

Abstract
INTRODUCTION:
Late-onset Alzheimer’s Disease (LOAD) manifests comorbid neuropsychiatric symptoms and posttraumatic stress disorder (PTSD) is associated with an increased risk for dementia in late-life, suggesting the two disorders may share genetic etiologies.
METHODS:
We performed genetic pleiotropy analysis using LOAD and PTSD GWAS datasets from Whites and African-American populations, followed by functional-genomic analyses.
RESULTS:
We found an enrichment for LOAD across increasingly stringent levels of significance with the PTSD GWAS association (LOAD|PTSD) in the discovery and replication cohorts and a modest enrichment for the reverse conditional association (PTSD|LOAD). LOAD|PTSD association analysis identified and replicated the MS4A genes region. These genes showed similar expression pattern in brain regions affected in LOAD, and across-brain-tissue analysis identified a significant association for MS4A6A. The African-American samples showed moderate enrichment, however, no FDR-significant associations.
Keywords: Late-onset Alzheimer’s Disease, Post traumatic stress disorder, PTSD, genetic pleiotropy, inflammation and immune-based pathways and Alzheimer’s disease, MS4A gene family
1. Introduction
Patients with late-onset Alzheimer’s Disease (LOAD) frequently manifest comorbid neuropsychiatric symptoms (NPS) and related conditions with depression and anxiety being most prevalent1–4. Posttraumatic stress disorder (PTSD) is one neuropsychiatric condition that is also highly comorbid with depression and anxiety and has been shown to prospectively increase the risk for developing dementia5. However, cross-sectional epidemiological studies have been unable to clarify the magnitude of the link between PTSD and dementia due to insufficient data6. While combat-related PTSD usually occurs early in life (ages 20–40 years) LOAD is a disease of late onset, ages greater than 65 years. Epidemiological data has shown that young individuals who are susceptible to psychiatric diseases including PTSD have an increased risk for LOAD as they age5,7,8. Therefore, genetic variation common to these two diseases could provide insights about the neuropsychiatric symptoms that are common to both and may be predisposing genetic factors for both diseases.
LOAD and PTSD are diseases that develop as a consequence of genetic and environmental factors. More than 30 genetic loci have been identified as associated with LOAD from a combination of linkage, GWAS and whole genome/exome sequencing9. The most recent 3-stage GWAS meta-analysis with 35,724 clinically diagnosed LOAD cases and 59,163 controls identified 25 LOAD risk loci (20 previously identified loci and 5 novel loci) and implicated pathways including immune-based, lipid metabolism, tau binding proteins and APP metabolism10. Genetic factors involved in susceptibility to PTSD have been identified in GWAS studies11,12 and in studies of male twins13. The first large GWAS results from the Psychiatric Genomics Consortium (PGC) GWAS of PTSD was published in 201811 and showed that no SNPs in this study exceeded genome-wide significance in a transethnic meta-analysis. Noteworthy, the results showed strong evidence for overlapping genetic risk between PTSD and schizophrenia and moderate evidence for overlapping genetic risk between PTSD and bipolar and major depressive disorder11. Here we aimed to examine whether overlap in the genetic etiologies underlying PTSD and LOAD, specifically to identify pleiotropic genetic variants, genes and biochemical pathways that comprise a shared molecular phenotype for LOAD and PTSD. Towards this goal we utilized genome-wide pleiotropy analysis, an approach that has been widely used to examine genetic pleiotropy between multiple diverse diseases and phenotypes including neuropsychiatric conditions and neurological diseases14–18,19–25. Our results provided clues for shared molecular phenotypes that are responsible, at least in part, for the heterogeneity of NPS symptoms in LOAD.
2. Materials and methods
2.1. GWAS datasets for the Discovery sample
GWAS summary statistics were obtained from publicly-accessible web sites for the LOAD GWAS and the PTSD GWAS (see Data Availability). Descriptive statistics for all of the GWASs used in this study are provided in Table 1.
Table 1.
Descriptive characteristics of the LOAD and PTSD samples.
| Disease/Trait | Phase | n | SNPs, n | Reference |
|---|---|---|---|---|
| LOAD, IGAP stage 1 | Discovery | 63,976 (21,982 LOAD cases, 41,994 controls) | 11,480,632 | Kunkle10 |
| PTSD, PGC (European American) | Discovery | 9,537 (2424 PTSD cases, 7,113 controls) | 13,207,411 | Duncan11 |
| PTSD, STRONG STAR | Replication | 2,056 (1,011 PTSD cases, 1,045 controls) | 5,086,096 | Nievergelt12 |
| LOAD, African American | Discovery | 5,896 (1,968 LOAD cases, 3,928 controls) | 17,332,474 | Reitz28 |
| PTSD, African American | Discovery | 9,223 (2,479 PTSD cases, 6,744 controls) | 21,225,122 | Duncan11 |
The LOAD GWAS dataset consisted of summary statistics of p values, beta coefficients and standard errors, effect alleles from the International Genomics of Alzheimer’s Disease Project (IGAP)10. IGAP is a large three-stage study based upon GWAS on individuals of European ancestry. Component studies include The Alzheimer Disease Genetics Consortium (ADGC); The European Alzheimer’s disease Initiative (EADI); The Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium (CHARGE); and The Genetic and Environmental Risk in AD Consortium Genetic and Environmental Risk in AD/Defining Genetic, Polygenic and Environmental Risk for Alzheimer’s Disease Consortium (GERAD/PERADES). Individuals with non-European ancestry according to principal components analysis of ancestry-informative markers were excluded from the primary GWAS analysis10.
The PTSD GWAS dataset consisted of summary statistics of p values, odds ratios and standard errors, reference allele, imputation quality score (INFO) and direction of effect in each cohort from the Psychiatric Genomics Consortium (PGC)11. Since the LOAD GWAS results were obtained from a sample that was limited to European ancestry, the PGC PTSD European ancestry dataset was used. This dataset consisted of individuals from 7 different cohorts as described in Duncan et al.11
The chromosome 19 region containing the APOE gene is well-recognized as a strong genetic risk factor for LOAD, with GWAS association p values on order of 10−238 to 10−12 in the IGAP discovery dataset. Since the objective of the study was to find pleiotropic variants for LOAD and PTSD and the PTSD datasets did not have any variants with the strong level of APOE association, variants in the APOE region on chromosome 19, defined as ±300Kb of the APOE epsilon coding SNPs were excluded from all datasets prior to analysis. The excluded region was chr19:45,111,942 – 45,711,941. Secondary analyses were performed including this region to assess the effect of the APOE SNPs.
Restricting variants to SNPs with high quality imputation scores (0.6≤INFO<1.06) resulted in a total of 9,154,389 SNPs in common for the two datasets (LOAD and PTSD) that were used for further analysis. Details for the genotyping procedure, quality control and GWAS analysis are provided in the primary manuscripts for the LOAD GWAS10 and the PTSD GWAS26. All genomic coordinates are based on NBCI Build 37/UCSC hg19.
2.2. Genome-wide association analysis for the replication sample
The replication sample for PTSD consisted of summary statistics of p values, beta coefficients and standard errors, effect alleles from the “Genetic and Environmental Predictors of Combat-Related PTSD” study (Douglas E. Williamson - PI). As part of the STRONG STAR Consortium (https://tango.uthscsa.edu/strongstar/research.asp) this study recruited 4,123 soldiers prior to deployment and followed over 2,200 post-deployment assessing lifetime psychiatric disorders including PTSD. For the present study, sample sizes were 1011 PTSD cases and 1045 trauma exposed controls. The Illumina PsychArray was used to genotype 593,260 markers and imputation was performed to constitute a set of 5,086,096 SNPs using the 1,000 Genomes Reference Panel, phase 3 in IMPUTE2). Genotype quality control and genome-wide association analysis is described in Nievergelt et al.27
2.3. GWAS dataset for the African American LOAD and PTSD samples
The Alzheimer’s Disease Genomics Consortium (ADGC) GWAS for Alzheimer’s disease28 and the PGC GWAS for PTSD11 in African American Individuals was used to study genetic pleiotropy between LOAD and PTSD in African Americans.
2.4. Pleiotropy analysis
The pleiotropy analysis strategy, based on conditional false discovery rates, fold-enrichment plots and conditional quantile-quantile (Q-Q) plots is described in detail elsewhere14,24 In brief, for two phenotypes A and B, pleiotropic enrichment of phenotype A conditional on phenotype B exists if the proportion of variants (SNPs) statistically significantly associated with phenotype A increases as a function of increased statistically significant SNP associations with phenotype B. The number of SNPs associated with phenotype A was determined for several thresholds of SNP association with phenotype B; the proportions are calculated relative to a baseline of all SNPs statistically significantly associated with phenotype A. For this study, the analysis was run in both directions, with phenotype A, the primary phenotype, late-onset Alzheimer’s disease (LOAD) and phenotype B, the conditional phenotype, PTSD followed by interchange of the primary and conditional phenotypes. Fold enrichment plots graphically depict pleiotropy by showing the relationship between empirical quantiles of nominal −log10(p) values for SNP association with the primary phenotype (LOAD or PTSD) for all SNPs relative to subsets of SNPs stratified by the nominal −log10(p) values for their association with the secondary phenotype. Specifically, the fold enrichment was calculated from the empirical cumulative distribution function (ECDF) of nominal p-values for a subset of SNPs with significance levels below the indicated cut-offs for the secondary phenotypes relative to the ECDF for all SNPs.
| (1) |
| (2) |
Enrichment for pleiotropy was detected if the deflection of the curve from the expected null distribution (horizontal line at 1-fold) was dependent on the association with the secondary phenotype. The nominal p-values −log10(p) for the primary phenotype are plotted on the x-axis, and fold enrichment for the primary phenotype as a function of the secondary phenotype is plotted on the y-axis.
Conditional quantile-quantile plots for the same data shown in the fold enrichment plots provided additional assessment of genetic pleiotropy for each set of GWAS results. Following the prior analysis strategy24, we focused the analysis for polygenic enrichment on SNPs below the standard GWAS Bonferroni-corrected p-value thresholds for association with cognitive impairment by using subsets of SNPs with a nominal −log10(p) < 9.0.
For identification of specific SNPs associated with LOAD conditional with an association with PTSD and/or SNPs associated with PTSD conditionally associated with LOAD, a conditional false discovery rate (cFDR) statistic was calculated as described in the prior implementation of this analysis strategy14,24 and other publications16–20,29,30. This framework was an extension of the standard analysis for FDR calculations and uses information from the secondary phenotype to re-rank the p-values for the primary phenotype. Of note, only GWAS summary statistics are required to calculate the cFDR and there is no requirement that the samples for the two phenotype are the same. For a set of SNPs, the FDR was interpreted as the probability that a SNP association with a phenotype was null given that the observed p value was as small or smaller than its observed P value. Ranking p values from a GWAS by observed p value or FDR would provide the same order by magnitude. The conditional FDR was defined as the probability that a SNP association was null in the primary phenotype given that the p values in both the primary and secondary phenotypes were as small or smaller than the observed p values. If the primary and secondary phenotypes were genetically-related, the cFDR reordered the SNPs with a different ranking than the order for the primary phenotype alone24. The value of the conditional FDR for each SNP was calculated in the case where LOAD is the principal phenotype conditioned on PTSD (LOAD|PTSD) as well as the reverse (PTSD|LOAD). We used a conditional FDR of Q<0.05 to show statistical significance. The significance threshold of Q = 0.05 for the conditional FDR31 corresponds to 5 false positives per 100 reported associations. Manhattan plots and stratified quantile-quantile (Q-Q) plots of the conditional FDRs for were used to summarize the data.
In order to detect common susceptibility loci for LOAD and PTSD after calculating the conditional FDRs (Q values) for each SNP under LOAD|PTSD and PTSD|LOAD, we computed the conjunction conditional FDR, which refers to the probability that a given SNP is null for both phenotypes when the p-values are less than the observed p values. The conjunction conditional FDR is the maximum value of the two conditional FDR (Q) values.
2.5. Functional genomics bioinformatics analysis
Functional bioinformatics analysis was performed to evaluate the biological significance of the SNPs that were found to be significantly associated with LOAD, conditional on association with PTSD. Three bioinformatics analysis tools were used to map the SNPs to genes by proximity, define the genomic context for the variant, annotate effects on phenotypes and identify relevant literature about the variant. The UCSC genome browser (http://genome.ucsc.edu/) was used to map each variant to proximate genes and to provide the first level of information about the genes and biological consequences of the genes32. The Variant Effect Predictor (VEP) that is part of the Ensembl genome database project (http://www.ensembl.org) was used to provide information about the effects of the SNPS on genes, transcripts and regulatory regions33. SNPnexus (http://www.snp-nexus.org/) was used to provide additional annotation on gene/protein consequences and phenotype- and disease- association for the variants34,35.
Gene set enrichment and pathway analysis was completed using i-Gsea4Gwas36. This analysis was run on all SNP association results for the IGAP discovery dataset and the PTSD replication dataset using the FDR q values Q(LOAD|PSTD). The SNP to gene mapping was limited to 500kb upstream and downstream of the gene. Candidate gene sets included canonical pathways, GO biological processes and GO molecular function.
Gene expression analysis and clustering was performed using the GENE2FUNC capability of the FUMA GWAS analysis suite37. Gene expression data was from GTEx v6, 53 tissue types38,39. The unified test for molecular signatures, UTMOST40, was utilized for statistical analysis of cross-brain tissue expression associated with SNPs identified as FDR significant from the pleiotropy analysis.
3. Results
3.1. Genome-wide association summary results for LOAD and PTSD
Prior to assessment of polygenetic overlap between LOAD and PTSD, the individual genome-wide association studies for each phenotype were compared for quality control and overall genetic association statistics. For the discovery sample, these results are reported in the primary publications for IGAP LOAD10 and for the Psychiatric Genomics Consortium PTSD11 GWAS. Genomic inflation was well controlled in both of these GWAS and the minor allele frequencies were limited to MAF > 0.01. For the replication sample, the supplement contains a Manhattan plot (Supplementary Fig. S1) and a q-q plot (Supplementary Fig. S2).
This study tested for polygenic effects below the standard GWAS significance threshold. Inspection of the Manhattan plots (Supplementary Fig. S1) show several regions of the genome with nominal levels of association (P≤1×10−5) for the different phenotypes. The q-q plots (Supplementary Fig. S2) showed that population stratification was accounted for in the association analysis.
3.2. Assessment of polygenic overlap between LOAD and PTSD
3.2.1. Discovery sample.
The fold enrichment plot demonstrated moderate (2.0 – 3.0 fold) enrichment for SNP association with LOAD across increasingly stringent levels of significance for SNP association with PTSD (Q(LOAD|PTSD)) (Fig. 1A). The reverse conditional association (Q(PTSD|LOAD)) showed enrichment of approximately 2.0 – 3.5 fold (Fig. 1B). These results support a moderate level of polygenic overlap between LOAD and PTSD.
Fig 1.
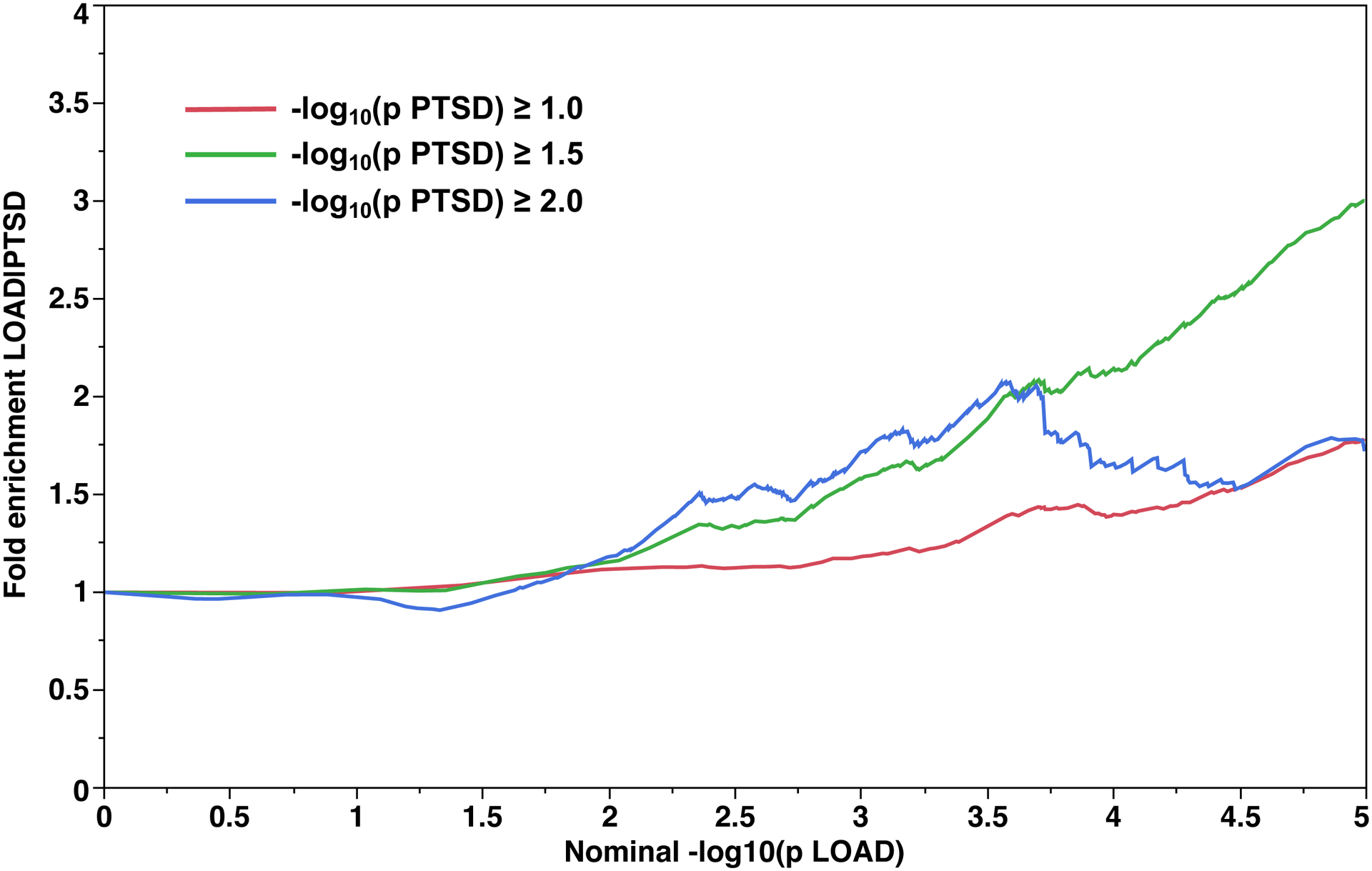

Fold-enrichment plots for the Discovery sample. Ordinate is fold-enrichment. Fig. 1A: Abscissa is nominal −log10(p) for SNP association with LOAD, curves are differentiated by the threshold for level of statistical significance for SNP association with the secondary phenotype (PTSD). Fig. 1B: Abscissa is nominal −log10(p) for SNP association with PTSD, curves are differentiated by the threshold for level of statistical significance for SNP association with the secondary phenotype (LOAD).
3.2.2. Replication sample.
The fold enrichment plot (Fig. 2A) demonstrates moderate (2.2 – 7.5 fold) enrichment for SNP association with LOAD across increasingly stringent levels of significance for SNP association with PTSD (Q(LOAD|PTSD)). The reverse conditional association Q(PTSD|LOAD) showed enrichment of approximately 1.3 – 1.8 fold (Fig. 2B). The lower fold enrichment seen for this direction of primary vs. secondary phenotypes is a consequence of the stronger genetic associations for LOAD (p ≥ 6.54×10−18) than for PTSD (p ≥ 1.83×10−8). These results support a moderate level of polygenic overlap between LOAD and PTSD.
Fig 2. Fold-enrichment plots for the Replication sample.
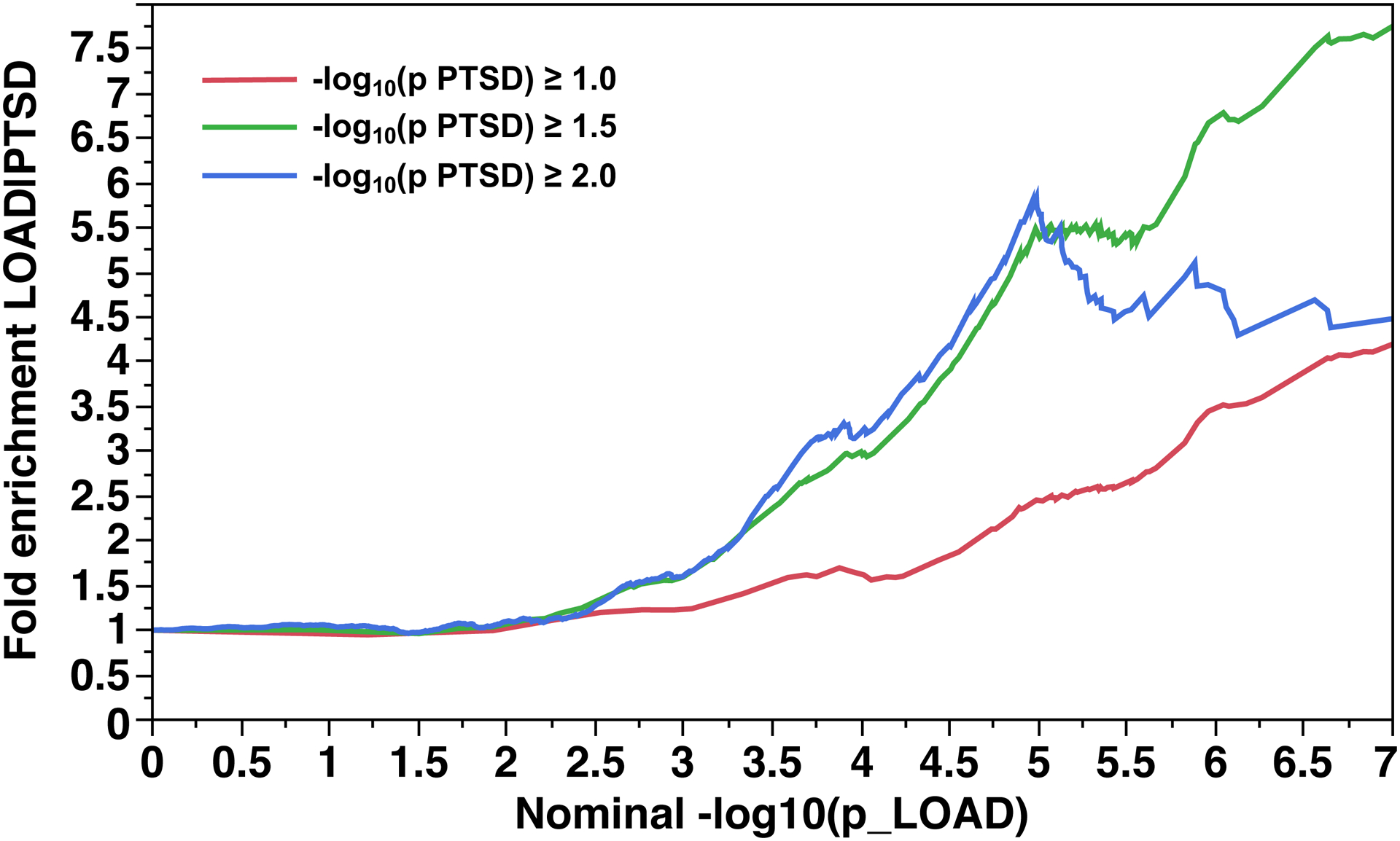
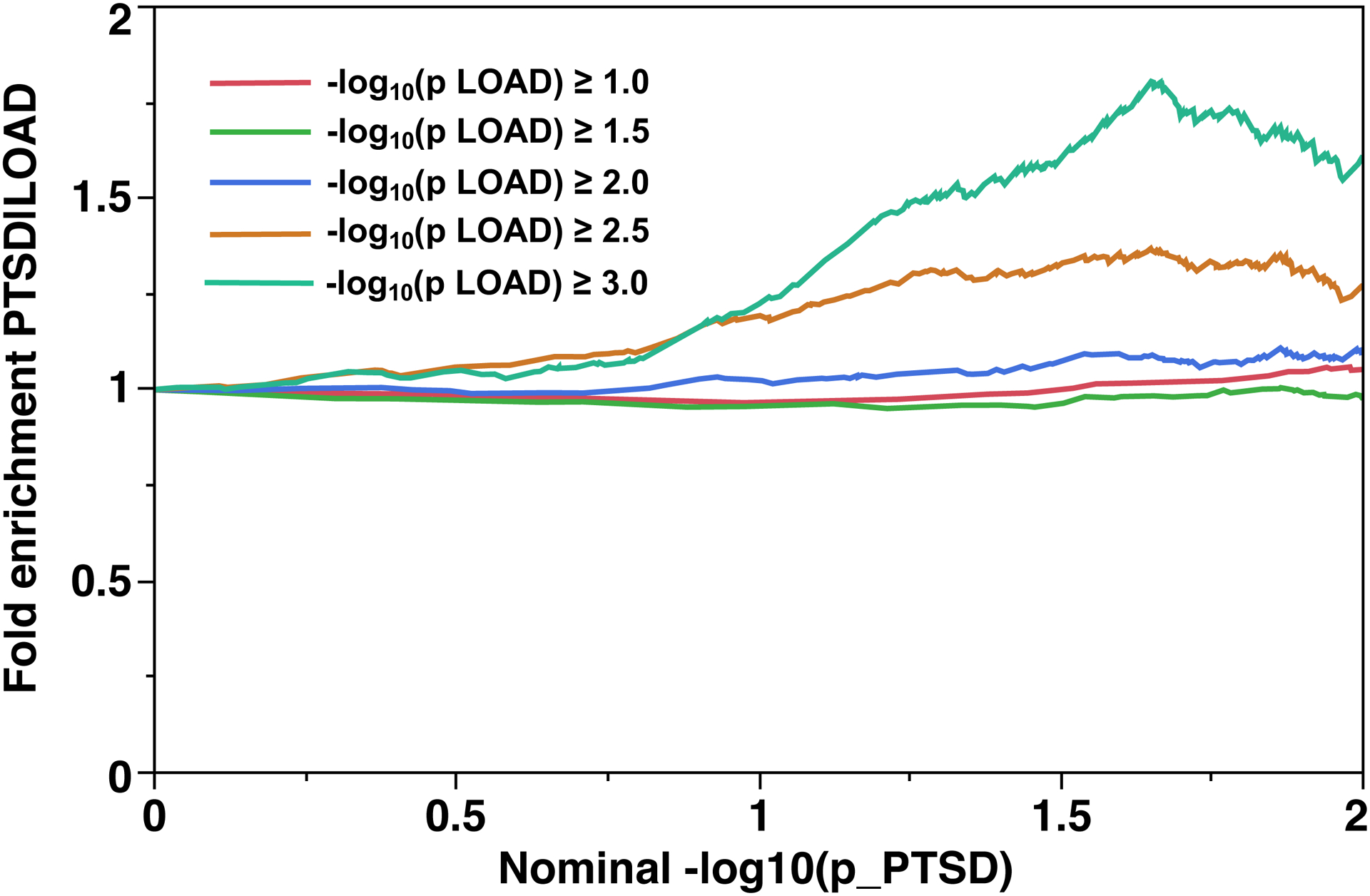
Ordinate is fold-enrichment. Fig. 2A: Abscissa is nominal −log10(p) for SNP association with LOAD, curves are differentiated by the threshold for level of statistical significance for SNP association with the secondary phenotype (PTSD). Fig. 2B: Abscissa is nominal −log10(p) for SNP association with PTSD, curves are differentiated by the threshold for level of statistical significance for SNP association with the secondary phenotype (LOAD).
3.2.3. African American sample.
The fold enrichment plot (Fig. 3A) demonstrates moderate (2.3 – 5.5 fold) enrichment for SNP association with LOAD across increasingly stringent levels of significance for SNP association with PTSD (Q(LOAD|PTSD)). The reverse conditional association Q(PTSD|LOAD) showed enrichment of approximately 1.0 – 1.3 fold (Fig. 3B). These results support a moderate level of polygenic overlap between LOAD and PTSD.
Fig 3. Fold-enrichment plots for the African American sample.

Ordinate is fold-enrichment. Fig. 3A: Abscissa is nominal −log10(p) for SNP association with LOAD, curves are differentiated by the threshold for level of statistical significance for SNP association with the secondary phenotype (PTSD). Fig. 3B: Abscissa is nominal −log10(p) for SNP association with PTSD, curves are differentiated by the threshold for level of statistical significance for SNP association with the secondary phenotype (LOAD).
3.3. Specific variants and genes identified by conditional false discovery rate analysis
Manhattan plots for the conditional FDR Q(LOAD|PTSD) are shown for the discovery sample (Fig. 4A) and the replication sample (Fig. 4B). For the discovery sample, there is a collection of SNPs on chromosome 11 and single SNPs on chromosome 2 and chromosome 19 where Q(LOAD|PTSD) < 1×10−5. There were 629 loci representing 46 genes SNPs where Q(LOAD|PTSD) < 0.05. For the replication sample, similar to the discovery sample, a strong conditional association signal Q(LOAD|PTSD) < 1×10−5 for SNPs was observed on chromosome 11. There were 467 SNPs mapping to 59 proximal genes where Q(LOAD|PTSD) < 0.05.
Fig 4. Conditional Manhattan plots of the conditional −log10 (FDR) values for SNP association with LOAD conditional with SNP association with PTSD.


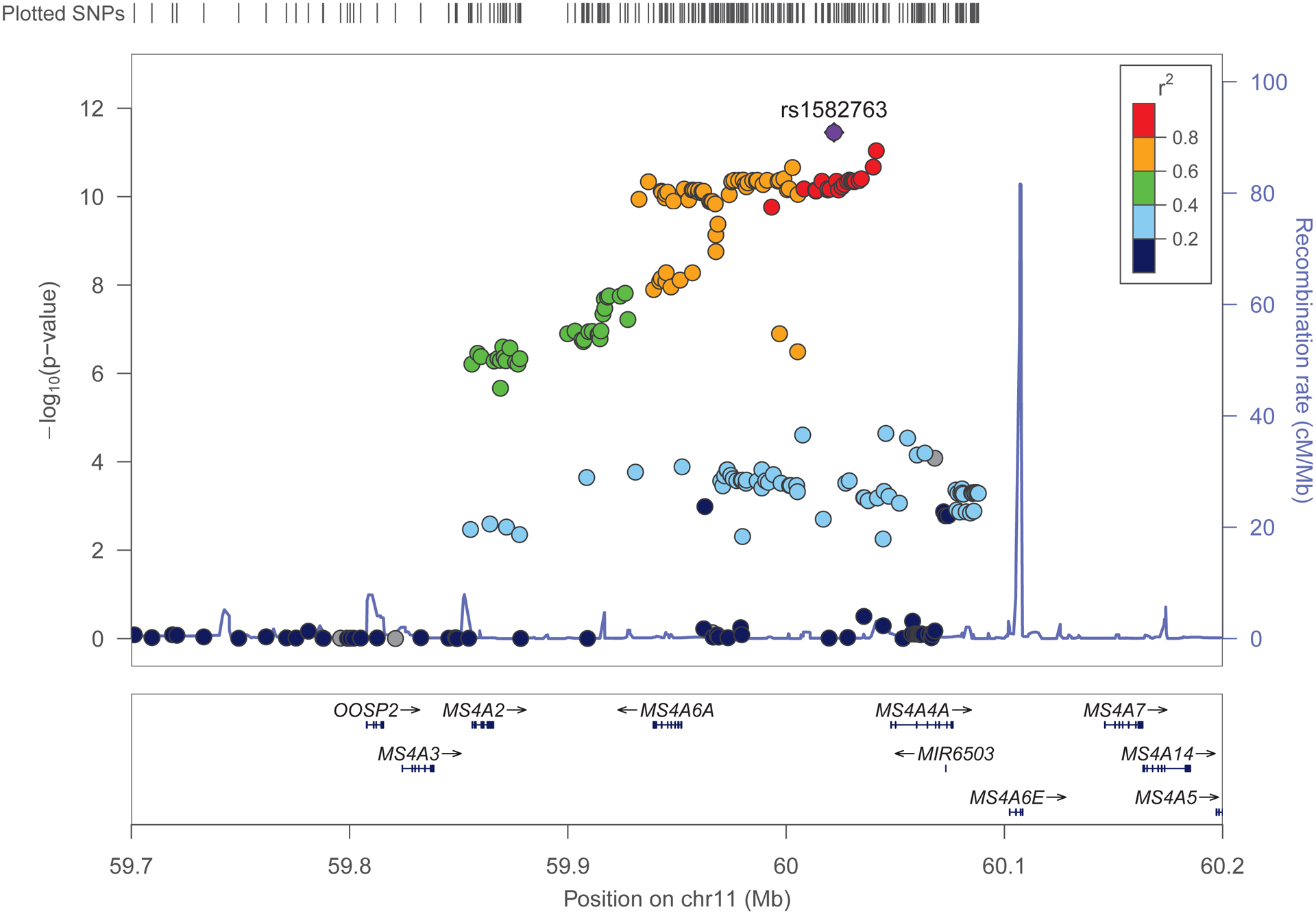
Conditional Manhattan plots show the FDR q value for SNP association with LOAD association conditional with SNP association for PTSD. Fig. 4A: Discovery sample. Fig. 4B. Replication sample. Genome-wide significant line (red) is drawn at −log10(1×10−5), suggestive line (blue) is drawn at −log10(0.05). Fig. 4C. Locus zoom plot of the region on chromosome 11 contains MS4A2 that was identified as associated with Q(LOAD|PTSD) for both the discovery and replication samples
FDR analysis identified SNPs associated ((q) ≤ 0.05) with LOAD conditional on association with PTSD Q(LOAD|PTSD) for both the discovery and the replication samples. Six loci were identified as significant for Q(LOAD|PTSD) located on 3 chromosomes, four of which were located in loci on chromosome 11, one SNP on chromosome 14 and one SNP on chromosome 17. These SNPs were characterized by minor allele frequency (MAF) as common SNPs with 0.02 ≤ MAF ≤ 0.46. Individual GWAS association results for these loci were, LOAD (IGAP): 0.90 ≤ OR ≤ 1.21, 7.25*×10−15 ≤ p ≤ 2.83×10−5; PTSD Discovery: 0.91 ≤ OR ≤ 1.07, 1.79×10−2 ≤ p ≤ 9.58×10−2, PTSD Replication: 0.41 ≤ OR ≤ 1.16, 1.14×10−3 ≤ p 3.13×10−2; these are consistent with effect sizes for GWAS studies of complex diseases. Mapping the SNPs in these loci to genes by proximity leads to a list of 6 genes. Two genes in this set have been identified as LOAD risk genes by GWAS: MS4A2 and SLC24A4. SNPs in the SPI1 gene were identified as significant (FDR(q) = 2.95×10−3) in the replication sample but not in the discovery sample. Other LOAD genes identified as FDR significant (FDR (q) ≤ 0.05) in the replication sample, but not in the discovery sample were CD2AP and CELF1. LOAD genes identified as FDR significant FDR (q) ≤ 0.05 in the discovery sample, but not in the replication sample were ABCA7, BIN1, INPP5D, PICALM and PTK2B. Figure 4C is the Locus Zoom plot for the region on chromosome 11 containing MS4A2 that was identified as associated with Q(LOAD|PTSD) for both the discovery and replication samples. From the plot, it is clear that this is a moderate to high linkage disequilibrium region for several MS4 family genes including: MS4A2, MS4A3, MS4A6A, MS4A4E and MS4A4A. The strongest LD region (r2 = 0.8 to 1.0) contains MS4A6A, MS4A4E and MS4A4A.
We did not identify any SNPs in the analysis of PTSD conditional on association with LOAD P(PTSD|LOAD) where FDR (q) ≤ 0.05 for both the discovery and the replication samples, therefore there were no SNPs observed that reached FDR (q) ≤ 0.05 for the conjunction conditional FDR.
The strong signal for the 6 replicated loci was also observed when the analysis included the APOE region. The conditional FDR results for the APOE SNP variants in the discovery and replication cohorts were substantially different, with FDR(q) < 10−140 for the discovery cohort and FDR(q) < 10−6 for the replication cohort. There were no FDR significant results in either direction Q(LOAD|PSTD) or Q(PTSD|LOAD) for the African American dataset. The African American LOAD dataset is substantially smaller (n = 9,223) than the IGAP dataset (n = 63,976), greatly limiting the statistical power to detect significant effects for the conditional analysis.
3.4. Mapping of genes to pathways and function
The six genes which were FDR significant for Q(LOAD|PTSD) in both the discovery and replication cohort share common biological function in processes involved in inflammation and immunity. The MS4 family genes are involved as chemosensory receptors9. These genes are expressed in microglia and regulate cell activation41.
Gene set enrichment was performed using the replication sample for the conditional FDRs in both directions, Q(LOAD|PTSD) and Q(PTSD|LOAD) for all SNPs with cFDR < 0.05. The SNP to gene mapping was limited to 500kb upstream and downstream of the gene. Candidate gene sets included canonical pathways, GO biological processes and GO molecular function. The strongest statistical confidence (enrichment FDR < 0.05) for pathway gene set association with the phenotype LOAD conditional on PTSD was for several general biological pathways involving regulation of transcription, RNA processing and splicing and nuclear receptors (Supplemental Table S1). Notably, the top pathway, negative regulation of transcription from RNA polymerase II promoter, results from a mapping to several LOAD-associated genes42 including SPI1, CUX1, GLI2, ZMYND11 and GLIS3. Two pathways associated with brain-specific function are identified: (1) the gene set “brain development” is highly associated (FDR = 0.005) and (2) axonogenesis is possibly associated (FDR = 0.10) with P(LOAD|PTSD).
For PTSD conditional with LOAD, there are numerous pathways identified as associated with high confidence as shown in Supplemental Table S2. These pathways comprise a broad set of regulatory pathways including apoptosis, P38 MAPK, tryptophan metabolism and also some cancer-related pathways. It is unclear why numerous cancer-related pathways are identified in the analysis and whether these are false positives. In this regard it is important to note that there were no SNPs above the FDR cutoff for Q(PTSD|LOAD) for both the discovery and replication cohorts. Interestingly, a signature for Alzheimer’s disease is identified as possibly associated (FDR = 0.15) with P(PTSD|LOAD). Significant genes in the gene set include APP, BACE1, MME, SNCA and IL1B which are involved in LOAD pathology.
3.5. Gene expression analysis
Tissue-specific gene expression in 53 GTEx tissue types for the 6 genes identified by proximity mapping for association with Q(LOAD|PTSD), common to both the discovery and replication samples are shown in heat map format in Figure 5. There are 13 brain-specific tissues highlighted in the heat map, which was clustered by both genes and tissues. The KCTD2 (potassium channel tetramerization domain-containing protein 2) gene showed high expression levels in the cerebellar hemisphere and cerebellum which were differentially more highly expressed than in other brain regions, including the cortex. Expression levels of KCTD2 were higher in brain tissue than expression levels for the MS4A genes and SLC24A4. The KCTD gene family has roles in functions including DNA transcription, degradation of ubiquitinated proteins, proteasome physiology, voltage-dependent potassium channel function and GABA neurotransmitter receptor B heteromultimeric composition43.
Fig. 5. Heat map of gene expression data for genes associated with LOAD conditional on association with PTSD in the discovery and replication samples.
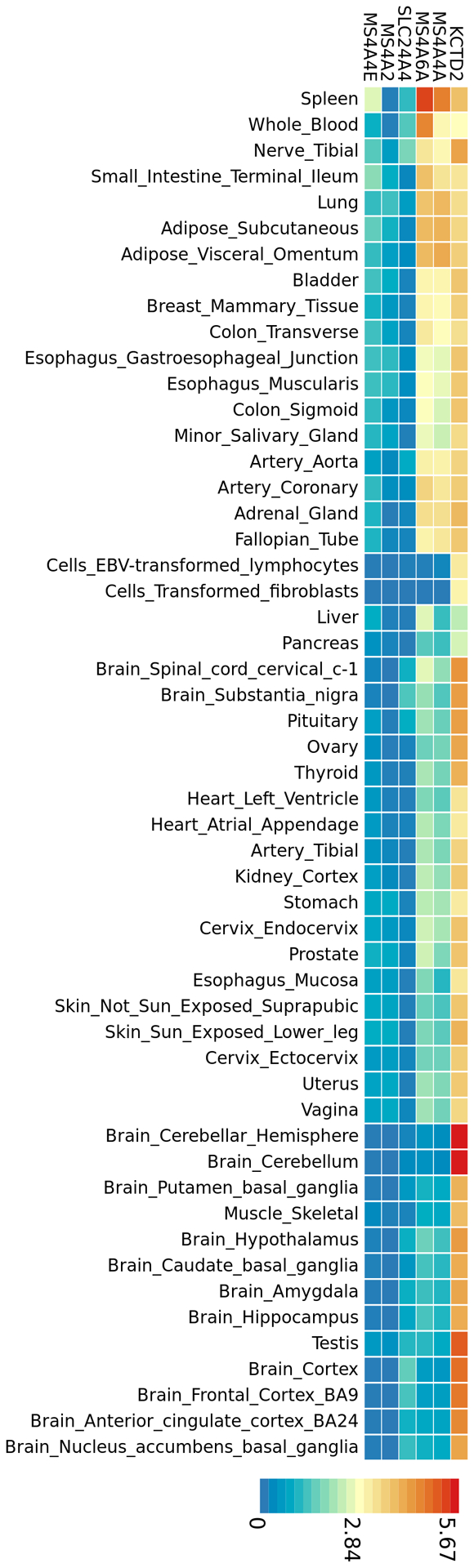
Heat map plot of tissue-specific gene expression in 53 GTEx tissue types for the 6 genes common to both the discovery and replication samples identified by proximity mapping for SNP association with LOAD, conditional with SNP association with PTSD. There are 13 brain-specific tissues highlighted in the heat map, which was ordered by both gene and tissue clustering.
A novel statistical analysis approach, Unified Test for Molecular Signatures (UTMOST)40 was utilized to quantitatively assess the association between gene expression in the brain with the SNPs that were identified as FDR significant for the conditional analysis Q(LOAD|PTSD) in both the discovery and replication cohorts. This approach utilizes cross-tissue imputation to improve estimation of effects and implements calculation of single tissue associations with genes and cross-tissue analysis of gene-trait association based on a generalized Berk-Jones test. The cross-tissue analysis combines statistical evidence across multiple tissues. The single tissue association results are provided in Supplemental Table S3. Three genes show single tissue association results with p < 3× 10−5, MS4A6A, MPEG1 and DTX4. The generalized Berk-Jones cross-tissue association test shows significance (p < 3×10−7) for these genes across the 9 GTEx brain tissues listed in Supplemental table S3.
3.6. Genomic analysis of the SPI1 locus
The replication sample identified SNPs in the SPI1 region as having a strong level of significance for Q(LOAD|PTSD). In addition, recent reports identify SPI1 as a causal gene for LOAD age of onset44. Thus, we analyzed the linkage disequilibrium region of the SPI1 locus for the conditional FDR statistics for Q(LOAD|PTSD). In the replication sample, the SNP rs3740686 in the SPI1 gene with the strongest association for Q(LOAD|PTSD) (FDR = 0.0034) is located only 83 base pairs from the SNP reported as causal for LOAD age on onset (rs1057233)44. Rs3740686 is therefore a proxy for this causal SNP with r2 = 1 and D’ = 1. Rs3740686 did not reach conditional FDR significance in the discovery sample for Q(LOAD|PTSD) or Q(PTSD|LOAD).
4. Discussion
In this study we demonstrated genetic pleiotropy between LOAD and PTSD and found a moderate level of polygenic overlap between LOAD and PTSD. We used two independent PTSD datasets for discovery and replication which increased the statistical rigor of the analysis and we focused on SNPs found in common between the two samples. Six loci replicated as FDR significant for Q(LOAD|PTSD) in both sample sets. Estimates of the phenotypic variance in LOAD case-control studies explained by common SNPs ranges from 24%45 to 33%46. SNP-based heritability estimate for PTSD is 15%11. While our study supports overlap between SNPs associated with both diseases, the amount of genetic association explained by the pleiotropic SNPs is likely not substantial as a consequence of each variant contributing a small portion to the overall genetic heritability. Nonetheless, the genetic pleiotropy may explain, in part, the interplay between the LOAD and PTSD. A recent systematic review suggested an intricate bidirectional relationship between PTSD and dementia47. Specifically the authors reviewed evidence that PTSD in mid-life may initiate and catalyze the pathological cascade of specific neurodegenerative disorders in late-life including LOAD and noted that individuals with PSTD and with AD show hypometabolism in the medial temporal lobe, hippocampus and cingulate cortex47,48.
The most significant shared loci belonged to several genes mapped on chromosome 11 from the MS4A gene family. The MS4A gene cluster encodes a family of cellular membrane spanning proteins spanning with a similar protein sequence and similar predicted topological structure, which have roles in signal transduction and regulation of cell activation and are highly expressed in microglia9. A recent study reported genome-wide significant associations for Alzheimer’s within 1.0 kb windows at MS4A6A and MS4A4A for CpG-related SNPs (CGSes) and identified a strong negative dosage effect of the CGSes on LOAD risk49. The window containing the MS4A4A CGSes was found to be associated with increased DNA methylation in brain and blood49. CGSes in the intergenic window between MS4A4A and MS4A4E were associated with increased expression of MS4A4A in brain and blood. The results of the study support a role for these genes in LOAD risk as suggested by earlier studies and implicated the potential role for epigenetic regulation of gene transcription. Thus, future studies that further explore the specific nature of epigenetic regulation of immune-related genes, such as the MS4A family, in the context of LOAD and PTSD are warranted.
Studies of biological mechanisms associated with the shared physiopathologies have considered the question of shared genes and mechanisms between the two diseases and highlighted the importance of innate immunity and associated regulatory genes50,51. LOAD functional genomic studies showed enrichment of risk alleles in active enhancers of monocytes, macrophages and microglia and further characterization of these GWAS signals with myeloid multi-omics data have pointed to specific causal genes, notably SPI144,52. For PTSD transcriptomic studies in blood found dysregulation associated with signaling cascades including innate immunity, cytokines and type I interferon pathways53,54. Additional studies have supported the involvement of the innate immune system at the molecular level with roles for genes including TNFAIP3, TRAFD1, and PML51 and the interferon genes IFI35, IFIH1, PARP14, RSAD2 and UBE2L650. Moreover, development of PTSD biomarkers have focused on the stress response, peripheral inflammation (inflammatory cytokines, c-reactive protein), neural-immune communication55 and response of the blood immune cells to glucocorticoid activation53. Collectively, the results of the current study support and extend the previously suggested role of the immune system and regulation of cell activation in both LOAD10,56–60 and PTSD61,62.
While shared genetic architecture between LOAD and NPS has been investigated, particularly for major depressive disorder (MDD)63, there is limited precedent on studies to investigate genetic pleiotropy and molecular mechanisms that are shared between LOAD and PTSD64,65. One mouse model study implicated actin-associated Formin 2 (FMN2) in both conditions and showed that downregulation of FMN2 in a PTSD mouse model accelerates LOAD related neuropathophysiology65,66. As far as we know, our study is the first to examine genetic pleiotropy between LOAD and PTSD based on human datasets. The two major findings of our study were the determination of the level of polygenic overlap between LOAD and PTSD and the biochemical characteristics of the set of genes and related pathways that were identified as significantly associated with LOAD conditional with PTSD. In all three sample cohorts (European ancestry, discovery and replication; African American ancestry) there was a moderate level (2–6-fold) of enrichment for SNPs associated with the two phenotypes in both directions, Q(LOAD|PTSD) and Q(PTSD|LOAD).
Bidirectional conditional associations at a pre-defined level of significance would be the strongest evidence for a shared genetic etiology. Our results showed that the genetic associations between SNPs and LOAD, however, had substantially lower P values than the genetic associations between SNPs and PTSD. Therefore, the conditional FDR calculations identified FDR significant results in the direction of LOAD conditional with PTSD and not the reverse. The conditional analysis is based on the individual disease GWAS and the LOAD GWAS was based on a sample size of over 63,000 individuals providing substantially more statistical power to detect genetic associations than the PTSD GWAS. Including two independent PTSD cohorts and focusing the analysis results only on SNPs that were identified as significant in both studies offers some mitigation of this limitation, however, as larger sample size PTSD GWAS become available, this difference in sample size and absence of bidirectional association likely will be addressed. The conceptual framework presents another strength as noted by Desikan et al.24. Specifically, the conditional FDR framework can detect genetic pleiotropy independent of directionality and the inclusion of two phenotypes increases statistical power to detect an association that may not be detected with a single phenotype GWAS24. Moreover, taking together the FDR significant results at the gene level, the gene expression results, and the pathway analysis provided clues regarding the potential biochemical characterization of the common biochemical mechanisms.
While the association of APOE and LOAD has been extensively studied, the association between APOE genotypes and PTSD has also been investigated in prior studies67–69. A meta-analysis that investigated the relationship between APOE genotype and susceptibility to combat-related PTSD found a significant association between the APOE ε4 variant and risk for PTSD. However there was heterogenity among the individual studies used in the meta-analysis for the association between the APOE ε4 variant and PTSD67. A study based on the National Health and Resilience in Veterans study concluded that carriage of the APOE e4 allele and PTSD were independently associated with small reductions in self-reported cognitive function68. A recent epigenetic study of APOE and PTSD concluded that the interaction between APOE ε4 genotype and PTSD was associated with DNA methylation at a specific APOE promoter CpG site69. Plasma ApoE concentration was also found to be associated with PTSD symptom severity69. Our study found significant associations for APOE SNP variants with LOAD conditional on association with PTSD for both the discovery and replication cohorts which is consistent with these reports. However, the substantial difference in cFDR for the two cohorts suggests that caution is needed to interpret the APOE findings from our study.
Several of the risk loci identified in LOAD GWAS cohorts of European ancestry were also identified in African American cohorts, however, different risk profiles by ancestry are evident as well, including differences in the amount of risk attributable to APOE28,70. The results of our study supported some level of genetic pleiotropy between LOAD and PTSD in African Americans at fold enrichment levels similar to individuals of European ancestry, although no individual locus reached a conditional FDR < 0.05. Future studies of LOAD and PTSD GWAS in individuals of African American ancestry are needed to further define specific loci that demonstrated association with either LOAD or PTSD conditional with the other phenotype.
This study had several strengths in design and methods. First, the conditional FDR framework controlled for the likelihood of false positive results and was based on the number of statistical tests; second, the use of two independent PTSD cohorts for the study design greatly increased the statistical rigor of the analysis. In spite of these strengths, several limitations are worth noting. Foremost, replication in additional well-powered cohorts would strengthen statistical support as the smaller sample size of the PTSD replication cohort limited the power of the original GWAS. Replication using an independent LOAD cohort would also increase statistical rigor. Also, the definition of the LOAD and PTSD phenotypes is complex and subtle differences in these definitions can influence the genetic association results. Not all of the LOAD datasets had diagnoses confirmed by autopsy, CSF or PET scans and therefore case/control status may reflect mixed pathologies. Although sex was included as a covariate in the original GWAS studies, since the analysis in this paper is based on the GWAS summary statistics, we are unable to include sex in the downstream analysis. Thus, future studies are required to investigate potential sex-based biological differences shared between LOAD and PTSD as well as the importance of other gene by environmental factors. Last, alternative statistical methods are available to test for genetic pleiotropy including factor analysis and correlated meta-analysis71, genetic correlation analysis72, Bayesian methods and multivariate mixed models73. Future studies using these methods will further test our LOAD|PTSD pleiotropic discoveries.
The overall approach used in this study has the potential to provide biologically relevant information about shared etiology and intersecting pathways between LOAD and PTSD. The shared genetic etiology and pathways establish a molecular phenotype that may extend to and explain, at least in part, some of the heterogeneity of the NPS in LOAD. Inflammation and immune related processes are associated with potentially modifiable risk factors that can be controlled through lifestyle interventions and/or medications and therefore have the potential to impact prevention and treatment of LOAD, PTSD and NPS symptoms in LOAD. The results suggest that genes involved in immune function may play a role in both LOAD and PTSD, implicating common biochemical pathways. Investigation of these pathways in cellular and animal models will test whether the pleiotropic associations reported in this study are causal at a biochemical level.
Our study advances the understanding of the possible genetics underlying the heterogeneity and comorbidity of LOAD and PTSD. Identifying a shared genetic overlap between LOAD and PTSD provides mechanistic insight about early biological substrates that may confer risk for the development of LOAD decades later. This is consistent with prior research showing an increased risk for developing dementia among individuals with PTSD5. Collectively, this knowledge points toward a potential new avenue to develop prophylactic interventions for individuals who experience PTSD early in life in an effort to delay the onset of LOAD.
Supplementary Material
Table 2.
Loci reaching statistical significance (FDR (q) ≤0.05) for association with LOAD conditional on association with PTSD in both the discovery and replication samples.
| SNP | Chr | Position | Nearest Gene | Minor allele | MAF | Effect Allele | Conditional FDR discovery | Conditional FDR replication | AD OR (95% CI) | P AD (recent IGAP) | PTSD association P (Discovery) | PTSD association P (Replication) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs71380858 | 17 | 73,029,301 | KCTD2 | G | 0.03 | C | 4.65E-02 | 4.60E-02 | 1.21 (1.09 – 1.34) | 2.83E-05 | 0.10 | 0.001 |
| rs1441586 | 11 | 59,856,028 | MS4A2 | C | 0.46 | T | 7.68E-07 | 6.65E-05 | 0.91 (0.89 – 0.94) | 1.09E-10 | 0.08 | 0.021 |
| rs12421663 | 11 | 60,035,550 | MS4A4A | G | 0.32 | A | 7.76E-04 | 3.75E-03 | 1.07 (1.04 – 1.12) | 3.14E-07 | 0.02 | 0.029 |
| rs10897011 | 11 | 59,961,427 | MS4A4E | A | 0.41 | A | 1.15E-10 | 1.58E-06 | 0.90 (0.88 – 0.93) | 7.25E-15 | 0.05 | 0.022 |
| rs2278867 | 11 | 59,943,109 | MS4A6A | T | 0.40 | A | 1.19E-10 | 1.13E-06 | 0.90 (0.88 – 0.93) | 7.63E-15 | 0.07 | 0.024 |
| rs8008388 | 14 | 92,936,690 | SLC24A4 | G | 0.19 | A | 7.63E-04 | 3.45E-03 | 1.09 (1.05 – 1.14) | 2.39E-07 | 0.09 | 0.031 |
Table 3.
Cross-tissue unified test for molecular signatures analysis for association of FDR significant loci for Q(LOAD|PTSD) with 9 brain regions.
| gene | test score | p value |
|---|---|---|
| DTX4 (Deltex E3 Ubiquitin Ligase 4) | 18.5 | 4.8E-09 |
| MPEG1 (Macrophage Expressed Gene 1) | 24.5 | 1.3E-11 |
| MS4A6A (Membrane Spanning 4-Domains A6A) | 14.6 | 2.9E-07 |
Acknowledgments
This work was funded in part by the National Institutes of Health/National Institute of Aging (NIH/NIA) [R56 AG062302-01to M.W. L and R01 AG057522 to O.C-F.]. The authors appreciate the assistance provided by Dr. Hongyu Zhao and Jerome Yu with the UTMOST software.
Footnotes
Disclosure statement
The authors report no relevant disclosures for this work.
DISCUSSION: We demonstrated common genetic signatures for LOAD and PTSD and suggested immune response as a common pathway for these diseases.
References
- 1.Lyketsos CG Neuropsychiatric Symptoms in Dementia: Overview and Measurement Challenges. J Prev Alzheimers Dis 2, 155–156 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lyketsos CG et al. Neuropsychiatric symptoms in Alzheimer’s disease. Alzheimers Dement 7, 532–9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao QF et al. The prevalence of neuropsychiatric symptoms in Alzheimer’s disease: Systematic review and meta-analysis. J Affect Disord 190, 264–271 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Hallikainen I et al. The Progression of Neuropsychiatric Symptoms in Alzheimer’s Disease During a Five-Year Follow-Up: Kuopio ALSOVA Study. J Alzheimers Dis 61, 1367–1376 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Yaffe K et al. Posttraumatic stress disorder and risk of dementia among US veterans. Arch Gen Psychiatry 67, 608–13 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuring JK, Mathias JL & Ward L Prevalence of Depression, Anxiety and PTSD in People with Dementia: a Systematic Review and Meta-Analysis. Neuropsychol Rev 28, 393–416 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Weiner MW et al. Military risk factors for Alzheimer’s disease. Alzheimers Dement 9, 445–51 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weiner MW et al. Effects of traumatic brain injury and posttraumatic stress disorder on Alzheimer’s disease in veterans, using the Alzheimer’s Disease Neuroimaging Initiative. Alzheimers Dement 10, S226–35 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pimenova AA, Raj T & Goate AM Untangling Genetic Risk for Alzheimer’s Disease. Biol Psychiatry 83, 300–310 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kunkle BW et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet 51, 414–430 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duncan LE et al. Largest GWAS of PTSD (N = 20 070) yields genetic overlap with schizophrenia and sex differences in heritability. Mol Psychiatry 23, 666–673 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nievergelt CM et al. Largest genome-wide association study for PTSD identifies genetic risk loci in European and African ancestries and implicates novel biological pathways. bioRxiv, 458562 (2018). [Google Scholar]
- 13.Koenen KC et al. Common genetic liability to major depression and posttraumatic stress disorder in men. J Affect Disord 105, 109–15 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang XF et al. Linking Alzheimer’s disease and type 2 diabetes: Novel shared susceptibility genes detected by cFDR approach. J Neurol Sci 380, 262–272 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smeland OB et al. Identification of Genetic Loci Jointly Influencing Schizophrenia Risk and the Cognitive Traits of Verbal-Numerical Reasoning, Reaction Time, and General Cognitive Function. JAMA Psychiatry (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andreassen OA et al. Improved detection of common variants associated with schizophrenia and bipolar disorder using pleiotropy-informed conditional false discovery rate. PLoS Genet 9, e1003455 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andreassen OA et al. Genetic pleiotropy between multiple sclerosis and schizophrenia but not bipolar disorder: differential involvement of immune-related gene loci. Mol Psychiatry 20, 207–14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le Hellard S et al. Identification of Gene Loci That Overlap Between Schizophrenia and Educational Attainment. Schizophr Bull 43, 654–664 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Witoelar A et al. Genome-wide Pleiotropy Between Parkinson Disease and Autoimmune Diseases. JAMA Neurol 74, 780–792 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andreassen OA et al. Identifying common genetic variants in blood pressure due to polygenic pleiotropy with associated phenotypes. Hypertension 63, 819–26 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu JZ et al. Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nat Genet 45, 670–5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andreassen OA et al. Shared common variants in prostate cancer and blood lipids. Int J Epidemiol 43, 1205–14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chung J et al. Genome-wide pleiotropy analysis of neuropathological traits related to Alzheimer’s disease. Alzheimers Res Ther 10, 22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Desikan RS et al. Polygenic Overlap Between C-Reactive Protein, Plasma Lipids, and Alzheimer Disease. Circulation 131, 2061–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lutz MW et al. Analysis of pleiotropic genetic effects on cognitive impairment, systemic inflammation and plasma lipids in the Health and Retirement Study.. Neurobiol Aging (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wray NR et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet 50, 668–681 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nievergelt CM et al. International meta-analysis of PTSD genome-wide association studies identifies sex- and ancestry-specific genetic risk loci. Nat Commun 10, 4558 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reitz C et al. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E 4, and the risk of late-onset Alzheimer disease in African Americans. JAMA 309, 1483–92 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andreassen OA et al. Abundant genetic overlap between blood lipids and immune-mediated diseases indicates shared molecular genetic mechanisms. PLoS One 10, e0123057 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andreassen OA et al. Improved detection of common variants associated with schizophrenia by leveraging pleiotropy with cardiovascular-disease risk factors. Am J Hum Genet 92, 197–209 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hochberg Y & Benjamini Y More powerful procedures for multiple significance testing. Stat Med 9, 811–8 (1990). [DOI] [PubMed] [Google Scholar]
- 32.Kent WJ et al. The human genome browser at UCSC. Genome Res 12, 996–1006 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McLaren W et al. The Ensembl Variant Effect Predictor. Genome Biol 17, 122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chelala C, Khan A & Lemoine NR SNPnexus: a web database for functional annotation of newly discovered and public domain single nucleotide polymorphisms. Bioinformatics 25, 655–61 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dayem Ullah AZ, Lemoine NR & Chelala C A practical guide for the functional annotation of genetic variations using SNPnexus. Brief Bioinform 14, 437–47 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Zhang K, Cui S, Chang S, Zhang L & Wang J i-GSEA4GWAS: a web server for identification of pathways/gene sets associated with traits by applying an improved gene set enrichment analysis to genome-wide association study. Nucleic Acids Res 38, W90–5 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watanabe K, Taskesen E, van Bochoven A & Posthuma D Functional mapping and annotation of genetic associations with FUMA. Nat Commun 8, 1826 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.GTEx. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–60 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.GTEx et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu Y et al. A statistical framework for cross-tissue transcriptome-wide association analysis. Nat Genet 51, 568–576 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eon Kuek L, Leffler M, Mackay GA & Hulett MD The MS4A family: counting past 1, 2 and 3. Immunol Cell Biol 94, 11–23 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Li MJ et al. GWASdb: a database for human genetic variants identified by genome-wide association studies. Nucleic Acids Res 40, D1047–54 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boada M et al. ATP5H/KCTD2 locus is associated with Alzheimer’s disease risk. Mol Psychiatry 19, 682–7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang KL et al. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat Neurosci 20, 1052–1061 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee SH et al. Estimation and partitioning of polygenic variation captured by common SNPs for Alzheimer’s disease, multiple sclerosis and endometriosis. Hum Mol Genet 22, 832–41 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ridge PG, Mukherjee S, Crane PK, Kauwe JS & Alzheimer’s Disease Genetics C Alzheimer’s disease: analyzing the missing heritability. PLoS One 8, e79771 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Desmarais P et al. The Interplay Between Post-traumatic Stress Disorder and Dementia: A Systematic Review. Am J Geriatr Psychiatry 28, 48–60 (2020). [DOI] [PubMed] [Google Scholar]
- 48.Tsolaki M, Eleftheriou M & Karavida N Alzheimer’s dementia and post-traumatic stress disorder differences and similarities in neuroimaging. Hell J Nucl Med 12, 41–6 (2009). [PubMed] [Google Scholar]
- 49.Ma Y et al. CpG-related SNPs in the MS4A region have a dose-dependent effect on risk of late-onset Alzheimer disease. Aging Cell, e12964 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Breen MS et al. Gene networks specific for innate immunity define post-traumatic stress disorder. Mol Psychiatry 20, 1538–45 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Novellino F et al. Innate Immunity: A Common Denominator between Neurodegenerative and Neuropsychiatric Diseases. Int J Mol Sci 21(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Novikova G et al. Integration of Alzheimer’s disease genetics and myeloid cell genomics identifies novel causal variants, regulatory elements, genes and pathways. bioRxiv, 694281 (2019). [Google Scholar]
- 53.Breen MS et al. Differential transcriptional response following glucocorticoid activation in cultured blood immune cells: a novel approach to PTSD biomarker development. Transl Psychiatry 9, 201 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Breen MS et al. PTSD Blood Transcriptome Mega-Analysis: Shared Inflammatory Pathways across Biological Sex and Modes of Trauma. Neuropsychopharmacology 43, 469–481 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baker DG, Nievergelt CM & O’Connor DT Biomarkers of PTSD: neuropeptides and immune signaling. Neuropharmacology 62, 663–73 (2012). [DOI] [PubMed] [Google Scholar]
- 56.Amlie-Wolf A et al. INFERNO: inferring the molecular mechanisms of noncoding genetic variants. Nucleic Acids Res 46, 8740–8753 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Broussard GJ, Mytar J, Li RC & Klapstein GJ The role of inflammatory processes in Alzheimer’s disease. Inflammopharmacology 20, 109–26 (2012). [DOI] [PubMed] [Google Scholar]
- 58.Efthymiou AG & Goate AM Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol Neurodegener 12, 43 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Holmes C Review: systemic inflammation and Alzheimer’s disease. Neuropathol Appl Neurobiol 39, 51–68 (2013). [DOI] [PubMed] [Google Scholar]
- 60.Jones L et al. Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer’s disease. PLoS One 5, e13950 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang Z, Caughron B & Young MRI Posttraumatic Stress Disorder: An Immunological Disorder? Front Psychiatry 8, 222 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mellon SH, Gautam A, Hammamieh R, Jett M & Wolkowitz OM Metabolism, Metabolomics, and Inflammation in Posttraumatic Stress Disorder. Biol Psychiatry 83, 866–875 (2018). [DOI] [PubMed] [Google Scholar]
- 63.Gibson J et al. Assessing the presence of shared genetic architecture between Alzheimer’s disease and major depressive disorder using genome-wide association data. Transl Psychiatry 7, e1094 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brady KT, Killeen TK, Brewerton T & Lucerini S Comorbidity of psychiatric disorders and posttraumatic stress disorder. J Clin Psychiatry 61 Suppl 7, 22–32 (2000). [PubMed] [Google Scholar]
- 65.Agis-Balboa RC et al. Formin 2 links neuropsychiatric phenotypes at young age to an increased risk for dementia. EMBO J 36, 2815–2828 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Graff J FORMINg a link between PTSD and AD. EMBO J 36, 2809–2811 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Roby Y Apolipoprotein E variants and genetic susceptibility to combat-related post-traumatic stress disorder: a meta-analysis. Psychiatr Genet 27, 121–130 (2017). [DOI] [PubMed] [Google Scholar]
- 68.Averill LA et al. Apolipoprotein E gene polymorphism, posttraumatic stress disorder, and cognitive function in older U.S. veterans: Results from the National Health and Resilience in Veterans Study. Depress Anxiety 36, 834–845 (2019). [DOI] [PubMed] [Google Scholar]
- 69.Nielsen DA, Spellicy CJ, Harding MJ & Graham DP Apolipoprotein E DNA methylation and posttraumatic stress disorder are associated with plasma ApoE level: A preliminary study. Behav Brain Res 356, 415–422 (2019). [DOI] [PubMed] [Google Scholar]
- 70.Hohman TJ et al. Global and local ancestry in African-Americans: Implications for Alzheimer’s disease risk. Alzheimers Dement 12, 233–43 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kraja AT et al. Pleiotropic genes for metabolic syndrome and inflammation. Mol Genet Metab 112, 317–38 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Traylor M et al. Shared genetic contribution to Ischaemic Stroke and Alzheimer’s Disease. Ann Neurol 79, 739–747 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hackinger S & Zeggini E Statistical methods to detect pleiotropy in human complex traits. Open Biol 7(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.