

Summary
Although upland cotton (Gossypium hirsutism L.) originated in the tropics, this early maturity cotton can be planted as far north as 46°N in China due to the accumulation of numerous phenotypic and physiological adaptations during domestication. However, how the genome of early maturity cotton has been altered by strong human selection remains largely unknown. Herein, we report a cotton genome variation map generated by the resequencing of 436 cotton accessions. Whole‐genome scans for sweep regions identified 357 putative selection sweeps covering 4.94% (112 Mb) of the upland cotton genome, including 5184 genes. These genes were functionally related to flowering time control, hormone catabolism, ageing and defence response adaptations to environmental changes. A genome‐wide association study (GWAS) for seven early maturity traits identified 307 significant loci, 22.48% (69) of which overlapped with putative selection sweeps that occurred during the artificial selection of early maturity cotton. Several previously undescribed candidate genes associated with early maturity were identified by GWAS. This study provides insights into the genetic basis of early maturity in upland cotton as well as breeding resources for cotton improvement.
Keywords: upland cotton, early maturity, genome‐wide association analysis, flowering time
Introduction
Cotton is considered a model species for the study of polyploidy in plants and provides an organizational framework and phylogenetic perspective to understand the patterns and mechanisms of gene and genome evolution (Wendel et al., 2012). In the Gossypium genus, two diploid species (Gossypium herbaceum and Gossypium arboreum) as well as two allotetraploid species (Gossypium hirsutum and Gossypium barbadense) were independently domesticated in the old and new worlds (Renny‐Byfield et al., 2016; Wendel and Cronn, 2003; Wendel et al., 2012). Upland cotton (Gossypium hirsutum L. 2n = 52) has replaced the other three species and become the leading species, dominating world cotton commerce with a global yield of 75 million tons in 2010 (Wendel and Cronn, 2003).
The early maturity cotton has been widely planted in the north of China, including the cotton growing area of the Yellow River region (YRR), north of the Northwest Inland region (NIR) and the Northern Specific Early Maturity region (NSER) (Figure S1). The environment of northern China differs tremendously from that of Mesoamerica, which is the origin of Gossypium hirsutum. The development of early maturity cotton, which is seeded directly after wheat or rapeseed harvest, is a remarkable achievement that has helped address the grain‐cotton balance caused by population growth and loss of farmland in China. For example, between the years 1989 and 1994, ‘CRI16’, an elite variety of early maturity cotton, was widely planted in China, accounting for more than 367 million ha of planting area and greatly increasing the cotton yield on limited land. The whole growth period (WGP) of wild Gossypium hirsutum, which was initially domesticated at least 5000 years ago (Smith and Stephens, 1971), is actually quite long (typically >180 days); in addition, the presence of photoperiod sensitivity also hampers the use of wild Gossypium hirsutum in breeding programmes (Zhu and Kuraparthy, 2014). Following millennia of direct selection, early maturity of upland cotton has been greatly improved; in particular, the WGP has been reduced to approximately 140 days. In fact, breeding efforts over the past decade have decreased WGPs even further. For example, ‘Zhong213’ has a WGP of less than 105 days, with good quality and high yield (Li et al., 2017). Due to the breeding of early maturity cotton, the planting area has expanded to 46°N in the north of Xinjiang province. Recently, many studies have utilized next‐generation sequencing to investigate population evolution and domestication by genomic analysis in crops such as soybean (Lam et al., 2010; Valliyodan et al., 2016; Zhou et al., 2015), sorghum (Zheng et al., 2011), maize (Hufford et al., 2012), rice (Xun et al., 2012), cucumber (Qi et al., 2013) and tomato (Lin et al., 2014), which have tremendously advanced our understanding of crop domestication. However, how strong selective pressure has altered the genome of upland cotton, particularly the genetic changes underlying the adaptation to environmental changes (e.g. from tropical to temperate latitudes), and improved the early maturity remains unknown.
In this study, to gain a better understanding of the genome‐wide variations and genetic architecture of early maturity cotton, we collected a total of 436 cotton accessions with diverse WGP for genomic resequencing analysis with more than 10 Tb sequence data. In particular, the samples included 136 elite early maturity cotton accessions that have been bred in recent decades and provide a large genome variation map, providing a valuable early maturity genetic resource for future genomic‐enabled breeding.
Results
Genomic resequencing and variation calling
A total of 436 Gossypium hirsutum accessions were collected worldwide for genomic sequence analysis, comprising 32 wild Gossypium hirsutum lines and 404 cultivars. These accessions originated from different countries and have wide geographical distribution in China, representing more than 100 years of upland cotton breeding around the world (Figure 1a and Table S1). Resequencing of the 436 upland cotton accessions on the Illumina platform generated a total of 75 149 billion paired‐end reads (10.82 Tb of sequence). The average depth for each line was approximately two‐fold greater than that of the three published genomic variation maps of Gossypium hirsutum (11 × versus 6.90 (Wang et al., 2017a), 6.55 (Ma et al., 2018), 5.0 (Fang et al., 2017b)) (Table S1). Reads were mapped to the upland cotton cultivar ‘TM‐1’ reference genome (Wang et al., 2019) using BWA software (Li and Durbin, 2009), with the mapping rate ranging from 62.10% to 89.30% and an average unique mapping rate of 78.23% (Table S1). After strict application of quality controls and filters, we detected 10 118 884 high‐quality SNPs and 864 132 Indels (insertion and deletions shorter than or equal to 10 bp), of which 96.64% SNPs and 97.63% Indels were located on 26 chromosomes, respectively (Tables S2, S3 and Figures S2‐S4). We then analysed the SNP position information by genomic annotation. Most SNPs (91.06%) were in intergenic regions, with only a small portion (0.66%) located in coding sequences corresponding to 19 318 genes (Figure S3). Of these, 42 052 nonsynonymous SNPs were identified among 15 006 genes, causing codon changes. The Ka/Ks (Ka, number of nonsynonymous substitutions per nonsynonymous site; Ks, number of synonymous substitutions per synonymous site) ratio was 1.82, which was higher than those found in rice (1.29) (Xun et al., 2012), soybean (1.61) (Lam et al., 2010) and pigeon pea (1.18) (Varshney et al., 2017), indicating positive selection during domestication. To validate the accuracy of the SNP results, we employed Sanger sequencing on 316 randomly selected SNPs from 8 accessions, along with specific‐locus amplified fragment sequencing (SLAF) based on previously reported SNP data (43 accessions with 20 206 SNPs; Su et al., 2016a). These methods estimated the accuracy to be 94.9% and 90.9% (Tables S4‐S6). These variants can be used for cotton functional genomic investigations in the future.
Figure 1.
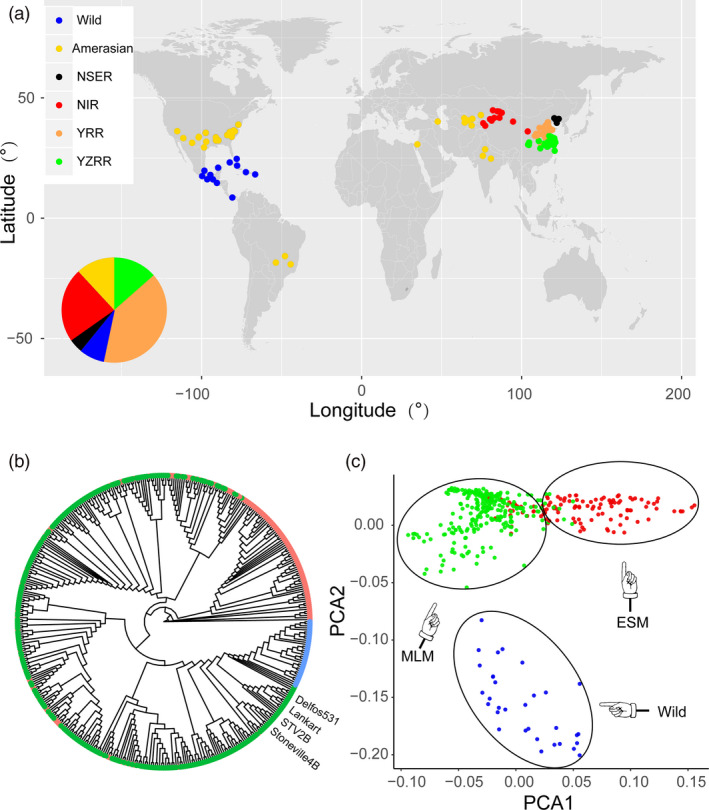
Overview of the SNP map of 436 upland cotton accessions. (a) The geographic distribution of the 436 accessions drawn using R (www.r‐project.org). Each accession is represented by a dot. Wild: 32 accessions originating from the islands in the Caribbean and Mesoamerica; Amerasian: 40 accessions primarily from central and southern Asia, America; NSER: Northern Specific Early Maturity region in China; NIR: Northwest Inland region in China; YRR: Yellow River region in China; YZRR: Yangtze River region in China. (b) Phylogenetic tree of 436 accessions inferred from 10 180 SNPs at fourfold‐degenerate sites, including three groups of wild (blue), MLM (green) and ESM (red). (c) PCA plots of 436 accessions.
Population structure and genetic diversity
The early maturity features of upland cotton were selected during domestication and improvement, resulting in significant variations among different populations (Figure S5). Based on their WGP, the 436 upland cotton accessions were assigned to three populations: early maturity and special early maturity cotton group (ESM), including 136 early maturity and special early maturity accessions developed in recent decades in China (WGP = 113.06 ± 10.85 days; Figure S1); medium and late maturity cotton group (MLM), including 268 medium and late maturity accessions (WGP = 134.66 ± 10.66 days); and Wild, including 32 wild Gossypium hirsutum lines (WGP > 180 days). To observe whether the phenotypic divergence among these three populations was supported at the genetic level, a rooted phylogenetic tree was generated using fourfold degenerate sites (4D SNPs) that represent neutral or near‐neutral variants in the complete set of 436 accessions using 10 180 SNPs (minor allele frequency (MAF) > 0.05; Figure 1b). Interestingly, the phylogenetic tree contained three major groups, corresponding roughly to the classification by phenotypic characterization. All the wild upland cotton lines clustered in an independent clade (blue), indicating that wild species exhibit not only considerable morphological but also genetic variations compared with cultivated species. Some landraces (older varieties representative of before the 1930s), such as ‘STV2B’, ‘STV4B’ and ‘Delfos531’ introduced from the US, were closer to the wild species. Similar observations were also consistent with previous research (Fang et al., 2017a). The remaining cultivars in the phylogenetic tree were clearly divided into separate clusters corresponding to ESM and MLM. Similar results were found via principal component analysis (PCA; Figure 1c). We then analysed the structure of the ancestral properties in each accession by increasing K (the number of populations) from 2 to 10 (Figure S6). The best suitable number of subpopulations was set to 2 by ‘chooseK’ packages (Raj et al., 2014) and revealed a very distinct divergence between wild Gossypium hirsutum and cultivated Gossypium hirsutum. However, for K = 3, the cultivar group revealed extensive admixture between ESM and MLM. Furthermore, to evaluate the genetic diversity among all samples, we further quantified variations in the nucleotide diversity and linkage disequilibrium (LD) for all three populations. Nucleotide diversity (π value) was significantly higher for the Wild population (1.01 × 10−3) than for the cultivars (MLM: 0.71 × 10−3 and ESM: 0.68 × 10−3; Figure S7). Earlier reports have suggested the presence of a high LD in cotton genomes (Fang et al., 2017b; Ma et al., 2018; Wang et al., 2017a). We used PLINK software (Purcell et al., 2007) to calculate correlation coefficient values (r 2) of alleles to measure the LD decay rate in the three populations. The cultivars (MLM: 288 kb and ESM: 320 kb) exhibited higher LD decay rates than the wild group (158 kb) when decreased to half its maximum value (Figure S8). The results of phylogenetic, PCA, population structure and LD analyses revealed a very distinct divergence between wild Gossypium hirsutum and cultivated Gossypium hirsutum, consistent with previous reports (Fang et al., 2017a; Wang et al., 2017a). We also found the ESM population showed a lower nucleotide diversity and higher LD and was enriched within a small clade of the phylogenetic tree, suggesting that ESM originated as an improved form of MLM but not highly structured and thus were suitable for genome‐wide association analysis (GWAS) (Yano et al., 2016). This interpretation is also supported by the pedigree (Figure S9).
Adaptation changes and increasing early maturity during domestication
Upland cotton originated in tropical and subtropical regions (Wendel et al., 2012) but can now be planted in Xinjiang, northern China (46°N). Selective breeding for earlier maturity may have caused the genetic structure to diverge during domestication and improvement. To address how the cotton adapts to the low‐temperature region, northern China, we first calculated population differentiation using F statistics. The differentiation between groups was further evaluated based on the F ST value. The highest population differentiation estimate (F ST) was observed between the wild lines and cultivars, with a pairwise F ST of 0.31 (MLM) and 0.32 (ESM) (Figure S7). In comparison, the F ST value (0.05) of the MLM and ESM populations was lower (Figure S7). Our results again confirm that the Wild population is the ancestral population and that ESM experienced adaptive changes over a long period of domestication and artificial selection in the breeding history.
Second, to identify genomic regions affected by selection that were important for adaptation during domestication and improvement, we examined whole‐genomic‐region signals of selective sweeps using a site frequency spectra (SFS)‐based method (Pavlidis et al., 2013; Figure 2a, b). A total of 357 selective sweeps were detected (Table S7), covering 4.94% (112 Mb) of the upland cotton genome and harbouring 5184 genes (Table S8). Among the sweep regions, 16.38% were intergenic, implying a potential regulatory role in domestication and breeding. Similarly, a study of maize inbred lines showed that many putative sweep regions were located in nongenic regions by CLR analysis (Jiao et al., 2012). Functional classification of the genes presents in the sweep regions showed that their products are predicted to be involved in four significantly enriched biological process GO terms, namely, flowering time, hormone catabolism, defence responses and ageing (false discovery rate [FDR] < 0.01; Table S9). Early maturity is well known to be accompanied by early senescence during the development of early maturity cotton (YU et al., 2005). The ageing process category included multiple genes (WRKY22, VIN2, PUB44, SAG21, UBA2c and SWEET15) (Gao et al., 2016; Kim et al., 2008; Veyres et al., 2008; Vogelmann et al., 2012; Yang et al., 2011b; Zhou et al., 2011) known to be highly expressed during leaf senescence in early maturity cotton (Lin et al., 2013). Hormone signalling also plays diverse and critical roles during plant development, and many well‐known genes were significantly enriched in the hormone catabolic process category (Table S9). For example, CKX3 encode proteins with sequences similar to that of cytokinin oxidase and plays an important role in regulating flower organ development (Ding et al., 2015). GA2OX2, GA2OX6 and GA2OX8 participate in the inactivation of gibberellin and affect flowering behaviour (Rieu et al., 2008; Schomburg et al., 2003). Notably, flowering time (FT) is an important early maturity‐related trait in cotton and has also been well characterized in soybean (Zhang et al., 2015). Several candidate FT genes within sweep regions were homologous to members of the photoperiod pathway (CUL1, TOC1, PHYA, ZTL and ELF4), vernalization pathway (VIP4), autonomous pathway (FVE and FLK), GA signalling (SPY and RGL2), and to integrators and downstream genes (ATJ3, AGL15, AGL24, EBS and ELF6; Grover et al., 2015; Komeda, 2004; Ratcliffe and Riechmann, 2002; Table S9). These biological processes suggest potential roles in the improved early maturity of domesticated cotton.
Figure 2.
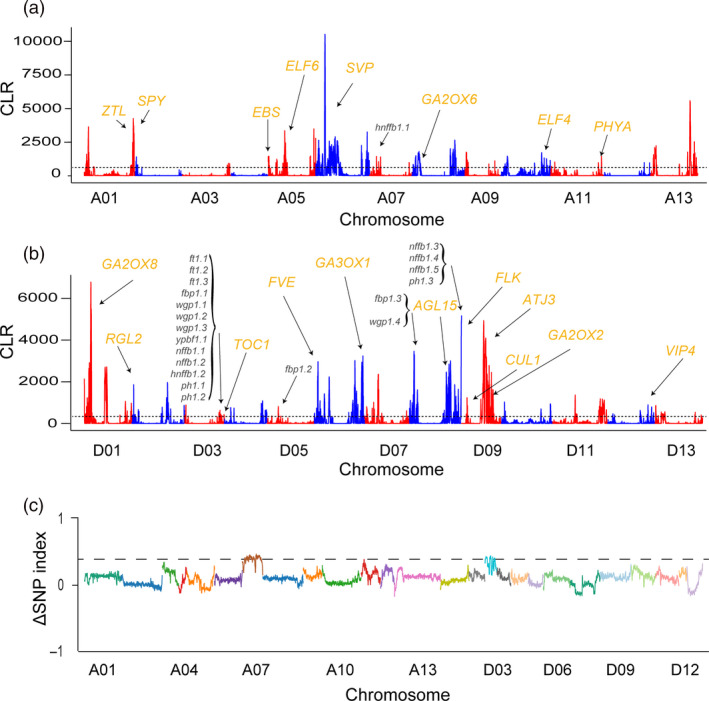
Candidate genomic regions under selection in early maturity cotton. (a,b) Selection sweeps in the A subgenome (At) (a) and the D subgenome (Dt) (b); the y‐axis represents the composite likelihood ratio, and the x‐axis represents the chromosome numbers. The solid black line indicates the cut‐off for the top 5% of windows. The 21 early maturity‐related trait QTL hotspots that overlap with selection sweeps are shown in black for each chromosome. Gibberellin catabolism (GA2OX6: Ghir_A08G004420; GA2OX8: Ghir_D01G006870; GA3OX1: Ghir_D06G021240; GA3OX1: Ghir_D06G021240; GA2OX2: Ghir_D09G008270) and flowering time (SPY: Ghir_A01G019780; EBS: Ghir_A05G017710; ELF6: Ghir_A05G030300; SVP: Ghir_A06G014970; ELF4: Ghir_A10G024000; PHYA: Ghir_A11G028330; RGL2: Ghir_D02G006710; TOC1: Ghir_D03G010390; FVE: Ghir_D06G007770; ZTL: Ghir_D08G002610; AGL15: Ghir_D08G015850; FLK: Ghir_D08G023290; ATJ3: Ghir_D09G010620; CUL1: Ghir_D09G006690; VIP4: Ghir_D12G019520) candidate genes are marked in orange. (c) x‐axis represents the position of 26 chromosomes and y‐axis represents the Δ SNP index (subtracting the SNP index of the early flowering bulk population from that of the late flowering bulk population).
Linkage analysis is an effective tool for identifying genes that control some of the most important morphological changes during domestication and improvement (Doebley et al., 2006). Over the past decade, many quantitative trait loci (QTLs) related to early maturity traits have been identified (Guo et al., 2008; Jia et al., 2016; Kushanov et al., 2017; Li et al., 2013a; Li et al., 2016; Li et al., 2017; Su et al., 2016b; Zhang et al., 2016). To assess whether the sweep regions associated with domestication colocalize with loci known to control early maturity‐related traits, we overlapped selection sweeps with the locations of known QTL hotspots. We detected a total of 21 QTLs in sweep regions, which likely played an important role during the remarkable improvement of maturity during long‐term selection (Figure 2a, b and Table S10). Interestingly, QTL hotspot regions on chromosome D03 were overlapped in selection sweeps. In this region, an ortholog of TOC1 (Ghir_D03G010390), which influences flowering time through the circadian clock‐regulated photoperiod pathway, promotes the expression of FLOWERING LOCUS T (FT) and SUPPRESSOR OF OVEREXPRESSION OF CO 1 (SOC1) by controlling CONSTANS (CO) (Jung and Müller, 2009). Moreover, previous studies have demonstrated that Ghir_D03G010390 expression differs significantly between early and late maturing varieties (Li et al., 2017). Therefore, Ghir_D03G010390 may be a key gene related to the control of flowering time during the breeding process.
We next chose an F2 population and performed bulked segregant analysis (BSA) using resequencing data from extreme FT lines to further verify selection sweeps related to early maturity (Table S11). The notable variations in FT among modern domesticated cotton are related to photoperiod sensitivity. The frequency distributions showed continuous variations, indicating that FT is a quantitatively inherited trait (Figure S10). Interestingly, three regions associated with FT have strong signals (A07: 20.53–21.60 Mb; D03: 35.49–36.49 Mb and 39.17–40.29 Mb) overlapped with selection sweeps that had experienced artificial selection during improvement (Figure 2c). This result verifies the accuracy of the identified selection sweeps associated with the domestication of early maturity in cotton.
GWAS for early maturity‐related traits in upland cotton
From 2015 to 2017, seven quantitative traits for early maturity, comprising FT, the period from first flower blooming to first boll opening (FBP), WGP, yield percentage before frost (YPBF), node of the first fruiting branch (NFFB), height of the node of the first fruiting branch (HNFFB) and plant height (PH), were investigated at three locations. Significant variation was observed for seven maturity‐related traits among the 355 upland cotton accessions (Figure S11). Pearson’s correlation coefficient analysis showed a significant negative correlation between YPBF and the other traits, all six of which showed significantly positive pairwise correlations (Figure S12). ANOVA revealed that early maturity‐related traits presented significant environmental and genetic effects (P < 0.001; Table S12). A broad sense heritability analysis was performed, and all traits ranged from 0.67 (WGP) to 0.79 (FT).
GWAS performed using 355 accessions from eight environments further identified a total of 307 significant SNP loci that were mainly distributed on chromosomes A01, A02, A03, A05, A06, A07, D01, D03 and D05 (Table S13 and Figures S13‐S20). Of all these significant loci, six (rsD03_37996318, rsD03_37952328, rsD03_38191576, rsD03_38175272, rsD03_38370420 and rsD03_39122594) shared more than four traits, indicating a genetic basis for pleiotropism that made it possible to simultaneously improve multiple early maturity traits during breeding. Notably, two distinct enrichment regions were located on chromosome A05 and chromosome D03, which accounted for more than 88.92% (273) of the loci.
Previous studies have indicated that chromosome D03 is rich in QTLs for early maturity traits (Jia et al., 2016; Li et al., 2013a; Li et al., 2017; Su et al., 2016b). In the present study, 43 significantly associated SNPs spanned a region of approximately 3.7 Mb on chromosome D03 (between 36.68 and 40.38 Mb). Of these, 88.37% (38) loci overlapped with our previously mapped early maturity QTLs and selection sweeps. These results strongly suggest that chromosome D03 is a major region and has potential functional genes related to early maturity. Interestingly, two associated SNPs lie within the most significant haplotype block, which is 82.17 kb long and contains three genes (Figure 3a, b). The RNA‐seq data showed that Ghir_D03G011310 had higher expression levels in the early maturity variety ‘CRI50’ than in the late maturity variety ‘TM‐1’ compared with the other two genes during flower development from 0 to 20 DPS (Figure 3c and Figure S21). Gene annotation analysis indicated that Ghir_D03G011310 encodes a cysteine protease, the closest known homologue in Arabidopsis thaliana of which is CEP1, which is expressed specifically in the tapetum and is involved in pollen development (Zhang et al., 2014). Furthermore, quantitative reverse‐transcription PCR (qRT‐PCR) showed that the expression of Ghir_D03G011310 in developing stages at three leaf growth stages and four leaf growth stages was significantly higher in the early maturity varieties (‘Zhong213’ and ‘CRI50’) than in the late maturity varieties (‘NDM8’ and ‘TM‐1’; P < 0.01; Figure S22). The SNP (rsD03_39122594) was located 1810 bp upstream of the start codon of Ghir_D03G011310 (Figure S23), which was found to have the strongest association with FT, WGP, YPBF and PH (average P value = 4.46E‐08). Varieties carrying the HapA (A allele) exhibited earlier maturity than carrying of the HapB (G allele) (Figure 3d). To confirm the practical utility of this locus, high‐resolution melting (HRM) analysis was used to genotype HapA and HapB in two recombinant inbred line (RIL) populations generated by crossing the early maturity line with the late maturity line from a previous report (Jia et al., 2016; Li et al., 2017; Figure 3e, f and Figure S24). The HRM assay showed that the mean WGP in the two RIL lines with the A allele was significantly shorter than that in the lines with the G allele, which is consistent with the GWAS results (Figure 3d). We further validated the function of Ghir_D03G011310 through virus‐induced gene silencing (VIGS) in early maturity cotton ‘CRI50’. The fruit branches or squares have not been observed in the silenced plants. However, the CK plants have yielded fruit branches with squares at the same growth status as shown in Figure 3g. And as shown in Figure S25, the drastically reduced expression of the Ghir_D03G011310 in the VIGS plants demonstrated that the gene had been successfully knocked down. We then analysed the nucleotide diversity, F ST values and Tajima’s D for the strongly LD block region (82.17 kb) that mentioned in the Figure 3b contained Ghir_D03G011310 in the flanking 150 kb region. Interestingly, it clearly showed that ESM group had lowest diversity than wild and MLM group and F ST values of ESM versus MLM was significantly higher than the flanking region (Figure S26). The Tajima’s D value in this strongly LD block region was −0.81 and biased from the balance. Thus, from the above results, we inferred that Ghir_D03G011310 is a previously undescribed gene that contributes to early maturity in cotton and involved in artificial selection in cotton.
Figure 3.
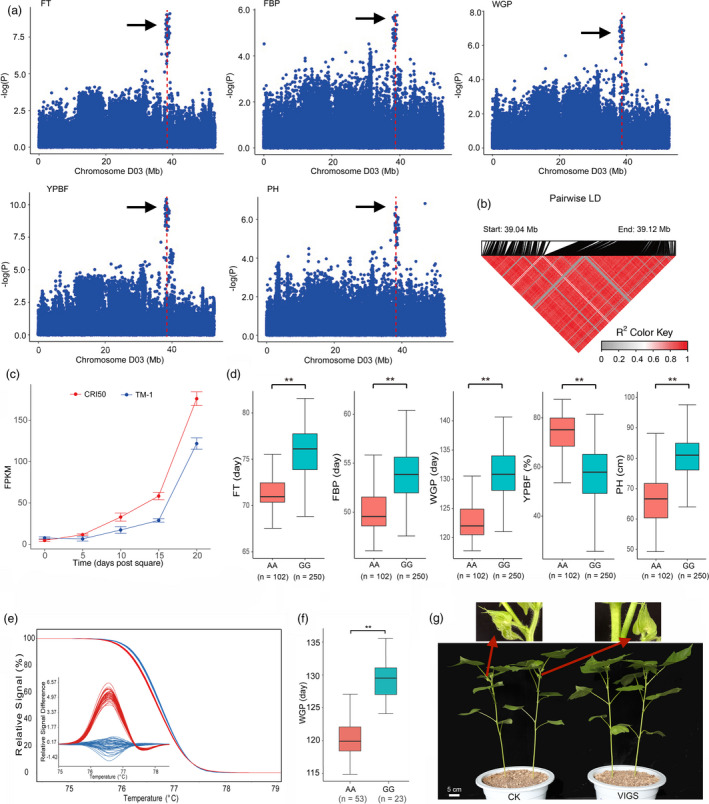
GWAS for early maturity‐related traits and identification of the candidate gene on chromosome D03. (a) Manhattan plots for flowering time (FT), period from the first flower blooming to the first boll opening (FBP), whole growth period (WGP), yield percentage before frost (YPBF) and plant height (PH) on chromosome D03; Arrowheads indicate the strongly loci rsD03_39122594 associated with the candidate gene Ghir_D03G011310. (b) LD heat map for the 82.17 kb long candidate region. The pairwise LD between the SNP markers is indicated as D′ values, where red indicates a value of 1 and grey indicates 0. (c) Expression profiles of Ghir_D03G011310. The x‐axis represents developmental stages (0, 5, 10, 15 and 20 DPS), and the y‐axis indicates the relative expression levels as determined by RNA‐seq. The error bars indicate standard deviation of three biological replicates. (d) Box plots for FT, FBP, WGP, YPBF and PH between two haplotypes mentioned above (** P < 0.01). (e) HRM analysis for SNP (rsD03_39122594) in recombinant inbred line population. The axis of the outside is original melting curves; the axis of the inside is melting curves after logarithm. Red and blue curves correspond to favourable alleles (A) and unfavourable alleles (G), respectively. (f) Box plots for two haplotypes in whole growth period at recombinant inbred line population mentioned above (** P < 0.01). (g) VIGS of Ghir_D03G011310 in early maturity cotton ‘CRI50’. ‘CRI50’ treated with an empty vector were used as a control group. Red arrows indicate the squares and fruit branches.
We then focused on chromosome A05 (Figure S13). These loci exhibited pleiotropic associations with PH and HNFFB. We identified 75.57% (232) peaks that were associated with the phenotypic variation. Notably, 237 loci were tightly linked within the candidate region (Table S14), a 16.30–16.94 Mb (630.69 kb) segment on chromosome A05 (Figure 4a, b) containing 57 candidate genes. Previous studies have shown that a majority (93%) of GWAS loci are located in nongenic regions and have regulatory functions (Maurano et al., 2012). However, in this 630.69 kb region, the annotated genetic variants of approximately 40% (134) of the identified loci were nonintergenic. These loci may directly control the relevant biological pathways involved in early maturity that are linked to causal polymorphisms in nearby genes. SNP rsA05_16453277 (G/T) was in the 3′ UTR regions of Ghir_A05G017290. A total 125 accessions exhibiting the T/T homozygous allele showed a shorter PH and HNFFB than those with the G/G homozygous allele (Table S15 and Figure S27). In our transcriptome analyses, Ghir_A05G017290 was more highly expressed in the early maturity variety ‘CRI50’ than the late maturity variety ‘TM‐1’ at the flowering development stage (Figure S28). In addition, Ghir_A05G017290 was a homologue of OsbHLH068 that encodes an MYB transcription factor in rice. Heterologous overexpression of OsbHLH068 in Arabidopsis regulates the expression of genes involved in the control of flowering time (Chen et al., 2017). These results suggest that Ghir_A05G017290 might determine early maturity in cotton.
Figure 4.
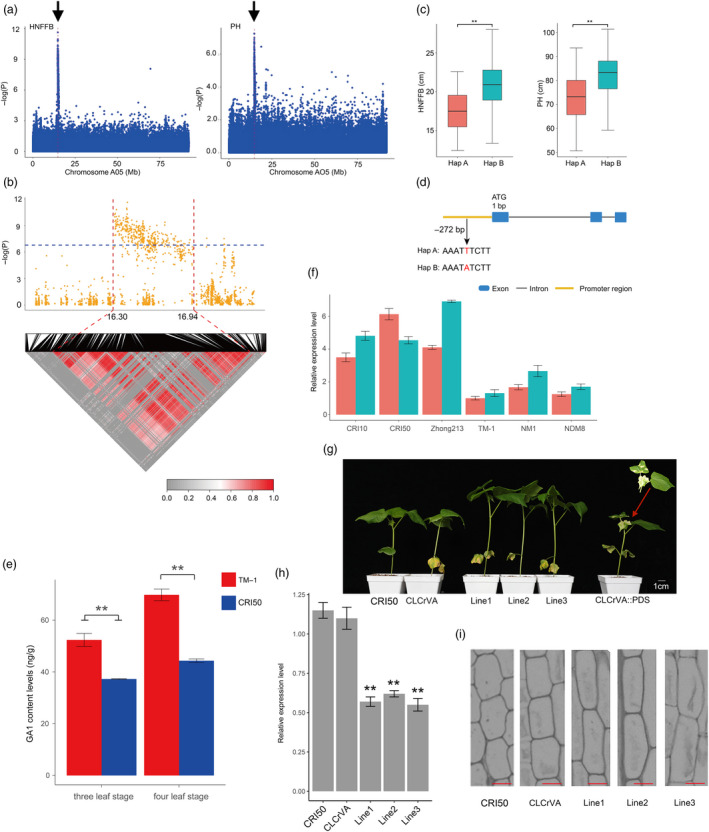
GWAS for early maturity‐related traits (HNFFB and PH) and identification of the candidate gene on chromosome A05. (a) Manhattan plots for height of the node of the first fruiting branch (HNFFB) and plant height (PH) on chromosome A05; Arrowheads indicate the strongly loci rsA05_16520008 associated with the candidate gene GhGA2OX8. (b) Local Manhattan plot (top) and LD heat map (bottom). The candidate region lies between the red dashed lines. (c) Box plots for rsA05_16520008 with two haplotypes in HNFFB and PH mentioned above (** P < 0.01). (d) Gene structure of GhGA2OX8 and the polymorphism in two haplotypes. (e) GA1 content measured in the apical bud of ‘CRI50’ and ‘TM‐1’ plants. (f) Comparison expression levels of GhGA2OX8 at stem apexes during three (red) and four (green) leaf stage. (g) Phenotype of VIGS GhGA2OX8 in early maturity cotton ‘CRI50’. ‘CRI50’ treated with an empty vector were used as a control group. (h) Expression level of GhGA2OX8 in ‘CRI50’, empty control and VIGS lines. (i) Longitudinal section from the first internode of ‘CRI50’, empty control and VIGS lines, scale bar is 25 μm.
Early maturity cotton is a short and compact plant (Figures S5 and S7). PH and HNFFB are very important early maturity traits that correlate significantly with FT, FBP, WGP and YPBF (Figure S12), and previous studies have also shown that maturity and plant architecture genes exhibit a strong correlation (Li et al., 2013a; Li et al., 2017). In the present study, GhGA2OX8 (Ghir_A05G017390), was significantly associated with PH and HNFFB in the promoter region of the 630.69 kb tight‐linkage region (Figure 4c, d and Table S15). Interestingly, we additionally observed that another gene, Ghir_D05G017200, also homologous to GhGA2OX8, was identified on chromosome D05 associated with HNFFB (Figure S29). Notably, GA biosynthesis and the role of GA signalling in controlling plant height have been investigated in rice, wheat and maize (Wang et al., 2017b). GhGA2OX8 is annotated as encoding a gibberellin 2‐oxidase involved in the biosynthesis of gibberellin. Overexpression GA2OX8 in Arabidopsis and tobacco has been shown to decrease gibberellin levels and create dwarf varieties (Schomburg et al., 2003). Interestingly, we discovered that the early maturity variety ‘CRI50’ (PH = 52.43 ± 6.88 cm) carrying the A allele contained significantly less active GA1 than the late maturity variety ‘TM‐1’ with the G allele (PH = 76.55 ± 8.49 cm; Figure 4e). The qRT‐PCR results indicated that GhGA2OX8 expression was higher in apical buds in early maturity varieties (PH = 59.25 ± 5.69 cm) than in those in late maturity varieties (PH = 87.96 ± 4.39 cm; Figure 4f). Overexpression of the GhGA2OX8 in Arabidopsis resulted in dwarf phenotypes compared with the wild type (Figure S30), and the height of each line was presented in Table S16. Furthermore, the resulting VIGS lines had significantly higher and drastically reduced expression of the GhGA2OX8 (Figure 4g, h) with parenchyma cells showing increased length compared with ‘CRI50’ and CLCrVA (empty vector; Figure 4i and Figure S31). From the above results, we inferred that GhGA2OX8 has a potential role in controlling cotton stature in the future.
Elite alleles for pyramid breeding in ESM
We selected 136 early maturity accessions (Table S1) and collected 27 elite loci associated with yield and fibre quality from previous studies (Fang et al., 2017b; Wang et al., 2017a; Table S17). Based on the release time, the 136 ESM lines used in this study were divided into three groups: before the 2000s, 2000s–2010s and after 2010. Analysis of the distribution of elite alleles indicated that the percentages of total elite alleles have gradually increased at the selected 27 SNP loci to 49.6%, 51.2% and 56.2% (Figure 5a).
Figure 5.
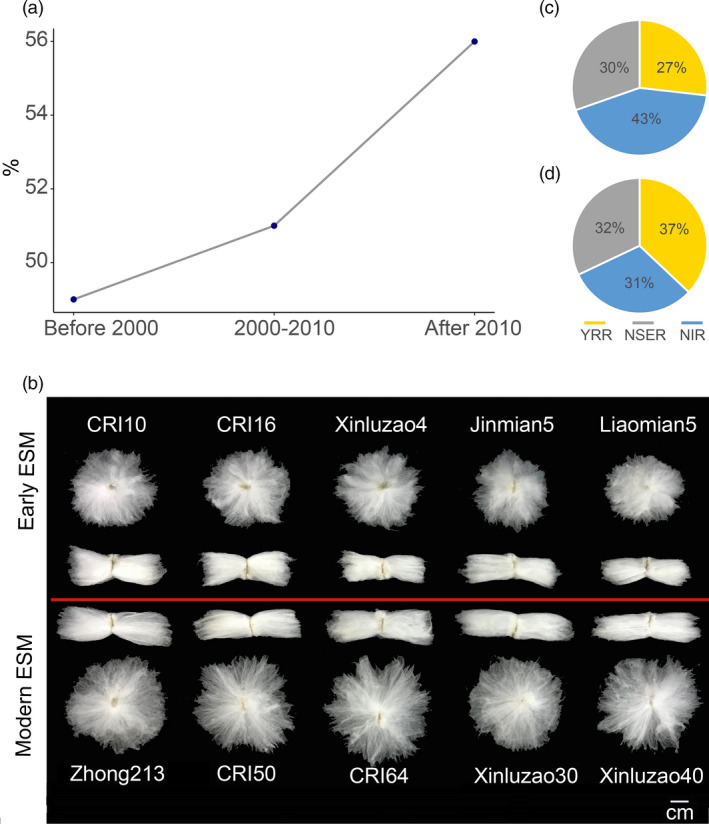
Elite alleles distributions for the fibre quality and yield traits in the early maturity cotton. (a) Percentages of elite alleles in early maturity cotton cultivars released before 2000s, 2000s–2010s and after 2010s. (b) Representative images of individual seeds with attached fibre between early ESM and modern ESM. (c, d) Percentages of elite alleles for fibre quality and yield traits in Yellow River region (YRR), Northern Specific Early Maturity region (NSER) and Northwest Inland region (NIR) in China, respectively.
Discussion
Here, we report a comprehensive genome variation map of upland cotton by deep resequencing that is more comprehensive than recent publications (436 versus 352 (Wang et al., 2017a), 318 (Fang et al., 2017b) and 419 (Ma et al., 2018) accessions). The large nature population represents a core cotton germplasm from the early 1900s to 2010s. Although 79% of the accessions are cultivated in different ecological cotton growing areas of China: Yellow River region, Yangtze River region, north of the Northwest Inland region and the Northern Specific Early Maturity region, we also included many early landraces such as ‘King’, ‘Deltapine’, ‘Stoneville’ and ‘Acala’ from the US cotton belt. Most importantly, we collected 136 elite early maturity breeding lines and cultivars bred in recent decades, of which 48 accessions having a WGP within 110 days in particular. The phenotypic diversity of early maturity‐related traits was more abundant (Table S12 and Figure S11), which suggested that the panel contained a greater variety of elite alleles associated with early maturity‐related traits than previous GWAS populations (Fang et al., 2017b; Ma et al., 2018; Wang et al., 2017a). In addition, GWAS in cotton based on whole‐genome resequencing has been focused more on fibre quality, yield and disease resistance (Fang et al., 2017b; Ma et al., 2018; Wang et al., 2017a), but the genetic mechanisms of early maturity‐related traits are unclear. Previous reports have indicated that mutation and favourable allele accumulation are probably two major routes to improve important agronomic traits in crops (Moose et al., 2004). Our results provide evidence for the latter hypothesis in the GWAS, in which we performed the first genome‐wide association analysis for seven early maturity‐related traits by resequencing.
After strict quality controls and filters of variations, we detected more high‐quality SNPs used genomic analysis (10 118 884 versus 8 621 073 (Fang et al., 2017b); 7 497 568 (Wang et al., 2017a) and 3 665 030 (Ma et al., 2018)) than previous research. The number of SNPs as well as the nucleotide diversity significantly decreased throughout the domestication process from wild to cultivated cotton (Figure S7), consistent with previous studies (Fang et al., 2017a; Wang et al., 2017a). Our data revealed that 2 002 869 private SNPs in wild cotton were not polymorphic in the modern cultivar (ESM and MLM). Previous studies have hypothesized that modern plant breeding reduces genetic diversity and jeopardizes future crop improvement (Li et al., 2013b). Although this concept seems correct for the majority of crop species (Lin et al., 2014; Qi et al., 2013; Valliyodan et al., 2016), our data showed limited effects of breeding on the reduction in nucleotide diversity between the MLM and ESM group. We found that the ESM gene pool harboured a high proportion of the SNPs (87.72%) presented in the MLM group and show a reduction in nucleotide diversity of 2.4%, possibly due to the shorter time for artificial selection from medium and late maturity to early maturity. In addition, the phylogenetic results were corresponding roughly to the whole growth period characteristics, whereas contrast with a previous study in other crops, in which the populations were structured geographically (Lam et al., 2010). However, similar result in cotton has been reported by Ma et al. (2018). These observations suggest that upland cotton has a relatively lower genetic diversity due to gene introgression, interspersed introduction or hybrid breeding among these very closely related individuals and narrow the introduction of resources from the US in the first 30 years of the twentieth century (Huang, 2007; Huang et al., 2017).
In this study, a gibberellin biosynthesis‐related pathway gene GhGA2OX8 associated with early maturity have been found by GWAS analysis. The overexpression of GhGA2OX8 showed consistent results, as observed in GA2OX8, revealing conserved functions between GhGA2OX8 and GA2OX8 in reducing plant height. Virus‐induced gene silencing cotton has significantly increased plant height when the expression of the GhGA2OX8 was suppressed. During the ‘Green Revolution’ of the 1960s and 1970s, the genetic manipulation of dwarfing genes led to the development of semidwarf rice and wheat varieties with improved lodging resistance and yield (Khush, 2001). In recent years, decreases in the cotton planting area in China have left cotton breeders struggling to maximize the yield from limited land. Increasing the planting density to raise the yield is much more effective than increasing yields from individual plants (Duvick, 2005). Planting early maturity cotton, which increases the lint yield by increasing the boll number per unit area, is an effective method to improve the cotton plant density and satisfy demand. In the present study, the GhGA2OX8 showed excellent potential for improving the plant architecture. Through the improvement of plant architecture by GhGA2OX8, which planting at a high plant density and make the canopy more compact, it could contribute to cotton yield under high‐density cultivation in fields and be a selection target for the genetic improvement of early maturity in cotton.
Although our genomic analyses identified many genes previously known to influence early maturity, we also identified a new candidate gene Ghir_D03G011310 was greatly upregulated during flower development from 0 to 20 DPS and more highly expressed in the early maturity variety by transcriptome and qRT‐PCR. VIGS of Ghir_D03G011310 in early maturity cotton also resulted in delayed formation of fruit branches and squares. Furthermore, with the widely application of next‐generation sequencing technology, the density of molecular markers is increasing. Investigating the artificial selection history of a specific gene is one way to decipher the history of crop and its impact on humans (He et al., 2015). We also found Ghir_D03G011310 is an artificial selection gene. The genetic diversity of ESM was lower than that of MLM and wild for the genomic regions of Ghir_D03G011310 and proved by F ST and Tajima’s D test. This artificially imposed selection pressure on the expression of Ghir_D03G011310 could be an important factor affecting the observed difference in early maturity in cotton, because the varieties carrying the HapA resulted in improved early maturity than carrying of the HapB. Therefore, the Ghir_D03G011310 should be useful for improving early maturity in upland cotton breeding via a molecular design approach, so as to further ensure stable cotton production.
Early maturity has always had a negative genetic correlation with yield and fibre quality. The early maturity breeding programme was launched in China in the 1970s; however, the yield and fibre quality of early maturity cotton have been greatly improved (Figure 5b). We also found that 22.1% (1146) of the genes were differentially expressed during ovule and fibre development in the selected regions (Table S18). This finding demonstrates that with an increase in early maturity through breeding, fibre yield and quality are also altered by artificial selection, an effect known as ‘domestication syndrome’ (Doebley et al., 2006). For example, the early stage of ‘CRI10’ has a poor yield and fibre quality compared with the modern early maturity variety ‘Zhong213’ (Li et al., 2017). How have the elite alleles associated with yield and fibre quality changed within early maturity accessions during the four decades of breeding development? To address this question, we found that with improvement of early maturity in cotton, elite loci associated with yield and quality became increasingly popular and were gradually integrated into the breeding process over time. Furthermore, the percentages of elite alleles related to fibre quality were much greater in the NIR than in the YRR and NESR populations, whereas the opposite trend was observed for yield (Figure 5c, d). This phenomenon was likely caused by different origins and regional breeding objectives.
In summary, our future work will not only examine particular loci (genes) but also characterize the regulatory networks/pathways underlying early maturity by utilizing functional genomic methodologies such as genome editing and genetic transformation to validate the effects of these candidate genes on the genetic improvement of modern cultivars.
Experimental procedures
Plant materials and resequencing
A total of 436 diversity upland cotton accessions were collected from multiple countries with a wide geographic distribution, including China, the United States, Uzbekistan, India, Brazil and other countries. The panel including 356 accessions that were newly produced in this study and 80 resequencing data sets that were previously analysed by Fang et al. (2017a); Wang et al. (2017a). Detailed information on the 436 accessions is listed in Table S1. The geographic distribution of upland cotton accessions was visualized with the R packages of ‘ggplot2’ and ‘maps’ (Brownrigg, 2013; Wickham, 2016). Young leaves were collected 4 weeks after planting and quickly frozen in liquid nitrogen for sequencing. Total DNA was extracted using the CTAB method (Paterson et al., 1993). For each accession, at least 5 μg of genomic DNA was used to construct paired‐end sequencing libraries.
Identification of variation and filtering
All paired‐end sequence reads were mapped to the Gossypium hirsutum cv. TM‐1 reference genome (Wang et al., 2019) using BWA software with default parameters (Li and Durbin, 2009). Only reads with unique mapping position in the TM‐1 reference genome and mapping quality value greater 30 were retained in BAM format by SAMtools (Li et al., 2009). The Picard programme was used to sort mapping results from name order into coordinate order and mark the PCR reads that were duplicated during library construction or sequencing. Additionally, we improved the alignment performance by realignment of reads around Indels from the BWA mapping results with the IdnelRealigner package in the Genome Analysis toolkit (GATK) (McKenna et al., 2010). SNP and Indel detection were performed using bcftools (Li, 2011) and GATK software. High‐quality SNPs and Indel variations were obtained according to the following criteria. (a) Only concordant sites identified by GATK and bcftools with the SelectVariants packages were retained. (b) SNPs within the 5‐bp range of Indels were filtered out. (c) The SNP quality value should be greater than 30. (d) SNPs and Indels with a MAF < 1% and missing rate < 10% were discarded. (e) The average sequencing depth had to be greater than 5 × and less than 30×. (f) Insertions and deletions with a maximum length 10 bp were taken into account. The annotation information of variants was obtained by ANNOVAR (Wang et al., 2010).
SNP validation
Two methods were used to validate the resequencing accuracy and quality. (1) We randomly selected 316 SNPs and carried out at least three replicates of direct PCR and Sanger sequencing to compare genotypes called from the resequencing data to evaluate the SNP accuracy rate (Tables S4 and S5). (2) We checked the previously published cotton SNP SLAF data (Su et al., 2016a) in 43 accessions with 20 206 SNPs (Table S6).
Population structure
To build a phylogenetic tree, we selected a total of 10 180 SNPs at 4D SNPs and filtered all accessions with a MAF> 5% and missing rate per site < 10%. An unrooted phylogenetic tree was constructed using SNPhylo (Lee et al., 2014) and visualized with the R package of ‘ggtree’ (Yu et al., 2017). LD was calculated for each subpopulation (Wild, ESM and MLM) using SNPs with a MAF> 5% and missing rate per site < 10% using PLINK (Purcell et al., 2007). Each chromosome was separately calculated with the following parameters: (‐‐r 2 ‐‐ld‐window‐r 2 0 ‐ld‐window‐kb 1000 ‐‐ld‐window 99999). The LD heat maps with surrounding peaks in the GWAS results were visualized using the R package of ‘LDheatmap’ (Shin et al., 2006). Using the same data set as LD, PCA was performed using GCAT (Yang et al., 2011a), and two‐dimensional coordinates were plotted for the 436 cotton accessions in R (www.r‐project.org) and the population structure was analysed with the fastSTRUCTURE (Raj et al., 2014) programme.
Population genetic analysis and identification of selective sweeps
F ST provides insights into the biology of evolutionary processes as a measure of population differentiation in genetic distance. To determine the pairwise F ST values in two subpopulations, VCFtools (Danecek et al., 2011) software was used with a step size of 20 kb and a 100‐kb sliding window. The π value was used to measure each 20‐kb interval across the genome with a 100‐kb sliding window. In addition, we also employed SweepFinder2 to detect selective sweeps using the CLR statistic (DeGiorgio et al., 2016), and windows with the highest 5% of values and windows with a distance of ≤ 50 kb were merged into a single selected region.
Bulk segregant analysis of the F2 population by whole‐genome resequencing
Two upland cotton accessions, ‘Zhong213’ and ‘Richmond6’, were used as parental lines to develop segregating populations for FT. Richmond6, a wild upland cotton with an FT of more than 100 days, was considered a line with extremely late flowering, and ‘Zhong213’, an excellent early maturing cultivar with an FT of approximately 65 days, was used as an early flowering cultivar (Li et al., 2017). In 2015, ‘Zhong213’ (female parent, P1) was crossed with Richmond6 (pollen donor, P2) in Sanya, Hainan, China (18°29′N, 109°52′E), to create F1 seeds from a single crossed plant. Then, the multiple F1 plants were planted during the winter and self‐pollinated to generate 500 F2 individuals in the same field. The F2 seeds were planted at the Cotton Research Institute of the Chinese Academy of Agricultural Sciences (CRICAAS), Anyang, Henan, China (36°08′N, 114°48′E), in 2017. Genomic DNA was isolated from young cotton leaves using the CATB method (Paterson et al., 1993). Bulked DNA samples were prepared by mixing an equal ratio from 30 individuals showing extremely early flowering and late flowering for sequencing on an Illumina HiSeq 4000 sequencer. A total of 278.61 Gb of sequence for 20 × in each parent and 30 × in each bulk sample was generated. Detailed information on the resequencing of parental lines and bulks is listed in Table S11. Short reads were aligned to the cotton reference genome (Wang et al., 2019) using BWA software (Li and Durbin, 2009), and SNP calling was performed using SAMtools (Li et al., 2009). SNPs with a base quality value >30 and read depth >10× were used for further analysis. The SNP index was calculated for all SNP positions using the method of Takagi et al. (2013). An average SNP index was calculated using a 1‐Mb sliding window and a step size of 10 kb, and a 95% confidence interval was used to obtain the average SNP index according to Lin et al. (2014).
Phenotyping and genome‐wide association study
A total of 436 cotton accessions were selected for this study. For phenotyping, 355 accessions were planted at Anyang, Henan, China (36°08′N, 114°48′E), Shihezi, Xinjiang, China (44°31’N, 86°01′E) and Huanggang, Hubei, China (30°57′N, 114°92′E). From 2015 to 2017, multiple environmental evaluations were conducted in the three different cotton planting areas (YRR: Yellow River region, the YZRR: Yangtze River region, NIR: Northwest Inland region) throughout China (Table S1). Seven early maturity‐related traits were investigated in this study, including FT (days), FBP (days), WGP (days), YPBF (%), NFFB, HNFFB (cm) and PH (cm), as described previously (Jia et al., 2016; Li et al., 2017; Su et al., 2016b). The values of the best linear unbiased prediction (BLUP) of seven early maturity‐related traits in the 355 accessions were calculated using the R package of ‘lme4’ (Bates et al., 2014). A total 4 521 969 high‐quality SNPs (MAF > 0.05 and missing rate per site < 10%) were used to perform the GWAS for early maturity‐related traits in 355 accessions. A linear mixed model was used for marker association analysis with phenotype with GEMMA software (Zhou and Stephens, 2012). The Bonferroni‐corrected significance threshold was set as 1/n (n = total SNP number used in the association analysis).
Gene expression analysis
qRT‐PCR: Total RNA was extracted using the Plant RNA Prep Pure Plant kit (Tiangen, Beijing, China). Then, cDNA was obtained using the SuperScript III First‐Stand Synthesis System (Takara, Dalian, China). Real‐time PCR was performed on an ABI 7500 Real‐Time PCR System (Foster City, CA). Gene expression levels were calculated using the 2−ΔΔCT method (Livak and Schmittgen, 2001), and three biological replicates were used for each sample. The primers used for qRT‐PCR analysis are listed in Table S19.
Transcriptome expression analysis
The flower bud was chosen to perform RNA‐seq at five developmental stages as follows: (1) square; (2) 5 days post‐square (5 DPS); (3) 10 DPS; (4) 15 DPS; (5) 20 DPS. Bracts were removed from each flower bud. Three biological replicates were collected for each stage. The libraries were constructed according to Zhu et al. (2018) and sequenced on the BGISEQ‐500 platform using 100‐bp paired‐end sequencing. The expression level of each gene was calculated using fragments per kilobase of exon model per million mapped reads (FPKM) determined by DESeq2 (Love et al., 2014) and visualized using custom R scripts. The RNA‐Seq data PRJNA248163 were obtained from NCBI (https://www.ncbi.nlm.nih.gov/bioproject/). The expression value of each gene was determined using GFOLD software (Feng et al., 2012). Differentially expressed genes were defined at P < 0.05 using Student’s t‐test, expression changes of at least twofold, and expression levels of at least 1 FPKM in at least one stage during ovule and fibre development.
Virus‐induced gene silencing in cotton
Approximately 300 bp of the gene‐specific region for Ghir_D03G011310 and GhGA2OX8 was inserted into the pCLCrVA vector and pCLCrVB as the helper vector (Gu et al., 2014). pCLCrVA::Ghir_D03G011310, pCLCrVA::GhGA2OX8 and pCLCrVB were transferred into cotton cotyledons of ‘CRI50’ through Agrobacterium tumefaciens strain LBA4404 as previously described (Gu et al., 2014). The primers used for construction of the VIGS vector are listed in Table S19.
Genetic transformation of Arabidopsis thaliana
The full‐length open reading frame of GhGA2OX8 was amplified by PCR using cDNAs synthesized from RNA that was isolated from ‘CRI50’ corresponding to the SNP allele. The amplified products were further inserted into the binary expression vector pBI121 driven by the 35S promoter to generate the 35S::GhGA2OX8 construct. The resulting construct was individually introduced into Agrobacterium tumefaciens strain GV3101 and transformed into the Arabidopsis ecotype Columbia. The homozygous T3 generation was further verified by PCR. The primers used for gene cloning are listed in Table S19.
Determination of endogenous GAs
‘CRI50’ and ‘TM‐1’ were grown under greenhouse conditions for approximately 6 weeks. To analyse GA content, the apical buds of ‘CRI50’ and ‘TM‐1’ were collected at the three leaf stage and four leaf stage, and they were analysed using both high‐performance liquid chromatography fluorescence and LC‐MS/MS according to Li et al. (2011). Each sample was mixed in equal amounts, and three biological and three technical replicates were evaluated.
Tissue sectioning analysis
For paraffin section observation, elongated mature tissue of the first internode was cut from ‘CRI50’, CLCrVA and VIGS lines and placed in formalin‐acetic acid‐alcohol (FAA) mixed solution for no less than 48 h, and then the samples were dehydrated with a graded ethanol series. The prepared sections were sequentially stained, infiltrated with xylene and finally mounted beneath a coverslip. The sections were then dewaxed twice in dimethylbenzene and each time for 10 min. Finally, light microscopy was performed using microscope and photographed.
Funding
This research was supported by the National Key Research and Development Program of China (2016YFD010401, 2016YED0101006), the China Agriculture Research System (No. CARS‐18) and the National Natural Science Foundation of China (31601346, 31621005).
Competing interests
The authors declare no competing interests.
Authors’ contributions
SY, JH and LL designed the experiments; LL and CZ performed most of the experiments; GL and LG provided technical assistance; LL and QL performed data analysis; HW and HW revised the language. SY and LL supervised and complemented the writing. All authors read and approved the final manuscript.
Availability of supporting data and materials
The raw data for resequencing, RNA‐seq and Sanger sequencing results have been deposited in the NCBI database under PRJNA389777, PRJNA431876, PRJNA509318 and MK847254‐MK844855. The accession numbers are summarized in Table S1. Phenotype information used in GWAS is summarized in Table S20.
Supporting information
Table S1 Summary of the 436 accessions for resequencing.
Table S2 Summary of SNPs in each chromosome.
Table S3 Summary of Indels in each chromosome.
Table S4 Summary of accessions used in PCR and Sanger sequencing.
Table S5 SNP loci selected for validation PCR and Sanger sequencing.
Table S6 SNP loci selected for validation by the SLAF SNP dataset.
Table S7 Putative regions identified to be under domestication selection sweeps.
Table S8 Genes within the putative domestication sweeps.
Table S9 Candidate genes involved in significantly enriched biological process GO terms, including flowering time, hormone catabolism, defence responses and ageing, in selection sweeps.
Table S10 Summary of known early maturity‐related QTLs during domestication.
Table S11 Resequencing information of parental lines and bulks for BSA analysis.
Table S12 ANNOVA and a broad sense heritability of early maturity‐related traits in the 355 accessions.
Table S13 Genome‐wide significant association signals for seven early maturity‐related traits in 355 accessions using the LMM method.
Table S14 237 SNPs in the GWAS peaks on the chromosome A05.
Table S15 GWAS peaks haplotype on chromosome A05 and D03.
Table S16 Comparison of the plant height for GhGA2OX8‐transgenic Arabidopsis.
Table S17 Elite loci associated with yield and fibre quality from previous studies used in the present study.
Table S18 Differentially expressed genes during fibre and ovule development.
Table S19 List of all primers used in this study.
Table S20 Seven early maturity‐related traits used for GWAS
Figure S1 Map of the early maturity cotton growing area and early maturity cotton used in this study, including the cotton growing area of the Yellow River region (YRR), north of the Northwest Inland region (NIR), and the Northern Specific Early Maturity region (NSER).
Figure S2 Circos plot showing SNP diversity across the 26 chromosomes of Gossypium hirsutum. The chromosomes are numbered. The blue circle represents SNP density; the purple circle shows Indel diversity; the orange and green colours represent gene density within SNP and Indel markers, respectively.
Figure S3 The SNP number on each chromosome and distributions at different regions.
Figure S4 The Indel number on each chromosome and distributions at different regions.
Figure S5 Morphological changes in early, medium and late maturity cotton and wild cotton during domestication and improvement. a: cotton at the stage when the node of the first fruiting branch flower is opening in early maturity cotton. b: cotton at the stage when all the balls are open in early maturity cotton.
Figure S6 Model‐based clustering analysis with different numbers of clusters (K = 2 and 3).
Figure S7 Nucleotide diversity (π) and population divergence (F ST) across the three population, and a diagram of early maturity and special early maturity (ESM) population and medium and late maturity (MLM) population.
Figure S8 Decay of linkage disequilibrium (LD) in Wild, early maturity and special early maturity (ESM) population and medium and late maturity (MLM) population.
Figure S9 Pedigree information for early maturity cotton breeding. The accessions in green are the founder cultivars of early maturity cotton; the accessions in red were collected and analysed in our study; the accessions in black were early maturity cotton but not analysed in our study.
Figure S10 The frequency of FT (flowering time) among the parents (‘Zhong213’ and ‘Richmondi 6’) and 500 F2 individuals in Anyang 2017.
Figure S11 Distributions of the mean values of seven early maturity‐related traits of 355 accessions in eight environments, including flowering time (FT), the period from first flower blooming to first boll opening (FBP), whole growth period (WGP), yield percentage before frost (YPBF), node of the first fruiting branch (NFFB), height of the node of the first fruiting branch (HNFFB) and plant height (PH).
Figure S12 Correlation analysis of seven early maturity traits in 355 natural populations, including flowering time (FT), the period from first flower blooming to first boll opening (FBP), whole growth period (WGP), yield percentage before frost (YPBF), node of the first fruiting branch (NFFB), height of the node of the first fruiting branch (HNFFB) and plant height (PH). **Indicates significance at 0.01.
Figure S13 Manhattan plots for BLUP of flowering time (FT), the period from first flower blooming to first boll opening (FBP), whole growth period (WGP), yield percentage before frost (YPBF), node of the first fruiting branch (NFFB), height of the node of the first fruiting branch (HNFFB) and plant height (PH). Significant trait‐associated SNPs are distinguished by red lines.
Figure S14 Manhattan plots for flowering time (FT) in separate environment.
Figure S15 Manhattan plots for the period from first flower blooming to first boll opening (FBP) in separate environment.
Figure S16 Manhattan plots for whole growth period (WGP) in separate environment.
Figure S17 Manhattan plots for yield percentage before frost (YPBF) in separate environment.
Figure S18 Manhattan plots for node of the first fruiting branch (NFFB) in separate environment.
Figure S19 Manhattan plots for height of the node of the first fruiting branch (HNFFB) in separate environment.
Figure S20 Manhattan plots for plant height (PH) in separate environment.
Figure S21 Expression profiles of Ghir_D03G011290 and Ghir_D03G011300. The x‐axis represents developmental stages (0, 5, 10, 15 and 20 DPS), and the y‐axis indicates the relative expression levels as determined by RNA‐seq. The error bars indicate standard deviation of three biological replicates.
Figure S22 Expression levels of Ghir_D03G011310 at three leaf growth stages and four leaf growth stages by qRT‐PCR (**indicates significance at the 0.01 probability level).
Figure S23 Gene structure of Ghir_D03G011310 and the polymorphism in two haplotypes, ‘A’ allele and ‘G’ allele.
Figure S24 (a) HRM analysis for SNP (rsD03_39122594) in recombinant inbred line population. The axis of the outside is original melting curves; the axis of the inside is melting curves after logarithm. Red and blue curves correspond to favourable alleles (A) and unfavourable alleles (G), respectively. (b) Box plots for two haplotypes in whole growth period at recombinant inbred line population mentioned above (** P < 0.01).
Figure S25 Expression level of Ghir_D03G011310 in empty control as CK and VIGS plants. **Indicates significance at 0.01.
Figure S26 Nucleotide diversity and population divergence (FST) on the chromosome D03 (red part is the strong LD block regions). (a) Nucleotide diversity across the three population. (b) Population divergence (FST) between early maturity and special early maturity (ESM) population and medium and late maturity (MLM) population.
Figure S27 Box plots for SNP rsA05_16453277 (G/T) in height of the node of the first fruiting branch (HNFFB) and plant height (PH).
Figure S28 Comparison of Ghir_A05G017290 expression levels between ‘CRI50’ (green) and ‘TM‐1’ (red) during developmental stages (0, 5, 10, 15, and 20 DPS) by RNA‐seq. **Indicates significance at 0.01.
Figure S29 Manhattan plots for HNFFB on chromosome D05.
Figure S30 Morphological phenotypes of wild‐type and Arabidopsis containing the 35S::GhGA2OX8 cDNA construct. WT, OE8 and OE9 represent wild‐type and transgenic lines, respectively.
Figure S31 Comparison of cell length of CRI50, CLCrVA and VIGS lines. **Indicates significance at 0.01.
Acknowledgements
We thank Prof. Xianlong Zhang and Dr. Maojun Wang from Huazhong Agriculture University for their kindly advisement in data analysis. We are grateful to Prof. Xiaoya Chen from SIPPE for his insightful suggestions. We thank Prof. Tianzhen Zhang from Zhejiang University for releasing resequencing data of wild cotton accessions.
Li, L. , Zhang, C. , Huang, J. , Liu, Q. , Wei, H. , Wang, H. , Liu, G. , Gu, L. and Yu, S. (2021) Genomic analyses reveal the genetic basis of early maturity and identification of loci and candidate genes in upland cotton (Gossypium hirsutum L.). Plant Biotechnol J, 10.1111/pbi.13446
References
- Bates, D. , Mächler, M. , Bolker, B. and Walker, S. (2014) Fitting linear mixed‐effects models using lme4. arXiv preprint arXiv:1406.5823. [Google Scholar]
- Brownrigg, M.R. (2013) Package ‘maps’. Citeseer. [Google Scholar]
- Chen, H.‐C. , Hsieh‐Feng, V. , Liao, P.‐C. , Cheng, W.‐H. , Liu, L.‐Y. , Yang, Y.‐W. , Lai, M.‐H. et al. (2017) The function of OsbHLH068 is partially redundant with its homolog, AtbHLH112, in the regulation of the salt stress response but has opposite functions to control flowering in Arabidopsis. Plant Mol. Biol. 94, 531–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. , Albers, C.A. , Banks, E. , DePristo, M.A. , Handsaker, R.E. et al. (2011) The variant call format and VCFtools. Bioinformatics, 27, 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGiorgio, M. , Huber, C.D. , Hubisz, M.J. , Hellmann, I. and Nielsen, R. (2016) SweepFinder2: increased sensitivity, robustness and flexibility. Bioinformatics, 32, 1895–1897. [DOI] [PubMed] [Google Scholar]
- Ding, L. , Yan, S. , Jiang, L. , Zhao, W. , Ning, K. , Zhao, J. , Liu, X. et al. (2015) HANABA TARANU (HAN) bridges meristem and organ primordia boundaries through PINHEAD, JAGGED, BLADE‐ON‐PETIOLE2 and CYTOKININ OXIDASE 3 during flower development in Arabidopsis. PLoS Genet. 11, e1005479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doebley, J.F. , Gaut, B.S. and Smith, B.D. (2006) The molecular genetics of crop domestication. Cell, 127, 1309–1321. [DOI] [PubMed] [Google Scholar]
- Duvick, D. (2005) Genetic progress in yield of United States maize (Zea mays L.). Maydica, 50, 193. [Google Scholar]
- Fang, L. , Gong, H. , Hu, Y. , Liu, C. , Zhou, B. , Huang, T. , Wang, Y. et al. (2017a) Genomic insights into divergence and dual domestication of cultivated allotetraploid cottons. Genome Biol. 18, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, L. , Wang, Q. , Hu, Y. , Jia, Y. , Chen, J. , Liu, B. , Zhang, Z. et al. (2017b) Genomic analyses in cotton identify signatures of selection and loci associated with fiber quality and yield traits. Nat. Genet. 49, 1089. [DOI] [PubMed] [Google Scholar]
- Feng, J. , Meyer, C.A. , Wang, Q. , Liu, J.S. , Shirley Liu, X. and Zhang, Y. (2012) GFOLD: a generalized fold change for ranking differentially expressed genes from RNA‐seq data. Bioinformatics, 28, 2782–2788. [DOI] [PubMed] [Google Scholar]
- Gao, S. , Gao, J. , Zhu, X. , Song, Y. , Li, Z. , Ren, G. , Zhou, X. et al. (2016) ABF2, ABF3, and ABF4 promote ABA‐mediated chlorophyll degradation and leaf senescence by transcriptional activation of chlorophyll catabolic genes and senescence‐associated genes in Arabidopsis. Mol. Plant, 9, 1272–1285. [DOI] [PubMed] [Google Scholar]
- Grover, C.E. , Gallagher, J.P. and Wendel, J.F. (2015) Candidate gene identification of flowering time genes in cotton. Plant Genome, 8, 1–13. [DOI] [PubMed] [Google Scholar]
- Gu, Z. , Huang, C. , Li, F. and Zhou, X. (2014) A versatile system for functional analysis of genes and micro RNA s in cotton. Plant Biotechnol. J 12, 638–649. [DOI] [PubMed] [Google Scholar]
- Guo, Y. , McCarty, J.C. , Jenkins, J.N. and Saha, S. (2008) QTLs for node of first fruiting branch in a cross of an upland cotton, Gossypium hirsutum L., cultivar with primitive accession Texas 701. Euphytica, 163, 113–122. [Google Scholar]
- He, Q. , Yu, J. , Kim, T.‐S. , Cho, Y.‐H. , Lee, Y.‐S. and Park, Y.‐J. (2015) Resequencing reveals different domestication rate for BADH1 and BADH2 in rice (Oryza sativa). PLoS One 10, e0134801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Z. (2007) Cotton Varieties and their Genealogy in China. Beijing: Chinese Agricultural Press. [Google Scholar]
- Huang, C. , Nie, X. , Shen, C. , You, C. , Li, W. , Zhao, W. , Zhang, X. et al. (2017) Population structure and genetic basis of the agronomic traits of upland cotton in China revealed by a genome‐wide association study using high‐density SNPs. Plant Biotechnol. J. 15, 1374–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hufford, M.B. , Xu, X. , Van Heerwaarden, J. , Pyhäjärvi, T. , Chia, J.‐M. , Cartwright, R.A. , Elshire, R.J. et al. (2012) Comparative population genomics of maize domestication and improvement. Nat. Genet. 44, 808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia, X. , Pang, C. , Wei, H. , Wang, H. , Ma, Q. , Yang, J. , Cheng, S. et al. (2016) High‐density linkage map construction and QTL analysis for earliness‐related traits in Gossypium hirsutum L. BMC Genom. 17, 909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao, Y. , Zhao, H. , Ren, L. , Song, W. , Zeng, B. , Guo, J. , Wang, B. et al. (2012) Genome‐wide genetic changes during modern breeding of maize. Nat. Genet. 44, 812. [DOI] [PubMed] [Google Scholar]
- Jung, C. and Müller, A.E. (2009) Flowering time control and applications in plant breeding. Trends Plant Sci. 14, 563–573. [DOI] [PubMed] [Google Scholar]
- Khush, G.S. (2001) Green revolution: the way forward. Nat. Rev. Genet. 2, 815. [DOI] [PubMed] [Google Scholar]
- Kim, C.Y. , Bove, J. and Assmann, S.M. (2008) Overexpression of wound‐responsive RNA‐binding proteins induces leaf senescence and hypersensitive‐like cell death. New Phytol., 180, 57–70. [DOI] [PubMed] [Google Scholar]
- Komeda, Y. (2004) Genetic regulation of time to flower in Arabidopsis thaliana. Annu. Rev. Plant Biol. 55, 521–535. [DOI] [PubMed] [Google Scholar]
- Kushanov, F.N. , Buriev, Z.T. , Shermatov, S.E. , Turaev, O.S. , Norov, T.M. , Pepper, A.E. , Saha, S. et al. (2017) QTL mapping for flowering‐time and photoperiod insensitivity of cotton Gossypium darwinii Watt. PLoS One, 12, e0186240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam, H.‐M. , Xu, X. , Liu, X. , Chen, W. , Yang, G. , Wong, F.‐L. , Li, M.‐W. et al. (2010) Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat. Genet. 42, 1053. [DOI] [PubMed] [Google Scholar]
- Lee, T.‐H. , Guo, H. , Wang, X. , Kim, C. and Paterson, A.H. (2014) SNPhylo: a pipeline to construct a phylogenetic tree from huge SNP data. BMC Genom. 15, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. (2011) A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics, 27, 2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25(14), 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. et al. (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Jiang, J. , Qian, Q. , Xu, Y. , Zhang, C. , Xiao, J. , Du, C. et al. (2011) Mutation of rice BC12/GDD1, which encodes a kinesin‐like protein that binds to a GA biosynthesis gene promoter, leads to dwarfism with impaired cell elongation. Plant Cell, 23, 628–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, C. , Wang, X. , Dong, N. , Zhao, H. , Xia, Z. , Wang, R. , Converse, R.L. et al. (2013a) QTL analysis for early‐maturing traits in cotton using two upland cotton (Gossypium hirsutum L.) crosses. Breed. Sci. 63, 154–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y.‐H. , Zhao, S.‐C. , Ma, J.‐X. , Li, D. , Yan, L. , Li, J. , Qi, X.‐T. et al. (2013b) Molecular footprints of domestication and improvement in soybean revealed by whole genome re‐sequencing. BMC Genom. 14, 579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, C.Q. , Xu, X.J. , Dong, N. , Ai, N.J. and Wang, Q.L. (2016) Association mapping identifies markers related to major early‐maturating traits in upland cotton (Gossypium hirsutum L.). Plant Breed. 135, 483–491. [Google Scholar]
- Li, L. , Zhao, S. , Su, J. , Fan, S. , Pang, C. , Wei, H. , Wang, H. et al. (2017) High‐density genetic linkage map construction by F2 populations and QTL analysis of early‐maturity traits in upland cotton (Gossypium hirsutum L.). PLoS One, 12, e0182918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, M. , Lai, D. , Pang, C. , Fan, S. , Song, M. and Yu, S. (2013) Generation and analysis of a large‐scale expressed sequence tag database from a full‐length enriched cDNA library of developing leaves of Gossypium hirsutum L. PLoS One, 8, e76443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, T. , Zhu, G. , Zhang, J. , Xu, X. , Yu, Q. , Zheng, Z. , Zhang, Z. et al. (2014) Genomic analyses provide insights into the history of tomato breeding. Nat. Genet. 46, 1220. [DOI] [PubMed] [Google Scholar]
- Livak, K.J. and Schmittgen, T.D. (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2−ΔΔCT method. Methods, 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Love, M.I. , Huber, W. and Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, Z. , He, S. , Wang, X. , Sun, J. , Zhang, Y. , Zhang, G. , Wu, L. et al. (2018) Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield. Nat. Genet. 50, 803. [DOI] [PubMed] [Google Scholar]
- Maurano, M.T. , Humbert, R. , Rynes, E. , Thurman, R.E. , Haugen, E. , Wang, H. , Reynolds, A.P. et al. (2012) Systematic localization of common disease‐associated variation in regulatory DNA. Science, 337, 1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , Garimella, K. et al. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moose, S.P. , Dudley, J.W. and Rocheford, T.R. (2004) Maize selection passes the century mark: a unique resource for 21st century genomics. Trends Plant Sci. 9, 358–364. [DOI] [PubMed] [Google Scholar]
- Paterson, A.H. , Brubaker, C.L. and Wendel, J.F. (1993) A rapid method for extraction of cotton (Gossypium spp.) genomic DNA suitable for RFLP or PCR analysis. Plant Mol. Biol. Reporter, 11, 122–127. [Google Scholar]
- Pavlidis, P. , Živković, D. , Stamatakis, A. and Alachiotis, N. (2013) SweeD: likelihood‐based detection of selective sweeps in thousands of genomes. Mol. Biol. Evol. 30, 2224–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell, S. , Neale, B. , Todd‐Brown, K. , Thomas, L. , Ferreira, M.A. , Bender, D. , Maller, J. et al. (2007) PLINK: a tool set for whole‐genome association and population‐based linkage analyses. Am. J. Human Genet. 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi, J. , Liu, X. , Shen, D. , Miao, H. , Xie, B. , Li, X. , Zeng, P. et al. (2013) A genomic variation map provides insights into the genetic basis of cucumber domestication and diversity. Nat. Genet. 45, 1510. [DOI] [PubMed] [Google Scholar]
- Raj, A. , Stephens, M. and Pritchard, J.K. (2014) fastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics, 197, 573–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratcliffe, O.J. and Riechmann, J.L. (2002) Arabidopsis transcription factors and the regulation of flowering time: a genomic perspective. Curr. Issues Mol. Biol. 4, 77–92. [PubMed] [Google Scholar]
- Renny‐Byfield, S. , Page, J.T. , Udall, J.A. , Sanders, W.S. , Peterson, D.G. , Arick, M.A. , Grover, C.E. et al. (2016) Independent domestication of two old world cotton species. Genome Biol. Evol. 8, 1940–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieu, I. , Eriksson, S. , Powers, S.J. , Gong, F. , Griffiths, J. , Woolley, L. , Benlloch, R. et al. (2008) Genetic analysis reveals that C19‐GA 2‐oxidation is a major gibberellin inactivation pathway in Arabidopsis. Plant Cell, 20, 2420–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schomburg, F.M. , Bizzell, C.M. , Lee, D.J. , Zeevaart, J.A. and Amasino, R.M. (2003) Overexpression of a novel class of gibberellin 2‐oxidases decreases gibberellin levels and creates dwarf plants. Plant Cell, 15, 151–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, J.‐H. , Blay, S. , McNeney, B. and Graham, J. (2006) LDheatmap: an R function for graphical display of pairwise linkage disequilibria between single nucleotide polymorphisms. J. Statist. Software, 16, 1–10. [Google Scholar]
- Smith, C.E. and Stephens, S. (1971) Critical identification of Mexican archaeological cotton remains. Econ. Bot., 25, 160–168. [Google Scholar]
- Su, J. , Li, L. , Pang, C. , Wei, H. , Wang, C. , Song, M. , Wang, H. et al. (2016a) Two genomic regions associated with fiber quality traits in Chinese upland cotton under apparent breeding selection. Scientific Rep. 6, 38496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, J. , Pang, C. , Wei, H. , Li, L. , Liang, B. , Wang, C. , Song, M. et al. (2016b) Identification of favorable SNP alleles and candidate genes for traits related to early maturity via GWAS in upland cotton. BMC Genom. 17, 687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi, H. , Abe, A. , Yoshida, K. , Kosugi, S. , Natsume, S. , Mitsuoka, C. , Uemura, A. et al. (2013) QTL‐seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 74, 174–183. [DOI] [PubMed] [Google Scholar]
- Valliyodan, B. , Qiu, D. , Patil, G. , Zeng, P. , Huang, J. , Dai, L. , Chen, C. et al. (2016) Landscape of genomic diversity and trait discovery in soybean. Scientific Rep. 6, 23598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney, R.K. , Saxena, R.K. , Upadhyaya, H.D. , Khan, A.W. , Yu, Y. , Kim, C. , Rathore, A. et al. (2017) Whole‐genome resequencing of 292 pigeonpea accessions identifies genomic regions associated with domestication and agronomic traits. Nat. Genet. 49, 1082. [DOI] [PubMed] [Google Scholar]
- Veyres, N. , Danon, A. , Aono, M. , Galliot, S. , Karibasappa, Y.B. , Diet, A. , Grandmottet, F. et al. (2008) The Arabidopsis sweetie mutant is affected in carbohydrate metabolism and defective in the control of growth, development and senescence. Plant J. 55, 665–686. [DOI] [PubMed] [Google Scholar]
- Vogelmann, K. , Drechsel, G. , Bergler, J. , Subert, C. , Philippar, K. , Soll, J. , Engelmann, J.C. et al. (2012) Early senescence and cell death in Arabidopsis saul1 mutants involves the PAD4‐dependent salicylic acid pathway. Plant Physiol. 159, 1477–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, K. , Li, M. and Hakonarson, H. (2010) ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res. 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M. , Tu, L. , Lin, M. , Lin, Z. , Wang, P. , Yang, Q. , Ye, Z. et al. (2017a) Asymmetric subgenome selection and cis‐regulatory divergence during cotton domestication. Nat. Genet. 49, 579. [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Zhao, J. , Lu, W. and Deng, D. (2017b) Gibberellin in plant height control: old player, new story. Plant Cell Rep. 36, 391–398. [DOI] [PubMed] [Google Scholar]
- Wang, M. , Tu, L. , Yuan, D. , Zhu, D. , Shen, C. , Li, J. , Liu, F. et al. (2019) Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nat. Genet. 51, 224. [DOI] [PubMed] [Google Scholar]
- Wendel, J.F. and Cronn, R.C. (2003) Polyploidy and the evolutionary history of cotton. Adv. Agronomy, 78, 139. [Google Scholar]
- Wendel, J.F. , Flagel, L.E. and Adams, K.L. (2012) Jeans, genes, and genomes: cotton as a model for studying polyploidy. In Polyploidy and Genome Evolution ( Soltis, P.S. and Soltis, D.E. , eds), pp. 181–207. Berlin, Heidelberg: Springer. [Google Scholar]
- Wickham, H. (2016) ggplot2: elegant graphics for data analysis. New York, NY: Springer. [Google Scholar]
- Xun, X. , Xin, L. , Song, G. , Jensen, J.D. , Fengyi, H. , Xin, L. , Yang, D. et al. (2012) Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat. Biotechnol. 30, 105–111. [DOI] [PubMed] [Google Scholar]
- Yang, J. , Lee, S.H. , Goddard, M.E. and Visscher, P.M. (2011a) GCTA: a tool for genome‐wide complex trait analysis. Am. J. Human Genet. 88, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, S.‐D. , Seo, P.J. , Yoon, H.‐K. and Park, C.‐M. (2011b) The Arabidopsis NAC transcription factor VNI2 integrates abscisic acid signals into leaf senescence via the COR/RD genes. Plant Cell, 23, 2155–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano, K. , Yamamoto, E. , Aya, K. , Takeuchi, H. , Lo, P.‐C. , Hu, L. , Yamasaki, M. et al. (2016) Genome‐wide association study using whole‐genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat. Genet. 48, 927. [DOI] [PubMed] [Google Scholar]
- Yu, S.X. , Song, M.Z. , Fan, S.L. , Wang, W. and Yuan, R.H. (2005) Biochemical genetics of short‐season cotton cultivars that express early maturity without senescence. J. Integr. Plant Biol. 47, 334–342. [Google Scholar]
- Yu, G. , Smith, D.K. , Zhu, H. , Guan, Y. and Lam, T.T.Y. (2017) ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8, 28–36. [Google Scholar]
- Zhang, D. , Liu, D. , Lv, X. , Wang, Y. , Xun, Z. , Liu, Z. , Li, F. et al. (2014) The cysteine protease CEP1, a key executor involved in tapetal programmed cell death, regulates pollen development in Arabidopsis. Plant Cell, 26, 2939–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Song, Q. , Cregan, P.B. , Nelson, R.L. , Wang, X. , Wu, J. and Jiang, G.‐L. (2015) Genome‐wide association study for flowering time, maturity dates and plant height in early maturing soybean (Glycine max) germplasm. BMC Genom. 16, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, S. , Lan, Q. , Gao, X. , Yang, B. , Cai, C. , Zhang, T. and Zhou, B. (2016) Mapping of genes for flower‐related traits and QTLs for flowering time in an interspecific population of Gossypium hirsutum × G.darwinii . J. Genet. 95, 197–201. [DOI] [PubMed] [Google Scholar]
- Zheng, L.‐Y. , Guo, X.‐S. , He, B. , Sun, L.‐J. , Peng, Y. , Dong, S.‐S. , Liu, T.‐F. et al. (2011) Genome‐wide patterns of genetic variation in sweet and grain sorghum (Sorghum bicolor). Genome Biol. 12, R114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, X. and Stephens, M. (2012) Genome‐wide efficient mixed‐model analysis for association studies. Nat. Genet. 44, 821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, X. , Jiang, Y. and Yu, D. (2011) WRKY22 transcription factor mediates dark‐induced leaf senescence in Arabidopsis. Mol. Cells, 31, 303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Z. , Jiang, Y. , Wang, Z. , Gou, Z. , Lyu, J. , Li, W. , Yu, Y. et al. (2015) Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nature Biotechnol. 33, 408. [DOI] [PubMed] [Google Scholar]
- Zhu, L. and Kuraparthy, V. (2014) Molecular genetic mapping of the major effect photoperiod response locus in Pima cotton (Gossypium barbadense L.). Crop Sci. 54, 2492–2498. [Google Scholar]
- Zhu, F.‐Y. , Chen, M.‐X. , Ye, N.‐H. , Qiao, W.‐M. , Gao, B. , Law, W.‐K. , Tian, Y. et al. (2018) Comparative performance of the BGISEQ‐500 and Illumina HiSeq4000 sequencing platforms for transcriptome analysis in plants. Plant Methods, 14, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Summary of the 436 accessions for resequencing.
Table S2 Summary of SNPs in each chromosome.
Table S3 Summary of Indels in each chromosome.
Table S4 Summary of accessions used in PCR and Sanger sequencing.
Table S5 SNP loci selected for validation PCR and Sanger sequencing.
Table S6 SNP loci selected for validation by the SLAF SNP dataset.
Table S7 Putative regions identified to be under domestication selection sweeps.
Table S8 Genes within the putative domestication sweeps.
Table S9 Candidate genes involved in significantly enriched biological process GO terms, including flowering time, hormone catabolism, defence responses and ageing, in selection sweeps.
Table S10 Summary of known early maturity‐related QTLs during domestication.
Table S11 Resequencing information of parental lines and bulks for BSA analysis.
Table S12 ANNOVA and a broad sense heritability of early maturity‐related traits in the 355 accessions.
Table S13 Genome‐wide significant association signals for seven early maturity‐related traits in 355 accessions using the LMM method.
Table S14 237 SNPs in the GWAS peaks on the chromosome A05.
Table S15 GWAS peaks haplotype on chromosome A05 and D03.
Table S16 Comparison of the plant height for GhGA2OX8‐transgenic Arabidopsis.
Table S17 Elite loci associated with yield and fibre quality from previous studies used in the present study.
Table S18 Differentially expressed genes during fibre and ovule development.
Table S19 List of all primers used in this study.
Table S20 Seven early maturity‐related traits used for GWAS
Figure S1 Map of the early maturity cotton growing area and early maturity cotton used in this study, including the cotton growing area of the Yellow River region (YRR), north of the Northwest Inland region (NIR), and the Northern Specific Early Maturity region (NSER).
Figure S2 Circos plot showing SNP diversity across the 26 chromosomes of Gossypium hirsutum. The chromosomes are numbered. The blue circle represents SNP density; the purple circle shows Indel diversity; the orange and green colours represent gene density within SNP and Indel markers, respectively.
Figure S3 The SNP number on each chromosome and distributions at different regions.
Figure S4 The Indel number on each chromosome and distributions at different regions.
Figure S5 Morphological changes in early, medium and late maturity cotton and wild cotton during domestication and improvement. a: cotton at the stage when the node of the first fruiting branch flower is opening in early maturity cotton. b: cotton at the stage when all the balls are open in early maturity cotton.
Figure S6 Model‐based clustering analysis with different numbers of clusters (K = 2 and 3).
Figure S7 Nucleotide diversity (π) and population divergence (F ST) across the three population, and a diagram of early maturity and special early maturity (ESM) population and medium and late maturity (MLM) population.
Figure S8 Decay of linkage disequilibrium (LD) in Wild, early maturity and special early maturity (ESM) population and medium and late maturity (MLM) population.
Figure S9 Pedigree information for early maturity cotton breeding. The accessions in green are the founder cultivars of early maturity cotton; the accessions in red were collected and analysed in our study; the accessions in black were early maturity cotton but not analysed in our study.
Figure S10 The frequency of FT (flowering time) among the parents (‘Zhong213’ and ‘Richmondi 6’) and 500 F2 individuals in Anyang 2017.
Figure S11 Distributions of the mean values of seven early maturity‐related traits of 355 accessions in eight environments, including flowering time (FT), the period from first flower blooming to first boll opening (FBP), whole growth period (WGP), yield percentage before frost (YPBF), node of the first fruiting branch (NFFB), height of the node of the first fruiting branch (HNFFB) and plant height (PH).
Figure S12 Correlation analysis of seven early maturity traits in 355 natural populations, including flowering time (FT), the period from first flower blooming to first boll opening (FBP), whole growth period (WGP), yield percentage before frost (YPBF), node of the first fruiting branch (NFFB), height of the node of the first fruiting branch (HNFFB) and plant height (PH). **Indicates significance at 0.01.
Figure S13 Manhattan plots for BLUP of flowering time (FT), the period from first flower blooming to first boll opening (FBP), whole growth period (WGP), yield percentage before frost (YPBF), node of the first fruiting branch (NFFB), height of the node of the first fruiting branch (HNFFB) and plant height (PH). Significant trait‐associated SNPs are distinguished by red lines.
Figure S14 Manhattan plots for flowering time (FT) in separate environment.
Figure S15 Manhattan plots for the period from first flower blooming to first boll opening (FBP) in separate environment.
Figure S16 Manhattan plots for whole growth period (WGP) in separate environment.
Figure S17 Manhattan plots for yield percentage before frost (YPBF) in separate environment.
Figure S18 Manhattan plots for node of the first fruiting branch (NFFB) in separate environment.
Figure S19 Manhattan plots for height of the node of the first fruiting branch (HNFFB) in separate environment.
Figure S20 Manhattan plots for plant height (PH) in separate environment.
Figure S21 Expression profiles of Ghir_D03G011290 and Ghir_D03G011300. The x‐axis represents developmental stages (0, 5, 10, 15 and 20 DPS), and the y‐axis indicates the relative expression levels as determined by RNA‐seq. The error bars indicate standard deviation of three biological replicates.
Figure S22 Expression levels of Ghir_D03G011310 at three leaf growth stages and four leaf growth stages by qRT‐PCR (**indicates significance at the 0.01 probability level).
Figure S23 Gene structure of Ghir_D03G011310 and the polymorphism in two haplotypes, ‘A’ allele and ‘G’ allele.
Figure S24 (a) HRM analysis for SNP (rsD03_39122594) in recombinant inbred line population. The axis of the outside is original melting curves; the axis of the inside is melting curves after logarithm. Red and blue curves correspond to favourable alleles (A) and unfavourable alleles (G), respectively. (b) Box plots for two haplotypes in whole growth period at recombinant inbred line population mentioned above (** P < 0.01).
Figure S25 Expression level of Ghir_D03G011310 in empty control as CK and VIGS plants. **Indicates significance at 0.01.
Figure S26 Nucleotide diversity and population divergence (FST) on the chromosome D03 (red part is the strong LD block regions). (a) Nucleotide diversity across the three population. (b) Population divergence (FST) between early maturity and special early maturity (ESM) population and medium and late maturity (MLM) population.
Figure S27 Box plots for SNP rsA05_16453277 (G/T) in height of the node of the first fruiting branch (HNFFB) and plant height (PH).
Figure S28 Comparison of Ghir_A05G017290 expression levels between ‘CRI50’ (green) and ‘TM‐1’ (red) during developmental stages (0, 5, 10, 15, and 20 DPS) by RNA‐seq. **Indicates significance at 0.01.
Figure S29 Manhattan plots for HNFFB on chromosome D05.
Figure S30 Morphological phenotypes of wild‐type and Arabidopsis containing the 35S::GhGA2OX8 cDNA construct. WT, OE8 and OE9 represent wild‐type and transgenic lines, respectively.
Figure S31 Comparison of cell length of CRI50, CLCrVA and VIGS lines. **Indicates significance at 0.01.