

Contents
| Chapter 1. Introduction | |
| Chapter 2. Methodology for guideline development | |
| Chapter 3. Referral and diagnostics | |
| Chapter 4. Perioperative care | |
| Chapter 5. Surgical treatment of isolated, non-syndromic craniosynostosis | |
| Chapter 6. Surgical treatment of multisuture and syndromic craniosynostosis – the cranial vault | |
| Chapter 7. Surgical treatment of syndromic craniosynostosis – FACE | |
| Chapter 8. Increased intracranial pressure | |
| Chapter 9. Hydrocephalus | |
| Chapter 10. Chiari | |
| Chapter 11. Visual, refractive and motility impairments | |
| Chapter 12. Respiratory disorders | |
| Chapter 13. Hearing impairments and speech/language development | |
| Chapter 14. Dentofacial abnormalities | |
| Chapter 15. (Neuro)cognitive, socio-emotional and behavioural functioning | |
| Chapter 16. Psychosocial functioning | |
| Chapter 17. Criteria for craniosynostosis expertise center and team members |
CHAPTER 1. INTRODUCTION
The first version of the Dutch guideline for care and treatment of patients with craniosynostosis was established in 2010 and published in 2015.1 The Dutch Society for Plastic and Reconstructive Surgery initiated the revision of this guideline, to update it according to the most recent scientific literature. This second version has been approved by all participating societies in 2020.
All chapters from the previous version were revised and new topics on prenatal detection and speech and language development were introduced.
REFERENCES
-
1.
Mathijssen IMJ. Guideline for care of patients with the diagnoses of craniosynostosis: Working group on craniosynostosis. J Craniofac Surg 2015:26:1735-1807.
CHAPTER 2. METHODOLOGY FOR GUIDELINE DEVELOPMENT
Input of Patient Perspective
The patient perspective was taken into account by asking the Patient Federation Netherlands for written input on bottlenecks and points for attention in the preparatory phase. They forwarded the request for the submission of bottlenecks to LAPOSA and Stichting Kind en Ziekenhuis (Child and Hospital Foundation). No bottlenecks were submitted. In addition, the chair of LAPOSA had a seat in the working group. During the comment phase, the draft guideline was also presented to the Patient Federation Netherlands, LAPOSA and Foundation Child and Hospital.
For information: A focus group meeting had been organized during the development of the guideline in 2010 (see Appendix 1). This report is still valid.
Method Employed by the Working Group Procedure in 2010
The content of this guideline is based on evidence from published scientific research. Relevant articles were identified using systematic searches in Medline, Embase, and the Cochrane Library. Existing guidelines were specifically searched for in online accessible (international and national guideline clearinghouses).
Searches were limited to the Dutch, English and French languages. In addition, articles were extracted from reference lists of relevant literature. This resulted for several basic questions in additional articles.
Care for children with craniosynostosis was introduced in the late 1960 s, and consequently the English literature from those years onwards was included. Searches were performed until 1 December 2009 and articles available as “Epub ahead of publication” at that date were included as well.
Regarding all basic questions, patient categories were defined in a uniform way.
The following search terms were used: craniofacial, craniosynostosis combined with: genetics, hydrocephalus, Chiari, cerebral pressure, otitis, hearing, vision, psychology, anesthesia, complications, infection, development, growth, maxilla, mandible, distraction, osteotomy, Fort, midface, RED, halo, monobloc, facial bipartition, median fasciotomy, hypertelorism.
The complete search strategies are available on request. Relevant articles extracted from reference lists of retrieved literature and several relevant publications until 1 November 2009 were included as well. Ongoing research was left out of consideration. Under the headings Summary of the literature / Conclusions only published studies / guidelines are discussed. Case reports and letters were excluded, unless they reported a complication.
The selected articles were assessed on methodological quality graded by level of evidence according to the standard classification: see Table 1. After selection, those articles remained that are listed to underpin the various conclusions. The articles are assessed under the heading ‘Summary of the literature’. Next, the scientific evidence was briefly summarized in a ‘Conclusion’. The main literature on which a conclusion is based was mentioned as well, including the level of evidence (see Table 2).
Other aspects than scientific evidence may be relevant to making a recommendation as well, such as patient preferences (derived from the results of the focus group sessions or relevant literature on the patient perspective), costs, availability, or organizational aspects. These kinds of aspects, provided they have not been subject of research, were mentioned under the heading ‘Considerations’. The experience and the opinion of the working group members have been key to the other considerations. The ‘Recommendation’ results from the combination of the available evidence and the other considerations.
TABLE 1. Classification of Methodological Quality of Individual Studies
| Intervention | Diagnostic Accuracy Assessment | Harm or Side Effects, Etiology, Prognosis∗ | |
| A1 | Systematic review of at least two mutually independent studies of A2-level | ||
| A2 | Randomized double-blind comparative clinical study of good quality and sufficient size | Study comparing with a reference test (a ‘gold standard’) with previously defined cut-off values and independent assessment of the results of study test and gold standard regarding a sufficiently large series of consecutive patients who all were administered the index and reference test | Prospective cohort study of sufficient size and follow-up, adequately controlled for confounding and with satisfactory exclusion of selective follow-up |
| B | Comparative study, but not possessing all qualities mentioned under A2 (this category also includes patient-checkup study, cohort study) | Study comparing with a reference test, but not possessing all qualities mentioned under A2 | Prospective cohort study, but not possessing all qualities mentioned under A2, or retrospective cohort study or patient-checkup study |
| C | Non-comparative study | ||
| D | Expert opinion |
This classification only applies to situations in which controlled trials are not feasible for ethical or other reasons. If they should be feasible, the classification for interventions applies.
TABLE 2. Level of Evidence for the Conclusion
| Conclusion based on: |
| 1. Level A1 study or at least two mutually independent Level A2 studies |
| 2 .One Level A2 study or at least two mutually independent Level B studies |
| 3 .One Level B or C study |
| 4. Expert opinion |
Procedure in 2017/2018
AGREE
This guideline has been revised in accordance with the requirements according to the report Medical Specialist Guidelines 2.0 of the Advisory Committee on Guidelines of the Council for Quality (www.kwaliteitskoepel.nl). This report is based on the AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II) (www.agreecollaboration.org), which is an internationally widely accepted instrument, and on ’guidelines for guideline’ for assessing the quality of guidelines (www.cvz.nl).
Bottleneck Analysis
During the preparatory phase, the chair and advisors of the working group made an inventory of the bottlenecks and drew up a draft framework (= new topics and topics to be revised). This draft was then presented to the working group with the request to provide for written input. During the first working group meeting, this draft version was discussed. At the same time, input was requested from the following stakeholder parties regarding perceived bottlenecks and points of attention of a medical, organizational and financial nature for the to be revised and updated guideline: Care Institute the Netherlands, Inspectorate for Health Care and Youth, Dutch Healthcare Authority, Patient Federation the Netherlands, LAPOSA, Stichting Kind en Ziekenhuis, Nederlands Huisartsen Genootschap, Zorgverzekeraars Nederland, Nederlandse Federatie van Universitair Medische Centra, and Samenwerkende topklinische ziekenhuizen.
Basic questions and outcome measures
On the basis of the results of the bottleneck analysis, the chair and the advisors drew up draft basic questions. These were discussed with the working group, after which the working group agreed on the final basic questions.
Regarding the New Basic Questions (3.1; 3.5 and 13.2)
The working group then inventoried for each basic question which outcome measures are relevant for the patient, looking at both desired and undesired effects.
Strategy for Searching and Selecting Literature
Existing international guidelines and systematic reviews were explored. The search strategy or used keywords of the search can be found in appendix 2a, to this chapter.
Regarding the New Basic Questions
Because it had been decided that a systematic literature search was not useful for questions 3.1 and 3.5, a systematic literature search was carried out only for 13.2. To this end, published scientific studies in (various) electronic databases were searched using specific search terms. In addition, studies were also searched for on the basis of the literature lists of the selected articles. In the first instance, studies with the highest degree of evidence in terms of study design were searched for. The working group members selected the articles found through the search on the basis of predetermined selection criteria. The selected articles served to answer the basic question. The databases in which the search was carried out, the search or keywords used in the search as well as the selection criteria used can be found in the chapter dealing with the particular basic question.
Regarding Updates of the Existing Basic Questions
One overarching systematic search was carried out in the Medline and Embase databases. On the basis of general selection criteria, the chair of the working group pre-selected the relevant literature for all basic questions. The members of the working group made a final selection on the basis of specific selection criteria. The selected articles were used to answer the basic question. The databases in which the search was carried out, the search strategy or keywords used in the search as well as the selection criteria used can be found in appendix 2b, to this chapter.
Quality Assessment of Individual Studies
Individual studies were systematically assessed on the basis of methodological quality criteria drawn up beforehand, in order to assess the risk of biased study results. These assessments can be found in the methodological checklists.
Summarizing the Literature
The relevant research data of all selected articles are presented in evidence tables (for the new baseline question) in a clear manner. The most important findings from the literature were described in the summary of the literature.
Assessing the Strength of the Scientific Evidence
Regarding the New Basic Questions (13.2)
A methodologist assessed the certainty of evidence using GRADE.1 GRADE is a method that assigns a grading to the quality of evidence for each outcome measure of an intervention, or for a risk factor or prognostic factor, based on the degree of confidence in the estimation of the effect size (Tables 3 and 4).
Regarding Updates of the Existing Basic Questions
The working group members themselves determined the strength of evidence of the conclusion according to the usual EBRO method in accordance with Tables 1 and 2 (van Everdingen et al, 2004). If necessary, the working group members were supported by a methodologist.
TABLE 3. Grading of the Certainty of Evidence According to GRADE
| High | The authors have a lot of confidence that the true effect is similar to the estimated effect. |
| Moderate | The authors believe that the true effect is probably close to the estimated effect, but may also substantially deviate from this. |
| Low | The authors have little confidence in the estimated effect: the true effect might be markedly different from the estimated effect. |
| Very low | The authors have little confidence in the estimated effect: the true effect is probably markedly different from the estimated effect. |
TABLE 4. The Certainty of the Confidence in the Effect on Estimate is Determined on the Basis of the following Criteria
| Type of evidence | For studies about interventions: | |
| RCT starts in category ’high’. Observational study starts in the category ’low’. | ||
| All other study types start in the category ’very low’ | ||
| For studies about a risk factor or prognostic factor: | ||
| Prospective or retrospective cohort study starts in the category ’high’. For other study designs, downgrading is done via ’risk of bias’. | ||
| Rating down | ‘Risk of bias’ | −1 serious |
| −2 very serious | ||
| Inconsistency | −1 serious | |
| −2 very serious | ||
| Indirect evidence | −1 serious | |
| −2 very serious | ||
| Imprecision | −1 serious | |
| −2 very serious | ||
| Publication bias | −1 probable | |
| −2 highly probable | ||
| Rating up | Large magnitude of effect | +1 large |
| +2 very large | ||
| Dose-response gradient | +1 evidence for gradient | |
| All plausible residual ‘confounding1 | +1 could decrease magnitude of effect | |
| +1 could suggest an opposite effect while the results show no effect | ||
This criterion is applied sporadically. Sometimes a situation occurs in which all plausible confounders (variables that distort results) for which no correction has been made in high-quality observational studies (residual confounders) would result in an underestimation of an apparent treatment effect. For example, if only sicker patients receive experimental treatment and they are better off, it is likely that the actual treatment effect is even greater than the data suggest. A similar situation occurs when observational studies show no treatment effect.
Formulating the conclusions
For the New Basic Questions
A conclusion does not refer to one or more studies, but is drawn on the basis of all the studies together (body of evidence) and per outcome measure.
Regarding Updates of the Existing Basic Questions
The scientific evidence is summarized in one or more conclusions, the level of which is based on the strongest evidence of the relevant studies.
In order to arrive at a recommendation, in addition to the quality of the scientific evidence about the desired and undesired effects of an intervention, or about the effect size of a risk or prognosis factor, other aspects are often important.
In addition to the consideration of beneficial and adverse effects, other aspects are:
costs;
values, preferences and experiences of patients and practitioners with regard to interventions and outcomes of care;
balance of desired and undesired effects of interventions;
feasibility of a recommendation.
These aspects are discussed after the ’conclusion’ under the heading ’considerations’.
Formulating Recommendations
The recommendations answer the basic question and are based on the best available scientific evidence and the most important considerations. The strength of the scientific evidence and the weight given to the considerations by the working group together determine the strength of the recommendation. In accordance with the EBRO and the GRADE methodologies, weak evidence of conclusions in the systematic literature analysis does not exclude a strong recommendation, and weak recommendations with strong evidence are also possible. The strength of the recommendation is always determined by weighing all relevant arguments together.
Preconditions (Organization of Care)
In the bottleneck analysis and in the development of the guideline, explicit account was taken of the organization of care: all aspects that are preconditions for the provision of care (such as coordination, communication, financial resources and other resources, human resources and infrastructure). Preconditions that are relevant to answering a specific basic question are part of the considerations associated with the basic question concerned. More general, overarching or additional aspects of the organization of care are dealt with in Chapter 17.
Indicator Development
Simultaneously with the development of the draft guideline, internal quality indicators were developed to monitor and strengthen the application of the directive in practice. For this purpose, the methodology described in Programm für Nationale VersorgungsLeitlinien von BÄK, KBV und AWMF Qualitätsindikatoren was used. Manual für Autoren: 6. Qualitätsindikatoren für Nationale VersorgungsLeitlinien.2
Knowledge Gaps
During the development of this guideline, a systematic search has been made for research whose results contribute to answering the basic questions. For each basic question, the working group checked whether (additional) scientific research was desirable. An overview of recommendations for further research can be found in the Knowledge gaps chapter.
Comment and Authorization Phase
The draft guideline was submitted to the relevant scientific associations for comments. In addition, it was submitted to the following organizations for comment: Care Institute Netherlands, Inspectorate for Health Care and Youth, Netherlands Healthcare Authority, Patient Federation The Netherlands, LAPOSA, Stichting Kind en Ziekenhuis, Nederlands Huisartsen Genootschap, Zorgverzekeraars Nederland, Nederlandse Federatie van Universitair Medische Centra and Samenwerkende topklinische ziekenhuizen. The comments were collected and discussed with the working group. In response to the comments, the draft guideline was adapted and definitively adopted by the working group. The final guideline was submitted to the relevant associations for authorization and authorized by them.
REFERENCES
-
1.
Guyatt GH, Oxman AD, Vist GE, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ 2008;336:924-6
-
2.
Programm für Nationale VersorgungsLeitlinien von BÄK, KBV und AWMF Qualitätsindikatoren. Manual für Autoren: 6. Qualitätsindikatoren für Nationale VersorgungsLeitlinien, 2009
CHAPTER 3 REFERRAL AND DIAGNOSTICS
Basic Questions
3.1. What are the implications for pregnancy care once craniosynostosis has been prenatally recognised?
3.2. What is the policy on recognition, referral and radiological diagnostics in primary/secondary care in children with suspected craniosynostosis?
3.3. What is the policy regarding genetic diagnostics in a child with confirmed or suspected craniosynostosis?
3.1 What are the implications for pregnancy care once craniosynostosis has been prenatally recognised?
Introduction
Craniosynostosis is rarely recognized prenatally. It does have implications for perinatal care, however. Possibly, recognition will increase due to improved knowledge of prenatal presentation and due to abnormal biometry of the head in the third trimester compared to the regular ultrasound examination. This offers the possibility to adjust the perinatal care trajectory for optimal care of the child during and after birth.
Search and Selection
No systematic literature analysis has been carried out. Relevant publications were used to answer the basic question.
Summary of the Literature
Not applicable. See professional perspective.
Considerations
Quality of Evidence
Not applicable, because no systematic literature analysis has been performed. See professional perspective.
• Values and Preferences
According to the working group, future parents/mothers are generally motivated to have their child referred in the best interests of the child.
• Costs and Resources
Because of the increased diagnostics, the referral to secondary care and the centre of expertise will increase, which will lead to an increase in costs. On the other hand, a cost reduction will be achieved by reducing acute care. As a whole, no major change in financial flows is expected here.
• Professional Perspective
Single-suture non-syndromic synostosis
A pregnant woman with suspected craniosynostosis in the unborn child should be referred to a tertiary care centre for further prenatal diagnostics. If craniosynostosis is diagnosed, the pregnant woman will be referred to the center of expertise for counselling. Counselling should involve at least a clinical geneticist, plastic surgeon/neurosurgeon/maxillofacial surgeon and gynaecologist. The gynaecologist should take over the care, because single-suture craniosynostosis increases the risk of non-natural childbirth.1–5
Multisuture or syndromic synostosis
A pregnant woman with suspected multisuture or syndromic craniosynostosis in the unborn child should be referred to a gynaecologist in a tertiary care centre for further prenatal diagnostics. If craniosynostosis is diagnosed, the pregnant woman is referred to the craniosynostosis expert team for counselling. It is necessary for the gynaecologist in the expertise centre to take over the care, because in the event of multisuture or syndromic craniosynostosis there is a high risk of a non-natural childbirth and of respiratory problems in the neonate.4–9
Rationale for the recommendation(s)
The guiding principle for the formulation of the recommendations is the increased risk of non-natural childbirth and the increased risk of respiratory problems in the neonate.
Recommendations
Single-suture non-syndromic craniosynostosis
In case of ultrasound suspicion of craniosynostosis, the general practitioner or midwife should refer the pregnant woman to a tertiary care centre for prenatal diagnostics. After confirmation of the diagnosis of craniosynostosis, the gynaecologist will take over the care.
After the diagnosis of craniosynostosis, refer the pregnant woman to the craniosynostosis expert team for counselling and information.
Multisuture or syndromic craniosynostosis
In case of ultrasound suspicion of multisuture or syndromic craniosynostosis, the general practitioner, midwife or obstetrician should refer the pregnant woman to a tertiary care centre for prenatal diagnostics. After confirmation of the diagnosis of multisuture or syndromic craniosynostosis, the gynaecologist takes over care at the centre of expertise for syndromic craniosynostosis.
After confirmation of the diagnosis of multisuture or syndromic craniosynostosis, refer the pregnant woman to the craniosynostosis expertise team for counselling and information.
When a child is prenatally diagnosed with other congenital conditions, in addition to craniosynostosis, which predominantly affect life expectancy or quality of life, such as spina bifida or diaphragmatic hernia, counselling focused on the dominant congenital condition may be provided by the clinical geneticist at the university medical centre.
Counselling should involve at least a clinical geneticist, plastic surgeon/neurosurgeon/maxillofacial surgeon and gynaecologist.
Literature
-
1.
Constantine S, David D, Anderson P. The use of obstetric ultrasound in the antenatal diagnosis of craniosynostosis: We need to do better. AJUM 2016;3:1-8
-
2.
Cornelissen MJ, Söfteland M, Apon I, et al. Perinatal complications in patients with single-suture craniosynostosis: An international multicenter retrospective cohort study. J Craniomaxillofac Surg 2017;45:1809-1814
-
3.
Heliövaara A, Vuola P, Hukk J, et al. Perinatal features and rate of cesarean section in newborns with non-syndromic sagittal synostosis. Childs Nerv Syst 2016;32:1289–1292
-
4.
Swanson J, Oppenheimer A, Al-Mufarrej F, et al. Maternofetal trauma in craniosynostosis. Plast Reconstr Surg 2015;136:214-222
-
5.
Weber B, Schwabegger AH, Oberaigner W, et al. Incidence of perinatal complications in children with premature craniosynostosis. J Perinat Med 2010;38:319-325
-
6.
Al-Saleh S, Riekstins A, Forrest CR, et al. Sleep-related disordered breathing in children with syndromic craniosynostosis. J Cranio-Maxillo-Fac Surg 2011 Apr;39:153-7
-
7.
Driessen C, Joosten KF, Bannink N, et al. How does obstructive sleep apnoea evolve in syndromic craniosynostosis? A prospective cohort study. Arch Dis Child 2013;98:538-43
-
8.
Inverso G, Brustowicz KA, Katz E, et al. The prevalence of obstructive sleep apnea in symptomatic patients with syndromic craniosynostosis. Int J Oral Maxillofac Surg 2016;45:167-9
-
9.
Zandieh SO, Padwa BL, Katz ES. Adenotonsillectomy for obstructive sleep apnea in children with syndromic craniosynostosis. Plast Reconstr Surg 2013;131:847-52
3.2 What is the policy on recognition, referral and radiological diagnostics in primary or secondary care in children with suspected craniosynostosis?
Introduction
Craniosynostosis should be recognized in time for optimal treatment. Craniosynostosis patients, however, often turn out not to be recognised or to be referred at a late stage. A complicating factor in the recognition of craniosynostosis is the high incidence of positional cranial deformities. Approximately 20% of all infants have a preferred position in the first few months and are referred to the paediatric physiotherapist for management of the preferred position, whether or not via referral by a general practitioner or a child health care centre doctor. Recognition of craniosynostosis or positional cranial deformity is largely done by physical examination, particularly skull shape, in combination with the medical history, which rarely indicates a radiological examination.1-4
Prior to tertiary care referral, often too many diagnostic procedures such as radiological imaging and genetic analysis are performed, with consequently further delay in referral, an additional burden and uncertainty for the patient and parents, and unnecessary costs. This should be kept to a minimum.
Search and Selection
For the following specific questions, original scientific studies or systematic reviews of scientific studies have been included:
-
1.
Is triage using a flowchart in primary and secondary care effective for the rapid recognition of craniosynostosis and correct referral?
-
2.
What are the causes of late referral to a centre of expertise?
-
3.
Which radiological diagnostics are used in tertiary care for the diagnosis of craniosynostosis?
In the Medline (OVID) and Embase databases, a single comprehensive search was carried out for studies on craniosynostosis. The search strategy is shown in appendix 2, to the guideline. After deduplication, the literature search yielded 2732 hits.
Given the large number of studies, the chair of the working group first selected those that met the following general selection criteria:
General Selection and Exclusion Criteria
| Study type | -original studies -systematic reviews of sufficient quality: - research question of systematic review corresponds (largely) to the basic question - search is performed in at least 2 relevant databases, e.g. Cochrane Library, Medline/PubMed - reporting of the complete search strategy - no relevant keywords/search terms are missing |
| Follow-up period | -minimum follow-up period of 12 months for therapeutic or prognostic studies. |
| Exclusion criteria | - Case-reports - Expert opinion - Letters -Editorials - Case control studies for diagnostic tests - Narrative reviews |
The pre-selected studies that met the specific selection criteria listed in the Table below are included in the literature summary of this chapter.
Specific Selection and Exclusion Criteria
| Selection criteria | - minimum study size: 20 patients for patient series, where no multivariate analysis was used to identify prognostic factors for a relevant outcome measure. - minimum study size: 35 patients for patient series, with multivariate analysis of possible predictive variables for the effect - minimum number of participants of studies with a direct comparative design: 20 per study arm. |
Summary of the Literature
1. Is triage using a flowchart in primary and secondary care effective for the rapid recognition of craniosynostosis and correct referral?
Ghizoni offers a review on synostotic and non-synostotic abnormalities of the skull shape, with a search in Pubmed, ScIELO and LILACS without limitation of time or language.4 In this article, the three questions as formulated by Bredero-Boelhouwer are mentioned as the only source for determining the distinction between synostotic and non-synostotic:3
-
1.
Was the abnormal skull shape present immediately after birth?
-
2.
Is there a preferred posture?
-
3.
Did the skull shape improve?
The article by Bredero-Boelhouwer describes the Dutch situation in which 18 children were evaluated as having craniosynostosis by the referrers; in 14 cases this diagnosis was confirmed in the tertiary centre.3 Of the 89 referrals with the initial diagnosis of non-synostotic occipital plagiocephaly (NSOP) made by the referrer, 10 patients were found to have craniosynostosis (false negative 11.2% and false positive 4/18 = 22.2%). A total of 14 out of 107 patients (13%) were thus misdiagnosed in primary and secondary care. Based on the flowchart, 39 children were assigned to the craniosynostosis group and this diagnosis was justified in 24; none of the patients assessed as NSOP had craniosynostosis (false negative 0%). The false positive level was 38.5%, which was reduced to 25% following a further anamnesis by phone call from the nurse specialist. The use of a flowchart at intake appears to be a safe method to make this distinction quickly and to avoid delay in the treatment process.3
Conclusion
| Level 3 | It is likely that triage using a flow diagram at intake of children with skull shape abnormalities is effective in recognizing craniosynostosis and quickly referring the children to the right specialist, given the false-negative score of 0%. B Bredero-Boelhouwer et al, 2009 C Ghizoni et al, 2016 |
2. What are the causes of late referral to a centre of expertise?
With the exception of the study by Bredero-Boelhouwer, there is no literature available on the referral pattern in the Netherlands.3 Early recognition is important; in case of incorrect or delayed referrals there is a risk of medical complications and less good surgical results.5 Timely referral to a specialised centre is recommended to prevent the use of incorrect diagnostic tools. The delay is often at the level of the paediatrician or general practitioner, because the abnormality is expected to resolve spontaneously.5 Additional diagnostics by a paediatrics department and general practitioners (e.g. CT scan) delays referrals and it is advised to refer immediately without further diagnostics.5 Gandolfi performed an analysis of the referral pattern of 477 referred children based on a suspicion of craniosynostosis, in 197 of whom the diagnosis was confirmed.6 Only 28% had been referred before the age of 3 months. The main risk factors for delayed referral were radiological examinations prior to referral. Other factors found were: having multisuture craniosynostosis, belonging to a racial minority, and having been referred by a healthcare provider other than a paediatrician.
The review by Ghizoni mentions, among other things, the Dutch guideline to refer patients with a suspicion of craniosynostosis directly to a centre of expertise, without additional diagnostics.4
Conclusion
| Level 3 | Delay of referral is usually caused by radiological diagnostics in the centre of the referrer. C Chatterjee et al, 2009; Ghizoni et al, 2016; Gandolfi et al, 2017 |
3. Are there differences in diagnostic accuracy between X-skull, ultrasound and 3D CT scan with regard to the detection or exclusion of craniosynostosis?
Plain skull radiography (skull X-ray) is generally regarded as the first radiological tool for diagnoses of craniosynostosis.1,2,5,7–11 However, recent literature shows a clear role of ultrasound as a primary imaging modality for the detection or exclusion of craniosynostosis in children up to the age of 8 to 9 months.12–14 The studies by Proisy and Hall show that ultrasound has both a high sensitivity of 100% (confidence interval 56.1–100%) and specificity of 100% (confidence interval 90.2–100%) for the detection or exclusion of premature closure of sutures.12,14 In the study by Pogliani, the sensitivity is 100% and the specificity 86%.13 In particular, the non-ionizing technique of ultrasound advocates the use of ultrasound as the first radiological diagnostic tool for suspicion of craniosynostosis. The choice between ultrasound and X-ray examination of the skull is partly determined by the expertise of the executive radiologist.
A 3D CT scan is the most reliable imaging modality for diagnosing craniosynostosis,7–9,15 with a higher diagnostic accuracy compared to ultrasound of the skull or skull X-ray.7–9 The 3D CT scan should preferably be made using low-dose CT techniques (e.g. low tube voltage and iterative reconstruction). Using these low-dose techniques, the effective radiation dose can be reduced to 0.02–0.05 mSv (comparable to the effective radiation dose of a plain skull radiography ranging from 0.01–0.04 mSv), while maintaining adequate diagnostic image quality.16,17 For the methods described above, experience in the preparation and evaluation of these imaging techniques gives higher reliability.9,13,14 In children with skull shape deformities suspected of craniosynostosis (medium risk craniosynostosis), an ultrasound scan of the skull or skull X-ray must first be performed, followed by a 3D CT scan if the primary screening modality raises suspicion of craniosynostosis.2,5,10,18–21 In the case of evident clinical suspicion of craniosynostosis (high-risk craniosynostosis), a 3D CT scan should be performed immediately, without an ultrasound scan of the skull or skull X-ray.9 Cerovac concludes from a retrospective study of 109 single-suture craniosynostoses that an experienced clinician can make the diagnosis with 100% certainty on clinical grounds.8 The diagnosis should be confirmed with an ultrasound of the skull or skull X-ray, even if it is less reliable (80–100% and 91% respectively). 3D CT scanning should be reserved for questionable diagnostic cases or for surgical preparation.8
MRI is performed on indication in syndromic craniosynostosis. Black Bone MRI is a promising alternative to 3D CT scan of the skull in children with syndromic craniosynostosis for whom an MRI examination to detect or exclude associated intracranial abnormalities is indicated.22 The disadvantage of Black Bone MRI is that the examination generally has to be performed under anaesthesia.
Conclusions
| Level 3 | Ultrasound scan of the skull or X-skull is considered the first radiological diagnostic tool for children with skull shape abnormalities suspected of craniosynostosis (medium risk craniosynostosis). C Proisy et al, 2017; Hall et al, 2017; Pogliani et al, 2017; Simanovsky et al, 2009; Komotar et al, 2006; Ridgway et al, 2004; White et al, 2010; Parameters ACPA, 2007; Gellad et al, 1985; Cerovac et al, 2002; Medina et al, 2002 |
| Level 3 | Low dose 3D-CT is considered the first radiological diagnostic tool for children with high clinical suspicion of craniosynostosis (high-risk craniosynostosis). C Chatterjee et al, 2009; Komotar et al, 2006; Bruce et al, 1996; Mathijssen et al, 2007; Parameters ACPA, 2007; Strauss et al, 1998; Medina et al, 2002; Ernst et al, 2016; Kaasalainen et al, 2015 |
Considerations
• Evidence of the Conclusions
Recognition of craniosynostosis in primary and secondary care
The number of articles is very low and actually limited to one article that is specific to the Netherlands and of good quality.
Referral
The number of articles is very low and of moderate quality.
Radiological diagnostics
There is a reasonable number of articles on the specificity and sensitivity of ultrasound, X-skull and CT scan for the diagnosis of craniosynostosis, but these are mostly from centres of expertise. There is no literature available on the specificity and sensitivity of these examinations after assessment in a non-expertise centre.
• Values and Preferences
Recognition of craniosynostosis in primary and secondary care
Most parents will prefer to get a quick and reliable assessment of their child's skull deformity in their own region. If there is any doubt on the side of the parents or care provider, consultation with a centre of expertise is preferred in order to obtain certainty and to enable early treatment.
Referral
Most parents of a child with a suspicion of craniosynostosis will preferably get an appointment at the centre of expertise quickly, so that certainty about the diagnosis can be obtained and treatment can be initiated in time.
Radiological diagnostics
With regard to radiological imaging, parents will preferably want to keep the number of uncomforTable examinations for the child as low as possible and keep the exposure to radiation from the radiological examinations for the child to a minimum. Therefore, in secondary care, ultrasound of the cranial sutures will be preferred as the first imaging diagnostic tool in case of suspected craniosynostosis, under the condition of sufficient expertise of the executing radiologist. If the executing radiologist has insufficient experience with ultrasound of the cranial sutures, X-skull is an alternative for which expertise is also required. The further development of Black Bone MRI (non-ionizing) for diagnosing craniosynostosis and the possible replacement of the 3D CT scan of the skull (ionizing) in the future is a task for the centres of expertise. Secondary care radiological imaging of children with suspected craniosynostosis should not delay any referral to an expertise centre.
• Costs and Resources
Recognition of craniosynostosis in primary and secondary care
The assessment of an abnormal skull shape rarely requires additional radiological imaging. If there is doubt about the nature of the abnormality, it is better to consult with the centre of expertise rather than proceed with additional diagnostics, because of radiation exposure and costs.
Referral
The use of radiological and/or genetic diagnostics by the referrer leads to costs and use of resources, while the research is not always indicated, might be of insufficient quality or needs to be expanded. For these reasons, in addition to the resulting delay in referral, this should be avoided.
Radiological diagnostics
Introducing Black Bone MRI for diagnosing craniosynostosis and possibly replacing the 3D CT scan of the skull in the future will increase the cost of radiological imaging; on the one hand because MRI is a more expensive radiological technique and, on the other hand, because MRI examination has to be performed under general anaesthesia in order to obtain adequate imaging quality.
• Professional Perspective
Recognition of craniosynostosis in primary and secondary care
The flow diagram distinguishing between positional cranial deformity and craniosynostosis should be the guiding principle for primary or secondary care assessment. If there is any doubt, consultation with a centre of expertise is recommended in order to prevent unnecessary diagnostics.
Referral
Many parents experience a long process between first contact with a doctor and the final referral to the expertise centre. The referral from primary care to a paediatrician contributes to a somewhat longer process, but the contribution of the paediatrician in the patient's own region at this stage and in the follow-up process is of great value. However, it is important to ensure a quick referral, preferably before the age of 3 months, in order to keep minimally invasive surgery open as an option, for example.
As reported in the annual reports of the craniosynostosis expert team of Erasmus University Medical Center, some children with craniosynostosis are still being referred late.
Radiological diagnostics
In a tertiary centre, additional medical imaging for the differentiation between positional cranial deformities and craniosynostosis is rarely performed. To avoid unnecessary radiological examination (with associated costs, radiation load, burden on patient and parents, lack of added value and required experience in interpreting the images), ultrasound scan of the skull or X-skull are not recommended if the clinical diagnosis concerns a positional cranial deformity. In case of doubt, consultation with a tertiary craniosynostosis expertise centre is indicated, in which case sending the ultrasound examination (dynamic recordings) or the X-skull examination (front view, side view, back view and top view) is often sufficient.
For surgical planning, the standard use of a 3D CT scan of the skull to objectify the abnormality is highly recommended, given the major implications of making the diagnosis of craniosynostosis incorrectly: only after surgical opening of the scalp will this error be recognised.
An additional MRI examination of the skull is of added value in children with syndromic craniosynostosis for the purpose of detecting or excluding associated defects of the brain and signs of increased intracranial pressure and the simultaneous assessment of aberrant venous vascular structures that may have implications for surgical planning.
• Balance of Anticipated Desired and Undesired Outcomes
Recognition of craniosynostosis in primary and secondary care
Direct referral of all children with a deviation of the skull shape leads to a capacity problem at the centres of expertise and an unnecessary burden on parents to visit these centres, which are often located outside their own regions. A wait-and-see policy in primary and secondary care with regard to skull shape abnormalities can lead to a referral that is too late to provide the most effective treatment and keeps parents insecure. In case of doubt, consultation should take place to avoid delay and unnecessary diagnostics.
Referral
Rapid determination of the diagnosis is desirable but can lead to use of diagnostic tools in primary and secondary that are not the right or desired for the practitioner in the centre of expertise. These diagnostics are therefore better performed in the expertise centre in order to guarantee correct use of staff and resources, avoid unnecessary radiation exposure and avoid delay in referral to the expertise centre.
Radiological diagnostics
Adequate and timely radiological diagnostics in children with skull deformities is of great importance. In secondary care, a trade-off must be made between the added value of and available expertise for carrying out the recommended radiological examination and the possible resulting delay in referral to an expertise centre.
With regard to the additional radiological diagnostics for pre-operative planning in the tertiary centers, the 3D CT scan of the skull will currently be the primary modality because of its superior diagnostic accuracy, short scan time (no anaesthesia required) and wide availability of the CT scanner. The advantages of MRI are the non-ionizing technique and the additional information on intracranial pathology, although currently the long duration of scanning (anaesthesia required) and moderate availability make the use of MRI in clinical practice even more difficult.
Rationale for the recommendation(s)
Recognition of craniosynostosis in primary and secondary care
The guiding principle is to timely differentiate children with craniosynostosis from children with non-synostotic deformities of the skull, with early referral to allow minimally invasive surgical techniques (before the age of 6 months). The initial assessment may well take place in the patient's own region, but in case of doubt on the side of parents or care provider, or in case of insufficient improvement, consultation with a centre of expertise is indicated.
Referral
The guiding principle is to have children with a suspicion of craniosynostosis assessed as soon as possible in the expertise centre, so that parents can be sure of the diagnosis and informed about the treatment. Timely referral also keeps the option of early surgical treatment open (before the age of 6 months).
Radiological diagnostics
The guiding principle is to make the correct diagnosis with regard to the detection or exclusion of craniosynostosis. In addition, it is important to minimize the use of ionizing radiological imaging in order to make the correct diagnosis, taking into account the available radiological facilities.
Recommendations
Triage in primary and secondary care
Use the flow diagram (Bredero-Boelhouwer, 2009) to optimize the detection of craniosynostosis in primary and secondary care.3
Referral
Refer a child with a suspicion of craniosynostosis to a craniosynostosis expertise centre as soon as possible, without additional diagnostics.
Radiological diagnostics in a craniosynostosis expertise centre
Perform an ultrasound scan of the skull or X-skull in children with abnormal skull shape and a moderate risk of craniosynostosis.
Always perform a low dose 3D CT scan in children with abnormal skull shape and a high clinical suspicion of craniosynostosis (high-risk craniosynostosis).
Research Gaps
The scientific literature lacks studies on the consequences of late referral of children with craniosynostosis.
Literature
-
1.
Ridgway EB, Weiner HL. Skull deformities. Pediatr Clin N Am 2004;51:359-87
-
2.
Komotar RJ, Zacharia BE, Ellis JA, et al. Pitfalls for the pediatrician: positional molding or craniosynostosis. Pediatr Ann 2006;35:365-75
-
3.
Bredero-Boelhouwer H, Treharne LJ, Mathijssen IMJ. A triage system for referrals of pediatric skull deformities. J Craniofac Surg 2009;20:242-5
-
4.
Ghizoni E, Denadai R, Raposo-Amaral CA, et al. Diagnosis of infant synostostic and non-synostotic cranial deformities: a review for paediatricians. Rev Paul Pediatr 2016;34:495-502
-
5.
Chatterjee JS, Mahmoud M, Karthikeyan S, et al. Referral pattern and surgical outcome of sagittal synostosis. J Plast Reconstr Aesthet Surg 2009;62:211-15
-
6.
Gandolfi BM, Sobol DL, Farjat AE, et al. Risk factors for delayed referral to a craniofacial specialist for treatment of craniosynostosis. J Pediatr 2017;186:165-71
-
7.
Gellad FE, Haney PJ, Sun JCC, et al. Imaging modalities of craniosynostosis with surgical and pathological correlation. Pediatr Radiol 1985;15:285-90
-
8.
Cerovac S, Neil-Dwyer JG, Jones BM, et al. Are routine preoperative CT scans necessary in the management of single suture craniosynostosis? Br J Neurosurg 2002;16:348-54
-
9.
Medina LS, Richardson RR, Crone K. Children with suspected craniosynostosis: a cost-effectiveness analysis of diagnostic strategies. Am J Roentgenol 2002;179:215-21
-
10.
Parameters for evaluation and treatment of patients with cleft lip/palate or other craniofacial anomalies. Official publication of the American Cleft Palate-Craniofacial Association. Revised edition November 2007. East Franklin Street, Chapel Hill, NC, USA, 2007
-
11.
White N, Warner RM, Noons P, et al. Changing referral patterns to a designated craniofacial centre over a four-year period. J Plast Reconstr Aesthet Surg 2010;63:921-5
-
12.
Hall KM, Besachio DA, Moore, MD, et al. Effectiveness of screening for craniosynostosis with ultrasound: a retrospective review. Pediatr Radiol 2017;47:606-12
-
13.
Pogliani L, Zuccotti GV, Furlanetto M, et al. Cranial ultrasound is a reliable first step imaging in children with suspected craniosynostosis. Child Nerv Syst 2017;33:1545-52
-
14.
Proisy M, Riffaud L, Chouklati K, et al. Ultrasonography for the diagnosis of craniosynostosis. Eur J Radiol 2017;90:250-255
-
15.
Parisi M, Mehsdizadeh HM, Hunter JC, et al. Evaluation of craniosynostosis with three-dimensional CT imaging. J Comput Assist Tomogr 1989;13:1006-12
-
16.
Ernst CW, Hulstaert TL, Belsack D, et al. Dedicated sub 0.1 mSv 3DCT using MBIR in children with suspected craniosynostosis: quality assessment. Eur Radiol 2016;26:892-9
-
17.
Kaasalainen T, Palmu K, Lampinen A, et al. Limiting CT radiation dose in children with craniosynostosis: phantom study using model-based iterative reconstruction. Pediatr Radiol 2015;45:1544-53
-
18.
Selber J, Reid RR, Chike-Obi CJ, et al. The changing epidemiologic spectrum of single-suture synostosis. Plast Reconstruct Surg 2008;122:527-33
-
19.
Bruce DE. Consensus: craniofacial synostoses. Childs Nerv Syst. 1996 Nov;12(11): 734-6.
-
20.
Mathijssen IMJ, Arnaud E. Benchmarking for craniosynostosis. J Craniofac Surg 2007;18:436-42
-
21.
Strauss RP. Cleft palate and craniofacial teams in the United States and Canada: a national survey of team organization and standards of care. Cleft Palate Craniofac J 1998;35:473-80
-
22.
Eley KA, Watt-Smith SR, Sheerin F, et al. “Black Bone” MRI: a potential alternative to CT with three-dimensional reconstruction of the craniofacial skeleton in the diagnosis of craniosynostosis. Eur Radiol 2014;24:2417-26
3.3 What is the policy regarding genetic diagnostics in a child with confirmed or suspected craniosynostosis?
Introduction
To an increasing extent, genetic causes are being identified in all types of craniosynostoses. Genetic diagnostics are relevant for counselling of parents and predicting the clinical course and management of the child.1
Search and Selection
No systematic literature analysis has been carried out. Relevant publications were used to answer the basic question.
Summary of the Literature
Not applicable.
Considerations
• Quality of Evidence
Not applicable, as no systematic literature analysis has been performed. See professional perspective.
• Values and Preferences
By performing diagnostics only after parents had received information and provided consent, room was left for the values and preferences of parents.
• Costs and Resources
Genetic diagnostics in single-suture craniosynostosis means an increase in costs compared to the previous guideline. Offering a craniosynostosis panel in children “with proven craniosynostosis without obvious phenotype” compared to multiple single gene testing is cost-efficient.
• Professional Perspective
Genetic diagnostics in a centre of expertise for craniosynostosis
Optimal use and assessment of genetic diagnostics will benefit from multidisciplinary input, as is done in a centre of expertise for craniosynostosis. Additional reasons are the prevention of either too few or unnecessary diagnostics, because sub-optimal recognition of craniosynostosis or related disorders is more likely outside the expertise centre. Moreover, this centralization of diagnostics makes the samples potentially available for scientific research.
In children with suspected craniosynostosis
If the diagnosis of craniosynostosis has not yet been proven, the use of clinical genetic diagnostics targeting craniosynostosis genes is not useful.
In children with proven craniosynostosis and evident phenotype
Focused diagnostics give these children faster results with fewer costs.
In children with proven craniosynostosis and other birth defects and/or developmental disorders
Diagnostic tests in these children are aimed at recognizing chromosomal abnormalities and syndromes associated with craniosynostosis. These diagnostic tests are: array analysis, targeted DNA diagnostics for a particular syndrome and Next Generation Sequencing. Next Generation Sequencing panels are particularly indicated if there is no specific syndrome diagnosis with possible occurrence of other congenital abnormalities and/or developmental disorders (craniosynostosis single package, multiple congenital anomalies trio analysis).1–6
In children with proven craniosynostosis without evident phenotype
NGS craniosynostosis panel gives the highest chance of identifying rare genetic causes of craniosynostosis. Trio analysis can increase the reliability of the genetic findings.2,7-10
Rationale for the recommendation(s)
Centralized care for the rare condition craniosynostosis from diagnosis to treatment and aftercare is leading in the preparation of the recommendations.
Recommendations
Genetic diagnostics are in principle performed in a centre of expertise for craniosynostosis.
In children with suspected craniosynostosis
Offer clinical genetic diagnostics only to children with proven craniosynostosis.
In children with proven craniosynostosis and evident phenotype
Offer targeted clinical genetic diagnostics.
In children with proven craniosynostosis and other birth defects and/or developmental disorders
Perform array analysis, targeted DNA diagnostics, Next Generation Sequencing craniosynostosis panel (single) or NGS Multiple Congenital Anomalies/Intellectual disability trio analysis, possibly followed by ’opening exome’.
In children with proven craniosynostosis without an evident phenotype
Perform NGS craniosynostosis panel.
Perform trio analysis on indication.
Literature
-
1.
Johnson D, Wilkie AO. Craniosynostosis. Eur J Hum Genet. 2011 Apr;19(4):369-76.
-
2.
Agochukwu NB, Solomon BD, Muenke M. Impact of genetics on the diagnosis and clinical management of syndromic craniosynostosis. Child Nerv Syst 2012;28:1447-63
-
3.
Azimi C, Kennedy SJ, Chitayat D, et al. Clinical and genetic aspects of trigonocephaly: a study of 25 cases. Am J Med Genet 2003;117A:127–35
-
4.
Clarke CM, Fok VT, Gustafson JA, et al. Single suture craniosynostosis: Identification of rare variants in genes associated with syndromic forms. Am J Med Genet 2018;176:290-300
-
5.
Ittleman BR, Mckissick J, Bosanko KA, et al. Less common underlying genetic diagnoses found in a cohort of 139 individuals surgically corrected for craniosynostosis. Am J Med Genet 2018;176:487-91
-
6.
Ye X, Guilmatre A, Reva B, et al. Mutation screening of candidate genes in patients with nonsyndromic sagittal craniosynostosis. Plast Reconstr Surg 2016;137:952-61
-
7.
Timberlake AT, Choi J, Zaidi S, et al. Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles. Elife 2016;5
-
8.
Miller KA, Twigg SR, McGowan SJ, et al. Diagnostic value of exome and whole genome sequencing in craniosynostosis. J Med Genet 2017;54:260-8
-
9.
Lattanzi W, Barba M, Di Pietro L, et al. Genetic advances in craniosynostosis. Am J Med Genet 2017;173A:1406-29
-
10.
Wilkie AOM, Johnson D, Wall SA. Clinical genetics of craniosynostosis. Curr Opin 2017;29:622-8
CHAPTER 4 PERIOPERATIVE CARE
4.1 What is the perioperative surgical management of craniosynostosis?
Introduction
The correction of craniosynostosis at childhood age, with the exception of minimally invasive techniques, can be associated with relatively high blood loss. This risk increases with extensive, open skull corrections. In addition to the surgical and anesthesiological challenge, the comorbidity associated with the syndromic conditions must be taken into account. For this reason, strict organisational conditions must be imposed on the surgical process, before, during and after the procedure.
The Dutch “Guideline on the Qualification of Paediatric Surgery” states that anesthesiological goals in complex care such as craniofacial surgery can only be guaranteed in specialised paediatric centres.
Open cranial surgery in children with craniosynostosis is a model for operations with relatively high blood loss. The conclusions and recommendations from this chapter can be extrapolated to all surgical treatments with relatively high blood loss, although the indications may vary according to the type of surgical procedure.
Search and Selection
For the following specific question, original scientific studies or systematic reviews of scientific studies have been included:
-
1.
Which substances (tranexamic acid, erythropoietin, fibrinogen, fresh frozen plasma, vitamin K1) or measures, such as inducing hypotension or use of the cell saver, are effective in reducing blood loss or the need for blood transfusion?
In the Medline (OVID) and Embase databases, a single comprehensive search was carried out for studies on craniosynostosis. The search strategy is shown in appendix 2, to the guideline. After deduplication, the literature search yielded 2732 hits.
Given the large number of studies, the chair of the working group first selected those that met the following general selection criteria:
General Selection and Exclusion Criteria
| Study type | -original studies -systematic reviews of sufficient quality: - research question of systematic review corresponds (largely) to the basic question - search is performed in at least 2 relevant databases, e.g. Cochrane Library, Medline/PubMed - reporting of the complete search strategy - no relevant keywords/search terms are missing |
| Follow-up period | -minimum follow-up period of 12 months for therapeutic or prognostic studies. |
| Exclusion criteria | -Case-reports - Expert opinion - Letters - Editorials - Case control studies for diagnostic tests - Narrative reviews |
The pre-selected studies that met the specific selection criteria listed in the Table below are included in the literature summary of this chapter.
Specific Selection and Exclusion Criteria
| -minimum study size: 20 patients |
Summary of the Literature
Which substances (tranexamic acid, erythropoietin, fibrinogen, fresh frozen plasma, vitamin K1) or measures, such as inducing hypotension or use of the cell saver, are effective in reducing blood loss or the need for blood transfusion?
One of the potential problems in the surgical correction of craniosynostosis is massive blood loss, which can occur during surgery in a relatively short period and in patients with a small circulating volume due to the young age.1 Predictive factors for blood loss are type of surgery (more blood loss with open skull corrections compared to minimally invasive surgery) and duration of surgery.2
Tranexamic acid Administration, Cell Saver and EPO Administration
In a systematic review, White investigated all different methods to reduce blood loss by searching Cochrane Central, Medline and EMBASE.3 Of the 696 articles, 18 were included, 14 case series with control group and 4 RCTs. Due to a large variation in patient characteristics, different definitions and outcome measures, there were all sorts of limitations that made a meta-analysis impossible. Only for the use of tranexamic acid, a meta-analysis of 3 RCTs was possible. The reduction in transfused blood volume was evident, but not all studies indicated whether this also involved a reduction in the number of units and therefore donors. The mean transfused blood volume of the tranexamic group versus the control group in these three studies were: 58 versus 133 cc; 185 versus 258 cc; 376 versus 655 cc. The value of using the cell saver was less obvious as no RCTs were done. Three of the four studies claimed a lower transfusion volume but the fourth study did not. Erythropoietin (EPO) administration was described in five non-randomized studies, but sometimes in combination with other interventions. All studies were of low quality, but all reported a reduction in transfusion requirements.
About the usefulness of the other methods such as haemodilution, aminocaproic acid, aprotinin, fibrin seals and fibrin glue, no reliable statement could be made.
Fibrinogen Administration
Haas describes a non-blinded RCT of fibrinogen administration in 31 children with craniosynostosis, with 14 children receiving fibrinogen at a FIBTEM MCF of < 8 mm and 17 children receiving an earlier administration as soon as the measurement was < 13 mm.4 This policy resulted in a significant decrease in transfused blood volume from a mean of 56 ml/kg to 28 ml/kg. This is clinically relevant as only 1 unit (from 1 donor) was averaged instead of 2 units (from 2 donors). Haas and Bolliger describe the shortcomings of the study, i.e. too low power due to insufficient inclusion of patients, insufficient power to perform safety analyses on the fibrinogen administration (especially thromboembolic events).4,5 Especially this last uncertainty makes that fibrinogen administration on FIBTEM MCF lower than 13 mm cannot be readily implemented. Bolliger concludes in his comment on Haas's article, and in light of related studies, that the prophylactic administration of fibrinogen is not recommended, but a concentration below 2.0 gr/l or a FIBTEC MCF < 10 mm is an accepTable trigger point for initiating haemostatic interventions in patients with a high bleeding risk.5
Fresh Frozen Plasma (FFP) Administration
In general, FFPs are administered as soon as there is evidence of abnormal coagulation during the procedure, such as diffuse blood loss, lack of clot formation or abnormal coagulation parameters. In an RCT, Pieters investigated whether prophylactic administration of FFPs would reduce transfused blood volume and donor exposure.6 The study included 81 children of whom 41 were in the prophylactic group. Despite the better laboratory parameters for coagulation, there was no difference in measured blood loss, transfused blood volume or donor exposition.
Hypotension
Fearon investigated in a randomised study in 100 children whether or not hypotension during surgery is effective in reducing blood loss.7 In one group a mean arterial blood pressure of 50 mm Hg was aimed for (n = 53), and in the other group 60 mm Hg (n = 47): the achieved blood pressure values were 55 and 65 mm Hg, respectively. The cell saver was used to collect 163 cc and 204 cc of blood, respectively. Postoperative Hb was not significantly different (8.8 versus 9.3), as was transfusion requirement (9/53 versus 6/47). The pursuit of hypotension within the mentioned values therefore does not seem to be of added importance.
Vitamin K1 Administration
Despite the seemingly good design of a placebo-controlled RCT towards the use of vitamin K1 administration at the start of surgery, the study appears to be of poor quality.8 Only 15 patients were included, six of whom received vitamin K1. However, the perioperative management was highly variable per anaesthetist, which means that there are hardly any conclusions to be drawn from the finding that both groups needed the same number of transfusions.
Conclusions
| Level 2 | The use of tranexamic acid is likely to result in a strong reduction of the transfused blood volume and possibly reduces the need for transfusion. B Duran et al, 2003; Dadure et al, 2011; Goobie et al, 2011 |
| Level 3 | The use of the cell saver and erythropoietin may result in a reduction of the transfused blood volume and reduced need for transfusion. C Deva et al, 2002; Jimenez and Barone, 1995; Duncan et al, 2008; Dahmani et al, 2000 |
| Level 3 | The use of the other strategies (haemodilution, aminocaproic acid, aprotinin, fibrin seals and fibrin glue) has an unproven effect on transfused blood volume and need for transfusion. White et al, 2015 |
| Level 3 | Fibrinogen administration on a FIBTEM MCF < 13 mm may result in a lower transfused blood volume compared to a threshold of < 8 mm. However, the safety of fibrinogen administration at a threshold of 13 mm has not yet been sufficiently demonstrated. B Haas et al, 2015, C Bolliger et al, 2015 |
| Level 3 | The prophylactic use of fresh frozen plasma may not lead to reduced blood loss and thus not lead to reduced need of transfusion compared to reactive use of fresh frozen plasma. B Pieters et al, 2015 |
| Level 3 | Within the range of a mean arterial blood pressure of 55 mm Hg to 65 mm Hg, there is no significant difference in the need of blood transfusion and the pursuit of hypotension may not be of added value. B Fearon et al, 2014 |
| Level 3 | It is unclear whether vitamin K1 administration to reduce blood loss or blood transfusion is useful. C Kicker et al, 2014 |
Considerations
• Evidence of the Conclusions
There is fairly strong evidence for the use of tranexamic acid. For the other measures, this evidence is weaker or even non-existent. According to the literature consulted, the dosage is not uniform.
• Values and Preferences
According to the working group, parents are generally aware of the possible drawbacks of transfusion and often agree with measures aimed at reducing the amount of blood transfused and the number of donors.
• Costs and Resources
Application of the recommendations is expected by the guideline committee to lead to little or no increase in costs, because the recommendations are in line with existing practice.
• Professional Perspective
Tranexamic acid is frequently used in craniosynostosis surgery and has hardly any complications.
• Balance of Anticipated Desired and Undesired Outcomes
The desired results from the use of tranexamic acid, such as a reduction in the volume of transfused blood and in the number of donor exposures, clearly outweigh the low-frequency side effects, such as thromboembolic disorders. In addition, tranexamic acid costs are low.
Use of the cell saver can be considered for extensive, open skull repairs where significant blood loss in relation to body weight is expected; for average blood loss, the yield of the cell saver is often insufficient to avoid a blood transfusion and the additional cost of the cell saver also weighs in.
Administration of EPO is not proven effective enough, requires a number of hospital visits and repeated blood tests on the child, and is expensive. The use of EPO is therefore not considered to be useful.
The use of fibrinogen and/or fresh frozen plasma is reserved for situations in which blood coagulation is considered abnormal in order to improve blood coagulation. Prophylactic use has no proven efficacy and is associated with higher costs.
Rationale for the recommendation(s)
The guiding principle for the perioperative treatment of a child with craniosynostosis is safety. For this reason, this type of surgery is only performed in a specialised paediatric centre. The aim is to reduce blood loss, provided that the measures taken are safe and effective and that the costs are in proportion.
Tranexamic acid is effective in limiting blood loss, has few side effects, and is relatively inexpensive. For open skull corrections, the cell saver is an additional option, but only if significant blood loss is expected. Only then is the yield of own blood volume for autotransfusion sufficient to justify the extra costs involved and is there a potential benefit by reducing donor exposures. With an average blood loss, these advantages are less evident and the use of the cell saver will often be dispensed with because of the costs.
Fresh frozen plasma and/or fibrinogen use may not lead to reduced blood loss if used prophylactically, while it does generate costs. These means are reserved to correct blood coagulation as soon as it becomes abnormal during the procedure in order to reduce blood loss.
Recommendations
Organisational condition(s) for safe perioperative care
Children with craniosynostosis should be treated in a specialized children's center.
Measures to reduce blood loss or need for blood transfusion in operations with expected high levels of blood loss
Use tranexamic acid during surgical correction to reduce blood loss.
Consider the use of a cell saver to reduce the volume of blood transfusion needed.
Use fresh frozen plasma and/or fibrinogen as soon as signs of abnormal coagulation develop during surgery.
Research Gaps
Of the perioperative measures described that may reduce blood loss, the effectiveness and/or safety of most of them has either not or hardly been proven.
Literature
-
1.
Koh JL, Gries H. Perioperative management of pediatric patients with craniosynostosis. Anesthesiol Clin 2007;25:465-81
-
2.
Meier PM, Zurakowski D, Goobie SM, et al. Multivariable predictors of substantial blood loss in children undergoing craniosynostosis repair: implications for risk stratification. Paediatr Anaesth 2016;26:960-9
-
3.
White N, Bayliss S, Moore D. Systematic review of interventions for minimizing perioperative blood transfusion for surgery for craniosynostosis. J Craniofac Surg 2015;26:26-36
-
4.
Haas T, Spielmann N, Restin T, et al. Higher fibrinogen concentrations for reduction of transfusion requirements during major paediatric surgery: A prospective randomised controlled trial. Br J Anaesth 2015;115:234-43
-
5.
Bolliger D, Tanaka KA. Haemostatic efficacy of fibrinogen concentrate: is it the threshold or the timing of therapy? Br J Anaesth 2015;115:158-61
-
6.
Pieters BJ, Conley L, Weiford J, et al. Prophylactic versus reactive transfusion of thawed plasma in patients undergoing surgical repair of craniosynostosis: a randomized clinical trial. Paediatr Anaesth 2015;25:279-87
-
7.
Fearon JA, Cook TK, Herbert M. Effects of hypotensive anesthesia on blood transfusion rates in craniosynostosis corrections. Plast Reconstr Surg 2014;133:1133-6
-
8.
Kicker JS, Willson DF, Kelly RL, et al. Impact of supplemental vitamin K1 administration on postoperative blood component requirements after craniosynostosis repair: a prospective, placebo-controlled, randomized, blinded study. J Craniofac Surg 2014;25:154-9
CHAPTER 5 SURGICAL TREATMENT OF ISOLATED, NON-SYNDROMIC CRANIOSYNOSTOSIS
5.1 What is the surgical management of non-syndromic craniosynostosis?
Introduction
The four most common forms of isolated, non-syndromic craniosynostosis are in order of prevalence: sagittal suture synostosis (scaphocephaly), metopic suture synostosis (trigonocephaly), unilateral coronal suture synostosis (frontal plagiocephaly) and unilateral lambdoid suture synostosis (pachycephaly). Unilateral coronal suture synostosis may be associated with a syndrome, such as Muenke or Saethre-Chotzen syndrome, and a genetic cause should be considered.
Definitions
-
-
Severe form of trigonocephaly: when the most prominent part of the forehead is in the midline of the forehead. In top view, the lateral orbital rims are clearly visible due to the retrusion of the lateral parts of the forehead.1
-
-
Moderate form of trigonocephaly: when the most prominent part of the forehead lies between the medial parts of both eyebrows. In top view, the lateral orbital rims are either just or not visible.1
-
-
Mild form of trigonocephaly: if the above definitions are not met. There is discussion about this definition among the experts as it is a sliding scale of deformation without a cut-off point. In top view, the lateral orbit rims are not or hardly visible.1
Search and Selection
For the following specific questions, original scientific studies or systematic reviews of scientific studies have been included:
-
1.
What is the indication for surgery in the different types of non-syndromic craniosynostosis?
-
2.
What are the patient-relevant effects of different surgical techniques, in particular minimally invasive surgery versus open surgery for the different types of non-syndromic craniosynostosis?
-
3.
What are the patient-relevant effects of different timing of surgery, i.e. ‘early’ (below 6 months of age) versus ‘late’ (above 6 months of age)?
In the Medline (OVID) and Embase databases, a single comprehensive search was carried out for studies on craniosynostosis. The search strategy is shown in appendix 2, of the guideline. After deduplication, the literature search yielded 2732 hits.
Given the large number of studies, the chair of the working group first selected those that met the following general selection criteria:
General Selection and Exclusion Criteria
| Study type | -original studies - systematic reviews of sufficient quality: - research question of systematic review corresponds (largely) to the basic question - search is performed in at least 2 relevant databases, e.g. Cochrane Library, Medline/PubMed - reporting of the complete search strategy - no relevant keywords/search terms are missing |
| Follow-up period | -minimum follow-up period of 12 months for therapeutic or prognostic studies. |
| Exclusion criteria | -Case-reports - Expert opinion - -Letters - -Editorials - -Case control studies for diagnostic tests - -Narrative reviews |
The pre-selected studies that met the specific selection criteria listed in the Table below are included in the literature summary of this chapter.
Specific Selection and Exclusion Criteria
| Selection criteria | - minimum study size: 20 patients for patient series, where no multivariate analysis was used to identify prognostic factors for a relevant outcome measure. - minimum study size: 35 patients for patient series, with multivariate analysis of possible predictive variables for the effect - minimum number of participants of studies with a direct comparative design: 20 per study arm. |
Summary of the Literature
1. What is the indication for surgery in the different types of non-syndromic craniosynostosis?
Indication for Surgical Treatment of Metopic Suture Synostosis
The two extremes of the spectrum of clinical presentation of metopic suture synostosis are a bone ridge on the metopic suture in an otherwise normal forehead and a classic trigonocephaly or wedge skull in which there is a narrow forehead, a prominent bone ridge at the site of the metopic suture, hypotelorism, lateral orbital hypoplasia, biparietal widening, epicanthic folds and upward eyebrows.1 There are few objective criteria that determine whether an operation is indicated. Only for the most pronounced presentation of trigonocephaly, surgery is associated with an undisputed gain of aesthetic result, but whether this is also achieved for milder presentations is unclear. Whether surgery is associated with gains in neurocognition by has not yet been determined, even for the extreme presentation. Birgfeld describes an analysis to objectify the surgical indication, using the phenotypic description by the doctor, photo analysis and CT scan measurements.1 In 34% of the photographs, the assessors disagreed with the classification as metopic ridge or metopic craniosynostosis (i.e. trigonocephaly). Characteristic for genuine trigonocephaly appears to be parental recognition of the abnormality at a younger age and the relationship between lateral parts of the forehead and the lateral orbital rim seen from above, with the frontal bone having a straight course and the lateral orbital rim becoming visible.
Cho describes a CT method to differentiate the metopic ridge from metopic craniosynostosis by curvature measurements of forehead and lateral orbit, although the cut-off point remains subjective in the determination of surgical indication.2 Anolik describe a similar scoring method for photographs and CTs.3 They, too, find poor consensus for the intermediate presentation of metopic craniosynostosis.
Indication for Surgical Treatment of Sagittal Suture Synostosis, Unilateral Coronal Suture Synostosis and Unilateral Lambdoidal Suture Synostosis
In the literature on sagittal, coronal or lambdoidal suture synostosis, there is no discussion about whether or not there is an indication for surgery. No spontaneous improvement of the abnormal skull shape is expected.
Conclusions
| Level 2 | Only for the most distinct form of metopic craniosynostosis is the indication for surgery undisputed. In one out of three patients there is doubt among experts about the diagnosis of mild or moderate metopic craniosynostosis and therefore about the indication for surgery. B Birgfeld et al, 2013; Cho et al, 2016; Anolik et al, 2016 |
| Level 4 | Surgical treatment of sagittal suture synostosis, unilateral coronal suture synostosis and unilateral lambdoidal suture synostosis is indicated since no spontaneous improvement of the abnormal skull shape is expected. D Expert opinion |
2. What are patient-relevant effect of different surgical techniques, in particular minimally invasive surgery versus open surgery for the different types of non-syndromic craniosynostosis?
Since the first operative intervention for craniosynostosis, many surgical techniques for the various types of craniosynostosis have been described. A broad distinction is made between minimally invasive surgery and open surgery. In the first group, the fused suture with surrounding bone is removed or an osteotomy is performed at the site of the fused suture or on either side of it. The procedure is usually followed by helmet therapy to remodel the shape of the skull, or by the insertion of springs or distractors to achieve active widening of the skull. With the open surgery, the desired cranial shape is immediately achieved by correcting a large part of the skull. In the Netherlands, two centres of expertise for craniosynostosis offer, besides the open cranial correction technique, also minimally invasive techniques.4,5 Randomized comparisons of the different surgical techniques have never been made. The overview of the literature below deals with the different techniques applied for the different types of non-syndromic craniosynostosis.
A) Sagittal Suture Synostosis
Minimally invasive techniques
The strip craniectomy is usually followed by helmet therapy for a few months to 1 year,6,7 or combined with spring distraction.8–10 Advantages of these minimally invasive methods are low blood volume losses, low transfusion requirements, short duration of surgery and a good cranial index (CI) postoperatively. Several studies have described these positive results for the minimally invasive methods, but rarely have provided information on the occurrence of elevated intracranial pressure (ICP) during follow-up, the long-term aesthetic outcome or on neurocognitive outcomes. Only Bonfield reports that 2.7% of patients had a reoperation because of a symptomatic growth limitation of the skull, i.e. proven elevated ICP or headache.11
Open cranial correction
In 2013, Van Veelen published a series of 79 children with sagittal suture synostosis undergoing an extended strip craniectomy.12 This is an open technique in which the fused sagittal suture is removed and the parietal bones are bent outwards. This technique has 4 variants in which the details vary. The average blood loss was 213 ml, with a range of 50 to 400 ml. After a follow-up of 2 years, 9% (7/79) of papilledema is diagnosed and 4 patients are operated again because of increased intracranial pressure. The skull circumference stabilized around 0.6 SD.
Van Veelen describes two variants of fronto-biparietal remodelling in sagittal suture synostosis with 7 years follow-up.13 The mean blood loss was 1042 ml (range 300 to 7000). The cranial index (CI) was mainly dependent on the preoperative CI rather than the surgical technique. The head circumference decreased slowly during the 4 years postoperatively. The head circumference was also particularly dependent on the preoperative value and stabilized at 0.9 SD. Seven percent of patients develop postoperative papilledema as a sign of increased ICP.
Minimally invasive techniques versus open cranial correction
Han in a retrospective study compares the operation parameters, complications and reoperation rate between 140 endoscopic (of which 94 sagittal suture) and 155 open (of which 76 sagittal suture) treated non-syndromic craniosynostosis children.14 The choice of technique was determined by age at referral (if six months or older, then always open technique), comorbidity and potentially expected problems with helmet therapy. The authors found the same complication rates for both techniques, with a lower reoperation rate for endoscopic surgery (3/140, of which two because of a persistent cranial defect and one because of disappointing aesthetic results; follow-up time averaged 25 months) than for open surgery (10/155, of which 1 because of a chronic wound, 2 because of a haematoma, 2 because of an implant removal, 2 because of a cranial defect and 3 because of disappointing aesthetic result; follow-up time averaged 37 months). Han also observed a clearly more favourable operating profile (shorter surgery duration, less blood loss, lower transfusion requirements) for the endoscopic technique.14 Arts describes similar data.15 Arts analyses a prospective complication registry in which 120 endoscopic (of which 63 sagittal suture) operations are compared with 66 open procedures (of which 30 sagittal suture).15 The authors also found similar complication rates with a clearly more favourable operating profile for the endoscopic procedure. There was one reoperation in each group for sagittal suture synostosis.
In a systematic review on the use of spring distraction, there is low to very low evidence that spring distraction would be a better technique than an open skull correction; the CI was not different, and factors such as operation time, duration of hospitalisation and blood loss were slightly lower than with open skull correction.9 Mundinger found no difference in these factors in the systematic review on the use of distraction osteogenesis for cranial corrections (including spring distraction) compared to open surgery.16
Only one study compares minimally invasive spring distraction with open surgery. This study covers the first 7 patients operated with the minimally invasive approach, compared to 7 retrospective patients.17 Due to the inclusion criteria of this guideline, it has been left out of consideration.
Three systematic reviews and meta-analyses compare open corrections with distractors/springs with open corrections without distractors/springs.
Gerety performed a meta-analysis of 3 surgical techniques, namely open cranial correction, strip craniectomy and open spring distraction.18 Twelve articles were included. The CI was significantly better after open cranial surgery compared to strip craniectomy and was not different after open cranial surgery compared to spring distraction. There was no difference in CI after strip craniectomy compared to spring distraction. Open cranial correction has a significantly longer surgical time, duration of hospitalisation, higher blood loss and higher costs compared to the other two techniques. In a systematic review on the use of open spring distraction, there is low to very low evidence that spring distraction would be a better technique than open cranial correction; the CI was not different and factors such as surgical time, duration of hospitalisation, and blood loss were slightly lower.9 In the systematic review by Mundinger on differences between mainly traditional distraction osteogenesis in single and multiple synostoses and open cranial surgery, no difference was found for surgical time, blood loss, intensive care unit admission or blood transfusions.16
In the systematic review of Chummun on CI and neuropsychological outcome of treatment of sagittal suture synostosis, six studies, of which four had a comparative population,19 were included for CI analysis. For neuropsychological outcome, only 2 articles were included. According to GRADE, the level of evidence for both outcomes was very low and therefore no conclusions can be drawn. Delye reports a CI of 0.72 after an unspecified follow-up period, but at least 6 months after endoscopic strip craniectomy in combination with helmet therapy.4 Van Veelen reports a CI of 0.74 after 2 years and 0.72 after 5 years following minimally invasive spring distraction.5
Other studies so far show varying results with respect to intracranial volume as an outcome between the open and minimally invasive techniques. Ghenbot finds no difference one year after surgery,20 whereas Van Veelen finds a small difference postoperatively at a mean age of 2 years, with a possible association with headache and papilledema.21 Bergquist describes a large decrease in intracranial volume over time for children who received surgery before the age of 6 months.22 Arab found a statistically significantly smaller postoperative volume only for open surgery using the Pi procedure, and not for strip craniectomy or spring distraction.23
Ridgway describes 56 patients with sagittal suture synostosis treated with a strip craniectomy and helmet therapy.24 Only two patients needed a blood transfusion (3.6%) and the authors describe an increase in head circumference from an average of 61% preoperatively to 89% at 3 years postoperatively. Arts reports a transfusion rate of 28% with a mean blood loss of 32 (±30) ml for 63 patients with sagittal suture synostosis who underwent endoscopic treatment.15
In a report of 83 consecutive patients with sagittal suture synostosis who underwent spring distraction and a follow-up period of almost three years, Van Veelen found a transfusion rate of 19%, average blood loss 70 (±50) ml for the combination of spring placement and spring removal, papilledema 2.4% (2/83), a result with regard to cranial shape comparable to the previously described open techniques and a head circumference that seems to stabilize at 0.7 SD.5,12,13 In comparison with the cohorts of open techniques reported earlier by Van Veelen, the prevalence of papilledema is lower, with comparable screening methods being used.
B) Metopic Suture Synostosis
Minimally invasive techniques
De Jong described the evolution of cranial shape and volume of 86 children after endoscopic treatment of metopic suture synostosis followed by helmet therapy in relation to the cranial shape and volume of unaffected children.25 Correction of the forehead was objectively evaluated; the cranial volume (both total and frontal) normalized after the procedure and was equal to that of non-affected children, and this until the age of almost 4 years. The shorter surgical time, shorter hospitalization and reduction in blood transfusions of the minimally invasive technique is also described for trigonocephaly.4
Cranial correction
Seruya describes in a follow-up study of 3 years after open skull correction a significant relapse of the width of the forehead over time.26 None of the 31 patients had signs of elevated ICP during follow-up. Wes describes similar results and found temporal hollowing and lateral orbital retrusion especially from 5 years follow-up or more.27 Long-term studies are therefore essential to provide a good assessment of the aesthetic outcome. Arab also find a smaller intracranial volume 3 years after surgery.23 This is in line with the study by Maltese in which 60 children with trigonocephaly are compared with 198 control children.28 Preoperatively, both groups have the same intracranial volume, but the frontal volume is smaller in trigonocephaly, while at the age of 3 years both the total volume and the frontal volume are smaller in trigonocephaly.
Minimally invasive techniques versus open cranial correction
In a comparative study into open versus endoscopic technique for metopic suture synostosis, Nguyen describes that a similar correction of the hypotelorism and wedge shape of the forehead is achieved after a short follow-up of up to 1 year.29
In a comparison of 67 microscopic minimally invasive operations and 113 open technique operations over 10 years for different types of craniosynostosis, a higher rate of extensive reoperations is found for the minimally invasive technique.30 For metopic suture synostosis, the reoperation rates are 2/8 patients (25%) for the minimally invasive technique versus 1/26 patients (3.8%) for the open technique. In comparison with the endoscopic minimally invasive surgeries that are regularly described, the technique used in this study differs on a number of crucial points: 1. to be able to use the microscope, considerably more incisions are made; 2. postoperatively, the redression helmet is only worn for 3 months, whereas in most centres the helmet is worn 10 to 12 months. This makes the comparison of this minimally invasive technique with an open technique specific to this surgeon and not generally applicable. Han compared in a retrospective study operation parameters, complications and reoperation rate between 140 endoscopic (of which 24 metopic suture) and 155 open (of which 31 metopic suture) treated non-syndromic craniosynostosis children.14 The choice of technique was determined by the child's age at referral (if 6 months or older then always an open procedure), comorbidities and possibly expected problems with helmet therapy. The authors found the same complication rates for both techniques, with a lower reoperation rate for endoscopic operations (3/140, of which 2 because of a persistent cranial defect and 1 because of a disappointing aesthetic result; follow-up averaged 25 months) than for open surgery (10/155, of which 1 because of a chronic wound, 2 because of a haematoma, 2 because of implant removal, 2 because of a cranial defect and 3 because of a disappointing aesthetic result; follow-up averaged 37 months). Furthermore, the authors found a clearly more favourable operating profile (shorter surgical time, less blood loss, fewer blood transfusions) for the endoscopic techniques. Arts described comparable data.15 Arts analysed a prospective complication registry in which 120 endoscopic (of which 35 metopic suture) operations are compared with 66 open procedures (of which 15 metopic suture).15 The authors also found similar complication rates with a clearly more favourable operating profile for the endoscopic procedure.
C) Coronal Suture Synostosis
Minimally invasive techniques
Tan reports on 11 patients early treated endoscopically and 11 patients treated late with an open procedure.31 The choice of technique is based on age at referral. The evaluation of orbital and facial symmetry by anthropometric measurements after a follow-up of 3 to 4 years shows better results for facial symmetry only for early endoscopic treatment. Jimenez describes the treatment as very good in terms of blood loss and duration of hospitalisation as well as aesthetic results.7 The orbital dystopia is well corrected in the majority: 51% of the patients even has a 100% correction. Delye also describes the short surgical time, shorter duration of hospitalisation and reduction in transfusion need for the minimally invasive technique for coronal suture synostosis.4
Open cranial correction
The 6-year follow-up results of fronto-orbital remodelling in 207 patients with coronal suture synostosis show that no reoperations were necessary because of elevated ICP.17 Supraorbital retrusion and temporal hollowing were particularly seen 5 years or more postoperatively; therefore, long-term studies are required to assess the ultimate outcome.
Minimally invasive techniques versus open cranial correction
The comparison of 21 and 22 patients who underwent respectively an endoscopic versus an open procedure for coronal suture synostosis mainly focuses on the remaining ophthalmic abnormalities.32 Better ophthalmic results were observed using the endoscopic technique, possibly because of the earlier timing of the surgery.
Han compared in a retrospective study 10 endoscopically and 28 openly treated coronal suture synostosis the operation parameters, complications and reoperation rate.14 The choice of technique was determined by age at referral (if 6 months or older then always open technique), comorbidity and expected problems with helmet therapy. The authors found a similar complication rate for both techniques, however a lower reoperation rate for the group of endoscopic surgeries (3/140) than for open surgeries (10/155) and a significantly more favourable surgical profile (shorter surgical time, less blood loss, lower transfusion need) for the endoscopic technique. Arts reported similar data comparing 120 endoscopic (of which 12 coronal suture) operations with 66 open (of which 14 coronal suture).15 The authors also found a similar complication rate with a clearly more favourable operating profile for the endoscopic procedure. Masserano compared the extent to which facial asymmetry and widening occurred in the temporal and orbital area one year after endoscopic coronal suture synostosis (24 patients) versus open-treated coronal suture synostosis (32 patients).33 These 56 patients are a selection of 120 patients, operated on coronal suture synostosis (95 open and 25 endoscopic) and selected based on the availability of a pre- and postoperative CT scan. The reason for a postoperative CT scan is unclear and may result in a bias. The measures achieved were also compared with age-matched controls (10 controls). The comparison showed that the results were the same for both techniques and that neither of the two techniques resulted in a significant improvement in temporal width.
D) Lambdoid Suture Synostosis
Minimally invasive techniques versus open cranial correction
The number of publications on lambdoid suture synostosis is very limited, probably due to the very low prevalence. The morphological changes are a facial asymmetry and an asymmetric basal occipital bone. Al-Jabri provides a systematic review of the results of lambdoid suture synostosis surgery and outcome from 1966 to 2013 and includes 17 studies with a total of 188 patients with lambdoid suture synostosis.34 Both minimally invasive surgery and open cranial correction are described. Facial asymmetry is an important factor with moderate postoperative improvement. Minimally invasive techniques are used as they are associated with less blood loss, and shorter surgical time and duration of hospitalisation.
Conclusions
| Level 2 | Sagittal suture synostosis Minimally invasive surgery is likely associated with significantly less blood loss, fewer blood transfusions, shorter surgical time and shorter duration of hospitalisation with a similar aesthetic result compared with open cranial correction. B Chummun et al, 2016 ; Maltese et al, 2015 ; Gerety et al, 2015; Arts et al, 2018 C Ridgway et al, 2011; Han et al, 2016; Van Veelen et al, 2018 |
| Level 3 | Sagittal suture synostosis Spring distraction is likely associated with a lower prevalence of papilledema (as a sign of increased intracranial pressure) in the follow-up compared with open cranial correction. C Van Veelen et al, 2013 ; Van Veelen et al, 2015 ; Van Veelen et al, 2018 |
| Level 3 | Metopic suture synostosis Minimally invasive surgery is likely to result in less blood loss, fewer blood transfusions, shorter surgical time and shorter duration of hospitalisation. Minimally invasive surgery for metopic suture synostosis may have the same aesthetic results as open cranial correction. B Arts et al, 2018; C Nguyen et al, 2015; Han et al, 2016; Seruya et al, 2014; Wes et al, 2014; Delye et al, 2016. |
| Level 2 | Metopic suture synostosis Open cranial correction for metopic suture synostosis results in a smaller frontal volume and intracranial volume at the age of 3 years compared to a control group. In the same comparative study for minimally invasive surgery for metopic suture synostosis with the same follow-up time, a normal frontal volume and intracranial volume were observed. B De Jong et al, 2017; Maltese et al, 2014 C Arab et al, 2016 |
| Level 3 | Coronal suture synostosis The risk of papilledema (as a sign of increased intracranial pressure) in a 6-year follow-up period after open cranial correction is possibly very low. C Taylor et al, 2015 |
| Level 3 | Coronal suture synostosis The ophthalmic outcomes of early minimally invasive surgery for coronal suture synostosis may be superior to those of late open cranial correction. B MacKinnon et al, 2013 |
| Level 3 | Coronal suture synostosis Minimally invasive surgery is likely associated with less blood loss, fewer blood transfusions, shorter surgical procedures and shorter duration of hospitalisation. Minimally invasive surgery for coronal suture synostosis may have the same aesthetic results as open cranial correction. B Arts et al, 2016; C Tan et al, 2011; Jimenez et al, 2013; Delye et al, 2016; Han et al, 2016; Masserano et al, 2018 |
| Level 3 | Lambdoid suture synostosis Minimally invasive surgery may give the same aesthetic results as open cranial correction for lambdoid suture synostosis. B Al-Jabri et al, 2014 |
3. What are patient-relevant effects of different timing of surgery, i.e. ‘early’ (below 6 months of age) versus ‘late’ (above 6 months of age)?
The timing depends, among other things, on the surgical technique used, in which minimally invasive techniques are usually performed before the age of 6 months. Open cranial corrections are mainly performed after the age of 6 months, but are also described as an early procedure.
A) Sagittal Suture Synostosis
Van Veelen shows a 10% incidence of papilledema in children with scaphocephaly with mean age 11 months versus 2.5% in these children with mean age 6 months.13 This is an objective reason to recommend early surgery.
Sun studied 36 patients with scaphocephaly treated with a minimally invasive technique with spring distraction.35 At a mean age of 3.9 months (range 1.9–9.2 months), the age at surgery strongly related to the change in cranial index: for each additional month in age the change in CI decreased by 1.3 (p = 0.03).
B) Metopic Suture Synostosis
Cornelissen shows that the prevalence of papilledema in children with trigonocephaly around the age of 11 months is below 2% and that it is therefore safe to wait until this age concerning the risk of increased ICP.36
C) Coronal Suture Synostosis
MacKinnon compares 21 and 22 patients with early, endoscopic versus late, open remodelling technique for coronal suture synostosis and finds better ophthalmic results with the endoscopic technique, possibly due to the earlier timing of surgery.32 If confirmed in follow-up studies, this could be a good reason for early surgery. If only children with milder forms of coronal suture synostosis underwent an endoscopic procedure, this may also explain the finding.
D) Lambdoid Suture Synostosis
Al-Jabri reports in a systematic review on conflicting results of surgery regarding the timing of surgery in a limited number of studies.34
Conclusions
| Level 3 | Sagittal suture synostosis The prevalence of papilledema in sagittal suture synostosis is likely to increase during the second half of the first year of life, and therefore surgery before the age of 6 months is recommended. B Van Veelen et al, 2015 |
| Level 3 | Sagittal suture synostosis For minimally invasive treatment with spring distraction, there is a relationship between age at surgery (within a range of 1.9 to 9.2 months) and change in cranial index: for each additional month in age, the change in CI decreases by 1.3. C Sun et al, 2018 |
| Level 3 | Metopic suture synostosis Considering the low prevalence of papilledema in metopic suture synostosis during the first year of life, there seems to be no compelling reason to perform the operation before the age of 6 months. B Cornelissen et al, 2017 |
| Level 3 | Coronal suture synostosis The ophthalmic outcomes of early minimally invasive surgery seem to be better for coronal suture synostosis than for late open cranial correction. It is unclear whether this difference is caused by severity of presentation, type of surgery or timing. B MacKinnon et al, 2013 |
| Level 3 | Lambdoid suture synostosis Early or late treatment for lambdoid suture synostosis may have a similar aesthetic result. C Al-Jabri et al, 2014 |
Considerations
• Evidence of the Conclusions
Indication for surgical treatment
The indication for surgical treatment of sagittal suture synostosis, unilateral coronal suture synostosis and unilateral lambdoid suture synostosis is based on expert opinion.
The indication for the most severe form of metopic suture synostosis is based on considerable scientific evidence because most observational studies had a direct comparative approach, and the design and execution were adequate (=no serious risk of bias).
Surgical technique
The results of minimally invasive surgery compared to open cranial correction in sagittal suture synostosis and metopic suture synostosis – i.e. a shorter surgical time, less blood loss, fewer blood transfusions, shorter duration of hospitalisation and a comparable aesthetic result – are based on considerable scientific evidence because most observational studies had a direct comparative set-up, and design and execution were adequate (=no serious risk of bias). The comparable aesthetic results of minimally invasive surgery and open cranial correction for metopic suture synostosis are based on weak evidence because most studies have a non-comparative setup.
The better ophthalmic results and the comparable aesthetic outcome of minimally invasive surgery and open cranial correction for corona suture synostosis are supported by weak evidence.
The comparable aesthetic outcome of minimally invasive surgery and open cranial correction for lambdoid suture synostosis is supported by weak evidence, i.e. only one observational study.
Timing of surgery
The outcome ’prevalence of papilledema’ in sagittal suture synostosis and metopic suture synostosis in the first year of life is supported by weak evidence, i. e. only one study.
The better ophthalmic results of early minimally invasive surgery for coronal suture synostosis compared to late open cranial correction are supported by weak evidence, i.e. only one observational study.
The comparable aesthetic outcome of early or late treatment for lambdoid suture synostosis is supported by weak evidence, i.e. from non-comparative studies with small numbers of patients.
• Values and Preferences
Indication for surgical treatment
Most parents of children with metopic suture synostosis, sagittal suture synostosis, coronal suture synostosis and lambdoid suture synostosis, according to the working group, find the aesthetic result, neuro-cognitive functions and sight, crucial outcome measures and can therefore agree with a surgical treatment.
Surgical technique
Most parents of children with isolated, non-syndromic craniosynostosis find the aesthetic result regarding cranial shape and scar visibility, minimal risk of reoperation, neuro-cognitive functions and sight crucial outcome measures, according to the working group. In case of similar results in treatment, there is a preference for surgery at an early age, as parents experience the prospect of surgery as a burden.
Timing of surgery
Most parents of children with isolated, non-syndromic craniosynostosis prefer surgery at an early age, according to the workgroup, because parents experience the prospect of surgery as a burden.
• Costs and Resources
The guideline committee expects that the implementation of the recommendations leads to little or no increase in costs, because the recommendations are in line with existing practice. This applies to all recommendations relating to:
-
-
indication for surgical treatment
-
-
surgical technique
-
-
timing of surgery
• Professional Perspective
Indication for surgical treatment
In the case of a metopic bone ridge and a milder type of trigonocephaly, spontaneous improvement of the deformed skull is expected. Spontaneous recovery does not occur in the other cranial deformities.
Considering the discussion on the definition of the mild/moderate type of trigonocephaly, monitoring of (predominantly non-surgical treated) patients with mild/moderate trigonocephaly on aesthetic outcome at the age of 5 years is recommended.
The most frequently used methods for aesthetic assessment are panel analysis of standardized photographs or 3D photogrammetry.
Surgical technique
In general, there is a preference for minimally invasive surgery because of 1. lower risk of blood transfusion need, and thus avoiding the associated risks; and 2. less prominent scars. For sagittal suture synostosis, the aesthetic results of the skull shape are almost identical to an open cranial correction and therefore minimally invasive surgery is preferred. For metopic, coronal and lambdoid suture synostosis, an increasing number of publications show that the aesthetic (and for coronal suture also functional in relation to vision problems) results of endoscopic minimally invasive techniques are the same (or possibly better in terms of vision for coronal suture) than those of open remodelling. Nevertheless, this remains a controversial issue in the (inter)national craniofacial field, where there is not yet sufficient ground to indicate a clear preference for endoscopic or open remodelling techniques.
A well-founded decision for the type of minimally invasive surgery, i.e. strip craniectomy with helmet therapy or spring distraction, cannot be given and depends mainly on the preference and experience of the surgeon.
Timing of surgery
Operating on before the age of 3 months is not recommended for anesthesiological safety reasons. Minimally invasive surgery after the age of 6 months is less effective than before that age because increased ossification makes the skull less deformable. An extensive open cranial correction before the age of 6 months seems to induce a growth limitation of the skull and is therefore mainly performed after that age. Surgical correction of the supra-orbital rim with an open technique (as done with metopic and coronal suture synostosis) is technically more difficult for the age from 6 to 9 months as the ossification of this bone is still limited.
• Balance of Anticipated Desired and Undesired Outcomes
Indication for surgical treatment
Only surgery can improve the aesthetic aspect and reduce neurocognitive deficits and vision problems. For the metopic bone ridge this does not apply because of spontaneous recovery of the skull shape. For the mild and moderate form of trigonocephaly, this is not so certain given the dependence of the definition used.
Surgical technique
Advantages of minimally invasive techniques are shorter surgical time and duration of admission, less blood loss and less chance of a blood transfusion. As a result, the balance is positive towards minimally invasive techniques as long as they achieve equally good results in terms of improved skull shape, neurocognition and vision as open remodelling. The aesthetic results are probably comparable in the short term (1 year postoperatively). Whether the long-term aesthetic results (5 years postoperatively) are the same is unclear, as is the relationship between the neurocognitive and visual results of the two techniques.
Timing of surgery
Increased intracranial pressure, identified by the presence of papilledema in fundoscopy, is detrimental to the vision and possibly to the neurocognitive outcomes. Due to the increasing prevalence of elevated intracranial pressure in sagittal suture synostosis in the first year of life, early treatment is desirable. This risk is not associated with metopic suture synostosis and it is safe to wait until a later point in time when the supraorbital rim is technically easier to correct if open treatment is chosen. For coronal suture synostosis and lambdoid suture synostosis, there are no obvious timing reasons, although a better outcome for vision problems may be observed for early minimally invasive treatment of coronal suture synostosis.
Surgical technique
The guideline committee expects that the will and ability to comply with the recommendations is accepTable to all stakeholders (plastic surgeons, neurosurgeons, maxillofacial surgeons, patients and their parents, members of LAPOSA, paediatricians, clinical geneticists, gynaecologists, prenatal physicians, youth physicians, general practitioners, physiotherapists, midwives, neurologists, radiologists, ZN, IGJ, NFU, VWS, ZIN, NZA, PFN), because the recommendations are already largely in line with existing practice.
Timing of surgery
The guideline committee expects that the will and ability to comply with the recommendations is acceptable to all stakeholders (plastic surgeons, neurosurgeons, maxillofacial surgeons, patients and their parents, members of LAPOSA, paediatricians, clinical geneticists, gynaecologists, prenatal physicians, youth physicians, general practitioners, physiotherapists, midwives, neurologists, radiologists, ZN, IGJ, NFU, VWS, ZIN, NZA, PFN), because the recommendations are already largely in line with existing practice.
Rationale for the recommendation(s)
Indication for surgical treatment
The guiding principle in the formulation of the recommendations is that in the mild type of trigonocephaly the chance of spontaneous recovery is high in contrast to the severe type of trigonocephaly. Evaluation of the aesthetic result of the unoperated mild or moderate type of trigonocephaly is only reliable at the age of 5 years. In sagittal, unilateral coronal and unilateral lambdoid suture synostosis no spontaneous recovery will occur.
Surgical technique
The guiding principle in the formulation of the recommendations is that both surgical techniques should be considered in the case of a referral before the age of 6 months. The main advantage of minimally invasive surgery is a lower risk of blood transfusion need. The aesthetic improved skull shape 1 year after surgery appears to be similar for both techniques in case of sagittal and possibly for metopic, coronal and lambdoid suture synostosis.
In case of a later referral, a minimally invasive technique is generally no longer possible and open remodelling is recommended.
Timing of surgery
Key in formulating the recommendations regarding timing of surgery is that aesthetic results of the skull shape, neurocognitive performance and vision are as optimal as possible. The only clear indication for early treatment is the increasing prevalence of papilledema in sagittal suture synostosis. For the other three types of craniosynostosis, there is no definite reason to give a timing recommendation at this moment.
Recommendations
Indication for surgical treatment
Surgically correct isolated, non-syndromic craniosynostosis, with the exception of metopic bone ridge.
Do preferably not operate on children with a mild type of trigonocephaly.
No recommendation is given for children with moderate trigonocephaly.
Plastic surgeon, neurosurgeon and/or maxillofacial surgeon
Evaluate the aesthetic result of children with mild and moderate type of trigonocephaly who have not undergone surgery, at the age of 5 years, using a panel analysis of standardized photographs or 3D photogrammetry.
Surgical technique
Perform minimally invasive surgery on patients with sagittal suture synostosis, younger than 5.5 months at referral, because of the lower transfusion needs.
Perform an open cranial correction if the patient is older than 6 months at referral.
For metopic, coronal and lambdoid suture synostosis, no recommendation is given regarding the surgical technique.
Timing of surgery
Remodel the skull of non-syndromic craniosynostosis patients in the first year of life.
Correct, if possible, sagittal suture synostosis before the age of 6 months.
For metopic, coronal and lambdoid suture synostosis, no recommendation is given regarding the timing of surgery.
Refer as a general practitioner, paediatrician or youth doctor patients well before the age of 6 months, so that the choice for minimally invasive surgery is possible.
Research Gaps
Indication for Surgical Treatment
Follow-up studies of at least 5 years should demonstrate whether a wait-and-see policy for mild/moderate trigonocephaly is justified with respect to aesthetic outcomes and neurocognitive outcome.
Surgical Technique
Follow-up studies of at least 5 years should demonstrate in a prospective direct comparison whether a minimally invasive technique provides aesthetic correction of the skull shape and neurocognitive outcome comparable to open skull correction.
Timing of Surgery
Prevalence studies for symptoms of elevated ICP such as papilledema prior to surgery are lacking for coronal and lambdoid suture synostosis. Ophthalmic outcomes in coronal suture synostosis should be specifically related to timing, severity of presentation and type of surgery.
Literature
-
1.
Birgfeld CB, Saltzman BS, Hing AV, et al. Making the diagnosis: Metopic ridge versus metopic craniosynostosis. J Craniofac Surg 2013;24:178-85
-
2.
Cho MJ, Kane AA, Seaward JR, et al. Metopic “ridge” vs. “craniosynostosis”. Quantifying severity with 3D curvature analysis. J Craniomaxillofac Surg 2016;44:1252-8
-
3.
Anolik RA, Allori AC, Pourtaheri N, et al. Objective assessment of the interfrontal angle for severity grading and operative decision-making in metopic synostosis. Plast Reconstr Surg 2016;137:1548-55
-
4.
Delye HHK, Arts S, Borstlap WA, et al. Endoscopically assisted craniosynostosis surgery (EACS): The craniofacial team Nijmegen experience. J Cranio-Maxillo-Fac Surg 2016;44:1029-36
-
5.
Van Veelen MLC, Touw C, Kamst N, et al. Minimally invasive, spring-assisted correction of sagittal suture synostosis. Technique, outcome and complications in 83 cases. Plast Reconstr Surg 2018;141:423-33
-
6.
Barone CM, Jimenez DF. Endoscopic craniectomy for early correction of craniosynostosis. Plast Reconstr Surg 1999;104:1965-73; discussion 1974-5
-
7.
Jimenez DF. Early treatment of coronal synostosis with endoscopy-assisted craniectomy and postoperative cranial orthosis therapy: 16-year experience. J Neurosurg Pediatr 2013;12:207-19
-
8.
Lauritzen CGK, Davis C, Ivarson A, et al. The evolving role of springs in craniofacial surgery: the first 100 clinical cases. Plast Reconstr Surg 2008;121:545-54
-
9.
Maltese G, Fischer S, Strandell A, et al. Springs-assisted surgery in the treatment of sagittal synostosis: A systematic review. J Plast Surg Hand Surg 2015;49:177-82
-
10.
Fischer S, Maltese G, Tarnow P, et al. Comparison of intracranial volume and cephalic index after correcton of sagittal synostosis with spring-assisted surgery or Pi-plasty. J Craniofac Surg 2016;27:410-3
-
11.
Bonfield CM, Lee PS, Adamo MA, et al. Surgical treatment of sagittal synostosis by extended strip craniectomy: Cranial index, nasofrontal angle, reoperation rate, and a review of the literature. J Cranio-Maxillo-Fac Surg 2014;42:1095-101
-
12.
Van Veelen ML, Eelkman Rooda OH, de Jong T, et al. Results of early surgery for sagittal suture synostosis: long-term follow-up and the occurrence of raised intracranial pressure. Childs Nerv Syst 2013;29:997-1005
-
13.
Van Veelen MLC, Mihajlovic D, Dammers R, et al. Frontobiparietal remodeling with or without a widening bridge for sagittal synostosis: comparison of 2 cohorts for aesthetic and functional outcome. J Neurosurg Pediatr 2015;16:86-93
-
14.
Han RH, Nguyen DC, Bruck BS, et al. Characterization of complications associated with open and endoscopic craniosynostosis surgery at a single institution. J Neurosurg Pediatr 2016;17:361-70
-
15.
Arts S, Delye H, van Lindert EJ. Intraoperative and postoperative complications in the surgical treatment of craniosynostosis: minimally invasive versus open surgical procedures. J Neurosurg Pediatr 2018;21:112-8
-
16.
Mundinger GS, Rehim SA, Johnson III O, et al. Distraction osteogenesis for surgical treatment of craniosynostosis: A systematic review. Plast Reconstr Surg 2016;138:657-69
-
17.
Taylor JA, Paliga JT, Wes AM, et al. A critical evaluation of long-term aesthetic outcomes of fronto-orbital advancement and cranial vault remodelling in nonsyndromic unicoronal craniosynostosis. Plast Reconstr Surg 2015;135:220-31
-
18.
Gerety PA, Basta MN, Fischer JP, et al. Operative management of nonsyndromic sagittal synostosis: A head-to-head meta-analysis of outcomes comparing 3 techniques. J Craniofac Surg 2015;26:1251-57
-
19.
Chummun S, McLean NR, Flapper WJ, et al. The management of nonsyndromic, isolated sagittal synostosis. J Craniofac Surg 2016;27:299-304
-
20.
Ghenbot RG, Patel KB, Skolnick GB, et al. Effects of open and endoscopic surgery on skull growth and calvarial vault volumes in sagittal synostosis. J Craniofac Surg 2015;26:161-4
-
21.
Van Veelen MLC, Jippes M, Carolina JCA, et al. Volume measurements on three-dimensional photogrammetry after extended strip versus total cranial remodelling for sagittal synostosis: A comparative cohort study. J Cranio-Maxillo-Fac Surg 2016;44:1713-8
-
22.
Bergquist CS, Nauta AC, Selden NR, et al. Age at the time of surgery and maintenance of head size in nonsyndromic sagittal craniosynostosis. Plast Reconstr Surg 2016;137:1557-65
-
23.
Arab K, Fischer S, Bahtti-Softeland M, et al. Comparison Between Two Different Isolated Craniosynostosis Techniques: Does It Affect Cranial Bone Growth? J Craniofac Surg 2016;27:e454-7
-
24.
Ridgway EB, Berry-Candelario J, Grondin RT, et al. The management of sagittal synostosis using endoscopic suturectomy and postoperative helmet therapy. J Neurosurg Ped 2011;7:620-6
-
25.
De Jong G, Tolhuisen M, Meulstee J, et al. Radiation-free 3D head shape and volume evaluation after endoscopically assisted strip craniectomy followed by helmet therapy for trigonocephaly. J Craniomaxillofac Surg 2017;45:661-71
-
26.
Seruya M, Shen SH, Wang LL, et al. Three patterns of fronto-orbital remodelling or metopic synostosis: Comparison of cranial growth outcomes. Plast Reconstr Surg 2014;134:787e-95e
-
27.
Wes AM, Paliga JT, Goldstein JA, et al. An evaluation of complications, revisions, and long-term aesthetic outcomes in nonsyndromic metopic craniosynostosis. Plast Reconstr Surg 2014;133:1453-64
-
28.
Maltese G, Tarnow P, Wikberg E, et al. Intracranial volume before and after surgical treatment for isolated metopic synostosis. J Craniofac Surg 2014;25:262-6
-
29.
Nguyen DC, Patel KB, Skolnick GB, et al. Are endoscopic and open treatments of metopic synostosis equivalent in treating trigonocephaly and hypotelorism? J Craniofac Surg 2015;26:129-34
-
30.
Teichgraeber JF, Baumgartner JE, Vivian SL, et al. Microscopic versus open approach to craniosynostosis: A long-term outcomes comparison. J Craniofac Surg 2014;25:1245-8
-
31.
Tan SPK, Proctor MR, Mulliken JB, et al. Early frontofacial symmetry after correction of unilateral coronal synostosis: Frontoorbital advancement vs endoscopic strip craniectomy and helmet therapy. J Craniofac Surg 2013;24:1190-4
-
32.
MacKinnon S, Proctor MR, Rogers GF, et al. Improving ophthalmic outcomes in children with unilateral coronal synostosis by treatment with endoscopic strip craniectomy and helmet therapy rather than fronto-orbital advancement. J AAPOS 2013;17:259-65
-
33.
Masserano B, Woo AS, Skolnick GB, et al. The temporal region in unilateral coronal craniosynostosis: Fronto-orbital advancement versus endoscopy-assisted strip craniectomy. Cleft Palate Craniofac J 2018;5:423-9
-
34.
Al-Jabri T, Eccles S. Surgical correction for unilateral lambdoid synostosis: a systematic review. J Craniofac Surg 2014;25:1266-72
-
35.
Sun J, Ter Maaten NS, Mazzaferro DM, et al. Spring-mediated cranioplasty in sagittal synostosis: does age at placement affect expansion? J Craniofac Surg 2018;29:632-5
-
36.
Cornelissen MJ, Loudon SE, van Doorn FE, et al. Very low prevalence of intracranial hypertension in trigonocephaly. Plast Reconstr Surg 2017;139:97e-104e
CHAPTER 6 SURGICAL TREATMENT OF MULTISUTURE AND SYNDROMIC CRANIOSYNOSTOSIS – THE CRANIAL VAULT
6.1 What is the policy on surgical treatment of the cranial vault in multisuture and syndromic craniosynostosis?
Introduction
The distinction between multisuture craniosynostosis and syndromic craniosynostosis is made based on phenotype. Multisuture craniosynostosis can occur in all variations of two or more affected cranial sutures. In this group, new genetic causes for craniosynostosis are still identified, such as the genes TCF12, ERF, IL11RA. In syndromic craniosynostosis, additional congenital defects and dysmorphisms are present.
The four most common forms of syndromic craniosynostosis are: Apert, Crouzon (including Pfeiffer syndrome), Saethre-Chotzen and Muenke syndrome.
Search and Selection
For the following specific questions, original scientific studies or systematic reviews of scientific studies have been included:
-
1.
What are the patient-relevant effects of different indications for surgical treatment of multisuture and syndromic craniosynostosis, i.e. routine treatment versus in response to signs of elevated ICP?
-
2.
What are the surgical specific outcomes of different surgical techniques, in particular minimally invasive surgery (endoscopic strip craniectomy with helmet therapy, or spring/conventional distraction of the occiput) versus open cranial correction (of forehead/ occiput)?
-
3.
What are the long-term results regarding cognition and aesthetics of a different timing of surgery, i.e. ’early’, defined as before the age of 12 months, versus ’late’, i.e. after the age of 12 months?
In the Medline (OVID) and Embase databases, a single comprehensive search was carried out for studies on craniosynostosis. The search strategy is shown in appendix 2, of the guideline. After deduplication, the literature search yielded 2732 hits.
Given the large number of studies, the chair of the working group first selected those that met the following general selection criteria:
General Selection and Exclusion Criteria
| Study type | - original studies - systematic reviews of sufficient quality: - research question of systematic review corresponds (largely) to the basic question - search is performed in at least 2 relevant databases, e.g. Cochrane Library, Medline/PubMed - reporting of the complete search strategy - no relevant keywords/search terms are missing |
| Follow-up period | - minimum follow-up period of 12 months for therapeutic or prognostic studies. |
| Exclusion criteria | - Case-reports - Expert opinion - Letters - Editorials - Case control studies for diagnostic tests - Narrative reviews |
The pre-selected studies that met the specific selection criteria listed in the Table below are included in the literature summary of this chapter.
Specific Selection and Exclusion Criteria
| Selection criteria | - minimum study size: 20 patients for patient series, where no multivariate analysis was used to identify prognostic factors for a relevant outcome measure. - minimum study size: 35 patients for patient series, with multivariate analysis of possible predictive variables for the effect - minimum number of participants of studies with a direct comparative design: 20 per study arm. |
Summary of the Literature
1. What are the patient-relevant effects of different indications for surgical treatment of multisuture and syndromic craniosynostosis, i.e. routine treatment versus in response to signs of elevated ICP?
Hayward questions in an opinion paper protocolized surgical treatment of all children with complex or syndromic craniosynostosis.1 His main argument for not doing this routinely is that the relationship between increased intracranial pressure (ICP) and neurocognitive deficits in craniosynostosis patients has not been proven. There is consensus that surgery is indicated as soon as there are signs of increased ICP. The Great Ormond Street team is one of the few with a policy of surgery only when there is increased ICP. They have published two studies on this subject: Marucci on Apert syndrome and Abu-Sittah on Crouzon syndrome.2,3 Screening in the research period was mainly done by repeated measurements of visual evoked potentials.
| Apert syndrome | ||
| Marucci2 | Spruijt4 | |
| Included patients (n) | 24 | 19 |
| Excluded patients (n) | 12∗ | |
| Inclusion period | 1992-2000 | 1999-2013 |
| Age at referral (mean; range) | 1.4 mos; 1–6 mos | |
| Patients with elevated ICP preop | 20/24 (83%) | 10% |
| Age at elevated ICP, i.e. operation (mean; range) | 18 mos; 1 mos–1yr 5mos | 12 mos; 5 mos – 2.5yrs |
| Initial treatment: | ||
| VP drain with/without endoscopic 3rd ventriculostomy | 2/24 (8%) | |
| OSA treatment | 2/24 (8%) | |
| Cranial remodelling | 16/24 (67%) | 95% |
| Follow up: | ||
| Age at last follow-up (mean/range) | 7 – 14 yrs | 5.7 yrs |
| Recurrence of elevated ICP | 7/20 (2 with drain) (35%) | 7 (37%) |
| Time between 1st and 2nd elevated ICP | 3yrs 4mos; | 3yrs; |
| 1yr 11mos – 5yrs 9mos | 1yr 6mos – 4yrs 10mos | |
| No cranial remodelling | 5/24 (21%) | 1/19 (5%) |
surgery for aesthetic reasons, 5 surgery elsewhere, 1 moved to other center, 1 deceased. mo(s)=month(s), yr(s)=year(s).
In summary, Marucci found in 20/24 patients with Apert syndrome (83%) a first episode of elevated ICP and that 7/20 (35%) had a second episode of elevated ICP.2 Three out of five patients (60%), who underwent cranial surgery elsewhere before the age of 12 months, still developed elevated ICP for which treatment was performed. As outcomes with respect to neurocognitive functioning, vision and prevalence of Chiari are lacking, it is not possible to compare the results of this wait-and-see policy with those of treatment as part of a protocol. In addition, it cannot be ruled out that the four patients who did not receive any treatment may have had elevated ICP, which remained unnoticed because none of the screening methods is 100% sensitive.
Spruijt describes 19 children with Apert syndrome, one of whom is not receiving surgery (reason not given) and the other 18 (95%) are undergoing routine cranial expansion at an average age of 1 year, with 2 patients suffering from papilledema.4 In the average follow-up period of 5.7 years, 7 patients developed papilledema. Prior to surgery there was no tonsillar herniation (defined as herniation of less than 5 mm through the foramen magnum) and in the follow-up tonsillar herniation of less than 5 mm was found in 3 patients.
In comparison, 12% (18/19 versus 20/24) more patients are operated on because of protocol-based treatment than because of treatment for demonstrated elevated ICP, where the prevalence of papilledema after initial cranial expansion is almost equal to the preoperative prevalence. Given the small numbers in both series and the exclusion of 12 patients in one of the studies, there is no strong evidence to support either option for surgical indication in children with Apert syndrome.
The study by Abu-Sittah included 49 Crouzon patients from the period 1985–2002; how many were excluded is not mentioned.3 The patients are divided into 3 groups: Group A. no signs of elevated ICP (n = 19), Group B. one episode of elevated ICP (n = 16) and Group C. two or more episodes of elevated ICP (n = 14). It is clear that the children in Group A are referred considerably later. Group A is referred between the age of 4 months and 10 years (average age of 3 years) and has therefore not been screened for signs of ICP throughout this time; Group B is referred between 1 month and 6 years (average age of 1.6 years), and Group C is referred between 2 months and 3 years (average age of 7 months). In 61% of the patients, elevated ICP was detected. The conclusion that 38.8% of the patients did not develop elevated ICP and therefore did not need treatment is dependent on the representativeness of Group A. The missing screening prior to the late referral can strongly influence this. Other data are lacking, such as neurocognitive functioning, vision and prevalence of Chiari. The age range at which elevated ICP first occurred was 4 months to 6 years 4 months for group B (mean age of 2.3 years) and 4 months to 4 years for group C (mean age of 1.5 years).
Spruijt describes 23 patients with Crouzon syndrome, four of whom do not undergo cranial expansion (reason not given) and the remaining 19 (83%) receive a routine cranial expansion at a mean age of 1.5 years (range 0.4–3.9), with 11 patients (57.9%) having papilledema.4 In the average follow-up of 5.7 years, 8 patients (35%) developed papilledema. Prior to surgery, tonsillar herniation was present in 7 patients and in the follow-up tonsillar herniation was found in 10 patients, of whom 2 patients had a Chiari (5 mm herniation or more). The presence of both papilledema and tonsillar herniation was found in 83% of patients.
Compared to operating only on indication of elevated ICP, 22% (19/23 versus 19/49) more patients are operated on because of protocol-based treatment, with the prevalence of papilledema after initial cranial expansion remaining almost the same. Given the small numbers in both series and incomplete, unclear screening in one series, there is no strong evidence to support a choice between the two options for surgical indication for children with Crouzon syndrome.
These studies are still lacking for the other multisuture and syndromic craniosynostosis. However, a 19% prevalence of increased ICP in patients with Saethre-Chotzen syndrome is reported (5/26, mean age of 14 months) and a 35% prevalence (24/68, mean age of 30 months) prior to cranial expansion.5,6 For multisuture craniosynostosis, a 58% prevalence of elevated ICP is reported and a 67% prevalence, and a 77% prevalence in a mixed group of syndromic and multisuture craniosynostosis patients (30/39, mean age not described) prior to cranial expansion.7–9 For Muenke syndrome, this risk was 0% (0/39) in the study by Kress and 4% (1/28) in De Jong where the indication for surgery in particular improvement of the abnormal skull shape.5,6
Conclusions
| Level 2 | Starting treatment not until signs of elevated ICP are observed results possibly in a surgical indication for 83% of children with Apert syndrome and an average age of 18 months. When routine expansion is applied, possibly 95% of children will be operated on at mean age 12 months. B Marucci et al, 2008; Spruijt et al, 2016 |
| Level 3 | In children (with Crouzon syndrome and an average age of 18 to 24 months), starting treatment for signs of elevated ICP may result in a surgical indication in at least 61%. With protocol-based treatment, regardless of the presence of elevated ICP, possibly 83% of patients will be operated on at mean age 18 months. B Spruijt et al, 2016; C Abu-Sittah et al, 2016 |
| Level 3 | For patients with Apert and Crouzon syndrome, there is probably no clinically relevant difference in the prevalence of elevated ICP during a 5-year follow-up between a routine cranial expansion regardless of the presence of elevated ICP in the first year of life and treatment once elevated ICP has been diagnosed. B Marucci et al, 2008; Spruijt et al, 2016; C Abu-Sittah et al, 2016. |
| Level 2 | For patients with Saethre-Chotzen syndrome, surgical treatment is indicated for both the cranial deformity and the risk of elevated ICP, given the prevalence of elevated ICP of 19–35% prior to cranial expansion around the age of 12 months. For patients with multisuture craniosynostosis, surgical treatment is indicated for both cranial deformity and the risk of increased ICP, given the prevalence of increased ICP of 58–67% prior to cranial expansion around the age of 12 months. For patients with Muenke syndrome, surgical treatment is mainly indicated for correction of the cranial deformity, given the low prevalence of elevated CPI of 0–4% prior to cranial expansion around the age of 12 months. For patients with Saethre-Chotzen and Muenke syndrome and multisuture craniosynostosis, it is unknown whether there is a clinically relevant difference in prevalence of elevated ICP during a 5-year follow-up between a routine cranial expansion in the first year of life and treatment once elevated ICP has been diagnosed. B Kress et al, 2008; De Jong et al, 2010; Thompson 1995, Renier 2000. |
2. What are the surgical specific outcomes of different surgical techniques, in particular minimally invasive surgery (endoscopic strip craniectomy with helmet therapy, or spring/conventional distraction of the occiput) versus open cranial correction (of forehead/ occiput)?
In a study by Rottgers between 2005 and 2012, 18 patients with bicoronal synostosis, 9 with non-syndromic craniosynostosis and 9 with syndromic craniosynostosis (i.e. 2 Apert, 1 Crouzon, 3 Saethre-Chotzen and 3 Muenke) were treated with an endoscopic strip craniectomy between the ages of 1 and 4 months.10 This treatment option was always offered if a child had been referred before the age of 5 months, except for severe cranial deformity, fingerprinting or the presence of bone spikes sticking into the gyri and sulci of the brain. After a follow-up of 37 months (range 6–102 months), 11% (1/9) of the non-syndromic versus 55.6% (5/9) of the syndromic patients had undergone fronto-orbital advancement. The reason for this was that 12 months after the first correction, slowed cranial growth and signs of increased ICP were observed, despite good morphological improvement, and subsequent to progressive fusion of other sutures or fusion of the opened defects in the coronal sutures. Fusion alone without a flattening of the growth curve of the head circumference was not a reason for reoperation. No details are given about the specific diagnosis of the patients who required reoperation. One patient developed a pseudo-meningocele as a result of a dural defect that required up to two times surgical repair. The reported outcome measures are limited to head circumference and cranial index. The authors do not clearly describe how the occurrence of elevated ICP was determined.
Much more positive about the aesthetic results achieved are Jimenez and Barone.11 They report on 21 patients with various types of non-syndromic, multisuture craniosynostosis, operated on at an average age of 3 months. Only aesthetic correction is mentioned, which is claimed to be excellent. During a 5-year follow-up, the cranial growth curve of all patients was expected to be normal. No measurements or other objective data are given, so that the quality of this study cannot be established reliably and no conclusions can be drawn from it.
Han compares operation parameters, complications and reoperation rate between 19 endoscopically (of which 10 syndromic and 9 non-syndromic multisuture) and 36 open (of which 23 syndromic and 13 multisuture) treated children with craniosynostosis.12 No endoscopic technique was performed in children who already had a VP-shunt or in whom 3 or more sutures were synostotic. The authors found a complication rate in the endoscopically treated syndromic group of 1/10 and 10/23 after an open surgical treatment. The number of reoperations in the syndromic group after endoscopic treatment was 4/10, of which 3 because of suboptimal aesthetic reasons and 1 because of persistent liquor leakage. Reoperations in the syndromic group after open surgery were performed in 10/23 because of implant removal in one patient, a skull defect in one patient, suboptimal aesthetic result in 6 patients and an abscess in two patients. Also in this group, the authors describe a clearly more favourable operating profile (shorter surgical time, less blood loss, fewer blood transfusions) for the endoscopic technique. Long-term outcome measures such as increased ICP or neurocognition are not reported.
Thomas describes the experience of occipital expansion with distraction in a group of 31 patients with syndromic craniosynostosis, for 23 of them this was the first craniofacial procedure.13 In 28 patients (90.3%) the target of 20 mm distraction was reached, with 27 patients showing a significant improvement in skull shape. Symptoms of increased ICP disappeared in all patients who showed these prior to surgery. Because of a haemorrhage, the technique was adapted in one patient during surgery. After surgery, there was persistent liquor leakage in two patients, for whom reoperation was performed, and an imminent wound dehiscence in one, and for these reasons, the distraction was stopped early. In 9 patients, a wound infection occurred, requiring the removal of one or both distractors in 3 patients; seven had skin necrosis due to the pressure of the distractor. The removal of the distractor resulted in persistent liquor leakage, partly due to underlying hydrocephalus, which required the placement of a VP-drain. Overall, complications occurred in 61.3%, with 19.4% requiring one or more corrective operations, but the technique is an effective method of correction for severe brachycephaly.
De Jong compared the increase in head circumference as approximate measure of intracranial volume and the prevention of complications between occipital expansion according to open cranial correction (n = 16) versus spring distraction (n = 15) in 31 patients with Apert or Crouzon syndrome.14 The head circumference increased significantly more after spring distraction (1.9 SD versus 0.8 SD). Specific complications were wound dehiscence and insufficient expansion in the open remodelling group and skin perforation by the spring in 2 patients in the latter group.
One systematic review reports on the use of distraction techniques (conventional and spring distraction) in craniofacial procedures.15 No significant difference was found for surgical time, blood loss and duration of hospitalisation between open cranial correction and distraction techniques. Conventional distraction results in a 21% increase in intracranial volume and spring distraction in an increase of 27%, but the increase after open craniofacial surgery is not given. Three selected studies provide evidence for greater intracranial volume gain after occipital distraction than after fronto-orbital advancement.
Spruijt compares patients with Apert and Crouzon syndrome according to type of initial surgery: 18 received a fronto-orbital advancement and 19 an occipital expansion (conventional or with springs).4 Outcome measures were head circumference, results of fundoscopy (presence of papilledema), sight and presence of tonsillar herniation, after an identical follow-up time of 5.7 years. Significantly better results after occipital expansion were found for head circumference (+1.09 SD after occipital expansion versus + 0.32 SD after fronto-orbital advancement), tonsillar herniation (3/11 Crouzon patients after occipital expansion versus 7/8 Crouzon patients after fronto-orbital advancement) and papilledema (4/19 after occipital expansion versus 11/18 Apert and Crouzon patients after fronto-orbital advancement) relative to a fronto-orbital advancement. The sight for both groups was comparable with 0.09 versus 0.13 (p = 0.28). Derderian found in a series of 30 patients that occipital distraction (142 cm3) was associated with a significantly greater increase in mean intracranial volume than fronto-orbital advancement (66 cm3).16
Conclusions
| Level 3 | For patients with Apert and Crouzon syndrome, occipital cranial expansion with distraction (conventional distraction or with springs) may result in a greater increase in head circumference (an approximate measure of intracranial volume), intracranial volume and a significantly lower prevalence of tonsillar herniation (3/11 versus 7/8) and papilledema (4/19 versus 11/18) compared to a fronto-orbital advancement or an occipital expansion without distraction after more than 5 years of follow-up. B Spruijt et al, 2016; C De Jong et al, 2013; Thomas et al, 2014; Derderian et al, 2015; Mundinger et al, 2016 |
| Level 3 | An endoscopic strip craniectomy with helmet therapy to treat syndromic bicoronal synostosis may have a higher risk of reoperation due to delayed cranial growth or signs of elevated ICP within 1 year after surgery compared to an open procedure. Minimally invasive endoscopic surgery is likely to involve less blood loss, fewer blood transfusions, shorter surgical and duration of hospitalisation than open cranial correction techniques. C Rottgers et al, 2016; Han et al, 2016 |
3. What are the long-term results regarding cognition and aesthetics of a different timing of surgery, i.e. ’early’, defined as before the age of 12 months, versus ’late’, i.e. after the age of 12 months?
In studies from Paris on cognitive outcome in syndromic craniosynostosis, the proportion of children with a normal IQ is greater if the operation was performed before the age of 1 year, but the age at which the analyses were performed is not mentioned. For patients with Apert syndrome, Crouzon syndrome and 99 patients with bicoronal synostosis, including 48 patients with Muenke syndrome, this difference has been demonstrated.7,17 Although there are methodological limitations to these studies, such as the bias of delayed referral resulting in a higher age at surgery. Renier describes that 17% of Apert children and 81% of Crouzon children had a normal IQ at surgery before age 1 versus 0% and 56% respectively at surgery after age 1.7 For bicoronal synostosis, the difference was an IQ of 99 (n = 59) for surgery before the age of 1 year versus an IQ of 89 (n = 13) for surgery after the age of 1 year.
Ridgway analyses the aesthetic result of the forehead in 20 patients with Muenke syndrome. Of these, 13 had an indication for an additional correction.18 The average age at which these 13 were operated on, was 5.9 months (range 2.5–10 months), while the age of the 7 patients without indication for correction was on average 39.4 months (range 5.9–112 months). An age-dependent cut-off point at which a less good result was achieved was not determined.
De Jong states that postponing fronto-orbital advancement until the age of 9 to 12 months for patients with Muenke syndrome is justified because of the low risk (4%) of increased intracranial pressure, in contrast to the risks in Apert, Crouzon and Saethre-Chotzen syndrome.5
Utria assess the results of surgery according to the Whitaker classification of 52 patients with syndromic craniosynostosis (Apert, Crouzon, Saethre-Chotzen and complex) in relation to their age at first surgery.19 Children younger than 6 months had a 4.1 fold higher odds on reoperation while the odds of children older than 9 months were 13.2 times higher. The optimum age for surgery is therefore suggested to be between 6 and 9 months, if there are no earlier signs of increased ICP.
Conclusions
| Level 3 | Patients with syndromic craniosynostosis in whom the cranial expansion is performed within the first year of life may have a better cognitive outcome: 17% of Apert children and 81% of Crouzon children have a normal IQ at surgery before the age of 1 versus 0% and 56% respectively at surgery after the age of 1. For bicoronal synostosis, the difference was an IQ of 99 at surgery before the age of 1 year versus 89 at surgery after the age of 1 year. Arnaud, 2002; Renier, 2000 |
| Level 3 | Timing of treatment of patients with Muenke syndrome from the age of 6 to 9 months may have a favourable effect on the aesthetic outcome compared to an earlier intervention and is justified in view of the low prevalence of elevated ICP. Timing of treatment of patients with Apert, Crouzon or Saethre-Chotzen syndrome between the ages of 6 to 9 months may have a favourable effect on the aesthetic outcome compared to an earlier or later operation and seems appropriate given the high prevalence of elevated ICP. B De Jong, 2010; C Utria, 2015; Ridgway, 2011 |
Considerations
• Evidence of the Conclusions
Indication for surgical treatment
Regarding the Apert and Crouzon syndromes, there is reasonable evidence for the outcome measure intracranial pressure as indication for surgical treatment (comparison over 5 years of follow-up routine cranial expansion in the first year of life versus treatment after elevated ICP has been detected).
Surgical technique
Regarding the Apert and Crouzon syndromes, there is weak to reasonable evidence for the outcome measures head circumference (as an approximate measure of intracranial volume), tonsillar herniation and papilledema (comparison occipital cranial expansion versus fronto-orbital advancement or occipital expansion without distraction after more than 5 years of follow-up), because most studies do not have a direct comparative design. Regarding multisuture or syndromic synostosis, there is weak evidence for the outcome measure reoperation. Only one study with a non-comparative design is available.
Timing of surgery
For syndromic synostosis, there is weak evidence for the outcomes in terms of aesthetic and mental outcome: the studies had no direct comparative design. To the extent that a multivariate analysis was applied to analyse the predictive effect of operating age on these outcome measures, this analysis was probably not conducted adequately.
• Values and Preferences
Indication for surgical treatment
According to the workgroup, most parents of children with multisuture or syndromic craniosynostosis find the final aesthetic result, neuro-cognitive functions and sight, crucial outcome measures and can therefore usually agree on a surgical recovery for the child. Sometimes parents are reluctant to accept the proposed cranial surgery and prefer intensive screening for signs of elevated ICP.
Surgical technique
According to the workgroup, most parents of children with multisuture or syndromic craniosynostosis find the aesthetic result in terms of skull shape and scar visibility, minimal risk of reoperation, neuro-cognitive functions and sight, crucial outcome measures and will opt for a technique that best fulfils these criteria. In case of a similar result of treatment, there is a preference for surgery at a young age, because parents experience the prospect of surgery as a burden.
Timing of surgery
According to the workgroup, most parents of children with multisuture or syndromic craniosynostosis have a preference for surgery at an early age, because parents experience the period with surgery in prospect as a burden. If the aesthetic result is better by postponing the operation slightly, with equal results for neuro-cognitive functions and vision, this will be preferred.
• Costs and Resources
The guideline committee expects that the implementation of the recommendations leads to little or no increase in costs, because the recommendations are in line with existing practice. This applies to all recommendations relating to:
-
-
indication for surgical treatment
-
-
surgical technique
-
-
timing of surgery
• Professional Perspective
Indication for surgical treatment
In multisuture and syndromic craniosynostosis, spontaneous improvement of the abnormal skull shape is not expected and may threaten neurocognition and vision if elevated ICP develops.
Elevated ICP is a hard indication to proceed to surgical treatment to prevent neurocognitive and vision impairment. The applied methods for monitoring predictors or symptoms of elevated ICP (following the growth curve of the head circumference, fundoscopy, optical coherence tomography (OCT), MRI and on indication direct ICP measurement) have their own specificity and sensitivity and can be false negative. If a cranial expansion is not protocolized, screening for increased ICP is essential. The frequency of monitoring will depend on the syndrome-specific prevalence of elevated ICP and will therefore be high for Apert and Crouzon syndrome and low for Muenke syndrome (see chapter 8). Since each screening tool can give false negative results, the guideline committee finds that there is still a valid fear that surgical indication based on elevated ICP (and therefore not protocol-based) may have a worse outcome for vision and neurocognition over the long term.
Surgical technique
The aim is to prevent or treat the occurrence of increased ICP with the lowest possible rate of intracranial surgery (open cranial correction and minimally invasive surgery) and to achieve the best possible results in terms of neurocognition, vision and aesthetics. Occipital expansion, especially in combination with distraction, seems to be more appropriate than fronto-orbital advancement for the treatment of Apert and Crouzon syndrome. Occipital expansion with spring distraction has the advantage over traditional distraction that no osteosynthetic material penetrates through the skin and therefore causes less inflammation. For Saethre-Chotzen and Muenke syndrome this may be different because of a lower risk of increased ICP and because a fronto-orbital advancement also offers aesthetic improvement.
Timing of surgery
Surgery before the age of 3 months is undesirable for anesthesiological safety reasons. Surgical correction of the supraorbital rim with an open technique (as is done with metopic and coronal suture synostosis) is technically more difficult for children aged 6 to 9 months, as the ossification of this bone is still limited. The advantage of surgery before or around the age of 1 year is that it achieves almost complete re-ossification of remaining cranial defects from the surgery.
• Balance of Anticipated Desired and Undesired Outcomes
Indication for surgical treatment
Only surgery can improve the aesthetic aspect and reduce elevated ICP, neurocognitive deficit and vision impairment. Screening for elevated ICP by using fundoscopy and/or OCT is not very stressful for the child and is the best available method for detection of elevated ICP. The detection of elevated ICP is predominantly described in children under the age of 6 years. After this age, screening seems to be indicated only in the presence of symptoms indicating elevated ICP.
Surgical technique
In general, there is a preference for minimally invasive surgery because of 1. a lower risk of blood transfusion and thus avoiding the associated risks and 2. less visible scars. For syndromic bicoronal synostosis, the risk of minimally invasive surgery for reoperation within 1 year seems too high.
In Apert and Crouzon syndrome and multisuture craniosynostosis involving at least both lambdoid sutures, occipital expansion with distraction is preferred due to the high risk of increased ICP and tonsillar herniation. Leaving the fronto-orbital region undisturbed during the first operation reduces the risk of complications in a later monobloc. In addition, a fronto-orbital advancement in Apert or Crouzon/Pfeiffer increases the facial disbalance by emphasizing the midface hypoplasia.
For Saethre-Chotzen and Muenke syndrome, the risk of increased ICP and tonsillar herniation is lower and cranial deformity is better corrected with fronto-orbital advancement. Moreover, these diagnoses often have no indication for midface advancement. For multisuture synostosis, the surgical technique chosen is based on the shape of the skull in order to achieve the most normal cranial shape possible.
Timing of surgery
Increased intracranial pressure is causing damage to the vision and possibly to the neurocognitive outcomes. Once increased intracranial pressure is detected, surgical treatment is indicated. Usually surgery is performed within the first year of life because of the high risk of increased ICP and possibly better neurocognitive outcomes with this timing. Occipital expansion with spring distraction is preferably performed around the age of 6 months as the cranium is more compliant and the ossification of the created cranial defect is more complete.
Rationale for the recommendation(s)
Indication for surgical treatment
The guiding principle in the formulation of the recommendations is that in multisuture and syndromic craniosynostosis there is no spontaneous improvement in cranial deformity and there is a significant syndrome-specific risk of increased ICP, which can lead to neurocognition and vision impairment in the patient. If protocol-based surgery is not performed in the first year of life, frequent monitoring for signs of elevated ICP is necessary.
Surgical technique
The guiding principle in the formulation of the recommendations is that elevated ICP should be prevented or treated rapidly, as soon as it occurs, in order to protect neurocognitive functions, and sight. In addition, the aim is to minimise the number of operations. Based on a high risk of elevated ICP and tonsillar herniation, an occipital expansion with distraction will be chosen in Apert and Crouzon syndrome and in multisuture craniosynostosis involving both lambdoid sutures. In the other multisuture and syndromic craniosynostosis, the risk of increased ICP is lower and the surgical technique is adapted to the cranial shape.
Timing of surgery
A guiding principle in the formulation of the recommendations is that aesthetic results of the cranial shape, neurocognitive functioning and vision are as optimal as possible. The neurocognitive results may be better if the operation is performed within the first year of life.
Recommendations
Indication for surgical treatment
Surgically correct multisuture and syndromic craniosynostosis.
Screen frequently for elevated ICP if surgery is waived. For specific advice on screening frequency, see chapter 8.
Treat as soon as an elevated CPI is detected.
Evaluate the neurocognitive functioning and vision of children with multisuture or syndromic craniosynostosis at the age of 7 years using standardized tests.
Surgical technique
Remodel the skull in Apert and Crouzon syndrome and multisuture craniosynostosis of at least both lambdoid sutures by occipital expansion with distraction.
Remodel the skull in Saethre-Chotzen and Muenke syndrome with fronto-supraorbital expansion.
Remodel the skull for other types of syndromic craniosynostosis based on the cranial deformity.
Consider minimally invasive treatment for non-syndromic bicoronal synostosis.
Correct the skull for other multisuture craniosynostosis based on cranial deformity. There is no scientific justification available for the choice between a minimally invasive or open technique.
Timing of surgery
Remodel the skull in multisuture and syndromic craniosynostosis between 6 and 9 months and in Muenke syndrome between 9 and 12 months.
Perform minimally invasive treatment for multisuture craniosynostosis as early as possible and at the latest before the age of 6 months.
Research Gaps
Indication for Surgical Treatment
Follow-up studies at the age of at least 6 years should demonstrate whether a wait-and-see policy for multisuture and syndromic craniosynostosis, in which surgery is performed only when elevated ICP is detected, is justified concerning aesthetic outcome, neurocognitive outcome, including tonsillar herniation, and sight.
Surgical Technique
The long-term outcome regarding aesthetics, neurocognition and vision of strip craniectomies in multisuture and syndromic craniosynostosis should be determined in studies with larger numbers of patients and in multiple centres, as current studies contradict each other.
The risk of increased ICP and tonsillar herniation, prior to surgery and for minimum 6 years of follow-up, is largely unknown for the different types of multisuture craniosynostosis, Saethre-Chotzen and Muenke syndrome.
Timing of Surgery
Further research should prove whether cranial expansion surgery within the first year of life indeed has better neurocognitive results than surgery after the age of 12 months and whether this period can be specified more precisely. This should be done for any type of multisuture and syndromic craniosynostosis.
Literature
-
1.
Hayward R, Britto J, Dunaway D, et al. Connecting raised intracranial pressure and cognitive delay in craniosynostosis: many assumptions, little evidence. J Neurosurg Pediatr 2016;18:242-50
-
2.
Marucci DD, Dunaway DJ, Jones BM, et al. Raised intracranial pressure in Apert syndrome. Plast Reconstr Surg 2009;123:1570-7
-
3.
Abu-Sittah GS, Jeelani O, Dunaway D, et al. Raised intracranial pressure in Crouzon syndrome: incidence, causes, and management. J Neurosurg Pediatr 2016;17:469-75
-
4.
Spruijt B, Rijken BFM, Den Ottelander BK, et al. First vault expansion in Apert and Crouzon-Pfeiffer syndromes: front or back? Plast Reconstr Surg 2016;137:112e-21e
-
5.
De Jong T, Bannink N, Bredero-Boelhouwer HH, et al. Long-term functional outcome in 167 patients with syndromic craniosynostosis; defining a syndrome-specific risk profile. J Plast Reconstr Aesthet Surg 2010;63:1635-41
-
6.
Kress W, Schropp C, Lieb G, et al. Saethre-Chotzen syndrome caused by TWIST 1 gene mutations: functional differentiation from Muenke coronal synostosis syndrome. Eur J Hum Genet 2006;14:39-48
-
7.
Renier D, Lajeunie E, Arnaud E, et al. Management of craniosynostoses. Childs Nerv Syst 2000;16:645–58
-
8.
Thompson DNP, Harkness W, Jones B, et al. Subdural intracranial pressure monitoring in craniosynostosis: its role in surgical management. Childs Nerv Syst 1995;11:269-75
-
9.
Greene AK, Mulliken JB, Proctor MR, et al. Phenotypically unusual combined craniosynostoses: presentation and management. Plast Reconstr Surg 2008;122:853-62
-
10.
Rottgers SA, Lohani S, Proctor MR. Outcomes of endoscopic suturectomy with postoperative helmet therapy in bilateral coronal craniosynostosis. J Neurosurg Pediatr 2016;18:281-6
-
11.
Jimenez DF, Barone CM. Multiple-suture nonsyndromic craniosynostosis; early and effective management using endoscopic techniques. J Neurosurg Pediatr 2010;5:223-31
-
12.
Han RH, Nguyen DC, Bruck BS, et al. Characterization of complications associated with open and endoscopic craniosynostosis surgery at a single institution. J Neurosurg Pediatr 2016;17:361-70
-
13.
Thomas GPL, Wall SA, Jayamohan J, et al. Lessons learned in posterior cranial vault distraction. J Craniofac Surg 2014:25:1721-7
-
14.
De Jong T, Van Veelen MLC, Mathijssen IMJ. Springs-assisted posterior vault expansion in multisuture craniosynostosis. Childs Nerv Syst 2013;29:815-20
-
15.
Mundinger GS, Rehim SA, Johnson III O, et al. Distraction osteogenesis for surgical treatment of craniosynostosis: A systematic review. Plast Reconstr Surg 2016;138:657-69
-
16.
Derderian CA, Wink JD, McGrath JL, et al. Volumetric changes in cranial vault expansion: Comparison of fronto-orbital advancement and posterior cranial vault distraction osteogenesis. Plast Reconstr Surg 2015;135:1665-72
-
17.
Arnaud E, Meneses P, Lajeunie E, et al. Plast Reconstr Surg 2002;110:6-12
-
18.
Ridgway EB, Wu JK, Sullivan SR, et al. Craniofacial growth in patients with FGFR3Pro250Arg mutation after fronto-orbital advancement in infancy. J Craniofac Surg 2011;22:455-61
-
19.
Utria AF, Mundinger GS, Bellamy JL, et al. The importance of timing in optimizing cranial vault remodelling in syndromic craniosynostosis. Plast Reconstr Surg 2015;135:1077-84
CHAPTER 7 SURGICAL TREATMENT OF SYNDROMIC CRANIOSYNOSTOSIS – FACE
7.1 What is the surgical management of face in syndromic craniosynostosis with midface hypoplasia?
Introduction
The Apert and Crouzon syndromes are associated with hypoplasia of the maxilla, exorbitism and hypertelorism and, to a lesser extent, hypoplasia of the mandible. The indication for surgical correction varies from an acute ophthalmic or respiratory-threatening problem to a relatively functional (non-acute) occlusal problem and/or aesthetic/psychological problem. Various different techniques are possible to correct these deformities, the timing of which has a major influence on the result.
Search and Selection
For the following specific questions, original scientific studies or systematic reviews of scientific studies have been included:
-
1.
What are the surgical specific factors that influence the choice between different surgical techniques (internal versus external distraction and Le Fort III osteotomy versus variations on Le Fort III osteotomy) for the treatment of midface hypoplasia?
-
2.
What are the long-term surgical specific results of different timing of surgery in the absence of a hard indication, i.e. ’early’, defined as before the age of 6 to 8 years, versus ’late’, i.e. after the age of 6 to 8 years?
In the Medline (OVID) and Embase databases, a single comprehensive search was carried out for studies on craniosynostosis. The search strategy is shown in appendix 2, to the guideline. After deduplication, the literature search yielded 2732 hits.
Given the large number of studies, the chair of the working group first selected those that met the following general selection criteria:
General Selection and Exclusion Criteria
| Study type | -original studies - systematic reviews of sufficient quality: - research question of systematic review corresponds (largely) to the basic question - search is performed in at least 2 relevant databases, e.g. Cochrane Library, Medline/PubMed - reporting of the complete search strategy - no relevant keywords/search terms are missing |
| Follow-up period | - minimum follow-up period of 12 months for therapeutic or prognostic studies. |
| Exclusion criteria | - Case-reports - Expert opinion - Letters -Editorials - Case control studies for diagnostic tests - Narrative reviews |
The pre-selected studies that met the specific selection criteria listed in the Table below are included in the literature summary of this chapter.
Specific Selection and Exclusion Criteria
| -minimum study size: 10 patients - minimum follow-up time: 36 months |
Summary of the Literature
1. What are the surgical specific factors that influence the choice between different surgical techniques (internal versus external distraction and Le Fort III osteotomy versus variations on Le Fort III osteotomy) for the treatment of midface hypoplasia?
A systematic review analyses midface relapse after internal versus external distraction in Le Fort III procedures.1 Of the 57 selected articles, 12 were included. The analysis shows that a Le Fort III with distraction results in more advancement than without distraction, i.e. 9–16 mm versus 8–12 mm. Moreover, a Le Fort III with distraction is associated with 14.4% or less than 10% long-term relapse, and a Le Fort III without distraction with 8.7–11.9% relapse (with one study even showing 50% relapse). The age of the patients at the time of surgery ranged from 2 to 24 years for the conventional Le Fort III and from 3 to 32 years for the Le Fort III with distraction.
Goldstein reports a retrospective analysis of complications in 23 patients with Apert syndrome and 29 patients with Crouzon syndrome who received a midface advancement using a Le Fort III or monobloc procedure with external or internal distraction.2 A total of 33 patients received 34 Le Fort III corrections and 21 patients received 21 monoblocs. Thirty operations were performed with external distraction (18 Le Fort III and 12 monobloc) and 25 with internal distraction (16 Le Fort III and 9 monobloc). Of the 19 distractor-related complications, 10 cases were in the external distractor group and 9 cases in the internal distractor group. Serious infections were observed more frequently in the internal versus the external distraction group (n = 8 versus n = 3) and 4 patients in the external distraction group needed reoperation due to dislocation of the distractor or transcranial migration of a pin. The choice for type of surgery was based on the present facial abnormalities; the choice for internal or external distraction was mainly based on a preference for external in the last years. The advantages mentioned are vector control with the possibility to adjust it during the distraction, better correction of facial concavity, fewer infections and better osteogenesis.
Hopper describe an alternative to the Le Fort III in patients with Apert syndrome, namely a Le Fort II with bilateral zygomatic advancement.3 This segmentation would contribute to a better facial correction. The study compares 5 Apert patients receiving a conventional Le Fort III to 4 Apert patients receiving a Le Fort II/zygomata and compares these with 5 unoperated Crouzon patients and 6 controls. The Le Fort II/zygomata provided more facial lengthening and more advancement of the central region compared to the conventional Le Fort III, with the facial ratios no longer deviating from normal, as was still the case after a conventional Le Fort III.
Greig describes a retrospective analysis of 20 patients (19 Apert, 1 Pfeiffer) in which a facial bipartition distraction with an external frame is performed at the age of 1.6 to 21 years.4 The authors indicate that the mean central advancement was 13.2 +/− 5.9 mm measured at the sella-nasion point and 11.7 +/− 5.4 mm measured at the sella-A point, while the lateral advancement was 4.7 +/− 2.8 mm. Facial bipartition was considered an efficient method of moving forward the central part of a face of an Apert patient.
In a series of 105 patients, Arnaud and Di Rocco describe the results of monobloc distraction with 4 internal distractors (performed at mean age 4.9 years, range 7 months-14 years).5 The main complication was liquor leakage in 19 patients, which was treated with conservative management in 11 and with temporary lumbar drainage in 8. In 9 patients, revision surgery was required due to distractor problems. In 4 of these 9 patients, the distraction could be completed. The authors conclude that monobloc distraction can lead to correction of airway problems and exorbitism at an early age.
Ahmad describes in a retrospective analysis 12 patients with monobloc distraction with an external frame.6 All patients are younger than 30 months with a mean age of 18 months. The mean forward displacement is 16.6 mm for the upper part of the face and 17 mm for the midface. In this series the following complications are reported: in 2 cases a liquor leak (16.7%), in 3 cases an infection of the skin around the pin (25%) and in 2 cases the frame had to be repositioned under general anaesthesia after the frame had shifted (16.7%; 1x due to trauma and 1x spontaneously).
Conclusions
| Level 3 | Le Fort III with distraction probably achieves more midface advancement than Le Fort III without distraction (9–16 mm versus 8–12 mm) and probably has less long-term relapse than Le Fort III without distraction (8.7–11.9% or 50% versus 14% or less than 10%). C Saltaji et al, 2014 |
| Level 3 | For Le Fort III distraction, an external frame is probably preferable over an internal frame because of optimal vector control. Possible other advantages of an external frame are better facial concavity correction and fewer wound infections. C Goldstein et al. 2015 |
| Level 3 | A monobloc with distraction at a young age can be performed with both internal and external distraction to correct respiratory problems and exorbitism, where complications such as liquor leakage and mechanical problems with the devices (9% versus 16.7%) are not substantially different. C Arnaud and Di Rocco, 2012; Ahmad et al, 2012 |
| Level 3 | In Apert syndrome, a facial bipartition with distraction is likely to be performed with an external frame because of the possibility of moving the loosened distraction fragments in the middle to the front of the face, thus changing the facial appearance from concave to convex. C Greig et al, 2013 |
| Level 3 | In Apert syndrome, a Le Fort II with distraction combined with bilateral zygomatic advancement (without distraction) probably results in a better correction of the facial abnormalities than does a Le Fort III with distraction. B Hopper et al, 2013 |
2. What are the long-term surgical specific results of different timing of surgery in the absence of a hard indication, i.e. ’early’, defined as before the age of 6 to 8 years, versus ’late’, i.e. after the age of 6 to 8 years?
Caterson follow 19 patients who have undergone a Le Fort III without distraction at the age of 3 to 5 years, until the age of complete skeletal development.7 All 19 patients turned out to need a reoperation, 12 of them a Le Fort III, because of relapse midface underdevelopment. The other 7 patients underwent a Le Fort I or II osteotomy.
Patel and Fearon evaluate 32 patients who have undergone a Le Fort III with external distraction and have now reached skeletal facial outgrowth.8 Factors associated with a second midface distraction were surgery before the age of 8 years and the lack of overcorrection.
Gwanmesia describes the 10-year follow-up of 20 patients who received a monobloc with external distraction.9 Striking is the fact that three patients died: one by accidental decannulation in the home situation and two by epileptic seizures. Skeletal advancement was found to remain stable; functional gains with respect to respiratory function decreased in 4 out of 17 patients, all of whom had had surgery at or before the age of 7 years (11 total). An increase in exorbitism was seen in 6 of the 11 patients undergoing surgery before the age of 7. In none of these cases was additional treatment provided.
Conclusions
| Level 3 | A Le Fort III without distraction, performed before the age of 6 years, probably leads to a high risk of recurrent midface hypoplasia in adulthood. C Caterson et al, 2013 |
| Level 3 | A Le Fort III with external distraction, performed before the age of 8 years and without overcorrection, probably increases the risk of recurrent midface hypoplasia in adulthood. C Patel and Fearon, 2015 |
| Level 3 | A monobloc with external distraction seems to provides Table midface advancement regardless of age at surgery. This procedure performed before the age of 8 years seems to lead to a higher risk of recurrence of respiratory problems. C Gwanmesia et al, 2015 |
Considerations
• Evidence of the Conclusions
Surgical technique
The strength of evidence for the conclusions is weak: most studies had a non-comparative design; the few studies with a comparative design had a serious risk of bias and the numbers of patients studied were low.
Timing of surgery
The strength of evidence for the conclusions is weak: all studies were non-comparative in design; the numbers of patients studied were low.
• Values and Preferences
Surgical technique
According to the workgroup, most parents of children with Apert or Crouzon/Pfeiffer syndrome consider the aesthetic result of the face and functional improvement of eye closure and breathing to be crucial outcome measures. Because of the better results achieved with distraction, parents will agree to this. However, the use of an external distraction frame may raise objections from parents because more cooperation of the child is necessary with adjustment of daily activities. Mental developmental deficit can be a relative contraindication for the use of an external frame.
Timing of surgery
According to the workgroup, parents often prefer surgery at a young age because they are afraid that their child will be bullied because of its abnormal appearance. Some parents prefer, if there is no functionally compelling reason for early treatment, to wait until the child is at an age to decide for himself or herself. In this case, there is a relative indication: research has shown that parents find it difficult to make a decision in this situation.10
• Costs and Resources
Surgical technique
The guideline committee expects that the implementation of the recommendations lead to little or no increase in costs, because the recommendations are in line with existing practice.
Timing of surgery
The guideline committee expects that the implementation of the recommendations lead to little or no increase in costs, because the recommendations are in line with existing practice.
• Professional Perspective
Surgical technique
A Le Fort III gives more reliable results with external distraction by better control of the distraction vector. A monobloc with distraction can be performed with both internal and external distraction.
The choice of surgical technique is determined by the preference and expertise of the surgeon and is tailored to the specific characteristics of the patient and the wishes of the child and parents.
The alternative technique of Le Fort II with distraction, combined with bilateral zygomatic advancement, can be a good alternative, depending on the facial abnormalities.
In the case of a monobloc distraction, there seems to be a preference to use internal distractors (instead of external distractors) because of fewer problems with the application of internal distractors. Because of the convenience and comfort for the patient, an internal distractor is always preferred.
In the case of a monobloc distraction at a very young age, the thin skull bone sometimes needs to be temporally reinforced with a titanium mesh plate (to prevent intracranial migration), which also needs to be removed after consolidation. This eliminates the advantage of the ease of removal of an external frame.
In the case of hypertelorism (with or without vertical dystopia), a facial bipartition can be considered to correct this. This technique also gives more advancement of the midline of the face, which is often desired in Apert syndrome. Distraction in facial bipartition is mainly performed with an external frame, because this allows pulling of the distraction fragment precisely in the midline, which can change the concave face into a convex face. This is technically not possible with internal distractors placed laterally.
For all techniques it is also possible to do a combination of external and internal distraction, where the external frame is only used during the distraction period and then removed, after which the internal distractors provide stability during the consolidation period. This limits the period during which a child walks with an external frame and is slightly more at risk of injury from the frame.
Timing of surgery
Functional indications such as moderate to severe respiratory problems, incomplete eye closure with risk of corneal damage (severe exorbitism) may require midface surgery at a very young age (before 4 years of age). This implies a high risk of reoperation due to recurrent midface hypoplasia due to lack of intrinsic growth.
Without a hard indication there is a possibility to perform midface surgery from the age of 8 years, as this can bring the orbital part of the face into a permanent position, a (temporary) improvement of the profile is given, which can be positive for the child. In addition, the risk of relapse is less than when the patient is operated on before the age of 8 years.
Given almost every adolescent's anxiety about his or her appearance, there is great reluctance to perform midface surgery between the ages of 12 to 17 years and preferably only after this period.
After midface advancement, regardless of the age at which it is performed, an additional surgical correction of the upper and lower jaw is often required to close the open bite.
If necessary, from the age of 17, a combined correction of the lower jaw can be performed to achieve a definitive occlusion.
Patients with Crouzon/Pfeiffer regularly show that despite the midface hypoplasia there is a normal nasal projection. A Le Fort III (including nose) advancement would lead to an undesirably long and large nose. In these cases, a Le Fort I including bilateral zygomatic advancement (Butterfly) can be chosen.
• Balance of Anticipated Desired and Undesired Outcomes
Surgical technique
Given the severe degree of midface hypoplasia in Apert and Crouzon/Pfeiffer syndrome, the use of distraction is almost always indicated to achieve sufficient advancement.
The choice for Le Fort III, monobloc, facial bipartition or Le Fort II with zygomatic advancement is determined by the individual facial abnormalities, in order to correct these in the best possible way.
The distraction in a Le Fort III and facial bipartition are almost always done with an external frame to have an optimal control over the vector of distraction, but can be combined with internal distraction to reduce the time that a child walks around with a frame, so there is less risk of trauma and comfort increases.
Timing of surgery
Midface surgery in the first two years of life is technically more difficult due to the lower ossification of the facial bones. At the same time, the degree of advancement is limited and recurrent midface hypoplasia occurs more often if the surgery is performed at a younger age, because there is no intrinsic growth of the midface. Timing of midface surgery is therefore determined in the first years of life by functional indications, in particular insufficient eye closure with risk of corneal damage and severe obstructive sleep apnea. If these hard indications do not occur, postponement until the age of 7 to 8 years is indicated, because the risk of recurrent midface hypoplasia decreases from that age. Due to psychological considerations, puberty is avoided to perform elective midface surgery and the procedure is postponed to the age of 17 years or older if surgery before the age of 12 years is waived.
Rationale for the recommendation(s)
Surgical technique
The guiding principle in the formulation of the recommendations is that there is little or no growth of the midface in Apert and Crouzon syndrome. A normal position of the midface can only be achieved with surgery. More advancement can be achieved with distraction. By individual difference in phenotype, midface correction can be done with different techniques.
Timing of surgery
As soon as hard indications for midface surgery arise, this surgery will be performed in the short term, regardless of age. In the absence of hard indications, the timing will be done in consultation with parents and can be chosen between 8 and 12 years of age or from 17 years of age.
Recommendations
Surgical technique
Combine a midface advancement in children with Apert and Crouzon/Pfeiffer syndrome in principle always with distraction.
Use preferably an external frame for Le Fort III distraction and facial bipartition.
Consider combining external distraction with internal distraction so that the frame can be removed after finishing the distraction.
Choose between Le Fort III, monobloc, facial bipartition, Le Fort II with zygomatic (Butterfly) advancement or Le Fort I with zygomatic (butterfly) advancement for each individual patient, tailored to its facial abnormalities.
Timing of surgery in the absence of a hard indication
Perform a midface advancement with distraction in children with Apert and Crouzon syndrome as a rule between the ages of 8 and 12 years or from the age of 17.
Advance this procedure in case of severe OSA and/or insufficient eye closure with risk of corneal damage.
Perform a midface advancement preferably not in children aged 12 to 17 because of the higher risk of psychosocial problems or unrealistic expectations.
Research Gaps
Indication for Surgical Treatment
There are no proper studies available that compare the degree of midface advancement of a conventional monobloc with a monobloc with distraction.
There are no studies available that compare the long-term results of monobloc with distraction with the two-stage procedure of fronto-orbital advancement and later Le Fort III with distraction.
There are no studies available that compare the correction of hypertelorism with an orbital box osteotomy and facial bipartition.
Literature
-
1.
Saltaji H, Altalibi M, Major MP, et al. Le Fort III distraction osteogenesis versus conventional Le Fort III osteotomy in correction of syndromic midfacial hypoplasia: a systematic review. J Oral Maxillofac Surg 2014;72:959-72
-
2.
Goldstein JA, Paliga JT, Taylor JA, et al. Complications in 54 frontofacial distraction procedures in patients with syndromic craniosynostosis. J Craniofac Surg 2015;26:124-8
-
3.
Hopper RA, Kapadia H, Morton T. Normalizing facial ratios in Apert syndrome patients with Le Fort II midface distraction and simultaneous zygomatic repositioning. Plast Reconstr Surg 2013;132:129-40
-
4.
Greig AVH, Britto JA, Abela C, et al. Correcting the typical Apert face: combining bipartition with monobloc distraction. Plast Reconstr Surg 2013;131:219e-30e
-
5.
Arnaud E, Di Rocco F. Faciocraniosynostosis: monobloc frontofacial osteotomy replacing the two-stage strategy? Childs Nerv Syst 2012;28:1557-64
-
6.
Ahmad F, Cobb ARM, Mills C, et al. Frontofacial monobloc distraction in the very young: a review of 12 consecutive cases. Plast Reconstr Surg 2012;129:488e-97e
-
7.
Caterson EJ, Shetye PR, Grayson BH, et al. Surgical management of patients with a history of early Le Fort III advancement after they have attained skeletal maturity. Plast Reconstr Surg 2013;132:592e-601e
-
8.
Patel N, Fearon JA. Treatment of the syndromic midface: A long-term assessment at skeletal maturity. Plast Reconstr Surg 2015;135:731e-42e
-
9.
Gwanmesia I, Jeelani O, Hayward R, et al. Frontofacial advancement by distraction osteogenesis: a long-term review. Plast Reconstr Surg 2015;135:553-60
-
10.
Bredero-Boelhouwer H. Joosten KFM, Van Veen-Van der Hoek M, et al. Family centred care during midface advancement with a rigid external device: What do families need? J Plast Reconstr Aesth Surg 2013;66:1103-8
CHAPTER 8 INCREASED INTRACRANIAL PRESSURE
8.1 What is the management of increased intracranial pressure (ICP) in craniosynostosis?
Introduction
The risk of increased ICP varies greatly depending on the type of craniosynostosis; the multisuture and the syndromic types are associated with a much higher risk than are the isolated non-syndromic types. However, the risk of these problems in the isolated non-syndromic group is much less recognised and therefore possibly underdiagnosed. It is important to timely detect and treat increased ICP. High intracranial pressure, for example, can lead to irreversible vision impairment.1 It is unclear which method is most suitable for detecting increased intracranial pressure, which cut-off values should be used and how often this examination should be carried out in order to detect problems in time. Increased ICP is caused by craniocerebral imbalance, aberrant venous drainage, OSA, tonsillar herniation and hydrocephalus. Depending on the affected cranial sutures, this is determined in increasing percentage for cranial decompression. However, increased ICP can also occur after or despite decompression, in relation to the severity of the condition and probably the type of surgery.
Search and Selection
For the following specific questions, original scientific studies or systematic reviews of scientific studies have been included:
-
1.
What is the prevalence of increased intracranial pressure in different types of craniosynostosis?
-
2.
What is the diagnostic accuracy of the following diagnostic modalities for detecting or excluding increased intracranial pressure?
-
-
(abnormal) head circumference growth curves;
-
-
the presence or absence of diffuse impressions detected by X-skull;
-
-
the presence or absence of additional coronal suture fusion detected by X-skull or CT;
-
-
optic nerve ultrasound;
-
-
the presence or absence of papilledema detected by fundoscopy;
-
-
optical coherence tomography (OCT)?
-
-
-
3.
What are craniosynostosis-specific factors that influence the choice between different surgical techniques for the treatment of increased ICP?
In the Medline (OVID) and Embase databases, a single comprehensive search was carried out for studies on craniosynostosis. The search strategy is shown in appendix 2, to the guideline. After deduplication, the literature search yielded 2732 hits.
Given the large number of studies, the chair of the working group first selected those that met the following general selection criteria:
General Selection and Exclusion Criteria
| Study type | - original studies - systematic reviews of sufficient quality: - research question of systematic review corresponds (largely) to the basic question - search is performed in at least 2 relevant databases, e.g. Cochrane Library, Medline/PubMed - reporting of the complete search strategy - no relevant keywords/search terms are missing |
| Follow-up period | -minimum follow-up period of 12 months for therapeutic or prognostic studies. |
| Exclusion criteria | - Case-reports -Expert opinion -Letters -Editorials -Case control studies for diagnostic tests -Narrative reviews |
The pre-selected studies that met the specific selection criteria listed in the Table below are included in the literature summary of this chapter.
Specific Selection and Exclusion Criteria
| -minimum study size: 20 patients -minimum follow-up period: up to age 2 years. |
Summary of the Literature
1. What is the prevalence of increased intracranial pressure in different types of craniosynostosis?
Review of the literature on the prevalence of increased intracranial pressure cannot be separated from the method used to measure ICP, and is therefore related to the next question (question 2).
Isolated Non-syndromic Craniosynostosis
The studies by Thompson, Renier, Mathijssen, Eley, and Wall describe invasive preoperative ICP measurements in isolated synostosis (Table 1).2–6 The studies of Van Veelen and of Cornelissen use fundoscopy for the detection of papilledema.7–9
TABLE 1. Prevalence of Increased Intracranial Pressure in Non-syndromic Craniosynostosis Prior to Cranial Correction (Number of Patients with Preoperative Increased ICP/Number of Patients Measured (%)).
| Scapho | Trigono | Plagio | |
| Thompson 1995–1#,2 | 6/25 (24) | 4/12 (33) | 3/37 (8) |
| Renier 20003 | 34/246 (14) | 3/39 (8) | 8/63 (13) |
| Mathijssen 20064 | 8/50 (16) | ||
| Eley 2012∗,5 | 3/7 (42) | ||
| Wall 2014∗,6 | 16/39 (44) | ||
| Van Veelen 2013 (max. 6 months old)7 | 2/79 (2.5) | ||
| Van Veelen 2015 (max. 11 months old)8 | 5/58 (10) | ||
| Cornelissen 20179 | 5/261 (2) |
ICP measurement only in patients who were suspected of increased ICP to determine timing of surgery.
only inclusion of patients in whom conservative management was considered due to mild phenotype or parental hesitation; no relationship was found with degree of cranial deformity.
One systematic review addressed the prevalence of increased intracranial pressure after surgical correction. The authors only searched in PubMed with the limited search terms “craniosynostosis” and “intracranial hypertension”, and increased ICP had to have been demonstrated with an invasive ICP measurement.10 Only 7 studies were included and only sagittal suture synostosis was mentioned, with prevalence estimated around 5%. The reliability of this review is low, since at least 1 of the 7 studies is not based on invasive ICP measurement but on fundoscopy.7 However, this 5% corresponds with the percentages of several studies given in Table 2. Of the two studies with a follow-up of more than 7 years, only Van Veelen provides the age at which papilledema was diagnosed in children with sagittal suture synostosis: at the ages of 3.5, 4 and 6 years.8,11
Table 1 Prevalence of increased intracranial pressure in non-syndromic craniosynostosis during follow-up (number of patients with preoperative increased ICP/number of patients measured (%)).
| Scapho | Trigono | Follow-up period | |
| Van Veelen 20137 | 7/79 (9) | 3 yrs 9 mos, after strip craniectomy (age 6 mos) | |
| Van Veelen 20158 | 3/44 (7) | 7 yrs, after frontobiparietal correction (age 11 mos) | |
| Van Veelen 201823 | 2/82 (2) | 4 yrs, after spring distraction (age 6 mos) | |
| Thomas 201511 | 15/217 (7) | 7 yrs 4 mos, after strip craniectomy (age < 1 yrs) | |
| Cornelissen 20179 | 3/196 (1.5) | 4 yrs, after frontorbital correction (age 11 mos) |
Multisuture and Syndromic Craniosynostosis
Both Thompson and Renier describe invasive preoperative ICP measurements.12,3
Marucci reports on 24 patients with Apert syndrome who were operated on after showing signs of increased ICP.13 Of these 24, 83% developed increased ICP at a mean age of 18 months. Of these treated patients, 35% experienced a second episode of increased ICP during follow-up. The increased ICP was defined as the presence of papilledema, an abnormal VEP, an ICP measurement > 15 mm Hg or more than 3 abnormal plateaus in 24 hours.
Kress reports a prevalence of 35% of increased ICP in Saethre-Chotzen syndrome based on papilledema or ICP measurement above 20 mm Hg.14 Papilledema was never found in 42 male patients.
Greene selected 39 patients with unusual combinations of synostotic sutures, excluding isolated and bicoronal synostosis and pansynostosis, 16 of whom have syndromic craniosynostosis (1 Apert, 8 Crouzon/Pfeiffer, 3 Saethre-Chotzen, 1 2q13 deletion, 1 9pdeletion/2p duplication, 1 Kabuki, 1 VACTERL).15 In this group, 30 out of 39 patients (77%) were diagnosed with increased ICP, defined as papilledema, deflecting head circumference curve or extensive endocortical erosion on CT images.
Woods evaluates 34 Saethre-Chotzen patients in whom after 1 year of follow-up 35% and after 5 years of follow-up 42% developed increased ICP.16 The definition of increased ICP was a baseline of 20 mm Hg or higher or 4 or more B waves. The indication for the measurement of the ICP was papilledema, a deflecting head circumference curve, headache or aggressive behaviour, impressions on the skull X-ray, narrow ventricles, Chiari, with elapsed basal cisterns and sulci spaces. De Jong gives the prevalence of increased ICP per syndrome, determined by fundoscopy.17 Abu-Sittah uses a set of symptoms to determine increased ICP, namely papilledema, an abnormal VEP scan, a tense fontanel, progressive ventriculomegaly and invasive ICP measurement.18
Table 3 Prevalence of increased intracranial pressure in multisuture and syndromic craniosynostosis.
| Apert | Crouzon | Saethre-Chotzen | Muenke | Multisuture | Bicoronal | |
| Thompson 199512 | ||||||
| preop | 5/13 (38) | 16/25 (64) | 6/14 (43) | 8/12 (67) | ||
| Reinier 20003 | ||||||
| Preop | ? (45) | ? (63) | 65/112 (58) | 10/32 (31) | ||
| Kress 200614 | ||||||
| preop | 24/68 (35) | 0/42 (0) | ||||
| postop | ? (18) | ? | ||||
| Greene 200815 | ||||||
| preop∗ | ||||||
| Marucci 200813 | ||||||
| preop | 20/24 (83) | |||||
| postop | 7/20 (35) | |||||
| De Jong 201017 | ||||||
| preop | 2/22 (9) | 24/45 (53) | 5/26 (19) | 1/28 (4) | ||
| postop | 11/31 (35) | 8/40 (20) | 4/24 (17) | 2/38 (5) | ||
| Woods 200916 | ||||||
| postop 1 yrs | 9/26 (35) | |||||
| postop 5 yrs | 8/19 (42) | |||||
| Abu-Sittah 201518 | ||||||
| preop | 30/49 (61) | |||||
| postop | 14/30 (47) |
In 30 out of 39 (77%) increased ICP is reported but without discriminating between the specific types of syndromic or multisuture craniosynostosis.
In summary, the prevalence of increased ICP in multisuture and syndromic craniosynostosis is significantly higher compared to non-syndromic craniosynostosis and it is syndrome specific.
Conclusions
| ----- | The prevalence of preoperatively increased ICP in sagittal suture synostosis probably ranges from 2.5 to 14%, and increases with age. The prevalence of preoperatively increased ICP in metopic suture synostosis probably ranges from 2 to 8%, and for isolated coronal suture synostosis around 16%. Thompson et al, 1995–1; Renier et al, 2000; Mathijssen et al, 2006; Eley et al, 2012; Wall et al, 2014; Van Veelen et al, 2013; Van Veelen et al, 2015; Cornelissen et al, 2017. |
| ----- | The prevalence of increased ICP during follow-up after cranial correction in isolated non-syndromic craniosynostosis probably varies from 2 to 9% for sagittal suture synostosis, and around 1.5% for metopic suture synostosis. Corresponding figures for isolated coronal suture synostosis are not known. Christian et al, 2015; Van Veelen et al, 2013; Van Veelen et al, 2015; Van Veelen et al, 2017; Thomas et al, 2015; Cornelissen et al, 2017. |
| ----- | The prevalence of increased ICP in syndromic craniosynostosis before cranial surgery is likely to be 9 to 83% for Apert, 53 to 64% for Crouzon, 19 to 43% for Saethre-Chotzen and 0 to 4% for Muenke syndrome. Kress et al, 2006; Marucci et al, 2008–1; De Jong et al, 2010; Woods et al, 2009 Abu-Sittah et al, 2015. |
| ----- | The prevalence of increased ICP in syndromic craniosynostosis after cranial surgery is likely to be 35 to 45% for Apert, 20 to 47% for Crouzon, 17 to 42% for Saethre-Chotzen and 0 to 5% for Muenke syndrome, 58 to 67% for multisuture craniosynostosis and around 31% for bicoronal synostosis. Thompson et al, 1995–2; Renier et al, 2000; Kress et al, 2006; Greene et al, 2008 Marucci et al, 2008–1; De Jong et al, 2010; Abu-Sittah et al, 2015. |
2. What is the diagnostic accuracy of the following diagnostic modalities for detecting or excluding increased intracranial pressure: (abnormal) head circumference growth curves; the presence or absence of diffuse impressions detected by X-skull; the presence or absence of additional coronal suture fusion detected by X-skull or CT; optic nerve ultrasound; the presence or absence of papilledema detected by fundoscopy; optical coherence tomography (OCT)?
For metopic suture synostosis, Cornelissen found a relation between the results of two screening methods, namely papilledema in fundoscopy and a deflecting head circumference growth curve.9 Given the very low incidence of papilledema in this type of craniosynostosis, follow-up by head circumference measurement is advised, whereby additional diagnostics by fundoscopy are only used as soon as the growth curve deflects. In the case of sagittal suture synostosis, Van Veelen described a deflecting head circumference growth curve for all types of surgery.19 A possible correlation is found with papilledema, but this is insufficient to use only the head circumference as a screening method.
In Apert and Crouzon syndrome, a deflected head circumference growth curve also appears to be an important predictor of increased ICP.20 Measurements of head circumference appear to correlate strongly with the intracranial volume, which can therefore be used as a screening method.21 Increased ICP in syndromic craniosynostosis can also have causes other than reduced cranial growth, such as obstructive sleep apnea, and for this reason, screening with a different method is recommended.
Tuite relates in 74 children with unoperated non-syndromic craniosynostosis (21 sagittal, 14 metopic, 36 unicoronal and 3 lambdoid suture synostosis) and 49 children with unoperated syndromic craniosynostosis (22 Saethre-Chotzen, 16 Crouzon, 8 Apert and 3 Pfeiffer syndrome) the presence of signs of increased pressure on a skull image with an invasive ICP measurement.22 In the non-syndromic group, the average age was 1.9 years ± 2.5 and in the syndromic group, the average age was 12 months (range 3 months to 11.3 years). For comparison, the same signs of increased pressure on a skull image were determined in children who had suffered moderate head trauma and did not need hospitalisation or additional radiological examination, matched to age and sex. The presence of impressions did not differ between patients and controls, but the severity of impressions was higher in craniosynostosis patients (2.6 versus 1.4). Children with elevated ICP were more likely to have a diffuse impression, diastatic sutures and erosion of the sella turcica. In both patients and controls younger than 18 months, impressions were hardly seen or only in the posterior part. With increasing age, more impressions were found in both groups. Of the 123 patients, 37 had severe impressions and a mean ICP of 14.8 mm Hg ± 5.4; the 86 with less severe impressions had a mean ICP of 12.0 mm Hg ± 4.7. Both impressions and ICP were age-related, and after correction, there was no correlation between these 2 elements. The sensitivity of diffuse impressions was 0% for age < 18 months, 64% for 18 months to 4 years, 60% for older than 4 years; the specificity was 98%, 50% and 20% respectively. With this low sensitivity, it is not a good screening method, but the presence of impressions in patients younger than 18 months is very predictive for increased ICP. In the study by Van Veelen, 83 patients were routinely followed up with skull images after spring distraction for sagittal suture synostosis.23 Shortly after the procedure, some impression was visible on the skull in 10% and this percentage gradually increased to 40% at the age of 5 years. The occurrence of impressions was not related to having papilledema, because of the very low prevalence of only 2 patients with papilledema.
A further suggested risk factor for the development of increased ICP is fusion of the remaining cranial sutures following the surgical correction.
In 2009, Arnaud published a large series of patients with sagittal suture synostosis, divided over 4 groups, who have been followed for at least 3 years.24 Group 1 (n = 193) received a so-called H-craniotomy of which 20 (10.4%) subsequently developed coronal suture synostosis, especially around the age of 2 years, and 18 of these 20 also had diffuse impressions; group 2 (n = 24) received a craniotomy including coronal suture removal and did not develop a coronal suture fusion according to the authors, but a new suture is said to have been formed; group 3 (n = 36) received an H-craniotomy without coronal suture removal and 4 (11%) developed an additional coronal suture fusion. In group 4 (n = 253), surgery was waived (mild presentation, refusal of parents) and coronal suture fusion was found in only 3 children (1.2%). Of the 20 children with coronal suture fusion in group 1, 2 developed papilledema in the follow-up and increased ICP was diagnosed with an invasive measurement followed by surgical intervention (2/20 = 10%). None of the other group 1 patients without coronal sutures (n = 173) developed symptoms of increased ICP (0%). Following a similar technique as described by Arnaud in group 1, Van Veelen found coronal suture fusion in 14 out of 69 (20%) patients 1 year after surgery.7 Four out of 14 patients with coronal suture fusion had papilledema (28.6%), while 3 out of 55 patients without coronal suture fusion had papilledema (5.5%).
In the study by Kuang, all patients receive a pre-operative, immediate postoperative and 2 years after surgery a CT scan and an annual fundoscopy.25 The data are complete for 37 of the 51 patients. They underwent a total cranial correction at an average age of 5.4 months (range 3 to 11 months). The surgical technique used involves multiple osteotomies, including in front of and behind the coronal and lambdoid sutures, after which the coronal and lambdoid sutures are separated from the dura. This technical aspect explains the high percentage of fusion: in 87%, bilateral coronal suture synostosis was found in the follow-up scan, with 15% also having a partial fusion of the lambdoid sutures. Only 1 patient (2.7%) was diagnosed with increased ICP (papilledema and high ICP at invasive measurement) for which reoperation was performed. The mean follow-up time was 3.75 years, ranging from 2 to 6 years.
Seruya evaluated routinely performed postoperative CT scans of 42 children 6 to 12 months after total cranial vault remodelling for sagittal suture synostosis.26 They found fusion of the right coronal suture (42.6%), left coronal suture (38.3%), right lambdoid suture (74.5%) and left lambdoid suture (74.5%). Increased ICP was not investigated.
Yarbrough assessed non-syndromic craniosynostosis patients for fusion of additional cranial sutures and found additional closure of cranial sutures in 3/145 (2.1%) patients after open correction and in 2/121 (1.7%) patients after endoscopic correction.27 This observation was found 16.4 and 15.3 months after the operation, respectively.
This observation was found 16.4 and 15.3 months after the operation, respectively. In the open remodelling for coronal suture synostosis, synostosis of the sagittal suture occurred in 1 patient and of the metopic suture in 2 patients; in the endoscopic group for sagittal suture synostosis both patients developed isolated coronal suture synostosis. However, there was no routine screening and on indication of physical examination, a new CT scan was made in these 5 children. It is therefore possible that the percentages mentioned are an underestimation.
The systematic review of Kim on additional closure of cranial sutures distinguishes between iatrogenic closure (because the cranial suture is manipulated during surgery) on the one hand and idiopathic closure (where the cranial suture was left intact during surgery) on the other hand.28 The review reports a significantly higher incidence of additional closure in studies that performed routine radiological screening during post-surgery follow-up compared to studies that did not report this (p = 0.01). Only 2 studies that performed routine imaging during follow-up describe idiopathic closure: the previously mentioned study by Arnaud reports 10.4% of 229 scaphocephaly patients and Agrawal 9.5% of 42 scaphocephaly patients.24,29
In the study by Van Veelen, 83 patients were routinely followed up with skull images after spring distraction for sagittal suture synostosis.23 Shortly after the procedure, the coronal sutures were no longer visible on the skull images in 10% of the patients and this percentage gradually increased to 30% at the age of 3 years and remained the same until the age of 5 years. The occurrence of additional fusion was not related to papilledema, which occurred in only 2 patients in this study.
Tuite compared 58 unoperated patients with non-syndromic synostosis (16 sagittal, 10 metopic, 29 unicoronal and 3 lambdoid), 55 with syndromic (22 Saethre-Chotzen, 23 Crouzon/Pfeiffer and 10 Apert) and 9 with multisuture synostosis in which fundoscopy and an invasive ICP measurement were performed.30 Their age ranged from 2.5 months–15 years, with an average of 2.4 years ± 3.2. Fifteen patients had papilledema and four also showed atrophy (11 syndromic, 2 multisuture, two isolated suture synostosis); 13 of them had an ICP> 15 and two had an ICP between 10 and 15 mmHg. Of the 15 children with papilledema, eight were younger than 1 year and seven were 5 years or older. Above the age of 8, detection of papilledema by fundoscopy was specific and sensitive. Sensitivity under the age of 8 was only 22%. In this young group, the absence of papilledema is therefore no guarantee for a normal ICP, but the presence of papilledema is an evident sign of increased ICP.
Striking in this study is that 41 out of 58 (70.7%) children had an ICP above 15 mmHg, and that this was almost equally distributed between isolated (n = 19, 32.8%) and multisuture/syndromic (n = 22, 34.4%). For isolated craniosynostosis, this is exceptionally high and can be explained by the used definition of increased ICP. Compared to more recent studies, this is a broad definition but there is still no consensus on normal values for children.6,31
Using ultrasound measurements of the thickness of the optic nerve, a good association between this measurement and the presence of papilledema was found in 128 patients with multisuture and syndromic craniosynostosis and with real-time ICP in 5 patients who received both measurements simultaneously.32,33 The thickness of the optic nerve was on average 3.3 mm ± 0.5 in patients with papilledema and on average 3.1 mm ± 0.5 in patients without papilledema. Since the ultrasound measurements in the daytime had a very low sensitivity of 11%, this method is not suitable for screening for increased ICP.
A relatively new method for detecting increased ICP is the use of optical coherence tomography (OCT), in which the thickness of the retina is determined. In 38 patients, 58 OCTs were performed (29 OCTs in 23 patients with sagittal suture synostosis and 29 OCTs in 15 patients with Crouzon syndrome). The total retinal thickness (TRT) was determined and was 410 microns in patients with normal fundoscopy and 525 microns in patients with abnormal fundoscopy, which was a statistically significant difference (p = 0.002). This method seems to be a good quantitative method to demonstrate increased ICP that can be applied as soon as the child is able to concentrate, which is usually around the age of 3 to 4 years.34 Other authors have described similar results.35,36 Dagi describes 54 patients with a wide variety of types of isolated and syndromic craniosynostosis.35 The sensitivity of OCT to detect papilledema was 60% and the specificity 90%. In the study by Swanson, 40 patients with craniosynostosis received an OCT, 5 patients with hydrocephalus and increased ICP and 34 healthy controls.36 In the first two groups an invasive ICP measurement was performed. Based on the reference values, the sensitivity is 89% and the specificity 62%.
Conclusions
| Level 2 | A deflecting growth curve of the head circumference is probably a useful screening method to detect increased ICP in metopic suture synostosis. In sagittal suture synostosis, this method is not sufficiently discriminating for the detection of increased ICP. For unilateral coronal suture synostosis, the use of this method is not described. Increased ICP in syndromic craniosynostosis due to reduced skull growth can probably also be detected by following the head circumference growth curve. B Rijken et al, 2015; Spruijt et al, 2015; Van Veelen et al, 2017; Cornelissen et al, 2017 |
| Level 3 | The presence or absence of diffuse impressions on X-skull in children under 18 months may be an unreliable screening method for increased ICP because of a sensitivity of 0%, but very suspicious for increased ICP due to a specificity of 98%. For children aged 18 months to 4 years the sensitivity was 64% and the specificity 50%, making it a reasonably reliable screening method. B Tuite et al, 1996–1 |
| Level 3 | The presence of additional coronal suture fusion on X-skull or CT in a child with sagittal suture synostosis younger than 2 years after surgical correction (with the coronal suture spared) may be a potentially relevant risk factor for increased ICP. In the presence of additional coronal suture fusion, the risk of papilledema is 10 to 28.6%; in absence, 0 to 5.5%. C Arnaud et al, 2009; Van Veelen et al, 2013; Kim et al. 2017 |
| Level 3 | Ultrasound measurement of the thickness of the optic nerve does not appear to be a reliable screening method for increased ICP because of its very low sensitivity of 11%. B Driessen et al, 2011; C Driessen et al, 2012 |
| Level 3 | Papilledema in fundoscopy for screening may be a certain sign of increased ICP, but its absence does not rule out increased ICP in children under 8 years of age. B Tuite et al, 1996–2 |
| Level 2 | OCT is probably a reliable quantitative method for screening for increased ICP, but requires patient cooperation. The sensitivity of OCT to predict papilledema is 60% and the specificity 90%; the sensitivity of OCT to invasive ICP measurement is 89% and the specificity 62%. B Driessen et al, 2014; Dagi et al, 2014; Swanson et al, 2017 |
3. What are craniosynostosis-specific factors that influence the choice between different surgical techniques for the treatment of increased ICP?
Increased ICP in isolated non-syndromic craniosynostosis during follow-up after a cranial correction mainly affects patients with sagittal suture synostosis and is mainly related to a too small intracranial volume. In 95 patients with sagittal suture synostosis, 6 of whom developed papilledema, the mean intracranial volume in z-score was 0.5 (SD 1.07) in children with papilledema and 1.4 (SD 1.16) in children without papilledema (p 0.16).37 For this reason, a second cranial expansion is usually performed if persistently increased ICP is detected.
In syndromic craniosynostosis, the cause of increased ICP is multifactorial, i.e. too small intracranial volume, moderate to severe obstructive sleep apnea (OSA), hydrocephalus and venous intracranial hypertension.38,39 A too small intracranial volume can be determined by continuing the head circumference growth curve and cranial expansion surgery is also an appropriate solution for this cause.19,20 Treatments for obstructive sleep apnea and hydrocephalus are discussed in the relevant chapters. There is no diagnostic test for determining venous intracranial hypertension other than excluding the other causes and transcranial Doppler, although this is mainly done in a research setting. Cranial expansion surgery also seems to improve venous intracranial hypertension.40
Conclusions
| Level 3 | For sagittal suture synostosis, the underlying cause of increased ICP is predominantly a too small intracranial volume. Treatment is therefore aimed at re-enlargement of the skull. For syndromic craniosynostosis, the causes may be too small intracranial volume, moderate to severe obstructive sleep apnea, hydrocephalus and venous intracranial hypertension or a combination of these factors. Treatment focuses on the cause, which appears to be clinically the major contributor to the increase in intracranial pressure. B Spruijt et al, 2015 Van Veelen et al, 2016; Hayward et al, 2016; Spruijt et al, 2016; Deschamps et al, 2011 |
Considerations
• Evidence of the Conclusions
Prevalence of increased ICP
The prevalence studies of increased ICP in sagittal suture synostosis have a reasonable level of evidence, whereas for the other types of isolated non-syndromic craniosynostosis the number of studies is particularly limited.
Prevalence studies of increased ICP in syndromic craniosynostosis have a limited evidence due to the low number of studies and low number of included patients.
Screening for increased ICP
The reliability of a head circumference growth curve for detection of increased CPI has only been investigated to a limited extent. The reliability of fundoscopy has been described in only one study. The newest method with OCT has a better level of evidence, but whether it is more sensitive or more specific than fundoscopy is not yet known.
Surgical treatment of increased ICP
There is hardly any scientific evidence to prove the effectiveness of the various treatments. In general, these are articles by craniofacial centres that present the results of their own treatment protocol, but do not make a comparison by type of treatment for increased ICP.
• Values and Preferences
Prevalence of increased ICP
Not applicable.
Screening for increased ICP
Screening by measuring the head circumference is not very stressful for the child and is a good method for metopic suture synostosis. For sagittal suture synostosis and syndromic craniosynostosis, fundoscopy for screening remains recommended, including OCT in children who are capable of being instructed. Regarding unicoronal synostosis, a recent study by Van de Beeten found papilledema in 1 patient out of 89 (1.1%) in the postoperative follow-up trajectory for which no surgery was necessary.41 Given this very low prevalence, screening in children with non-syndromic unicoronal synostosis does not seem to be of any value.
Surgical treatment of increased ICP
According to the workgroup, parents will prefer rapid treatment, once increased ICP has been diagnosed, by the least invasive operation possible with the greatest chance of success in normalising ICP. If there are several options for treatment, for example cranial expansion surgery, a ventriculoperitoneal (VP) drain or an endoscopic third ventriculostomy, it is important to explain the pros and cons to parents and come to a joint decision.
• Costs and Resources
Prevalence of increased ICP
Not applicable.
Screening for increased ICP
A reduction in the number of fundoscopies for screening in patient groups where the benefit is very low has a cost-saving effect and reduces the staff required for this screening. This can be applied in metopic suture synostosis and non-syndromic unicoronal synostosis.
Surgical treatment of increased ICP
The guideline committee expects that the implementation of the recommendations leads to little or no increase in costs, because the recommendations are in line with existing practice.
• Professional Perspective
Prevalence of increased ICP
Not applicable.
Screening for increased ICP
Routine fundoscopy in metopic suture synostosis and non-syndromic unicoronal synostosis does not seem to be indicated, as the prevalence of papilledema is very low. These patients can also be detected by annual follow-up of the head circumference growth curve. This selection of high-risk patients prevents a significant number of examinations that cause discomfort in small children. For sagittal suture synostosis and syndromic craniosynostosis, routine fundoscopy and/or OCT is chosen, depending on the availability of an OCT device. At the moment, OCT is still predominantly used in research.
There is no scientific justification for the frequency of screening and up to what age this is meaningful, other than that detection of papilledema in annual fundoscopy has been reported in children with sagittal suture synostosis up to the age of 9 and most commonly between 3 and 6 years. At the moment there is no evidence that less or more frequent screening than annually is as good or better and the workgroup therefore follows what is common practice in current scientific studies.
For children with sagittal suture synostosis, annual screening is usually conducted until the age of 6, as this is the age at which the main increase in brain volume has occurred. The other causes of increased ICP in syndromic craniosynostosis can also occur at a slightly later age and therefore screening until later in life is preferred. For syndromic craniosynostosis, a higher screening frequency is initially assumed because there is a higher risk and because the degree of ICP elevation is often more severe, which means that a delay in the diagnosis and subsequent treatment of increased ICP has more consequences for the patient. For Crouzon syndrome, this is most evident, followed by Apert and Saethre-Chotzen syndrome. This is a reason to maintain a specific protocol for each syndrome for the first years of life.
Surgical treatment of increased ICP
If, in the follow-up period after cranial surgery, increased ICP is detected and there is a strong suspicion that the intracranial volume is too small, a cranial expansion operation will be the preferred option. In case of increased ICP due to hydrocephalus, a ventriculocisternostomy or a VP-drain can be chosen. The disadvantage of the VP-drain is that the patient often depends on the functioning of the drain for the rest of his/her life (see chapter hydrocephalus).
• Balance of Anticipated Desired and Undesired Outcomes
Prevalence of increased ICP
Not applicable.
Screening for increased ICP
Considering the potential damage, that untreated increased ICP can cause, screening is advised, adjusted to the risk. For metopic suture synostosis, it is sufficient to follow the head circumference growth curve, while for sagittal suture synostosis, this is insufficient and fundoscopy and/or OCT is chosen. For non-syndromic unicoronal suture synostosis, the reliability of the growth curve is unclear and fundoscopy and/or OCT is also chosen. Given the very low frequency of increased ICP in unicoronal synostosis, screening via a (slightly) stressful examination such as fundoscopy/OCT is not meaningful. Possibly, the head circumference curve can be used for this, but its reliability has not yet been clearly established.
Surgical treatment of increased ICP
Screening for sagittal suture synostosis is carried out until the age of 6 years and after that only on indication, as the prevalence decreases considerably after that age.
In syndromic craniosynostosis, head circumference measurement is relevant as a predictor of increased CPI, but fundoscopy and/or OCT is required to detect increased ICP with other causes than reduced skull growth.
Rationale for the recommendation(s)
Prevalence of increased ICP
Not applicable.
Screening for increased ICP
The potential damage that can be caused by untreated increased ICP is reason to identify patients at risk for this, i.e. sagittal suture synostosis and syndromic craniosynostosis. After the age of 6 years, increased ICP occurs much less frequently in isolated non-syndromic craniosynostosis, because the brain has increased in volume the most; in syndromic craniosynostosis, increased ICP can occur later in life due to the multiple causes.
Only for metopic suture synostosis, screening with just the follow-up of the head circumference growth curve seems reliable, thus avoiding a rather stressful eye examination for the child. Given the very low frequency of increased ICP in non-syndromic unicoronal synostosis, screening with a stressful examination such as fundoscopy/OCT is not recommended. Possibly, the head circumference growth curve can be used for this, but its reliability has not yet been clearly established. For other types, fundoscopy and/or OCT is desirable, for which cooperation is necessary.
Surgical treatment of increased ICP
In isolated non-syndromic craniosynostosis, the cause is mainly a too small intracranial volume. In syndromic craniosynostosis, other causes may also be responsible. Increased ICP due to a too small intracranial volume is easy to treat by cranial expansion surgery. The presence of other causes should be investigated and, if present, the treatment should be adjusted accordingly.
Recommendations
Screening for increased intracranial pressure
Screen annually for increased ICP in patients with sagittal suture synostosis by fundoscopy and/or OCT during a follow-up up until the age of 6 years.
Screen annually for increased ICP in metopic, unicoronal and unilambdoid suture synostosis by measurement of the head circumference. If the growth curve deflects, fundoscopy and/or OCT is still indicated.
-
Screen for increased ICP in syndromic and multisuture craniosynostosis by fundoscopy and/or OCT during a follow-up until at least the age of 6 years. Because of the difference in prevalence of increased ICP, the frequency of screening will be adjusted to the specific diagnosis:
-
∘
Crouzon syndrome: screen 4-monthly until the age of 2 years, screen 6-monthly until the age of 4 years and then annually;
-
∘
Apert syndrome, Saethre-Chotzen multisuture craniosynostosis: screen 6-monthly;
-
∘
Muenke syndrome: screen annually.
-
∘
Patient-specific factors that should be taken into account when choosing between different surgical techniques for the treatment of increased ICP.
Treat increased ICP based on patient-specific causal factor(s), i.e. a too small intracranial volume, moderate to severe obstructive sleep apnea, hydrocephalus and/or venous intracranial hypertension.
Research Gaps
There is lack of knowledge about what a normal ICP is in children and therefore there is no consensus on the definition of increased ICP. There is almost no literature available on the prevalence of elevated ICP after cranial correction in non-syndromic, unicoronal and unilambdoid suture synostosis, whereas for syndromic craniosynostosis it is limited in number and size of patients included. The reliability of the different screening methods for increased ICP in unicoronal synostosis is not known.
Comparative (multicenter) studies on different treatment methods for increased ICP are lacking.
Literature
-
1.
Stavrou P, Sgouros S, Willshaw HE, et al. Visual failure caused by raised intracranial pressure in craniosynostosis. Childs Nerv Syst 1997;13: 64-7
-
2.
Thompson DNP, Malcolm GP, Jones BM, et al. Intracranial pressure in single-suture craniosynostosis. Pediatr Neurosurg 1995;22:235-40
-
3.
Renier D, Lajeunie E, Arnaud E, et al. Management of craniosynostoses. Childs Nerv Syst 2000;16:645-58
-
4.
Mathijssen I. Arnaud E, Lajeunie E, et al. Postoperative cognitive outcome for synostotic frontal plagiocephaly. J Neurosurg 2006;105(1 suppl):16-20
-
5.
Eley KA, Johnson D, Wilkie AOM, et al. Raised intracranial pressure is frequent in untreated nonsyndromic unicoronal synostosis and does not correlate with severity of phenotypic features. Plast Reconstr Surg 2012;130:690e-7e
-
6.
Wall SA, Thomas GPL, Johnson D, et al. The preoperative incidence of raised intracranial pressure in nonsyndromic sagittal craniosynostosis is underestimated in the literature. J Neurosurg Pediatr 2014;14:674-81
-
7.
Van Veelen MLC, Eelkman Rooda OHJ, De Jong T, et al. Results of early surgery for sagittal suture synostosis: long-term follow-up and the occurrence of raised intracranial pressure. Childs Nerv Syst 2013:29:997-1005
-
8.
Van Veelen MLC, Mihajlovic D, Dammers R, et al. Frontobiparietal remodeling with or without a widening bridge for sagittal synostosis: comparison of 2 cohorts for aesthetic and functional outcome. J Neurosurg Pediatr 2015;16:86-93
-
9.
Cornelissen MJ, Loudon SE, Van Doorn FEC, et al. Very low prevalence of intracranial hypertension in trigonocephaly. Plast Reconstr Surg 2017;139:97e-104e
-
10.
Christian EA, Imahiyerobo TA, Nallapa S, et al. Intracranial hypertension after surgical correction for craniosynostosis: a systematic review. Neurosurg Focus 2015;38:1-6
-
11.
Thomas GPL, Johnson D, Byren JC, et al. The incidence of raised intracranial pressure in nonsyndromic sagittal craniosynostosis following primary surgery. Neurosurg Pediatr 2015;15:350-60
-
12.
Thompson DNP, Harkness W, Jones B, et al. Subdural intracranial pressure monitoring in craniosynostosis: its role in surgical management. Childs Nerv Syst 1995;11:269-75
-
13.
Marucci DD, Dunaway DJ, Jones BM, et al. Raised intracranial pressure in Apert syndrome. Plast Reconstr Surg 2008;122:1162-8
-
14.
Kress W, Schropp C, Lieb G, et al. Saethre–Chotzen syndrome caused by TWIST 1 gene mutations: functional differentiation from Muenke coronal synostosis syndrome. Eur J Hum Genet 2006;14:39-48
-
15.
Greene AK, Mulliken JB, Proctor MR, et al. Phenotypically unusual combined craniosynostosis: presentation and management. Plast Reconstr Surg 2008;122:853-62
-
16.
Woods RH, Ul-Haq E, WIlkie AOM, et al. Reoperation for intracranial hypertension in TWIST1-confirmed Saethre-Chotzen syndrome: A 15-year review. Plast Reconstr Surg 2009;123:1801-10
-
17.
De Jong T, Bannink N, Bredero-Boelhouwer HH, et al. Long-term functional outcome in 167 patients with syndromic craniosynostosis; defining a syndrome-specific risk profile. J Plast Reconstr Aesthet Surg 2010;63:1635-41
-
18.
Abu-Sittah GS, Jeelani O, Dunaway D, et al. Raised intracranial pressure in Crouzon syndrome: incidence, causes, and management. J Neurosurg Pediatr 2015;17:469-75
-
19.
Van Veelen MLC. Thesis Sagittal suture synostosis. 2017. Chapter 10. General discussion.
-
20.
Spruijt B, Joosten KFM, Driessen C, et al. Algorithm for the management of intracranial hypertension in children with syndromic craniosynostosis. Plast Reconstr Surg 2015;136:331-40
-
21.
Rijken BFM, Den Ottelander BK, Van Veelen MLC, et al. The occipitofrontal circumference: reliable prediction of the intracranial volume in children with syndromic and complex craniosynostosis. Neurosurg Focus 2015;38:1-6
-
22.
Tuite GF, Evanson J, Chong WK, et al. The beaten copper cranium: a correlation between intracranial pressure, cranial radiographs and computed tomographic scans in children with craniosynostosis. Neurosurg 1996;39:691-9
-
23.
Van Veelen MLC, Touw C, Kamst N, et al. Minimally invasive, spring-assisted correction of sagittal suture synostosis. Technique, outcome and complications in 83 cases. Plast Reconstr Surg 2018;141:423-33
-
24.
Arnaud E, Capon-Degardin N, Michienzi J, et al. Scaphocephaly part II: secondary coronal synostosis after scaphocephalic surgical correction. J Craniofac Surg 2009;20(suppl2):1843-50
-
25.
Kuang AA, Jenq T, Didier R, et al. Benign radiographic coronal synostosis after sagittal synostosis repair. J Craniofac Surg 2013;24:937-40
-
26.
Seruya M, Tan SY, Wray AC, et al. Total cranial vault remodelling for isolated sagittal synostosis: part I. Postoperative cranial suture patency. Plast Reconstr Surg 2013;132:602e-10e
-
27.
Yarbrough CK, Smyth MD, Holekamp TF, et al. Delayed synostosis of uninvolved sutures after surgical treatment of nonsyndromic craniosynostosis. J Craniofac Surg 2014;25:119-23
-
28.
Kim SY, Shin HJ, Lim SY. Determining the fate of cranial sutures after surgical correction of non-syndromic craniosynostosis. J Craniomaxillofac Surg 2017;45:1801-8
-
29.
Agrawal D, Steinbok P. Cochrane DD. Reformation of the sagittal suture following suture for isolated sagittal craniosynostosis. J Neurosurg 2006;105:115-7
-
30.
Tuite GF, Chong WK, Evanson J, et al. The effectiveness of papilledema as an indicator of raised intracranial pressure in children with craniosynostosis. Neurosurg 1996;38:272-8
-
31.
Hayward R, Britto JA, Dunaway D, et al. Raised intracranial pressure and nonsyndromic sagittal craniosynostosis. Letter to the Editor. Neurosurg Forum 2015;16:346-7
-
32.
Driessen C, Bannink N, Lequin M, et al. Are ultrasonography measurements of optic nerve sheath diameter an alternative to fundoscopy in children with syndromic craniosynostosis? J Neurosurg Pediatr 2011;8:329-34
-
33.
Driessen C, Van Veelen MLC, Lequin M, et al. Nocturnal ultrasound measurements of optic nerve sheath diameter correlate with intracranial pressure in children with craniosynostosis. Plast Reconstr Surg 2012;130:448e-51e
-
34.
Driessen C, Eveleens J, Bleyen I, et al. Optical coherence tomography: a quantitative tool to screen for papilledema in craniosynostosis. Childs Nerv Syst 2014;30:1067-73
-
35.
Dagi LG, Tiedemann LM, Heidary G, et al. Using spectral-domain optical coherence tomography to detect optic neuropathy in patients with craniosynostosis. J AAPOS 2012;18:543-9
-
36.
Swanson JW, Aleman TS, Xu W, et al. Evaluation of optical coherence tomography to detect elevated intracranial pressure in children. JAMA Ophthalmol 2017;135:320-8
-
37.
Van Veelen MLC, Jippes M, Carolina JCA, et al. Volume measurements on three-dimensional photogrammetry after extended strip versus total cranial remodelling for sagittal synostosis: A comparative cohort study. J Cranio-Maxillo-Fac Surg 2016;44:1713-8
-
38.
Hayward R, Britto J, Dunaway D, et al. Connecting raised intracranial pressure and cognitive delay in craniosynostosis: many assumptions, little evidence. J Neurosurg Pediatr 2016;18:242-50
-
39.
Spruijt B, Mathijssen IMJ, Bredero-Boelhouwer HH, et al. Sleep architecture linked to airway obstruction and intracranial hypertension in children with syndromic craniosynososis. Plast Reconstr Surg 2016;138:1019e-1029e
-
40.
Deschamps-Braly J, Hettinger P, El Amm C, et al. Volumetric analysis of cranial vault distraction for cephalocranial disproportion. Pediatr Neurosurg 2011;47:396-405
-
41.
Van de Beeten SDC, Cornelissen MJ, Van Seeters RM, et al. Papilledema in unicoronal synostosis: A rare finding. J Neurosurg Pediatr 2019;17:1-6
CHAPTER 9 HYDROCEPHALUS
9.1 What is the surgical management of hydrocephalus in craniosynostosis?
Introduction
Hydrocephalus is defined as a progressive increase in ventricular size, accompanied by signs of increased pressure. This should be distinguished from ventriculomegaly without increased intracranial pressure.
In isolated craniosynostosis the prevalence of hydrocephalus is very low. Cinalli found a prevalence of 0.88% in a large study with 1447 non-syndromic craniosynostosis patients.1 Given this extremely low frequency, this group will not be discussed in rest of the chapter.
Patients with syndromic or multisuture craniosynostosis may develop hydrocephalus, and this risk appears to be syndrome-dependent. Because hydrocephalus develops gradually in craniosynostosis and the classical signs of hydrocephalus are often absent, specific screening methods have to be used. Treatment of hydrocephalus is also discussed, as it includes specific aspects for this patient population. Untreated hydrocephalus results in a decline in neurocognitive function.
Search and Selection
For the following specific questions original scientific studies or systematic reviews of scientific studies have been included:
-
3.
How common is hydrocephalus in children with craniosynostosis and what diagnostics are indicated to detect it?
-
4.
What are the anatomical factors that influence the choice between different surgical techniques for the treatment of hydrocephalus?
In the Medline (OVID) and Embase databases, a single comprehensive search was carried out for studies on craniosynostosis. The search strategy is shown in appendix 2, of the guideline. After deduplication, the literature search yielded 2732 hits.
Given the large number of studies, the chair of the working group first selected those that met the following general selection criteria:
General Selection and Exclusion Criteria
| Study type | -original studies -systematic reviews of sufficient quality: -research question of systematic review corresponds (largely) to the basic question -search is performed in at least 2 relevant databases, e.g. Cochrane Library, Medline/PubMed -reporting of the complete search strategy -no relevant keywords/search terms are missing |
| Follow-up period | -minimum follow-up period of 12 months for therapeutic or prognostic studies. |
| Exclusion criteria | -Case-reports -Expert opinion -Letters -Editorials -Case control studies for diagnostic tests -Narrative reviews |
The pre-selected studies that met the specific selection criteria listed in the Table below are included in the literature summary of this chapter.
Specific Selection and Exclusion Criteria
| -minimum study size: 20 patients -minimum follow-up time: up to. 2 years of age |
Summary of the Literature
1. How common is hydrocephalus in children with craniosynostosis and what diagnostics are indicated to detect it?
TABLE 2. Prevalence of Hydrocephalus
| Apert | Crouzon/Pfeiffer | Saethre-Chotzen | Muenke | multisuture | |
| Cinalli 19981 | 30/77 (39%) | 14/104 (13%) | |||
| Collmann 19882 | 6/13 (46%) | 15/27 (56%) | 5/30 (17%) | 4/17 (24%) | |
| Collmann 20053 | 32/45 (71%) | 32/78 (41%) | 3/37 (8%) | 2/24 (8%) |
TABLE 1. Prevalence of Ventriculomegaly
| Apert | Crouzon/Pfeiffer | Saethre-Chotzen | Muenke | multisuture | |
| Cinalli 19981 | 5/77 (6%) | 27/104 (26%) | 0/40 (0%) | 2/43 (5%) | |
| Collmann 19882 | 0/13 (0%) | 2/32 (6%) | 0/30 (0%) | 2/17 (12%) | |
| Collmann 20053 | 0/45 (0%) | 12/78 (15%) | 0/37 (0%) | 0/24 (0%) |
De Jong carried out measurements of brain volume and ventricular volume on MRI scans in 13 patients with Apert syndrome, 31 with Crouzon syndrome, 15 with Muenke syndrome, 10 with Saethre-Chotzen syndrome and 15 with multisuture craniosynostosis, ranging in age from 0 to 18 years.4 Brain volume did not deviate significantly from the normal values reported in the literature. These normal values are missing for ventricular volume. In multivariate analysis, patients with Apert syndrome and patients with a Chiari were found to have a significantly higher ventricular volume compared to the other syndromes. Of the 12 patients with Chiari, 10 were diagnosed with Crouzon. In this series, 3 patients with Crouzon were excluded for having a ventriculoperitoneal shunt; whether they were placed because of hydrocephalus was not mentioned. No serial MRI results have been described, making the prevalence of hydrocephalus an estimate.
No recommendations are published in the literature for systematic screening of hydrocephalus. Collmann proposes a pragmatic approach in which based on present risk factors, i.e. multisuture or syndromic craniosynostosis, in particular Crouzon/Pfeiffer, lambdoid suture synostosis, “crowded posterior fossa” and Chiari, a routine MRI and MR-venography are performed.3 De Jong advises screening with MRI in Apert and Crouzon syndrome but does not mention frequency.4
Conclusion
| ----- | The prevalence of ventriculomegaly is possibly 13-56% in Crouzon/Pfeiffer and 39-71% in Apert, 8-17% in Saethre-Chotzen, 8% in Muenke and 24% in multisuture craniosynostosis. Risk factors for ventriculomegaly are the diagnosis of Apert and Chiari syndrome. The prevalence of hydrocephalus is possible 6–26% in Crouzon/Pfeiffer, 0–6% in Apert, 5–12% in multisuture craniosynostosis and negligible in Saethre-Chotzen and Muenke syndrome. Cinalli et al, 1998; Collmann et al, 1988; Collmann et al, 2005; De Jong et al, 2012. |
2. What are the anatomical factors that influence the choice between different surgical techniques for the treatment of hydrocephalus?
Placing a shunt for hydrocephalus has an opposite effect to the main goal of craniosynostosis treatment, i.e. expansion of the skull. Sequence and timing of treatment have never been systematically investigated. Renier advises, as long as the clinic permits, to first perform the cranial expansion and only then to place the shunt.5 Collmann points to the increase in ventricular size that occurs after each cranial expansion and that must be distinguished from a real hydrocephalus.2,3 Only if the increased ICP persists and, despite adequate cranial expansion, persists after several weeks a shunt is indicated.
Alternative treatment methods for hydrocephalus include fossa posterior decompression and third ventriculocisternostomy. However, posterior fossa decompression does not always have a good effect on the hydrocephalus. Renier calls it a difficult procedure that should be reserved for those cases that have obvious symptoms of tonsillar herniation.5
The choice of surgical treatment seems to vary considerably from centre to centre:
Di Rocco describes the results of 11 endoscopic 3rd ventriculostomies (8 Crouzon patients and 3 multisuture craniosynostosis), in which the choice of this technique was made when the MRI showed an obstruction of the liquor circulation at the level of the posterior fossa, at the level of the aqueduct or at the level of the cisterna magna and foramen magnum. In the end 7 of the 11 procedures were successful and a shunt had to be placed in the other 4 patients.6
Abu Sittah (from London) describes 30 Crouzon patients who had to be treated for hydrocephalus in which nine were given a shunt and one was given an endoscopic 3rd ventriculostomy.7 In the follow-up period of 5 months to 5 years, 6 patients with a shunt still needed cranial expansion due to increased ICP.
The team in Rotterdam has a preference for initial cranial expansion as reported for all 19 Crouzon patients.8 In only 1 patient this treatment was followed by the placement of a shunt.
There are no comparative studies on the long-term results of these differences in treatment.
Conclusion
| Level 3 | Predictive anatomical factors for a successful treatment are not yet known. Hydrocephalus in craniosynostosis may be successfully treated with a cranial expansion, placement of a ventriculoperitoneal shunt, an endoscopic 3rd ventriculostomy or decompression of the foramen magnum. Of all these treatments, good and bad results have been described, so that other follow-up surgical treatment of the hydrocephalus and/or increased ICP may still be necessary. C Collmann et al, 1988; Collmann et al, 2005; Renier et al, 2006; Di Rocco et al, 2010; Abu Sittah et al, 2016; Spruijt et al, 2016. |
Considerations
• Evidence of the Conclusions
Screening
The evidence of the conclusion is limited: there is only one study by Cinalli in which a sufficiently large group of patients with sequential MRIs has been analysed to differentiate actual hydrocephalus from ventriculomegaly.1
Surgical technique
The evidence of the conclusion is weak: subgroup analyses of studies with a direct comparative design and multivariate analyses of potential prognostic variables in studies with a non-comparative design were completely lacking.
• Values and Preferences
Screening
For evaluation of hydrocephalus, a short MRI protocol (e.g. single-shot T2-weighted sequence) is always chosen, which can be done without anaesthesia. If the MRI is indicated for additional diagnostics, such as determination of tonsillar herniation, anaesthesia is usually indicated because the MRI takes considerably more time and especially young children cannot lie still for that long.
Surgical technique
The disadvantage of a shunt treatment for hydrocephalus is that the patient is dependent on this shunt for life; this can mean that in case of a malfunctioning shunt (disconnection, infection, blockage) acute symptoms arise that require surgical treatment. This risk does not exist for the other options. Shunt placement and endoscopic 3rd ventriculostomy have the advantage that they are much less invasive procedures for the patient compared to cranial expansion surgery. According to the workgroup, parents may have a specific preference for a particular technique.
• Costs and Resources
Screening
The guideline committee expects that the implementation of the recommendations leads to little or no increase in costs, because the recommendations are in line with existing practice.
Surgical technique
The guideline committee expects that the implementation of the recommendations leads to little or no increase in costs, because the recommendations are in line with existing practice.
• Professional Perspective
Screening
Screening for hydrocephalus requires imaging of the initial situation by CT or MRI and follow-up scans to determine the change. How quickly the second scan should follow depends on the clinical course of the patient: in general, hydrocephalus develops within the first 2 years of life and can be clinically detected by a tense fontanel, a head circumference growth curve that exceeds its own SD and symptoms associated with hydrocephalus. MRI is not associated with radiation exposure and is also the best method for determining tonsillar herniation, which is often associated with hydrocephalus. An MRI examination takes longer and therefore often requires anaesthesia to obtain a good image in children who are not (or cannot be) cooperative. A CT scan is generally more accessible than an MRI, which can determine the choice between the two techniques. However, an MRI examination is strongly preferred because of the importance of radiation limitation in this young patient population.
Surgical technique
The disadvantage of a shunt treatment for hydrocephalus is that the patient is dependent on this shunt for life; this can mean that in case of a malfunctioning shunt (disconnection, infection, blockage) acute symptoms arise that require surgical treatment. This risk does not exist for the other options. Shunt placement and endoscopic 3rd ventriculostomy have the advantage that they are much less invasive procedures for the patient compared to cranial expansion surgery, such as posterior fossa decompression. The shape and size of the skull plays a role in the decision to perform a cranial expansion operation: if the procedure leads to a deteriorated shape or an abnormally large head, a shunt or endoscopic 3rd ventriculostomy is preferred. The surgeon's preference is determined in particular by personal experience with these techniques, anatomical presentation, symptoms and the patient's medical history.
• Balance of Anticipated Desired and Undesired Outcomes
Screening
Given the syndrome-specific high prevalence of hydrocephalus, protocol-based screening is indicated in Crouzon syndrome and multisuture craniosynostosis. For the other syndromic types of craniosynostosis, the benefit of screening is too low relative to the associated burden.
Surgical technique
Considering the severe consequences of untreated hydrocephalus on neurocognitive functions, treatment is indicated by definition. A choice of technique in a specific patient does not guarantee adequate treatment of the hydrocephalus. Parents should be informed in advance of this uncertainty and the possible need for additional treatment.
Rationale for the recommendation(s)
Screening
Early detection of hydrocephalus in order to start treatment as soon as possible and to prevent neurocognitive decline is a key part of the screening process.
Surgical technique
The leading factor in the choice of surgical technique is the likelihood that the chosen technique will resolve the hydrocephalus in the specific patient, taking into account individual factors such as skull shape, skull size, MRI observations and the preference of parents and surgeon. The need for additional treatment is not uncommon, as the initial treatment does not always seem to be sufficient to correct the hydrocephalus.
Recommendations
Screening for hydrocephalus
Screen all patients with Crouzon syndrome and multisuture craniosynostosis by MRI upon referral. Patients with ventriculomegaly should be followed by a second MRI to exclude hydrocephalus, the timing of which depends on clinical course.
Set up a treatment plan as soon as hydrocephalus is detected.
Patient-specific factors that should be taken into account when choosing between different surgical techniques for the treatment of increased ICP.
Treat hydrocephalus by cranial expansion with or without foramen magnum decompression, by placing a ventriculoperitoneal shunt or by endoscopic 3rd ventriculostomy. The choice is made per patient and depends on the MRI observations and patient-specific factors.
After treatment, the effect of the treatment is monitored, for example with MRI, and, if necessary, additional treatment is initiated if the hydrocephalus persists.
Research Gaps
There are no studies describing the long-term results of hydrocephalus treatment in craniosynostosis. In particular, a comparative study of the different treatment options is lacking.
Literature
-
1.
Cinalli G, Sainte-Rose C, Kollar EM, et al. Hydrocephalus and craniosynostosis. J Neurosurg 1998;88:209-14
-
2.
Collmann H, Sorensen N, Krauss JJ, et al. Hydrocephalus in craniosynostosis. Childs Nerv Syst 1988;4:279-85
-
3.
Collman H, Sorenson N, Krauss J. Hydrocephalus in craniosynostosis: a review. Childs Nerv Syst 2005;21:902-12
-
4.
De Jong T, Rijken BFM, Lequin MH, et al. Brain and ventricular volume in patients with syndromic and complex craniosynostosis. Childs Nerv Syst 2012;28:137-40
-
5.
Renier D, Arnaud E, Marchac D. Craniosynostosis: functional and morphologic postoperative results (French). Neurochirurgie 52:302-10, 2006
-
6.
Di Rocco F, Juca CE, Arnaud E, et al. The role of endoscopic third ventriculostomy in the treatment of hydrocephalus associated with faciocraniosynostosis. J Neurosurg Pediatr 2010;6:17-22
-
7.
Abu-Sittah GS, Jeelani O, Dunaway D, et al. Raised intracranial pressure in Crouzon syndrome: incidence, causes, and management. J Neurosurg Pediatr 2015;17:469-75
-
8.
Spruijt B, Rijken BFM, Den Ottelander BK, et al. First vault expansion in Apert and Crouzon-Pfeiffer syndromes: front or back? Plast Reconstr Surg 2016;137:112e-21e
CHAPTER 10 CHIARI
10.1 What is the management of Chiari in craniosynostosis?
Introduction
The prevalence of Chiari varies considerably depending on the type of craniosynostosis. The prevalence, causes, consequences and need for treatment are often unclear. Chiari is best visualized with an MRI-scan, but there is uncertainty about how often it should be made for the different types of craniosynostosis and when which treatment is indicated.
Search and Selection
For the following specific questions original scientific studies or systematic reviews of scientific studies have been included:
-
1.
How common is Chiari in children with craniosynostosis and what diagnostics are needed to detect it?
-
2.
What are Chiari-specific factors that are considered in the indication for treatment?
-
3.
What are determining factors with regard to the choice of surgical technique, indication and/or timing for the treatment of Chiari?
In the Medline (OVID) and Embase databases, a single comprehensive search was carried out for studies on craniosynostosis. The search strategy is shown in appendix 2, to the guideline. After deduplication, the literature search yielded 2732 hits.
Given the large number of studies, the chair of the working group first selected those that met the following general selection criteria:
General Selection and Exclusion Criteria
| Study type | -original studies -systematic reviews of sufficient quality: -research question of systematic review corresponds (largely) to the basic question -search is performed in at least 2 relevant databases, e.g. Cochrane Library, Medline/PubMed -reporting of the complete search strategy -no relevant keywords/search terms are missing |
| Follow-up period | -minimum follow-up period of 12 months for therapeutic or prognostic studies. |
| Exclusion criteria | -Case-reports -Expert opinion -Letters -Editorials -Case control studies for diagnostic tests -Narrative reviews |
The pre-selected studies that met the specific selection criteria listed in the Table below are included in the literature summary of this chapter.
Specific Selection and Exclusion Criteria
| -minimum study size: 20 patients -minimum follow-up time: max. 5 years of age |
Summary of the Literature
1. How common is hydrocephalus in children with craniosynostosis and what diagnostics are indicated to detect it?
Isolated, non-syndromic craniosynostosis
In the study by Leikola, all 121 patients with isolated, non-syndromic craniosynostosis (90 sagittal, 13 metopic, 11 coronal, 7 lambdoid suture synostosis) had undergone MRI prior to surgery.1 On analysis, 9 patients (7 sagittal and 2 coronal suture synostosis) had a tonsillar herniation, ranging from 6 to 12 mm, none of whom showed symptoms consistent with a tonsillar herniation. Seven patients received an MRI postoperatively for 27 to 81 months, after which the tonsillar herniation decreased in 4 patients and remained stable in 3 patients. The remaining 2 did not receive an MRI because of a contraindication due to a metal implant and a persistent upper respiratory tract infection. All remained symptom free and no one developed a syrinx.
Strahle conducted a retrospective analysis of CT and MRI scans in 343 patients with isolated, non-syndromic craniosynostosis: 183 sagittal, 71 metopic, 80 coronal and 9 lambdoid suture synostosis.2 Of these patients, 5 (2.8%), 0 (0%), 5 (6.3%) and 5 (55.6%) showed tonsillar herniation of > 5 mm (Chiari).
Engel presents the results of a retrospective study of routine pre-operative MRI in 69 patients with isolated, non-syndromic craniosynostosis, including 9 MRIs from after this period made on indication only and 11 MRIs made at the referring centre.3 Of these 89 children, 42 have sagittal, 29 metopic, 14 coronal, and 4 lambdoid suture synostosis. Only once a Chiari has been diagnosed, namely in a child with lambdoid suture synostosis, who does not have any symptoms of this.
In the series of Fearon, 12 out of 20 (60%) children with a unilateral lambdoid suture synostosis had a Chiari.4
Multisuture and syndromic craniosynostosis
Cinalli published an analysis on Chiari of 44 Crouzon and 51 Apert patients detected by MRI and finds a prevalence of 72.7% for Crouzon and 1.9% for Apert.5 In a review article by Cinalli the prevalence is given per diagnosis as 70% in Crouzon, 75% in oxycephaly, 50% in Pfeiffer, 100% in cloverleaf skull and rare in Apert.6 Chiari is seen in 88% of syndromic craniosynostosis patients with hydrocephalus, while 53% of children with a Chiari do not have hydrocephalus.
Fearon found a Chiari in 23 out of 28 (82%) Pfeiffer patients and in 29% in his study on Apert syndrome .7,8 Czerwinski (2011) reports that 40% of the patients with multisuture craniosynostosis developed a Chiari; this percentage was only 7% in the absence of lambdoid suture synostosis, but 70% in the presence of lambdoid suture synostosis.9 The latter finding is described differently by Fearon, with 12 out of 17 (70.6%) children with either one-sided or two-sided lambdoid suture synostosis having a Chiari.4 In these last four studies from Dallas, it is unclear whether all patients had undergone an MRI.4,7–9
Strahle performed a retrospective analysis of CT and MRI scans in 86 patients with multisuture or syndromic craniosynostosis (n = 40): 16 Crouzon, 9 Pfeiffer, 9 Saethre-Chotzen, 2 craniofacial dyssynostosis and 1 Norman-Roberts syndrome.2 Of the 40 patients with multisuture craniosynostosis, 19 have no involvement of the lambdoid sutures, of whom 2 (10.5%) have a tonsillar herniation of > 5 mm (Chiari). Of the 21 patients with involvement of the lambdoid sutures, 12 patients (57.1%) have a Chiari.
In Cinalli, only 19% (6/44) of Crouzon patients with tonsillar herniation were symptomatic.5 In their review of 2005, they describe that more than 1/3 of the patients develop symptoms of Chiari or develop a syringomyelia later in life.6 Symptoms may be suboccipital pain, painful torticollis, syringomyelia syndrome, apnea, life-threatening brainstem dysfunction and axial hypotonia. Cinalli recommend an MRI in the syndromic craniosynostosis group with a high risk of Chiari, without specifying the age and frequency of screening.6 The four studies from Dallas also describe the use of MRI for the detection of Chiari.4,7–9
Conclusions
| ----- | The prevalence of Chiari in isolated, non-syndromic craniosynostosis may be 3–8% for sagittal suture synostosis; 0% for metopic suture synostosis; 6–18% for coronal suture synostosis and 25–60% for lambdoid suture. Strahle et al, 2011; Engel et al, 2012; Leikola et al, 2010; Fearon et al, 2016. |
| Level 3 | Chiari in isolated, non-syndromic craniosynostosis may often be asymptomatic and can only be diagnosed by screening with radiological diagnostics, preferably MRI. C Strahle et al, 2011; Engel et al, 2012; Leikola et al, 2010; Fearon et al, 2016. |
| ----- | The prevalence of Chiari in Crouzon/Pfeiffer may be 70–82%, in Apert syndrome 2–29%. The prevalence of Chiari in multisuture craniosynostosis with involvement of lambdoid sutures may be 57–71% and 7–11% if lambdoid sutures are not affected. For Saethre-Chotzen and Muenke syndrome this prevalence is not known. Cinalli et al, 1995; Cinalli et al, 2005; Fearon et al, 2009; Czerwinski et al, 2011; Strahle et al, 2011; Fearon et al, 2013; Fearon et al, 2016 |
| Level 3 | Chiari in multisuture and syndromic craniosynostosis is possibly predominantly asymptomatic and can only be diagnosed by screening with radiological diagnostics, preferably MRI. B Cinalli et al, 1995; C Cinalli et al, 2005; Fearon et al, 2009; Czerwinski et al, 2011; Strahle et al, 2011; Fearon et al, 2013; Fearon et al, 2016. |
2. What are Chiari-specific factors that are considered in the indication for treatment?
Six out of 32 (19%) Crouzon patients with Chiari developed symptoms, and for them treatment was initiated.5 In a study by Fearon, 10 out of 21 (48%) Pfeiffer patients with Chiari received surgical decompression of the Chiari because of symptoms, i.e. swallowing and coordination problems, headache when coughing, development of syrinx and central apnea.7 None of the patients with multisuture craniosynostosis and Chiari initially showed symptoms.4 During 6 years of follow-up, 2 out of 12 (17%) patients became symptomatic for which surgical treatment was performed. In the series on Apert patients it is not mentioned whether patients become symptomatic, however one patient with a syrinx is reported.8
Conclusion
| Level 3 | It is possible that 17–50% of patients with Crouzon-Pfeiffer or multisuture craniosynostosis with a Chiari become symptomatic and thus have an indication for surgical treatment. Cinalli et al, 1995; Fearon et al, 2009; Fearon et al, 2013; Fearon et al, 2016 |
3. What are determining factors with regard to the choice of surgical technique, indication and/or timing for the treatment of Chiari?
Cinalli describes posterior fossa decompression in 3 patients, cranial expansion in one and shunt revision in another patient to treat a Chiari, with good results in all 5, while the 6th patient improved without treatment.5
Later, Cinalli describes a surgical technique combining occipital expansion with suboccipital decompression, but do not report any results.6 The study by Strahle describes cranial expansion with and without decompression of the Chiari, but does not describe on which criteria this choice was based and whether there were symptoms.2 The degree of tonsillar herniation improved in a number of patients, but no conclusion can be drawn from this study about which technique gave better results.
Fearon states that foramen magnum decompression in children younger than 1 year often results in reossification and therefore this intervention should preferably be postponed, but they do not provide any data on this.7
Scott evaluates the results of occipital cranial expansion in combination with suboccipital decompression as a routine technique for children with craniosynostosis and Chiari.10 Thus, no selection was made on the basis of symptomatology and 20 out of 43 patients were free of symptoms prior to surgery. At follow-up of 71% of the patients the Chiari was radiologically resolved in 35%, worsened in 35% and stable in 30%. During follow-up, 2 patients required reoperation because of increasing central apnea in one patient and syrinx in another. Fearon described a similar combined method, also performed regardless of the presence of symptoms of Chiari.4 Of the 17 patients with multisuture craniosynostosis, none showed an increase in tonsillar herniation on MRI or symptoms of Chiari.
Conclusion
| Level 3 | Relevant factors for the results of treatment for the various techniques, the indication or timing are unknown. Various surgical options are described for the treatment of Chiari (foramen magnum decompression immediately following or after occipital expansion; preventive or in case of symptoms), with varying indications and timing of surgery, and with varying results. C Cinalli et al, 1995; Fearon et al, 2009; Scott et al, 2013; Fearon et al, 2016. |
Considerations
• Evidence of the Conclusions
Screening
Most studies show serious flaws in design and execution and/or come from only a few research groups, which limits the generalizability of the results.
Indication for treatment
The evidence for the conclusion is weak: due to, among other things, the small number of patients studied, the estimated percentages of patients with Crouzon-Pfeiffer or multisuture craniosynostosis in combination with a Chiari becoming symptomatic vary widely.
Surgical technique
The evidence for the conclusion is weak: subgroup analyses of studies with a direct comparative design and multivariate analyses of potential prognostic variables in studies with a non-comparative design were completely absent.
• Values and Preferences
Screening
Considering the possible consequences of Chiari and its difficult recognition based on clinical research, there is a good reason to perform screening.
Indication for treatment
Development or progression of clinical symptoms of Chiari is a good indication for treatment.
Surgical technique
The choice of treatment for Chiari is determined, among other things, by the abnormalities on the MRI scan and individual factors, such as the presence or absence of a ventriculoperitoneal (VP) shunt and skull shape. In consultation with the patient and parents, a treatment method is chosen and the result is evaluated.
• Costs and Resources
Screening
The guideline committee expects that application of the recommendations leads to little or no increase in costs, because the recommendations are in line with existing practice.
Indication
The guideline committee expects that application of the recommendations leads to little or no increase in costs, because the recommendations are in line with existing practice.
Surgical technique
The guideline committee expects that application of the recommendations leads to little or no increase in costs, because the recommendations are in line with existing practice.
• Professional Perspective
Screening
A Chiari is sometimes already present on an MRI scan made in the first year of life, or develops during the following years. A significant part of the Chiari appears to be asymptomatic, but this can change over time. Knowledge about this is still limited, and therefore an MRI is performed for pragmatic reasons after the referral and at the ages of 2 and 4 years. The clinical course determines the follow-up.
Screening for Chiari is more reliable with an MRI scan than with a CT scan and also does not expose to radiation. Assessment of an MRI scan is negatively influenced by the presence of an orthodontic brace. For this reason, an MRI for screening prior to placing braces should be considered. Clinical experience shows that symptoms of Chiari and/or syrinx sometimes do not occur until the age of 18. Screening for high-risk patients (non-syndromic, uni-lambdoid suture synostosis, Crouzon and multisuture craniosynostosis involving one or both lambdoid sutures) should take place until that age, and after that age in case of symptoms. In the case of proven Chiari on MRI that increases in severity and/or becomes symptomatic, an additional MRI of the cervical, thoracic and lumbar spinal cord is indicated to demonstrate or rule out syrinx.
Indication for treatment
As the surgical treatment of Chiari is not without risk, the indication for treatment is reserved for patients with symptomatic Chiari. If there are no symptoms (yet), but the Chiari further develops with possible risk of a syrinx, then a wait-and-see policy can be agreed upon with the patient and parents.
Surgical technique
The choice of surgical technique will be made according to the most probable cause. In the case of reduced skull growth, an (occipital) expansion seems indicated. Depending on the symptoms and MRI findings, this procedure can be combined with foramen magnum decompression, with or without duraplasty. If hydrocephalus is present, the treatment can also consist of an endoscopic 3rd ventriculostomy or VP-drain.
• Balance of Anticipated Desired and Undesired Outcomes
Screening
Screening is especially intended for patients with the highest risk of Chiari, namely Crouzon syndrome and multisuture craniosynostosis involving the lambdoid sutures. In order to determine presence and progression over time, an MRI scan following the referral is recommended. As soon as symptoms of Chiari occur, a repeat MRI scan is indicated. An MRI at the age of 18, when skull growth is complete, seems to be indicated because symptoms may still develop.
Indication for treatment
In a non-symptomatic Chiari, the balance of surgery risk versus health gain is insufficient to proceed with surgery.
Surgical technique
When choosing the surgical technique, it is important to weigh the risks of the different methods for the individual patient. For example, the presence of venous collaterals may be a reason to choose not foramen magnum decompression but a VP shunt.
• Rationale for the Recommendation(s)
Screening
The rationale for screening is to identify Chiari in the at-risk groups (non-syndromic unilambdoid suture synostosis, Crouzon syndrome and multisuture craniosynostosis with lambdoid suture synostosis) so that the follow-up can be tailored to this. To assess possible progression of the Chiari, a baseline MRI is indicated at the time of the first referral. How often it should be repeated in the absence of symptoms is unclear. However, it is known that neurological abnormalities can still present at a late age (after 18 years of age), which can be improved by surgical interventions.
Indication for treatment
The rationale of the indication for treatment is to achieve health gains and currently this can only be achieved in symptomatic Chiari.
Surgical technique
The technique to be chosen should, on the one hand, give the best chance of improvement of the Chiari symptoms and, on the other hand, minimize the risk of complications. This consideration will have to be made per patient.
Recommendations
-
Screen by means of an MRI on initial contact with the center of expertise the following patients for the presence of Chiari:
-
-
non-syndromic unilambdoid suture synostosis
-
-
Crouzon/Pfeiffer syndrome
-
-
multisuture craniosynostosis with involvement of the lambdoid sutures.
-
-
-
Repeat the MRI:
-
-
at the age of 4 years
-
-
at the age of 18 years
-
-
in case of a clinical suspicion on a symptomatic Chiari.
-
-
Screen by means of an MRI of the cervical, thoracic and lumbar spinal cord for the presence of a syrinx if the demonstrated Chiari increases and/or becomes symptomatic.
Perform surgical treatment of Chiari only if the patient has symptoms.
-
Otherwise, follow an active follow-up policy by the pediatric neurosurgeon or pediatric neurologist:
-
-
with annual check-up for neurological symptoms or signs
-
-
carry out an MRI when indicated.
-
-
give instructions to the parents.
-
-
Research Gaps
Screening
There is no reliable information available on the prevalence of Chiari in Saethre-Chotzen syndrome or Muenke syndrome. There is no study on the age at which Chiari develops, becomes symptomatic or a syrinx develops. Thus, it is not clear when screening is most effective.
Indication
It is unclear whether treatment of asymptomatic Chiari will result in long-term health gains. Nor whether this depends on the age at which a preventive procedure is performed, in light of rapid reossification in small children.
Surgical Technique
Comparative studies on different surgical techniques are lacking as well as long-term studies of the outcomes of the different techniques.
Literature
-
1.
Leikola J, The incidence of Chiari malformation in nonsyndromic, single suture craniosynostosis. Childs Nerv Syst 2010;26:771-4
-
2.
Strahle J, Muraszko KM, Buchman SR, et al. Chiari malformation associated with craniosynostosis. Neurosurg Focus 2011;31:1-8
-
3.
Engel M, Castrillon-Oberndorfer G, Hoffmann J, et al. Chiari malformation in nonsyndromic single craniosynostosis – Much ado about nothing? Acta Neurochir 2012;154:1803-7
-
4.
Fearon JA, Dimas V, Ditthakasem K. Lambdoid craniosynostosis: The relationship with Chiari deformations and an analysis of surgical outcomes. Plast Reconstr Surg 2016;137:946-51
-
5.
Cinalli G, Renier D, Sebag G, et al. Chronic tonsillar herniation in Crouzon's and Apert's syndromes: the role of premature synostosis of the lambdoid suture. J Neurosurg 1995;83:575–82
-
6.
Cinalli G, Spennato P, Sainte-Rose C, et al. Chiari malformation in craniosynostosis. Childs Nerv Syst 2005;21: 889–901
-
7.
Fearon JA, Rhodes J. Pfeiffer syndrome: a treatment evaluation. Plast Reconstr Surg 2009;123: 1560-9
-
8.
Fearon JA, Podner C. Apert syndrome: Evaluation of a treatment algorithm. Plast Reconstr Surg 2013;131:132-42
-
9.
Czerwinski M, Kolar JC, Fearon JA. Complex craniosynostosis. Plast Reconstr Surg 2011;128:955-61
-
10.
Scott WW, Fearon JA, Swift DM, et al. Suboccipital decompression during posterior cranial vault remodeling for selected cases of Chiari malformations. J Neurosurg Pediatr 2013;12:166-70
CHAPTER 11 VISUAL, REFRACTIVE AND MOTILITY IMPAIRMENTS
11.1 What screening is necessary to detect visual and motility impairments in the different types of non-syndromic and syndromic craniosynostosis in a timely manner?
Introduction
Loss of vision in craniosynostosis is caused by optic atrophy secondary to papilledema in elevated ICP, primary optician atrophy, corneal abnormalities by lagophthalmos, or amblyopia secondary to strabismus or refractive abnormalities. Timely recognition and treatment are essential for vision retention.
Search and Selection
For the following specific questions, original scientific studies or systematic reviews of original scientific studies have been included:
-
1.
What is the prevalence of vision and motility disorders in the different types of non-syndromic and syndromic craniosynostosis?
-
2.
Which screening tests are most accurate?
In the Medline (OVID) and Embase databases, one overall search was conducted for studies on craniosynostosis. The search strategy is given in the annex to the guideline. After deduplication, the literature search yielded 2732 hits.
Given the large number of studies, the chair of the working group first selected those that met the following general selection criteria:
General Selection and Exclusion Criteria
| Study type | -original studies -systematic reviews of sufficient quality: - research question of systematic review corresponds (largely) to the basic question - search is performed in at least 2 relevant databases, e.g. Cochrane Library, Medline/PubMed - reporting of the complete search strategy - no relevant keywords/search terms are missing |
| Follow-up period | -minimum follow-up period of 12 months for therapeutic or prognostic studies. |
| Exclusion criteria | -Case-reports -Expert opinion -Letters -Editorials -Case control studies for diagnostic tests -Narrative reviews |
The pre-selected studies that met the specific selection criteria listed in the Table below are included in the literature summary of this chapter.
Specific Selection and Exclusion Criteria
| -minimum study size: 20 patients -minimum follow-up time: up to 2 years of age |
Summary of the Literature
1. What is the prevalence of vision and motility disorders in the different types of non-syndromic and syndromic craniosynostosis?
Non-syndromic craniosynostosis
Four studies describe eye abnormalities in sagittal, metopic and unilateral coronal suture synostosis and cover small patient groups, shown in Table 1.1–4 The patients from Vasco's article had no refractive abnormalities and all had a normal vision postoperatively for 12 months.2
TABLE 1. Prevalence of Strabismus and Refractive Abnormalities in Non-syndromic Craniosynostosis.
| Sagittal | Metopic | Coronal | References | |
| Refractive deviations | ||||
| Hyperopia | 1/7 (14%) | 9/27 (33%) | Chung 20154 | |
| 5/91 (6%) | Nguyen 20143 | |||
| Myopia | 0/7 (0%) | 0/27 (0%) | Chung 20154 | |
| 5/91 (6%) | Nguyen 20143 | |||
| Astigmatism | 2/7 (29%) | 13/27 (48%) | Chung 20154 | |
| 19/91 (21%) | Nguyen 20143 | |||
| 7/29 (24%) | 3/7 (43%) | 2/7 (29%) | Gupta 20031 | |
| Anisometropy | 0/7 (0%) | 7/27 (26%) | Chung 20154 | |
| 5/91 (6%) | Nguyen 20143 | |||
| Strabismus | ||||
| Exodeviation | 1/7 (14%) | 6/27 (22%) | Chung 20154 | |
| 1/30 (3%) | 0/8 (0%) | 2/7 (29%) | Gupta 20031 | |
| Esodeviation | 0/7 (0%) | 5/27 (19%) | Chung 20154 | |
| 0/12 (0%) | 1/10 (10%) | 3/7 (34%) | Vasco 20082 | |
| 0/30 (0%) | 0/8 (0%) | 0/7 (0%) | Gupta 20031 | |
| Vertical deviation | 1/7 (14%) | 1/27 (4%) | Chung 20154 | |
| 1/12 (8%) | 0/10 (0%) | 2/7 (29%) | Vasco 20082 | |
| H + V deviation | 1/7 (14%) | 2/27 (7%) | Chung 20154 | |
In a series of 64 children with trigonocephaly from 2011 by Macintosh, 20 children (31%) have abnormal visual function.5 Eighteen of these 20 children have a refractive abnormality for which glasses are indicated; 10 have strabismus. The refractive abnormality is mainly based on hyperopia.
Nguyen found amblyopia in 8 (9%) patients with trigonocephaly, which is a consequence of strabismus in 5 patients and anisometropy in 3 patients.3
In a systematic review of ophthalmic outcomes of fronto-orbital advancement in children with unicoronal synostosis, Gencarelli describes a prevalence ranging from 15 to 92% for astigmatism, 19 to 48% for anisometropy and 3 to 56% for amblyopia.6
Syndromic craniosynostosis
Of the various syndromic forms, the most detailed and complete ophthalmic publications concern Apert's syndrome. The studies on the other syndromes are limited in number but of good quality and give few contradictory results.
Jadico compared 18 Apert patients, of whom 11 with S252W mutation and 7 with P253R mutation (Table 2).7 The S252W mutation causes more serious eye problems with significant differences with regard to strabismus, astigmatism and tear duct obstruction.
TABLE 2
| Type of mutation | ||
| Eye defect (%) | P253R | S252W |
| Strabismus | 85 | 91 |
| Ptosis | 71 | 73 |
| Amblyopia | 43 | 73 |
| Tear duct obstruction | 14 | 100 |
| Myopia | 14 | 36 |
| Hypermetropia | 14 | 9 |
| Astigmatism | 14 | 82 |
Khong makes the same comparison for Apert's syndrome between 20 S252W patients and 9 P253R patients (Table 3).8
TABLE 3
| Type of mutation | ||
| Eye defect (%) | P253R | S252W |
| Vision < 6/12 in best eye | 20 | 12.5 |
| Vision < 6/12 in min. 1 eye | 60 | 12.5 |
| Vision < 6/12 (per eye) | 40 | 12.5 |
| Pale papilla | 16 | 29 |
| Amblyopia | 56 | 20 |
| Corneal scarring and keratopathy (per eye) | 25 | 21 |
| Strabismus (per eye) | 47 | 39 |
Khong describes ophthalmic findings in 61 patients with Apert syndrome (Table 4).9,10 The most common cause of vision loss was amblyopia (prevalence 35%), followed by corneal scarring (8%) and optic atrophy (5%).
TABLE 4
| Eye defect (%) | Apert syndrome |
| Vision < 6/12 in best eye | 19 |
| Vision < 6/12 in min. 1 eye | 54 |
| Strabismus | 63 |
| Ametropia (hypermetropia; myopia) | 69 (42; 27) |
| Anisometropia (≥ 0.75 diopter) | 50 |
Gray found in 71 patients with Crouzon syndrome that a decline in vision is mainly caused by amblyopia (21%) (Table 5).11
TABLE 5
| Eye defect (%) | Crouzon syndrome |
| Loss of vision in min. 1 eye | 35 |
| Ametropia (hypermetropia; myopia) | 77 (57; 20) |
| Keratopathy | 15 |
| Amblyopia | 21 |
Jadico compared 10 patients with a TWIST mutation (Saethre-Chotzen syndrome) with 11 patients with P250R FGFR3 mutation (Muenke syndrome) (Table 6).12
TABLE 6
| Type of mutation | ||
| Eye defect (%) | TWIST | FGFR3 |
| Ptosis | 90 | 36 |
| Amblyopia | 70 | 18 |
| Horizontal strabismus | 70 | 55 |
| Vertical strabismus | 60 | 36 |
| Tear duct obstruction | 60 | 0 |
| Astigmatism | 50 | 9 |
| Inferior oblique overactivity | 40 | 45 |
| Hypermetropia | 40 | 27 |
| Myopia | 30 | 18 |
| Nystagmus | 30 | 18 |
| Optic nerve abnormalities | 30 | 27 |
De Jong report the following refractive abnormalities per syndrome in 132 patients: Apert 22/29 (76%), Crouzon/Pfeiffer 16/41 (39%), Muenke 17/35 (49%), Saethre-Chotzen 14/27 (52%).13 Tables 7 and 8 give an overview of the reported prevalences of strabismus and astigmatism by the different authors, with Khan describing 141 children with syndromic craniosynostosis, Sharma 22 patients with Pfeiffer syndrome and Kruszka 106 patients with Muenke syndrome from 71 families.14-16
In a review by Lehman, frequent assessment of refractive abnormalities and motility disorders with adequate treatment is recommended for the prevention of amblyopia and retention of vision.17
TABLE 7. Prevalence of Strabismus as a Percentage (Esotropia/Exotropia)
| Type of craniosynostosis | |||||
| First author and year | Apert | Crouzon | Pfeiffer | Saethre-Chotzen | Muenke |
| Khan 200314 | 82.9 (48.8/34.1) | 66.7 (20.0/46.7) | 94.7 (15.8/78.9) | 53.4 (29.3/24.1) | |
| Gray 200511 | 39 | ||||
| Jadico 200612 | 70 horizontal | 55 | |||
| 60 vertical | 36 | ||||
| Jadico 20067 | 85 P253R 91 S252W | ||||
| Khong 20069 | 65 (36/19) | ||||
| Lehman 200617 | 39 | 63 horizontal | |||
| De Jong 201013 | 93 | 63 | 37 | 39 | |
| Sharma 201615 | 55 | ||||
| Kruszka 201616 | 45 | ||||
TABLE 8. Prevalence of Astigmatism (>1D) as a Percentage (Right Eye/Left Eye)
| Type of craniosynostosis | |||||
| First author and year | Apert | Crouzon | Pfeiffer | Saethre-Chotzen | Muenke |
| Khan 200314 | 52.4 (54.8/50.0) | 43.4 (40.0/46.7) | 44.8 (40.0/46.7) | 30.2 (28.6/31.7) | |
| Jadico 200612 | 50 | 9 | |||
| Jadico 20067 | 14 P253R 82 S252W | ||||
| Khong 20069 | 42 | ||||
| Sharma 201615 | 18 | ||||
| Kruszka 201616 | 14 | ||||
Conclusions
| _____ | Visual and motility abnormalities may be frequent in metopic suture and unilateral coronal suture synostosis. Gupta et al, 2003; Vasco et al, 2008; Macintosh et al, 2011; Nguyen et al, 2014 Chung et al, 2015; Gencarelli et al, 2016. |
| _____ | Visual, refractive and motility abnormalities are likely to be very frequent in all types of syndromic craniosynostosis. Frequent assessment for these abnormalities with appropriate treatment can contribute to the prevention of amblyopia and to vision retention. Jadico et al, 2006–1; Jadico et al, 2006–2; Khong et al, 2007; Gray et al, 2005; De Jong et al, 2010; Khan et al, 2003; Khong et al, 2006–1; Khong et al, 2006–2; Lehman et al, 2006; Sharma et al, 2016; Kruszka et al, 2016. |
2. Which screening tests are most accurate?
The literature is limited to general advice, such as ’early’ referral to the ophthalmologist and ’follow-up’ by the ophthalmologist, without mentioning specific ages or tests for screening. For non-syndromic, single-suture craniosynostosis, these recommendations specifically address unicoronal synostosis and trigonocephaly.3–6 For syndromic craniosynostosis, identical advices are described.12,13,15,17
Conclusion
| _____ | The literature lacks evidence about screening tests for vision and motility disorders. |
Considerations
• Evidence of the Conclusions
Grading of the conclusions does not apply.
Despite the fact that there are few publications on this subject, there is a general agreement on the prevalence for both non-syndromic and syndromic craniosynostosis. Evidence on how to screen and at what moment is lacking in the literature.
• Values and Preferences
According to the working group, most parents consider an optimal vision to be very important for their child and will therefore agree to screening for disorders that can negatively influence the vision.
• Costs and Resources
The guideline committee expects that application of the recommendations leads to little or no increase in costs, because the recommendations are in line with existing practice.
• Professional Perspective
Screening for refractive abnormalities is possible from infancy, vision assessment from 3 years of age, and assessment of motility from 4 to 5 years of age. The child healthcare center screens children between the ages of 3.75 and 4 years. Experience shows that most parents do not take their children to the child healthcare center other than for vaccinations, because protocolized checkups are performed in the center of expertise. For this reason, it is recommended that the initial referral to the ophthalmologist takes place at the time of the first contact with the center of expertise.
Depending on the initial findings by the ophthalmologist in the center of expertise and in consultation with the parents, it may be decided to have the ophthalmological checkups carried out by an ophthalmologist in his or her own region, with the results being communicated to the center of expertise.
• Balance of Anticipated Desired and Undesired Outcomes
Incomplete screening for problems of vision, refraction and motility can lead to unnecessary loss of function. The screening is generally not very burdensome for the child, so the advantages outweigh the disadvantages.
Rationale of the recommendation(s)
The guiding principle in the preparation of the recommendations is maintaining optimal vision by early detection and treatment of conditions that can negatively affect the vision. There are virtually no reasons to refrain from screening and thus run the risk of a poorer visual outcome.
Recommendations
Screening for vision, refraction and motility abnormalities
Screen children with metopic suture synostosis, unilateral coronal suture synostosis, multisuture craniosynostosis involving a single coronal suture and all syndromic forms of craniosynostosis for
vision, refraction and motility abnormalities. Referral to the ophthalmologist is made at the first consultation in the centre of expertise
Depending on the results of the screening, follow-up examinations are agreed upon by the ophthalmologist (in accordance with guideline NOG).
Research Gaps
Screening for Vision, Refraction and Motility Abnormalities
Little has been described about the final results on vision per type of craniosynostosis and the influence of timing of both screening and treatment on this. In particular, gaining insight into intrinsic factors that are part of the condition versus factors that can be improved by treatment is useful to know which visual outcomes are realistically achievable.
Literature
-
1.
Gupta PC, Foster J, Crowe S, et al. Ophthalmologic findings in patients with nonsyndromic plagiocephaly. J Craniofac Surg 2003;14:529-32
-
2.
Vasco G, Baranello G, Ricci D, et al. Longitudinal assessment of visual development in non-syndromic craniosynostosis: a 1-year pre- and post-surgical study. Arch Dis Child 2008;93:932-5
-
3.
Nguyen TB, Shock LA, Missoi TG, et al. Incidence of Amblyopia and Its Risk Factors in Children With Isolated Metopic craniosynostosis. Cleft Palate Craniofac J. 2014 Dec 1:1545-69
-
4.
Chung SA, Yun IS, Moon JW, et al. Ophthalmic findings in children with nonsyndromic craniosynostosis treated by expansion cranioplasty. J Craniofac Surg 2015;26:79-83
-
5.
Macintosh C, Wells R, Johnson D, et al. What are the effects of metopic synostosis on visual function? J Craniofac Surg 2011;22:1280-3
-
6.
Gencarelli JR, Murphy A, Samargandi OA, et al. Ophthalmologic outcomes following fronto-orbital advancement for unicoronal craniosynostosis. J Craniofac Surg 2016;27:1629-35
-
7.
Jadico SK, Young DA, Huebner A, et al. Ocular abnormalities in Apert syndrome: Genotype/phenotype correlations with fibroblast growth factor receptor type 2 mutations. J AAPOS 2006;Dec 10:521-27
-
8.
Khong JJ, Anderson PJ, Hammerton M, et al. Differential effects of FGFR2 mutation in ophthalmic findings in Apert syndrome. J Craniofac Surg 2007;18:39-42
-
9.
Khong JJ, Anderson P, Gray TL, et al. Ophthalmic findings in Apert's syndrome after craniofacial surgery. Ophthalmology 2006;113:347–52
-
10.
Khong JJ, Anderson P, Gray TL, et al. Ophthalmic findings in Apert syndrome prior to craniofacial surgery. Am J Ophthalmol 2006;142:328-30
-
11.
Gray TL, Casey T, Selva D, et al. Ophthalmic sequelae of Crouzon syndrome. Ophthalmology 2005;112:1129–34
-
12.
Jadico SK, Huenber A, McDonald-McGinn DM, et al. Ocular phenotype correlations in patients with TWIST versus FGFR3 genetic mutations. J AAPOS 2006;Oct 10:435-44
-
13.
De Jong T, Bannink N, Bredero-Boelhouwer HH, et al. Long-term functional outcome in 167 patients with syndromic craniosynostosis; defining a syndrome-specific risk profile. J Plast Reconstr Aesthet Surg 2010;63:1635-41
-
14.
Khan SH, Nischal KK, Dean F, et al. Visual outcomes and amblyogenic risk factors in craniosynostotic syndromes: a review of 141 cases. Br J Ophthalmol 2003;87:999-1003
-
15.
Sharma N, Greenwell T, Hammerton M, et al. The ophthalmic sequelae of Pfeiffer syndrome and the long-term visual outcomes after craniofacial surgery. J AAPOS 2016;20:315-19
-
16.
Kruszka P, Addissie YA, Yarnell CMP, et al. Muenke syndrome: an international multicenter natural course study. Am J Med Genet Part A 2016;170A:918-29
-
17.
Lehman S. Strabismus in craniosynostosis. Curr Opin Ophthalmol 2006;17:432–4
CHAPTER 12 RESPIRATORY DISORDERS
12.1 What is the policy on respiratory disorders in syndromic craniosynostosis?
Introduction
Children with craniosynostosis syndromes belong to the risk groups for sleep-related respiratory disorders characterized by upper airway obstruction. This disrupts normal breathing and sleep. The respiratory disorders are obstructive sleep apnea and hypopnea (OSA), central sleep apnea and hypopnea (CSA) and upper airway resistance (UAR).
The clinical symptoms of respiratory disorders are diverse and can be divided into symptoms at night: restless sleep, snoring, apnea, bedwetting and sweating, and during the day: dry mouth when getting up, fatigue, impaired cognitive functioning, reduced school performance and behavioral disorders. In the long term, growth disorders may occur. In addition, respiratory disorders may contribute to an increase in ICP, probably due to the cerebral vasodilation that occurs when CO2 accumulates, causing more blood to flow to the brain and resulting in an increase in ICP. Since children with syndromic craniosynostosis often have a somewhat higher ICP, respiratory disorders in them may result in too high an ICP. In view of the seriousness of the respiratory disorders and the good treatment possibilities, early recognition is very important.
The treatment of respiratory disorders can be pharmacological, e.g. nasal corticosteroid spray or antibiotics, surgical, e.g. adenotonsillectomy or midface surgery, or non-surgical, such as nocturnal supplemental oxygen or continuous / bi-level positive airway pressure (CPAP or BiPAP). In the articles in which the European Respiratory Society presents a 'state-of-the-art’ overview, a flowchart is given for the treatment of children up to 23 months old and for the treatment of older children with OSA.1,2 There is one systematic review specific to craniosynostosis, but because it also includes case reports with only 2 patients it is not always of high quality.3 These case reports, deal with treatment by nasopharyngeal tube (NPT), CPAP or BIPAP and tracheotomy. The NPT can be used to bridge a period pending a more definitive treatment. Respiratory support through non-invasive ventilation with CPAP or BIPAP is an accepted treatment in children with good results, but in which compliance is essential.3 The systematic review by Nash describes that many of the 23 articles included describe a tracheal cannula for treatment of severe OSA. Eventually, surgical treatment will be necessary to eliminate the causes of the respiratory disorders.
Definitions
In accordance with the Dutch guideline ’OSAS in children’, the following definitions are used:
The use of the apnea-hypopnea index (AHI) and the oxygenation-desaturation index (ODI) is recommended to characterize respiratory disorders during sleep.
Obstructive apnea: the presence of chest and/or abdominal movements associated with absence of oronasal airflow. At least 2 breathing cycles are missing.
An obstructive apnea is defined if, in the presence of chest and/or abdominal movements, there is an absence of oronasal airflow with a duration of > 2 breathing cycles.
Central apnea: the absence of chest and/or abdominal movements associated with absence of oronasal airflow with a duration of > 20 sec or a shorter apnea with at least 2 breathing cycles and associated with a desaturation ≥3% or an arousal or an awakening.
Hypopnea: ≥50% decrease in the amplitude of the oronasal airflow or pressure signal with a desaturation ≥3% or an arousal or an awakening.
AHI = apnea/hypopnea index
Severity of respiratory disorder:
| Mild: | AHI 1–5/hr |
| Moderate: | AHI > 5–10/hr |
| Severe: | AHI > 10/hr |
Search and Selection
For the following specific questions, original scientific studies or systematic reviews of original scientific studies have been included:
-
1.
Which respiratory disorders occur with craniosynostosis, in what frequency and in what severity?
-
2.
What are the OSA-specific factors that weigh in the indication system for treatment, especially in the case of mild OSA?
-
3.
What are the anatomical factors influencing the choice of surgical treatment to be used?
In the Medline (OVID) and Embase databases, one overall search was conducted for studies on craniosynostosis. The search strategy is given in the annex to the guideline. After deduplication, the literature search yielded 2732 hits.
Given the large number of studies, the chair of the working group first selected those that met the following general selection criteria:
General Selection and Exclusion Criteria
| Study type | -original studies -systematic reviews of sufficient quality: research question of systematic review corresponds (largely) to the basic question search is performed in at least 2 relevant databases, e.g. Cochrane Library, Medline/PubMed reporting of the complete search strategy no relevant keywords/search terms are missing |
| Follow-up period | -minimum follow-up period of 12 months for therapeutic or prognostic studies. |
| Exclusion criteria | -Case-reports -Expert opinion -Letters -Editorials -Case control studies for diagnostic tests -Narrative reviews |
The pre-selected studies that met the specific selection criteria listed in the Table below are included in the literature summary of this chapter.
Specific Selection and Exclusion Criteria
| Selection criteria for indication | - Minimum study size: 20 patients for patient series, where no multivariate analysis was used to identify prognostic factors for a relevant outcome measure. - Minimum study size: 35 patients for patient series with multivariate analysis of possible predictive variables for the effect - minimum number of participants of studies with a direct comparative design: 20 per study arm. |
Summary of the Literature
1. Which respiratory disorders occur with craniosynostosis, in what frequency and in what severity?
Symptoms and Signs of OSA and Detection Instruments
Based on literature review, the European Respiratory Society (ERS) statement, describes symptoms that parents report as fitting for OSA in their child aged 1 to 23 months: 1. Snoring or audible breathing in the first 2 years of life, apnea, frequent movements during sleep, mouth breathing and repeated waking up; 2. Anamnesis with seemingly life-threatening events.1 Delayed growth may be a presentation of OSA. Nutritional problems and otitis media are associated with having OSA. Screening for OSA is done by polysomnography. In a state-of-the-art review of OSA in children from the same group, maxillary hypoplasia in craniosynostosis syndromes and mandibular hypoplasia are mentioned as disorders related to having OSA.4 Alternative methods for polysomnography, such as home respiratory polygraphy, oximetry or sleep questionnaires, have a limited predictive value (positive predictive value 82% and negative predictive value 79%).5
Prevalence of OSA
Al-Saleh retrospectively examined the sleep studies of 35 patients (14 Apert, 20 Crouzon, 1 Saethre-Chotzen syndrome) and found a prevalence of 74% of sleep-related respiratory disorders.6 This concerned in 7/26 mild OSA, in 7/26 moderate OSA and in 10/26 severe OSA. In addition, 2 patients with moderate to severe OSA were also diagnosed with central apnea.
The prevalence of respiratory problems was prospectively determined in the study of Driessen in 97 children with multisuture and syndromic craniosynostosis by means of an ambulatory level III sleep study, and amounted to 68%.7 Respiratory problems were moderate to severe in 26% and mostly concerned children with midface hypoplasia (Apert or Crouzon syndrome). Comparison of the sleep studies with results of MRI scans in 71 patients showed no relationship between obstructive sleep apnea and the presence or absence of tonsillar herniation.8 The prevalence of central sleep apnea in outpatient sleep measurements in 138 patients with syndromic craniosynostosis was found to be only 3.6% and to decrease further with age.9
Luna-Paredes describes the results of the Pediatric Sleep Questionnaire and sleep studies in 44 patients with a craniofacial disorder, of whom 30 with syndromic craniosynostosis.10 The questionnaire gives indications for respiratory problems in 82% and the sleep study in 45%, but no distinction is made by diagnosis, so the percentage for syndromic craniosynostosis cannot be determined.
Analysis of sleep studies in 110 children with Apert or Crouzon syndrome and symptomatic of respiratory disorders resulted in a prevalence of 74%.11
A similar prevalence of 83% is described by Zandieh, who studied 36 out of 87 patients with polysomnography (23 Apert, 13 Crouzon) and found a normal measurement at 17%, mild OSA at 22%, moderate OSA at 19% and severe OSA at 42%.12 There seems to be some selection because only 36 out of 87 patients were tested, so that both the overall prevalence and that of moderate OSA appear to be higher than in the other studies.
Conclusion
| ____ | In children with multisuture or syndromic craniosynostosis, there may be a high prevalence of obstructive sleep apnea, around 70%. The prevalence and severity are highest in patients with Apert, Crouzon and Pfeiffer syndrome. Central apneas have a prevalence of about 4%, and this decreases with age. Al-Saleh et al, 2011; Driessen et al, 2013–1; Driessen et al, 2013–2; Driessen et al, 2012; Inverso et al, 2016; Zandieh et al, 2013 |
2. What are the OSA-specific factors that weigh in the indication system for treatment, especially in the case of mild OSA?
The indication for treatment of respiratory disorders in craniosynostosis is determined by the severity of the symptoms and is aimed at improving upper airway accessibility. The presence of elevated ICP may be an indication for treatment of OSA if this has been demonstrated. Spruijt shows that only moderate and severe OSA are associated with elevated ICP and mild OSA are not.13
The sleep architecture is disturbed by the presence of moderate or severe OSA, but not by mild OSA.14 Thus, mild OSA in itself does not seem to be a sufficient reason for treatment, unless the patient has symptoms. As soon as moderate or severe OSA is treated, recovery of the sleep architecture occurs.
Conclusion
| Level 3 | The presence of moderate and severe OSA may be related to elevated ICP and disturbed sleep architecture and is therefore an indication for treatment. The presence of mild OSA may not be related to elevated ICP and disturbed sleep architecture. Mild OSA are treated if the patient experiences many symptoms. B Spruijt et al, 2015; Spruijt et al, 2016 |
3. What are the anatomical factors influencing the choice of surgical treatment to be used?
The method of palate surgery in case of palatoschisis in syndromic craniosynostosis is not discussed here, as there is hardly any evidence for this.3
Adenotonsillectomy
In 25 children (Antley-Bixler, Apert, Crouzon and Saethre-Chotzen syndrome) with mild (n = 7), moderate (n = 11) to severe (n = 7) OSA, an adenotonsillectomy (ATE) was performed.15 This resulted in a reduction in the number of incidents with a decrease in saturation of 4%/h or more, with no significant change in mean saturation, pulse rate increases/h, or the percentage of time with a SaO2 below 90%. In 15 (60%) children there was a reduction in severity of the respiratory disorder after treatment. It is not mentioned how long after surgery the repeated measurements were taken, so it is not clear how long the improvement persists. The authors indicate that an ATE can be considered as a temporary improvement in breathing, but is not an alternative to other treatment of OSA, such as CPAP and midface surgery.
Zandieh describes that 29 children (18 Apert, 11 Crouzon) underwent an ATE because of proven OSA at an average age of 5.6 years (+ 14.2 years).12 A postoperative sleep measurement was also performed on 13, showing that the obstructive apnea/hypopnea index did not change significantly (even deteriorated in 3) and that OSA persisted in 11 of the 13 children.
Craniofacial Surgery
The systematic review only mentions midface advancement surgery (especially Le Fort III and monobloc) in 12 articles with predominantly positive results.3 This adds little to the evidence for these treatments.
Arnaud prospectively investigated the effect of frontofacial monobloc advancement with internal distraction in a group of 36 children with syndromic craniosynostosis (mean age 5.2 yrs).16 Sixteen children had upper respiratory tract problems, for which 6 needed a tracheostoma, while the remaining 10 children had regular < 95% saturation decreases. After the operation, 4 of 6 children could be decannulated and the desaturations disappeared in 8 of the 10 children.
Witherow retrospectively investigated the long-term results (mean follow-up 24 months, range 6 months–4 years) of a monobloc in 20 children (mean age 7.8 yrs, range 2–16 yrs).17 Seventeen children had upper airway obstruction; polysomnography results are not mentioned. Of the 7 children who needed a tracheostoma, 5 could be decannulated and in 2 of the 5 children who had CPAP, this could be stopped.
Flores retrospectively investigated airway changes after a Le Fort III distraction in 20 children with syndromic craniosynostosis.18 In children with airway problems a standard polysomnography was performed pre-operatively. The severity of respiratory disturbance during sleep was classified using a respiratory disturbance index (RDI): mild 2–5, moderate 5–10 and severe > 10. Ten children had severe respiratory problems, for which two needed a tracheostoma. After surgery, one child could be decannulated, in the second one this was not possible due to a subglottic stenosis. Of the other 8 children with severe airway problems, three underwent postoperative polysomnography, with a decrease in RDI. The 5 remaining children showed subjective improvement in OSA symptoms.
Examination of the upper airway by the otorhinolaryngologist by means of endoscopy in patients with Apert and Crouzon syndrome shows that the obstruction can be present in several places, in the nose, rhino-, oro-, and hypopharynx, but also in the larynx or trachea.19–21 When the location of the obstruction is known, a more targeted treatment can be instituted. Doerga (2016) describes improvement of the polysomnography study after midface advancement in 8 patients combined with mandibula advancement on guided upper airway endoscopy in 2 patients: severe OSA in 4 patients was reduced to moderate (n = 2) and mild (n = 2); moderate OSA in 3 was reduced to mild (n = 1) and no OSA (n = 2); mild OSA in 1 patient was reduced to no OSA21. Better results appear to be achieved by considering all levels of obstruction when choosing surgery, as the addition of mandibular advancement resulted in only mild or no OSA.
Conclusions
| Level 3 | If adenotonsillar hypertrophy is diagnosed in children with syndromic craniosynostosis in combination with OSA, adenotonsillectomy may reduce the severity of respiratory disorders. However, this rarely suffices as a definitive treatment. C Amonoo et al, 2009 Zandieh et al, 2013 |
| Level 3 | If patients with syndromic craniosynostosis have been shown to have moderate to severe OSA, upper airway obstruction may be present at different and multiple levels. Upper airway obstruction levels can be identified by endoscopy. C Fujimoto et al, 2011; Anton-Pacheco et al, 2012; Doerga et al, 2016 |
| Level 3 | If midface hypoplasia is present, midface advancement may reduce moderate to severe respiratory failure to mild or none. If obstruction is present at the level of the tongue base, mandibular advancement can reduce moderate to severe respiratory failure to mild or none. C Arnaud et al, 2007; Witherow et al, 2008; Flores et al, 2009; Doerga et al, 2016 |
Considerations
• Evidence of the Conclusions
Indication for treatment
The evidence of the conclusions on elevated ICP and disturbed sleep architecture for children with syndromic craniosynostosis is reasonable to weak, as only one research group has published on the subject.
The treatment to be applied
The evidence of the conclusions on the treatment method for respiratory disorders in syndromic craniosynostosis is weak, because the studies are often narrative in nature and little supported by objective data.
• Values and Preferences
Screening for respiratory disorders
According to the working group, parents may have a strong preference for an ambulatory measurement in order to reduce the number of hospital visits and because, according to their experience, the child sleeps better in the own bed. Preferably, an upper airway endoscopy will be performed in combination with an already planned procedure under anesthesia, so that there is no need for separate admission and procedure under anesthesia.
Indication for treatment
In view of the considerable consequences that moderate and severe OSA can have for the child, according to the working group parents will often agree with treatment.
For moderate OSA, this is less clear, and together with the parents it is considered whether the symptoms indicate treatment. In general, surgeries such as A(T)E are sufficient.
The treatment to be applied
Depending on the symptoms, the severity of the OSA and other symptoms (such as severe exorbitism), a choice of treatment is made in consultation with the parents and, if possible, with the patient. This involves weighing up the burden of the intervention on the patient and parents, how definitive the result is likely to be, how feasible the treatment is in terms of patient cooperation, and what the risks of the treatment are.
• Costs and Resources
Screening for respiratory disorders
The guideline committee expects that application of the recommendations leads to little or no increase in costs, because the recommendations are in line with existing practice.
Indication for treatment
The guideline committee expects that application of the recommendations leads to little or no increase in costs, because the recommendations are in line with existing practice.
The treatment to be applied
The guideline committee expects that application of the recommendations leads to little or no increase in costs, because the recommendations are in line with existing practice.
• Professional Perspective
Screening for respiratory disorders
In general, the degree of OSA determined during an initial measurement strongly determines the natural course of OSA and a spontaneous deterioration does not occur frequently. However, other factors such as respiratory infection can cause a deterioration. Preferably, a type 1 polysomnography is performed, which is a fully observed polysomnography, by a laboratory technician or video-recording, performed in a sleep laboratory. This allows optimal monitoring of breathing, heart action, leg movements, sleep and also visual diagnostics (guideline OSA in children). In view of the considerable consequences that moderate to severe OSA can have in children with syndromic craniosynostosis, screening is desirable at regular intervals and on indication if there are respiratory problems in the medical history. After the age of 6 years, the prevalence of tonsillar hypertrophy decreases, and an increase in OSA is less likely.
Since moderate and severe OSA is often caused by a multi-level upper airway obstruction, endoscopy in a spontaneously breathing child is indicated. The high prevalence of upper airway obstruction at the level of the tongue base seems to explain remaining respiratory disturbances after midface advancement. This endoscopy is preferably performed prior to the midface advancement, so that any mandibular correction can be performed simultaneously with the midface advancement.
Apert syndrome has a low prevalence of palatoschisis, especially of the palatum molle. Closure of the palatum molle may aggravate the severity of OSA. Preliminary polysomnography type 1 measurement, possibly combined with a custom palatum plate mimicking the postoperative situation, is required to predict the safety of palate closure.
Indication for treatment
In case of upper airway resistance or mild OSA, the pattern of symptoms is decisive for whether or not to treat, with a preference for non-invasive treatment, such as short-term use of a corticosteroid nasal spray or, in special circumstances, oxygen therapy for the night, or an A(T)E if there is an indication for this.
The treatment to be applied
The choice of treatment for moderate or severe OSA is determined by the patient's age, the anatomical cause of the OSA and the preference of parents and, if possible, the patient. In a patient under the age of 2, midface advancement using a monobloc procedure with distraction is technically more difficult and requires the use of a facial pin; if there is obstruction at the level of the base of the tongue, a combined mandibula distraction at this age is not recommended due to a high risk of complications. For these patients, a tracheal cannula may be considered, and to postpone the monobloc with mandibula distraction until, for example, the age of 3 to 4 years. In the opinion of the working group, NPT as a bridging therapy is not desirable, because of the need for frequent changes (often under sedation or anesthesia) to prevent nasal septum necrosis. Sometimes CPAP or BiPAP can be an intermediate solution, but it can be very difficult to make a suitable face mask and prevent the protruding eyes from being damaged by the airflow. Cooperation can also be a problem, and the mask fixation bands that run over the occiput can interfere with an occiput expansion. From adulthood, septum surgery can provide additional improvement in nasal accessibility and OSA-related complaints, as experience has shown.
• Balance of Anticipated Desired and Undesired Outcomes
Screening for respiratory disorders
Given the very high prevalence of respiratory disorders in syndromic craniosynostosis and the consequences of leaving it untreated, screening is indicated. In order to reduce costs and burden, a home measurement instead of a clinical measurement should be considered when there is low suspicion of respiratory disorders, or for follow-up screening.
Theoretically, an upper airway scopy examination carries a risk of airway damage and laryngospasm. In practice, however, this appears to be a low and acceptable risk in experienced hands, which outweighs the benefits of this screening, namely a more targeted treatment on the anatomical cause of respiratory problems.
Indication for treatment
Failure to establish indication for treatment as a result of not performing polysomnography will eventually result in undesirable consequences for the child's development and quality of life.
The treatment to be applied
An undesirable outcome may be that the respiratory disorders do not recover sufficiently and a (different) treatment is needed. This risk is particularly associated with corticosteroid nasal spray and ATE.
Rationale of the recommendation(s)
Screening for respiratory disorders
The guiding principle is the fact that sleep-related respiratory disorders have a very high prevalence in children with syndromic craniosynostosis and, if left untreated, can have lasting severe consequences. Shortly after the first referral, a polysomnography should be performed and repeated annually until at least the age of 6 years. In the event of interim complaints indicating OSA, the polysomnography should be performed earlier.
Indication for treatment
The guiding principle is to prevent damage to the child's health and quality of life by identifying moderate or severe OSA at an early stage and proceeding to the most appropriate treatment.
The treatment to be applied
The guiding principle is to prevent damage to the child's health and quality of life as a result of untreated moderate or severe OSA and thus to proceed to the most appropriate treatment as soon as possible.
Recommendations
Screening for respiratory disorders
Refer children with syndromic craniosynostosis suspected of OSA to a specialized center for polysomnography screening.
Screen children with syndromic craniosynostosis annually by type 1 polysomnography at a center of expertise until at least the age of 6 years.
Carry out a type 1 polysomnography if the medical history shows complaints indicating respiratory disorders.
Perform an upper airway endoscopy to determine levels of obstruction if moderate or severe OSA
has been detected
Treatment of respiratory disorders
Start treatment of mild OSA if matching symptoms are present. Non-invasive or minimally invasive surgeries such as adenotonsillectomy are preferred.
Select treatment for OSA based on severity of OSA, age of patient, causal factors, feasibility of treatment and possibly other functional problems (such as increased ICP or exorbitism).
Consider Le Fort III surgery or monobloc advancement in children with syndromic craniosynostosis and severe respiratory OSA to treat the respiratory problems. If necessary, combine this procedure with a mandibula advancement.
Consider septum surgery from adulthood for additional improvement of nasal accessibility and OSA-related complaints.
Research Gaps
Screening for Respiratory Disorders
More studies on the anatomical levels of airway obstruction and its consequences for treatment are desirable.
Indication for Treatment
More studies on the specific effects of mild, moderate and severe OSA in syndromic craniosynostosis are desirable with respect to increased ICP and sleep quality.
The Treatment to be Applied
For all the different treatments, there is a virtual absence of studies presenting objective data before and after treatment, especially in the long term (more than 2 years follow-up).
Literature
-
1.
Kaditis AG, Alonso Alvarez ML, Boudewyns A, et al. ERS statement on obstructive sleep disordered breathing in 1- to 23-month-old children. Eur Respir J 2017; Dec 7;50:1-22
-
2.
Kaditis AG, Alonso Alvarez ML, Boudewyns A, Alexopoulos EI, Ersu R, Joosten K, Larramona H, Miano S, Narang I, Trang H, Tsaoussoglou M, et al. Obstructive sleep disordered breathing in 2- to 18-year-old children: diagnosis and management. Eur Respir J 2016;47:69-94
-
3.
Nash R, Possamai V, Manjaly J, et al. The management of obstructive sleep apnea in syndromic craniosynostosis. J Craniofac Surg 2015;26:1914–6
-
4.
Joosten KF, Larramona H, Miano S, et al. How do we recognize the child with OSAS? Pediatr Pulmonol 2017;52:260-271
-
5.
Bannink N, Mathijssen IMJ, Joosten KFM. Use of ambulatory polysomnography in children with syndromic craniosynostosis. J Craniofac Surg 2010;21:1365-8
-
6.
Al-Saleh S, Riekstins A, Forrest CR, et al. Sleep-related disordered breathing in children with syndromic craniosynostosis. J Cranio-Maxillo-Fac Surg 2011;39:153-7
-
7.
Driessen C, Joosten KF, Bannink N, et al. How does obstructive sleep apnoea evolve in syndromic craniosynostosis? A prospective cohort study. Arch Dis Child 2013;98:538-43
-
8.
Driessen C, Joosten KF, Florisson JM, et al. Sleep apnoea in syndromic craniosynostosis occurs independent of hindbrain herniation. Childs Nerv Syst 2013;29:289-96
-
9.
Driessen C, Mathijssen IM, De Groot MR, et al. Does central sleep apnea occur in children with syndromic craniosynostosis? Respir Physiol Neurobiol 2012;181:321-5
-
10.
Luna-Paredes C, Antón-Pacheco JL, García Hernández G, et al. Screening for symptoms of obstructive sleep apnea in children with severe craniofacial anomalies: assessment in a multidisciplinary unit. Int J Pediatr Otorhinolaryngol 2012;76:1767-70
-
11.
Inverso G, Brustowicz KA, Katz E, et al. The prevalence of obstructive sleep apnea in symptomatic patients with syndromic craniosynostosis. Int J Oral Maxillofac Surg 2016;45:167-9
-
12.
Zandieh SO, Padwa BL, Katz ES. Adenotonsillectomy for obstructive sleep apnea in children with syndromic craniosynostosis. Plast Reconstr Surg 2013;131:847-52
-
13.
Spruijt B, Joosten KF, Driessen C, et al. Algorithm for the management of intracranial hypertension in children with syndromic craniosynostosis. Plast Reconstr Surg 2015;136:331-40
-
14.
Spruijt B, Mathijssen IM, Bredero-Boelhouwer HH, et al. Sleep architecture linked to airway obstruction and intracranial hypertension in children with syndromic craniosynostosis. Plast Reconstr Surg 2016;138:1019e-29e
-
15.
Amonoo-Kuofi K, Phillips SP, Randhawa PS, et al. Adenotonsillectomy for sleep-disordered breathing in children with syndromic craniosynostosis. J Craniofac Surg 2009;20:1978-80
-
16.
Arnaud E, Marchac D, Renier D. Reduction of morbidity of the frontofacial monobloc advancement in children by the use of internal distraction. Plast Reconstr Surg 2007;120:1009-26
-
17.
Witherow H, Dunaway D, Evans R, et al. Functional outcomes in monobloc advancement by distraction using the rigid external distractor device. Plast Reconstr Surg 2008;121:1311-22
-
18.
Flores RL, Shetye PR, Zeitler D, et al. Airway changes following Le Fort III distraction osteogenesis for syndromic craniosynostosis: a clinical and cephalometric study. Plast Reconstr Surg 2009;124:590-601
-
19.
Fujimoto T, Imai K, Matsumoto H, et al. Tracheobronchial anomalies in syndromic craniosynostosis with 3-dimensional CT image and bronchoscopy. J Craniofac Surg 2011;22:1579-83
-
20.
Antón-Pacheco JL, Luna Paredes C, Martínez Gimeno A, et al. The role of bronchoscopy in the management of patients with severe craniofacial syndromes. J Pediatr Surg 2012;47:1512-5
-
21.
Doerga PN, Spruijt B, Mathijssen IM, et al. Upper airway endoscopy to optimize obstructive sleep apnea treatment in Apert and Crouzon syndromes. J Craniomaxillofac Surg 2016;44:191-6
CHAPTER 13 HEARING IMPAIRMENTS AND SPEECH/LANGUAGE DEVELOPMENT
Basic Questions
13.1 What is the policy on hearing impairments and speech/language development in craniosynostosis?
13.2 What is the indication for screening for speech-language development?
13.1 What is the policy on hearing impairments and speech/language development in craniosynostosis?
Introduction
There are several reasons why patients with craniosynostosis have hearing impairments and/or a delay in language/speech development. Hearing loss can be an additional cause of developmental delay in children who are already at increased risk.
Search and Selection
For the following specific question, original scientific studies or systematic reviews of original scientific studies have been included:
-
1.
What type of hearing loss occurs in patients with craniosynostosis and at what frequency?
In the Medline (OVID) and Embase databases, one overall search was conducted for studies on craniosynostosis. The search strategy is given in appendix 2, to the guideline. After deduplication, the literature search yielded 2732 hits.
Given the large number of studies, the chair of the working group first selected those that met the following general selection criteria:
General Selection and Exclusion Criteria
| Study type | -original studies -systematic reviews of sufficient quality: - research question of systematic review corresponds (largely) to the basic question - search is performed in at least 2 relevant databases, e.g. Cochrane Library, Medline/PubMed - reporting of the complete search strategy - no relevant keywords/search terms are missing |
| Follow-up period | -minimum follow-up period of 12 months for therapeutic or prognostic studies. |
| Exclusion criteria | -Case-reports -Expert opinion -Letters -Editorials -Case control studies for diagnostic tests -Narrative reviews |
The pre-selected studies that met the specific selection criteria listed in the Table below are included in the literature summary of this chapter.
Specific Selection and Exclusion Criteria
| Selection criteria for indication | - Minimum study size: 20 patients for patient series, where no multivariate analysis was used to identify prognostic factors for a relevant outcome measure. - Minimum study size: 35 patients for patient series with multivariate analysis of possible predictive variables for the effect - minimum number of participants of studies with a direct comparative design: 20 per study arm. |
Summary of the Literature
What type of hearing loss occurs in patients with craniosynostosis and at what frequency?
Since hearing loss is scarcely more common in children with single-suture non-syndromic craniosynostosis than the norm, this section is limited to multisuture and syndromic craniosynostosis.
In the systematic review of Agochukwu, the occurrence of hearing loss in 7 FGFR-related craniosynostosis syndromes was investigated.1 In Muenke syndrome, hearing loss was found in 61% of patients, in Apert syndrome in 80% of patients, in Pfeiffer syndrome in 92% and in Crouzon syndrome in 74% of patients. Usually it concerns conductive hearing losses, only in patients with Muenke syndrome there is a sensorineural hearing loss in the majority of cases and in patients with Crouzon's syndrome there is a (partial) sensorineural hearing loss in almost half of the cases. Information is lacking about how old patients were at the time of the hearing measurement, about the size of the measured hearing loss, uni- or bilaterality, about the type of hearing test that was done, and about the nature of the measured conductive loss. In addition, any interventions and their effects are not reported.
In the cross-sectional study by de Jong, the hearing was mapped of 132 children and young adults with Apert syndrome (n = 25), Crouzon syndrome (n = 42), Muenke syndrome (n = 29), Saethre-Chotzen syndrome (n = 21) and complex craniosynostosis (n = 15).2 Hearing tests that had already been carried out were requested and, in the absence of hearing tests, these were still carried out. The hearing of the best hearing ear was noted. Mild to moderate hearing losses were found in 44% of Apert syndrome patients, 28.5% of Crouzon syndrome patients, 62.1% of Muenke syndrome patients, 28.6% of Saethre-Chotzen syndrome patients and 6.7% of complex craniosynostosis patients. In the majority of cases there was conductive hearing loss, mostly caused by otitis media with effusion; only in the patients with Muenke syndrome the hearing losses were perceptive in nature. Treatment with middle ear aeration tubes and in some cases with hearing aids is discussed. Measuring only the hearing of the best hearing ear ignores the fact that being functionally one-eared also can be a handicap for speech/language development.
In the retrospective case study by Rosen, 29 patients with Saethre-Chotzen syndrome whose hearing tests were available were reported.3 Most patients had an abnormal hearing test somewhere in their childhood, but in the majority of these patients, in 71% of cases, the last hearing test was normal. Of the patients whose last hearing test was abnormal, 2 were younger than 3 years of age. Because otitis media with effusion appears to be the main cause of hearing loss in this group, there is a good chance that in these cases the hearing will return to normal later. The average age at which a hearing test was taken for the first time differed widely from 0.7 to 24.5 yrs, mean 6.7 yr. There is no description of which type of hearing test was done in which patient, and in the summary Table the characterization of the hearing loss does not always match the data from tympanometry.
In the prospective cohort study by Kruszka, hearing was tested in 106 patients with Muenke syndrome (genetically confirmed).4 In 70.8% of the patients a form of hearing loss was found. Most of these losses are sensorineural in nature. It is not clear from the summary Table how these numbers were arrived at; there is no mention of how the hearing was tested, nor of the size of the losses.
The definitions of degrees of hearing loss differ in the various studies, as do the frequencies measured when determining the pure tone average.
Conclusions
| Level 3 | Hearing loss should be considered for all children with syndromic craniosynostosis: for Muenke syndrome at 61–71%, for Apert syndrome at 44–80%, for Pfeiffer syndrome at 92%, for Crouzon syndrome at 29–74% and for Saethre-Chotzen syndrome at 29%. Children with multisuture craniosynostosis had a hearing loss of approximately 7%. Hearing loss in children with syndromic or multisuture craniosynostosis mainly is mild to moderate conductive hearing loss, with the exception of Muenke syndrome, in which mainly sensorineural hearing loss occurs. Agochukwu et al, 2014; de Jong et al, 2011; Rosen et al, 2011; Kruszka et al, 2016 |
Considerations
• Evidence of the Conclusions
Not applicable.
• Values and Preferences
According to the working group, parents will usually be sympathetic to a screening of the child's hearing, so that speech/language development has the best chances.
• Costs and Resources
The guideline committee expects that application of the recommendations leads to little or no increase in costs, because the recommendations are in line with existing practice.
• Professional Perspective
In most forms of craniosynostosis, initial routine screening at the child healthcare center, the neonatal hearing screening, is sufficient. Only in the case of Muenke syndrome is additional hearing screening recommended, because it is known that children with Muenke syndrome may have normal neonatal screening while later on there appears to be a sensorineural hearing loss. Due to the frequent occurrence of otitis media in all children with syndromic craniosynostosis, regular, preferably annual, monitoring of hearing is recommended, at least for the first 4 years of life and thereafter on indication. Depending on the child's age, hearing can be tested by means of otoacoustic emission measurement and tympanometry, and from about 4 years of age by means of tone radiometry and tympanometry. If these forms of audiometry are not feasible, hearing can be tested using BERA (brain stem audiometry) or free field studies. These tests can be done in an audiological center, in the treating center of expertise, or locally.
• Balance of Anticipated Desired and Undesired Outcomes
Failure or late detection of hearing loss can have a significant impact on language/speech development, while the hearing test is hardly burdensome for the child or parent.
Rationale of the recommendation(s)
The guiding principle for the recommendations is that screening and more extensive testing of hearing should be proportionate to the expected risks of hearing loss. These depend on the age of the patient and the type of craniosynostosis. A hearing test is therefore indicated annually for young children whose speech/language development is ongoing.
Recommendations
For children with craniosynostosis up to age 4 years
Neonatal hearing screening takes place as with all newborns. If necessary, further hearing tests are carried out in an audiological centre.
Otorhinolaryngologist performs annual otoscopy and age- and developmentally-appropriate audiometry.
For children with craniosynostosis from the age of 4 years (or slightly earlier depending on level of development and instructibility)
Screen hearing on indication by means of age and developmentally-appropriate tone audiometry.
Audiometry can be performed at the audiological centre of the treating craniofacial centre of expertise or at a local audiological centre. In the case of audiometry at a local audiological centre, the report is shared with the craniofacial centre of expertise.
Research Gaps
The available literature focuses mainly on the prevalence of hearing loss but provides little information on the severity of the hearing loss, on type of treatment and on treatment results.
Literature
-
1.
Agochukwu NB, Solomon BD, Muenke M. Hearing loss in syndromic craniosynostoses: otologic manifestations and clinical findings. Int J Pediatr Otorhinolaryngol 2014;78:2037-47
-
2.
De Jong T, Toll MS, de Gier HH, Mathijssen IM. Arch Otolaryngol Head Neck Surg 2011;137:775-8
-
3.
Rosen H, Andrews BT, Meara JG, Stoler JM, Mulliken JB, Rogers GF. Audiologic findings in Saethre-Chotzen syndrome. Plast Reconstr Surg 2011;127:2014-20
-
4.
Kruszka P, Addissie YA, Yarnell CM, Hadley DW, Guillen Sacoto MJ, Platte P, Paelecke Y, Collmann H, Snow N, Schweitzer T, Boyadjiev SA, Aravidis C, Hall SE, Mulliken JB, Roscioli T, Muenke M. Muenke syndrome: An international multicenter natural history study. Am J Med Genet A 2016;170A:918-29
13.2 What is the indication for screening for speech and language development?
Introduction
Several studies concluded that craniosynostosis was associated with a delay in cognitive development in early childhood and with an increased risk of learning and language problems at school age.1 However, most studies had serious limitations in design and implementation: no control group was used and the study size was very small. In these uncontrolled studies, the results of children with craniosynostosis were compared with test norms or prevalence rates of learning and language problems. Thus, norms or prevalence rates related to the general population. However, children diagnosed with craniosynostosis may differ in a number of respects from children in whom the test norms were developed or the prevalence of learning and language problems was measured, for example in socio-economic background.
Studies with a direct comparative design, in which the control group matches the group of patients with craniosynostosis in a number of aspects (age, sex, socio-economic background, intelligence quotient of the mother), give a more reliable picture of possible differences in speech and language development. Therefore, only studies with a direct comparative design have been included for this review.
Search and Selection
To be able to answer the basic question, the working group carried out a systematic review with the following PICO question:
-
1.
Do children, adolescents and adults with single-suture or multisuture craniosynostosis, or with syndromic craniosynostosis, have an increased risk of speech and language problems compared to children without craniosynostosis?
Selection and Exclusion Criteria
| Type or studies | - SRs of good quality in terms of design and execution |
| - observational studies with a direct comparative design (patient control study; cohort study with control group) | |
| Type of patients | - adults, adolescents and children with single-suture or syndromic craniosynostosis and adults, adolescents and children without craniosynostosis |
| Types of outcome measures | - Preschool Language Scale, Third Edition (PLS-3) (AC: Auditory Comprehension. EC: Expressive Communication), |
| - Test of Word Reading Efficiency (TOWRE), | |
| - Wide Range Achievement, Test, Fourth Edition (WRAT-4), | |
| - Comprehensive Test of Phonological Processing (CTOPP), | |
| - A Developmental Neuropsychological Assessment (NEPSY-II), | |
| - Children's Communication Checklist-2 (CCC-2) (Parent, Teacher), | |
| - Token Test for Children, Second Edition (Token Test-II); | |
| - Wechsler Intelligence Scale for Children-IV (WISC-IV verbal comprehension). | |
| Types of setting | - secondary care, tertiary care |
| Exclusion criteria | - editorials |
| - narrative reviews | |
| - observational studies without direct comparative design |
The literature search yielded 133 hits. The above selection and exclusion criteria were used for the selection (appendix 3). Eight studies were selected on the basis of title and abstract.2–9 After reading of the full articles, five of these studies were finally included in the literature analysis.2,3,6,8,9 Appendix 3, shows the reasons for exclusion of the other three studies.
Summary of the Literature
Description of Studies
The five studies included are prospective cohort studies with a control group of children without craniofacial anomaly.2,3,6,8,9 The controls were matched on sex, age, ethnicity and family socioeconomic status. The analysis always examined whether differences in IQ of the mothers between the cohort of children with craniosynostosis and the controls influenced the study results. The five studies are multicenter studies, with partly the same cohorts and control groups, and all come from the same research group in the US. These studies all concerned children with single-suture, non-syndromic craniosynostosis. No direct comparative studies were found on adolescents and adults with single-suture, non-syndromic craniosynostosis. With respect to multisuture or syndromic craniosynostosis, no studies with a direct comparative design were found.
Study Characteristics
The studies examined patients with sagittal suture synostosis, metopic suture synostosis, unilateral coronal suture synostosis, or unilateral lambdoid suture synostosis. The diagnosis had been confirmed by a CT scan. The children's mean age in the five studies ranged from 6 months to over 7 years, and about 60% of the children were boys (see Tables 2a and 2b). The PLS-3 tests had been administered by trained psychological staff. The other outcome measures of speech and language development had been scored by a second psychological staff member on the basis of video recordings (i.e. independently).
TABLE 2a. Inclusion and Exclusion Criteria of Studies
| Inclusion criteria | Exclusion criteria | |
| Starr 2007,220126 Speltz 2007,320158 Kapp-Simon 20169 | Cohort: children who: 1) had sagittal suture synostosis, metopic suture synostosis, unilateral coronal suture synostosis and unilateral lambdoid suture synostosis confirmed by a CT scan; 2) had not yet undergone a surgical correction; 3) were not more than 30 months old Control: - children without craniofacial anomaly | Cohort and control: 1) Premature birth (< 34 weeks gestational age); 2) presence of serious medical or neurological conditions (for example: (cardiac defects, epileptic disorder, conditions that require surgical correction 3) presence of three or more extracranial mild malformations as defined by Leppig and co-authors, or 4) presence of serious malformations |
TABLE 2b. Other Study Characteristics
| Starr 20072 | Type of craniosynostosis: • sagittal suture: n = 86 • metopic suture: n = 35 • unilateral coronal suture: n = 36 • unilateral lambdoid suture: n = 11 Control: n = 115 Median age: 18.4 months (cohort and control) Sex (%female): 36–41% (cohort-control) Ethnicity (%white): 73–81% (cohort-control) |
| Speltz 20073 | Type of craniosynostosis: • sagittal suture: n = 62 • metopic suture: n = 27 • unilateral coronal suture: n = 28 • unilateral lambdoid suture: n = 8 Control: n = 125 Mean age: 6.5–6.6 months (cohort-control) Sex (%female): 39% (cohort and control) Ethnicity (%white): 75–78% (cohort-control) |
| Starr 20126 | Type of craniosynostosis: • sagittal suture: n = 94 • metopic suture: n = 50 • unilateral coronal suture: n = 52 • unilateral lambdoid suture: n = 10 Control: n = 222 Median age: 36 months (cohort and control) Sex (%female): 34.9–37.4% (cohort-control) Ethnicity (%white): 76.6–71.2% (cohort-control) |
| Speltz 20158 | Type of craniosynostosis: • sagittal suture: n = 76 • metopic suture: n = 48 • unilateral coronal suture: n = 46 • unilateral lambdoid suture: n = 12 Control: n = 183 Mean age: 7.5–7.4 years (cohort-control) Sex (%female): 38–37% (cohort-control) Ethnicity (%white): 74–80% (cohort-control) |
| Kapp-Simon 20169 | Type of craniosynostosis: • sagittal suture: n = 76 • metopic suture: = 48 • unilateral coronal suture: n = 46 • unilateral lambdoid suture: n = 12 Control: n = 183 Mean age: 7.5–7.4 years (cohort-control) Sex (%female): 37% (cohort and control) Ethnicity (%white): 74–79% (cohort-control) |
Differences in Speech and Language Development between Children with Craniosynostosis and Children without Craniofacial Anomaly
The differences in speech and language development between, on the one hand, the cohort of children with single-suture, non-syndromic craniosynostosis and, on the other hand, the control group of children without craniofacial anomaly, vary from small to moderate (Table 3). The effect sizes vary from 0 to 0.4. Of the 16 effect sizes, 9 are ≤0.20, 5 are ≥0.21 and ≤0.30 and 2 are ≥0.31 and ≤ 0.41. Effect sizes ≤ 0.20 are considered a small effect, and effect sizes ≤ 0.5 are considered a small to moderate effect.10
Effect of an Intervention Measured as Covariate
Differences in speech and language development between the four single-suture forms of craniosynostosis Speech and language development appear to be particularly lagging behind in children with unilateral coronal and lambdoid suture synostosis, while children with sagittal suture synostosis (aged 7 years) score similarly to children without craniofacial anomaly (Table 4). Children with metopic suture synostosis seem to be slightly behind in speech and language (Table 4).
Effect of Surgical Correction of Single-suture Craniosynostosis
Starr reported differences in mean standard scores with the control group of -0.43 (95% BI -0.74;- 0.12) and 0.03 (95% BI -0.36;0.43), respectively, for the PLS-AC and PLS-EC measured before surgery.2 After surgery, when children were on average 18 months old, the differences with the control group were -0.53 (95% BI -1.18;0.13) and -0.62 (95% BI -1.30; 0.05), respectively.
TABLE 3. Outcomes for all Types of Single-suture Non-syndromic Craniosynostoses Taken Together
| Outcome measure | Cohort with Single-suture Craniosynostosis (A) | Control without Craniofacial Anomaly (B) | Difference: A-B (95% BI)I | Effect size |
| Age: 6–7 mos | ||||
| PLS 3-AC (norm: average 100; SD 15) | 91.97 | 94.46 | −2.50 (−5.19; 0.19) | −0.20 |
| PLS 3-EC (norm: average 100; SD 15) | 96.65 | 97.31 | −0.65 (−3.92; 2.60) | −0.05 |
| Age: 18 mos | ||||
| PLS 3-AC (scale not indicated; raw scores) | 13.62∗ | 14.21∗ | −0.71 (−1.37; -0.04)§ | −0.28II |
| PLS 3-EC | 13.11∗ | 13.59∗ | −0.63 (−1.30; 0.05)§ | −0.25II |
| Age: 36 months | ||||
| PLS 3-AC (norm: average 100; SD 15) | 97.6 | 106.5 | −5.9 (−8.8;−3.1)§ | −0.41II |
| PLS 3-EC (norm: average 100; SD 15) | 94.5 | 101.4 | −3.8 (−6.3;−1.3)§ | −0.30II |
| Age: 7 years | ||||
| WRAT-4 (reading) (norm: average 100; SD 15) | 105.4 | 109.3 | −2.0 (−5.2;1.2)∗∗ | −0.13II |
| WRAT-4 (spelling) (norm: average 100; SD 15) | 105.2 | 107.2 | −0.9 (−3.8;2.0)∗∗ | −0.07II |
| TOWRE (norm: average 100; SD 15) | 104.0 | 106.6 | −0.9 (−3.9;2.1)∗∗ | −0.06II |
| CTOPP (phonological awareness) (norm: average 100; SD 15) | 107.1 | 111.1 | −2.5 (−5.4;0.4)∗∗ | −0.18II |
| CTOPP (rapid naming) (norm: average 100; SD 15) | 100.3 | 101.0 | 0.3 (−2.1;2.7)∗∗ | 0.03II |
| NEPSY-II (semantic) (standaard score 10; SD 3) | 10.8 | 12.0 | −0.8 (−1.5;−0.2)∗∗ | −0.26II |
| Tokentest-II (norm: average 100; SD 15) | 101.9 | 104.8 | −1.1 (−3.5;1.4)∗∗ | −0.10II |
| CCC-2 Parent (norm: average 100; SD 15) | 102.7 | 104.7 | −0.4 (−3.2;2.4)∗∗ | −0.03II |
| CCC-2 Teacher (norm: average 100; SD 15) | 104.3 | 107.9 | −3.2 (−6.8;0.5)∗∗ | −0.23II |
| WISC-IV verbal comprehension (norm: average 100; SD 15) | 101.6 | 110.0 | −5.3 (−8.4;−2.1)∗∗ | −0.36II |
AC, Auditory Comprehension; CCC-2, Children's Communication Checklist-2, CTOPP, Comprehensive Test of Phonological Processing, EC, Expressive Communication; NEPSY-II, A Developmental Neuropsychological Assessment, PLS-3, Preschool Language Scale, Third Edition; Token Test-II, Token Test for Children, Second Edition; TOWRE, Test of Word Reading Efficiency, WISC-IV, Wechsler Intelligence Scale for Children-IV; WRAT-4, Wide Range Achievement, Test, Fourth Edition.
Notes:.
I. In a number of cases, these differences were corrected for confounding variables by linear or logistic regression. As a result, the differences are not always equal to the difference of the averages given in columns A and B.
II. The calculation was carried out by the working group as follows. The size of the 95% confidence interval was divided by 4 to obtain the standard error of the difference. This standard error is equal to the root of (2∗MSE/n); and represents the number of observations over which the difference has been calculated. MSE represents the estimated variance of the difference. The standard error of the difference is the square root of the variance. The effect size is equal to the difference divided by the standard deviation.
∗rough scores. ∗∗: corrected by logistic or linear regression for age, sex, SES, IQ of the mother. § Corrected using logistic or linear regression for age, SES; ethnicity; maternal IQ, center where children were recruited. Bold are the confidence intervals that exclude a difference of zero, i.e. where the difference was statistically significant.
TABLE 4. Results for Speech and Language Development by Type of Single-suture, Non-syndromic Craniosynostosis
| Outcome measure | Sagittal Suture | Metopic Suture | Unilateral Coronal Suture | Unilateral Lambdoid Suture |
| Age: 6–7 months | ||||
| PLS 3-AC (norm: average 100; SD 15) | 93.65 | 90.37 | 89.06 (right) 92.67 (links) | 89.50 |
| PLS 3-EC (norm: average 100; SD 15) | 97.39 | 97.78 | 90.00 (left) 102.67 (left) | 91.50 |
| Age: 18 months | ||||
| PLS 3-AC∗∗ (scale not indicated; raw scores) | 14.07 | 13.40 | 13.25 (right) 13.01 (left) | 12.71 |
| PLS 3-EC∗∗ (scale not indicated; raw scores) | 13.35 | 12.94 | 13.06 (right) 13.11 (left) | 11.66 |
| Age: 36 months (difference compared to reference category) [effect size] | ||||
| PLS 3-AC (norm: average 100; SD 15)§∗∗ | reference category | −0.9 (−5.8;4.0) [−0.07]I | −5.9 (−11.5;−0.3) [−0.42]I | −10.8 (−22.4;0.8) [−0.83]I |
| PLS 3-EC (norm: average 100; SD 15)§∗∗ | reference category | −1.5 (−6.2;3.2) [−0.13]I | −5.1 (−9.8;−0.4) [−0.43]I | −8.5 (−17.5;0.5) [−0.84]I |
| Age: 7 years (difference compared to reference category) [effect size] | ||||
| WRAT-4 (reading) (norm: average 100; SD 15)§∗ | reference category | −3.0 (−8.4;2.4) [−0.23]I | −11.7 (−16.8,−6.7) [−0.98]I | −14.8 (−25.8;−3.7) [−1.09]I |
| WRAT-4 (spelling) (norm: average 100; SD 15)§∗ | reference category | −2.9 (−7.9;2.2) [−0.23]I | −10.1 (−15.4;−4.8) [−0.80]I | −10.8 (−22.2;0.6) [−0.77]I |
| TOWRE§∗ | reference category | −2.7 (−7.9;2.4) [−0.18]I | −10.0 (−15.2;−4.7) [−0.81]I | −10.5 (−23.1;2.0) [−0.68]I |
| CTOPP (phonological awareness) (norm: average 100; SD 15)§∗ | reference category | −1.5 (−7.0;4.0) [−0.11]I | −4.7 (−9.9;0.6) [−0.38]I | −11.6 (−21.2;−1.9) [−0.98]I |
| CTOPP (rapid naming) (norm: average 100; SD 15)§∗ | reference category | 1.0 (−3.5;5.5) [0.09]I | −1.5 (−6.2;3.1) [−0.14]I | 1.4 (−10.2;12.9) [0.11]I |
| Age: 7 jaar (verschil ten opzichte van controlegroep [effectgrootte] | ||||
| NEPSY-II (semantic) (standard score 10; SD 3)§§∗ | −0.6 (−1.5;0.3) [−0.22]I | −0.8 (−1.7;−0.05) [−0.41]I | −1.2 (−2.3;−0.1) [−0.46]I | −0.9 (−3.5;1.7) [−0.28]I |
| Tokentest (norm: average 100; SD 15)-II§§∗ | 0.09 (−2.8;3.0) [0.01]I | −0.3 (−4.9;4.2) [−0.03]I | −3.7 (−7.4;0.1) [−0.42]I | −1.6 (−10.1;7.0) [−0.37]I |
| CCC-2 Parent (norm: average 100; SD 15)§§∗ | 1.8 (−1.8;5.3) [0.01]I | −2.3 (−6.6;2.0) [−0.24]I | −2.5 (−7.2;2.2) [−0.24]I | −0.2 (−7.9;7.6) [−0.02]I |
| CCC-2 Teacher (norm: average 100; SD 15)§§∗ | 0.2 (−4.1;4.3) [0.02]I | −5.4 (−11.1;0.4) [−0.49]I | −6.1 (−14.0;1.9) [−0.42]I | −6.5 (−14.4;1.4) [−0.74]I |
| WISC-IV verbal comprehension (norm: average 100; SD 15)§§∗ | −2.3 (−5.7;1.1) [−0.22]I | −3.6 (−8.6;1.4) [−0.30]I | −11.5 (−16.9; −6.2) [−0.90]I | −4.5 (−18.1;9.0) [-0.27]I |
see Table 3.
Notes:.
I. See note 2 to Table 3.
§ differences with respect to sagittal suture synostosis; §§ differences with respect to control group; ∗adjusted for mother's age, sex, SES and IQ. ∗∗adjusted for age, SES; ethnicity; IQ of mother, center where children were recruited.
Quality of evidence.
To assess the quality of evidence for the different outcome measures, a distinction has been made between comparing:
-
-
the outcomes of children with single-suture craniosynostosis versus those of children without craniofacial anomaly, and
-
-
the results of children with sagittal suture synostosis versus those of children with metopic suture synostosis versus those of children with unilateral coronal suture synostosis versus those of children with unilateral lambdoid suture synostosis (subgroup analysis).
A) To Compare Outcomes of Children with Craniosynostosis and Children without Craniofacial Anomaly
PLS-3 (AC and EC)
For the PLS-AC and PLS-EC in children aged 6 to 18 months, there was a serious risk of bias in the results, mainly due to doubtful validity and reliability of the PLS-3 for this age group (Appendix 3).2,3 In addition, all PLS scores, except the PLS-AC of children aged 18 months, were downgraded by one level due to inaccuracy: the 95% confidence intervals also include a difference of zero.
The PLS-AC and PLS-EC in children aged 36 months were downgraded for restrictions in design and execution due to not blinding of outcome evaluator (appendix 3).6
WRAT-4 (reading), WRAT-4 (spelling), TOWRE, CTOPP (phonological awareness), CTOPP (rapid naming)
The study by Speltz mentioned a serious risk of distortion of the results due to the high drop-out rate (appendix 3).8 In addition, all outcome measures were downgraded for inaccuracy: the 95% confidence intervals also include a difference of zero.
NEPSY-II (semantic), tokentest-II, CCC-2 parent, CCC-2 teacher, WISC-IV verbal com-prehension
In the study by Kapp-Simon, there was a serious risk of distortion of the results due to the high drop-out rate (Appendix 3).9 In addition, all outcome measures except NEPSY-II (semantic) and WISC-IV verbal comprehension were downgraded for inaccuracy: the 95% confidence intervals also include a difference of zero.
B) For Subgroup Analyses (Differences between Single-suture Types of Craniosynostoses)
PLS-3 (AC and EC), WRAT-4 (read), WRAT-4 (games), TOWRE, CTOPP (phonological awareness), CTOPP (rapid naming)
For all these outcome measures, the quality of evidence is low. The studies did not report the percentages of dropouts by type of single-suture craniosynostosis. Therefore, it is not possible to determine whether there was selective dropout. To what extent this has influenced the matching on sex, age, etc. cannot be determined either. In addition, the results are often inaccurate: many confidence intervals include a difference of zero.
Conclusions
| PLS 3-AC, PLS 3-EC | |
| Moderate to low GRADE | Children with single-suture craniosynostosis aged 6 to 18 months appear to have a slightly increased risk of speech and language problems compared to children without a craniofacial anomaly. Sources: Starr et al, 2007; Speltz et al, 2007 |
| PLS 3-AC, PLS 3-EC | |
| Low GRADE | Children with sagittal suture synostosis at the age of 6 to 18 months appear to have little or no increased risk of speech and language problems compared to children without craniofacial anomaly. Children with metopic suture, lambdoid suture or coronal suture synostosis aged 6 to 18 months appear to have a slightly increased risk of speech and language problems compared to children without craniofacial anomaly. Sources: Starr et al, 2007; Speltz et al, 2007 |
| PLS 3-AC, PLS 3-EC | |
| Moderate GRADE | Children with single-suture craniosynostosis at the age of 36 months probably have a slightly to moderately increased risk of speech and language problems compared to children without a craniofacial anomaly. Source: Starr et al, 2012 |
| PLS 3-AC, PLS 3-EC | |
| Low GRADE | Children with metopic suture synostosis at the age of 36 months may not have an increased risk of speech and language problems compared to children with sagittal suture synostosis. Children with unilateral coronal or lambdoid suture synostosis may have an increased risk of speech and language problems compared to children with sagittal suture synostosis. Source: Starr et al, 2012 |
| WRAT-4 (reading), WRAT-4 (spelling), TOWRE, CTOPP (phonological awareness), CTOPP (rapid naming) | |
| Low GRADE | Children with single-suture craniosynostosis at the age of 7 years appear to have a little or no increased risk of speech and language problems compared to children without a craniofacial anomaly. Source: Speltz et al, 2015 |
| NEPSY-II (semantic), Tokentest-II, CCC-2 Parent, CCC-2 Teacher, WISC-IV verbal comprehension | |
| Moderate to low GRADE | Children with single-suture craniosynostosis at the age of 7 years are likely to have a slightly increased risk of speech and language problems compared to children without craniofacial anomaly. Source: Kapp-Simon et al, 2016 |
| WRAT-4 (reading), WRAT-4 (spelling), TOWRE, CTOPP (phonological awareness), CTOPP (rapid naming) | |
| Low GRADE | Children with metopic suture synostosis at the age of 7 years may have a slightly higher risk of speech and language problems than children of the same age with sagittal suture synostosis. Children with unilateral lambdoid or coronal suture synostosis at the age of 7 years may have a moderately increased risk of speech and language problems compared to children of the same age with sagittal suture synostosis. Source: Speltz et al, 2015 |
| NEPSY-II (semantic), Tokentest-II, CCC-2 Parent, CCC-2 Teacher, WISC-IV verbal comprehension | |
| Low GRADE | Children with sagittal suture synostosis at the age of 7 do not seem to have an increased risk of speech and language problems compared to children without craniofacial anomaly. Children with metopic suture, lambdoid suture or coronal suture synostosis at the age of 7 years appear to have a slightly to moderately increased risk of speech and language problems compared to children without craniofacial anomaly. Source: Kapp-Simon et al, 2016 |
Considerations
• Quality of Evidence
The quality of evidence varied from low to moderate for the outcome measures discussed above.
• Values and Preferences
From the perspective of parents/carers and patients, there are probably no essential differences in the relevance of outcome measures for the different aspects of speech and language development.
• Professional Perspective
With regard to single-suture craniosynostosis
Based on the results of the literature review, a distinction is made between children with sagittal or metopic suture synostosis and children with coronal or lambdoid suture synostosis. Children with one of the latter two types of single-suture craniosynostosis have a higher risk of (written) speech and language problems from 36 months onwards.
Critical moments in speech and language development occur around the age of 5 years, and between 6 and 8 years. Phonological awareness should start around the age of 5 years. At the age of 6 to 8 years the reading and spelling process should start.
With Regard to Multisuture Craniosynostosis
No studies on children with multisuture craniosynostosis were found that met the selection criteria. These children form a heterogeneous group. This is mainly due to the variation in the sutures that are closed, and whether additional congenital abnormalities are visible. As far as the sutures are concerned, the coronal and/or lambdoid sutures often have an impact. If no additional congenital abnormalities are visible, these children can probably be compared to children with single-suture craniosynostosis in which the coronal or lambdoid suture is involved. If additional congenital abnormalities are visible, these children are more likely to be considered children with syndromic craniosynostosis as regards speech and language problems.
With Regard to Syndromic Craniosynostosis
Speech and language development are related to hearing, among other things, and for this reason children with syndromic craniosynostosis have an increased risk of speech and language problems. For example, Muenke syndrome gives a specific sensorineural hearing loss, which is an additional risk factor. Children with Apert syndrome may have a cleft palate, which increases the risk of hearing problems and speech development. They often also have intraoral abnormalities such as thickened gingival puffiness, palatal mucosa and a gothic shape of the palate, which can contribute to speech problems. All syndromic forms have a higher risk of a narrow upper jaw with a gothic shape of the palate. There may also be a reduced muscle tone, for example, which leads to the tendency to push the tongue always between the front teeth, resulting in an open bite and abnormal tongue positioning in speech.
Children with Saethre-Chotzen syndrome often have variable hearing under the influence of otitis media with effusion. They have narrowed ear canals, which can make assessment of the eardrum or placement of eardrum tubes more difficult. In addition, they have a risk of lower intelligence, which also increases the risk of speech and language problems.
• Balance of Desired and Undesired Effects
With regard to single-suture craniosynostosis (sagittal suture or metopic suture synostosis)
These children probably have a small additional risk of speech and language problems. Speech and language testing are therefore best carried out on indication, i.e. when there are concerns among parents/carers and/or care professionals. Prior to any speech and language testing, it is recommended to first use a validated screening instrument, i.e. the SNEL questionnaire by Luinge, Post, & Goorhuis-Brouwer, 2007.11 The choice of one validated screening instrument is based on the better possibility of comparing the results of the different centers.
The speech therapist in the craniosynostosis expert team will initiate this diagnostic procedure in consultation with the other team members.
With Regard to Single-suture Craniosynostosis (Coronal or Lambdoid Suture Synostosis) and Multisuture Craniosynostosis (without Additional Congenital Abnormalities)
These children are likely to have an increased risk of speech and language problems from the age of 36 months. From this age, screening using the FAST is useful. If case of failure on this screening, additional speech therapy by a speech therapist from a craniosynostosis expert team is indicated. In view of the increased risk with regard to (written) language skills (phonological awareness and initial reading and spelling process), supervision is advised. For the children in primary school second grade and fourth grade, it is advised to bring a printout of the school tracking system to the consultation with the craniosynostosis assessment team. In case of problems with (initial) reading and spelling, speech and language testing is indicated, followed, if necessary, by a psychological examination.
With Regard to Syndromic Craniosynostosis
From the moment of referral to the center of expertise, these children are actively followed by an otorhinolaryngologist and speech therapist because of an increased risk of hearing problems and the (partly as a result of this) increased risk of speech and language development problems.
However, cognitive limitations are one of the possible causes. Other causes for poor speech-language development include cleft palate, impairment of the auditory chain, more frequent occurrence of otitides, and intraoral abnormalities. These require speech therapy and medical intervention.
• Costs and Resources
The recommendations are expected to lead to a slight increase in care costs: children with multisuture or syndromic craniosynostosis (nationwide approximately 25 new children per year, 10 of whom are multisuture and 14 syndromic) are currently not subject to periodic speech and language testing. Based on three longitudinal speech and language testing moments per child (2 years, 5 years and 8 years) within the craniosynostosis expertise team and € 440 per test, this would mean an estimated increase of 75 ∗ € 440, or € 33,000.
Rationale of the recommendation(s)
A guiding principle for the recommendations is that screening and speech and language development testing should be in proportion to the expected risks of speech and language development problems. These depend on the type of craniosynostosis. In the case of a low risk of speech and language development problems, such as in children with sagittal suture or metopic suture synostosis, screening is only recommended if there are concerns among parents/carers and/or care professionals. In children with coronal or lambdoid suture synostosis or multisuture synostosis who do have an increased risk of speech and language problems, screening is more appropriate from the age of 36 months. In the case of a greatly increased risk, such as for children with syndromic craniosynostosis, speech and language testing from the age of 36 months onwards is more appropriate.
Recommendations
Single-suture craniosynostosis (sagittal suture or metopic suture synostosis)
If parents or healthcare professionals have concerns about speech and language development, ask parents/carers to fill in the validated screening instrument FAST.4
In the event of failure on of this screening, perform an additional speech and language test (preferably within the craniosynostosis expertise team).
Single-suture craniosynostosis (coronal suture synostosis and lambdoid suture synostosis) and multisuture craniosynostosis
Younger than 36 months:
If parents or care professionals have concerns about the child's speech and language development, ask parents/carers to fill in the validated screening instrument FAST.
In the event of failure of this screening, perform an additional speech and language test.
From 36 months:
Ask parents/carers to fill in the validated screening instrument FAST. In the event of failure on this screening, perform an additional speech and language test.
5–6 years of age:
Ask parents/carers of children in primary school grade 2 to send or bring a printout of the school tracking system to the craniosynostosis expert team in view of the increased risk of problems with regard to phonological awareness.
In the event of failure in the school tracking system with regard to reading and spelling, perform additional speech and language tests and, in the event of suspicion of a sustaining role of neuropsychological factors (attention, IQ), also perform neuropsychological tests.
7–8 years of age:
Ask parents/carers of children in primary school grade 4 to send or bring a printout of the school tracking system to the craniosynostosis expert team in view of the increased risk of problems with reading and spelling.
In the event of failure in the school tracking system with regard to reading and spelling, perform additional speech and language tests and, in the event of suspicion of a sustaining role of neuropsychological factors (attention, IQ), also perform neuropsychological tests.
Syndromic craniosynostosis
Perform, from referral to the craniosynostosis expert team onwards, periodic∗ speech and language development tests.
In case of suspicion of (neuro-)psychological factors that sustain the identified speech and language problems, also perform a (neuro-)psychological examination.
∗frequency at least conform that in children with unisutural craniosynostosis
Knowledge Gap
Studies conducted in the Netherlands on speech and language development in children with craniosynostosis in which a direct comparative design and measurement instruments validated for the Netherlands were applied, are lacking.
Literature
-
1.
Speltz ML, Kapp-Simon KA, Cunningham M, et al. Single-suture craniosynostosis: A review of neurobehavioral research and theory. J Pediatr Psychol 2004;29:651-68
-
2.
Starr JR, Kapp-Simon KA, Cloonan YK, et al. Presurgical and postsurgical assessment of the neurodevelopment of infants with single-suture craniosynostosis: comparison with controls. J Neurosurg 2007;107(2 Suppl):103-10
-
3.
Speltz ML, Kapp-Simon K, Collett B, et al. Neurodevelopment of infants with single-suture craniosynostosis: presurgery comparisons with case-matched controls. Plast Reconstr Surg 2007;119:1874-81
-
4.
Toth K, Collett B, Kapp-Simon KA, et al. Memory and response inhibition in young children with single-suture craniosynostosis. Child Neuropsychol 2008;14:339-52
-
5.
Knudsen E, Maltese G, Tarnow P, et al. Parental estimation of early psychological development in children operated on for single suture synostosis. J Plast Surg Hand Surg 2012;46(3-4):152-4
-
6.
Starr JR, Collett BR, Gaither R, et al. Multicenter study of neurodevelopment in 3-year-old children with and without single-suture craniosynostosis. Arch Pediatr Adolesc Med 2012;166:536-42
-
7.
Knight SJ, Anderson VA, Spencer-Smith MM, et al. Neurodevelopmental outcomes in infants and children with single-suture craniosynostosis: a systematic review. Dev Neuropsychol 2014;39:159-86
-
8.
Speltz ML, Collett BR, Wallace ER, et al. Intellectual and academic functioning of school-age children with single-suture craniosynostosis. Pediatrics 2015;135:e615-23
-
9.
Kapp-Simon KA, Wallace E, Collett BR, et al. Language, learning, and memory in children with and without single-suture craniosynostosis. J Neurosurg Pediatr 2016;17:578-88
-
10.
Cohen J. Statistical power analysis for the behavioral sciences (2nd ed.). Hillsdale, NJ: Lawrence Earlbaum Associates, 1988
-
11.
Luinge MR, Post WJ, Goorhuis-Brouwer SM. Het identificeren van mijlpalen in de taalontwikkeling van kinderen van 1 tot 6 jaar. Stem-, spraak- en taalpathologie. (Dutch) 2007;15, 33-52
CHAPTER 14 DENTOFACIAL ABNORMALITIES
14.1 What is the policy on orthodontic care for syndromic craniosynostosis?
Introduction
Dentofacial abnormalities are a characteristic part of syndromic craniosynostosis. Typical for orthodontic intervention are multiple phases in which each treatment has specific objectives. Orthodontic interventions must always take into account that the final correction must be matched with a future jaw surgical correction.
Search and Selection
The relevant item is:
Which dentofacial abnormalities occur in patients with syndromic craniosynostosis and in what frequency?
In the Medline (OVID) and Embase databases, one overall search was conducted for studies on craniosynostosis. The search strategy is given in appendix 2, to the guideline. After deduplication, the literature search yielded 2732 hits.
Given the large number of studies, the chair of the working group first selected those that met the following general selection criteria:
General Selection and Exclusion Criteria
| Study type | -original studies -systematic reviews of sufficient quality: - research question of systematic review corresponds (largely) to the basic question - search is performed in at least 2 relevant databases, e.g. Cochrane Library, Medline/PubMed - reporting of the complete search strategy - no relevant keywords/search terms are missing |
| Follow-up period | -minimum follow-up period of 12 months for therapeutic or prognostic studies. |
| Exclusion criteria | -Case-reports -Expert opinion -Letters -Editorials -Case control studies for diagnostic tests -Narrative reviews |
The pre-selected studies that met the specific selection criteria listed in the Table below are included in the literature summary of this chapter.
Specific Selection and Exclusion Criteria
| -minimum study size: 20 patients |
Summary of the Literature
Skull Growth
Abnormal skull growth is characteristic and is caused by the premature fusing of skull sutures. It has been shown that the spheno-occipital synchondrosis (SOS) merges significantly earlier in 38 patients with Apert's syndrome compared to 38 patients with age- and sex-appropriate controls.1 Also in patients with Crouzon syndrome (n = 30), the SOS merged earlier compared to controls (n = 112).2 in a retrospective case-control study of 30 FGFR2-mutation patients with Crouzon syndrome compared to 235 controls, Coll found that in addition to the SOS, all other skull base synchondroses also merge prematurely.3
Maxillary Growth
As a result of disturbed growth in the neurocranium, basicranium and middle face, the maxillary growth is abnormal in vertical, transversal and sagittal directions. Kreiborg showed (26 patients with Apert syndrome, 153 adult control patients) that the maxilla is underdeveloped in all dimensions, leading to a marked shortening of the upper facial height.4 At the same time, the width of the nasopharynx, and height and depth of the nasopharyngeal airway are clearly smaller.
Goldstein describes a disturbed growth of the basicranium in a retrospective case-control study (54 syndromic craniosynostosis patients, 206 control patients matched on age and sex) in which a premature fusion of the SOS was found and a significant positive correlation with an underdevelopment of the mid-face.5 Tahiri also shows how an accelerated fusion of the SOS is associated with hypoplasia of the maxilla.2 in 30 patients with Crouzon syndrome compared to 112 age and sex-matched controls. Reitsma demonstrated in 25 patients with Apert syndrome and 27 patients with Crouzon syndrome a significant difference in growth in both sagittal and vertical direction compared to 482 controls.6 However, maxillary growth in syndromic patients is more limited in sagittal than in vertical direction. Presumably, the lack of sutural growth of the maxilla and an abnormal remodelling pattern of the bone result in underdevelopment of the maxilla.6
Mandibular Growth
Not only the maxilla but also the mandibula is affected by the premature fusion of the cranial sutures. Two retrospective case-control studies (n1 = 26 Apert patients, 153 controls; n2 = 37 Crouzon patients, 25 Apert patients and 482 controls) showed that the mandibula in syndromic patients shows normal sagittal growth but is accompanied by an anterior rotation compared to control patients.4,6 This anterior rotation leads to a mesial malocclusion. In addition, a fluctuating asymmetric growth of the mandibula is statistically significantly more often reported in 35 Crouzon and 24 Apert patients compared to 327 controls, indicating instability during development.7
Integrity, Shape and Width of the Palate
A study of 136 Apert patients documented a soft palate cleft or a bifid uvula in 75% of these patients.8 Another study in 21 patients with Muenke showed a low cleft incidence of 5% and found a characteristic high-arched palate in 67% of these patients.9 Reitsma showed a palatal constriction (high-arched palate) in 28 Apert and 40 Crouzon patients as compared to 457 control patients, in addition to hypoplasia of the maxilla.10 The dental arch did not widen during growth, and furthermore, the abnormal growth pattern was more pronounced in Apert than in Crouzon patients. Large lateral gingival swellings were also found in these patients, which increased with age.8
Malocclusion
Cohen and Kreiborg studied 136 Apert patients (no control group) and described a class III malocclusion (68%), accompanied by an anterior cross bite (81%), anterior open bite (73%), bilateral posterior cross bite (63%), unilateral cross bite (22%), ectopic first maxillary permanent molars (50%) and a deviation of the middle of the dental arch (57%).8
Dental Development
Dental development is delayed and probably causes abnormal eruption patterns. This is a greater problem in Apert than in Crouzon patients (28 Apert patients, 39 Crouzon patients, 284 control) and girls are more affected than are boys.11 However, another study (26 Apert patients, 29 control) suggests that dental development is normal but deviates to the late side of the normal spectrum compared to controls.12 Local factors such as thickened gingiva, ectopic positioning of elements and severe crowding can explain the clinical picture of delayed tooth development. Possibly, this contradiction can be explained by differences in methodological characteristics of the different studies.
Agenesis
Tooth agenesis is more common in patients with Apert syndrome (46.4%) or Crouzon syndrome (35.9%) than in control patient groups (27.5%).13 The prevalence of second mandibular premolar agents is higher in syndromic patients than in a normal population.13,14
Oral Hygiene
In 57 children with craniosynostosis and controls of equal age, a comparison was made of caries, bacterial dental plaque, gingivitis and enamel abnormalities. These abnormalities were found significantly more frequently in the craniosynostosis group. The possibility for these patients to obtain good oral hygiene is reduced due to lack of space but also due to syndactyly in Apert patients.15
Conclusions
| Level 2 | It is likely that the cause of orthodontic and dental problems in patients with Apert's and Crouzon's syndrome lies in the abnormal growth of the maxilla in vertical, transversal and sagittal directions, leading to a maxilla that is hypoplastic in all dimensions. B Coll et al, 2018 B Goldstein et al, 2014 B Tahiri et al, 2014 B Reitsma et al, 2012 B McGrath et al, 2012 C Kreiborg et al, 1999 |
| Level 2 | Hypoplasia of the maxilla and palatal constriction are likely to occur frequently in patients with syndromic craniosynostosis. B Reitsma et al, 2012 C Kreiborg et al, 1999 B Tahiri et al, 2014 B Goldstein et al, 2014 |
| Level 3 | It is possible that the growth of the mandibula is influenced by the premature fusion of the cranial sutures, leading to abnormalities in both the sagittal and the vertical skeletal relationship. B Reitsma et al, 2012 C Kreiborg et al, 1999 B McGrath et al, 2012 C Kreiborg et al, 1999 |
| Level 3 | Asymmetry in growth of the mandibula may be seen more frequently in patients with Apert or Crouzon syndrome than in control groups. C Elmi et al, 2015 |
| Level 3 | A soft palate cleft or a bifid uvula may occur in 75% of patients with Apert syndrome and in 5% of patients with Muenke syndrome. C Cohen and Kreiborg 1996 B Agochukwu et al, 2012 |
| Level 3 | In patients with Apert or Crouzon syndrome, the dental arch may not widen during growth. Large lateral gingival swellings are also found in these patients, which may increase with age. C Reitsma et al, 2013 B Agochukwu et al, 2012 C Cohen en Kreiborg 1996 |
| Level 3 | Hypoplasia of the maxilla in patients with Apert and Crouzon syndrome may lead to a class III malocclusion (68%), accompanied by an anterior cross bite (81%), anterior open bite (73%), bilateral posterior cross bite (63%), unilateral cross bite (22%), ectopic first maxillary permanent molars (50%) and a deviation of the middle of the dental arch (57%). B Cohen and Kreiborg 1996 |
| Level 3 | Dental development in patients with Apert and Crouzon syndrome may be delayed or at the late stages of normal development, causing a delay in eruption and abnormal eruption patterns of the teeth. B Woods et al, 2015 C Reitsma et al, 2014a |
| Level 3 | Tooth agenesis may be more common in patients with Apert syndrome (46.4%) or Crouzon syndrome (35.9%) than in a control group (27.5%). The prevalence of second mandibular premolar tooth loss is higher in syndromic patients than in a normal population. B Reitsma et al, 2014b C Stavropoulos et al, 2011 |
| Level 3 | In craniosynostosis patients, more caries, plaque, gingivitis and enamel defects may be found compared to the normal population. C Mustafa et al, 2001 |
Considerations
• Evidence of Conclusions
The evidence of the conclusions varies from moderate to weak, largely due to the small study sizes.
• Values and Preferences
Most parents of children with syndromic craniosynostosis find it important to facilitate oral hygiene and obtain reasonable dental function. They therefore agree with multi-stage orthodontic interventions.
• Costs and Resources
The guideline committee expects that the implementation of the recommendations leads to little or no increase in costs, because the recommendations are in line with existing practice.
• Professional Perspective
Dental and dental abnormalities are common (see summary of the literature). Without intervention, improvements are not to be expected. The abnormalities can lead to caries, periodontal and oral hygiene problems. Interceptive orthodontics focuses on dental eruption support and creating a good starting situation for future orthodontic-dental surgical correction. In doing so, the burden of care must always be taken into account. A dentist or orthodontist who is not part of the craniosynostosis expertise team will perform treatments that have a definite character only after consultation with the craniosynostosis expertise team.
Consultation between orthodontist - treating surgeon
Typical for orthodontic intervention are multiple phases in which each treatment has specific objectives. Orthodontic interventions must always take into account that the orthodontic preparation forms the foundation for a future dental surgical correction. Close consultation between the orthodontist, maxillofacial surgeon and plastic surgeon of the craniosynostosis expert team prevents pursuing conflicting goals, limits the number and duration of treatments, and promotes the efficiency of treatment. If the orthodontic treatment is not performed in the craniosynostosis centre of expertise, the orthodontist of the craniosynostosis expertise team will communicate the policy to be followed with the external orthodontist.
Oral hygiene
The Dutch guideline oral care for young people recommends a visit to an oral care provider before the 2nd year of life. During orthodontic treatment, patients with syndromic craniosynostosis find it more difficult to achieve an adequate level of oral hygiene. This is partly caused by developmental and behavioural problems, but also by physical problems such as hand abnormalities. Regular support from one's own dentist and/or dental hygienist is desirable.
Around the age of 4 years
A global dentofacial assessment is made of the position and function of the milk teeth. In case of insufficient oral hygiene, additional support from a dentist, dental hygienist or a centre for special dentistry is recommended.
Around the age of 6 to 9 years
During the first teeth changing phase and during the intertransitional period, several treatment strategies are possible, such as dental or skeletal maxillary broadening, space management, eruption guidance and series extraction or a combination. Possible extractions of permanent elements should take into account future dental surgical correction. Regular orthodontic check-ups are important due to delayed dental development and abnormal eruption patterns.
Around the age of 12 to 15 years
During the 2nd teeth changing phase and in case of permanent dentition, orthodontic treatment is aimed at obtaining a functional bite and facilitating oral hygiene. During these treatments, one will always take into account that a definitive jaw correction will follow as soon as the patient has grown.
Around the age of 17 years
Around this age the end of craniofacial growth is approaching and the definitive jaw correction can be planned. Orthodontic-surgical intervention requires careful planning in consultation with the surgeon. Orthodontics during the pre-surgical pre-treatment is focused on the alignment of the dental arches. Post-surgical treatment is aimed at stabilizing the jaw correction and creating a functional occlusion.
Retention
Permanent retention is required by means of wire splints, if oral hygiene permits, and removable retention equipment. Sometimes additional support is required from a retention device that stabilizes the arches in relation to each other. Regular retention checks are necessary.
Treatment strategy - treatment plan
Typical for orthodontic intervention are the multiple phases in which each treatment has specific objectives. Orthodontic interventions will always have to take into account that the final correction is matched with a future jaw correction. This is due to contradictory goals sometimes, the limitation of the number of treatments, and the limitation of the duration of the treatment. This ensures the efficiency of care.
• Balance of Anticipated Desired and Undesired Outcomes
Following the opinion of the working group members, orthodontic pre-treatment in children with craniosynostosis will facilitate good oral hygiene, thus reducing the risk of periodontal problems. A good occlusion also ensures that no abnormal wear and tear and/or overloading of elements will occur due to abnormal tooth positions. Multiple interventions at different ages are regularly required.
Rationale of the recommendation(s)
The guiding principle in drawing up the recommendations is that very lengthy orthodontic treatments should be avoided. In almost all cases there will be several treatment phases, each with a specific goal. Each phase will be complementary and a preparation for the final jaw correction.
Recommendations
Syndromic craniosynostosis
A dentist or orthodontist who is not part of the craniofacial team never treats a patient with craniosynostosis without consulting the craniofacial team.
In addition to the advice to visit an oral care provider before the second year of life (see oral care guideline for adolescents), the orthodontist advises parents to visit the dentist, paediatric dentist or dental hygienist regularly if the oral hygiene is inadequate
-
Perform orthodontic checks within the craniosynostosis expert team in children with syndromic craniosynostosis around age:
-
-
4 years of age (milk teeth)
-
-
6 years (1st teeth changing phase)
-
-
9 years (intertransitional phase)
-
-
12 years (2nd teeth changing phase)
-
-
15 years (permanent dentition)
-
-
17 years (start orthodontic treatment in preparation for surgical jaw correction)
-
-
The craniosynostosis expert team draws up a long-term treatment strategy at the first contact at the patient's age of 4 years. The team adjusts this strategy on the basis of the findings of the orthodontic follow-up examinations.
The orthodontist of the craniosynostosis expert team develops this treatment strategy into a treatment plan for each stage of the patient's development in consultation with the treating maxillofacial and plastic surgeons.
The implementation of the orthodontic treatment plan does not have to take place in the craniofacial centre, but then still under the supervision of the orthodontist of the craniosynostosis expertise team.
Research Gaps
Dentofacial abnormalities occur in almost all syndromic craniosynostosis abnormalities. However, there is still insufficient knowledge about the type and prevalence of dentofacial abnormalities and the effectiveness of orthodontic treatments in syndromic craniosynostosis patients. The extent to which patients with other syndromes such as Saethre-Chotzen and Muenke are as severely affected as Apert and Crouzon syndromes is unclear, as is a possible difference in type and prevalence of dentofacial abnormalities between single-suture and multisuture. For this reason, it is of great importance that treatment centres, both nationally and internationally, maintain a standard schedule of documentation that allows for data pooling and a better understanding of the course of the syndromic craniosynostosis during growth as well as the effect of surgery on it. Therefore, the conclusions will have to be interpreted with caution.
Literature
-
1.
McGrath J, Gerety PA, Derderian CA, et al. Differential closure of the spheno-occipital synchondrosis in syndromic craniosynostosis. Plast Reconstr Surg 2012;130:681e-9e
-
2.
Tahiri Y, Paliga JT, Vossough A, et al. The spheno-occipital synchondrosis fuses prematurely in patients with crouzon syndrome and midface hypoplasia compared with age- and gender-matched controls. J Oral Maxillofac Surg 2014;72:1173-9
-
3.
Coll G, Sakka L, Botella C, et al. Pattern of closure of skull base synchondroses in crouzon syndrome. World Neurosurg 2018109:e460-7
-
4.
Kreiborg S, Aduss H, Cohen MM, Jr. Cephalometric study of the apert syndrome in adolescence and adulthood. J Craniofac Genet Dev Biol 1999;19:1-11
-
5.
Goldstein JA, Paliga JT, Wink JD, et al. Earlier evidence of spheno-occipital synchondrosis fusion correlates with severity of midface hypoplasia in patients with syndromic craniosynostosis. Plast Reconstr Surg 2014;134:504-10
-
6.
Reitsma JH, Ongkosuwito EM, Buschang PH, et al. Facial growth in patients with apert and crouzon syndromes compared to normal children. Cleft Palate Craniofac J 2012;49:185-93
-
7.
Elmi P, Reitsma JH, Buschang PH, et al. Mandibular asymmetry in patients with the crouzon or apert syndrome. Cleft Palate Craniofac J 2015;52:327-35
-
8.
Cohen MM, Jr., Kreiborg S. A clinical study of the craniofacial features in Aapert syndrome. Int J Oral Maxillofac Surg 1996;25:45-53
-
9.
Agochukwu NB, Solomon BD, Doherty ES, et al. Palatal and oral manifestations of Muenke syndrome (FGFR3-related craniosynostosis). J Craniofac Surg 2012;23:664-8
-
10.
Reitsma JH, Elmi P, Ongkosuwito EM, et al. A longitudinal study of dental arch morphology in children with the syndrome of Crouzon or Apert. Eur J Oral Sci 2013;121:319-27
-
11.
Reitsma JH, Balk-Leurs IH, Ongkosuwito EM, et al. Dental maturation in children with the syndrome of Crouzon and Apert. Cleft Palate Craniofac J 2014;51:639-44
-
12.
Woods E, Parekh S, Evans R, et al. The dental development in patients with Aperts syndrome. Int J Paediatr Dent 2015;25:136-43
-
13.
Reitsma JH, Ongkosuwito EM, van Wijk AJ, et al. Patterns of tooth agenesis in patients with crouzon or apert syndrome. Cleft Palate Craniofac J 2014;51:178-83
-
14.
Stavropoulos D, Bartzela T, Bronkhorst E, et al. Dental agenesis patterns of permanent teeth in apert syndrome. Eur J Oral Sci 2011;119:198-203
-
15.
Mustafa D, Lucas VS, Junod P, et al. The dental health and caries-related microflora in children with craniosynostosis. Cleft Palate Craniofac J 2001;38:629-35
CHAPTER 15 (NEURO)COGNITIVE, SOCIO-EMOTIONAL AND BEHAVIOURAL FUNCTIONING
15.1 What is the policy on (neuro)cognitive, social-emotional and behavioural problems in patients with craniosynostosis?
Introduction
The results of studies into cognitive, neurocognitive, socio-emotional and behavioural functioning in children with craniosynostosis vary widely. These varying results can often be explained by methodological limitations, such as different composition of the research group regarding the types of craniosynostosis, which characterizes many of these studies. The varying can also be explained by differences in informant (parents/teacher/child itself). Lastly, cognitive, neurocognitive, and social-emotional functioning, as well as behaviour may be broad concepts that consist of many different components which may be measured with different instruments.
Search and Selection
Relevant item:
Which (neuro)cognitive, social-emotional and behavioral problems occur in children with single-suture non-syndromic craniosynostosis, multisuture craniosynostosis or syndromic craniosynostosis and at what frequency?
In the Medline (OVID) and Embase databases, one overall search was conducted for studies on craniosynostosis. The search strategy is given in appendix 2, to the guideline. After deduplication, the literature search yielded 2732 hits.
Given the large number of studies, the chair of the working group first selected those that met the following general selection criteria:
General Selection and Exclusion Criteria
| Study type | -original studies -systematic reviews of sufficient quality: - research question of systematic review corresponds (largely) to the basic question - search is performed in at least 2 relevant databases, e.g. Cochrane Library, Medline/PubMed - reporting of the complete search strategy - no relevant keywords/search terms are missing |
| Follow-up period | -minimum follow-up period of 12 months for therapeutic or prognostic studies. |
| Exclusion criteria | -Case-reports -Expert opinion -Letters -Editorials -Case control studies for diagnostic tests -Narrative reviews |
The pre-selected studies that met the specific selection criteria listed in the Table below are included in the literature summary of this chapter.
Specific Selection and Exclusion Criteria
| Selection criteria for indication | - Minimum study size: 20 patients for patient series, where no multivariate analysis was used to identify prognostic factors for a relevant outcome measure. - Minimum study size: 35 patients for patient series with multivariate analysis of possible predictive variables for the effect - minimum number of participants of studies with a direct comparative design: 20 per study arm. - Studies with a good design and execution: for example, mentioning the response rate, describing inclusion and exclusion criteria, comparing the results of the research group with a control group or the norm group, taking into account the parents’ SES or IQ, describing the measuring instruments used. Not mentioning one or more of these items is not a strict exclusion criterion, but is taken into account in the quality assessment. |
Outcome measures:
Development and/or intelligence tests; Bayley Scales of Infant Development (BSID-II), Brunet-Lézine scale, Wechsler Preschool and Primary Scale of Intelligence (WPPSI), Wechsler Intelligence Scale for Children (WISC).
Tasks related to the executive function: Test of Everyday Attention in Children (TEACH), NEPSY (A Developmental NEuroPSYchological Assessment), WISC-IV: (working) memory, attention tasks
Factor Verbal Understanding (WISC-V)
Verbal Memory: Children's Memory Scale (CMS), NEPSY Task Verbal Fluency
Test for assessing school skills (e.g. reading, spelling, arithmetic): Wide Range Achievement Test (WRAT), Test of Word Reading Efficiency (TOWRE).
Visual working memory and response inhibition: A not B task with visible displacement (AB) and with invisible displacement (ABID)
Outcome measures on social-emotional functioning and behaviour:
Questionnaires completed by parents and/or teacher, such as Child Behavior CheckList (CBCL), Teacher Report Form (TRF), Social Communication Questionnaire (SCQ).
Interviews with parents: e.g. Diagnostic Interview Schedule IV Parent version (DISC-IV-P)
Summary of the Literature
Which (neuro)cognitive, social-emotional and behavioural problems occur in children with single-suture non-syndromic craniosynostosis, multisuture craniosynostosis or syndromic craniosynostosis, and at what frequency?
(A) Single-suture, Non-syndromic Craniosynostosis
Baby and Toddler Age (< 4 Years): Table 1
Development level
The studies by Da Costa and Speltz describe the level of development of very young children (average age < 12 months) who have not yet been operated on.1–3 In both studies, the level of development was measured with the BSID-II. In Da Costa's study, the results of children with craniosynostosis are compared with those of the norm group, while Speltz uses a control group matched on, among other things, age, sex, SES, ethnicity and mother's IQ.1–3 In both studies, the mean scores of children with craniosynostosis are significantly lower than those of the control group and the norm group, with differences between the craniosynostosis group and the control group being smaller than those between the craniosynostosis group and the norm group. The score on the mental scale is always higher than that on the motor scale (MDI > PDI). There are few differences between the craniosynostosis groups. Speltz reports that the child's sex, ethnicity and mother's IQ variables have no predictive value on the child's BSID-II scores.3
In the study by Mathijssen in a large group of children (144) with unilateral coronary aneurysm, the mean preoperative score on the Brunet-Lézine scale does not deviate from that of the norm group: 99.7 + 10.7.4 After surgery the score is 103 + 13.4. This article does not describe data on SES.
Starr compared pre-operative and post-operative BSID-II scores of children with craniosynostosis and related these to those of a control group matched to age at the time of inclusion, sex, family SES and ethnicity.5 The mean scores of the patients are lower than those of the control group, both preoperatively and postoperatively. Furthermore, the craniosynostosis group has both preoperative and postoperative MDI and PDI scores below 85. Preoperatively, 15% of the craniosynostosis group has an MDI score < 85 versus 10% of the control group. Postoperatively, this percentage increases to 30% in the craniosynostosis group compared to 19% in the control group. These proportions are much higher on the PDI score. Preoperatively, 45% of the children in the craniosynostosis group have a score < 85 versus 30% of the control group. Postoperatively, these proportions increase to 56% and 47%, respectively. Furthermore, a below-average score on T1 (preoperative) is predictive of a below-average score on T2 for both the craniosynostosis group and the control group.
Da Costa, too, compared the preoperative and postoperative BSID-II scores of children with craniosynostosis.2 Postoperatively, the mean MDI score of the craniosynostosis group is significantly lower than the mean preoperative MDI score; and this postoperative MDI score is also significantly lower than the mean score in the norm group. The authors explain the difference by the fact that the test administered at a later age is more reliant on speech and language, which is to the disadvantage of the craniosynostosis group. In this study, the postoperative PDI score does not differ significantly from the preoperative PDI score. Furthermore, 31.9% of children with craniosynostosis have a below-average MDI score and 40.1% a below-average PDI score postoperatively, while in the general population this is about 14.8% and 12.6%, respectively.
Gray has developed a prediction model that would accurately predict a developmental delay at the age of 3 years, using information collected at earlier ages (preoperative age 7 months and postoperative age 18 months).6 Because it is often not possible to test children with craniosynostosis preoperatively, only the BSID-II data collected at the age of 18 months were used in the model. In this model, the MDI score at the age of 18 months is predictive for functioning at the age of 3 years. Other predictors are sex of the child, SES and age at surgery.
Toth investigated visual working memory and response inhibition in young children (mean 18 months; range 17–24 months) with craniosynostosis, after surgical correction.7 These children achieved scores comparable to those of the control group.
Socio-emotional functioning and behaviour
Kapp-Simon used the CBCL and TRF to investigate the behaviour of children with craniosynostosis at the ages of 18 and 36 months with that of a matched control group.8 At the age of 18 months, there is little difference in CBCL and TRF scores between the craniosynostosis group and the control group. However, at the age of 36 months, parents of children with craniosynostosis clearly assign more scores in the clinical range on the Externalization scale than do parents of the children in the control group (14.5% versus 7.6%).
TABLE 1. Studies in Children < 4 Years:
| Suture | N | Response | Test age | Pre-op | Post-op | BSID-II: MDI | BSID-II: PDI | Other tests | Study |
| Sagittal | 26 | Consecutive sample | 8 mos | X | 96.4 ± 8.0, 80–111) | 87.7 ± 11.2, 58–106) | Da osta 20121 | ||
| Da osta 20132 | |||||||||
| 62 | 89% | 6.5 | X | 90.85 (9.05) | 84.27 (10.58) | Speltz 20073 | |||
| 86 | 55% | 18.4 mos | XX | 93.90 | 84.20 | Starr 20075 | |||
| Metopic | 20 | Consecutive sample | 10 mos | X | 99.1 ± 4.9, 86–106) | 90.1 ± 14.2, 50–108) | Da osta 20121 | ||
| Da osta 20132 | |||||||||
| 27 | 89% | 6.5 | X | 94.52 (7.32) | 86.63 (13.56) | Speltz 20073 | |||
| 35 | 55% | 18.4 mos | XX | 92.33 | 84.24 | Starr 20075 | |||
| Coronal (uni) | 144 | 100% | 12 mos | X | - | - | 99.7 ± 10.7) Brunet-Lézine scale | Mathijssen20064 | |
| Coronal right | 16 | 89% | 6.5 | X | 88.25 (13.57) | 81.06 (13.32) | Speltz 20073 | ||
| 20 | 55% | 18.4 mos | XX | 88.69 | 83.29 | Starr 20075 | |||
| Coronal left | 12 | 89% | 6.5 | X | 94.17 (5.98) | 85.42 (6.13) | Speltz 20073 | ||
| 16 | 55% | 18.4 mos | XX | 86.67 | 80.55 | Starr 20075 | |||
| Coronal (uni) | 10 | Consecutive sample | 9 mos | X | 98.7 ± 6.3, 87–109) | 82.6 ± 15.0, 55–101) | Da osta 20121 | ||
| Da osta 20132 | |||||||||
| Coronal (bi) | |||||||||
| Lambdoid | 8 | 89% | 6.5 | X | 95.38 (6.46) | 78.00 (19.32) | Speltz 20073 | ||
| 11 | 55% | 18.4 mos | XX | 83.98 | 80.15 | Starr 20075 |
Primary School Age: Table 2
Intelligence
Studies with the WISC on the intelligence of children with single-suture craniosynostosis at primary school age show the same picture as with younger children. In the studies by Speltz and Bellew, their intelligence scores, measured with the WISC-IV, are comparable to or slightly lower than those of the control group and norm group.9,10 However, the differences between the craniosynostosis group and the control group are usually small. The Verbal IQ is usually higher than the Normal IQ. Looking at the diagnostic subgroups, children with metopic suture, coronal suture, and lambdoid suture synostosis achieve on average lower IQ scores than children with sagittal suture synostosis and the percentages of children with a lower IQ (under 80 or 85) in these groups are clearly higher than in the norm group and in the group of children with sagittal suture synostosis: 21% in trigonocephaly versus 16% for the norm group (IQ< 85)11; 10.6% for scaphocephaly, 26.7% for plagiocephaly, 30.8% for trigonocephaly versus 9.3% for the norm group (IQ < 80)10; 4.9% for single-suture non-syndromic craniosynostosis versus 2.7% in the control group (IQ< 80).9
Research by Kapp-Simon shows that children with craniosynostosis on the factor Verbal Understanding of the WISC-V score significantly lower on average than children in the control group.12 In the craniosynostosis group 23% of the children score lower than 90 compared to 11% in the control group.
Reading, Spelling, Arithmetic
A study by Speltz into reading, spelling and arithmetic found that the craniosynostosis group scores only slightly lower than the control group on reading and spelling tests.9 The frequency of learning problems is also comparable in both groups. Children with craniosynostosis do score significantly lower than the control group in a math test.
Within the diagnostic subgroups, children with metopic suture, coronal suture, and lambdoid suture synostosis score lower on average on reading, spelling, and calculation tasks than children with sagittal suture stenosis.9 Children with coronal suture stenosis and lambdoid suture stenosis appear to be most vulnerable to learning difficulties.
Social-emotional functioning and behaviour
When looking at behaviour, the study by Speltz using the CBCL and TRF shows that parents and teachers of children with craniosynostosis indicate more problems on the Total Problems, Internalize and Externalize scales than do parents and teachers of the children in the control group, but these differences are small.13 However, children with craniosynostosis more often have a T-score > 60 (borderline and clinical range) on the Total Problems scale than children in the control group: 33% versus 21%. In children with metopic suture synostosis, 41% have a score in the borderline or clinical range on the Total Problems scale, while in children with sagittal suture synostosis it is 29%.
In the study by Van der Vlugt, parents report characteristics of autism on the SCQ (Social Communication Questionnaire) in 14% of children with metopic suture synostosis, and characteristics of ADHD (Attention Deficit Hyperactivity Disorder), ODD (Oppositional Deficit Hyperactivity Disorder) or CD (Conduct Disorder) on the DISC-IV-P (Diagnostic Interview Schedule IV Parent version) in 27%.11 Behavioural problems appear to be clearly correlated with IQ; children with a lower IQ are more likely to have behavioural problems than children with a higher IQ.
Executive Functioning and Memory
Research by Collet into executive functioning (EF) and attention shows that children with single-suture non-syndromic craniosynostosis score lower than the control group on almost all EF and attention tasks, although most differences are small and predominantly not significant.14 On tasks that assess Inhibition and Divided Attention, children with craniosynostosis scored significantly lower than children in the control group (mean corrected difference between the craniosynostosis group and control group respectively -0.91 (95% CI -1.71, -0.11) and -0.68 (95% CI -1.34, -0.02).
Kapp-Simon reports that children with single-suture non-syndromic craniosynostosis score consistently lower than children in the control group on verbal memory tasks, although the differences are modest, ranging from 0 to -0.4SD.12
Risk Factors for Neurocognitive, Socio-emotional and Behavioural Problems
Metopic suture synostosis: children with a dilated ventricular system (central part of the lateral ventricles) or a combination of a dilated ventricular system and additional congenital abnormalities have a greatly increased risk of a low IQ.15
Sex: In a group of 7-year-olds, girls with craniosynostosis as a group score higher than boys with craniosynostosis on, among other things, Total IQ (FSIQ) and tasks that measure school skills (reading, spelling, arithmetic). This difference is identically present in the control group. Of the girls, 30% achieve a low score (<25th percentile) on one or more reading, spelling or arithmetic tasks. Among boys with craniosynostosis this is 50%, especially boys with metopic suture synostosis (51%) or unicoronal synostosis (86%).16
Please note: The studies by Speltz, Kapp-Simon, Cradock refer to children with mean age 7 years; the study by Bellew to children with mean age 10 years.10,12,13,16 In the study by Speltz, Kapp-Simon, Cradock, both the craniosynostosis group and the control group at the age of 7 years are characterised by relatively many highly educated parents and intact families, which may paint a too rosy picture in terms of cognitive functioning. The response rate for the total group of 7-year-olds is ± 56%. The study by Van der Vlugt does not describe data on SES.11
Test Age > 4 Years:
| Suture | N | Response | Test age | Learning difficulties/ NPO | autism | ADHD/ODD/CD | IQ | Behaviour | Study |
| Entire group | 179 | 57% | 7 | Verbal memory: cranio group: 23% < 90 on verbal comprehension factor | Kapp-Simon 201612 | ||||
| Sagittal | 76 | 7 | FSIQ = 105.8 (14.7) | Speltz, 2015 | |||||
| 76 | 7 | CBCL/TRF:T > 60 Int:32% | Speltz, 2016 | ||||||
| Ext: 28% Tot. Pr: 29% | |||||||||
| 47 | 53% | 10 | VIQ = 104.1 (18.46) PIQ = 96.5 (15.60) FSIQ = 100.5 (17.32) | Bellew, 2015 children with and without surgery | |||||
| Metopic | 48 | 7 | FSIQ = 102.2 (17.8) | Speltz, 2015 | |||||
| 44 | 7 | CBCL/TRF:T > 60 Int:39% Ext:32% | Speltz, 2016 | ||||||
| Tot. Pr: 41% | |||||||||
| 82 | 94% | ? | SCQ: 14% deviant score: autistic characteristics reported by parents. Increased risk of ASD characteristics. With lower IQ. | DISC-IV-P: 27% deviant score. Reported by parents. Increased risk of behavioral problems at lower IQ. | 101.3 (±21.0, 50–147): Mullen, WPPSI, WISC, WAIS. 9% had IQ < 70 | Vd Vlugt, 2012 | |||
| 13 | 75% | 10 | VIQ = 90.9 (20.43) PIQ = 89.9 (14.16) FSIQ = 89.2 (17.56) | Bellew 201510children with and without surgery | |||||
| Coronal (uni) | 46 | 7 | FSIQ = 100.4 (16.3) | Speltz 20159 | |||||
| 44 | 7 | CBCL/TRF:T > 60 Int:46% | Speltz 201613 | ||||||
| Ext: 32% | |||||||||
| Tot. Pr: 34% | |||||||||
| 15 | 89% | 10 | VIQ = 94.3 (17.06) PIQ = 87.4 (12.52) FSIQ = 89.8 (15.10) | Bellew 201510children with and without surgery | |||||
| Lambdoid | 12 | 7 | FSIQ = 101.7 (14.3) | Speltz 20159 | |||||
| 12 | 7 | CBCL/TRF:T > 60 Int:50% | Speltz 201613 | ||||||
| Ext: 33% Tot. Pr: 33% |
B) Multisuture and Syndromic Craniosynostosis: Table 3
With regard to children with syndromic or multisuture craniosynostosis, only 3 studies of reasonable quality have been published concerning (neuro)cognitive, social-emotional functioning and behaviour: Bellew, 2015; Maliepaard, 2014 and Bannink, 2010.10,17,18 All other studies lack data on measuring instruments used, inclusion criteria, response, etc., as a result of which the studies cannot be easily compared or interpreted.
Intelligence
The studies of Bellew and Maliepaard investigate the cognitive functioning of children with syndromic or multisuture craniosynostosis.10,17 As a group, they generally score in the mean range on intelligence tests (FSIQ 102.9, SD 21.4 and 96.6, SD 21.6 respectively) and thus do not score significantly lower than the children in the norm group. However, they are almost twice as likely to have an IQ score lower than 85 than children in the norm group (30% versus 15.9%). There are, however, clear differences between the diagnostic groups. Maliepaard reports that children with Apert's syndrome have a very high chance (67%) of an IQ lower than 85, followed by children with Muenke syndrome (39%) and children with multisuture craniosynostosis (30%).17 In children with Crouzon/Pfeiffer syndrome or Saethre-Chotzen syndrome this chance is hardly increased compared to the norm group. Bellew also found an average IQ score in a group of 9 children with multisuture craniosynostosis; however, 33% of this group also had an IQ score lower than 90.10
Socio-emotional Functioning and Behaviour
In the study by Maliepaard, parents of children with syndromic or multisuture craniosynostosis report more social problems, attention problems and internalizing problems than do parents of children in the norm group (questionnaires: CBCL, DBD).17 Furthermore, a diagnostic interview with parents (DISV-IV-P) found a higher prevalence of ADHD (ADHD-any type and ADHD-hyperactive-impulsive type) than in the norm group. Broken down by diagnostic groups, parents of children with Apert's or Muenke's syndrome more often report social problems and attention problems in their child. In addition, parents of children with Muenke's syndrome also more often see internalising problems (T-scores > 60). Behavioural and emotional problems are associated with intelligence: the prevalence of behavioural and emotional problems is two to three times higher in children with IQ < 85, just as in the general population.
Quality of Life
Parents of children with syndromic or multisuture craniosynostosis report a significantly lower quality of life in their child than in the norm population, with scores of children with Apert's syndrome and multisuture craniosynostosis within the diagnostic groups lower than in the other groups in children under 4 years of age.18 In children aged 4 years and older, the scores of children with Apert syndrome and Muenke syndrome differ most significantly from the norm, indicating that parents attribute them a lower quality of life.18
| N (boys) | Response | Test age | Health Related Quality of Life | Behaviour | VIQ (SD) | PIQ (SD) | FSIQ (SD) | Study | |
| bicoronal | 7 (4) | 96% | 10 yrs | 87.4 (14.64) | 87.7 (17.43) | 85.7 (16.9) | Bellew 201510 | ||
| multisuture | 9 (4) | 86% | 10 yrs | 101.7 (21.65) | 103.7 (18.53) | 102.9 (21.39): 33%: IQ < 90 | Bellew 201510 | ||
| 20 | 6-13 | 93.9 (22.0) 30%: IQ< 85 | Maliepaard 201417 | ||||||
| Apert | 6 | 76.7 (13.3) 67%: IQ< 85 | Maliepaard 201417 | ||||||
| Crouzon/ Pfeiffer | 23 | 103.0 (20.1) 22%: IQ< 85 | Maliepaard 201417 | ||||||
| Saethre-Chotzen | 14 | 100.0 (26.6) 21%: IQ< 85 | Maliepaard 201417 | ||||||
| Muenke | 13 | 95.2 (16.4) 39%: IQ,85 | Maliepaard 201417 | ||||||
| Syndromic and multisuture craniosynostosis | 111 | 82% | 2–18 yrs | ITQoL (2–4 yrs, n = 23) | Bannink, 201018 | ||||
| CHQ-PF50 | |||||||||
| (4–18 yrs, n = 87) | |||||||||
| 82 (39) | 85% | 6-13 | CBCL: T-score Tot.Pr: 59.1(18.0) | 96.6 (21.6) 30%: IQ < 85 | Maliepaard, 201417 | ||||
| Int: 56.6(16.8) | |||||||||
| Ext: 51.0 (12.0) | |||||||||
| Soc.Pr68.4(23.8) | |||||||||
| Att.Pr59.2(15.8) | |||||||||
| DBD: Inatt. 55.1(11.9) | |||||||||
| Hyp/Imp 50.7 (8.6) | |||||||||
| DISC-IV: proportion/n | |||||||||
| Any DSM: 0.28 (19) | |||||||||
| Any Int: 0.30 (9) | |||||||||
| Any Ext: 0.21 (14) | |||||||||
| Soc ph: 0.01 (1) | |||||||||
| ADHDAny:0.30(9) | |||||||||
| ADHD comb 0.03 (27) | |||||||||
| ADHD inatt: 0.06 (27) | |||||||||
| ADHDhyper0.04(3) |
Conclusions
| Level 2 | Development level/intelligence: In young children with single-suture non-syndromic craniosynostosis, there is probably a significantly higher prevalence of developmental delay than in the control group or norm population, both on the mental scale and the motor scale. Young children (<4 years) with unilateral non-syndromic craniosynostosis probably score significantly lower on average in developmental research (BSID-II) than in the control group and the norm group, where the score on the mental scale is higher than that on the motor scale (MDI > PDI: range 83–99 versus 78–90). There is no evidence for a significant difference in cognitive functioning between the different types of single-suture craniosynostosis. B Mathijssen et al, 2006; Speltz et al, 2007; Starr et al, 2007; Kapp-Simon et al, 2012 C Da Costa et al, 2012 |
| Level 2 | Development level/intelligence: At primary school age, the average IQ scores of children with single-suture non-syndromic craniosynostosis (FSIQ 90–106) are probably comparable or slightly lower than those of children in the control or norm group. The VIQ is usually higher than the PIQ (range 91–104 versus 87–97). Verbal Meaning is significantly lower in children with single-suture, non-syndromic craniosynostosis than in the control group (23% below the score of 90 versus 11%). IQ scores <80–85 are more common in children with metopic suture (21–31% versus 9–16%), coronal suture (27% versus 9%) and lambdoid suture synostosis than in the norm group. B van der Vlugt et al, 2012; Kapp-Simon et al, 2016; Speltz et al, 2017 C Bellew et al, 2015 |
| Level 3 | Behaviour: At the child's age of 3, parents of children with single-suture non-syndromic craniosynostosis may report a higher prevalence of borderline or clinical range scores on the CBCL Externalize scale (14.5%) than parents of the control group (7.6%). B Kapp-Simon et al, 2012 |
| Level 2 | Behaviour: At the child's age of 7 years, parents of children with single-suture non-syndromic craniosynostosis are likely to report a higher prevalence of borderline or clinical range scores on the CBCL scale Total Problem Score than parents of control children: 33% and 21%, respectively. Children with metopic suture synostosis have the most behavioural problems (41% compared to the norm) and children with sagittal suture synostosis the least (29% compared to the norm). B Speltz et al, 2016; Van der Vlugt et al, 2012 |
| Level 3 | Executive functioning and memory: At the age of 18 months, children with single-suture non-syndromic craniosynostosis may score the same on tasks examining visual working memory and response inhibition as children in the control group. B Toth et al, 2008 |
| Level 3 | Executive functioning and memory: At the age of 7 years, children with single-suture non-syndromic craniosynostosis may score significantly lower on tasks investigating inhibition and distributed attention than children in the control group. B Collett et al, 2017 |
| Level 2 | The differences in cognitive functioning and school skills at 7 years of age between boys and girls with non-syndromic single-suture craniosynostosis may be similar to the differences in the norm group. At the age of 7 years, children with single-suture non-syndromic craniosynostosis probably score significantly lower on a numerical test than do children in the control group. B Speltz et al, 2015; Cradock et al, 2015 |
| Level 3 | A dilated ventricular system, whether or not in combination with additional birth defects, may increase the risk of a lower IQ in trigonocephaly. B Van der Vlugt et al, 2017 |
| Level 3 | Intelligence: Children with Apert's syndrome, Muenke's syndrome and children with multisuture craniosynostosis seem to have a (greatly) increased risk of an intellectual disability. C Maliepaard et al, 2014; Bellew et al, 2015 |
| Level 3 | Behavior: Parents of children with syndromic or multisuture craniosynostosis may report more social problems, attentional and attentional disorders, and internalizing problems in their child compared to the norm group. Children with Apert's syndrome or Muenke's syndrome show the most problems. Social-emotional and behavioural problems are strongly associated with intelligence. C Maliepaard et al, 2014 |
| Level 3 | Quality of life: Parents of children with syndromic or multisuture craniosynostosis report a potentially significantly lower quality of life in their child than in the norm population. Of children < 4 years of age, those with Apert syndrome and those with multisuture craniosynostosis appear to be the most vulnerable in this respect, and of children older than 4 years of age, those with Apert syndrome or Muenke syndrome. B Bannink et al, 2010 |
Considerations
• Evidence of the Conclusions
The evidence of most of the conclusions is weak. Besides limitations in study size, the reasons for this are limitations in the extent to which confounders are taken into account in analyses, in the description of measuring instruments, or the lack of a comparison with a control or norm group.
• Values and Preferences
According to the working group, the willingness of parents to have their child screened for (neuro)cognitive, social-emotional and behavioural problems varies greatly, and partly depends on the extent to which parents experience problems in this area with their child. If parents do not experience any worries or doubts about the child's development, they will see less need for screening. At the same time, acceptance of having a child with craniosynostosis may still be difficult for parents – and screening may then be perceived as stressful or stigmatising. As the child gets older and more concerns are identified in, for example, the home situation, the pre-school playgroup or school, the willingness to undergo screening may change. Screening opens up the possibility of detecting problems with regard to (neuro)cognitive, social-emotional and behavioural functioning so that further diagnostics and treatment can be offered. In this way, unnecessary developmental delays in the child can be overcome and pathological patterns can be prevented or limited. It can also enable parents to optimise the upbringing of their child.
• Costs and Resources
The recommendations are expected to lead to a slight increase in healthcare costs:
-
-
Children with single-suture, non-syndromic craniosynostosis (approximately 75 new cases per year nationwide) are currently not yet psychologically screened on a regular basis, but only when problems arise.
-
-
For children with metopic, coronal or lambdoid sutures, protocol screening may result in an earlier referral, but not so much in an increase in referrals.
-
-
For children with sagittal suture synostosis, however, protocol screening will result in an increase in costs and resources. Children with syndromic and multisuture craniosynostosis are already screened periodically.
On the other hand, a medical-psychological intervention can lead to an improvement in the child's and family's functioning, improving social participation, and potentially reducing care consumption in the long term.
• Professional Perspective
Gaining insight into (neuro)cognitive, socio-emotional and behavioural functioning is important for the timely deployment of interventions to stimulate development and for an appropriate choice of school at the age of 4 years. When the child is in primary school group 4 or 5 (7 or 8 years old), screening for possible learning disorders and behavioural problems is possible. Speech and language development, reading, spelling and arithmetic, intelligence, behaviour and socio-emotional functioning are (strongly) interrelated. In order to get a good picture of a child, it is important to look at all these factors in relation to each other, instead of focusing on just one aspect. In the literature on problems with (neuro)cognitive, socio-emotional and behavioural functioning in children with craniosynostosis, much has not yet been sufficiently proven, such as the prevalence and severity of the various types of craniosynostosis and the influence of timing and type of surgery on this. The great importance of gaining this knowledge, in addition to the personal interest for the child, is a reason to perform protocol screening for problems. More extensive (neuro)psychological research is reserved for children for whom screening gives reason to do so. The diagnostics and treatment can also be carried out in accordance with the “Guideline for aetiological diagnostics in children with a developmental delay/intellectual disability” from 2018.
• Balance of Anticipated Desired and Undesired Outcomes
Children with single-suture non-syndromic craniosynostosis are likely to have a slightly increased risk of (neuro)cognitive, social-emotional and behavioural problems, which increases with age and may interfere with every-day functioning and school functioning. The higher VIQ than the PIQ in combination with the significantly lower scores on Verbal comprehension compared to the norm group may entail the risk that too much is asked from the children. A standard follow-up in which the children are screened in time for these problems is therefore desirable. On indication, more extensive diagnostics can then be used.
Children with syndromic craniosynostosis probably have an increased risk of intellectual disability, social-emotional and behavioural problems and a lower quality of life as reported by parents. This applies especially to children with Apert or Muenke syndrome and children with multisuture craniosynostosis. Follow-up examinations for these disorders are therefore desirable with more extensive diagnostic research on indication.
A possibly undesirable outcome of (extensive) diagnostic examination is that a psychiatric diagnosis or a suspected psychiatric diagnosis is made, as this can have an influence in later life, for example when taking out insurance policies. On the other hand, (extensive) diagnostic testing provides insight into a child's possible strong and weak skills. Based on such a profile, targeted counselling can be used to support a child (and the family) to develop further as well as possible. This last argument weighs more heavily in the opinion of the working group.
Rationale of the recommendations
Screening for (neuro)cognitive, socio-emotional and behavioural functioning must be proportionate to the expected risks of problems in these areas. These risks differ per type of craniosynostosis. When there is a lower risk of problems, such as in children with sagittal suture synostosis, screening is of a more limited nature and focuses in particular on motor developmental delay and verbal comprehension. In children with metopic suture, coronal suture or lambdoid suture synostosis, Apert or Muenke syndrome or multisuture craniosynostosis, who have an increased risk of developmental delay, cognitive, social-emotional and/or behavioural problems, screening is indicated from the age of 18 months. If screening reveals problems, more extensive psychological tests are used.
Recommendations
For children with single-suture non-syndromic craniosynostosis
Screen these children aged between 18 months and 4 years for motor developmental delays, (neuro)cognitive, social-emotional and behavioural problems. In the event of abnormal screening, further psychological and/or paediatric physiotherapy tests should be carried out.
For children with metopic suture, coronal suture or lambdoid suture synostosis
Screen these primary school age children (group 4 or 5) for (neuro)cognitive, social-emotional and behavioural problems. In the event of an abnormal screening, further psychological tests should be carried out.
For children with sagittal suture synostosis
Screen these primary school age children (group 4 or 5) for verbal comprehension, numeracy skills, inhibition and divided attention. In the case of an anomalous screening, carry out further psychological tests.
For children with syndromic craniosynostosis or multisuture craniosynostosis
-
Screen these children at least for (neuro)cognitive, social-emotional and behavioural problems.
-
-
at least at an early age (around 2 to 3 years)
-
-
around the time of (primary) school choice
-
-
and if the child is in primary school group 4 or 5.
-
-
Always perform psychodiagnostic examinations in these children in case of (neuro)cognitive, socio-emotional and behavioural problems.
If necessary, refer for additional psychodiagnostics and treatment.
Measure the quality of life of these children by means of standardized questionnaires for parents or, if possible, for themselves (from 12 years of age). If possible, implement policy on the items on which a low score is achieved.
General about screening
Psychological screening and psychological examination of children with craniosynostosis is preferably done by the psychologist of the craniosynostosis expert team where the child is under treatment.
Additional diagnostics and treatment in case of a developmental delay can be carried out in accordance with the “guideline for aetiological diagnostics in children with a developmental delay/intellectual disability”.
Research Gaps
Few studies with a good design have been described, and the research gaps for the group of children with syndromic or multisuture craniosynostosis are greater than for the group of children with the single-suture, non-syndromic type.
Literature
-
1.
Da Costa AC, Anderson VA, Savarirayan R, et al. Neurodevelopmental functioning of infants with untreated single-suture craniosynostosis during early infancy. Childs Nerv Syst 2012;28:869-77
-
2.
Da Costa AC, Anderson VA, Holmes AD, et al. Longitudinal study of the neurodevelopmental characteristics of treated and untreated nonsyndromic craniosynostosis in infancy. Childs Nerv Syst 2013;29: 985-95
-
3.
Speltz ML, Kapp-Simon K, Collett B, et al. Neurodevelopment of infants with single-suture craniosynostosis: Presurgery comparisons with case-matched controls. Plast Reconstr Surg 2007;119:e1874-81
-
4.
Mathijssen I, Arnaud E, Lajeunie E, et al. Postoperative cognitive outcome for synostotic frontal plagiocephaly. J Neurosurg 2006;105(1 suppl):16-20
-
5.
Starr JR, Kapp-Simon K, Keich Cloonan Y, et al. Presurgical assessment of the neurodevelopment of infants with single-suture craniosynostosis: comparison with controls. J Neurosurg 2007;107:103-10
-
6.
Gray KE, Kapp-Simon K, Starr JR, et al. Predicting developmental delay in a longitudinal cohort of preschool children with single-suture craniosynostosis: is neurobehavioral assessment important? Dev Med Child Neurol 2015;57:456-62
-
7.
Toth K, Collett B, Kapp-Simon KA, et al. Memory and response inhibition in young children with single-suture craniosynostosis. Child Neuropsychol 2008;14:339-52
-
8.
Kapp-Simon KA, Collett BR, Barr-Schinzel MA, et al. Behavioral adjustment of toddler and preschool-aged children with single-suture craniosynostosis. Plast Reconstr Surg 2012;130:635-47
-
9.
Speltz ML, Collett BR, Wallace ER, et al. Intellectual and academic functioning of school-age children with single-suture synostosis. Pediatr 2015;135;e615-23
-
10.
Bellew M, Chumas P. Long-term developmental follow-up in children with nonsyndromic craniosynostosis. J Neurosurg Pediatr 2015;16:445-51
-
11.
Vlugt JJB van der, Meulen JJNM van der, Creemers HE, et al. Cognitive and behavioral functioning in 82 patients with trigonocephaly. Plast Reconstr Surg 2012;130:e885-93
-
12.
Kapp-Simon KA, Wallace E, Collett BR, et al. Language, learning, and memory in children with an without single-suture craniosynostosis. J Neurosurg Ped 2016;17:578-8
-
13.
Speltz ML, Collett, BR, Wallace ER, et al. Behavioral adjustment of school-age children with and without single suture craniosynostosis. Plast Reconstr Surg 2016;138:e435-45
-
14.
Collett BR, Kapp-Simon A, Wallace E, et al. Attention and executive function in children with and without single-suture craniosynostosis. Child Neuropsychol 2017;23:83-98
-
15.
Vlugt JJB van der, Meulen, JJMN van der, Coebergh van den Braak, RRJ, et al. Insight into the pathophysiologic mechanisms behind cognitive dysfunction in trigonocephaly. Plast Reconstr Surg 2017;139:e954-64
-
16.
Cradock MM, Gray KE, Kapp-Simon KA, et al. Sex differences in the neurodevelopment of school-age children with and without single-suture craniosynostosis. Childs Nerv Syst 2015;31:1103-11
-
17.
Maliepaard M, Mathijssen IMJ, Oosterlaan J, et al. Intellectual, behavioural, and emotional functioning in children with syndromic craniosynostosis. Pediatr 2014;133:e1608-15
-
18.
Bannink N, Maliepaard, M, Raat H, et al. Health-related quality of life in children and adolescents with syndromic craniosynostosis. J Plast Reconstr Aesthet Surg 2010;63:1972-81
CHAPTER 16 PSYCHOSOCIAL FUNCTIONING
16.1 What is the policy on psychosocial functioning of a child with craniosynostosis and the family members?
Introduction
Psychosocial functioning concerns the psychological, relational and social aspects of life. When a child is born with a (syndromic) craniosynostosis, not only does the condition itself influence the psychosocial functioning of the child and the family, but the medical treatment of the condition and the interaction with the outside world also influence the psychosocial functioning of the child, his or her parents, and siblings.
A whole system is involved in the treatment of a child with a craniofacial disorder.
In craniofacial care, there is a clear difference in the treatment of syndromic craniosynostosis and single-suture, non-syndromic craniosynostosis. A syndromic craniosynostosis is generally more visible than a non-syndromic craniosynostosis and needs a much longer and more intensive treatment with often multiple operations. Syndromic craniosynostosis therefore has a longer lasting effect on the life of the patient and family members, which may lead to more psychosocial problems.
Search and Selection
For the following specific questions, original scientific studies or systematic reviews of original scientific studies have been included:
Which psychosocial problems are involved in the patient and family? In what frequency do these problems occur and what are risk factors for the occurrence of these problems?
In the Medline (OVID) and Embase databases, one overall search was conducted for studies on craniosynostosis. The search strategy is given in appendix 2, to the guideline. After deduplication, the literature search yielded 2732 hits.
Given the large number of studies, the chair of the working group first selected those that met the following general selection criteria:
General Selection and Exclusion Criteria
| Study type | -original studies -systematic reviews of sufficient quality: - research question of systematic review corresponds (largely) to the basic question - search is performed in at least 2 relevant databases, e.g. Cochrane Library, Medline/PubMed - reporting of the complete search strategy - no relevant keywords/search terms are missing |
| Follow-up period | -minimum follow-up period of 12 months for therapeutic or prognostic studies. |
| Exclusion criteria | -Case-reports -Expert opinion -Letters -Editorials -Case control studies for diagnostic tests -Narrative reviews |
The pre-selected studies that met the specific selection criteria listed in the Table below are included in the literature summary of this chapter.
Specific Selection and Exclusion Criteria
| Selection criteria for indication | - Minimum study size: 20 patients for patient series, where no multivariate analysis was used to identify prognostic factors for a relevant outcome measure. - Minimum study size: 35 patients for patient series with multivariate analysis of possible predictive variables for the effect - minimum number of participants of studies with a direct comparative design: 20 per study arm. |
Summary of the Literature
Which psychosocial problems affect parents and patients, in what frequency and what are risk factors?
Clinical experience with regard to the psychosocial care of children with craniosynostosis and their families shows that the following themes often play a role in these families:
-
-
parents’ prolonged uncertainty around diagnosis;
-
-
anxiety about a next pregnancy;
-
-
uncertainty about the child's expected development;
-
-
uncertainty with regard to school choice and/or worries about learning performance;
-
-
child and parents’ coping with the visible disorder, the being ’different’;
-
-
how to shape the upbringing of a child with a visible disorder that requires long-term medical treatment;
-
-
how to deal with the information and/or different statements by the different care providers.
Having a child with a (syndromic) craniosynostosis is an extra stress factor in the relationship between parents. However, in 60% of divorced parents, the relationship was already in danger before the child's birth. If the relationship is good, the impact of this stress factor is less. Within ’Early Intervention’ it is wise to pay attention, too, to the relationship of the parents.1
Research by Bronner shows that 1 in 10 children and parents develop post-traumatic stress syndrome (PTSS) after the child has been admitted to intensive care.2 Stress reactions of the parents (especially of the mothers) are the most important predictor for PTSS in the child. The most important predictor of PTSS in parents is their mental vulnerability and the way in which they deal with the ICU admission.
Gray (2015) compared the reporting of stress by fathers and mothers of children with single-suture, non-syndromic craniosynostosis aged 6, 18 and 36 months, and children of these ages without a disorder.3 This was investigated by means of the Parenting Stress Index (PSI). In both groups (cases & controls) mothers reported significantly more stress in themselves than did fathers, and this was similar for the three age groups. The higher stress in mothers was independent of whether or not they had a child with single-suture, non-syndromic craniosynostosis. Possible explanations for the lack of a difference between mothers of affected and unaffected children are the provision of multi-disciplinary care in which attention is paid to psychosocial functioning, and the overrepresentation of well-to-do, white, intact families in both research groups. Despite the absence of a difference, it is advised to further follow families with a high score on PSI, as this was predictive of problems during childhood for other conditions.
Bannink studied quality of life in children and adolescents (2–18 years) with syndromic craniosynostosis with the Infant Toddler Quality of Life Questionnaire (ITQoL), the Child Health Questionnaire Parental Form 50 (CHQ-PF50), the Child Health Questionnaire Child Form 87 (CHQ-CF87) and the Short-Form Health Survey (SF-36), and compared these with the Dutch norm scores.4 The response to the questionnaires was 111 out of 136 children (81.6%). Parents of children/adolescents with syndromic craniosynostosis reported significantly lower quality of life in their child than did parents of children in the norm group. Parents of children with Apert syndrome reported the lowest quality of life for their child in the various domains, such as physical functioning, emotional impact parent and family activities. The parents reported a reduced health-related quality of life for themselves, especially at the psychosocial level with a lower perception of their general health.
In another Dutch study in a group of children with syndromic craniosynostosis that largely overlapped with Bannink, the health-related quality of life measured with the HUI-3 was lower than that of the Dutch norm group.4,5 In total, 131 out of 173 children between the ages of 4 and 18 years were included on the basis of questionnaires completed by the parents, as was the case for the norm group. The HUI consists of 8 items (vision, hearing, speech, ambulance, agility, emotion, cognition and pain) that are each scored from 0 (no limitation at all) to a maximum of 6 (severely limited). The composite HUI-3 score was 0.91 for the norm group; 0.44 for Apert syndrome; 0.76 for Crouzon syndrome; 0.87 for Saethre-Chotzen syndrome; 0.81 for Muenke syndrome; and 0.83 for multisuture craniosynostosis. Only for Apert syndrome the difference was statistically significant (p< 0.05). Vision, hearing and speech were the items with a statistically significant lower score. For Apert syndrome, the cognitive score was also significantly lower than the norm.
More than one-third of young people with a visible craniofacial disorder have appearance-related problems.6 It is important to provide psychological support to these young people. The counselling focuses on the psychosocial adaptation and self-understanding, social skills and self-image of these young people.6,7
Lefebvre interviewed 250 patients (age 6 weeks–39 years) with a severe craniofacial disorder (congenital or acquired) and their parents as part of the standard preoperative assessment: a semi-structured interview was conducted with parents and patient, a score was asked for the disorder on Hay's Scale, and the Piers-Harris Self-esteem Inventory was administered.8 One and two years after surgery, the same protocol was followed. The most significant predictive factors of postoperative psychosocial improvement were patient age, pre-operative expectation of the surgery, and who had made the decision for surgery (especially in adolescents).
Conclusions
| Level 3 | In syndromic craniosynostosis there is a greater risk of psychosocial problems. C St. John et al, 2003 |
| Level 3 | The health-related quality of life in children with syndromic craniosynostosis, measured with the ITQoL, CHQ-PF50, CHQ-CF87, SF-36 and HUI-3, was lower than that in the norm group. In particular, scores with regard to vision, hearing and speech were lower. For Apert's syndrome, scores on physical functioning, emotional impact, family activities and cognition were also significantly lower than the norm. C Bannink et al, 2010; De Jong et al, 2012 |
| Level 3 | Parents of children with multisuture or syndromic craniosynostosis have a reduced health-related quality of life, especially at the psychosocial level, compared to the norm group. C Bannink et al, 2010 |
| Level 3 | PTSS is seen in around 10% of the children who had been admitted to an ICU and their parents. C Bronner et al, 2008 |
| Level 3 | Stress responses of parents (especially mothers) are the most important predictors of PTSS in the child. C Bronner et al, 2008; Gray et al, 2015 |
| More than one-third of young people with a craniofacial disorder have appearance-related problems. Kapp-Simon et al, 2005; Strauss et al, 2007 |
| Level 3 | The most significant predictive factors of postoperative psychosocial were were patient age, pre-operative expectation of the operation and who had made the decision for surgery (especially in adolescents). C Lefebvre et al, 1982 |
Considerations
• Evidence of the Conclusions
The evidence of all the conclusions is weak. The main reasons for this are limitations in the statistical analysis carried out and limited generalizability of the research results due to lack of diversification of the research group.
• Values and Preferences
According to the working group, parents’ wishes to receive counselling on the psychosocial level vary greatly. This wish is influenced by each parent's own values and preferences and by whether or not problems develop in the child or within the family. Therefore, the need for psychosocial counselling can change over time. That is why shared decision-making is certainly appropriate here.
• Costs and Resources
The guideline committee expects that the implementation of the recommendations leads to little or no increase in costs, because the recommendations are in line with existing practice.
A medical-psychological intervention can lead to an improvement in patient and system functioning, thereby improving social participation and potentially reducing care consumption in the long term.
• Professional Perspective
Psychosocial functioning of child and family is a key point of attention. To be able to offer timely support, if necessary, it is important to examine the factors that influence psychosocial functioning (e.g. coping strategies, social support network, peer group, level of functioning of child and parents, support of parents).
A syndromic craniosynostosis is ’forever’ and therefore requires a lot of adaptation and flexibility from both the child and family. Having a child with syndromic craniosynostosis has a major impact on parenthood and family functioning. How do parents ensure balance, how can they shape their own meaning-making process, how to take care of the other children, how to organise the upbringing, how to deal with their own emotions, and how to organise the combination with work and family and care? It requires a lot of adaptability and flexibility from parents and family.
Parents of children with syndromic or complex craniosynostosis often mention that it is difficult to deal with possible reactions from the outside world to their child's visible condition, and want to protect their child from negative reactions to his or her appearance. At the same time, they find it very important that their child becomes resilient and can find his or her way in the outside world. Support to parents can sometimes be desirable in this respect, to think along with parents about how they can give their child the opportunity to explore the outside world, to gain sufficient social experiences, and to allow them to experience which way of responding to questions and comments about their condition best suits the child and parents. Parents also regularly report difficulties in raising their child with syndromic craniosynostosis because of all the medical treatments that the child, often from a very young age, has to undergo. Turning a blind eye is often quite understandable when the child has just had surgery, or is in hospital, or in the first period after admission at home. Sometimes it is difficult to make the transition back to ’normal’ upbringing afterwards, and it is important that parents can ask for support in this.
Feelings of fear about the child's visible condition, doubts and questions such as “can we do this” or “what do we do to our child”, can influence how confident or insecure parents feel in their parenting role. It can cause feelings of fear and insecurity to ’let go’ of the child and allow him or her to practice with the challenges of life. Here, too, it is important to offer parents support so that they can shape parenting in the best possible way (for all children in the family), despite their child's syndromic craniosynostosis and the associated treatment.
Adolescents do not always have real expectations regarding the outcome of surgical craniofacial treatments. This should be taken into account when preparing for these treatments. It is also not always clear how motivated they are for certain long-term medical treatments, which can sometimes lead to sub-optimal results.
Members of the craniosynostosis expert team do not always recognize possible psychosocial problems. It is important to have knowledge of development/education/parenting tasks in order to be able to seek help in a timely manner.
• Balance of Anticipated Desired and Undesired Outcomes
Failure to detect and treat psychosocial problems can have a disruptive effect within the family. If parents do not receive the necessary help in raising and supporting their child, this can negatively affect the child's self-confidence and resilience. In addition, some parents find it difficult to find the right way to appropriate advice and possibly guidance around school choices. The extent to which parents need and allow support is highly individual and can change over time. For this reason, regular and accessible help should be offered.
Rationale of the recommendation(s)
Having a child with craniosynostosis can have great impact on the psychosocial functioning of the family. Giving support can contribute to a better psychosocial functioning of all family members.
Recommendations
Support to parents and family from the craniosynostosis expertise team
Prevention of psychosocial problems
Inform patients and their parents about Patients and Parents Association LAPOSA.
Inform parents about the possibility of referral to a social worker/psychologist for support in raising the child.
Repeatedly offer parents with a child with syndromic craniosynostosis contact with a social worker/psychologist – mainly around the child's transitional phases, such as the moment of (primary) school choice.
Screen the family for the presence of psychosocial problems and symptoms of PTSS regularly throughout the course of treatment.
On indication
Refer the family to a social worker/psychologist in case of psychosocial problems.
Refer parents or child with PTSS or suspected PTSS to the psychologist of the craniosynostosis expert team or a psychologist in or near the place of residence.
Support to a child with craniosynostosis from the craniosynostosis expertise
Offer psychosocial care from the team throughout the treatment process.
Undertake psychosocial screening for long-term treatments that demand much from a patient's motivation. If necessary, offer support to improve the feasibility of treatment.
Offer counselling focused on psychosocial adaptation, self-understanding, social skills and self-image for young people experiencing problems in these areas.
Offer adolescents with a desire for surgical treatment, at least one contact with a specialised psychosocial counsellor to assess their expectations and motivation.
Research Gaps
The number of articles concerning psychosocial functioning in single-suture, non-syndromic craniosynostosis is very low, and for syndromic craniosynostosis very limited.
Literature
-
1.
St. John D, Pai L, Belfer M, et al. Effects of a child with a craniofacial anomaly on stability of the parental relationship. J Craniofac Surg 2003;14:704–8
-
2.
Bronner MB, Knoester H, Bos AP, et al. Posttraumatic stress disorder (PTSD) in children after paediatric intensive care treatment compared to children who survived a major fire disaster. Child Adolesc Psychiatry Ment Health 2008;2:9
-
3.
Gray KE. Cradock MM, Kapp-Simon KA, et al. Cleft Palate Craniofac J 2015;52:3-11
-
4.
Bannink N, Maliepaard M, Raat H. et al. Health-related quality of life in children and adolescents with syndromic craniosynostosis. J Plast Reconstr Aesthet Surg 2010;63:1972-81
-
5.
De Jong T, Maliepaard M, Bannink N, et al. Health-related problems and quality of life in patients with syndromic and complex craniosynostosis. Childs Nerv Syst 2012;28:879-82
-
6.
Strauss RP, Ramsey BL, Edwards TC, et al. Stigma experiences in youth with facial differences: a multi-site study of adolescents and their mothers. Orthod Craniofac Res 2007;10:96-103
-
7.
Kapp-Simon K, McGuire D, Long B, et al. Addressing quality of life issues in adolescents: social skills interventions. Cleft Palate Craniofac J 2005;42:45-50
-
8.
Lefebvre A, Barclay S. Psychosocial impact of craniofacial deformities before and after reconstructive surgery. Can J Psychiatry 1982;27:579-84
CHAPTER 17 CRITERIA FOR CRANIOSYNOSTOSIS EXPERTISE CENTRE AND TEAM MEMBERS
17.1 What are the minimum requirements for a craniosynostosis expertise centre and its team members?
Introduction
With an incidence of 7.2 out of 10,000 live births (Cornelissen, 2016), craniosynostosis is a rare disease.1 Care for these patients requires the deployment of multiple medical, dental and healthcare-allied specialties, in which coordination of care is essential. The multidisciplinary and long-term care of craniosynostosis makes demands on a hospital's organisation of, the craniosynostosis expertise team and the team members.
Search and Selection
No systematic literature analysis was carried out to answer the basic question.
Summary of the Literature
Not applicable
Considerations
• Evidence of the Conclusions
Not applicable
• Values and Preferences
Shared care and shared decision-making will have a more prominent role in health care, especially in child care. This concept includes an active participation of parents and child in the care and decision-making on treatments. This requires good communication between the team members and parents and child, whereby appointing a central contact person from the craniosynostosis expertise team is highly desirable. This role can, for example, be fulfilled by the nurse specialist or physician assistant. Giving parents and child access the medical file is also desirable, but is not sufficient to realise joint care and shared decision-making.
• Costs and Resources
The guideline committee expects that application of the recommendations leads to little or no increase in costs, because the recommendations are in line with existing practice.
• Professional Perspective
Verifiability of the quality of care provided by a craniosynostosis expertise center and the team members is related to the following items:
-
-
Composition of craniosynostosis expertise team
-
-
Cooperation within and outside the craniosynostosis expertise centre
-
-
Division of tasks within the craniosynostosis expertise centre
-
-
Centralisation
-
-
Reporting of results and activities
Re: Composition of craniosynostosis expertise team
Craniosynostosis is characterized by problems that may present in various domains, such as vision, hearing or behavioural problems. This makes multidisciplinary, well-coordinated care essential.
The management of craniosynostosis is largely surgical. It usually involves intracranial surgery. The contribution of neurosurgery is therefore a prerequisite. Plastic surgery and/or oral, maxillofacial and facial surgery is essential to facial corrections and skull shape correction. That is why these three specialties are the core specialties for the management of craniosynostosis. The contribution of all other specialties depends on the patient's specific diagnosis and individual presentation.
Cooperation within and outside the craniosynostosis expertise centre
The associated problems with craniosynostosis present at varying moments in the child's development. Screening at the right moments with appropriate examinations is therefore necessary. What screening, when screening and possible treatments are brought together in a care pathway. This ensures optimal coordination between the various specialties.
When working together in a multidisciplinary setting, it is necessary to clearly agree on the division of tasks and responsibilities.
Joint consultations of the various specialties enable optimal coordination of the care plan. Joint consultations result in fewer hospital visits for both child and parents.
Some of the treatments that a child with craniosynostosis has to undergo can be offered outside the craniosynostosis expertise centre, provided a number of conditions are met, such as normal anatomy of the upper respiratory tract in case anaesthesia is required. The assessment of whether a specific treatment outside the craniosynostosis centre is safe care is made by the craniosynostosis expertise centre and coordinated with the caregiver in the patient's own region.
Specific parts of the care trajectory can be carried out in the patient's own region on request and under coordination of the craniosynostosis expertise centre.
Division of tasks within the craniosynostosis expertise centre
Multidisciplinary cooperation makes it necessary for someone to take overarching responsibility. This responsibility lies with the team chair of one of the core specialties.
The multidisciplinary collaboration makes it necessary for someone 1) to supervise the timely execution of the various studies both within and outside the expertise centre, and 2) to ensure mutual communication between the various parties. This task is assigned to the care coordinator of the craniosynostosis expertise team.
Centralisation
Given the rarity of craniosynostosis and the complexity of treatments, centralisation for non-syndromic, and syndromic craniosynostosis has been agreed on. Underlying this decision is, among other things, the guideline “Anaesthesiology in children” of the Dutch Society for Anaesthesiology from 2017. Syndromic craniosynostosis is treated in only one centre of expertise as this type of craniosynostosis is the rarest and the most complex in terms of facial operations (e.g. Le Fort III, monobloc and facial bipartition). Given the complexity of pathology and treatment of syndromic craniosynostosis and its very low incidence, centralisation of this treatment in one centre in the Netherlands is desirable.
Reporting of outcomes and activities
A centre of expertise has the responsibility to facilitate the verifiability of quality of care and to ensure continuous quality monitoring within the team. This can be achieved by reporting the outcomes of care in an annual report and conducting an annual internal audit.
• Balance of Anticipated Desired and Undesired Outcomes
Opposite to the centralisation of expertise in a limited number of centres, this means more travel time for a number of patients. Because a large part of the care for craniosynostosis involves elective care, this rarely leads to medically undesirable situations. Where possible, parts of the care are organised in the patient's own region, under supervision of the expertise centre.
Rationale of the recommendation
The guiding principle in drawing up the criteria to be met by a craniosynostosis expertise centre and how care should be organised in the Netherlands is to provide the best possible care to children with craniosynostosis and their parents. The assessment and treatment of this condition is complex and has a lifelong impact on the children. This requires a dedication of caregivers to continuously train themselves in all aspects of this care.
Recommendations
Composition of craniosynostosis expertise team
Care for patients with craniosynostosis should be provided from a multidisciplinary setting.
A craniosynostosis expertise center has at least the following care providers and facilities:
| Care provider/facility | Single-suture non-syndromic | Multisuture or syndromic |
| Paediatrician | x | x |
| Clinical geneticist | x | x |
| Paediatric anaesthesiologist | x | x |
| Paediatric intensivist | x | x |
| Neurosurgeon | x | x |
| Paediatric neurologist | x | |
| Ophthalmologist | x | x |
| Paediatric radiologist | x | x |
| Plastic surgeon | x | x |
| Oral and maxillofacial surgeon | x | x |
| Orthodontist | x | |
| Otolaryngologist | x | |
| Psychologist | x | x |
| Social worker | x | x |
| Speech therapist | x | x |
| Educational worker | x | x |
| Team chair (one of the core specialists) | x | x |
| Care coordinator | x | x |
| (3D-) photogrammetry, X-ray, ultrasound, CT | x | x |
| MRI | x | |
| Pediatric-IC | x | x |
| Polysomnography | x |
Back up of the basic specialties (thus, at least 2 specialists for neurosurgery, plastic surgery, oral and maxillofacial surgery) is advised to guarantee continuity of care.
Collaboration within the craniosynostosis expertise center
Care for patients with craniosynostosis should be provided from a multidisciplinary setting. A care path should have been established.
The team roles should be clearly defined.
Joint consultations are held with the presence of the core specialists and availability of the other specialists.
Collaboration outside the craniosynostosis expertise center
Patients with craniosynostosis are only treated in an accredited craniosynostosis center of expertise center. Specific parts of the care program can be performed in the own region on request and under coordination of the craniosynostosis center of expertise.
Task division within the craniosynostosis expertise center
Care is provided on the basis of established protocols that are reviewed annually.
The multidisciplinary care per individual patient is coordinated between the care providers and communicated to patient and parents and any care providers from outside the team.
A practitioner from a core specialty is team leader. He or she is ultimately responsible for ensuring that the craniosynostosis expertise center meets all criteria.
The care coordinator (usually a nursing specialist) is responsible for coordinating care and is the point of contact for patients and co-treatment providers from outside the team.
Centralisation
Care for non-syndromic, unisuture craniosynostosis is centered in two craniosynostosis expertise centers.
Care for syndromic craniosynostosis is centered in one center.
The minimum number of intracranial operations for craniosynostosis is 20 per surgeon per year.
Reporting of results and activities
At least once a year an internal audit takes place.
Every craniosynostosis expert team issues an annual report:
| Item | Single-suture non-syndromic | Multisuture or syndromic |
| Number of operations per diagnosis | x | x |
| Number of procedures per type of operation | x | x |
| Number of patients treated by protocol | x | x |
| Perioperative dura and brain injury | x | x |
| Excessive blood loss | x | x |
| Infections | x | x |
| Unscheduled redo-operations | x | x |
| Material problems (springs, distractors, helmet) | x | x |
| Quality of life/patient-related outcome measure (PROM) | x | x |
| Appearance/aesthetic result | x | x |
| Behaviour | x | |
| Neurocognition and behaviour | x | x |
| OSA | x | |
| Increased ICP | x | x |
| Hydrocephalus | x | |
| Hearing | x | |
| Speech/language | x | x |
| Vision | x | x |
Research Gaps
Not applicable
Literature
-
1.
Cornelissen MJ, Den Ottelander B, Rizopoulos D, et al. Increase of prevalence of craniosynostosis. J Cranio-Maxillo-Fac Surg 2016;44:1273-9
CHAPTER 18 FLOWCHART
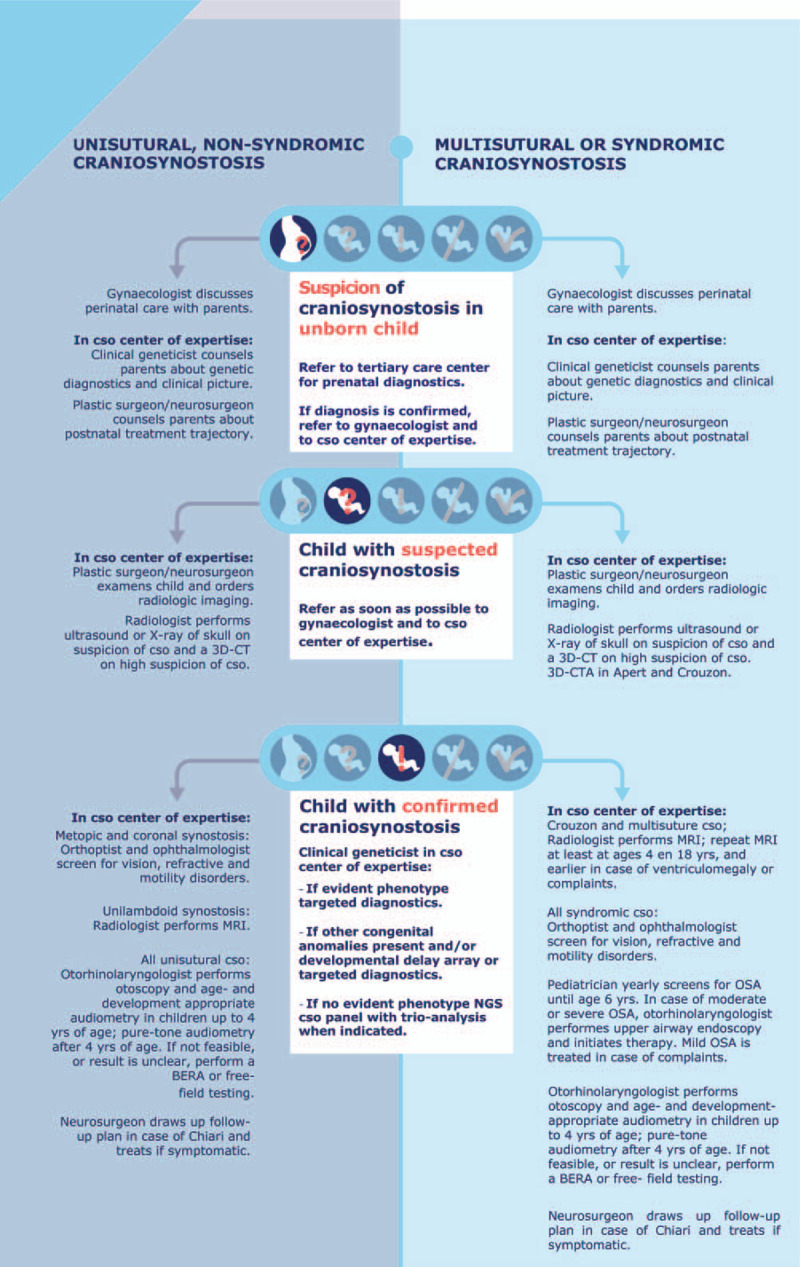


Supplementary Material
Supplementary Material
Supplementary Material
Footnotes
FUNDING: The revision of the guideline was financially supported by the Stichting Kwaliteitsgelden Medisch Specialisten (SKMS). The translation and publication was financially supported by the European Reference Network Craniofacial Anomalies and ENT Disorders (ERN CRANIO).
The authors have no conflicts of interest.
Contributor Information
Collaborators: Working Group Guideline Craniosynostosis
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.