

Abstract
Purpose of review
Hyperphosphatemia, iron deficiency and anemia are powerful stimuli of FGF23 production, and are highly prevalent complications of Chronic Kidney Disease (CKD). In this manuscript, we put in perspective the newest insights on Fibroblast Growth Factor 23 (FGF23) regulation by iron and phosphate and their effects on CKD progression and associated outcomes. We especially focus on new studies aiming to reduce FGF23 levels, and we present new data that suggest major benefits of combined corrections of iron, phosphate and FGF23 in CKD.
Recent findings
New studies show that simultaneously correcting iron deficiency and hyperphosphatemia in CKD reduces the magnitude of FGF23 increase. Promising therapies using iron based phosphate binders in CKD might mitigate cardiac and renal injury and improve survival.
Summary
New strategies to lower FGF23 have emerged, and we discuss their benefits and risks in the context of CKD. Novel clinical and pre-clinical studies highlight the effects of phosphate restriction and iron repletion on FGF23 regulation.
Keywords: iron, phosphate, anemia, chronic kidney disease, cardiovascular disease
INTRODUCTION
Chronic kidney disease (CKD) is a public health threat affecting approximately 37 million individuals in the United States (US)1 and 750 million individuals2 worldwide. Complex interactions between multiple biological mechanisms contribute to consequences of CKD, including disordered bone and mineral metabolism, and to associated risks of CKD progression and cardiovascular disease.
Elevated levels of the bone secreted hormone fibroblast growth factor 23 (FGF23) are a pervasive and early complication of CKD. While increased FGF23 levels help maintaining circulating phosphate in check in the early stages of the disease, FGF23 excess may contribute to kidney disease progression, cardiovascular events and death in CKD patients. This supports the rationale to design novel therapeutic strategies to lower FGF23 in CKD, but this approach requires more thorough understanding of the unknown mechanisms that cause FGF23 elevation in CKD, as well as the spectrum of disorders associated with excess FGF23.
Hyperphosphatemia contributes to higher FGF23 levels in the late stages of CKD, but does not drive the initial increase in FGF23, because increase in FGF23 occurs prior to changes in serum phosphate. Data from our group and others suggest that iron deficiency (ID) and anemia, which are common and early consequences of CKD, are novel mechanisms that stimulate FGF23 production3–6. In addition, hyperphosphatemia and iron deficiency can act in tandem with FGF23 to accelerate disease progression and development of negative outcomes7–9. Thus therapies that simultaneously reduce serum phosphate and treat iron deficiency may have a better chance of success in reducing circulating levels of FGF23 and improve clinical outcomes in CKD.
We recently showed that administration of ferric citrate, an iron based phosphate binder, to Col4a3KO mouse model of progressive CKD had the potential to lower serum phosphate, correct iron deficiency and anemia and lead to a significant decrease in circulating FGF23 levels10.
In addition, simultaneous corrections in phosphate, iron and FGF23 in CKD improved cardiac and kidney function, delayed CKD progression and resulted in prolonged lifespan10. This strongly suggests that combined management of iron and phosphate reduces FGF23 and is an effective therapy to improving clinical outcomes in CKD.
In this review, we will summarize recent findings highlighting the multifactorial roles of FGF23 in CKD, and the consequent multifactorial improvement that patients could benefit from FGF23 correction associated with phosphate and iron.
Physiological and pathological effects of FGF23
Under physiological conditions, FGF23 stimulates phosphaturia and reduces the efficiency of phosphate absorption in the gut by impairing production and accelerating the degradation of 1,25-dihyroxyvitamin D levels, thereby maintaining normal phosphate homeostasis11–14. Although in CKD elevated FGF23 maintains normal serum phosphate, this compensation is ultimately maladaptive leading to vitamin D deficiency and secondary hyperparathyroidism14. In addition, excess FGF23 is associated with increased risk of mortality across all stages of CKD and is independently associated with increased risk of heart failure15. Indeed, FGF23 has a pathogenic role in the development of left ventricular hypertrophy (LVH)16, 17, and/or impaired cardiac function10, 18. These findings underscore the necessity of understanding both FGF23 function and regulation which should lead to therapies designed to reduce FGF23 excess in CKD.
Balance of FGF23 transcription and cleavage
Circulating FGF23 levels are regulated by a balance between Fgf23 transcription and FGF23 cleavage4, 5. In the circulation, FGF23 is detected as a full-length intact bioactive protein (iFGF23) and as cleaved N- and C-terminal FGF23 derived peptides. Poorly defined subtilisin-like proprotein convertases cleave FGF2319, and GalNAc transferase 3 (GALNT3) protects newly synthesized FGF23 from cleavage, by O-glycosylation of the cleavage site20. FGF23 transcription and cleavage can be indirectly assessed non-invasively in vivo by using two different commercially available assays: the C-terminal FGF23 assay (cFGF23), which captures both intact FGF23 and FGF23 C-terminal peptides, and the intact FGF23 assay (iFGF23), which exclusively detects iFGF2321. The ratio between iFGF23 and cFGF23 can be used as a surrogate marker for FGF23 cleavage4 and at homeostasis this ratio is approximately 40%4, 18.
Regulation of FGF23 production
FGF23 production is regulated by local bone factors that modulate turnover and mineralization and systemic factors, such as phosphate, that control mineral metabolism22, increase Fgf23 transcription and lead to elevated levels of iFGF2323, 24. In addition to these classical factors, we and others recently discovered that iron deficiency and EPO also increase transcription of Fgf234–6, 25, 26. In sharp contrast to classical FGF23 stimuli, in patients and animals with preserved kidney function, these novel regulators also increase FGF23 cleavage4–6, 25, 26, leading to secretion of FGF23-derived peptides. However, in CKD, where FGF23 cleavage is impaired, iron deficiency may contribute to the rise of biologically active iFGF23 and increase the burden of the disease4, 6, 10 (Figure 1).
Figure 1: FGF23 regulation by phosphate and iron.
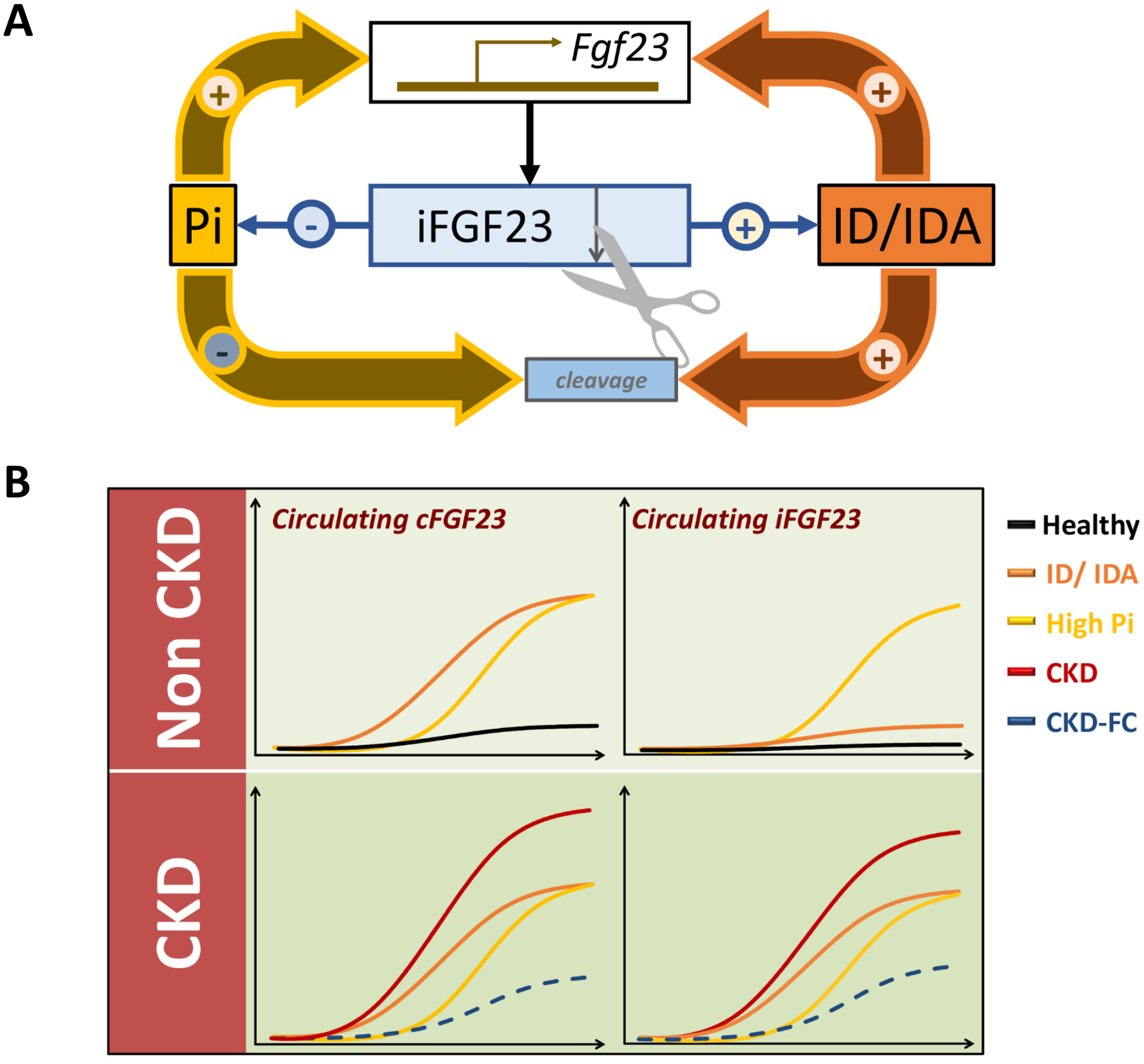
(A) Increased phosphate (Pi), iron deficiency (ID) and iron deficiency anemia (IDA) increase transcription of Fgf23 resulting in increased production of intact FGF23 (iFGF23). Phosphate increases stabilization of iFGF23 and minimizes its cleavage, while ID and IDA increase FGF23 cleavage. (B) In consequence, in animals and patients without impaired kidney function, increased phosphate levels trigger an increase in total FGF23, assessed by cFGF23, and iFGF23 levels. In contrast, iron deficiency and anemia lead to an increase in cFGF23 but only mild elevations of iFGF23. In CKD, where FGF23 cleavage is reduced, hyperphosphatemia, iron deficiency and anemia increased circulating levels of both total cFGF23 and iFGF23. Administration of ferric citrate (FC), which decreases intestinal phosphate absorption, increases circulating iron and corrects anemia, results in reductions of both total and intact FGF23.
Phosphate and FGF23: an old relationship
Extracellular Pi is a signaling molecule27, but the direct effects of phosphate on FGF23 production in bone and bone cells remain unclear and are context dependent. The sodium phosphate co-transporter Slc20a2 or Pit2 may be the phosphate sensing mechanism in bone cells28. Alternatively, phosphate may regulate FGF23 production by increasing FGFR1 signaling29, already known to increase Fgf23 transcription30, 31. In these settings, phosphate may also increase FGF23 protection against proteolytic cleavage, by increasing Galnt3 expression and FGF23 O-glycosylation at the cleavage site29. However, suppression of Slc20a2 increases Fgf23 expression28 and animals with global deletion of Galnt3, can still modulate Fgf23 transcription and even FGF23 processing in response to phosphate32. While these mechanisms illustrate several phosphate related effects, they fail to capture the full picture of FGF23 induction by phosphate, which likely combines multiple local, bone related and distant physiological effects.
Nevertheless, phosphate loading increases FGF23 levels in both humans and animals23, 33. Thus, reducing phosphate levels in CKD, should lower FGF23 by addressing a root cause of its elevation. Indeed, lowering FGF23 without sacrificing normal phosphate homeostasis is imperative given that patients with elevated serum phosphate levels are at high risk of developing end stage kidney disease (ESKD), coronary artery calcification, cardiovascular disease and mortality. FGF23 phosphaturic effects are conserved in early CKD, where increased FGF23 maintains serum phosphate levels by increasing urinary phosphate excretion34. In later stages of CKD, although FGF23 cannot maintain serum phosphate balance, FGF23 mitigates hyperphosphatemia, and studies showed that deletion of FGF23 or administration of anti-FGF23 antibodies to animals precipitated severe hyperphosphatemia that resulted in diffuse arterial calcification and early mortality35, 36.
Focusing on addressing phosphate alterations only, in order to reduce FGF23 in CKD has led however to mixed results. Reduction of the phosphate-to-protein ratio in the diet decreased serum phosphate but failed to correct FGF23 levels in ESKD patients over a short time period37. Nicotinamide, which inhibits the intestinal NPT2b expression in vivo and lowers serum phosphate levels in ESKD patients38–42, has only led to minor reductions in FGF23 in short term studies or showed no benefits in lowering phosphate or FGF23 in larger, long term studies43. Several phosphate binders, such as sevelamer and lanthanum carbonate, currently used to correct hyperphosphatemia in patients with advanced CKD, also failed to effectively decrease intact FGF23 in stage 3–4 CKD patients43–45. Thus solely targeting phosphate has had inconsistent and limited impact in preventing FGF23 elevations and FGF23 related outcomes in CKD.
Anemia, iron and FGF23
Like phosphate, iron is a critical element in mammal physiology, mostly for the synthesis of hemoglobin, myoglobin, and iron-containing enzymes. The predominant clinical manifestation of iron deficiency is anemia and at homeostasis, a tight control of iron levels is critical to maintain normal erythropoiesis and red blood cells (RBC) function. A link between iron and FGF23 regulation was first reported in 20113, 5, 46. In iron deficient patients and mice with normal kidney function, iron deficiency increased FGF23 production, but this was associated with increased proteolytic cleavage to yield FGF23 cleavage peptides3, 5, 46. Several studies have also shown that reduced iron leads to similar effects in culture4, 5. In addition to iron deficiency per se, erythropoietin (EPO) which is secreted in vivo in response to anemia, also increases FGF23 production and cleavage25, 26, triggering a response similar to iron deficiency. However, in patients and animals harboring mutations at the FGF23 cleavage site47, which stabilize FGF23 and cause autosomal dominant hypophosphatemic rickets (ADHR), iron deficiency increases FGF23 transcription and results in excess iFGF23 that is protected from cleavage, leading to functional consequences of hypophosphatemia, vitamin D deficiency, rickets and osteomalacia.3, 5, 46
FGF23 and anemia of CKD
True iron deficiency (a deficiency of total body iron) and functional iron deficiency (inadequate circulating iron despite normal or elevated body iron stores) are prevalent in CKD48. Anemia is also a common complication of CKD, which becomes nearly universal as CKD progresses.49 Although anemia is of multifactorial origin, limited iron bioavailability and an insufficient production of endogenous EPO by the injured kidneys to meet the erythropoietic demands are the major drivers. Given that CKD is analogous to ADHR in that it leads to a state of impaired FGF23 cleavage4, 50, ID, anemia or the use of EPO or EPO-stimulating agents (ESA) to cope with the relative EPO deficiency in CKD, have the potential to increase not only total cFGF23 but also iFGF23 in CKD.
Efforts to correct iron deficiency and/or anemia have also had mixed benefits on FGF23 production in CKD, and similar to phosphate interventions, may also be context dependent. Studies of intravenous (IV) iron administration reported a large variability in FGF23 response to iron therapies51–57. These effects may depend on compound, dose, duration, tolerability of the compound, and comorbidities.
Something old, something new: combined effects of phosphate and iron on FGF23 production
New emerging iron-based phosphate binders for the simultaneous management of hyperphosphatemia, iron deficiency and anemia in CKD might achieve a better control of FGF23 in CKD. One such a compound is ferric citrate (FC), which is commercialized in the United States as Auryxia (Akebia Biopharmaceuticals, Boston, MA, USA), and is effective in correcting hyperphosphatemia and iron deficiency in CKD10, 58. By targeting these independent mechanisms of FGF23 production, FC treatment reduced both cFGF23 and iFGF23 levels in two different experiments in Col4a3KO mice with CKD10. Our studies also suggest that reducing FGF23, in addition to correcting iron and phosphate balance, might be more beneficial to correct anemia. Indeed, FGF23 is also a risk factor for CKD patients to develop anemia59 and FGF23 excess could explain the relative EPO deficiency seen in patients with CKD60. Given FGF23 pro-inflammatory effects61, which accentuate functional iron deficiency in CKD62, reducing FGF23 might also lead to better management of iron stores, thus breaking a potential feed-forward loop of FGF23 stimulation. Consistent with these hypotheses, we found that, animals fed a 5% FC enriched diet, showed lower inflammatory markers and increased circulating EPO10.
Cardiovascular impact of ferric citrate treatment in CKD
As terminally differentiated cells, cardiac myocytes’ primary response to stress is hypertrophy. Initially adaptive, chronic myocardial stress leads to pathological left ventricular hypertrophy, which is the major CKD-related CVD event, a key mechanism of heart failure and ultimately death63, 64. Hyperphosphatemia has long been recognized as a risk factor for CVD in patients with ESKD and earlier stages of CKD8, 65, however, more recently, FGF23 has emerged as the major molecular mechanism linking phosphate to increased CVD risk16, 17.
FGF23 induces hypertrophic growth of cardiac myocytes in vitro and LVH in rodents16, 17 suggesting that FGF23 represents a novel therapeutic target for attenuating LVH and heart failure in CKD18, 66. Consistent with these observations, CKD mice treated with FC which resulted in reduced FGF23 also show a net improvement in cardiac function10, but corrections of phosphate and iron may also contribute to this improvement. Indeed, hyperphosphatemia alone, independently of FGF23, results in arterial calcification and early mortality67. Conversely, iron deficiency and anemia contribute to the development of cardiovascular outcomes68–70. Thus, whether the cardiovascular benefits of FC treatment are partially mediated by the increase in circulating iron, correction of anemia or reduced phosphate requires further study. Nonetheless animal studies show that FGF23 reduction in CKD, consistently leads to improvement of heart disease10, 66. The pro-hypertrophic effects of FGF23 on cardiac myocytes are mediated by FGFR4/PLCγ/calcineurin/NFAT signaling16, 17, and our studies suggest that activation of Ras/mitogen-activated protein kinase (MAPK) by FGF23 might lead to development of systolic dysfunction in mice10. A pathological FGF23 signaling through FGFR3, the highly expressed FGFR3 isoform in CKD mice18, 71, might be responsible for the onset and progression of systolic dysfunction. These effects are also potentiated by phosphate, given that administration of phosphate to mice rapidly induces cardiac MAPK activity and increases FGFR3 expression10. Thus FGF23 and phosphate reduction following FC treatment attenuates CKD associated systolic dysfunction10. It is also probable that increased iron and correction of anemia might also play a role in the improvement of cardiac function72, 73 (Figure 2) but additional studies are needed to confirm these effects.
Figure 2: Kidney and cardiovascular effects of impaired phosphate and iron metabolism in CKD.
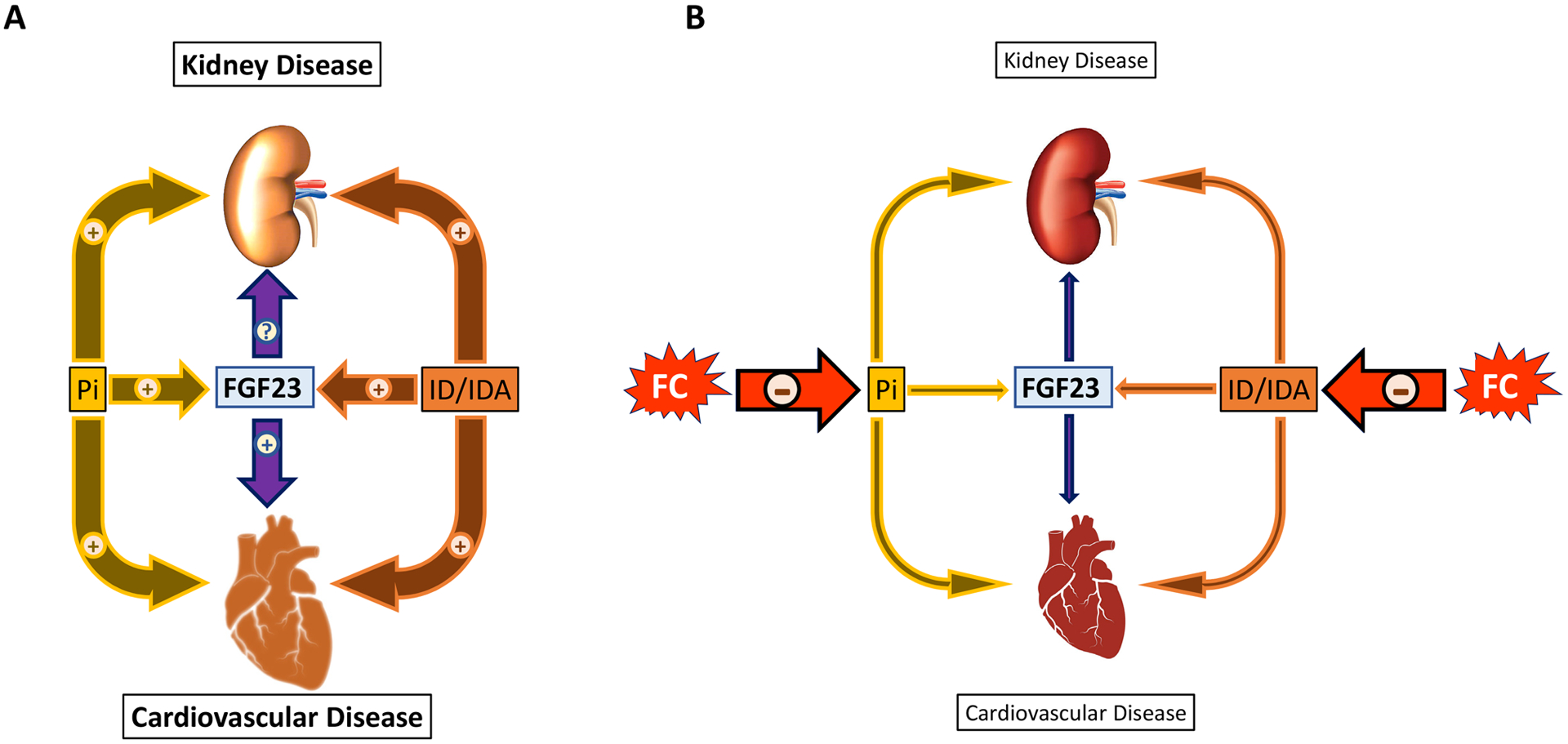
(A) In CKD, hyperphosphatemia, iron deficiency and anemia lead to progressive alterations in kidney and heart morphology and function and induce the production of FGF23. Excess FGF23 also targets the heart and contributes to development of cardiovascular disease and mortality may aggravate kidney disease progression. (B) Simultaneous reductions in phosphate and correction of iron balance and anemia by iron based phosphate binders, such as ferric citrate (FC), decrease FGF23 production and lead to improvement of kidney and cardiovascular disease.
Clinical perspectives: a silver bullet for CKD progression?
Despite current therapies, CKD patients remain at high risk for progression toward ESKD, and alternative renoprotective treatments that prevent further kidney function decline or reverse the course of the disease are needed. Most interestingly, FC treatment of mice with early CKD also resulted in marked improvement of kidney morphology and function, but had no effect on CKD progression when treatment was initiated later in the course of CKD10. Hyperphosphatemia74, 75 and excess FGF2376, 77 have long been recognized as risk factors for CKD progression. FGF23 is an independent predictor of progression of kidney disease in both adults and children with CKD78, 79, but several animal studies show that decrease of FGF23 in CKD has no immediate benefit66 or further reduces kidney function in rodents35. In contrast, phosphate restriction in several experimental models80–82 reduces markers of tubular injury and fibrosis, slowing the progression of the renal damage and delaying the appearance of ESKD. Thus, by aggravating hyperphosphatemia, reduced FGF23 might accentuate the onset and severity of CKD, and higher FGF23 might have a renoprotective effect in CKD35. However, alternative mechanisms may explain the underlying association between FGF23 levels and CKD progression. FGF23 may promote renal sodium absorption83 and suppression of angiotensin-converting enzyme 2 (ACE2)61, increasing blood pressure and the risk of faster CKD progression84. Finally, FGF23 also increases the total kidney phosphate burden, by increasing phosphaturia, which may also predict kidney disease progression85. Thus, this further emphasizes the need of simultaneous phosphate and FGF23 control, in early CKD, in order to slow CKD progression.
Correction of anemia and iron deficiency could also have an impact CKD progression86, but the renoprotective effects are more subtle87 and disputed, compared to the impact on cardiovascular disease. While reduced oxygen delivery could further impair kidney function, correction of anemia does not profoundly impact the rate of CKD progression88. However, it is unknown if the lack of robust and reproducible benefits on renal function, are due to the either lack of effects of increased hemoglobin (Hb) or adverse effects of high dose ESAs89, 90. Similarly, iron supplementation shows lack of effects on kidney function of either oral or IV iron administration in CKD91, 92, despite increased levels of kidney injury markers observed in infants and children with IDA93, 94 which are corrected by iron therapy. Nevertheless, increase in transferrin saturation following treatment of CKD mice with FC might have an important impact in slowing kidney disease progression10. Thus, combined reductions of phosphate, FGF23 and correction of anemia and iron balance (Figure 2) might explain why FC treatment resulted in lower rates of death or the progression to dialysis, lower rates of hospitalization, and fewer hospital days in a single-center pilot study of patients with advanced CKD95. Additional large multi-center clinical trials are needed to confirm these exciting preliminary findings.
CONCLUSION
CKD is a public health epidemic requiring the development of new ways to prevent and treat CKD progression and associated CVD, which is critical for reducing the overwhelming mortality in affected patients. Comprehensive investigative efforts in clinical and basic research established that reductions of FGF23, a mediator of CVD in CKD, combined with correction of hyperphosphatemia and iron deficiency may provide a potential therapeutic approach to delay CKD progression, improve cardiac function and ultimately increase lifespan in CKD. Although further studies are required to determine the individual mechanisms of end-organ injury inflicted by exposure to FGF23, phosphate and iron, including dose and duration, the functional effects of these findings can serve as the foundation for novel scalable multifactorial therapeutic strategies to be tested in future studies aimed at improving outcomes in patients with CKD.
KEY POINTS.
Greater long-term exposure to elevated FGF23 levels leads to increased risks of cardiovascular disease and death in CKD.
Hyperphosphatemia, iron deficiency and anemia in CKD contribute to excess circulating FGF23, development of cardiac disease and premature death.
Corrections of disordered phosphate and iron metabolism in CKD dramatically reduce FGF23 levels.
Simultaneous corrections of hyperphosphatemia, iron, anemia and FGF23 result in marked improvement in kidney and cardiac function and result in prolonged lifespan in animal models of CKD.
Financial support and sponsorship
This study was supported by American Heart Association grant 19POST34380583 to G.C and by grants from National Institute of Health (R01DK102815, R01DK114158) to V.D.
Footnotes
Conflicts of interest
VD receives research funding from Akebia and has received research funding from Vifor Pharma and consulting honoraria from Keryx Biopharmaceuticals, Vifor Pharma, Luitpold and Amgen.
All other authors have nothing to disclose.
REFERENCES AND RECOMMENDED READING
- 1.CDC. Chronic Kidney Disease in the United States, 2019. CDCgov; 2019. [Google Scholar]
- 2.DALYs GBD, Collaborators H. Global, regional, and national disability-adjusted life-years (DALYs) for 315 diseases and injuries and healthy life expectancy (HALE), 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016; 388: 1603–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clinkenbeard EL, Farrow EG, Summers LJ, et al. Neonatal iron deficiency causes abnormal phosphate metabolism by elevating FGF23 in normal and ADHR mice. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 2014; 29: 361–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.David V, Martin A, Isakova T, et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney international 2016; 89: 135–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farrow EG, Yu X, Summers LJ, et al. Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor-23 (Fgf23) knock-in mice. Proceedings of the National Academy of Sciences of the United States of America 2011; 108: E1146–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanudel MR, Chua K, Rappaport M, et al. Effects of dietary iron intake and chronic kidney disease on fibroblast growth factor 23 metabolism in wild-type and hepcidin knockout mice. American journal of physiology Renal physiology 2016; 311: F1369–f1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Isakova T, Xie H, Yang W, et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. Jama 2011; 305: 2432–2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kestenbaum B, Sampson JN, Rudser KD, et al. Serum phosphate levels and mortality risk among people with chronic kidney disease. Journal of the American Society of Nephrology : JASN 2005; 16: 520–528. [DOI] [PubMed] [Google Scholar]
- 9.Levin A, Thompson CR, Ethier J, et al. Left ventricular mass index increase in early renal disease: impact of decline in hemoglobin. American journal of kidney diseases : the official journal of the National Kidney Foundation 1999; 34: 125–134. [DOI] [PubMed] [Google Scholar]
- 10**.Francis C, Courbon G, Gerber C, et al. Ferric citrate reduces fibroblast growth factor 23 levels and improves renal and cardiac function in a mouse model of chronic kidney disease. Kidney international 2019; 96: 1346–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article shows that combined corrections of phosphate and iron metabolism in a mouse model of CKD, reduces FGF23, corrects anemia, prevents the development of cardiac disease, slows the progression of kidney disease and improves survival.
- 11.Shimada T, Urakawa I, Yamazaki Y, et al. FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem Biophys Res Commun 2004; 314: 409–414. [DOI] [PubMed] [Google Scholar]
- 12.Shimada T, Hasegawa H, Yamazaki Y, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 2004; 19: 429–435. [DOI] [PubMed] [Google Scholar]
- 13.Liu S, Tang W, Zhou J, et al. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. Journal of the American Society of Nephrology : JASN 2006; 17: 1305–1315. [DOI] [PubMed] [Google Scholar]
- 14.Pool LR, Wolf M. FGF23 and Nutritional Metabolism. Annu Rev Nutr 2017; 37: 247–268. [DOI] [PubMed] [Google Scholar]
- 15.Scialla JJ, Xie H, Rahman M, et al. Fibroblast growth factor-23 and cardiovascular events in CKD. Journal of the American Society of Nephrology : JASN 2014; 25: 349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Faul C, Amaral AP, Oskouei B, et al. FGF23 induces left ventricular hypertrophy. The Journal of clinical investigation 2011; 121: 4393–4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grabner A, Amaral AP, Schramm K, et al. Activation of Cardiac Fibroblast Growth Factor Receptor 4 Causes Left Ventricular Hypertrophy. Cell metabolism 2015; 22: 1020–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neuburg S, Dussold C, Gerber C, et al. Genetic background influences cardiac phenotype in murine chronic kidney disease. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association 2018; 33: 1129–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bhattacharyya N, Wiench M, Dumitrescu C, et al. Mechanism of FGF23 processing in fibrous dysplasia. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 2012; 27: 1132–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kato K, Jeanneau C, Tarp MA, et al. Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O-glycosylation. J Biol Chem 2006; 281: 18370–18377. [DOI] [PubMed] [Google Scholar]
- 21.Wolf M, Koch TA, Bregman DB. Effects of iron deficiency anemia and its treatment on fibroblast growth factor 23 and phosphate homeostasis in women. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 2013; 28: 1793–1803. [DOI] [PubMed] [Google Scholar]
- 22.Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiological reviews 2012; 92: 131–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perwad F, Azam N, Zhang MY, et al. Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice. Endocrinology 2005; 146: 5358–5364. [DOI] [PubMed] [Google Scholar]
- 24.Meir T, Durlacher K, Pan Z, et al. Parathyroid hormone activates the orphan nuclear receptor Nurr1 to induce FGF23 transcription. Kidney international 2014; 86: 1106–1115. [DOI] [PubMed] [Google Scholar]
- 25.Clinkenbeard EL, Hanudel MR, Stayrook KR, et al. Erythropoietin stimulates murine and human fibroblast growth factor-23, revealing novel roles for bone and bone marrow. Haematologica 2017; 102: e427–e430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rabadi S, Udo I, Leaf DE, et al. Acute blood loss stimulates fibroblast growth factor 23 production. American journal of physiology Renal physiology 2018; 314: F132–F139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khoshniat S, Bourgine A, Julien M, et al. The emergence of phosphate as a specific signaling molecule in bone and other cell types in mammals. Cellular and molecular life sciences : CMLS 2011; 68: 205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28**.Bon N, Frangi G, Sourice S, et al. Phosphate-dependent FGF23 secretion is modulated by PiT2/Slc20a2. Molecular metabolism 2018; 11: 197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article shows that phosphate effects of FGF23 production and secretion are partially mediated by sodium phosphate cotrasporter, PIT2 in bone cells.
- 29*.Takashi Y, Kosako H, Sawatsubashi S. Activation of unliganded FGF receptor by extracellular phosphate potentiates proteolytic protection of FGF23 by its O-glycosylation. 2019; 116: 11418–11427. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article shows that phosphate actives FGFR1 in bone cells and leads to FGF23 stabilization, via activation of GALNT3 dependent glycosylation of FGF23 cleavage site.
- 30.Martin A, Liu S, David V, et al. Bone proteins PHEX and DMP1 regulate fibroblastic growth factor Fgf23 expression in osteocytes through a common pathway involving FGF receptor (FGFR) signaling. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 2011; 25: 2551–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiao Z, Huang J, Cao L, et al. Osteocyte-specific deletion of Fgfr1 suppresses FGF23. PloS one 2014; 9: e104154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ichikawa S, Austin AM, Gray AK, et al. Dietary phosphate restriction normalizes biochemical and skeletal abnormalities in a murine model of tumoral calcinosis. Endocrinology 2011; 152: 4504–4513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burnett SM, Gunawardene SC, Bringhurst FR, et al. Regulation of C-terminal and intact FGF-23 by dietary phosphate in men and women. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 2006; 21: 1187–1196. [DOI] [PubMed] [Google Scholar]
- 34.Isakova T, Wahl P, Vargas GS, et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney international 2011; 79: 1370–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35**.Clinkenbeard EL, Noonan ML, Thomas JC, et al. Increased FGF23 protects against detrimental cardio-renal consequences during elevated blood phosphate in CKD. JCI insight 2019; 4. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article shows that massive reductions of FGF23 in a mouse model of CKD, without phosphate corrections, leads to severe hyperphosphatemia which aggravates both kidney and cardiac disease.
- 36.Shalhoub V, Shatzen EM, Ward SC, et al. FGF23 neutralization improves chronic kidney disease-associated hyperparathyroidism yet increases mortality. The Journal of clinical investigation 2012; 122: 2543–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37*.Tsai WC, Wu HY, Peng YS, et al. Short-Term Effects of Very-Low-Phosphate and Low-Phosphate Diets on Fibroblast Growth Factor 23 in Hemodialysis Patients: A Randomized Crossover Trial. Clinical journal of the American Society of Nephrology : CJASN 2019; 14: 1475–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that reductions of phosphate intake in pateints with ESKD, corrects hyperphosphatemia but does not impact circulating levels of FGF23.
- 38.Cheng SC, Young DO, Huang Y, et al. A randomized, double-blind, placebo-controlled trial of niacinamide for reduction of phosphorus in hemodialysis patients. Clinical journal of the American Society of Nephrology : CJASN 2008; 3: 1131–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ginsberg C, Ix JH. Nicotinamide and phosphate homeostasis in chronic kidney disease. Current opinion in nephrology and hypertension 2016; 25: 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lenglet A, Liabeuf S, El Esper N, et al. Efficacy and safety of nicotinamide in haemodialysis patients: the NICOREN study. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association 2017; 32: 870–879. [DOI] [PubMed] [Google Scholar]
- 41.Sabbagh Y, O’Brien SP, Song W, et al. Intestinal npt2b plays a major role in phosphate absorption and homeostasis. Journal of the American Society of Nephrology : JASN 2009; 20: 2348–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takahashi Y, Tanaka A, Nakamura T, et al. Nicotinamide suppresses hyperphosphatemia in hemodialysis patients. Kidney international 2004; 65: 1099–1104. [DOI] [PubMed] [Google Scholar]
- 43**.Ix JH, Isakova T, Larive B, et al. Effects of Nicotinamide and Lanthanum Carbonate on Serum Phosphate and Fibroblast Growth Factor-23 in CKD: The COMBINE Trial. Journal of the American Society of Nephrology : JASN 2019; 30: 1096–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article shows that administration of non iron-based phosphate binder lanthanum carbonate and/or nicotinamide to patients with advanced CKD fails to correct serum FGF23 levels.
- 44.Bouma-de Krijger A, van Ittersum FJ, Hoekstra T, et al. Short-term effects of sevelamer-carbonate on fibroblast growth factor 23 and pulse wave velocity in patients with normophosphataemic chronic kidney disease Stage 3. Clinical kidney journal 2019; 12: 678–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruggiero B, Trillini M, Tartaglione L, et al. Effects of Sevelamer Carbonate in Patients With CKD and Proteinuria: The ANSWER Randomized Trial. American journal of kidney diseases : the official journal of the National Kidney Foundation 2019; 74: 338–350. [DOI] [PubMed] [Google Scholar]
- 46.Imel EA, Peacock M, Gray AK, et al. Iron modifies plasma FGF23 differently in autosomal dominant hypophosphatemic rickets and healthy humans. The Journal of clinical endocrinology and metabolism 2011; 96: 3541–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Consortium ADHR. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet 2000; 26: 345–348. [DOI] [PubMed] [Google Scholar]
- 48.Zumbrennen-Bullough K, Babitt JL. The iron cycle in chronic kidney disease (CKD): from genetics and experimental models to CKD patients. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association 2014; 29: 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Foundation NK. KDOQI Clinical Practice Guidelines and Clinical Practice Recommendations for Anemia in Chronic Kidney Disease. American journal of kidney diseases : the official journal of the National Kidney Foundation 2006; 47: S11–145. [DOI] [PubMed] [Google Scholar]
- 50.Smith ER, Cai MM, McMahon LP, et al. Biological variability of plasma intact and C-terminal FGF23 measurements. The Journal of clinical endocrinology and metabolism 2012; 97: 3357–3365. [DOI] [PubMed] [Google Scholar]
- 51.Takeda Y, Komaba H, Goto S, et al. Effect of intravenous saccharated ferric oxide on serum FGF23 and mineral metabolism in hemodialysis patients. American journal of nephrology 2011; 33: 421–426. [DOI] [PubMed] [Google Scholar]
- 52.Prats M, Font R, Garcia C, et al. Effect of ferric carboxymaltose on serum phosphate and C-terminal FGF23 levels in non-dialysis chronic kidney disease patients: post-hoc analysis of a prospective study. BMC Nephrol 2013; 14: 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schaefer B, Wurtinger P, Finkenstedt A, et al. Choice of High-Dose Intravenous Iron Preparation Determines Hypophosphatemia Risk. PloS one 2016; 11: e0167146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54*.Fukao W, Hasuike Y, Yamakawa T, et al. Oral Versus Intravenous Iron Supplementation for the Treatment of Iron Deficiency Anemia in Patients on Maintenance Hemodialysis-Effect on Fibroblast Growth Factor-23 Metabolism. Journal of renal nutrition : the official journal of the Council on Renal Nutrition of the National Kidney Foundation 2018; 28: 270–277. [DOI] [PubMed] [Google Scholar]; This article shows that oral iron and IV iron administration to dialysis patients reduces total cFGF23. In contrast, oral iron has no effect on iFGF23 while IV iron increases it.
- 55.Honda H, Tanaka K, Michihata T, et al. Differential Impacts of Intravenous Iron Administration and Iron-Containing Phosphate Binders on Serum Intact Fibroblast Growth Factor 23 Levels. Blood purification 2019; 47 Suppl 2: 63–69. [DOI] [PubMed] [Google Scholar]
- 56*.Wolf M, Chertow GM, Macdougall IC, et al. Randomized trial of intravenous iron-induced hypophosphatemia. JCI insight 2018; 3. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article confirms that slow-release iron compounds are efficient in correcting anemia but paradoxically increase circulating levels of intact FGF23, and its phosphaturic effects, leading to hypophosphatemia.
- 57.Roberts MA, Huang L, Lee D, et al. Effects of intravenous iron on fibroblast growth factor 23 (FGF23) in haemodialysis patients: a randomized controlled trial. BMC Nephrol 2016; 17: 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fishbane S, Block GA, Loram L, et al. Effects of Ferric Citrate in Patients with Nondialysis-Dependent CKD and Iron Deficiency Anemia. Journal of the American Society of Nephrology : JASN 2017; 28: 1851–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nam KH, Kim H, An SY, et al. Circulating Fibroblast Growth Factor-23 Levels are Associated with an Increased Risk of Anemia Development in Patients with Nondialysis Chronic Kidney Disease. Scientific reports 2018; 8: 7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Agoro R, Montagna A, Goetz R, et al. Inhibition of fibroblast growth factor 23 (FGF23) signaling rescues renal anemia. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 2018; 32: 3752–3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dai B, David V, Martin A, et al. A comparative transcriptome analysis identifying FGF23 regulated genes in the kidney of a mouse CKD model. PloS one 2012; 7: e44161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Singh S, Grabner A, Yanucil C, et al. Fibroblast growth factor 23 directly targets hepatocytes to promote inflammation in chronic kidney disease. Kidney international 2016; 90: 985–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Silberberg JS, Barre PE, Prichard SS, et al. Impact of left ventricular hypertrophy on survival in end-stage renal disease. Kidney international 1989; 36: 286–290. [DOI] [PubMed] [Google Scholar]
- 64.Glassock RJ, Pecoits-Filho R, Barberato SH. Left ventricular mass in chronic kidney disease and ESRD. Clinical journal of the American Society of Nephrology : CJASN 2009; 4 Suppl 1: S79–91. [DOI] [PubMed] [Google Scholar]
- 65.Block GA, Klassen PS, Lazarus JM, et al. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. Journal of the American Society of Nephrology : JASN 2004; 15: 2208–2218. [DOI] [PubMed] [Google Scholar]
- 66**.Dussold C, Gerber C, White S, et al. DMP1 prevents osteocyte alterations, FGF23 elevation and left ventricular hypertrophy in mice with chronic kidney disease. Bone research 2019; 7: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]; This manuscript demonstrates that correction of FGF23 in mice with CKD, prevents the development of cardiovascular disease, despite persistent hyperphosphatemia and increased blood pressure.
- 67.Tonelli M, Sacks F, Pfeffer M, et al. Relation between serum phosphate level and cardiovascular event rate in people with coronary disease. Circulation 2005; 112: 2627–2633. [DOI] [PubMed] [Google Scholar]
- 68.Abramson JL, Jurkovitz CT, Vaccarino V, et al. Chronic kidney disease, anemia, and incident stroke in a middle-aged, community-based population: the ARIC Study. Kidney international 2003; 64: 610–615. [DOI] [PubMed] [Google Scholar]
- 69.Chang JM, Chen SC, Huang JC, et al. Anemia and left ventricular hypertrophy with renal function decline and cardiovascular events in chronic kidney disease. The American journal of the medical sciences 2014; 347: 183–189. [DOI] [PubMed] [Google Scholar]
- 70.Nair D, Shlipak MG, Angeja B, et al. Association of anemia with diastolic dysfunction among patients with coronary artery disease in the Heart and Soul Study. The American journal of cardiology 2005; 95: 332–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Touchberry CD, Green TM, Tchikrizov V, et al. FGF23 is a novel regulator of intracellular calcium and cardiac contractility in addition to cardiac hypertrophy. American journal of physiology Endocrinology and metabolism 2013; 304: E863–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Silverberg DS, Wexler D, Blum M, et al. The use of subcutaneous erythropoietin and intravenous iron for the treatment of the anemia of severe, resistant congestive heart failure improves cardiac and renal function and functional cardiac class, and markedly reduces hospitalizations. J Am Coll Cardiol 2000; 35: 1737–1744. [DOI] [PubMed] [Google Scholar]
- 73.Ponikowski P, Filippatos G, Colet JC, et al. The impact of intravenous ferric carboxymaltose on renal function: an analysis of the FAIR-HF study. Eur J Heart Fail 2015; 17: 329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Scialla JJ, Astor BC, Isakova T, et al. Mineral metabolites and CKD progression in African Americans. Journal of the American Society of Nephrology : JASN 2013; 24: 125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schwarz S, Trivedi BK, Kalantar-Zadeh K, et al. Association of disorders in mineral metabolism with progression of chronic kidney disease. Clinical journal of the American Society of Nephrology : CJASN 2006; 1: 825–831. [DOI] [PubMed] [Google Scholar]
- 76.Titan SM, Zatz R, Graciolli FG, et al. FGF-23 as a predictor of renal outcome in diabetic nephropathy. Clinical journal of the American Society of Nephrology : CJASN 2011; 6: 241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kendrick J, Cheung AK, Kaufman JS, et al. FGF-23 associates with death, cardiovascular events, and initiation of chronic dialysis. Journal of the American Society of Nephrology : JASN 2011; 22: 1913–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fliser D, Kollerits B, Neyer U, et al. Fibroblast growth factor 23 (FGF23) predicts progression of chronic kidney disease: the Mild to Moderate Kidney Disease (MMKD) Study. Journal of the American Society of Nephrology : JASN 2007; 18: 2600–2608. [DOI] [PubMed] [Google Scholar]
- 79.Portale AA, Wolf MS, Messinger S, et al. Fibroblast Growth Factor 23 and Risk of CKD Progression in Children. Clinical journal of the American Society of Nephrology : CJASN 2016; 11: 1989–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Koizumi T, Murakami K, Nakayama H, et al. Role of dietary phosphorus in the progression of renal failure. Biochem Biophys Res Commun 2002; 295: 917–921. [DOI] [PubMed] [Google Scholar]
- 81.Kusano K, Segawa H, Ohnishi R, et al. Role of low protein and low phosphorus diet in the progression of chronic kidney disease in uremic rats. Journal of nutritional science and vitaminology 2008; 54: 237–243. [DOI] [PubMed] [Google Scholar]
- 82.Omede F, Zhang S, Johnson C, et al. Dietary phosphate restriction attenuates polycystic kidney disease in mice. American journal of physiology Renal physiology 2020; 318: F35–f42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Andrukhova O, Slavic S, Smorodchenko A, et al. FGF23 regulates renal sodium handling and blood pressure. EMBO molecular medicine 2014; 6: 744–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Warady BA, Abraham AG, Schwartz GJ, et al. Predictors of Rapid Progression of Glomerular and Nonglomerular Kidney Disease in Children and Adolescents: The Chronic Kidney Disease in Children (CKiD) Cohort. American journal of kidney diseases : the official journal of the National Kidney Foundation 2015; 65: 878–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Santamaria R, Diaz-Tocados JM, Pendon-Ruiz de Mier MV, et al. Increased Phosphaturia Accelerates The Decline in Renal Function: A Search for Mechanisms. Scientific reports 2018; 8: 13701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kuriyama S, Tomonari H, Yoshida H, et al. Reversal of anemia by erythropoietin therapy retards the progression of chronic renal failure, especially in nondiabetic patients. Nephron 1997; 77: 176–185. [DOI] [PubMed] [Google Scholar]
- 87.Elliott S, Tomita D, Endre Z. Erythropoiesis stimulating agents and reno-protection: a meta-analysis. BMC Nephrol 2017; 18: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Drueke TB, Locatelli F, Clyne N, et al. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. The New England journal of medicine 2006; 355: 2071–2084. [DOI] [PubMed] [Google Scholar]
- 89.Covic A, Nistor I, Donciu MD, et al. Erythropoiesis-stimulating agents (ESA) for preventing the progression of chronic kidney disease: a meta-analysis of 19 studies. American journal of nephrology 2014; 40: 263–279. [DOI] [PubMed] [Google Scholar]
- 90.Thavarajah S, Choi MJ. The Use of Erythropoiesis-Stimulating Agents in Patients With CKD and Cancer: A Clinical Approach. American journal of kidney diseases : the official journal of the National Kidney Foundation 2019; 74: 667–674. [DOI] [PubMed] [Google Scholar]
- 91.Kim SM, Lee CH, Oh YK, et al. The effects of oral iron supplementation on the progression of anemia and renal dysfunction in patients with chronic kidney disease. Clinical nephrology 2011; 75: 472–479. [DOI] [PubMed] [Google Scholar]
- 92.Macdougall IC, Bock AH, Carrera F, et al. Renal function in patients with non-dialysis chronic kidney disease receiving intravenous ferric carboxymaltose: an analysis of the randomized FIND-CKD trial. BMC Nephrol 2017; 18: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.El-Shimi MS, El-Farrash RA, Ismail EA, et al. Renal functional and structural integrity in infants with iron deficiency anemia: relation to oxidative stress and response to iron therapy. Pediatric nephrology (Berlin, Germany) 2015; 30: 1835–1842. [DOI] [PubMed] [Google Scholar]
- 94.Hassan RH, Kandil SM, Zeid MS, et al. Kidney injury in infants and children with iron-deficiency anemia before and after iron treatment. Hematology (Amsterdam, Netherlands) 2017; 22: 565–570. [DOI] [PubMed] [Google Scholar]
- 95**.Block GA, Block MS, Smits G, et al. A Pilot Randomized Trial of Ferric Citrate Coordination Complex for the Treatment of Advanced CKD. Journal of the American Society of Nephrology : JASN 2019; 30: 1495–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]; This pilot randomized single-center clinical trial shows that treatment of advanced CKD patients with fixed doses of ferric citrate improves biochemical endpoints, slows CKD progression, and improves other hard clinical end points.