex using Quantifoil R2/2 grids covered with a 1 nm continuous carbon film. Excess liquid was blotted for 5 seconds at 5°C and 95 % humidity, followed by plunge freezing of the grids in liquid ethane/propane (1:2) using a Vitrobot Mk IV (FEI Company, Hillsboro). Data collection was performed using a Titan Krios cryo-transmission electron microscope (FEI Company, Hillsboro) operated in EFTEM mode at 300 kV. Images were collected with a K3 direct electron detector (Gatan Inc., Pleasanton) with zero loss filtering using a slit width of 20 eV at a magnification of 46,300x and defocus values from −700 nm to −3300 nm for Hoxa9 IRES P4–40S complex sample and −700 nm to −3000 nm for 40S control sample. For automated data collection the SerialEM was used (Mastronarde, 2005) to image 2 spots per hole. Images were acquired in a movie mode using 32 frames with an exposure time of 2.5 seconds, which resulted in an electron dose of 76 e−/Å2.
CryoEM: Image processing of the Hoxa9 IRES P4–40S complex and 40S control
After dose-weight and drift correction using MotionCor2 (Zheng et al., 2017), followed by CTF estimation with GCTF (Zhang, 2016) poor quality micrographs were discarded. By employing projections of a mammalian 40S ribosomal subunit as a reference (Kobayashi et al., 2018) within RELION (Scheres, 2012) a total of 1,664,622 particles and 506,457 were extracted from 4,666 and 1,598 micrographs from Hoxa9 IRES P4–40S complex and 40S control datasets, respectively. An initial 2D classification in RELION (Scheres, 2012) was performed with 80 classes at a pixel size of 6.88 Å on the object scale (80-pixel frames). 23 classes containing 40S ribosomal subunits were selected in the Hoxa9 IRES P4–40S complex dataset, which yielded 1,147,333 particles, whereas 226,243 particles were retained from 17 classes of the 40S control dataset.
We randomly selected 225,000 particles after 2D classification for the P4–40S dataset and processed the similar sized data sets of the control and P4–40S complex with the same refinement and classification parameters in RELION (Scheres, 2012). Only homogeneous 40S classes were selected, resulting in 107,467 particles from the Hoxa9 IRES P4–40S complex dataset and 131,996 particles from the 40S control dataset. Images were then subjected to a 3D refinement. By subtracting the generated volumes in Chimera (Pettersen et al., 2004) we could observe additional density in the Hoxa9 IRES P4–40S complex density which allowed us to create a double-sphere mask for a local 3D classification without particle alignment in RELION (Scheres, 2012). Only in the Hoxa9 IRES P4–40S complex dataset we could observe classes with a rod-shaped density at the 40S head, whereas no such classes were observed in the 40S control dataset.
For high-resolution reconstruction of 40S control dataset we continued by re-extracting the particles with the pixel size of 1.376 Å using the initial 3D refined map and subjecting the images to another round of 3D refinement and postprocessing in RELION (Scheres, 2012).
To obtain a high-resolution structure of the Hoxa9 IRES P4–40S complex we processed all 1,147,333 particles images from 2D classification. Similar to processing of the reduced dataset, the images were subjected to a 3D classification with 8 classes. We selected 687,028 particles from 4 classes containing 40S ribosomal subunits and refined the structure in RELION (Scheres, 2012). After 3D classification with the double-sphere mask and the 3D refined map from previous step 95,519 particles from 1 class out of 8 showed prominent rod-shaped density. We re-extracted these particles at a pixel size of 1.376 Å using the 3D refined map and performed another round of 3D refinement (Scheres, 2012).
To improve the resolution of the rod-shaped density we used a Multi Body refinement strategy (Nakane et al., 2018) implemented in RELION (Scheres, 2012). The high-resolution reconstruction of human 40S ribosomal subunit was split into head and body to create separate masks using a previously obtained atomic model of the subunit (Quade et al., 2015) (pdb 5a2q). A new, tighter mask was created around the rod-shaped density, to refine this structural feature. Re-centered subtracted image stack was written out from the Multi-Body and subjected to a 3D classification with 8 classes using the tighter mask. 40S subunit head particle images from a class that revealed a rod-shaped density with helical features were refined and postprocessed using RELION (Scheres, 2012). 40S subunit body was postprocessed immediately after Multibody refinement step.
The final resolutions for the entire Hoxa9 IRES P4–40S complex map, Hoxa9 IRES P4–40S head map, Hoxa9 IRES P4–40S body map and 40S control map according to the Fourier shell correlation (FSC) = 0.143 criterion were 3.42 Å, 4.14 Å, 3.11 Å and 3.24 Å, respectively. The local resolution calculation of all maps and low-pass filtering were performed using RELION (Scheres, 2012).
Western Blot Analysis and Antibodies
Proteins were resolved on 4–20% polyacrylamide gradient Tris-glycine gels SDS-PAGE gels (Biorad, 567–1095, 456–1096) and transferred onto 0.2 μm pore size PVDF membranes (Biorad) using the semi-dry Trans-Blot Turbo system (Biorad, 170–4273). Membranes were then blocked in 1x PBS-0.1% Tween-20 containing 5% non-fat milk powder for 1 hour, incubated with antibodies diluted in the same solution for 1 hour at RT or overnight at 4°C, and washed four times for 5 min in 1x PBS-0.1% Tween-20, incubated with secondary antibodies for 1 hour in 1x PBS-0.1% Tween-20 and washed four times for 15 min in 1x PBS-0.1% Tween-20. Horseradish peroxidase-coupled secondary antibodies (anti-mouse and anti-rabbit, GE Healthcare; anti-rat, Jackson Immunoresearch) in combination with Clarity Western ECL Substrate (Biorad, 170–5061) and imaging on a ChemiDoc MP (Biorad, 17001402) were used for detection. Antibodies were diluted in 1x PBS-0.1% Tween-20 at 1:1000 dilution either in 5% BSA (w/v) or 5% non-fat milk. The following primary antibodies were used for Western blot analysis: mouse monoclonal anti-PGK1 (Thermo-Fisher, Novex, 459250); rabbit polyclonal anti-RPS6/eS6 (Cell Signaling, 2217), anti-RPL10A/uL1 (yeast: Santa Cruz, sc-100827, mouse: RPL10A/uL1 (Abcam, ab174318)), anti-RPS5/uS7 (Abcam, ab58345); rabbit monoclonal anti-eIF4A (Cell Signaling, C32B4); and goat monoclonal anti-eIF3B (eif3-eta (N-20), Santa Cruz, sc-16377). Rabbit polyclonal anti-RPL10A antibody was kindly provided by Mary Ann Handel (The Jackson Laboratory, Bar Harbor, ME, USA).
Yeast Ribosomal Subunit Purification
Purification of yeast ribosomes was performed according to (Acker et al., 2007) with the following adjustments. In particular, stationary yeast cultures of NOY890 strains expressing WT or hES9S rRNA were cultured to OD600 = 0.05 in 2 L SD-Leu drop-out media and grown at 30 °C until mid-log phase (OD600 = 0.5). Cells were harvested and cell pellets were snap frozen in liquid nitrogen. A cell pellet of a 2 L culture was powderized in liquid nitrogen using mortar and pestle. Powderized yeast lysates were dissolved in 30 ml of lysis buffer (20 mM HEPES-KOH (pH 7.4), 100 mM KCl, 20 mM MgCl2, 2 mM DTT, 1 mg/ml heparin, 20 U/ml RNaseOUT (Thermo Fisher, 10777019), 1x Complete EDTA free protease inhibitor cocktail (Roche), 1 mM PMSF, 2 mM benzamidine, 2 μM pepstatin A, 0.6 μM leupeptin) and lysates were cleared by centrifugation for 30 min at 15000 rpm at 4 °C. For ribosome sedimentation, cleared lysates were layered on top of a 5 ml sucrose cushion (1 M sucrose, 20 mM HEPES-KOH (pH 7.4), 100 mM KCl, 15 mM MgCl2, 2 mM DTT, 2 mg/ml heparin, 20 U/ml RNaseOUT, 1x Complete EDTA free protease inhibitor cocktail, 1 mM PMSF, 2 mM benzamidine, 2 μM pepstatin A, 0.6 μM leupeptin) and centrifuged at 55000 rpm for 125 min at 4 °C in a type 70Ti rotor (Beckman Coulter). Ribosome pellets were resuspended in 1 ml of resuspension buffer (20 mM HEPES-KOH (pH 7.4), 100 mM KCl, 12 mM MgCl2, 2 mM DTT, 2 mg/ml heparin, 20 U/ml RNaseOUT, 1 mM PMSF, 2 mM benzamidine, 2 μM pepstatin A, 0.6 μM leupeptin) and loaded on top of a 10–45% sucrose gradient (50 mM HEPES-KOH (pH 7.4), 100 mM KCL, 12 mM MgCl2, 2 mM DTT, 2 mg/ml heparin) and centrifuged 22500 rpm for 510 min in a SW35Ti rotor (Beckman Coulter). Fractions were collected by the Density Gradient Fraction System (Brandel, BR-188). 80S ribosome fractions were combined for each strain and centrifuged at 55000 rpm for 125 min at 4 °C in a type 70Ti rotor to pellet 80S ribosomes. Ribosomes were resuspended in 250 μl of resuspension buffer (50 mM HEPES-KOH (pH 7.4), 500 mM KC L, 2 mM MgCl2, 2 mM DTT). To separate 80S ribosomes into 40S and 60S subunits, 1 mM puromycin (Sigma, P8833) was added to ribosomes and incubated on ice for 15 min followed by incubation at 37 °C for 10 min. To separate 40S and 60S ribosomal subunits by fractionation, ribosomes were loaded onto a 5–20% sucrose gradient (50 mM HEPES-KOH (pH 7.4), 500 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, 2 mM DTT) and centrifuged at 22500 rpm for 10 hours at 4 °C in a SW35Ti rotor. Ribosomal subunits were fractionated and fractions for each subunit were combined and centrifuged at 30000 rpm for 14 hours at 4 °C in a type 70.1Ti rotor. Each subunits were resuspended in 50 μl of storage buffer (20 mM HEPES-KOH (pH 7.4), 100 mM KOAc (pH 7.6), 2.5 mM Mg(OAc)2, 250 mM sucrose, 2 mM DTT) and aliquots of subunits were snap frozen in liquid nitrogen.
Yeast Ribosome Co-Sedimentation Assay
For co-sedimentation assays, a 322 nt long a9 IRES FL RNA was in vitro transcribed from linearized pSP73-a9(IRES)-4xS1m(MCS) plasmids linearized with EcoRV between the full-length a9 IRES and the 4xS1m aptamers to only generate a9 IRES FL RNA by run off SP6 in vitro transcription as described in the 4xS1m pulldown section. For IRES-ribosome co-sedimentation reactions, 0.5 μM of purified WT or hES9S rRNA-containing 40S subunit were mix with 1μM in vitro transcribed, refolded RNA in a volume of 25 μl in reaction buffer (20 mM HEPES (pH7.6), 150 mM KCl, 5 mM MgCl2, 2 mM DTT) and incubated for 5 min at 37°C. Ribosome-RNA complexes were loaded on a 5 ml 5–20% sucrose gradient (20 mM HEPES (pH7.6), 150 mM KCl, 5 mM MgCl2, 2 mM DTT) and centrifuged at 35000 rpm for 3 hours in a SW60 rotor. Fractions were collected by the Density Gradient Fraction System (Brandel, BR-188). To each fraction, 500 pg of a spike-in RNA (here: UTR-Nluc) was added to internally control for RNA extraction efficiency by RT-qPCR. RNA was extracted from each fraction by the phenol/chloroform method followed by isopropanol precipitation. Purified RNAs were reverse transcribed using iScript Supermix (Bio-Rad, 1708840) and the mRNA amount for the spike-in and IRES RNAs (Nluc KL585/86; native KL641/42, 125 nt PCR product) in WT and hES9S samples was quantified by qPCR as given in the RT-qPCR section.
Generating CRISPR/Cas9-edited mouse embryonic stem cell lines
Genome editing of E14 ESCs were achieved by using CRISPR/Cas9 nuclease-mediated recombination (Doudna and Charpentier, 2014). A mouse ES cell lines harboring a homozygous P4(M5) allele at the respective endogenous Hoxa9 locus was generated by CRISPR/Cas9-mediated genome engineering (Zuris et al., 2015). Guide RNAs (gRNAs) were designed for cleavage as close as possible to the position of the 4-nt M5 mutation in P4 using the CRISPR design tool in benchling (http://benchling.com). Individual gRNAs were subcloned into the pX459-pSpCas9(BB)-2A-Puro-V2.0 plasmid (Ran et al., 2013) (Addgene plasmid #62988) encoding Cas9 from S. pyogenes a 2A-puromycin cassette as a selectable marker. Low passage number mESCs were transfected with 1.5 μg of pX459-gRNA plasmid and 0.5 μg of Single-Stranded Oligo Donor (ssODN, 5 μM) in 12-well plates with 1.0 × 106 cells per well using 5 μL Lipofectamine 2000 (Invitrogen, 11668027) following manufacturer’s instructions. The ssODNs were designed to have 30 or 60 nt overhangs 5’ and 3’ of the editing site. sgRNA guide sequences and ssODN repair templates used are detailed in Table S3. 24 hours after transfection, the cells were subjected to puromycin-selection at final 1 μg/ml and 24 hours later, the media was changed to new puro-containing media. The next day, cells were recovered for a day in media without puromycin. 24 hours later, the cells were seeded at low density (500 and 3000 cells) on 10 cm plates. 6–7 days after plating, single colonies were picked and replica plated onto two 96-well plates. One of the two plates was used for subsequent genotyping and sequencing analyses. After 5–6 days, PCR genotyping was performed to identify the isogenic cell lines that are homozygous for the P4(M5) mutation. The primers for genotyping are listed in Table S3. The confirmed clones were expanded and used for RA-treatment and polysome-fractionation/RT-qPCR experiments.
Sucrose Gradient Fractionation Analysis in mESCs
For sucrose gradient fractionation of mESC lysates of cells treated with RA for 60 hours, a similar protocol as for yeast and in (Simsek et al., 2017). The selected homozygous clones for M5 and WT cells were subjected to a total of 60 hours (2.5 days) of 33 nM retinoic acid (RA) treatment, with replacement of fresh RA-containing media every 12 hours. Briefly, ca. 2.5 × 106 mES cells (WT and M5-edited clone D6) were seeded into 0.1% gelatin pre-coated 10 cm dishes. 4 hours after seeding, cells were treated with 33 nM retinoic acid (RA, Sigma, R2625) in DMSO in ESC media 60 hours prior to harvest and polysome analysis. Fresh media with RA was provided every 12 hours. For Cycloheximide treatment (CHX) (Sigma-Aldrich, C7698–1G) treatment, ESCs were incubated at 0.1 mg/ml for 3 min at 37°C. ESCs were lysed in polysome lysis buffer (25 mM Tris-HCl pH 7.5 (Ambion, AM9850G, and Ambion, AM9855G), 150 mM NaCl (Ambion, AM9759), 15 mM MgCl2 (Ambion, AM9530G), 1 mM DTT (Ambion, 10197777001), 8% glycerol (Sigma-Aldrich, G5516), 1% Triton X-100 (Sigma-Aldrich, T8787), 0.2 mg/ml Cycloheximide (CHX) (Sigma-Aldrich, C7698–1G), 100 U/ml SUPERase In RNase Inhibitor (Ambion, AM2694), 25 U/ml TurboDNase (Ambion, AM2238), Complete Protease Inhibitor EDTA-free (Sigma-Aldrich, 11836170001) in nuclease-free water (Thermo Fisher Scientific, 10977015)). For ca. 10 × 106 ESCs, 400 μL of lysis buffer was used to lyse the cells for 30 min on a rotator at 4°C vortexed 3x every 10 min. After lysis, nuclei were removed by two consecutive centrifugations at 800 g, 5 min at 4°C followed by one centrifugation at 8000 g, 5 min, and one centrifugation at 20817 g, 5 min. RNA concentrations were measured using Nanodrop UV spectrophotometer (Thermo Fisher Scientific) and normalized amounts of RNA were layered onto a linear sucrose gradient (10%–45% sucrose (Fisher Scientific, S5–12) (w/v), 25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 15 mM MgCl2, 1 mM DTT, 100 mg/ml CHX in nuclease-free water and centrifuged in a SW41Ti rotor (Beckman) for 2.5 hr at 40,000 rpm at 4°C. Fractions were collected by Density Gradient Fraction System (Brandel, BR-188) with continuous A260 measurement. After collection of polysome fractions in 2 ml safe-lock tubes (Eppendorf), RNA of all fractions was individually extracted using Acid-Phenol:Chloroform, pH 4.5 (with IAA, 125:24:1) (Ambion, AM9722). To each fraction we added 500 pg in vitro transcribed Rluc-Fluc RNA as a spike-in control and detected using Rluc-specific primers. 100 ng of total RNA from each fraction was reverse-transcribed to cDNA using iScript Reverse Transcription Supermix kit (Bio-Rad, 1708841) and qPCR was performed as described in the qPCR section. Relative mRNA levels were summed across all fractions analyzed and presented as percentage of this total. All RT-qPCR primers are listed in Table S3. In addition, we quantified the area under the curve between free fraction, light and heavy polysomes to assess the translation rate of an mRNA based on its distribution in the gradient.
Data Sources
For the 40-way multiple sequence alignment (MSA) and conservation analysis of ES9S and surrounding 18S rRNA sequence, the following GenBank 18S rRNA sequences were retrieved for eukaryotic species from the NCBI database as data sources and aligned by Multiple Alignment using Fast Fourier Transform (MAFFT, MView, EMBL-EBI webtools) with default settings: mouse (Mus musculus; accession number NR_003278.3), human (Homo sapiens; M10098.1), chimpanzee (Pan troglodytes; Sequence Read Archive (SRA): PRJNA189439 and SRP018689 (Prado-Martinez et al., 2013)), gorilla (Gorilla gorilla; PRJNA189439 (Prado-Martinez et al., 2013)), orangutan (Pongo abelii; XR_002913762.1, predicted (pred.), and PRJNA189439 (Prado-Martinez et al., 2013)), bonobo (Pan paniscus; PRJNA189439 (Prado-Martinez et al., 2013)), macaque (Macaca mulatta; NR_146166.1), guinea pig (Cavia porcellus; XR_002788481.1 (pred.)), sheep (Ovis aries; KY129860.1), cat (Felis catus; XR_002738341.1 (pred.)), flying fox (Pteropus vampyrus; XR_002778881.1), horse (Equus caballus; NR_046271.1), swine (Sus scrofa; NR_046261.1), rabbit (Oryctolagus cuniculus; NR_033238.1), naked mole-rat (Heterocephalus glaber; PRJNA72441 and SRP007995); chinese hamster (Cricetulus griseus; NR_045132.1), marmoset (Callithrix jacchus; NR_146325.1); cow (Bos taurus; NR_036642.1), rat (Rattus norvegicus; NR_046237.1), chicken (Gallus gallus; AF173612.1), Clownfish (Amphiprion ocellaris; XR_002748043.1 (pred.)), Japanese rice fish (Oryzias latipes; XR_002874070.1 (pred.)), Southern platyfish (Xiphophorus maculatus; XR_002752479.1 (pred.)), Rainbow trout (Oncorhynchus mykiss; XR_002474306.1 (pred.)), African clawed frog (Xenopus laevis; X02995.1), zebrafish (Danio rerio; NR_145818.1), lancelet (Branchiostoma floridae; M97571.1), sea squirt (Ciona intestinalis; AB013017.1), tick (Ixodes cookei; L76351.1), sea urchin (Strongylocentrotus purpuratus; L28055.1), purple false brome or stiff brome, grass (Brachypodium distachyon; XR_002961462.1 (pred.)); wheat (Triticum aestivum; AY049040.1); clementine (Citrus clementina; XR_002904493.1 (pred.)); thale cress (Arabidopsis thaliana; NR_141642.1); sea anemone (Nematostella vectensis; AF254382.1); hydra (Hydra magnipapillata; HQ392522.1); yeast (Saccharomyces cerevisiae; J01353.1, Saccharomyces pombe; X58056.1); red tree sponge (Amphimedon compressa; EU702409.1); and roundworm (Caenorhabditis elegans; NR_000054.1).
QUANTIFICATION AND STATISTICAL ANALYSIS
In all figures, data was presented as mean, SD or SEM as stated in the figure legends, and *p ≤ 0.05 was considered significant (ns: p > 0.05; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001). ^: given that two continuous datasets are different enough such that they are not overlapping, we cannot calculate p-value with Brunner-Munzel test. Blinding and randomization were not used in any of the experiments. Number of independent biological replicates used for the experiments are listed in the figure legends. Tests, two-tailed unpaired Student’s t-test if not stated otherwise, and specific p-values used are indicated in the figure legends. In all cases, multiple independent experiments were performed on different days to verify the reproducibility of experimental findings. For mouse experiments, embryos from multiple litters were used to avoid litter-specific bias.
Supplementary Material
Relative protein quantification for proteins identified in 4xS1m pulldown from C3H/10T1/2 cells in two biological replicates. Fold change is presented as the ratio of P3 or P4 to the 4xS1m aptamer control per replicate. Proteins were categorized into gene sets according to identity.
Relative protein quantification for proteins identified in 4xS1m pulldown from FVB stage E11.5 mouse embryos in three biological replicates. Fold change is presented as the ratio of a9 IRES180 or HCV IRES to the 4xS1m aptamer control per replicate. Proteins were categorized into gene sets according to identity.
KEY RESOURCES TABLE
| Reagent or Resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-PGK1 | Thermo, Novex | Cat# 459250, RRID:AB_2532235 |
| Mouse monoclonal anti-RPS5/uS7 | Abcam | Cat# ab58345; RRID: AB_2180899 |
| rabbit monoclonal anti-RPS6/eS6 | Cell Signaling | Cat# 2217, RRID:AB_331355 |
| Mouse monoclonal anti-RPL10A/uL1 (for yeast) | Santa Cruz | Cat# sc-100827, RRID:AB_2285311 |
| Rabbit monoclonal anti-RPL10A/uL1 (for mouse) | Abcam | Cat# ab174318 |
| Goat monoclonal anti-eIF3B (eif3-eta, N-20) | Santa Cruz | Cat# sc-16377, RRID: AB_671941 |
| Rabbit monoclonal anti-eIF4A (C32B4) | Cell Signaling | Cat# 2013, RRID:AB_2097363 |
| Rat monoclonal anti-Mouse IgG-HRP (eB144) | Rockland | Cat#18–8817-31; RRID: AB_2610850 |
| Mouse monoclonal anti-Rabbit IgG-HRP (eB182) | Rockland | Cat#18–8816-31; RRID: AB_2610847 |
| Sheep Anti-Mouse IgG, HRP Conjugated | GE Healthcare | Cat# NXA931, RRID:AB_772209 |
| Donkey Anti-Rabbit IgG, HRP Conjugated | GE Healthcare | Cat# NA934, RRID:AB_772206 |
| Donkey Anti-Rat IgG, HRP Conjugated | Jackson ImmunoResearch | Cat# 712–036-150, RRID:AB_2340640 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Puromycin dihydrochloride | Sigma-Aldrich | Cat# P8833 |
| RNase A | Invitrogen | Cat# AM2271 |
| RNA PureLink columns | Ambion | Cat# 12183018 |
| RNA Clean and Concentrator-5 columns | Zymo Research | Cat# R1016 |
| TMTsixplex Isobaric Label Reagent Set | Thermo Fisher Scientific | Cat# 90066 |
| Sequencing-grade modified trypsin | Promega | Cat# V5111 |
| TURBO DNase | Ambion | Cat# AM2238 |
| SUPERase In RNase Inhibitor | Ambion | Cat# AM2696 |
| RNaseOUT | Thermo Fisher | Cat# 10777019 |
| RNasin Plus RNase inhibitor | Promega | Cat# N261A |
| TRIzol | Invitrogen | Cat# 15596 |
| AccuPrime Pfx DNA Polymerase | Invitrogen | Cat# 12344024 |
| KOD Xtreme Hot Start DNA Polymerase | EMD Millipore | Cat# 71975 |
| iScript Supermix | Bio-Rad | Cat# 1708840 |
| SsoAdvanced SYBR Green supermix | Bio-Rad | Cat# 1725270 |
| CFX384 Touch qPCR machine | Bio-Rad | Cat# 1855485 |
| 5-Fluoroorotic Acid (5-FOA) | Fisher Scientific | Cat# F10501–5.0 |
| Geneticin | Gibco | Cat# 11811–031 |
| Amino acid supplements (Complete Supplement Mixture, CSM) | Sunrise Science Products | https://sunrisescience.com/products/growth-media/amino-acid-supplement-mixtures/csm-formulations/ |
| Salmon sperm DNA | Sigma | Cat# D1626–5G |
| Poly ethylene glycol (PEG) – MW 8000 | Millipore Sigma | Cat# 6510-OP |
| cOmplete Protease Inhibitor Cocktail, EDTA-free | Roche | Cat# 11836145001 |
| cOmplete Mini Protease Inhibitor Cocktail, EDTA-free | Roche | Cat# 11836170001 |
| Streptavidin Sepharose High Performance | GE Healthcare | Cat# 17–5113-01 |
| Avidin Agarose | Thermo, Pierce | Cat# 20219 |
| SDS-PAGE gels | Bio-Rad | Cat# 567–1095, 456–1096 |
| Semi-dry Trans-Blot Turbo system | Bio-Rad | Cat# 170–4273 |
| Clarity Western ECL Substrate | Bio-Rad | Cat# 170–5061 |
| ChemiDoc MP | Bio-Rad | Cat# 17001402 |
| Tissue Lyser (QIAgen TissueLyser II) | Qiagen | Cat# 85300 |
| Dulbecco’s Modified Eagle’s Medium | Gibco | Cat# 11965–118 |
| Fetal calf serum | EMD Millipore | Cat# TMS-013-B |
| EmbryoMax ES Cell Qualified Penicillin-Streptomycin Solution 100X | EMD Millipore | Cat# TMS-AB2-C |
| Opti-MEM | Gibco | Cat# 11058–021 |
| Lipofectamine 2000 | Invitrogen | Cat# 11668–019 |
| Lipofectamine MessengerMAX | Invitrogen | Cat# LMRNA001 |
| Knockout-DMEM | Gibco | Cat# 10829018 |
| Embryomax FBS | EMD Milipore | Cat# ES-009-B |
| Non-essential amino acids | EMD Milipore | Cat# TMS-001-C |
| L-Glutamine | EMD Milipore | Cat# TMS-002-C |
| 2-mercaptoethanol | Gibco | Cat# 21985023 |
| LIF | EMD Millipore | Cat# ESG1107 |
| Retinoic acid | Sigma | Cat# R2625 |
| 1x PBS | Gibco | Cat# 14190–250 |
| SYBR Gold | Invitrogen | Cat# S11494 |
| GlycoBlue | Ambion | Cat# LSAM9516 |
| 100 kDa MWCO concentrators | Amicon®Ultra, Merck | Cat# UFC910008 |
| SYPRO Ruby Gel stain | Thermo Fisher | Cat# S12000 |
| Sucrose | Fisher Scientific | Cat# S5–12 |
| Density Gradient Fraction System | Brandel | Cat# BR-188 |
| Acid-Phenol:Chloroform, pH 4.5 | Ambion | Cat# AM9722 |
| Dimethyl sulfoxide (DMSO, cell culture grade) | Sigma | Cat# D2650 |
| 4EGI-1 (eIF4E/eIF4G interaction inhibitor) | Millipore Sigma | Cat# 324517 |
| Critical Commercial Assays | ||
| ProteoExtract Protein Precipitation Kit | EMD Millipore | Cat#539180 |
| Dual-Luciferase Reporter Assay System | Promega | Cat# E1980 |
| Nano-Glo Dual-Luciferase Reporter Assay System | Promega | Cat# N1610 |
| GloMax-Multi | Promega | Cat# E7081 |
| OMIX C18 pipette tips column | Agilent | Cat# A57003100 |
| MEGAscript T7 Transcription Kit | Ambion | Cat# AM1333 |
| MEGAscript SP6 Transcription Kit | Ambion | Cat# AM1330 |
| G(5’)ppp(5’)A RNA Cap Structure Analog | NEB | Cat# S1406L |
| ScriptCap m7G Capping System | CellScript | Cat# C-SCCE0625 |
| A-Plus Poly(A) Polymerase Tailing Kit | CellScript | Cat# C-PAP5104H |
| MasterPure Yeast RNA Purification Kit | Epicentre | Cat# MPY03100 |
| QIAquick Gel Extraction Kit | QIAgen | Cat# 28706 |
| Monarch Gel Extraction Kit | NEB | Cat# T1020S |
| NEBuilder HiFi DNA Assembly Master Mix | NEB | Cat# E2621S |
| QIAquick PCR Purification Kit | QIAgen | Cat# 28106 |
| G-50 Mini Quick Spin Sephadex RNA columns | Roche | Cat# 11814427001 |
| Deposited Data | ||
| Raw mass spec data | ProteomeXchange | PXD021678 |
| Cryo-EM data | EMDB | EMD 11562 - EMD 11568 |
| Mendeley Data | This paper | http://dx.doi.org/10.17632/45zmm935g2.1 |
| Experimental Models: Cell Lines | ||
| C3H/10T1/2 mouse cells | ATCC | Cat# CCL-226 |
| E14Tg2a.4 mouse ESCs | (Smith and Hooper, 1987) | N/A |
| a9(M5)-edited mouse embryonic stem cells | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Yeast (S. cerevisiae) strains used: see Table S2 | This paper | N/A |
| Oligonucleotides | ||
| Oligonucleotides for genome editing, cloning, RT-qPCR analysis, in vitro transcription, see Table S3 | This paper | N/A |
| Synthesized oligonucleotides | Twist Bioscience | N/A |
| Recombinant DNA | ||
| Plasmids used and generated, see Table S1 | This paper | N/A |
| Software and Algorithms | ||
| MAFFT, MView | EMBL-EBI webtools | https://www.ebi.ac.uk/Tools/msa/mafft/ |
| Chimera | (Pettersen et al., 2004) | https://www.cgl.ucsf.edu/chimera/ |
| RELION | (Scheres, 2012) | N/A |
| RELION 3 | (Nakane et al., 2018) | N/A |
| CTFFIND | (Mindell and Grigorieff, 2003) | N/A |
| Batchboxer | (Ludtke et al., 1999) | N/A |
| COOT | (Emsley et al., 2010) | N/A |
| SerialEM | (Mastronarde, 2005) | N/A |
| MotionCor2 | (Zheng et al., 2017) | N/A |
| GCTF | (Zhang, 2016) | N/A |
| MAFFT, MView | EMBL-EBI webtools | https://www.ebi.ac.uk/Tools/msa/mafft/ |
| Vienna RNAfold | RNAfold WebServer | http://rna.tbi.univie.ac.at |
| VARNA | RNA structure visualization | http://varna.lri.fr |
| R | R Foundation | https://www.r-project.org/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Proteome Discoverer 1.4 | Thermo Fisher Scientific | https://portal.thermo-brims.com/ |
| Prism | GraphPad Software Inc. | Version 8.0 |
Highlights:
A Hoxa9 5’ UTR stem-loop, P4, selectively binds to the rRNA expansion segment ES9S
“Humanized” ES9S yeast ribosomes reveal species-specific binding of ribosomes t o P4
Disrupting Hoxa9-ES9S binding in neural stem cells reduces Hoxa9 mRNA translation
Selective mRNA recruitment via ESs is a novel gene-regulatory role for rRNA
ACKNOWLEDGEMENTS
We would like to thank the Barna lab members for support and constructive criticism of the work, Zhen Shi for technical assistance with mass spectrometry experiments, Adele F. Xu for technical advice on genome editing experiments, all lab members and Anupam K. Chakravarty for critical reading of and helpful comments on the manuscript. We also thank the ETH Zürich scientific center for optical and electron microscopy (ScopeM) for access to electron microscopy equipment, Peter Tittmann for technical support, and Martin Itten for support with data processing. We are grateful to Katsura Asano (Kansas State University, KS, USA) and Makoto Kitabatake (Kyoto University, Japan) for providing the rDNA deletion yeast strains and the rDNA plasmids; Mary Ann Handel (The Jackson Laboratory, Bar Harbor, ME, USA) for the RPL10A antibody; and Georg Stoecklin (DKFZ-ZMBH Alliance, Heidelberg University and Mannheim University, Germany), Conor J. Howard and Davide Ruggero (both UCSF, San Francisco, CA, USA) for kindly sharing plasmids. This work was supported by the New York Stem Cell Foundation (M.B.), Alfred P. Sloan Research Fellowship (M.B), Mallinckrodt Foundation Award (M.B.), Pew Scholars Award (M.B.), NIH grant R01HD086634 (M.B.), the Swiss National Science Foundation grant (310030B_163478) and via the National Centre of Excellence in RNA and Disease to N.B., and a Human Frontier Science Program Fellowship (K.F.). K.L. is supported by an EMBO Long-Term Fellowship (ALTF 539–2015), is the Layton Family Fellow of the Damon Runyon Cancer Research Foundation (DRG-2237–15), and is supported by the Katharine McCormick Advanced Postdoctoral Scholar Fellowship to Support Women in Academic Medicine (2019). M.B. is a New York Stem Cell Foundation Robertson Investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental Information includes 7 Supplemental Figures and 5 Supplemental Tables.
DECLARATION OF INTEREST
K.L. and M.B. are inventors on patents and submitted provisional patent applications related to the Hoxa9 P4 stem-loop and RNA therapeutics and their various uses.
REFERENCES
- Acker MG, Kolitz SE, Mitchell SF, Nanda JS, and Lorsch JR (2007). Reconstitution of Yeast Translation Initiation. In Methods in Enzymology, pp. 111–145. [DOI] [PubMed]
- Anger AM, Armache JP, Berninghausen O, Habeck M, Subklewe M, Wilson DN, and Beckmann R (2013). Structures of the human and Drosophila 80S ribosome. Nature 497, 80–85. [DOI] [PubMed] [Google Scholar]
- Armache J-P, Jarasch A, Anger AM, Villa E, Becker T, Bhushan S, Jossinet F, Habeck M, Dindar G, Franckenberg S, et al. (2010). Cryo-EM structure and rRNA model of a translating eukaryotic 80S ribosome at 5.5-A resolution. Proc. Natl. Acad. Sci. U. S. A 107, 19748–19753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrame M, Hendry Y, and Tollervey D (1994). Mutational analysis of an essential binding site for the U3 snoRNA in the 5′ external transcribed spacer of yeast pre-rRNA. Nucleic Acids Res 22, 5138–5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen E, Sharma MR, Shi X, Agrawal RK, and Joseph S (2014). Fragile X mental retardation protein regulates translation by binding directly to the ribosome. Mol. Cell 54, 407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CY, Chou F-C, Kladwang W, Tian S, Cordero P, and Das R (2015). Consistent global structures of complex RNA states through multidimensional chemical mapping. Elife 4, 1–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon RA, and Rossant J (1992). Exogenous retinoic acid rapidly induces anterior ectopic expression of murine Hox-2 genes in vivo. Development 116, 357–368. [DOI] [PubMed] [Google Scholar]
- Dahlberg AE (1989). The functional role of ribosomal RNA in protein synthesis. Cell 57, 525–529. [DOI] [PubMed] [Google Scholar]
- Doudna JA, and Charpentier E (2014). The new frontier of genome engineering with CRISPR-Cas9. Science 346, 1258096. [DOI] [PubMed] [Google Scholar]
- Dresios J, Chappell S. a, Zhou W, and Mauro VP (2006). An mRNA-rRNA base-pairing mechanism for translation initiation in eukaryotes. Nat. Struct. Mol. Biol 13, 30–34. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii K, Kitabatake M, Sakata T, Miyata A, and Ohno M (2009). A role for ubiquitin in the clearance of nonfunctional rRNAs. Genes Dev 23, 963–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii K, Susanto TT, Saurabh S, and Barna M (2018). Decoding the function of expansion segments in ribosomes. Mol. Cell 72, 1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genuth NR, and Barna M (2018). The Discovery of Ribosome Heterogeneity and Its Implications for Gene Regulation and Organismal Life. Mol. Cell 71, 364–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerbi SA (1996). Expansion segments: regions of variable size that interrupt the universal core secondary structure of ribosomal RNA In Ribosomal RNA: Structure, Evolution, Processing and Function in Protein Synthesis, Eds: Zimmermann RA and Dahlberg AE. Telford - CRC Press, Boca Raton, FL, pp. 71–87. [Google Scholar]
- Ghyselinck NB, and Duester G (2019). Retinoic acid signaling pathways. Dev 146, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton C, and Maden M (1995). Endogenous distribution of retinoids during normal development and teratogenesis in the mouse embryo. Dev. Dyn 202, 312–323. [DOI] [PubMed] [Google Scholar]
- Hundsdoerfer P, Thoma C, and Hentze MW (2005). Eukaryotic translation initiation factor 4GI and p97 promote cellular internal ribosome entry sequence-driven translation. Proc. Natl. Acad. Sci. U. S. A 102, 13421–13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RJ, Hellen CUT, Pestova TV, and Thompson SR (2010). The mechanism of eukaryotic translation initiation and principles of its regulation. Wiley Interdiscip. Rev. RNA 3, 697–705. [Google Scholar]
- Jeeninga RE, Van Delft Y, de Graaff-Vincent M, Dirks-Mulder A, Venema J, and Raué HA (1997). Variable regions V13 and V3 of Saccharomyces cerevisiae contain structural features essential for normal biogenesis and stability of 5.8S and 25S rRNA. RNA 3, 476–488. [PMC free article] [PubMed] [Google Scholar]
- Kmita M, and Duboule D (2003). Organizing axes in time and space; 25 years of colinear tinkering. Science 301, 331–333. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Jomaa A, Lee JH, Chandrasekar S, Boehringer D, Shan SO, and Ban N (2018). Structure of a prehandover mammalian ribosomal SRP·SRP receptor targeting complex. Science 360, 323–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov N, Pusic A, Stumpf CR, Shimizu K, Hsieh AC, Xue S, Ishijima J, Shiroishi T, and Barna M (2011). Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell 145, 383–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumlauf R (1994). Hox genes in vertebrate development. Cell 78, 191–201. [DOI] [PubMed] [Google Scholar]
- De Kumar B, Parrish ME, Slaughter BD, Unruh JR, Gogol M, Seidel C, Paulson A, Li H, Gaudenz K, Peak A, et al. (2015). Analysis of dynamic changes in retinoid-induced transcription and epigenetic profiles of murine Hox clusters in ES cells. Genome Res 25, 1229–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo BA, Gonzalez IL, Gillespie DA, and Sylvester JE (1996). Human ribosomal RNA variants from a single individual and their expression in different tissues. Nucleic Acids Res 24, 4817–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leffers H, and Andersen AH (1993). The sequence of 28S ribosomal RNA varies within and between human cell lines. Nucleic Acids Res 21, 1449–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppek K, and Stoecklin G (2014). An optimized streptavidin-binding RNA aptamer for purification of ribonucleoprotein complexes identifies novel ARE-binding proteins. Nucleic Acids Res 42, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppek K, Schott J, Reitter S, Poetz F, Hammond MC, and Stoecklin G (2013). Roquin promotes constitutive mRNA decay via a conserved class of stem-loop recognition motifs. Cell 153, 869–881. [DOI] [PubMed] [Google Scholar]
- Leppek K, Das R, and Barna M (2017). Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol 19, 158–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppek K, Byeon GW, Fujii K, and Barna M (2020). VELCRO-IP RNA-seq explores ribosome expansion segment function in translation genome-wide. bioRxiv 10.1101/2020.07.01.179515. [DOI] [PMC free article] [PubMed]
- Ludtke SJ, Baldwin PR, and Chiu W (1999). EMAN: Semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol 128, 82–97. [DOI] [PubMed] [Google Scholar]
- Luo Z, Rhie SK, and Farnham PJ (2019). The enigmatic hox genes: Can we crack their code? Cancers (Basel) 11, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallo M, and Alonso CR (2013). The regulation of Hox gene expression during animal development. Development 140, 3951–3963. [DOI] [PubMed] [Google Scholar]
- Malygin AA, Kossinova OA, Shatsky IN, and Karpova GG (2013). HCV IRES interacts with the 18S rRNA to activate the 40S ribosome for subsequent steps of translation initiation. Nucleic Acids Res 41, 8706–8714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin F, Ménétret J-F, Simonetti A, Myasnikov AG, Vicens Q, Prongidi-Fix L, Natchiar SK, Klaholz BP, and Eriani G (2016). Ribosomal 18S rRNA base pairs with mRNA during eukaryotic translation initiation. Nat. Commun 7, 12622, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol 152, 36–51. [DOI] [PubMed] [Google Scholar]
- Matsuda D, and Mauro VP (2014). Base pairing between hepatitis C virus RNA and 18S rRNA is required for IRES-dependent translation initiation in vivo. Proc. Natl. Acad. Sci. U. S. A 111, 15385–15389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauro VP, and Edelman GM (2002). The ribosome filter hypothesis. Proc. Natl. Acad. Sci. U. S. A 99, 12031–12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon R, Zaborowska I, and Walsh D (2011). Noncytotoxic Inhibition of Viral Infection through eIF4F-Independent Suppression of Translation by 4EGi-1. J. Virol 85, 853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mindell JA, and Grigorieff N (2003). Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol 142, 334–347. [DOI] [PubMed] [Google Scholar]
- Moerke NJ, Aktas H, Chen H, Cantel S, Reibarkh MY, Fahmy A, Gross JDD, Degterev A, Yuan J, Chorev M, et al. (2007). Small-Molecule Inhibition of the Interaction between the Translation Initiation Factors eIF4E and eIF4G. Cell 128, 257–267. [DOI] [PubMed] [Google Scholar]
- Musters W, Venema J, van der Linden G, van Heerikhuizen H, Klootwijk J, and Planta RJ (1989). A system for the analysis of yeast ribosomal DNA mutations. Mol. Cell. Biol 9, 551–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakane T, Kimanius D, Lindahl E, and Scheres SH (2018). Characterisation of molecular motions in cryo-EM single-particle data by multi-body refinement in RELION. Elife 7, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natchiar SK, Myasnikov AG, Kratzat H, Hazemann I, and Klaholz BP (2017). Visualization of chemical modifications in the human 80S ribosome structure. Nature 551, 472–477. [DOI] [PubMed] [Google Scholar]
- Nemoto N, Singh CR, Udagawa T, Wang S, Thorson E, Winter Z, Ohira T, Ii M, Valásek L, Brown SJ, et al. (2010). Yeast 18 S rRNA is directly involved in the ribosomal response to stringent AUG selection during translation initiation. J. Biol. Chem 285, 32200–32212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogi Y, Yano R, and Nomura M (1991). Synthesis of large rRNAs by RNA polymerase II in mutants of Saccharomyces cerevisiae defective in RNA polymerase I. Proc. Natl. Acad. Sci 88, 3962–3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte C, De Kumar B, and Krumlauf R (2019). Hox genes: Downstream “effectors” of retinoic acid signaling in vertebrate embryogenesis. Genesis 57, 1–17. [DOI] [PubMed] [Google Scholar]
- Osuna BA, Howard CJ, KC S, Frost A, and Weinberg DE (2017). In vitro analysis of RQC activities provides insights into the mechanism and function of CAT tailing. Elife 6, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozgur S, Chekulaeva M, and Stoecklin G (2010). Human Pat1b connects deadenylation with mRNA decapping and controls the assembly of processing bodies. Mol. Cell. Biol 30, 4308–4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pánek J, Kolář M, Vohradský J, and Shivaya Valášek L (2013). An evolutionary conserved pattern of 18S rRNA sequence complementarity to mRNA 5′ UTRs and its implications for eukaryotic gene translation regulation. Nucleic Acids Res 41, 7625–7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panopoulos P, and Mauro VP (2008). Antisense masking reveals contributions of mRNA-rRNA base pairing to translation of Gtx and FGF2 mRNAs. J. Biol. Chem 283, 33087–33093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papalopulu N, Lovel-Badage R, and Krumlauf R (1991). The expression of murine Hox-2 genes id dependent on the differenation pathway and displays a collinear sensitivity to retinoic acid in F9 cells and Xenopus embroys. Nucleic Acids Res 19, 5497–5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardi N, Hogan MJ, Porter FW, and Weissman D (2018). mRNA vaccines-a new era in vaccinology. Nat. Rev. Drug Discov 17, 261–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MS, Balasubramaniam A, Sallee FR, and Parker SL (2018). The expansion segments of 28S Ribosomal RNA extensively match human messenger RNAs. Front. Genet 9, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks MM, Kurylo CM, Dass RA, Bojmar L, Lyden D, Vincent CT, and Blanchard SC (2018). Variant ribosomal RNA alleles are conserved and exhibit tissue-specific expression. Sci. Adv 4, eaao0665, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins DN, Pappin DJC, Creasy DM, and Cottrell JS (1999). Probability-based protein identification by searching sequence databases using mass spectrometry data. In Electrophoresis, pp. 3551–3567. [DOI] [PubMed]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera - A visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Plank TDM, and Kieft JS (2012). The structures of nonprotein-coding RNAs that drive internal ribosome entry site function. Wiley Interdiscip. Rev. RNA 3, 195–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado-Martinez J, Sudmant PH, Kidd JM, Li H, Kelley JL, Lorente-Galdos B, Veeramah KR, Woerner AE, O’Connor TD, Santpere G, et al. (2013). Great ape genetic diversity and population history. Nature 499, 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quade N, Boehringer D, Leibundgut M, van den Heuvel J, and Ban N (2015). Cryo-EM structure of Hepatitis C virus IRES bound to the human ribosome at 3.9-Å resolution. Nat. Commun 6, 7646, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh M, and Woolford JL (2016). Eukaryote-specific rRNA expansion segments function in ribosome biogenesis. RNA 22, 1153–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanova L, Korobova F, Noniashvilli E, Dyban A, and Zatsepina O (2006). High resolution mapping of ribosomal DNA in early mouse embryos by fluorescence in situ hybridization. Biol. Reprod 74, 807–815. [DOI] [PubMed] [Google Scholar]
- Scheres SHW (2012). RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S, Brown A, Santhanam B, and Hegde RS (2015). Structure and assembly pathway of the ribosome quality control complex. Mol. Cell 57, 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh BN, Downer NL, Kueh AJ, Thomas T, and Voss AK (2014). Excessive versus physiologically relevant levels of retinoic acid in embryonic stem cell differentiation. Stem Cells 32, 1451–1458. [DOI] [PubMed] [Google Scholar]
- Shi Z, Fujii K, Kovary KM, Genuth NR, R?st HL, Teruel MN, Barna M, Segal E, Mohammed AK, Hamon C, et al. (2017). Heterogeneous ribosomes preferentially translate distinct subpools of mRNAs genome-wide. Mol. Cell 67, 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shine J, and Dalgarno L (1974). The 3’-terminal sequence of Escherichia coli 16S ribosomal RNA: complementarity to nonsense triplets and ribosome binding sites. Proc. Natl. Acad. Sci. U. S. A 71, 1342–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeone A, Acampora D, Arcioni L, Andrews PW, Boncinelli E, and Mavilio F (1990). Sequential activation of HOX2 homeobox genes by retinoic acid in human embryonal carcinoma cells. Nature 346, 763–766. [DOI] [PubMed] [Google Scholar]
- Simsek D, Tiu GC, Flynn RA, Xu AF, Chang HY, Barna M, Simsek D, Tiu GC, Flynn RA, Byeon GW, et al. (2017). The mammalian ribo-interactome reveals ribosome functional diversity and heterogeneity. Cell 169, 1051–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AG, and Hooper ML (1987). Buffalo rat liver cells produce a diffusible activity which inhibits the differentiation of murine embryonal carcinoma and embryonic stem cells. Dev. Biol 121, 1–9. [DOI] [PubMed] [Google Scholar]
- Sonenberg N, and Hinnebusch AG (2009). Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136, 731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steitz JA, and Jakes K (1975). How ribosomes select initiator regions in mRNA: base pair formation between the 3’ terminus of 16S rRNA and the mRNA during initiation of protein synthesis in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A 72, 4734–4738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney R, Chen L, and Yao MC (1994). An rRNA variable region has an evolutionarily conserved essential role despite sequence divergence. Mol. Cell. Biol 14, 4203–4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira FK, and Lehmann R (2018). Translational Control during Developmental Transitions. Cold Spring Harb. Perspect. Biol a032987, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A, Schäfer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, and Hamon C (2003). Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem 75, 1895–1904. [DOI] [PubMed] [Google Scholar]
- Tranque P, Hu MC-Y, Edelman GM, and Mauro VP (1998). rRNA complementarity within mRNAs: A possible basis for mRNA-ribosome interactions and translational control. Proc. Natl. Acad. Sci 95, 12238–12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng H, Chou W, Wang J, Zhang X, Zhang S, and Schultz RM (2008). Mouse ribosomal RNA genes contain multiple differentially regulated variants. PLoS One 3, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai HH, Vu L, Oakes M, and Nomura M (2000). Complete deletion of yeast chromosomal rDNA repeats and integration of a new rDNA repeat: use of rDNA deletion strains for functional analysis of rDNA promoter elements in vivo. Nucleic Acids Res 28, 3524–3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue S, Tian S, Fujii K, Kladwang W, Das R, and Barna M (2015). RNA regulons in Hox 5’ UTRs confer ribosome specificity to gene regulation. Nature 517, 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon A, Peng G, Brandenburg Y, Zollo O, Xu W, Rego E, and Ruggero D (2006). Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science 312, 902–906. [DOI] [PubMed] [Google Scholar]
- Zhang K (2016). Gctf: Real-time CTF determination and correction. J. Struct. Biol 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuris JA, Thompson DB, Shu Y, Guilinger JP, Bessen JL, Hu JH, Maeder ML, Joung JK, Chen ZY, and Liu DR (2015). Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol 33, 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Relative protein quantification for proteins identified in 4xS1m pulldown from C3H/10T1/2 cells in two biological replicates. Fold change is presented as the ratio of P3 or P4 to the 4xS1m aptamer control per replicate. Proteins were categorized into gene sets according to identity.
Relative protein quantification for proteins identified in 4xS1m pulldown from FVB stage E11.5 mouse embryos in three biological replicates. Fold change is presented as the ratio of a9 IRES180 or HCV IRES to the 4xS1m aptamer control per replicate. Proteins were categorized into gene sets according to identity.
Data Availability Statement
Mass Spectrometry data are available in Table S4 and S5 and have been uploaded to the ProteomeXchange database via the PRIDE repository; data are available via ProteomeXchange with the identifier PXD021678 (PRIDE database: PXD021678). Cryo-EM map data have been deposited in the Electron Microscopy Data Bank (EMDB) under accession numbers EMD 11562 - EMD 11568: mouse Hoxa9 IRES-like element bound to the human 80S ribosome EMD-11562; mouse Hoxa9 IRES-like element bound to the human 40S ribosomal subunit head - IRES binding site EMD-11563; mouse Hoxa9 IRES-like element bound to the human 40S ribosomal subunit - 40S head EMD-11564; mouse Hoxa9 IRES-like element bound to the human 40S ribosomal subunit - 40S body EMD-11565; Hoxa9 IRES P4 stem-loop bound to the 40S ribosomal subunit - 40S head EMD-11566; Hoxa9 IRES P4 stem-loop bound to the 40S ribosomal subunit - 40S body EMD-11567; human 40S ribosomal subunit (control) EMD-11568. Original/source data for figures in the paper is available at Mendeley Data: http://dx.doi.org/10.17632/45zmm935g2.1.