

Abstract
Objective:
We evaluated the effects of the BAFF targeting antibody, Belimumab, on human nonmemory B cell pools. Human B cell pools were identified using surface markers adapted from mouse studies that specifically assessed reductions in immature B cells due to BAFF-depletion. SLE patients have high levels of both BAFF and immature B cells. Mechanistic mouse studies provide a framework for understanding human responses to therapies that target B cells.
Methods:
PBMCS were isolated from healthy donors and SLE patients on Belimumab or standard-of-care therapy (SCT). Cells were stained for flow cytometry to identify B cell subsets based on CD21/CD24. Differences in subset proportions were determined by one way ANOVA and Tukey’s post hoc test.
Results:
Patients treated with Belimumab show alterations in the nonmemory B cell pool characterized by a decrease in the Transitional 2 (T2) subset (p=0.002), and an increase in the proportion of Transitional 1 (T1) cells (p=0.005) as compared to healthy donors and SCT patients. The naïve B cell compartment showed no significant differences between the groups (p = 0.293).
Conclusion:
Using a translational approach, we show that Belimumab-mediated BAFF-depletion reduces the T2 subset in patients, similar to observations in mouse models with BAFF-depletion.
Keywords: Belimumab, transitional B cells, translational, Systemic Lupus Erythematosus, human B cell production
Background
Understanding the applicability of lupus mouse models to mechanisms that impact human B cell production will provide insights for predicting clinical outcomes of B cell-targeted therapies (1–3). Mouse studies have begun to evaluate the dynamics of B cell subsets and their relationship to factors that influence B cell production. One focus of study has been the relationship of B cell activating factor (BAFF) to dynamic changes in transitional and naïve B cell subsets within the B cell pool. These dynamics are maintained in spite of the decreased production of immature B cells in the bone marrow that occurs in aged mice (4, 5), thus underscoring their potential utility for understanding mechanisms of B cell production and dynamics across the human life-span.
Abnormal B cell production has been reported in systemic lupus erythematosus where patients have higher levels of BAFF and autoreactive immature B cells (6, 7). In mice, BAFF levels are important, as a reduction compromises transitional B cell maturation and excess promotes survival of auto-reactive immature B cells (8). To facilitate the translation of information gained from mouse studies, we developed a schema for phenotypically identifying human B cells based on CD21/CD24 co-expresion (2, 8). This model identifies analogous populations of nonmemory B cells in mice and humans (2).
Our goal was to determine whether targeting BAFF in lupus patients produces alterations in the the nonmemory B cells pool that are predictable based on what has been observed in mouse studies, thus providing mechanistic insights into the use of BAFF-targeting antibodies for treatment of autoimmune diseases, such as lupus, where autoreactive B cells contribute to pathology. We used the CD21/CD24 schema to identify transitional and naïve B cells in human peripheral blood from healthy controls and from patients with systemic lupus erythematosus (SLE). We assessed B cell subsets from patients on Belimumab, (Benlysta™), a monoclonal antibody that blocks BAFF activity and the first drug in 30 years approved by the US Food and Drug Administration for the treatment of SLE (9). We hypothesized that the nonmemory B cell pool in Belimumab patients would be altered resulting in diminished proportions of B cells within the late transitional, T2, subset.
Materials and Methods
Patients and Samples.
This study was comprised of 37 adult patients who fulfilled American College of Rheumatology Classification criteria for SLE (10). Disease activity was determined using SLE Disease activity 2000 (SLEDAI-2K) score and damage due to SLE using SLE Damage Index (SDI). Belimumab (Table 1) and SCT patients were actively undergoing treatment and none of them were on treatment time out at the time of the study. Subjects on Belimumab received 10 mg/kg dose per infusion on a monthly basis; samples for flow cytometry were collected at a routine clinical visit at the time of monthly administration of Belimumab (Supplemental Table 1). Subjects not on Belimumab were designated as standard of care therapy (SCT) (Supplemental Table 2).
Table 1.
SLE Patient Demographics and Baseline Characteristics
| Total | SCT | Belimumab | ||
|---|---|---|---|---|
| n = 37 | n = 24 | n = 13 | ||
| Age (years), mean ±SD | 41.76±15.48 | 41.50±14.29 | 42.23±18.0 | |
| Disease duration (years), mean ±SD | 10.94±11.40 | 10.17±10.40 | 12.38±13.39 | |
| Sex | ||||
| Female, No. (%) | 35(95) | 23 (95) | 12 (92) | |
| Male | 2 (<1) | 1 (<1) | 1 (<1) | |
| Race | ||||
| Caucasian, No (%) | 12 (32) | 8 (33) | 4 (31) | |
| African-American | 6 (16) | 3 (13) | 3 (23) | |
| Asian | 1 (<1) | 0 | 1 (<1) | |
| Hispanic | 18 (49) | 13 (54) | 5 (38) | |
| Prednisone (mg) | ||||
| <10, No. (%) | 28 (76) | 19 (79) | 9 (69) | |
| 11-20 | 7 (19) | 4 (17) | 3 (23) | |
| 21-30 | 1 (<1) | 1 (<1) | 0 | |
| 31-40 | 0 | 0 | 0 | |
| > 40 | 1 (<1) | 0 | 1 (<1) | |
| ANA | ||||
| Positive, No. (%) | 34 (92) | 22 (92) | 12 (92) | |
| Negative | 3 (<1) | 2 (<1) | 1 (<1) | |
| dsDNA | ||||
| Positive, No (%) | 13 (35) | 7 (29) | 6 (46) | |
| Negative | 24 (65) | 17 (71) | 7 (54) | |
| C3 | ||||
| Normal, No (%) | 28 (76) | 19 (68) | 9 (69) | |
| Low | 6 (16) | 2 (<1) | 4 (31) | |
| High | 3 (<1) | 3 (<1) | 0 | |
| C4 | ||||
| Normal, No (%) | 11 (30) | 9 (38) | 2 (<1) | |
| Low | 26 (70) | 15 (63) | 11 (85) | |
| High | 0 | 0 | 0 | |
| WBC (bil/L) | ||||
| <2000, No (%) | 1 (<1) | 1 (<1) | 0 | |
| 2000-3000 | 4 (<1) | 1 (<1) | 3 (<1) | |
| 3001-4000 | 10 (27) | 7 (29) | 3 (<1) | |
| >4000 | 22 (59) | 15 (63) | 7 (54) | |
| Lymphs | ||||
| Normal, No (%) | 33 (89) | 21 (88) | 12 (92) | |
| Low | 1 (<1) | 0 | 1 (<1) | |
| High | 3 (<1) | 3 (<1) | 0 | |
| Platelets (bil/L) | ||||
| ≤100, 000, No. (%) | 1 (<1) | 1 (<1) | 0 | |
| >100, 000 | 36 (97) | 23 (96) | 13(100) | |
| Hematuria* | ||||
| Present, No. (%) | 4 (11) | 2 (<1) | 2 (<1) | |
| None | 31(84) | 22 (92) | 9 (69) | |
| Proteinuria* | ||||
| Present, No (%) | 2 (<1) | 2 (<1) | 0 | |
| None | 32 (86) | 21 (88) | 11 (85) | |
| Pyuria* | ||||
| Present, No (%) | 8 (22) | 5 (21) | 3 (<1) | |
| None | 27 (73) | 19 (79) | 8 (62) | |
| SLEDAI-2K, Mean ±SD | 4.3 ±3.4 | 4.1 ±3.1 | 4.0 ±4.1 | |
| SDI, Mean ±SD | 1.3 ±1.5 | 0.7 ±0.8 | 2.3 ±2.0 | |
Categories with an (*) denote missing values.
Patients were consented in accordance with regulations under the Loma Linda University Health Institutional Review Board (LLU IRB). Peripheral blood (PB) was drawn for this study once during a routine clinic visit. Healthy PB was obtained from Leuko-pak leukocyte filters (Fenwal Laboratories, Lake Zurich, IL) donated by Lifestream (San Bernardino, CA). White blood cell (WBC) and other counts shown in Table 1 were obtained from the medical record of patients based on blood draws prior to that for flow cytometry. All human tissues were obtained and handled according to protocols approved by the LLU IRB. Blood samples were collected from SLE patients and processed to isolate peripheral blood mononuclear cells (PBMCs) using the Ficoll Paque method as previously described (2).
Flow Cytometry.
Cells were thawed and stained with antibodies against CD19 Pacific Blue, CD21 FITC, IgD PE, CD24 PEcy7, CD27 APC Cy7 (Biolegend); IgM FITC, IgD FITC, and IgM PE (BD Biosciences); CD38 APC and IgD APC (Miltenyi Biotec), and with 7AAD or FVD 506 (eBioscience) as viability markers. After staining, cells were fixed with a 1% paraformaldehyde/PBS solution before analysis on a MACS-Quant Analyzer (Miltenyi Biotec). Flow cytometry data analysis was performed using Flowjo data analysis software (Tree Star, Ashland, OR).
Statistical Analysis.
Nonmemory B cell and PBMC subset percentages were normalized to 100%. Differences were graphed using Prism software (GraphPad Software, La Jolla, CA) and evaluated using IBM SPSS Statistic Software version 23.0 (Armonk, NY). Differences were assessed by one-way ANOVA and Tukey’s honest significant differences (HSD) post-hoc test (SPSS). Linear regression was used to evaluate direct relationships between SLEDAI score and transformed B cell subset proportions (SPSS).
Results
Application of CD21/CD24 translational model
B cell development and production are altered in SLE patients. These alterations arise due to a breakdown in tolerance mechanisms which may include inadequate negative or positive selection (Fig. 1A). BAFF has the potential to contribute to this process by providing pro-survival signals that prevent negative selection or promote positive selection of autoreactive cells (Fig. 1B) (5, 8).
Figure 1.
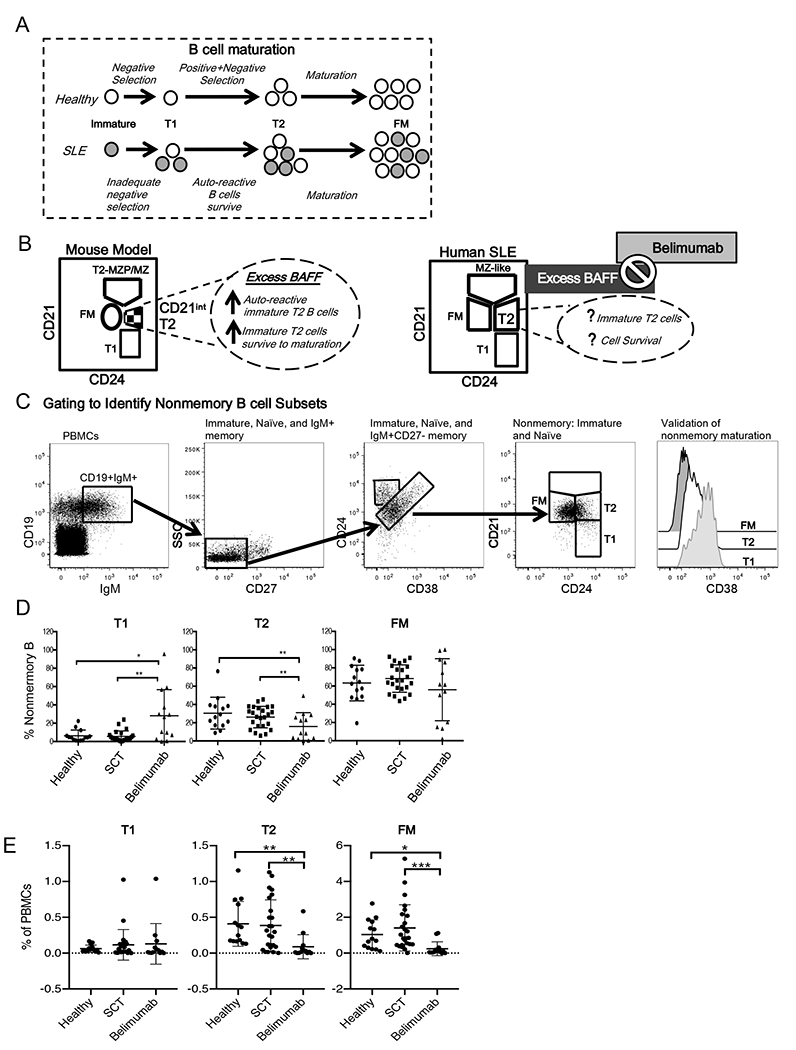
A. Diagram depicting alterations in mouse and human B cell development in healthy individuals vs. SLE. B. Diagram of analogous mouse (10) and human developmental B cell subsets identified by conserved surface markers, CD21/CD24. In mice, T2-MZP/MZP are marginal zone cells and their precursors (MZP) while CD21IntT2 cells are analogous to human T2 cells. C. Flow cytometry plots showing gating strategy to identify and validate human CD21/CD24 nonmemory B cell subsets. D-E. Human CD21/CD24 subsets were identified in peripheral blood from healthy individuals and patients on SCT and Belimumab. Percentages of CD21/CD24 subsets within the D. nonmemory B cell pool and E. peripheral blood mononuclear cells (PBMCs) are graphed. * p <0.05.; ** p<.01; *** p<.001. Healthy n=14, SCT =24, Belimumab=13.
The CD21/CD24 schema (Fig. 1B) identifies, within the nonmemory B cell pool, analogous B cell subsets in human and mouse tissues based on CD21/CD24 surface expression (2). We adapted this strategy from mouse studies that evaluated the effects of BAFF on transitional, follicular mature (FM) and marginal zone (MZ) subsets. MZ B cells are an antomically defined population of mouse B cells (based on their localization to the marginal zone of the spleen) that express unmutated IgM genes. Humans lack an anatomically defined MZ population, however there are circulating MZ-like B cells in humans, often referred to as IgM memory B cells, that phenotypically resemble mouse MZ B cells, but with mutated IgM genes (11). The human MZ-like population is excluded in our flow cytometry gating strategy (described below) to identify the nonmemory pool. We show this gate for reference in Fig. 1B because it was used in developing the CD21/CD24 schema in mouse (2, 8).
To identify the nonmemory B cell pool for evaluation using the CD21/CD24 schema, our strategy was to first gate on CD19+IgM+ cells (Fig. 1C first panel) to exclude class-switched (IgA+ IgG+, IgE+) memory B cells. We then gated on CD27– cells to exclude IgM+ memory B cells that that express CD27 (Fig 1C, second panel). Next, to exclude CD27– IgM+ memory B cells we used a gating strategy developed by Carsetti et al. that is based on co-expression of CD24 and CD38. In Carsetti’s strategy the memory cells, including CD27– memory B cells, are found within the CD24lo CD38bright population (Fig 1C, third panel) (7, 12, 13). Thus, our strategy identifies the human nonmemory B cell pool (Fig 1C, fourth panel) by excluding class switched memory B cells, as well as CD27+ and CD27– IgM+ memory B cells. Expression of CD38 (Fig. 1C, fifth panel) provided a means of validating, based on Carsetti’s criteria (12), the T1, T2 and FM subsets that we identified based on our successive gating and the CD21/CD24 co-staining.
To evaluate whether Belimumab altered nonmemory B cell subset proportions, we used the CD21/CD24 schema (Fig. 1C) to identify nonmemory T1, T2, and FM cells in healthy individuals, in patients on SCT, and in patients on Belimumab (Table 1).
Patient heterogeneity
Demographics and clinical characteristics of our diverse patient cohort on Belimumab and on SCT were noted (Table 1). Patients on Belimumab tended to have longer duration of disease and higher SDI scores compared to those on SCT. All patients had mild-moderate disease activity and no significant differences were noted between the two groups (Supplemental Table 2). Prior to assessment of statistically significant differences between groups, we evaluated the B cell subset proportions for normal distribution and skewedness (Supplemental Table 3).
Transitional subsets are altered in Belimumab patients
We examined the distribution of B cell subsets within the nonmemory B cell pool to determine whether Belimumab changed the distribution of these subsets in SLE patients in a manner predictable based on mouse models of BAFF depletion. Significant differences were observed in the T1 (p= 0.0021) and T2 (p= 0.0058) subsets, but not in the FM subsets (p= 0.2932) (Fig. 1D) within the nonmemory pool across the three groups. As predicted based on the mouse model, the proportion of T2 in the Belimumab group was significantly decreased when compared to either the SCT (p= 0.0127) or healthy (p= 0.0091) groups (Fig. 1 D). However, with respect to T1 subsets, the Belimumab group showed a significantly increased proportion of T1 cells compared to both patients on SCT (p= 0.0020) and healthy donors (p= 0.0133).
To gain insights into whether the change in proportions that we observed was due to an increase in number of T1, a decrease in number of T2, or both, we examined the percentage of T1, T2 and FM among PBMCs (Fig. 1E). We found no significant differences (p = 0.8213) within the T1 populations from the three groups. Belimumab patients showed a significant decreases in the their T2 (p = 0.0003) and FM (p < 0.0001) percentages compared to healthy and SCT. These data suggest that production of T1 cells is not altered following Belimumab treatment, but that T2 cells and their downstream progeny, FM cells, are reduced.
B cell subsets are associated with disease scores in SCT but not Belimumab patients
We then addressed whether alterations in B cell subsets within the nonmemory B cell pool were were associated with SLEDAI-2K score (Fig. 2). We used linear regression analysis to determine if SLEDAI-2K had a direct association with nonmemory subset proportions (Fig. 2). In the SCT group, we found that increasing SLEDAI-2K scores were correlated with a decrease in the proportion of both transitional T1 and T2 cells and an increase in FM cells within the nonmemory B cell pool (Fig 2 A−C).
Figure 2.
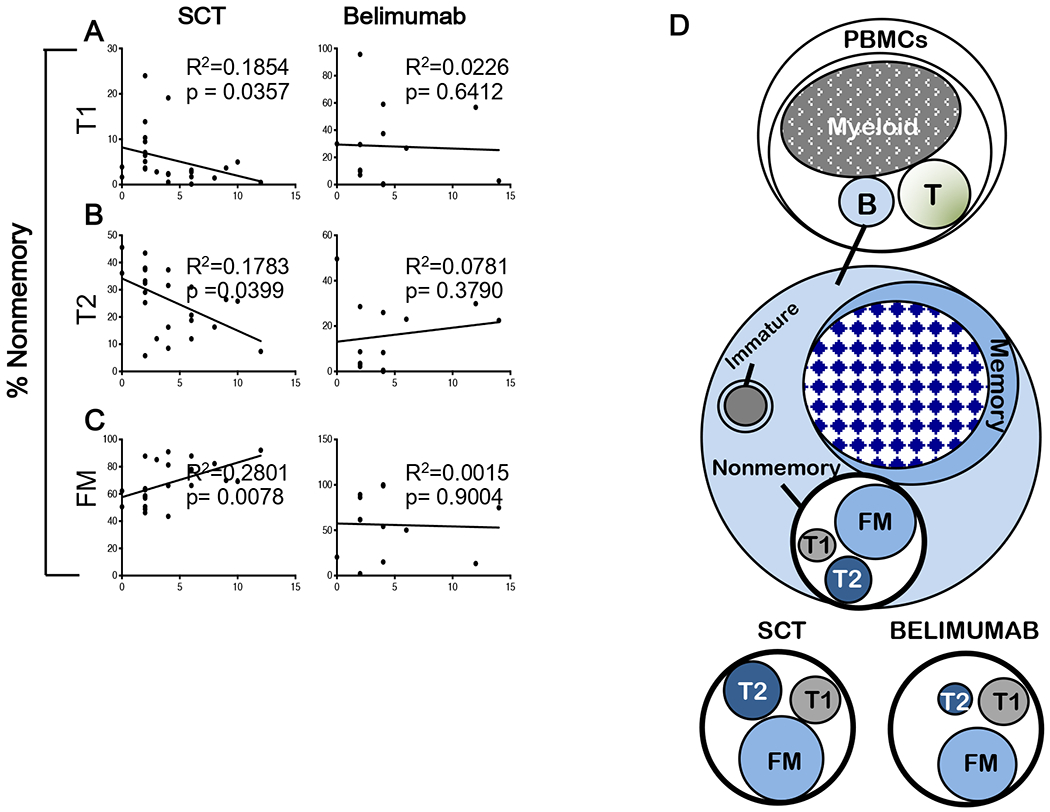
A-C. Linear regression analysis was performed for SCT and Belimumab patients to predict if there was a direct relationship between their nonmemory subset proportions and SLEDAI-2K scores. Slope was significantly different from non-zero when p < 0.05. D. Cartoon depicting changes in the nonmemory B cell pool in Belimumab vs Standard of Care (SCT) patients.
Discussion
Translational models can predict some outcomes from B cell targeted therapies at a cellular level, but responses to therapies can be heterogeneous (9). We set out to evaluate whether Belimumab specifically targets the T2 subset by reducing its proportion in relation to the nonmemory pool. Mouse studies show that BAFF-depletion causes a decrease in this population (8). Our study is novel in that it uses a strategy adapted from mouse studies of BAFF-induced alterations in B cell populations to asses for similar, clinically relevant, alterations in human B cell populations. Other human studies primarily use human-specific markers to identify nonmemory B cell subsets (7, 8, 13–15). Our data show that patients treated with the BAFF-targeting antibody, Belimumab, experienced a significant decrease in the proportion of T2 B cells within the nonmemory pool and PBMCs, when compared to healthy donors and patients on SCT. Mouse T1 cells are not as reliant on BAFF for maturation (2, 13). Consistent with these findings from mouse studies, the percentage of T1 cells in the PBMC pool did not change with Belimumab treatment suggesting that they were less dependent on BAFF than T2 B cells. Thus, Belimumab may be targeting specific transitional subsets, analogous to those targeted by BAFF-depletion in mice, regardless of disease activity in SLE patients.
The cartoon in Figure 2D depicts the differences we observed in SCT and Belimumab patients. B cells contribute a small percentage to the PBMC pool compared to myeloid lineage cells, thus cell numbers will be lower. Subset proportions within nonmemory B cells reflect regulatory mechanisms such as cell to cell interaction and access to growth factors crucial for development and maintenance of the naïve B cell pool (8).
A direct correlation was noted between SCT nonmemory subsets and SLEDAI-2K score, but not in Belimumab patients. We attribute these findings to broad non-targeted effects of SCT therapies that do not significantly alter B cell developmental subsets as opposed to a more targeted effect expected of Belimumab, based on mouse studies. The specific targeting of immature B cell subsets by Belimumab as demonstrated here provides a rationale for its use in SLE patients exhibiting abnormal B cell function due to loss of B cell tolerance as indicated by seropositivity for antinuclear antibody or double stranded DNA (9).
Supplementary Material
Acknowledgments
Funding
Transplantation Institute Loma Linda University Health Care, 2013 Grants to Promote Collaborative and Translational Research (GCAT) Award from Loma Linda University School of Medicine, (KJP and MED), NIH P20 MD006988, NIH 2R25 GM060507 (KJP), Bevra-Hahn Scholarship Award (MN), and Loma Linda University School of Medicine - Rheumatology Division Research Endowment Fund” (KT and MN).
Footnotes
Declaration of Conflicting Interests
Kimberly J. Payne is CEO, owns stock and is on the board of directors of Elf Zone, Inc. a startup company developing therapies for B cell acute lymphoblastic leukemia. Elf Zone, Inc. did not provide support for this work. The remaining authors have no commercial support or financial conflict of interest to report that pertains to this manuscript.
References
- 1.Rottman JB, Willis CR. Mouse models of systemic lupus erythematosus reveal a complex pathogenesis. Veterinary pathology. 2010;47(4):664–76. [DOI] [PubMed] [Google Scholar]
- 2.Benitez A, Weldon AJ, Tatosyan L, Velkuru V, Lee S, Milford TA, et al. Differences in mouse and human nonmemory B cell pools. Journal of immunology. 2014;192(10):4610–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vale AM, Schroeder HW Jr. Clinical consequences of defects in B-cell development. The Journal of allergy and clinical immunology. 2010;125(4):778–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mackay F, Figgett WA, Saulep D, Lepage M, Hibbs ML. B-cell stage and context-dependent requirements for survival signals from BAFF and the B-cell receptor. Immunological reviews. 2010;237(1):205–25. [DOI] [PubMed] [Google Scholar]
- 5.Vincent FB, Saulep-Easton D, Figgett WA, Fairfax KA, Mackay F. The BAFF/APRIL system: emerging functions beyond B cell biology and autoimmunity. Cytokine & growth factor reviews. 2013;24(3):203–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scholz JL, Oropallo MA, Sindhava V, Goenka R, Cancro MP. The role of B lymphocyte stimulator in B cell biology: implications for the treatment of lupus. Lupus. 2013;22(4):350–60. [DOI] [PubMed] [Google Scholar]
- 7.Kaminski DA, Wei C, Qian Y, Rosenberg AF, Sanz I. Advances in human B cell phenotypic profiling. Frontiers in immunology. 2012;3:302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meyer-Bahlburg A, Andrews SF, Yu KO, Porcelli SA, Rawlings DJ. Characterization of a late transitional B cell population highly sensitive to BAFF-mediated homeostatic proliferation. J Exp Med. 2008;205(1):155–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stohl W, Hilbert DM. The discovery and development of belimumab: the anti-BLyS-lupus connection. Nature biotechnology. 2012;30(1):69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis and rheumatism. 1997;40(9):1725. [DOI] [PubMed] [Google Scholar]
- 11.Weller S, Reynaud CA, Weill JC. Splenic marginal zone B cells in humans: where do they mutate their Ig receptor? European journal of immunology. 2005;35(10):2789–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carsetti R, Rosado MM, Wardmann H. Peripheral development of B cells in mouse and man. Immunological reviews. 2004;197:179–91. [DOI] [PubMed] [Google Scholar]
- 13.Palanichamy A, Barnard J, Zheng B, Owen T, Quach T, Wei C, et al. Novel human transitional B cell populations revealed by B cell depletion therapy. Journal of immunology. 2009;182(10):5982–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suryani S, Fulcher DA, Santner-Nanan B, Nanan R, Wong M, Shaw PJ, et al. Differential expression of CD21 identifies developmentally and functionally distinct subsets of human transitional B cells. Blood. 2010;115(3):519–29. [DOI] [PubMed] [Google Scholar]
- 15.Jacobi AM, Huang W, Wang T, Freimuth W, Sanz I, Furie R, et al. Effect of long-term belimumab treatment on B cells in systemic lupus erythematosus: extension of a phase II, double-blind, placebo-controlled, dose-ranging study. Arthritis and rheumatism. 2010;62(1):201–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.