

Abstract
Expanding the repertoire of reactions available to enzymes is an enduring challenge in biocatalysis. Due to the synthetic versatility of transition metals, metalloenzymes have been favored targets for achieving new catalytic functions. While less well explored, enzymes lacking metal centers can also be effective catalysts for non-natural reactions, providing access to reaction modalities that compliment those available to metals. By understanding how these activation modes can reveal new functions, strategies can be developed to access novel biocatalytic reactions. This review will cover discoveries in the last two years which access catalytic reactions that go beyond the native repertoire of metal-free biocatalysts.
Introduction
Biocatalysis is a powerful synthetic tool for the construction of complex molecules, developing from an area of purely academic interest into a vital tool in specialty chemical manufacturing. Enzymes offer several attractive synthetic advantages over traditional small molecules catalysts, such as high selectivities, low catalyst loadings, and a unique catalyst optimization strategy via protein engineering. Consequently, numerous examples exist of enzymes replacing traditional synthetic methods thereby increasing the efficiency of chemical manufacturing processes.1,2,3 While these are exciting developments for the field, it is also clear that there is still room for improvement, particularly in the realm of chemical reactivity, where biocatalysis is dwarfed by the sheer number of chemical reactions available to small molecule catalysts. This places certain limits on what can be made using solely a ‘biocatalytic retrosynthetic’ approach.4 For these reasons, there is great interest in exploring methods for augmenting the repertoire of chemistry that is currently possible using enzymes.5,6,7
Strategies for increasing the synthetic repertoire of biocatalytic reactions can be divided into two general approaches: discovery or design. The first aims to find new enzymes via reactions that occur in nature, typically in the context of natural product biosynthesis. By investigating enzymes within the biosynthetic gene clusters of natural products with unique chemical scaffolds, it is possible to discover new enzymatic reactions that can later be incorporated into a synthetic context.8,9,10 A notable example is the recent discovery of PLP-dependent enzymes catalyze the biosynthesis of alkynes in nature.11 The second strategy aims to build new reactivity into proteins, either via computational design or chemical principles. In this vein, a dominant approach has been to combine the known catalytic versatility of transition metal catalysis with enzymes either via the creation of artificial metalloenzymes or repurposing existent metalloenzymes.12,13,14 Using the enzyme as a scaffold for transition metal complexes, it becomes possible to carry out chemical transformations with selectivities and activities that would be impossible for either method on its own.15,16 An underexplored alternative is to devise strategies in which proteins lacking metal centers can catalyze reactions beyond their role in nature. While the best explored examples of this approach exploit hydrolases, studies in the last two years have demonstrated that other protein scaffolds can be used to similar effect. This review will cover these advances.
Computational Design with Directed Evolution
A longstanding goal in protein science is to design functional enzymes from scratch using only physical principles and computer guided modeling. While this remains a challenge, developments in high-throughput protein engineering via directed evolution have brought us much closer to that reality by producing new proteins than can start from a conceptual design in silico and quickly become useful catalysts on the lab bench. A recent example of this is in the engineering of a computationally designed retro-aldolase.17,18,19 By incorporating key catalytic residues, including a lysine required for Schiff-base formation, within a designed protein scaffold, a novel enzyme was achieved that provided low, but measurable activity, retro-aldolase function. The Hilvert and Griffiths groups exploited the power of directed evolution to take an initial hit and turn this into a robust catalyst. By using a high throughput microfluidic screening platform, they could to analyze ~2,000 variants per second (roughly 1 million times faster than conventional plate screening). After 13 rounds of engineering the researchers were able to arrive at an enzyme with a 3×107 fold rate enhancement, making the artificial aldolase competitive with naturally occurring enzymatic catalysts. In the course of evolution, the optimized aldolase (RA95.5–8F) developed an intricate catalytic tetrad (Lys/Arg/Tyr/Tyr) to facilitate proton transfers required for function (Figure 1a).
Figure 1.
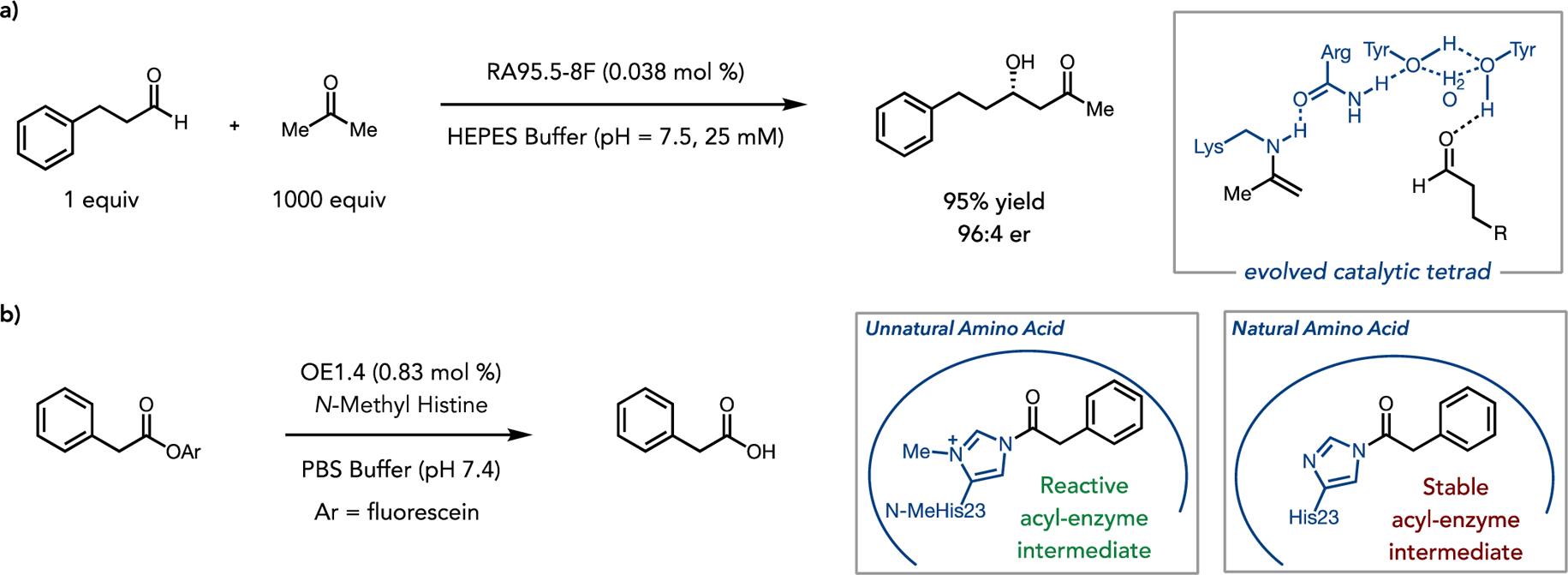
(a) Design and evolution of the artificial aldolase RA95.5–8F. Reactivity of the evolved enzyme (left) and the enzyme substrate interactions leading to enhanced reactivity (right).(b) Incorporation of a non-natural amino acid coupled with evolution leads to the creation of the novel esterase OE1.4. The unnatural amino acid facilitates hydrolysis of the acyl enzyme intermediate.
An alternative approach to bottom-up design of new enzyme function is to take advantage of unnatural amino acids (UAAs). Incorporation UAAs into proteins is an established tool for enzymologists to study enzyme structure-function relationships.20,21 Only recently has UAA incorporation technology emerged as a method for enhancing and expanding enzyme function.22,23 Green et al. recently used an unnatural histidine analogue, bearing the N δ-methyl histidine (NMH) moiety to participate directly in enzymatic catalysis, enabling an enzyme originally designed to catalyze a Morita-Baylis-Hillman reaction to catalyze ester hydrolysis (Figure 1b).24,25 Using NMH allows for the generation of a positively charged acyl enzyme intermediate, which rapidly undergoes hydrolysis, thereby enhancing what was previously the rate limiting step of the protein containing native histidine. The chemical design principles here mirror similar strategies that had been used in traditional organocatalysis, highlighting how strategies from small molecule catalysis can be used to inform and design improved biocatalysts.26,27 While the initial activity was modest, three rounds of directed evolution lead to a new biocatalyst with a 15-fold enhancement over the parent enzyme containing the unnatural amino acid and with kinetic characteristics on par with naturally occurring hydrolases. Importantly, NMH was crucial for this enzyme to remain highly active even after engineering, demonstrating the ability of directed evolution to augment non-natural functionality within the enzyme scaffold. This approach represents an emerging strategy by which novel enzymatic reactions may be discovered and rapidly improved upon using directed evolution. When this approach is applied to modes of reactivity with no parallel in nature, entirely new classes of biocatalysts can be designed and quickly applied to longstanding challenges in chemical synthesis.
Repurposing catalytic machinery
The ability of one enzyme to perform a variety of mechanistically distinct chemical transformations can broadly be classified as catalytic promiscuity.28 Hydrolases have been shown to be versatile catalysts in this regard, as these enzymes to be used to catalyze a variety of reactions via stabilizing anionic intermediates with their canonical oxy-anion hole.29,30 He et. al. has recently demonstrated that promiscuous lipase activity can be exploited in a concurrent reaction with photocatalysis to enable the direct enantioselective synthesis of 2,2-disubstituted indol-3-ones.31 Photocatalytic oxidation of 2-aryl indoles mediated by Ru(bpy)3 and O2 allow for the in situ generation of indole-3-ones, which can be bound and activated via protonation by wheat germ lipase (WGL). WGL then catalyzes non-natural C-C bond formation via addition of an enzymatically stabilized enolate into the newly generated iminium. This work constitutes the first example of using the non-natural activity of lipases with photocatalysis to enable an asymmetric synthesis (Figure 2a).
Figure 2.
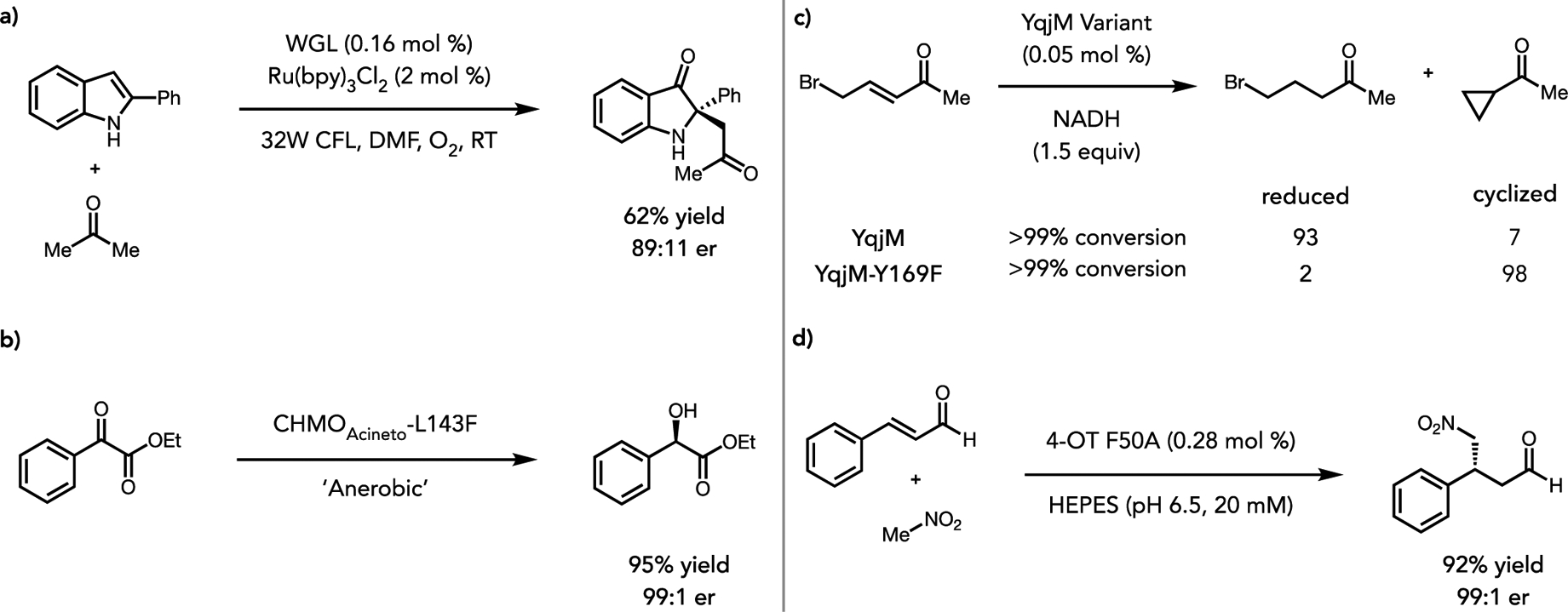
(a) Merger of photocatalysis with non-natural lipase reactivity using wheat germ lipase (WGL). (b) Cyclohexanone Monooxygenase (CHMO) catalyzed ketone reduction. (c) ‘Ene’-reductase (YqjM) catalyzed reductive carbocyclization. (d) 4-oxalocrotonate tautomerase (4-OT) catalyzed coupling of nitromethane with Michael acceptors via an iminium intermediate.
Xu et. al. recently showed that Baeyer–Villiger monooxygenases (BVMO), commonly used for oxidative biocatalytic reactions can be repurposed to perform highly selective carbonyl reductions.32 By excluding oxygen from the reaction, the typical BVMO pathway leading to the formation of the peroxyflavin cofactor is prevented, allowing for the reduced FAD cofactor to participate in hydride transfer chemistry instead. Cyclohexane monooxygenase from Acinetobacter sp. NCIMB 9871 (CHMOAcineto) was best suited for this promiscuous activity, reducing the model α-ketoester substrate with 99% e.e. albeit in low yield. Computational simulation and protein engineering were used to improve the non-natural carbonyl reduction, leading to a CHMO variant with an expanded active site and roughly 4-fold greater activity. This report highlights another way in which commonly used biocatalysts can be repurposed via cofactor versatility (Figure 2b).
Enolate Intermediates in ‘Ene’-Reductases
Another strategy for unlocking new biocatalytic functions is to leverage mechanistic understanding to divert well understood reactive intermediates toward new transformations. By understanding the innate reactivity of a given reaction intermediate, it becomes possible to use chemical principles to find new biocatalytic transformations. Recently, Breinbauer et al. demonstrated that carbocyclization reactions could be catalyzed by flavin-dependent ene-reductases (EREDs)33. In the natural mechanism, hydride is transferred from the flavin hydroquinone to the β-position of an activated olefin, resulting in the formation of an enolate that is rapidly protonated by a conserved tyrosine to furnish the reduced product.34 By mutating tyrosine to phenylalanine, the enolate intermediate can engage in an SN2 type reaction with pendant halides to afford chiral cyclopropanes with good yields and selectivities. By cleverly intercepting this well understood reaction pathway the researchers were able to enable this enzyme to catalyze a very different reaction than what would be seen observed in nature. Being able to reliably generate enolate species of this type could foreseeably open a path towards many other types of classic reactions (Figure 2c).
Iminium intermediates in 4-oxalocrotonate tautomerase
In nature, 4-oxalocrotonate tautomerase (4-OT) catalyzes an isomerization using an N-terminal proline as a base to deprotonate at the α-position of the natural substrate. Poelarends et. al. recognized that this proline could react with aldehydes and ketones to form an enamine, the intermediate responsible for some of the diverse reactivity available to amine organocatalysts.35 Recently, they found that 4-OT can also exploit iminium ions to catalyze the asymmetric addition of nitromethane to α,-unsaturated aldehydes.36 This method enabled the synthesis of chiral gamma nitro aldehydes, which are useful precursors to gamma amino butyric acids. This dual nature of nucleophilic catalysis mirrors strategies that were used in early organocatalysis.37 This enzyme provides a clear example of how a single enzymatic intermediate can allow access to a variety of new transformations (Figure 2d).
Radical Intermediates in ‘Ene’-Reductases
Unlocking new reactivity paradigms with natural redox cofactors is an emergent strategy for forming new reactive intermediates. In this context, flavin is an attractive because of its inherent ‘redox promiscuity’. Flavoenzymes are ubiquitous in biochemistry for using flavin cofactors (FMN or FAD) to function as either one or two electron reductants, dependent on the enzyme structure and biological context. In biocatalysis, however, they operate primarily in a closed shell 2-electron manifold when reacting with organic substrates (i.e. hydride transfers, or after the formation of hydroperoxy-flavin species).38 Single electron transfers (SET) to or from organic substrates to create open shell intermediates are less common and have not been used to access novel reactivity.39,40,41 As controlling the stereochemical outcome of radical reactions represents a long-standing challenge in asymmetric catalysis, if flavoenzymes can be used to catalyze these reactions it could have a significant impact on chemical synthesis. By using organic substrates with appropriate redox potentials, Sandoval et al. demonstrated that both FMN-dependent EREDs and FAD-dependent BVMO enzymes catalyze asymmetric dehalogenations that proceed via formation of open shell intermediates within their active sites.42 This reaction occurs via direct reduction of α-bromo esters (Ered=−750 mV vs saturated calomel electrode) by flavin hydroquinone located with the active site. Electron transfer is followed by rapid mesolytic cleavage of the substrate, forming Br− and an enzyme bound α-acyl radical. Notably, the substrate radical is quenched in a stereoselective manner to generate enantiomerically enriched α-aryl esters from racemic starting materials. Deuterium incorporation studies with the most selective enzyme support the hypothesis that after SET, the resultant neutral flavin semiquinone acts as a hydrogen atom source for the α-acyl radical (Figure 3a).
Figure 3.
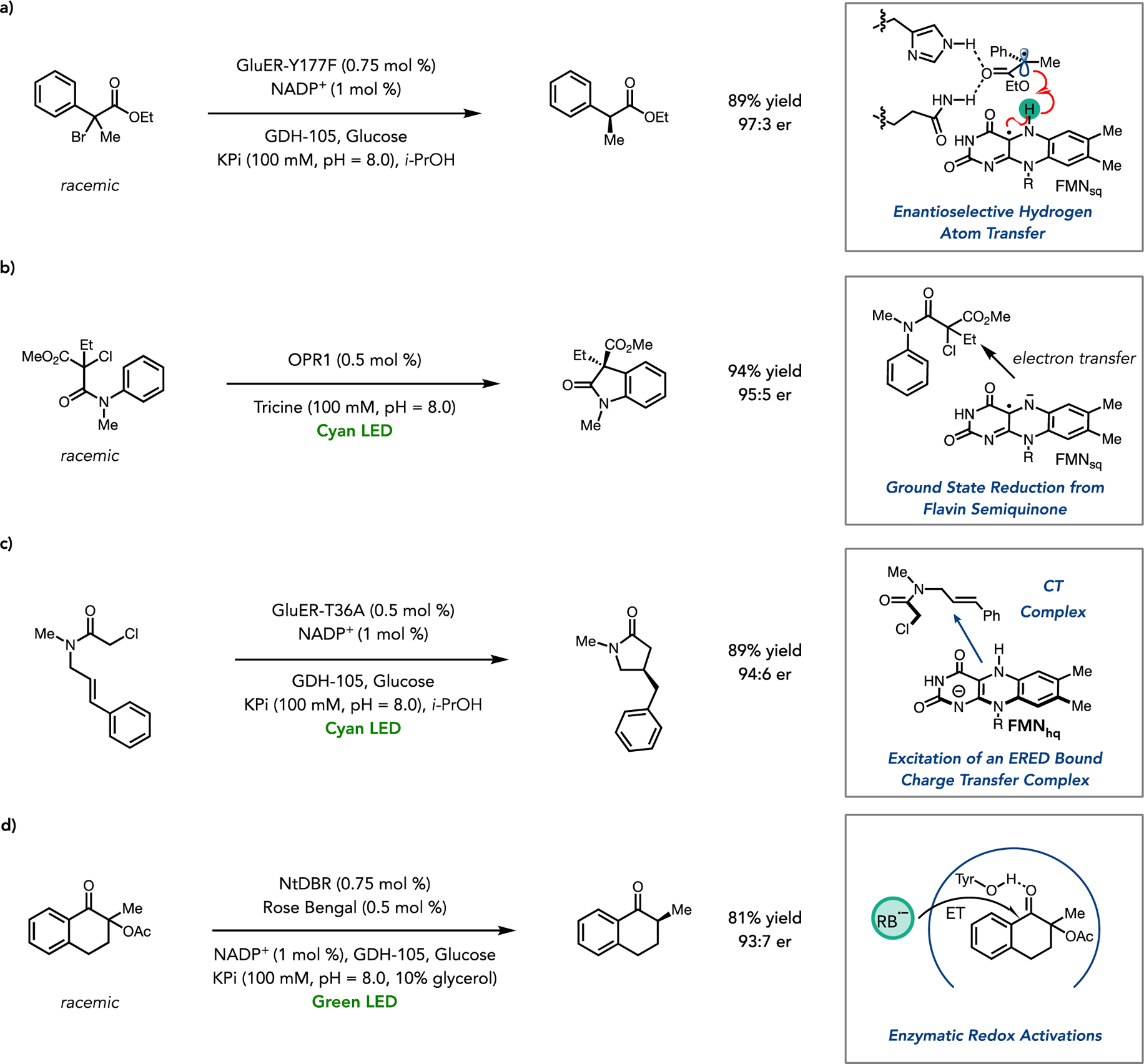
(a) ERED catalyzed radical dehalogenation using Gluconobacter oxydans ene-reductase (GluER). (b) Semiquinone enabled redox neutral radical cyclization with 12-oxophytodienoate reductase 1 (OPR1). (c) Photoinduced reductive cyclization with GluER. (d) Deacetoxylation via enzymatic redox activation using the NADPH dependent double bond reductase from Nicotiana tabacum (NtDBR).
Black et al. recently demonstrated that EREDs can also initiate radical reactions from the semiquinone oxidation state, resulting in a novel enzymatic C-C bond forming reaction.43 As demonstrated in pioneering work by Massey, shining visible light on OYE enzymes results in their photoreduction, which proceeds slowly through two sequential single electron transfers, with different enzymes stabilizing this intermediate to varying degrees.44,45 Consequently, by using light, it becomes possible to directly access the anionic flavin semiquinone and use it for catalysis. Using this strategy with EREDs enabled the catalytic asymmetric synthesis of oxindoles, via a redox neutral catalytic cycle. In this reaction the flavin semiquinone initiates the radical dehalogenation of enzyme bound α-chloro amides to form an α-acyl radical along with fully oxidized flavin. Unlike the semiquinone form of flavin, FMNox cannot function as a reductant allowing the substrate radical to cyclize onto a pendant arene, and upon oxidation of the resultant vinylogous amido radical, form product and regenerate the semiquinone. This further demonstrates how accessing new intermediates within well studied enzymes, has allowed access to atypical biocatalytic chemical transformations, namely radical C-C bond formation reactions (Figure 3b).
Recently studies have also demonstrated that EREDs can also function as photoenzymes. Biegasiewicz el al. found that these enzymes can catalyze the reductive cyclization of α-chloroamides to afford a variety of chiral lactams when irritated with cyan light.46 UV-vis analysis of the reduced enzyme in the presence of starting material results in the formation of a new absorption feature with a maximum absorption at 500 nm, corresponding to the formation of a charge transfer complex between the substrate and the flavin hydroquinone. Importantly, this species is only observed in the presence of the ERED, indicating the protein’s importance in gating electron transfer. In many respects, this mechanism mirrors the one responsible for enabling nicotinamide-dependent KREDs to catalyze the dehalogention of α-bromo lactones.47 As the cyclization occurs within the active site, the enzyme controls the facial selectivity for the radical addition into the olefin as well as the terminal hydrogen atom transfer event, both steps being a challenge for small molecule catalysts (Figure 3c).
Radical Intermediates via Enzymatic Redox Activation
Substrate binding to enzyme active sites often involve the formation of hydrogen bonds, where the protein serves as a hydrogen bond donor while the substrate serves as the acceptor. The net effect of this interaction is to render the substrate more electrophilic, enabling the enzyme’s native reactivity. The Hyster group recognized that the same interactions would also make substrates easier to reduce by exogenous single electron reductants. By coupling this ‘enzymatic redox activation’ (ERA) with photocatalysts, it would be possible to generate radicals from less activated substrates exclusively within enzyme active sites. This allows access to reduction potentials that would exceed what is possible with biological cofactors alone. This concept has been demonstrated now in three different classes of enzyme and may prove to be a general solution to achieving new radical mediated transformations in enzyme active sites (Figure 3d).
In an initial study, Biegasiewicz et al. demonstrated an enantioselective deacetoxylation of tetralones derivatives using the photocatalytic dye rose bengal with the double bond reductase NtDBR.48 Hydrogen bonding interactions in the active site are used in the natural reaction pathway to orient and activate the substrate for hydride transfer from NADPH. Binding in this way however also results in an increase the substrate’s reduction potential by circa 157 mV as calculated by DFT. This allows for the enzyme bound substrate to be reduced by an external photoredox catalyst more readily than the free substrate, creating an enzyme bound radical which is quenched via enantioselective hydrogen atom transfer from the NADPH cofactor. This reactivity was also generalized to other NADPH dependent enzymes such as KREDs, allowing for radical dehalogenation of α-bromo amides. In both cases, the enzyme plays a crucial role in accelerating the transformation, preventing undesired racemic background reactions and therefore maintaining high enantioselectivity.
Enzymatic redox activation was also found to compatible with flavin dependent EREDs. Using light with Ru(bpy)3Cl2 as a reductant enabled the photoenzymatic reduction of acetophenone to the corresponding enantioenriched alcohols, constituting the first example of this type of reactivity using the flavin cofactor.49 Notably binding to the enzyme is essential for this reaction to occur, and no product is formed using the photocatalyst alone. It was also demonstrated that the native reactivity of the biocatalyst is not lost during the transformation, allowing the enzyme to perform concurrent natural and unnatural reactions, in order to achieve new reaction products that would be inaccessible to either the enzyme of photocatalyst alone.
Conclusions
With the recent and rapid growth of both computational power and biotechnology, biocatalysis appears to be well poised to reap the benefits of both. Synthetic chemists will no doubt continue to incorporate biocatalytic steps and will benefit from novel retrosynthetic disconnections with high efficiencies and selectivities. With an increasing emphasis on new reactivity in biocatalysis, it appears future chemists might have a much larger pool of biocatalytic reactions to choose from. We anticipate that directed evolution will continue to be a very important tool for protein designers to take initial ‘hits’ and turn them into full-fledged biocatalysts. Flavoenzymes, in particular, look to be emergent scaffolds for finding new chemistry owing to their ability to bind a multitude of organic substrates and stabilize both anionic and radical intermediates. The recent demonstration of these enzymes abilities to catalyze asymmetric C-C bond forming reactions with little to no mutations present opportunities for further development of these enzymes into versatile catalysts.
Acknowledgements
Financial support was provided by the NIHGMS (R01 GM127703), Searle Scholar Award (SSP-2017-1741), the Alfred P. Sloan Foundation, and Princeton University.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Choi JM, Han SS, Kim HS, Industrial applications of enzyme biocatalysis: current status and future aspects, Biotechnol. Adv 33 (2015) 1443–1454. [DOI] [PubMed] [Google Scholar]
- 2.Adams JP, Brown MJB, Diaz-Rodriguez A, Lloyd RC, Roiban G, Biocatalysis: A Pharma Perspective, Adv. Synth. Catal 361 (2019) 2421. [Google Scholar]
- 3.Sheldon RA, Woodley JM, Role of Biocatalysis in Sustainable Chemistry, Chem. Rev 118 (2018) 801–838. [DOI] [PubMed] [Google Scholar]
- 4.Turner NJ, O’Reily E, Biocatalytic retrosynthesis, Nat. Chem. Biol 9 (2013) 285–288. [DOI] [PubMed] [Google Scholar]
- 5.Bornscheuer UT, Kazlauskas RJ, Catalytic promiscuity in biocatalysis: using old enzymes to form new bonds and follow new pathways, Angew. Chem. Int. Ed 43 (2004) 6032–6040. [DOI] [PubMed] [Google Scholar]
- 6.Renata H, Wang ZJ, Arnold FH, Expanding the enzyme universe: accessing non-natural reactions by mechanism-guided directed evolution, Angew. Chem. Int. Ed 54 (2015) 3351–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arnold FH, Directed evolution: bringing new chemistry to life, Angew. Chem. Int. Ed 57 (2018) 4143–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hager HH, Balskus EP, Biocatalytic Friedel–Crafts alkylation using a promiscuous biosynthetic enzyme, Angew. Chem. Int. Ed 58 (2019) 3151–3155. [DOI] [PubMed] [Google Scholar]
- 9.McKinnie SMK, Miles ZD, Jordan PA, Awakawa T, Pepper HP, Murray LAM, George JH, Moore BS, Total enzyme syntheses of napyradiomycins A1 and B1, J. Am. Chem. Soc 140 (2018) 17840–17845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dockrey SAB, Lukowski AL, Becker MR, Narayan ARH, Biocatalytic site- and enantioselective dearomatization of phenols, Nat. Chem 10 (2018) 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marchand JA, Neugubauer ME, Ing MC, Lin C-I, Pelton JG, Chang MCY, Discovery of a pathway for terminal-alkyne amino acid biosynthesis, Nature 567 (2019) 420–424. [DOI] [PubMed] [Google Scholar]
- 12.McIntosh JA, Farwell CC, Arnold FH, Expanding P450 catalytic reaction space through evolution and engineering, Curr. Opin. Chem. Biol 19 (2015) 126–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwizer F, Okamoto Y, Heinisch T, Gu Y, Pellizzoni MM, Lebrun V, Reuter R, Köhler V, Lewis JC, Ward TR, Artificial metalloenzymes: reaction scope and optimization strategies, Chem. Rev 118 (2018) 142–231. [DOI] [PubMed] [Google Scholar]
- 14.Lewis JC, Artificial metalloenzymes and metallopeptide catalysts for organic synthesis, ACS Catal. 3 (2013) 2954–2975. [Google Scholar]
- 15.Davis HJ, Ward TR, Artificial Metalloenzymes: challenges and opportunities. ACS Cent. Sci 5 (2019) 1120–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang K, Huang X, Arnold FH, Selective C-H bond functionalization with engineered heme proteins: new tools to generate complexity. Curr. Opin. Chem. Biol 49 (2019) 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang L, Althoff EA, Clemente FR, Doyle L, Röthlisberger DA, Zanghellini A, Gallaher JL, Betker JL, Tanaka F, Barbas III CF, Hilvert D, Houk KN, Stoddard BL, Baker D. De novo computational design of retro-aldol enzymes. Science 7 (2008) 1387–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giger L, Caner S, Obexer R, Kast P, Baker D, Ban N, Hilvert D, Evolution of a designed retro-aldolase leads to complete active site remodeling. Nat. Chem. Biol 9 (2013) 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **19.Obexer R, Godina A, Garrabou X, Mittl PRE, Baker D, Griffiths AD, Hilvert D, Emergence of a catalytic tetrad during evolution of a highly active artificial aldolase. Nat. Chem 9 (2017), 50–56. [DOI] [PubMed] [Google Scholar]; Highthroughput screening enables a computationally designed enzyme with low activity to become as active as naturally occurring enzymes.
- 20.Xie J, Schultz PG, A chemical toolkit for proteins – an expanded genetic code. Nat. Rev. Mol. Cell Biol 7 (2006) 775–782. [DOI] [PubMed] [Google Scholar]
- 21.Torrice MM, Bower KS, Lester HA, Dougherty DA, Probing the role of the cation-π interaction in the binding sites of GPCRs using unnatural amino acids. Proc. Natl. Acad. Sci 106 (2009) 11919–11924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewis JC, Metallopeptide catalysts and artificial metalloenzymes containing unnatural amino acids. Curr. Opin. Chem. Biol 25 (2015) 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hayashi T, Hilvert D, Green AP, Engineered metalloenzymes with non-canonical coordination environments, Chem. Eur. J 24 (2018), 11821–11830. [DOI] [PubMed] [Google Scholar]
- 24.Bjelic S, Nivón LG, Çelebi-Ölçüm N, Kiss G, Rosewall CF, Lovick HM, Ingalls EL, Gallaher JL, Seetharaman J, Lew S, Montelione GT, Hunt JF, Michael FE, Houk KN, Baker D, Computational design of enone-binding proteins with catalytic activity for the Morita–Baylis–Hillman reaction, ACS Chem. Biol 8 (2013), 749–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *25.Burke AJ, Lovelock SL, Frese A, Crawshaw R, Ortmayer M, Dunstan M, Levy C, Green AP. Design and evolution of an enzyme with a non-canonical organocatalytic mechanism. Nature 570 (2019) 219–223. [DOI] [PubMed] [Google Scholar]
- 26.Miller SJ, In search of peptide-based catalysts for asymmetric organic synthesis. Acc. Chem. Res 37 (2004) 601–610. [DOI] [PubMed] [Google Scholar]
- 27.Fance S, Guerin DJ, Miller SJ, Leckta T, Nucleophilic chiral amines as catalysts in asymmetric synthesis. Chem Rev. 103 (2003) 2985–3012. [DOI] [PubMed] [Google Scholar]
- 28.López-Iglesias M, Gotor-Fernández V, Recent advances in biocatalytic promiscuity: hydrolase-catalyzed reactions for nonconventional transformations, Chem. Rec 15 (2015) 743–759. [DOI] [PubMed] [Google Scholar]
- 29.Busto E, Gotor-Fernández V, Gotor V, Hydrolases: catalytically promiscuous enzymes for non-conventional reactions in organic synthesis. Chem. Soc. Rev 39 (2010) 4504–4523. [DOI] [PubMed] [Google Scholar]
- 30.Guan Z, Li L, He Y, Hydrolase-catalyzed asymmetric carbon-carbon bond formation in organic synthesis. RSC Adv. 5 (2015), 16801–16814. [Google Scholar]
- 31.Ding X, Dong C, Guan Z, He Y, Concurrent asymmetric reactions combining photocatalysis and enzyme catalysis: direct enantioselective synthesis of 2,2-disubstitued indol-3-ones from 2-arylindoles, Angew. Chem. Int. Ed 58 (2019) 118–124. [DOI] [PubMed] [Google Scholar]
- 32.Xu J, Peng Y, Wang Z, Hu Y, Fan J, Zheng H, Lin X, Wu Q., Exploiting cofactor versatility to convert FAD-dependent Baeyer-Villiger monooxygenase into a ketoreductase, Angew. Chem. Int. Ed (2019) doi: 10.1002/anie.201907606. [DOI] [PubMed] [Google Scholar]
- 33.Heckenbichler K, Schweiger A, Brandner LA, Binter A, Toplak M, Macheroux P, Gruber K, Breinbauer R, Asymmetric reductive carbocyclization using engineered ene reductases, Angew. Chem. Int. Ed 57 (2018) 7240–7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kohli RM, Massey VJ, The oxidative half-reaction of old yellow enzyme the role of tyrosine 196, J. Biol. Chem 273 (1998) 32763–32770. [DOI] [PubMed] [Google Scholar]
- 35.Poddar H, Rahimi M, Geertsema EM, Thunnissen AW, Poelarends GJ, Evidence for the formation of an enamine species during aldol and Michael‐type addition reactions promiscuously catalyzed by 4‐oxalocrotonate tautomerase. ChemBioChem, 16 (2015) 738–741. [DOI] [PubMed] [Google Scholar]
- 36.Guo C, Saifuddin M, Saravanan T, Sharifi M, Poelarends GJ, Biocatalytic asymmetric michael additions of nitromethane to α,β-unsaturated aldehydes via enzyme-bound iminium ion intermediates, ACS Catalysis 9 (2019) 4369–4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ahrendt KA, Borths CJ, MacMillan DWC, New strategies for organic catalysis: the first highly enantioselective organocatalytic Diels-Alder reaction, J. Am. Chem. Soc 122 (2000), 4243–4244. [Google Scholar]
- 38.B Dockrey SA, Narayan ARH, Flavin-dependent biocatalysts in synthesis, Tetrahedron 75 (2019) 1115–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu J, Chuenchor W, Rokita SE, A switch between one- and two-electron chemistry of the human flavoprotein iodotyrosine deiodinase is controlled by substrate, J. Biol. Chem 290 (2015), 590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamano T, Kuroda K, Fujii S, Miura R, Characterization of the electron acceptors of old yellow enzyme: mechanistic approach to the mode of one electron transfer from the enzyme to menadione or dyestuffs, J Biochem. 114 (1993) 879–884. [DOI] [PubMed] [Google Scholar]
- 41.Silverman RB, Zieske PA, Mechanism of inactivation of monoamine oxidase by 1-phenylcyclopropylamine. Biochemistry 1985. 24 (9), 2128–2138. [DOI] [PubMed] [Google Scholar]
- *42.Sandoval BA, Meichan AJ, Hyster TK, Enantioselective hydrogen atom transfer: discovery of catalytic promiscuity in flavin-dependent ‘ene’-reductases. J. Am. Chem. Soc 139 (2017), 11313–11316. [DOI] [PubMed] [Google Scholar]
- 43.Black MJ, Biegasiewicz KF, Meichan AJ, Oblinsky DG, Kudish B, Scholes GD, Hyster TK, Asymmetric redox-neutral radical cyclization catalyzed by flavin-dependent ‘ene’-reductases. Nat. Chem (2019) doi: 10.1038/s41557-019-0370-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Massey VJ, Stankovich M, Hemmerich P, Light-mediated reduction of flavoproteins with flavins as catalysts. Biochemistry 17 (1978) 1–8. [DOI] [PubMed] [Google Scholar]
- 45.Massey VJ, Hemmerich P, Knappe WR, Duchstein HJ, and Fenner H. Photoreduction of flavoproteins and other biological compounds catalyzed by deazaflavins. appendix: photochemical formation of deazaflavin dimers. Biochemistry 17 (1978) 9–17. [PubMed] [Google Scholar]
- **46.Biegasiewicz KF, Cooper SJ, Gao X, Oblinsky DG, Kim JH, Garfinkle SE, Joyce LA, Sandoval BA, Scholes GD, Hyster TK, Photoexcitation of flavoenzymes enables a stereoselective radical cyclization. Science 21 (2019) 1166–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]; EREDs enable the asymmetric synthesis of chiral lactams using a radical mechanism. Light is used to initiate the radical chemistry within the enzyme active site.
- 47.Emmanuel MA, Greenberg NR, Oblinsky DG, Hyster TK, Accessing non-natural reactivity by irradiating nicotinamide-dependent enzymes with light. Nature 540 (2016) 414–417. [DOI] [PubMed] [Google Scholar]
- 48.Biegasiewicz KF, Cooper SJ, Emmanuel MA, Miller DC, Hyster TK. Catalytic promiscuity enabled by photoredox catalysis in nicotinamide-dependent oxidoreductases. Nat. Chem 10 (2018)770–775. [DOI] [PubMed] [Google Scholar]
- 49.Sandoval BA, Kurtoic SI, Chung MM, Biegasiewicz KF, Hyster TK, Angew. Chem. Int. Ed 2019, 58, 8714. [DOI] [PMC free article] [PubMed] [Google Scholar]