

Abstract
Background
Patients heterozygous for COL4A3 or COL4A4 mutations show a wide spectrum of disease, extending from familial isolated microscopic haematuria, as a result of thin basement membranes (TBMs), to autosomal dominant Alport syndrome (ADAS) and end-stage renal disease (ESRD). Many patients are mentioned in the literature under the descriptive diagnosis of TBM nephropathy (TBMN), in which case it actually describes a histological finding that represents the carriers of autosomal recessive Alport syndrome (ARAS), a severe glomerulopathy, as most patients reach ESRD at a mean age of 25 years.
Methods
We performed a systematic literature review for patients with heterozygous COL4A3/A4 mutations with the aim of recording the spectrum and frequency of pathological features. We searched three databases (PubMed, Embase and Scopus) using the keywords ‘Autosomal Dominant Alport Syndrome’ OR ‘Thin Basement Membrane Disease’ OR ‘Thin Basement Membrane Nephropathy’. We identified 48 publications reporting on 777 patients from 258 families.
Results
In total, 29% of the patients developed chronic kidney disease (CKD) and 15.1% reached ESRD at a mean age of 52.8 years. Extrarenal features and typical Alport syndrome (AS) findings had a low prevalence in patients as follows: hearing loss, 16%; ocular lesions, 3%; basement membrane thickening, 18.4%; and podocyte foot process effacement, 6.9%. Data for 76 patients from 54 families emphasize extensive inter- and intrafamilial heterogeneity, with age at onset of ESRD ranging between 21 and 84 years (mean 52.8).
Conclusions
The analysis enabled a comparison of the clinical course of patients with typical ARAS or X-linked AS with those with heterozygous COL4A mutations diagnosed with TBMN or ADAS. Despite the consequence of a potential ascertainment bias, an important outcome is that TBM poses a global high risk of developing severe CKD, over a long follow-up, with a variable spectrum of other findings. The results are useful to practicing nephrologists for better evaluation of patients.
Keywords: Alport syndrome; chronic kidney disease; focal segmental glomerulosclerosis, gene dosage; phenotypic heterogeneity; thin basement membrane nephropathy
INTRODUCTION
Persistent or recurrent familial microscopic haematuria (MH) of glomerular origin is a very frequent finding caused by the most common hereditary renal pathological thinning of the glomerular basement membrane (GBM), referred to in most of the existing literature as thin basement membrane nephropathy (TBMN). The estimated population prevalence is ∼1% [1, 2] and in 40–50% of cases it is caused by defective synthesis of collagen type IV due to heterozygous mutations in the COL4A3 or COL4A4 genes. However, beyond haematuria, most of the patients have a preserved renal function, at least during the early stages of the disease, and minimal proteinuria or extrarenal manifestations.
TBMN has been reported in different ethnic groups, including Caucasians, Asians and African Americans. The age of detection ranges from 1 to 86 years, with a median of 7 years in children and 37 years in adults. Many patients remain undiagnosed due to the subclinical course of the disease, which makes it difficult to determine its accurate prevalence in the population, while there is incomplete penetrance [1, 3].
The GBM of the nephron is the extracellular matrix lying between the layer of capillary endothelial cells and the podocytes in the glomerulus. Along with its complex interconnections with those cells it comprises the glomerular filtration barrier, through which plasma is filtered for further processing into urine. A major GBM component is collagen type IV, interacting with laminin and other important constituents. While in embryonic life the GBM contains α1α1α2 trimers in a meshwork configuration, at maturity after birth it switches predominantly to the α3α4α5 collagen protomers synthesized and secreted by the podocytes [4, 5].
Alport syndrome (AS) is a severe hereditary nephritis caused by defective collagen IV synthesis due to mutations in COL4A3, COL4A4 or COL4A5, leading to the disrupted architecture of the GBM [6]. The majority of cases (85%) comprise X-linked AS (XLAS) caused by mutations in COL4A5. About 14% of cases represent the autosomal recessive Alport syndrome (ARAS), caused by homozygous or compound heterozygous mutations in COL4A3 or COL4A4. These two clinical entities share similar clinical and pathological features due to the complete absence of wild-type collagen IV, both complicated with a marked compromise of renal function during early childhood [7, 8].
A minority of ∼1% of patients are diagnosed with autosomal dominant AS (ADAS), carrying heterozygous COL4A3/A4 mutations. In addition to presenting with MH, they also progress to severe kidney function decline and ESRD later in life in the presence of ultrastructural AS-like features, including alternate thinning and thickening of the GBM. They may or may not demonstrate extrarenal features. There is some controversy around the terminology among experts as to whether they should all be diagnosed with ADAS, especially when no extrarenal findings are seen, or with TBMN, discussed in two recent counter-arguing consensus reports [9, 10]. In case the term ADAS is adopted universally for all patients with COL4A3/A4 heterozygous mutations, the autosomal dominant form of AS will be much more frequent than the ARAS or X-linked form [11, 12]. Recently it was reported that among 90 patients with COL4A mutations, 47 were heterozygous for a COL4A3 or COL4A4 mutation and 43 had mutations in COL4A5 [13]. It is interesting though that in his recent review Kashtan [11] mentions that synonyms for AS are familial nephritis, hereditary nephritis, thin basement membrane disease and thin basement membrane nephropathy. These synonyms apparently reflect the different terms several authors around the world have used to refer to patients with the histological feature of thin basement membranes, regardless of the term they finally adopt for naming this pathological condition.
In this analysis we steer clear of the controversy regarding terminology. With that noted, several reports have described heterozygous patients with a progressive course, frequently associated with focal and segmental glomerulosclerosis (FSGS), developing high-grade proteinuria and chronic renal failure, even leading to ESRD in later life [14–20]. Although several hundreds of patients with heterozygous COL4A3/A4 mutations have been studied, there has been no systematic analysis to derive a thorough picture of the clinical course or define the prevalence of key pathognomonic features. Despite the consequent ascertainment bias associated with the published works, we present all 777 reported patients and compare the prevalence of key features with those in patients with XLAS and ARAS.
MATERIALS AND METHODS
In this systematic review we searched three distinct databases: PubMed (www.ncbi.nlm.nih.gov/pubmed), as the major database for our, Embase (www.embase.com) and Scopus (www.scopus.com). We used the keywords ‘Autosomal Dominant Alport Syndrome’ OR ‘Thin Basement Membrane Disease’ OR ‘Thin Basement Membrane Nephropathy’ and the following search fields: all fields in PubMed; title/abstract/keywords in Embase; and title/abstract/keywords in Scopus. As of 10 August 2018, the search results included 1905 records: 361 records in Embase, 589 in PubMed and 955 in Scopus. Additionally, two records were identified through other sources. After determining the overlap of the results between the three databases and the removal of the duplicates and triplicates, 1093 records in total were pooled for screening (Figure 1).
FIGURE 1.

Venn diagram representing the overlap between the results of the three databases.
From the pool of records we isolated those reporting one or more clinical cases with molecularly documented heterozygous mutations in COL4A3 and/or COL4A4. Reportedly some patients were relatives of patients with documented ARAS. All records that included irrelevant subjects, or relevant subjects lacking clinical cases, or relevant subjects with clinical cases lacking genetic data, as well as records written in languages other than English were excluded. The records that included clinical cases with molecularly documented digenic inheritance of heterozygous mutations in COL4A3 or COL4A4 were processed separately.
Although some patients had features more typical for AS, not all had undergone comprehensive DNA sequencing in search for a second non-allelic COL4A mutation that would justify the phenotype, most probably due to technology limitations. For those in which sequencing identified a second non-genic mutation, there is a separate mention under digenic inheritance.
Finally, 49 records satisfied the set criteria, but 1 more was excluded as it had complete overlap with a larger subsequent publication from the same authors. Therefore 48 publications were deemed eligible and included in the qualitative synthesis. These publications reported on 258 families with 777 patients that satisfied our criteria. Importantly, we distinguished the records with shared data from the same clinical cases and we considered those duplicate cases as single ones. A Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) flow diagram presents the process of study selection (Figure 2).
FIGURE 2.

PRISMA flow diagram representing the process of study selection.
Each article reports one or more clinical cases of TBM due to heterozygous COL4A3 or COL4A4 mutations, which the authors had diagnosed with TBMN or ADAS. In each case, the clinical course and the results of the paraclinical examinations, including renal biopsies and genetic testing, are described in varying levels of detail in the published articles. Many articles report data from living or deceased relatives of the patients, either healthy or diseased. All data were recorded and processed by family, with each family considered as the patient alone or with his/her relatives. Due to inherent limitations, we preferred this approach rather than classifying the data by individual, because some articles present data classified by family without corresponding specific findings to specific members. On one occasion, families and patients were mentioned in more than one publication; they were carefully selected to avoid multiple inclusion.
Not all papers gave strict definitions for CKD, proteinuria or renal failure. We can only mention that patients followed by our clinical colleagues considered proteinuria as clinically significant when >500 mg/day. Similarly, few papers had details about criteria for assessing hearing loss or ocular findings. Authors were very laconic in just mentioning ‘hearing loss’, ‘sensorineural hearing’, ‘high tone bilateral sensorineural hearing loss’, ‘ocular findings’ or ‘anterior lenticonus’ or variations of these, but none had clear definitions and there was no common definition applied to all 48 papers studied. Otosclerosis was never formally excluded.
Notwithstanding this limitation, for our critical analysis we extracted the data exactly as reported and had no contacts with the authors for clarifications. Therefore we refer interested readers to the original 48 publications in Supplementary material Table S1.
In each family, the individuals were numerically classified into multiple categories regarding their sex, race, presence of haematuria, presence of proteinuria, renal function, hearing function, ocular findings, renal biopsy and genotype. Further details were collected for renal function (age at onset of ESRD), ultrastructural pathology and molecular findings. The age of diagnosis was not always available. For patients reported with incomplete information in publications of the group of the senior author (C.D.) we went back to our database and found the missing data, if available (Table 1). Statistical evaluation was with the use of the t-test and log-rank test.
Table 1.
Results for all data items reported for each family
| Data categories | Data items | Number of patients | Percentage of patients (against all patients) | Percentage of patients (against patients with reported data) |
|---|---|---|---|---|
| Sex | Males | 304 | 39.1 | 47.4 |
| Females | 337 | 43.4 | 52.6 | |
| Sex unknown | 136 | 17.5 | – | |
| Race | White or Caucasians | 531 | 68.3 | 89 |
| Black or African Americans | 5 | 0.6 | 0.8 | |
| Asians | 61 | 7.9 | 10.2 | |
| Race unknown | 180 | 23.2 | – | |
| Haematuria | Microhaematuria alone | 630 | 81.1 | 86.3 |
| Microhaematuria plus macrohaematuria | 62 | 8 | 8.5 | |
| Absence of haematuria | 38 | 4.9 | 5.2 | |
| Haematuria unknown | 47 | 6 | – | |
| Proteinuria | Proteinuria | 323 | 41.6 | 46.4 |
| Absence of proteinuria | 373 | 48 | 53.6 | |
| Proteinuria unknown | 81 | 10.4 | – | |
| Renal function | CKD (including ESRD) | 199 | 25.6 | 29 |
| ESRD | 104 | 13.4 (52.3)a | 15.1 | |
| Normal renal function | 488 | 62.8 | 71 | |
| Renal function unknown | 90 | 11.6 | – | |
| Hearing function | Hearing impairment | 101 | 13 | 15.6 |
| Absence of hearing impairment | 548 | 70.5 | 84.4 | |
| Hearing function unknown | 128 | 16.5 | – | |
| Ocular findings | Ocular lesions | 16 | 2.1 | 3 |
| Absence of ocular lesions | 509 | 65.5 | 97 | |
| Ocular lesions unknown | 252 | 32.4 | – | |
| Renal biopsy | Abnormal findings on renal biopsy | 193 | 24.9 | 86.9 |
| Normal findings on renal biopsy | 29 | 3.7 | 13.1 | |
| Renal biopsy not done | 555 | 71.4 | – | |
| Genotype | Mutated COL4A3 | 407 | 52.4 | 53.5 |
| Mutated COL4A4 | 354 | 45.6 | 46.5 | |
| Mutated gene unknown | 16 | 2 | – |
Percentages in the right-most column are calculated against the number of patients for which there were reported data available for each separate data item. Data mentioned as ‘unknown’ means that no results were reported, as there was no testing. The term ‘ocular findings’ represents a heterogeneous list of mentions in the various publications, including ocular lesions, ocular abnormalities, anterior lenticonus.
The number in parenthesis represents the percentage of the patients with CKD who reached ESRD.
RESULTS
In this systematic review, 777 patients from 258 families were recorded. The 48 articles used and the exact number of patients and families studied are presented in Supplementary material Table S1. Table 1 lists the data categories and data items recorded for each family, along with cumulative results for each data category.
Demographic features
Among all patients, 43.4% were females and 39.1% males and in 17.5% of the patients the sex remains unknown. The female:male ratio is 1.1.
TBMN and ADAS are reported substantially more frequently in Caucasian patients, representing 89% of those with known data available, presumably reflecting the fact that most published works are of European origin. For 23.2% of patients, the race remained unknown. Asian and African American patients are reported at 10.2 and 0.8%, respectively (Table 1).
Clinical features
Among 777 patients, the presence/absence of haematuria remained unknown in 6%. Haematuria is the predominant finding, in 94.8% of the patients with available data, including MH (86.3%) and MH with episodes of macrohaematuria (8.5%). Haematuria is reportedly absent in 5.2% of the patients, thus confirming incomplete penetrance (Table 1).
The second most common finding is proteinuria, a decisive manifestation for prognosis and disease course. Proteinuria remained unknown in 10.4% of the patients. Among patients with available data, 46.4% were proteinuric. The amount of protein loss was not provided in most papers, and no definition of proteinuria was ever available. In the authors’ group, clinically significant proteinuria was considered when protein loss exceeded 500 mg/day.
Renal function remained unknown in 11.6% of patients. For those with relevant data, renal function was reduced in 29%. Among patients with CKD, 52.3% (15.1% of all patients) had reached ESRD (Table 1). The age at onset of ESRD was specified in 76 of 104 patients with ESRD. These 76 patients are distributed in 54 families, with impressive inter- and intrafamilial heterogeneity regarding the age at onset, which ranged from 21 to 84 years, with a median of 55 years (mean 52.8) (Figure 3).
FIGURE 3.

(A) and (B) Age at onset of ESRD in 76 patients distributed in 54 families. The range of age at onset of ESRD is 21–84 years. The mean age at onset of ESRD is 52.8 years. As shown, the age at onset of ESRD is spectacularly variable, demonstrating intra- and interfamilial heterogeneity.
A less common manifestation, hearing impairment, was reported in 15.6% of the patients with available data, while it was missing (unknown) in 16.5% of the patients. The least common clinical feature was ocular findings (in different variations), reported at a frequency of only 3% among examined patients. It was unknown and not reported in 32.4% of patients.
Renal biopsy
The vast majority of patients (555; 71.4%) did not undergo renal biopsy, as in most centres this invasive procedure is indicated only under strict clinical criteria. Additionally, in more recent years, the genetic diagnosis obviates the need for a renal biopsy. This inherent difficulty prevents us from knowing if some patients who did not undergo a biopsy had coincidental causes of renal impairment, such as immunoglobulin A (IgA) disease or tubulointerstitial nephritis. Consequently we concentrated on those who did have a biopsy.
Among 222 patients who underwent a renal biopsy (28.6% of all), the vast majority [193 (86.9%)] had pathological findings, including thin membranes (Table 1). All samples were examined with light microscopy and in selected cases with electron microscopy (EM), immunofluorescence and immunohistochemistry (Table 2). A total of 193 patients had pathological findings with at least one of the four methods. Among the biopsies examined with light microscopy, in 77 (39.9%) the pathology included FSGS, membranoproliferative glomerulonephritis (MPGN) and IgA nephropathy in one patient.
Table 2.
Pathologic features on renal biopsies of 193 patients
| Method | Pathologic features | Number of patients | Percentage of patients |
|---|---|---|---|
| Light microscopy | FSGS | 68/193 | 35.2 |
| MPGN | 10/193 | 5.2 | |
| IgA nephropathy | 1/193 | 0.5 | |
| Electron microscopy | Thinning of GBM | 141/174 | 81 |
| Thickening of GBM | 32/174 | 18.4 | |
| Splitting of GBM | 28/174 | 16.1 | |
| Lamination of GBM | 12/174 | 6.9 | |
| Wrinkling of GBM | 3/174 | 1.7 | |
| Basket weaving of GBM | 10/174 | 5.8 | |
| Foot process effacement | 12/174 | 6.9 | |
| Immunofluorescence | Positive for IgA, IgG, IgM, C1q, C3, Fib | 17/41 | 41.5 |
| Immunohistochemistry | Positive for α5 | 28/31 | 90.3 |
fib, fibrinogen.
From a total of 174 biopsies examined with EM, diffuse GBM thinning was a prevailing feature (81%), while other typical AS features were much less prominent, compared with classical AS biopsies, with thickening reported in only 18.4% (Table 2). The vast majority had membrane thinning either alone [94 (54%)] or along with other findings [47 (27%)]. In 33 cases (19%) there was no thinning of the GBM, but the authors reported other findings or an unremarkable renal biopsy. In some papers, there was a vague mention to ‘Alport syndrome findings’ in 23/174 (13.2%) patients.
From a total of 41 biopsies examined with immunofluorescence, 17 (41.5%) showed pathology that included positivity for IgA, IgG and IgM, complement component (C3), complement component (C1q), fibrinogen or their combinations. Of 31 biopsies examined with immunohistochemistry, 28 (90.3%) were positive for collagen IV α5 chain, a finding that agrees with the heterozygous status of the mutation. The other three biopsies concerned two patients with mutations in the COL4A4 gene and one patient with a mutation in the COL4A3. It is unknown why they scored negative for α5 chain staining, considering the heterozygosity.
Mutations
The COL4A3 gene is found more commonly mutated compared with COL4A4 (53.5% versus 46.5%). Founder mutations may account for this, as 152 patients of 777 had mutation COL4A3-p.Gly1334Glu and another 29 had mutation COL4A3-Gly871Cys [16, 19]. The mutant gene remained unknown in 16 patients. Fifty-one patients had mutations on both COL4A3 and COL4A4, due to digenic inheritance [21–23]. The patients with digenic inheritance were not included in the statistical calculations vis-à-vis the TBMN features (Table 1).
Among 76 patients with known age at onset of ESRD, 38 inherited mutations in COL4A3 and 36 in COL4A4, while in two patients it was not specified, as it was examined by linkage analysis (Table 3). In the 74 patients with an identified mutation, 54 harbour missense mutations, mostly glycine substitutions, with a mean age of ESRD of 55.2 years. Importantly, the other 20 patients harbour non-missense mutations resulting in premature termination of translation (deletions, duplications, splice-site mutations) and their mean age of ESRD of 47.1 years. The difference in age at onset of ESRD is statistically significant (t-test, P = 0.03; log-rank test, P = 0.02) (Figure 4).
Table 3.
List of mutations found in patients who reached ESKD
| Patient | Article | Age at ESRD (years) | Mutated gene | cDNA position | Protein position | HGMD |
|---|---|---|---|---|---|---|
| 1 | Jefferson et al. [24] | 35 | COL4A3 | c.4421T>Ca | p.Leu1474Pro | CM1310319 |
| 2 | van der Loop et al. [25] | 35 | COL4A3 | c. 1315G>Ab (leading to exon 21 skipping) | p.Gly439Serb (including exon 21 skipping) | CS003734 |
| 3 | Ciccarese et al. [26] | 60 | COL4A4 | c.975G>T | p.Lys325Asn | CM014171 |
| 4 | Ciccarese et al. [26] | 66 | COL4A4 | c.975G>T | p.Lys325Asn | CM014171 |
| 5 | Pescucci et al. [27] | 41 | COL4A4 | c.2869G>C | p.Gly957Arg | CM044600 |
| 6 | Pescucci et al. [27] | 52 | COL4A4 | c.2869G>C | p.Gly957Arg | CM044600 |
| 7 | Pescucci et al. [27] | 84 | COL4A3 | c.3134G>T | p.Gly1045Val | CM044599 |
| 8 | Pescucci et al. [27] | 45 | COL4A3 | c.3655G>T | p.Gly1219Cys | CM044598 |
| 9 | Pescucci et al. [27] | 70 | COL4A3 | c.3655G>T | p.Gly1219Cys | CM044598 |
| 10 | Marcocci et al. [28] | 46 | COL4A4 | c.1580delG or c.1580_1580delG | p.Gly527Valfs*126 | CD095038 |
| 11 | Marcocci et al. [28] | 67 | COL4A4 | c.1580delG or c.1580_1580delG | p.Gly527Valfs*126 | CD095038 |
| 12 | Marcocci et al. [28] | 42 | COL4A4 | c.4494delG or c.4494_4494delG | p.Gln1499Lysfs*53 | CD095039 |
| 13 | Marcocci et al. [28] | 60 | COL4A4 | c.2383 + 2T>G (ivs28 + 2T>G) | NA (splicing mutation) | CS095029 |
| 14 | Marcocci et al. [28] | 45 | COL4A4 | c.3289 + 1G>A (ivs35 + 1G>A) | NA (splicing mutation) | CS073457 |
| 15 | Marcocci et al. [28] | 60 | COL4A4 | c.3289 + 1G>A (ivs35 + 1G>A) | NA (splicing mutation) | CS073457 |
| 16 | Pierides et al. [3] | 57 | COL4A3 | c.4001G>A | p.Gly1334Glu | CM014047 |
| 17 | Pierides et al. [3] | 64 | COL4A3 | c.4001G>A | p.Gly1334Glu | CM014047 |
| 18 | Pierides et al. [3] | 58 | COL4A3 | c.4001G>A | p.Gly1334Glu | CM014047 |
| 19 | Pierides et al. [3] | 68 | COL4A3 | c.4001G>A | p.Gly1334Glu | CM014047 |
| 20 | Pierides et al. [3] | 55 | COL4A3 | c.4001G>A | p.Gly1334Glu | CM014047 |
| 21 | Pierides et al. [3] | 53 | COL4A4 | c.3648delG (previously mentioned 3854delG) | p.Ser1217Alafs*71 | CD982552 |
| 22 | Pierides et al. [3] | 40 | COL4A4 | c.3648delG (previously mentioned 3854delG) | p.Ser1217Alafs*71 | CD982552 |
| 23 | Pierides et al. [3] | 67 | COL4A3 | c.4001G>A | p.Gly1334Glu | CM014047 |
| 24 | Pierides et al. [3] | 78 | COL4A3 | c.4001G>A | p.Gly1334Glu | CM014047 |
| 25 | Pierides et al. [3] | 62 | COL4A3 | c.4001G>A | p.Gly1334Glu | CM014047 |
| 26 | Pierides et al. [3] | 52 | COL4A3 | c.2611G>T | p.Gly871Cys | CM077179 |
| 27 | Pierides et al. [3] | 62 | COL4A3 | c.4001G>A | p.Gly1334Glu | CM014047 |
| 28 | Pierides et al. [3] | 45 | COL4A3 | c.4001G>A | p.Gly1334Glu | CM014047 |
| 29 | Pierides et al. [3] | 83 | COL4A3 | c.4001G>A | p.Gly1334Glu | CM014047 |
| 30 | Pierides et al. [3] | 55 | COL4A3 | c.4001G>A | p.Gly1334Glu | CM014047 |
| 31 | Pierides et al. [3] | 55 | COL4A3 | c.4001G>A | p.Gly1334Glu | CM014047 |
| 32 | Pierides et al. [3] | 65 | COL4A3 | c.2611G>T | p.Gly871Cys | CM077179 |
| 33 | Artuso et al. [29] | 24 | COL4A4 | c.4749_4751delGTC c.5044C>T | p.(Gln1583_Ser1584delinsHis) p.Arg1682Trpc | CD120076 CM120075 |
| 34 | Fallerini et al. [30] | 70 | COL4A3 | c.2083G>A | p.Gly695Arg | CM040407 |
| 35 | Fallerini et al. [30] | 72 | COL4A3 | c.3239G>C | p.Gly1080Ala | CM148115 |
| 36 | Fallerini et al. [30] | 60 | COL4A4 | c.217C>T | p.Gln73* | CM148117 |
| 37 | Fallerini et al. [30] | 43 | COL4A4 | c.1109G>A | p.Gly370Glu | CM148118 |
| 38 | Fallerini et al. [30] | 53 | COL4A4 | c.1109G>A | p.Gly370Glu | CM148118 |
| 39 | Fallerini et al. [30] | 59 | COL4A4 | c.1109G>A | p.Gly370Glu | CM148118 |
| 40 | Fallerini et al. [30] | 54 | COL4A4 | c.2752G>A | p.Gly918Arg | CM148122 |
| 41 | Fallerini et al. [30] | 38 | COL4A4 | c.4444delC | p.Leu1482Trpfs*70 | CD148126 |
| 42 | Fallerini et al. [30] | 52 | COL4A4 | c.4749_4751delGTC | p.Gln1583_Ser1584delinsHis | |
| 43 | Papazachariou et al. [31] | 37 | COL4A4 | c.3648delG | p.Ser1217Alafs*71 | CD982552 |
| 44 | Papazachariou et al. [31] | 69 | COL4A3 | c.4001G>A | p.Gly1334Glu | CM014047 |
| 45 | Papazachariou et al. [31] | 45 | COL4A3 | c.4001G>A | p.Gly1334Glu | CM014047 |
| 46 | Papazachariou et al. [31] | 39 | COL4A3 | c.4001G>A | p.Gly1334Glu | CM014047 |
| 47 | Papazachariou et al. [31] | 55 | COL4A3 | c.4001G>A | p.Gly1334Glu | CM014047 |
| 48 | Papazachariou et al. [31] | 40 | COL4A3 | c.4001G>A | p.Gly1334Glu | CM014047 |
| 49 | Papazachariou et al. [31] | 51 | COL4A3 | c.1450G>A | p.Gly484Arg | CM1413855 |
| 50 | Papazachariou et al. [31] | 55 | COL4A3 | c.1450G>A | p.Gly484Arg | CM1413855 |
| 51 | Papazachariou et al. [31] | 69 | COL4A4 | c.428G>T | p.Gly143Val | CM1413857 |
| 52 | Papazachariou et al. [31] | 43 | COL4A4 | c.623G>A | pGly208Asp | CM1413858 |
| 53 | Lin et al. [32] | 63 | COL4A4 | c.G2636A (;) c.C4715T | p.Gly879Glu(;) p.Pro1572Leu | CM1412497(;) CM980400 |
| 54 | Lin et al. [32] | 55 | COL4A4 | c.2990G>Ad | p.Gly997Glu | NA |
| 55 | Ramzan et al. [33] | 55 | COL4A4 | c.2420delG | p.Gly807Valfs*62 | CD1313492 |
| 56 | Rosado et al. [34] | 22 | COL4A3 | c.345delG | p.Pro116Leufs*37 | CD150451 |
| 57 | Rosado et al. [34] | 31 | COL4A3 | c.4235G>T | p.Gly1412Val | CM034407 |
| 58 | Rosado et al. [34] | 34 | COL4A3 | c.4235G>T | p.Gly1412Val | CM034407 |
| 59 | Kamiyoshi et al. [20] | 63 | COL4A4 | c.1733G>T | p.Gly578Val | CM143835 |
| 60 | Deng et al. [35] | 56 | COL4A4 | c.3213delA or c.3213_3213delA | p.Gly1072Glufs*69 | CD1617449 |
| 61 | Deng et al. [35] | 60 | COL4A4 | c.3213delA or c.3213_3213delA | p.Gly1072Glufs*69 | CD1617449 |
| 62 | Papazachariou et al. [14] | 55 | COL4A3 | c.3751G>A | p.Gly1251Ser | CM1714507 |
| 63 | Papazachariou et al. [14] | 61 | COL4A3 | c.3751G>A | p.Gly1251Ser | CM1714507 |
| 64 | Papazachariou et al. [14] | 65 | COL4A3 | c.3751G>A | p.Gly1251Ser | CM1714507 |
| 65 | Papazachariou et al. [14] | 23 | COL4A4 | c.2242G>A | p.Gly748Ser | CM1714512 |
| 66 | Papazachariou et al. [14] | 63 | COL4A4 | c.489 + 1G>C (ivs7 + 1G>C) | NA (splicing mutation) | CS1714514 |
| 67 | Imafuku et al. [36] | 60 | COL4A4 | c.1323_1340del18bp | p.(Pro444_Leu449del)e | CD982550 |
| 68 | Imafuku et al. [36] | 58 | COL4A4 | c.827G>C | p.Gly276Ala | CM1720200 |
| 69 | Imafuku et al. [36] | 42 | COL4A3 | c.2863G>A | p.Gly955Arg | CM1720204 |
| 70 | Bullich et al. [22] | 21 | COL4A3 | c.2125 + 1_2224-1)_(2980 + 1_3071-1)dup | Duplication of ex. 29-30_36 | CN1811341 |
| 71 | Bullich et al. [22] | 42 | COL4A3 | c.2275G>A | p.Gly759Arg | CM1811343 |
| 72 | Bullich et al. [22] | 56 | COL4A4 | c.1460G>T | p.Gly487Val | CM1414118 |
| 73 | Bullich et al. [22] | 40 | COL4A4 | c.4508delA or c.4508_4508delA | p.His1503Profs*49 | CD1811330 |
| 74 | Bullich et al. [22] | 41 | COL4A4 | c.3817G>A | p.Gly1273Arg | CM1811348 |
| 75 | Kharrat et al. [37] | 40 | COL4A3 or COL4A4f | ND | ND | ND |
| 76 | Kharrat et al. [37] | 53 | COL4A3 or COL4A4f | ND | ND | ND |
HGMD, Human Gene Mutation Database. In the original publication by Jefferson et al. [24], this mutation is referred to as p.Leu36Pro, as the counting in those days, was from the 3′-end of the gene and was corresponding to exon 5.
Described as p.Gly493Ser, which leads to a splicing mutation, skipping of exon 21, 55aa, from 439 to 493 (also coded as p.439. aGGT>AGT, Gly439Ser. A mutated chain that lacks 55 amino acids in the collagenous domain, between Gly438 and Gly493).
Glutamine and serine are deleted, and histidine is inserted.
In the original publication, it was erroneously referred to as c.Gly2290Ala.
Deletion of six amino acids (Gly-Lys-Pro-Gly-Ala-Pro) from Gly442 to Pro448.
Pathogenicity was defined by linkage analysis, specific gene not defined (ND), NA, not applicable.
FIGURE 4.
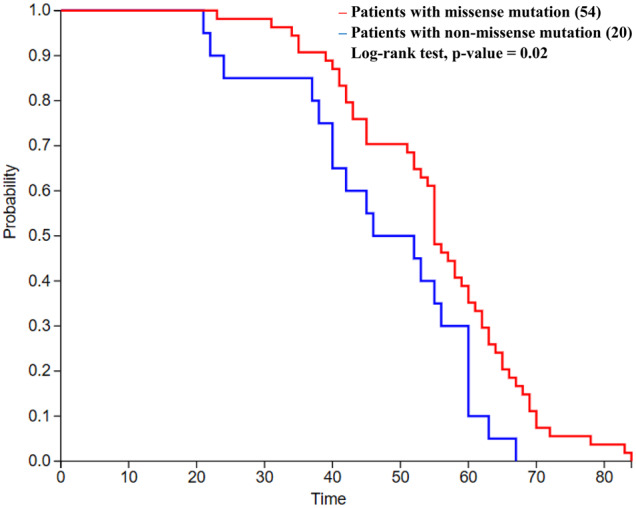
ESRD-free survival analysis comparing the age at ESRD of 74 patients with missense or non-missense mutations. There is a statistically significant difference between the ages at onset of the two groups of patients (log-rank test, P-value = 0.02).
DISCUSSION
Patients who inherit two mutant alleles of the COL4A3 or COL4A4 gene, present with typical ARAS, while heterozygous patients are not symptomless, as happens with most true recessive disorders. Based on the clinical or histological findings, these heterozygous patients were previously diagnosed with TBMN or ADAS by the reporting authors. Apparently the mutant gene dosage is what makes the difference in the clinical outcome between ARAS and TBMN/ADAS. While individuals with bi-allelic mutations present with early-onset ARAS, the most prominent feature of heterozygous patients is autosomal dominant MH. The MH can be either isolated until late ages or, in most cases, accompanied by non-clinically significant proteinuria. In fact, for features beyond haematuria, penetrance is highly variable and age dependent. As shown initially in a large Hellenic cohort, many of these patients with heterozygous COL4A mutations developed FSGS and progressed to proteinuria and other Alport-like features on long follow-up [19]. This histological feature of FSGS has been the reason for misdiagnosis, which was revealed only upon molecular testing, impacting treatment [18, 38–40].
But what are the similarities between heterozygous and homozygous patients for COL4A3/A4 mutations that prompted some authors to collectively diagnose them all with AS? We attempted to answer this question by analysing all described patients with heterozygous mutations previously published under the diagnosis of TBMN or ADAS. Many reports were variably incomplete but nevertheless permitted the evaluation of 777 patients.
Molecular findings
This is the first time that a genotype–phenotype correlation has been attempted in patients with heterozygous COL4A3/A4 mutations. Among 74 patients, not surprisingly, mutations resulting in premature termination of translation were associated with a lower mean age at ESRD (47.1 versus 55.2 years, log-rank test, P = 0.02) (Figure 4). Previous reports were published for patients with XLAS and ARAS and documented that non-missense mutations are associated with more severe disease and earlier age of ESRD [41–45]. In comparison, the age of onset of ESRD in male patients with XLAS was 37 years for those with missense mutations, 28 years for those with splice-site mutations and 25 years for those with truncating mutations [42]. Also, mutations that result in a substitution of glycine with bulkier amino acids demonstrate a correlation of decreasing age of ESRD onset, with an increasing number of side chain carbon atoms in the substituting residue [46].
Clinical characteristics and critical comparisons
Table 4 presents a comparison of the major clinical features of patients with TBMN/ADAS and those with ARAS or XLAS. Also, it provides information for a small number of patients with digenic inheritance. Haematuria remains the cardinal manifestation, affecting 94.8% of heterozygous patients and corroborating incomplete penetrance. In classical AS, there is 100% prevalence of haematuria and macroscopic haematuria in a substantial number of patients. Proteinuria is the second most common finding, in 46.4% of the patients, usually of non-nephrotic range (<3.5 g protein in urine per 24 h).
Table 4.
Comparison of features between patients with heterozygous mutations of COL4A3/A4 and patients with ARAS, XLAS (males) and digenic inheritance of COL4A3/A4 (all numbers are rounded up)
| Features | TBMN/ADAS with a single mutation (COL4A3/COL4A4) | ARAS | XLAS | Digenic inheritance (COL4A3 and COL4A4) |
|---|---|---|---|---|
| Sex (males, females), n (%) | 304, 337 (47, 53) | 71, 74 (49, 51) | 401, 0 (100, 0) | 25, 25 (50, 50) |
| Race (White, Black, Asian), n (%) | 531, 5, 61 (89, 1, 10) | 53, 2, 60 (46, 2, 52) | 401, 0, 0 (100, 0, 0) | NA |
| Haematuria, n/N (%) | 692/730 (95) | 93/93 (100) | 401/401 (100) | 43/51 (84) |
| Proteinuria, n/N (%) | 323/696 (46) | 89/89 (100) | Total 401 (95) | 25/51 (49) |
| CKD, n/N (%) | 199/687 (29) | NA | Total 360 (78) | 22/50 (44) |
| ESRD | 104/687 (15) | 59/95 (62) | Total 360 (70) | 14/50 (28) |
| Mean [median] age at onset of ESRD (years) | 53 [55] (76 cases) | [21] (59 cases) | [25] (233 cases) | <53 [50] (13 cases) |
| Hearing impairment, n/N (%) | 101/649 (16) | 82/129 (64) | Total 303 (79) | 5/40 (13) |
| Ocular findings, n/N (%) | 16/525 (3) | 15/88 (17) | Total 162 (35) | 0/16 (0) |
| Alport syndrome pathognomonic EM findings, n/N (%) | 75/174 (43.1)a | 42/49 (86) | Total 98 (88) | 0/2 (0) |
| Mutated gene (COL4A3, COL4A4), n (%) | 407, 354 (53, 47) | 96, 52 (65, 35) | NA | 46, 46 (100) |
| COL4A mutation type in ESRD cases (missense, non-missense), n (%) | 54, 20/total 74 (73, 27) | 25, 34/59 (42, 58) | NA | NA |
| Mean [median] age (years) at onset of ESRD regarding mutation type (missense, non-missense) | Missense: 55 [55], non-missense: 47 [49] (74 patients) | Missense: [26], non-missense: [19] (59 patients) | [25]b (282 male patients) | NA |
The data were extracted from a total number of 777 patients with TBMN/ADAS (this report), 148 patients with ARAS [30], 401 male patients with XLAS [29] and 48 patients with digenic inheritance [21–23]. The actual numbers and percentages for each feature (in parentheses) were based on those patients for whom solid known data were available in each case. NA: not available.
The percentage of 43.1% in the analysed TBMN/ADAS cohort reflects a very heterogeneous distribution, as each patient only had some of the pathognomonic AS features. For details, see Table 2.
For males with XLAS, the median age of ESRD refers to all kinds of mutations in the COL4A5 gene. Importantly, 76.5% of male patients with XLAS reached ESRD before the age of 31 years [29].
A crucial differentiating point between cohorts of patients with typical AS and TBMN/ADAS is severe kidney function impairment. CKD manifests in 29% of the reported TBMN/ADAS cases across all ages, while the point prevalence of ESRD is 15.1% compared with 62 and 70% for ARAS and XLAS male patients, respectively. The most obvious explanation is that the dose makes the difference, as TBMN/ADAS patients still synthesize 50% of normal collagen IV. Importantly, ESRD presents an age-dependent penetrance, as only four patients reached ESRD before 30 years of age (Figure 3). Two of these patients were described by the authors’ group, one had co-occurrence of vesico-ureteral reflux and the other was extremely obese [14–16].
There is a striking difference in the percentage of patients reaching ESRD. It should be mentioned that renal survival analysis performed by the authors’ lab on 228 Hellens with TBMN showed that by the age of 70 years ∼30–35% reach ESRD, while up to 90% of patients with AS do so by the age of 40 years [15, 16, 43].
The heterogeneity of age at ESRD onset, even among members οf the same family, suggests that the full phenotypic spectrum is multifactorial, implicating primary genes, modifier genes and environmental factors. Candidate genetic modifiers have been described in several reports and include functional variants in the NPHS2 gene, NEPH3, MYH9 and a single-nucleotide polymorphism in the 3′-UTR of the heparin-binding epidermal growth factor gene, which is part of an miRNA seed region [47–53]. For the mentioned candidate modifiers, genetic and functional supportive studies were published. The hypothesis is that modifier genes function over long follow-up in the background of another primary mutation and exacerbate the phenotype. Evidently, more genetic factors remain to be identified [54].
In heterozygous patients diagnosed with TBMN or ADAS, hearing impairment is not a main feature, as it manifests in only 15.6% of the reported cases. Apparently, ophthalmic examination is not done routinely. The ocular examination was reported in nearly two-thirds of the patients and only 3% demonstrated some lesions. Nearly none of the publications offered details on methods of hearing or eye examinations.
The documentation of thin basement membranes depends on a renal biopsy. However, due to its potential complications, its application is dependent on strict clinical criteria. The onset of robust next-generation sequencing (NGS) technologies has made molecular testing the method of choice for investigating patients with familial haematuria. Consequently, only a minority of 28.6% were submitted to renal biopsy (222/777). In most cases, it was remarkable for GBM structural aberrations. However, it must be stressed that there might be some bias, as these cases tended to be more serious and were referred to tertiary care centres.
Under light microscopy, 35.2% of the patients had FSGS, a finding that is indicative of greater severity [16, 18, 55]. It was reported in a few patients since the 1980s and it was well documented in genetically studied cohorts of patients who were initially misdiagnosed with autosomal dominant FSGS, only to discover that this histology-based diagnosis was on the background of TBMN and heterozygous COL4A3/A4 mutations [3, 16, 18, 19, 40].
Importantly, remarkable findings in EM included diffuse thinning of the GBM in 81% of the cases (in 54% of the cases this was the only finding) as well as other findings more congruent with typical AS, i.e. GMB thickening, splitting, foot processes effacement, etc., although less prevalent (Table 2). The different ultrastructural pathology between heterozygous patients and patients with AS, with a milder disturbance of the GBM architecture in heterozygous patients, accounts, at least partly, for the milder clinical course.
Cumulatively the data from multiple authors around the world confirm that TBMN is not a benign disease for all patients of disparate ethnic groups. It certainly does not have the severe course of classical AS or does it demonstrate severe kidney function decline in childhood or even early adulthood. Overall, in a total of 777 patients with ages ranging from early childhood to late adulthood, 15.1% reach ESRD (52.3% of all patients with CKD), a figure similar to what was reported in a previous study [3]. The average age of ESRD, 52.8 years, is also similar to what was published previously on a smaller number of patients [15, 20].
The heterozygosity for COL4A3/A4 mutations is highly prevalent in the general population and consequently many more patients are expected to reach ESRD due to later onset of Alport-like nephropathy than with classical AS, a rare disease [56, 57]. Finally, for the small number of patients with digenic inheritance, the clinical features seem to be between those with a single heterozygous mutation (TBMN/ADAS) and typical AS or closer to those with a single mutation (Table 4). For instance, although the percentage of patients reaching ESRD is higher than for TBMN/ADAS patients (28% versus 15.1%), the mean age at ESRD is nearly the same.
Limitations
We assume that the families examined were referred to tertiary care centres because of an affected proband and therefore enriched in severe phenotypes. This leads to an inevitable ascertainment bias, as probably there are families without severely affected patients and hence they have never been studied. Unfortunately, our systematic analysis has no way to remedy this and the reader should be aware of this potential limitation. However, it is worth noting that in one study where the proband of each family was excluded in order to avoid selection bias, almost half of the cases (47%) presented haematuria associated with proteinuria. Hearing loss and ocular abnormalities were observed at rates of 63 and 12.5%, respectively [30]. A limitation of these figures is that there were not adequate data to document these abnormalities were always due to a later-onset Alport-like nephropathy.
It is unknown if patients with severe disease at younger ages and typical features of AS actually had a second mutation that was never detected. This refers mostly to patients studied in the pre-NGS era. We have no way to resolve this, but based on the data (Table 4) and available literature, they could not account for >10%. No analysis was attempted for heterozygous women with XLAS.
Most studies analysed included Caucasian patients. However, findings presented for Asian patients in China and Japan did not have major differences compared with Europeans with respect to clinical presentation and conclusions. In one large Japanese study of 72 patients diagnosed with ADAS, the median renal survival was 70 years, while only one patient had hearing loss and another had ocular lesions. Also, in 16 biopsies performed, they noticed GBM thinning, but none had lamellation [20].
Another inevitable limitation relates to the pool of the clinical cases, which includes a wide range of ages without determination of the age at onset of each finding. In many studies the data are processed by family rather than by individual and thus it is impossible to associate the findings in each patient separately.
Despite the huge intrafamilial variability, a family history of progressive CKD is one risk factor that should alert doctors, while blood pressure and urine protein should be closely monitored. In conclusion, there is a pressing unmet need to find trustworthy robust biomarkers that can predict with confidence who, of all patients with COL4A heterozygous mutations, is highly predisposed to severe CKD at later ages.
SUPPLEMENTARY DATA
Supplementary data are available at ckj online.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank Dr G. Nikolopoulos and Dr K. Voskarides for advice during the preparation of the manuscript and D. Hadjipanagi, a PhD candidate, who assisted with correcting and recording mutation annotations.
FUNDING
This work was funded by internal sources of the University of Cyprus, specifically from chapter 3/311 of CD.
AUTHORS’ CONTRIBUTIONS
C.D. conceived, designed and supervised the study and assisted in writing and completing the manuscript. A.M. and T.P. searched the literature, analysed and processed the data, prepared the figures and tables and drafted the manuscript.
CONFLICT OF INTEREST STATEMENT
None declared.
REFERENCES
- 1. Savige J, Rana K, Tonna S et al. Thin basement membrane nephropathy. Kidney Int 2003; 64: 1169–1178 [DOI] [PubMed] [Google Scholar]
- 2. Gregory MC. The clinical features of thin basement membrane nephropathy. Semin Nephrol 2005; 25: 140–145 [DOI] [PubMed] [Google Scholar]
- 3. Pierides A, Voskarides K, Athanasiou Y et al. Clinico-pathological correlations in 127 patients in 11 large pedigrees, segregating one of three heterozygous mutations in the COL4A3/COL4A4 genes associated with familial haematuria and significant late progression to proteinuria and chronic kidney disease from focal segmental glomerulosclerosis. Nephrol Dial Transplant 2009; 24: 2721–2729 [DOI] [PubMed] [Google Scholar]
- 4. Plevova P, Gut J, Janda J. Familial hematuria: a review. Medicina (Kaunas) 2017; 53: 1–10 [DOI] [PubMed] [Google Scholar]
- 5. Abrahamson DR, Hudson BG, Stroganova L et al. Cellular origins of type IV collagen networks in developing glomeruli. J Am Soc Nephrol 2009; 20: 1471–1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Miner JH. Pathology vs. molecular genetics: (re)defining the spectrum of Alport syndrome. Kidney Int 2014; 86: 1081–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kashtan CE. Alport syndrome. An inherited disorder of renal, ocular, and cochlear basement membranes. Medicine (Baltimore) 1999; 78: 338–360 [DOI] [PubMed] [Google Scholar]
- 8. Gross O, Kashtan CE, Rheault MN et al. Advances and unmet needs in genetic, basic and clinical science in Alport syndrome: report from the 2015 International Workshop on Alport Syndrome. Nephrol Dial Transplant 2017; 32: 916–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kashtan CE, Ding J, Garosi G et al. Alport syndrome: a unified classification of genetic disorders of collagen IV alpha345: a position paper of the Alport Syndrome Classification Working Group. Kidney Int 2018; 93: 1045–1051 [DOI] [PubMed] [Google Scholar]
- 10. Savige J, Ariani F, Mari F et al. Expert consensus guidelines for the genetic diagnosis of Alport syndrome. Pediatr Nephrol 2019; 34: 1175–1189 [DOI] [PubMed] [Google Scholar]
- 11. Kashtan CE. Alport syndrome In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A. (eds). GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2019 [Google Scholar]
- 12. Torra R, Furlano M. New therapeutic options for Alport syndrome. Nephrol Dial Transplant 2019; 34: 1272–1279 [DOI] [PubMed] [Google Scholar]
- 13. Groopman EE, Marasa M, Cameron-Christie S et al. Diagnostic utility of exome sequencing for kidney disease. N Engl J Med 2019; 380: 142–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Papazachariou L, Papagregoriou G, Hadjipanagi D et al. Frequent COL4 mutations in familial microhematuria accompanied by later-onset Alport nephropathy due to focal segmental glomerulosclerosis. Clin Genet 2017; 92: 517–527 [DOI] [PubMed] [Google Scholar]
- 15. Deltas C, Pierides A, Voskarides K. Molecular genetics of familial hematuric diseases. Nephrol Dial Transplant 2013; 28: 2946–2960 [DOI] [PubMed] [Google Scholar]
- 16. Deltas C, Savva I, Voskarides K, Papazachariou L, Pierides A. Carriers of autosomal recessive Alport syndrome with thin basement membrane nephropathy presenting as focal segmental glomerulosclerosis in later life. Nephron 2015; 130: 271–280 [DOI] [PubMed] [Google Scholar]
- 17. Xie J, Wu X, Ren H et al. COL4A3 mutations cause focal segmental glomerulosclerosis. J Mol Cell Biol 2014; 6: 498–505 [DOI] [PubMed] [Google Scholar]
- 18. Gast C, Pengelly RJ, Lyon M et al. Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrol Dial Transplant 2016; 31: 961–970 [DOI] [PubMed] [Google Scholar]
- 19. Voskarides K, Damianou L, Neocleous V et al. COL4A3/COL4A4 mutations producing focal segmental glomerulo-sclerosis and renal failure in thin basement membrane nephropathy. J Am Soc Nephrol 2007; 18: 3004–3016 [DOI] [PubMed] [Google Scholar]
- 20. Kamiyoshi N, Nozu K, Fu XJ et al. Genetic, clinical, and pathologic backgrounds of patients with autosomal dominant Alport syndrome. Clin J Am Soc Nephrol 2016; 11: 1441–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mencarelli MA, Heidet L, Storey H et al. Evidence of digenic inheritance in Alport syndrome. J Med Genet 2015; 52: 163–174 [DOI] [PubMed] [Google Scholar]
- 22. Bullich G, Domingo-Gallego A, Vargas I et al. A kidney-disease gene panel allows a comprehensive genetic diagnosis of cystic and glomerular inherited kidney diseases. Kidney Int 2018; 94: 363–371 [DOI] [PubMed] [Google Scholar]
- 23. Li A, Cui YX, Lv X et al. The COL4A3 and COL4A4 digenic mutations in cis result in benign familial hematuria in a large Chinese family. Cytogenet Genome Res 2018; 154: 132–136 [DOI] [PubMed] [Google Scholar]
- 24. Jefferson JA, Lemmink HH, Hughes AE et al. Autosomal dominant Alport syndrome linked to the type IV collagen alpha 3 and alpha 4 genes (COL4A3 and COL4A4). Nephrol Dial Transplant 1997; 12: 1595–1599 [DOI] [PubMed] [Google Scholar]
- 25. van der Loop FT, Heidet L, Timmer ED et al. Autosomal dominant Alport syndrome caused by a COL4A3 splice site mutation. Kidney Int 2000; 58: 1870–1875 [DOI] [PubMed] [Google Scholar]
- 26. Ciccarese M, Casu D, Ki Wong F et al. Identification of a new mutation in the alpha4(IV) collagen gene in a family with autosomal dominant Alport syndrome and hypercholesterolaemia. Nephrol Dial Transplant 2001; 16: 2008–2012 [DOI] [PubMed] [Google Scholar]
- 27. Pescucci C, Mari F, Longo I et al. Autosomal-dominant Alport syndrome: natural history of a disease due to COL4A3 or COL4A4 gene. Kidney Int 2004; 65: 1598–1603 [DOI] [PubMed] [Google Scholar]
- 28. Marcocci E, Uliana V, Bruttini M et al. Autosomal dominant Alport syndrome: molecular analysis of the COL4A4 gene and clinical outcome. Nephrol Dial Transplant 2009; 24: 1464–1471 [DOI] [PubMed] [Google Scholar]
- 29. Artuso R, Fallerini C, Dosa L et al. Advances in Alport syndrome diagnosis using next-generation sequencing. Eur J Hum Genet 2012; 20: 50–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fallerini C, Dosa L, Tita R et al. Unbiased next generation sequencing analysis confirms the existence of autosomal dominant Alport syndrome in a relevant fraction of cases. Clin Genet 2014; 86: 252–257 [DOI] [PubMed] [Google Scholar]
- 31. Papazachariou L, Demosthenous P, Pieri M et al. Frequency of COL4A3/COL4A4 mutations amongst families segregating glomerular microscopic hematuria and evidence for activation of the unfolded protein response. Focal and segmental glomerulosclerosis is a frequent development during ageing. PLoS One 2014; 9: e115015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lin F, Bian F, Zou J et al. Whole exome sequencing reveals novel COL4A3 and COL4A4 mutations and resolves diagnosis in Chinese families with kidney disease. BMC Nephrol 2014; 15: 175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ramzan K, Imtiaz F, Taibah K et al. COL4A4-related nephropathy caused by a novel mutation in a large consanguineous Saudi family. Int J Pediatr Otorhinolaryngol 2014; 78: 427–432 [DOI] [PubMed] [Google Scholar]
- 34. Rosado C, Bueno E, Felipe C et al. Study of the true clinical progression of autosomal dominant Alport syndrome in a European population. Kidney Blood Press Res 2015; 40: 435–442 [DOI] [PubMed] [Google Scholar]
- 35. Deng S, Xu H, Yuan J et al. Identification of a novel collagen type IV alpha-4 (COL4A4) mutation in a Chinese family with autosomal dominant Alport syndrome using exome sequencing. Indian J Med Res 2016; 144: 200–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Imafuku A, Nozu K, Sawa N et al. Autosomal dominant form of type IV collagen nephropathy exists among patients with hereditary nephritis difficult to diagnose clinicopathologically. Nephrology (Carlton) 2018; 23: 940–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kharrat M, Makni S, Makni K et al. Autosomal dominant Alport's syndrome: study of a large Tunisian family. Saudi J Kidney Dis Transpl 2006; 17: 320–325 [PubMed] [Google Scholar]
- 38. Wu Y, Hu P, Xu H et al. A novel heterozygous COL4A4 missense mutation in a Chinese family with focal segmental glomerulosclerosis. J Cell Mol Med 2016; 20: 2328–2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Braunisch MC, Buttner-Herold M, Gunthner R et al. Heterozygous COL4A3 variants in histologically diagnosed focal segmental glomerulosclerosis. Front Pediatr 2018; 6: 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Malone AF, Phelan PJ, Hall G et al. Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int 2014; 86: 1253–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gross O, Netzer KO, Lambrecht R et al. Meta-analysis of genotype-phenotype correlation in X-linked Alport syndrome: impact on clinical counselling. Nephrol Dial Transplant 2002; 17: 1218–1227 [DOI] [PubMed] [Google Scholar]
- 42. Bekheirnia MR, Reed B, Gregory MC et al. Genotype-phenotype correlation in X-linked Alport syndrome. J Am Soc Nephrol 2010; 21: 876–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jais JP, Knebelmann B, Giatras I et al. X-linked Alport syndrome: natural history in 195 families and genotype- phenotype correlations in males. J Am Soc Nephrol 2000; 11: 649–657 [DOI] [PubMed] [Google Scholar]
- 44. Lee JM, Nozu K, Choi DE et al. Features of autosomal recessive Alport syndrome: a systematic review. J Clin Med 2019; 8: pii: E178. doi: 10.3390/jcm8020178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Storey H, Savige J, Sivakumar V et al. COL4A3/COL4A4 mutations and features in individuals with autosomal recessive Alport syndrome. J Am Soc Nephrol 2013; 24: 1945–1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tsiakkis D, Pieri M, Koupepidou P et al. Genotype-phenotype correlation in X-linked Alport syndrome patients carrying missense mutations in the collagenous domain of COL4A5. Clin Genet 2012; 82: 297–299 [DOI] [PubMed] [Google Scholar]
- 47. Stefanou C, Pieri M, Savva I et al. Co-inheritance of functional podocin variants with heterozygous collagen IV mutations predisposes to renal failure. Nephron 2015; 130: 200–212 [DOI] [PubMed] [Google Scholar]
- 48. Voskarides K, Arsali M, Athanasiou Y et al. Evidence that NPHS2-R229Q predisposes to proteinuria and renal failure in familial hematuria. Pediatr Nephrol 2012; 27: 675–679 [DOI] [PubMed] [Google Scholar]
- 49. Tonna S, Wang YY, Wilson D et al. The R229Q mutation in NPHS2 may predispose to proteinuria in thin-basement-membrane nephropathy. Pediatr Nephrol 2008; 23: 2201–2207 [DOI] [PubMed] [Google Scholar]
- 50. Voskarides K, Stefanou C, Pieri M et al. A functional variant in NEPH3 gene confers high risk of renal failure in primary hematuric glomerulopathies. Evidence for predisposition to microalbuminuria in the general population. PLoS One 2017; 12: e0174274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Voskarides K, Demosthenous P, Papazachariou L et al. Epistatic role of the MYH9/APOL1 region on familial hematuria genes. PLoS One 2013; 8: e57925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Papagregoriou G, Erguler K, Dweep H et al. A miR-1207-5p binding site polymorphism abolishes regulation of HBEGF and is associated with disease severity in CFHR5 nephropathy. PLoS One 2012; 7: e31021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Flamant M, Bollee G, Henique C, Tharaux PL. Epidermal growth factor: a new therapeutic target in glomerular disease. Nephrol Dial Transplant 2012; 27: 1297–1304 [DOI] [PubMed] [Google Scholar]
- 54. Deltas C. Digenic inheritance and genetic modifiers. Clin Genet 2018; 93: 429–438 [DOI] [PubMed] [Google Scholar]
- 55. Deltas C, Pierides A. COL4A3/COL4A4 heterozygous mutations with TBMN presenting as focal segmental glomerulosclerosis. Kidney Int 2015; 87: 859. doi: 10.1038/ki.2015.38 [DOI] [PubMed] [Google Scholar]
- 56. Levy M, Feingold J. Estimating prevalence in single-gene kidney diseases progressing to renal failure. Kidney Int 2000; 58: 925–943 [DOI] [PubMed] [Google Scholar]
- 57. Pajari H, Kaariainen H, Muhonen T, Koskimies O. Alport's syndrome in 78 patients: epidemiological and clinical study. Acta Paediatr 1996; 85: 1300–1306 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.