

Abstract
C1q/TNF-related protein 9 (CTRP9) is implicated in diverse cardiovascular diseases, but its role in viral myocarditis (VMC) is not well explored. This study is aimed at investigating the role and potential mechanism of CTRP9 in VMC. Herein, we found that the peripheral blood collected from children with VMC had lower CTRP9 levels than that from children who had recovered from VMC. H9c2 cardiomyocytes treated with coxsackievirus B3 (CVB3) were applied to establish a VMC model in vitro, and the expression of CTRP9 was significantly decreased in CVB3-induced H9c2 cells. The overexpression of CTRP9 attenuated CVB3-induced apoptosis, inflammation, and fibrosis reactions in H9c2 cells by promoting cell proliferation, reducing the cell apoptosis rate, and inhibiting inflammatory cytokine levels and fibrosis-related gene expression. Moreover, we found that thrombospondin 1 (THBS1) levels were increased in children with VMC, and CTRP9 negatively regulated THBS1 expression by interacting with THBS1. The downregulation of THBS1 inhibited CVB3-induced apoptosis, inflammation, and fibrosis in H9c2 cells. In addition, our mechanistic investigation indicated that the overexpression of THBS1 impaired the inhibitory effect of CTRP9 on CVB3-induced H9c2 cells. The results further revealed that the CVB3-induced NF-κB and TGF-β1/Smad2/3 signaling pathways of H9c2 cells were blocked by CTRP9 yet activated by THBS1. In conclusion, CTRP9 protected H9c2 cells from CVB3-induced injury via the NF-κB and TGF-β1/Smad2/3 signaling pathways by modulating THBS1.
1. Introduction
Viral myocarditis (VMC) is a common cardiovascular disease in the clinic [1]. It is localized or diffuse myocarditis caused by viral infection, which can cause localized or extensive myocardial interstitial inflammatory infiltration and myocardial cell necrosis or apoptosis, eventually leading to irreversible cardiac dysfunction [2]. In recent years, the incidence of VMC in our country has gradually increased [3, 4]. In addition, the incidence of VMC in children is higher than in adults, which is an important cause of sudden childhood death [5]. Therefore, clarifying the pathogenesis of VMC in children is of great significance for breakthroughs in clinical treatment.
Coxsackievirus B3 (CVB3) is the most common pathogenic virus causing VMC [2, 3, 6]. Studies have shown that CVB3 can induce the NF-κB of cardiomyocytes to enter the nucleus from the cytoplasm, leading to a significant increase in the secretion of inflammatory factors and others, thereby regulating inflammation and immune responses and promoting the development of VMC [7, 8]. Transforming growth factor- (TGF-) β1 is one of the most important precipitating factors in tissue fibrosis [9]. It affects the expression of extracellular matrix proteins by activating the downstream Smad protein family [9, 10]. The sustained high expression of TGF-β1 is closely related to the increase in collagen deposition in the chronic phase of VMC [11]. Studies have reported that CVB3 induces excessive activation of the TGF-β1/Smad signaling pathway to aggravate the pathological process of VMC myocardial fibrosis [12]. Moreover, downregulating the expression of myocardial TGF-β1 can reduce viral myocardial inflammatory damage [11–13].
C1q/TNF-related protein 9 (CTRP9) belongs to the CTRP superfamily, and its spherical domain (active region) has the highest homology with adiponectin (APN) [14, 15]. CTRP9 is a type of adipokine that plays an important protective role in regulating metabolism, inhibiting liver steatosis, and improving insulin resistance [16]. Recent studies have found that CTRP9 is closely related to various cardiovascular diseases [17]. Serum CTRP9 levels are significantly increased in patients with type 2 diabetes and coronary heart disease, and they are closely related to inflammation levels [18]. The overexpression of CTRP9 can inhibit myocardial fibrosis in diabetic rats [19]. In addition, other studies have found that CTRP9 reduces myocardial ischemia/reperfusion injury by inhibiting cardiomyocyte apoptosis [15, 20]. What is more, recent studies have indicated that human-derived recombinant CTRP9 can attenuate isoproterenol-induced myocardial injury in mice [14]. These findings suggest that CTRP9 has a protective effect on the heart, but its specific mechanism of action in VMC is still unclear.
The thrombospondin (THBS) family is a class of calcium-related glycoproteins with multiple domains, which are mainly located on the surface of cell membranes [21]. Due to their special trimeric structure, THBS are widely involved in many biological processes such as angiogenesis, cell movement and apoptosis, cytoskeletal composition, and extracellular matrix (ECM) reaction, through interactions with various target proteins [22, 23]. Moreover, previous papers have shown that the inhibition of THBS1 ameliorates fibrotic remodeling processes in aging cardiomyocytes, and the loss of THBS1 regulates the fibroblast phenotype and matrix remodeling to aggravate chamber dilation [24, 25]. However, its role in the development of VMC remains unclear.
This study attempted to demonstrate the role of CTRP9 in a CVB3-induced VMC cell model. Interestingly, it is reported that APN can directly interact with THBS1 [26], but whether CTRP9 interacts with this protein remains unknown. In addition, THBS1 modulates the inflammatory response via NF-κB signaling, and it can also act as an activator of TGF-β1 signaling [27, 28]. Therefore, our work investigated the relationship between CTRP9, THBS1, and the downstream NF-κB and TGF-β1/Smad2/3 pathways, to determine whether the THBS1 and NF-κB and TGF-β1/Smad2/3 pathways are involved in CTRP9-mediated protective effects on VMC. This finding might provide a potential strategy for VMC treatment.
2. Materials and Methods
2.1. Patients
A total of 35 cases of VMC children aged 9 months to 11 years old who were admitted to our hospital from July 2015 to March 2019 were included in this study. Since 7 patients with other diseases were excluded, only 28 cases were included in the VMC group. All children with VMC received symptomatic treatment, myocardial nutrition and antiviral treatment, and the protection of important organ functions. After the treatment, 24 cases (aged 9 months to 11 years old) finally recovered and were included in the control group; they were followed up regularly after rehabilitation. When fasting early in the morning, peripheral blood was collected from all sick and control children and stored in EDTA anticoagulation tubes at -20°C. All procedures were approved by the Medical Ethics Committee of Xi'an Children's Hospital. All patients or families provided written informed consent.
2.2. Cell Culture and Treatment
H9c2 cells, obtained from the American Type Culture Collection, were plated at a density of 5 × 105 cells/well into 6-well plates. The cells were cultured in Dulbecco's modified Eagle's medium (Invitrogen Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal calf serum (Invitrogen Life Technologies, Carlsbad, CA, USA) in 95% air and 5% CO2 at 37°C for incubation for 24 h. Then, H9c2 cells were infected with 2 × 104 pfu/mL CVB3 (Centre for Endemic Disease Control, Beijing, China) for 48 h.
2.3. Transfection
Full-length rat CTRP9 (PubMed No. NM_001191891.1) and full-length rat THBS1 (PubMed No. NM_001013062.1) were synthesized by Sangon Biotech Co., Ltd. (Shanghai, China) and inserted into recombinant adenovirus vectors, using the same construction protocol as the previous study [29]. The small interfering RNA against THBS1 (si-THBS1) was synthesized by GenePharma Co., Ltd. (Shanghai, China). The vectors were transfected into H9c2 cells by using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA). After transfection for 48 h, the transfection efficiency was more than 90%, which proved that the virus transfection was successful. Then, H9c2 cells were treated with CVB3 (2 × 104 pfu/mL) for 48 h.
2.4. Quantitative Real-Time PCR (qRT-PCR)
The total RNA from serum samples or H9c2 cells was extracted using the TRIzol reagent (Invitrogen, Shanghai, China) according to the manufacturer's protocol. The isolated RNA (2 μg) was reverse-transcribed to cDNA using a SuperScript® III First-Strand Synthesis System (Invitrogen, Shanghai, China). qRT-PCR was performed using the Real-Time Quantitative PCR SYBR Green Kit (Takara, Tokyo, Japan). The relative mRNA levels were normalized to GAPDH and calculated using the 2−ΔΔCt method.
2.5. Enzyme-Linked Immunosorbent Assay (ELISA)
After centrifugation, serum was collected from the peripheral blood of the patients, and the supernatant culture medium of H9c2 cells was also collected for analysis. The concentrations of CTRP9, THBS1, TNF-α, IL-6, and IL-1β were detected using ELISA kits (R&D Systems, Minneapolis, USA) according to the manufacturer's instructions.
2.6. Cell Proliferation Assay
H9c2 cell viability was detected using the Cell Counting Kit-8 (CCK-8, Dojindo, Japan). Briefly, cells were seeded at a density of 2 × 103 cells per well in 96-well plates. After being cultured for 48 h, cells were incubated with 10 μL CCK-8 solution per well at room temperature for 2 h, and the optical density (OD) absorbed by the tested objects was measured using a microplate reader (Bio-Rad, CA, USA) at 450 nm.
2.7. Cell Apoptosis Assay
The apoptosis rate of H9c2 cells was analyzed with an Annexin V-fluorescein isothiocyanate (FITC) conjugate combined with propidium iodide (PI) assay (Dojindo, Japan) and evaluated by fluorescence-activated cell sorting (BD Biosciences, San Diego, USA).
2.8. Western Blot and Coimmunoprecipitation (Co-IP)
After treatment, H9c2 cells were collected, and the total protein was extracted after being lysed with the RIPA lysis buffer (Beyotime Biotechnology, Inc., Shanghai, China). Part of the total protein samples quantified by the BCA method was electrophoresed on an SDS-PAGE gel, and the electrophoresis was completed after the target protein was separated. After transferring the proteins to a PVDF membrane, membranes were blocked by 5% nonfat milk for 2 h. Next, the primary antibody solution was added and samples were incubated for 2 h at room temperature; then, the secondary antibody was added for 1.5 h at room temperature. After Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific, Massachusetts, USA) treatment, the ImageJ software (National Institutes of Health, Maryland, USA) was used to analyze the relative expression level of the target protein with GAPDH as an internal control. For the Co-IP assay, after the extracted protein was quantified by the BCA method, the relevant antibodies and protein A/G beads (Thermo Fisher Scientific, Massachusetts, USA) were added to the protein lysates and then incubated at 4°C overnight. Afterwards, the immunocomplex samples were washed with the precold IP lysis buffer three times, and the supernatant was collected for Western blot.
2.9. Statistical Data Analysis
Experiments were performed in triplicate with at least three repetitions. Statistical analysis was conducted using SPSS version 19.0 statistical software and GraphPad Prism 5.0 software. Pearson's χ2 test was used to examine the correlation between the CTRP9 and THBS1 levels in the serum of children with viral myocarditis (VMC). Student's t-test and one-way ANOVA were used to compare the significance of differences between groups. The data are expressed as mean ± SD, and all statistical significances were set at P < 0.05.
3. Results
3.1. CTRP9 Was Poorly Expressed in Viral Myocarditis
To clarify the role of CTRP9 in pediatric viral myocarditis (VMC), the expression of CTRP9 mRNA and protein in the serum of children with VMC was detected by qRT-PCR and ELISA. There was no significant difference in serum CTRP9 mRNA expression between the different age groups in VMC children, suggesting that growth does not affect CTRP9 levels in children (SFig. 1). Also, our data showed that children with VMC have lower CTRP9 levels than children who have recovered from VMC, suggesting that CTRP9 might play a role in VMC (Figures 1(a) and 1(b), P < 0.05). In addition, we established the VMC cell model by H9c2 cardiomyocytes infected with CVB3. As shown in Figures 1(c) and 1(d), CVB3 infection significantly inhibited cell proliferation, while it enhanced the cell apoptosis of H9c2 cells (P < 0.05). The ELISA data indicate that CVB3 elevated the levels of the inflammatory cytokines TNF-α, IL-6, and IL-1β in H9c2 cells, and the qRT-PCR data further revealed that CVB3 promoted the mRNA expression of the fibrosis-related genes fibronectin, collagen I, and collagen III in H9c2 cells, suggesting the successful establishment of CVB3-induced myocarditis (Figures 1(e) and 1(f), P < 0.05). Then, the expression of CTRP9 in CVB3-induced H9c2 cells was determined, and the results demonstrated that CTRP9 was low expressed in H9c2 cells after CVB3 infection (Figures 1(g) and 1(h), P < 0.05).
Figure 1.
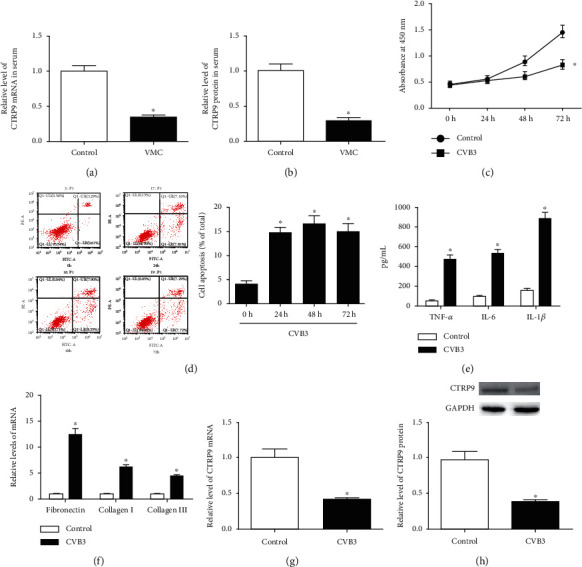
CTRP9 was poorly expressed in viral myocarditis. (a, b) Children with VMC were included in the VMC group (n = 28), and those who have recovered from VMC were included in the control group (n = 24). qRT-PCR was used to measure the expression of CTRP9 mRNA in serum, while ELISA was used to determine CTRP9 protein contents in serum. (c) H9c2 cells were infected with CVB3 (2 × 104 pfu/mL) for 24 h, 48 h, or 72 h; H9c2 cardiomyocytes that didn't infect with CVB3 were. Cell proliferation was measured by CCK-8 assay. (d) Apoptosis rate was detected by flow cytometry. (e) After treated with CVB3 for 48 h, the secretion levels of inflammatory cytokines, including TNF-α, IL-6, and IL-1β, in the medium of H9c2 cells were measured by ELISA; nontreated H9c2 cells performed as the control. (f) The mRNA expression of fibrosis-related genes, including fibronectin, collagen I, and collagen III, was detected by qRT-PCR. (g, h) The expression of CTRP9 mRNA and protein was measured by qRT-PCR and Western blot, respectively. ∗P < 0.05 vs. the control group. All the experiments were repeated at least three times. VMC: viral myocarditis; CVB3: coxsackievirus B3; CCK-8: Cell Counting Kit-8; TNF-α: tumor necrosis factor α; IL-6: interleukin 6; IL-1β: interleukin 1β; ELISA: enzyme-linked immunosorbent assay.
3.2. CTRP9 Protected H9c2 Cells against CVB3-Induced Apoptosis, Inflammation, and Fibrosis
To assess the effect of CTRP9 on CVB3-induced apoptosis, recombinant adenovirus vectors containing full-length rat CTRP9 (Ad-CTRP9) were transfected into H9c2 cells before CVB3 infection. The data of qRT-PCR and Western blot indicated that Ad-CTRP9 conspicuously improved the expression of CTRP9 mRNA and protein in CVB3-induced H9c2 cells (Figures 2(a) and 2(b), P < 0.05). The cell viability of CVB3-induced H9c2 cells was significantly increased, while the cell apoptosis rate was significantly decreased after Ad-CTRP9 transfection (Figures 2(c) and 2(d), P < 0.05). Moreover, the protein expression of Bax/Bcl-2 and cleaved caspase 3 of CVB3-induced H9c2 cells also showed a decrease following Ad-CTRP9 transfection (Figures 2(e) and 2(f), P < 0.05). The effect of CTRP9 on CVB3-induced inflammation and fibrosis in H9c2 cells was further explored. Our data showed that Ad-CTRP9 prominently attenuated the increase in CVB3 on mRNA expression and secretion levels of TNF-α, IL-6, and IL-1β in H9c2 cells (Figures 2(f) and 2(g), P < 0.05). With respect to fibrosis, the mRNA and protein expression of fibronectin, collagen I, and collagen III in CVB3-induced H9c2 cells was prominently reduced by Ad-CTRP9 transfection (Figures 2(h) and 2(i), P < 0.05). These data suggest that CTRP9 protected H9c2 cells from CVB3-induced apoptosis, inflammation, and fibrosis.
Figure 2.
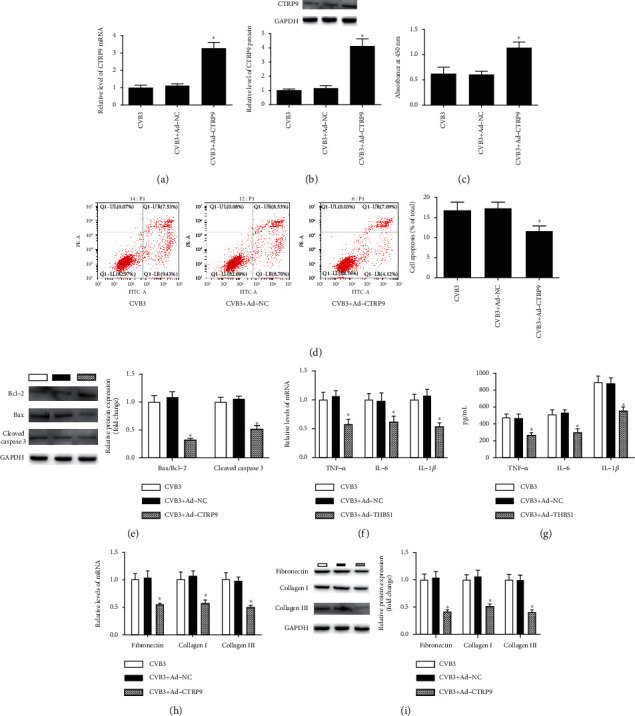
CTRP9 protected H9c2 cells against CVB3-induced apoptosis, inflammation, and fibrosis. (a, b) H9c2 cells were transfected with recombinant adenovirus vectors containing full-length rat CTRP9 (Ad-CTRP9) or its control vectors (Ad-NC) for 48 h before infected with CVB3. Then, the expression of CTRP9 mRNA and protein of H9c2 cells was measured. (c) The cell viability and (d) the apoptosis rate were detected after Ad-CTRP9 transfection. (e, f) The protein expression of apoptosis-related factors, including Bcl-2, Bax, and cleaved caspase 3, was measured after Ad-CTRP9 transfection. (f, g) The mRNA expression and secretion levels of proinflammatory factors TNF-α, IL-6, and IL-1β were measured after Ad-CTRP9 transfection. (h, i) The mRNA and protein expression of pro-fibrosis-related genes fibronectin, collagen I, and collagen III was detected after Ad-CTRP9 transfection. ∗P < 0.05 vs. the CVB3 group. n = 6 in each group. Bcl-2: B-cell lymphoma-2; Bax: B-cell lymphoma-2-associated X; TNF-α: tumor necrosis factor α; IL-6: interleukin 6; IL-1β: interleukin 1β.
3.3. CTRP9 Negatively Regulated THBS1 Expression in CVB3-Infected H9c2 Cells
The ELISA data showed that THBS1 protein levels in the serum of children with VMC were higher than those of children who had recovered from VMC (Figure 3(a), P < 0.05). Interestingly, Pearson's correlation scatterplot showed that CTRP9 levels were negatively correlated with THBS1 levels in children with VMC (Figure 3(b), P < 0.05). Furthermore, the Co-IP assay was applied to determine whether CTRP9 interacts with the THBS1 protein. As expected, Western blot successfully identified THBS1 protein in anti-CTRP9 rather than IgG pretreated immunocomplex samples, implying that CTRP9 interacted with THBS1 (Figure 3(c), P < 0.05). In addition, the data in Figure 4(d) indicate that THBS1 protein expression was negatively regulated by CTRP9 in CVB3-infected H9c2 cells (Figure 3(d), P < 0.05).
Figure 3.
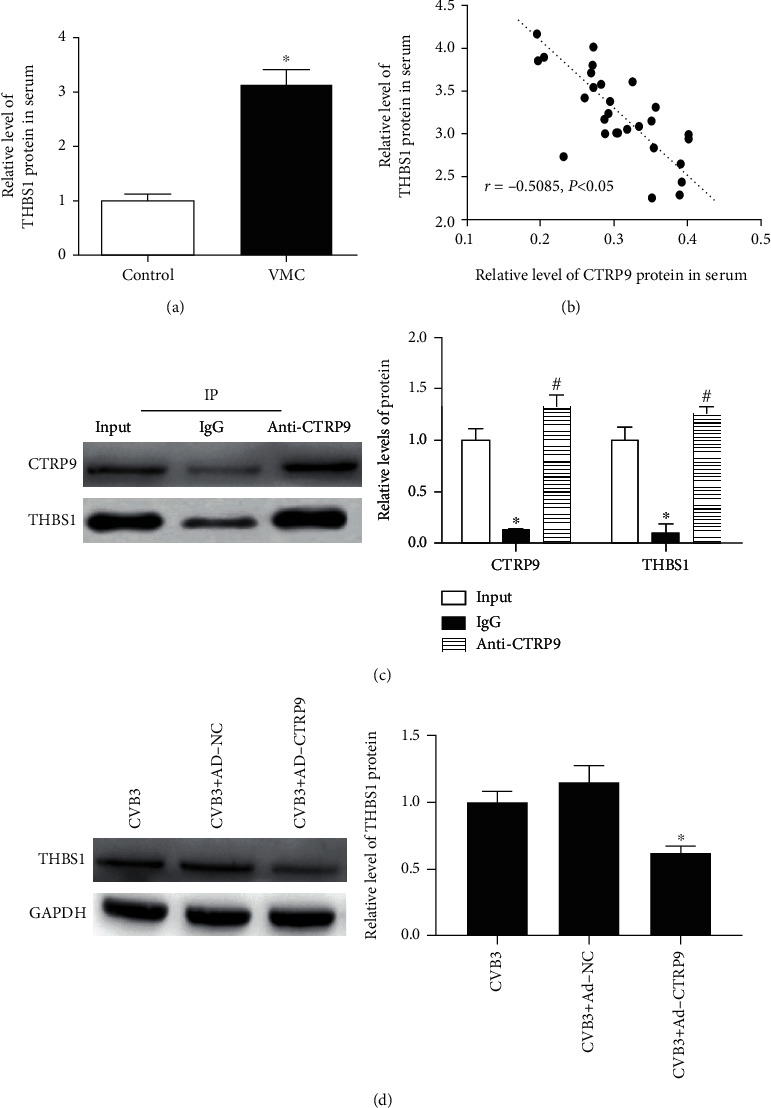
CTRP9 negatively regulated THBS1 expression in CVB3-infected H9c2 cells. (a) ELISA was used to determine THBS1 protein contents in the serum of the control and VMC groups. ∗P < 0.05 vs. the control group. n = 6 in each group. (b) The correlations among CTRP9 protein contents and THBS1 protein contents in the VMC group were determined by Pearson's correlation analysis (r = −0.5085, P < 0.05). (c) The interaction between CTRP9 protein and THBS1 protein was determined by coimmunoprecipitation (Co-IP) in H9c2 cells, with IgG as a negative control. ∗P < 0.05 vs. input. #P < 0.05 vs. IgG. n = 6 in each group. (d) After Ad-CTRP9 transfection, the protein expression of THBS1 of H9c2 cells was detected by Western blot. ∗P < 0.05 vs. the CVB3 group. n = 6 in each group.
Figure 4.
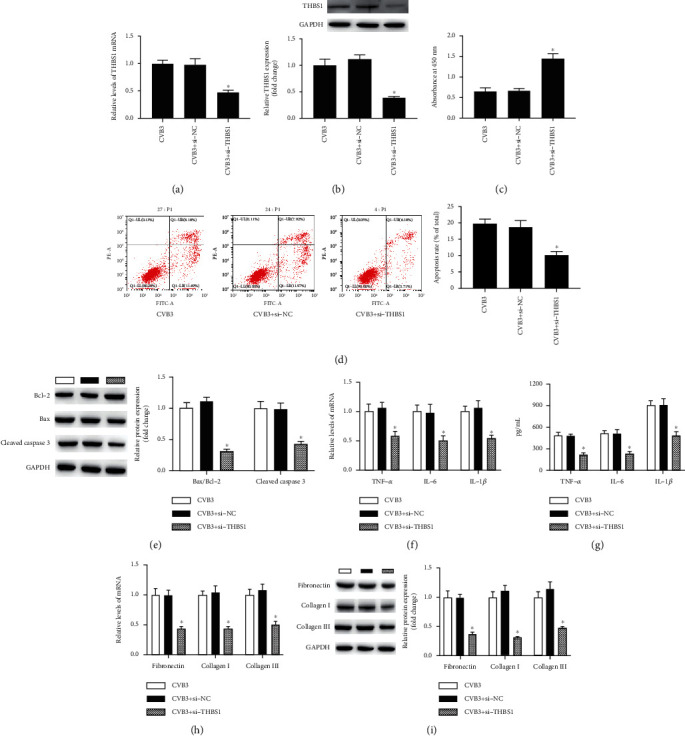
Inhibition of THBS1 protected H9c2 cells against CVB3-induced apoptosis, inflammation, and fibrosis. (a, b) H9c2 cells were transfected with the small interfering RNAs against THBS1 (si-THBS1) or its control vectors (si-NC) for 48 h before infected with CVB3. Then, the expression of THBS1 mRNA and protein of H9c2 cells was detected. (c) The cell viability and (d) the apoptosis rate were detected after si-THBS1 transfection. (e, f) The protein expression of Bcl-2, Bax, and cleaved caspase 3 of CVB3-induced H9c2 cells was determined after si-THBS1 transfection. (f, g) The mRNA expression and the secretion levels of TNF-α, IL-6, and IL-1β were measured after si-THBS1 transfection. (h, i) After si-THBS1 transfection, the mRNA and protein expression of fibronectin, collagen I, and collagen III was detected. ∗P < 0.05 vs. the CVB3 group. n = 6 in each group.
3.4. Inhibition of THBS1 Protected H9c2 Cells against CVB3-Induced Apoptosis, Inflammation, and Fibrosis
To investigate whether the inhibition of THBS1 plays a role in the pathogenesis of VMC, we inhibited THBS1 expression in CVB3-induced H9c2 cells through its small interfering RNA (si-THBS1). As indicated in Figures 4(a) and 4(b), si-THBS1 significantly reduced the expression of THBS1 mRNA and protein in CVB3-induced H9c2 cells (P < 0.05). Then, functional experiments revealed that si-THBS1 increased cell viability while decreasing the apoptosis rate and the expression of proapoptotic proteins in H9c2 cells after CVB3 stimulation (Figures 4(c)–4(e), P < 0.05). Further data showed that CVB3-induced proinflammatory factors and pro-fibrosis-related gene expression were inhibited after the transfection of si-THBS1 in H9c2 cells (Figures 4(h) and 4(i), P < 0.05). Therefore, the inhibition of THBS1 negatively regulated CVB3-induced apoptosis, inflammation, and fibrosis in H9c2 cells.
3.5. THBS1 Attenuated the Effect of CTRP9 on CVB3-Induced H9c2 Cells
Subsequently, the present study investigated the association between THBS1 and CTRP9 in CVB3-induced H9c2 cells. The recombinant adenovirus vectors containing full-length rat THBS1 (Ad-THBS1) were shown to significantly increase THBS1 mRNA and protein expression in CVB3-induced H9c2 cells (Figures 5(a) and 5(b), P < 0.05). Afterwards, THBS1 showed a promotional role in CVB3-induced apoptosis, inflammation, and fibrosis by inhibiting cell viability, enhancing the cell apoptosis rate, inflammatory cytokine levels, and fibrosis-related gene expression (Figures 5(c)–5(f), P < 0.05). Moreover, THBS1 diminished the inhibitory influence of CTRP9 on CVB3-induced apoptosis, inflammation, and fibrosis, suggesting that CTRP9 played a protective role in CVB3-infected H9c2 cells by interacting with THBS1.
Figure 5.
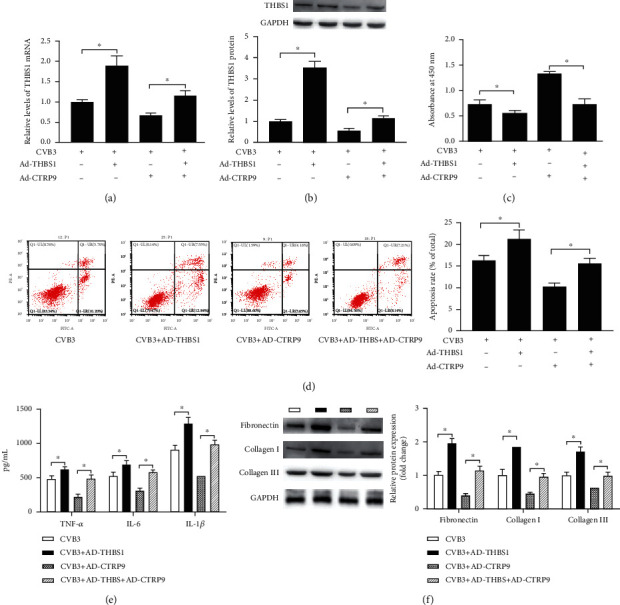
THBS1 attenuated the effect of CTRP9 on CVB3-induced H9c2 cells. (a, b) After transfected with recombinant adenovirus vectors containing full-length rat THBS1 (Ad-THBS1) or/and Ad-CTRP9, the mRNA and protein expression of THBS1 in CVB3-infected H9c2 cells was detected. (c) The cell viability, (d) apoptosis rate, (e) inflammatory cytokine levels, and (f) fibrosis-related gene expression were measured after Ad-THBS1 or/and Ad-CTRP9 transfection. ∗P < 0.05. n = 6 in each group.
3.6. CTRP9 Blocked NF-κB and TGF-β1/Smad2/3 Signaling Pathways via Modulating THBS1
It has been reported that the nuclear factor-κB (NF-κB) and transforming growth factor-β1 (TGF-β1)/Smad2/3 signaling pathways are regulated by THBS1 or CTRP9 in various cardiology diseases [19, 28, 30]; thus, we hypothesized that CTRP9 regulated these signaling pathways via modulating THBS1 in CVB3-infected H9c2 cells. The Western blot data determined that Ad-CTRP9 blocked the activation of the NF-κB and TGF-β1/Smad2/3 signaling pathways by suppressing the protein expression of p-IκBα/IκBα, p-p65/p65, TGF-β1, and p-Smad2/3 in CVB3-infected H9c2 cells, while THBS1 impaired this effect (Figure 6, P < 0.05).
Figure 6.
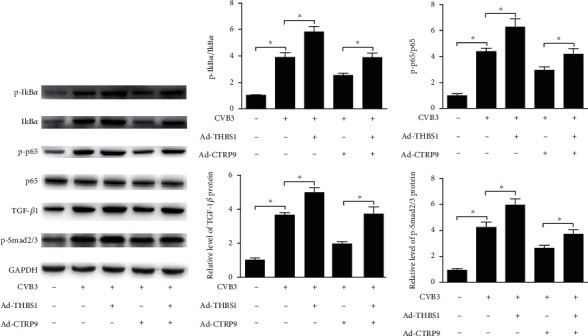
CTRP9 blocked NF-κB and TGF-β1/Smad2/3 signaling pathways via modulating THBS1. After transfected with Ad-CTRP9 or/and Ad-THBS1, the protein expression of NF-κB and TGF-β1/Smad2/3 signaling pathway-related factors, including p-IκBα, IκBα, p-p65, p65, TGF-β1, and p-Smad2/3, in CVB3-infected H9c2 cells was determined by Western blot. ∗P < 0.05. n = 6 in each group.
4. Discussion
VMC is a systemic inflammatory cardiovascular disease, with a high mortality rate among adolescents [2]. Since CVB3-induced VMC is very similar to the disease characteristics of human myocarditis, it has become a widely used model of VMC [3, 7]. Consistent with previous observations in VMC [31–34], our study also indicated that CVB3 infection can significantly promote apoptosis, inflammation, and fibrosis reactions, as demonstrated by the increased expression of the apoptosis-related genes Bax and cleaved caspase 3, increased levels of TNF-α, IL-6, and IL-1β, and increased levels of the fibrosis-related genes fibronectin, collagen I, and collagen III. In addition, we found that CTRP9 expression was downregulated in children with VMC and CVB3-induced H9c2 cells.
The involvement of CTRP9 was frequently observed in ischemic cardiac diseases and may play an important protective role in the cardiovascular system [15, 20]. It has been reported that CTRP9 gene knockout mice show increased areas of myocardial infarction and promote cardiac dysfunction after myocardial ischemia-reperfusion (IR) injury [15, 35]. In addition, CTRP9 gene knockout mice can aggravate left ventricular systolic and diastolic dysfunction, myocardial cell apoptosis, and inflammation in the ischemic heart [20], while the overexpression of CTRP9 can reduce the area of myocardial infarction after myocardial IR injury in normal or diabetic mice, and supplementing wild-type mice with the CTRP9 protein can improve cardiac function, apoptosis, and fibrosis after myocardial infarction [36, 37]. Furthermore, CTRP9 can inhibit hypoxia-induced apoptosis and LPS-induced inflammation by activating AMPK or cAMP in cardiomyocytes [17]. In this study, we found that the overexpression of CTRP9 protected H9c2 cells against CVB3-induced apoptosis, inflammation, and fibrosis by interacting with THBS1.
THBS1 is a matricellular protein, which was initially identified in the extracellular matrix (ECM) [38]. Normally, THBS1 shows a low expression within ECM, but in the tissue injury or changed cellular environment, THBS1 expression is dramatically elevated. Increased THBS1 levels can stimulate the expression of the components of ECM, including fibronectin and collagens such as types I and III [24, 39]. It has been well demonstrated that the excessive accumulation of ECM components is responsible for the development of cardiac fibrosis; thus, THBS1 plays a key role in human fibrosis diseases [24, 38]. Moreover, THBS1 contributes to vascular inflammation in the abdominal aortic aneurysm through activation of TGF-β1 [40, 41], and THBS1 mediates endothelial cell apoptosis by activating caspase [42]. Our findings indicated that THBS1 is involved in CVB3-induced apoptosis, inflammation, and fibrosis reactions in H9c2 cells. It has been reported that the spherical domain (active region) of CTRP9 has the highest homology with adiponectin (APN) [14, 15], and APN can directly interact with THBS1 [26]. Our Co-IP results showed that CTRP9 can bond with THBS1, and CTRP9 repressed CVB3-induced injury in H9c2 cells via interacting with THBS1.
TGF-β1 is a versatile polypeptide that plays an important role in regulating cell growth, differentiation, and the repair of various tissues [9]. In addition, TGF-β1 can participate in the regulation of myocardial fibroblast proliferation, transformation, migration, and ECM production via the TGF-β/Smad signaling pathway [9, 10]. A number of studies have shown that inhibition of the TGF-β1/Smad2/3 signaling pathway relieves the cardiomyocyte apoptosis and fibrosis reactions in myocarditis [43, 44]. In addition, the TGF-β1/Smad2/3 signaling pathway has a proinflammatory role in multiple injured tissues and can mediate mitochondrial apoptosis via regulating Bcl-2 expression [45, 46]. It is now clear that THBS1 is one of the most important physiological activators of TGF-β1, and research has shown that the THBS1-activated TGF-β1/Smad2/3 signaling pathway may play an important role in the development of myocardial interstitial fibrosis in diabetic cardiomyopathy [47]. NF-κB is a protein molecule with multiphase regulation, plays an important role in inflammatory response and immune regulation, and is regarded as the central regulator of cardiomyopathy [8]. Besides, the inactivation of NF-κB has been shown to alleviate the injuries of CVB3-induced VMC in vivo and in vitro [48, 49]. THBS1 can regulate inflammatory cytokine secretion and angiogenesis by activating NF-κB, and CTRP9 attenuates atrial inflammation and fibrosis by suppressing the NF-κB and Smad2/3 signaling pathways [19, 28, 30]. The present study shows that CTRP9 attenuated CVB3-induced injury via the NF-κB and TGF-β1/Smad2/3 signaling pathways by negatively modulating THBS1 expression in H9c2 cells.
5. Conclusions
The present study found that CTRP9 was downregulated and THBS1 was upregulated in children with VMC and H9c2 cells after CVB3 stimulation. Our research further demonstrated that CTRP9 interacted with THBS1 to alleviate CVB3-induced injury by blocking the NF-κB and TGF-β1/Smad2/3 signaling pathways, which could provide an effective strategy for VMC treatment. There are some limitations to this study. Since mammalian cardiomyocytes may not be able to fully replicate human VMC, the role of CTRP9 and THBS1 in the VMC process in vivo needs further investigation. In addition, this study only shows the interaction between CTRP9 and THBS1, and studying changes in this interaction in clinical samples will bring greater value to VMC treatment.
Acknowledgments
The study was funded by the Shaanxi Province Science Technology Research and Development Program (2011K12-05-11). The study was funded by the Health and Family Planning Commission of Xi'an (J201603049).
Data Availability
The data used to support the findings of this study are available from the corresponding author upon request.
Conflicts of Interest
The authors declare no conflict of interest.
Supplementary Materials
The CTRP9 expression in different age groups of VMC and the VP1 expression in different treated groups of H9c2 cells.
References
- 1.Moritz B. P., Frauke G., Karin K., et al. Cardiac function remains impaired despite reversible cardiac remodeling after acute experimental viral myocarditis. Clinical & Developmental Immunology. 2017;2017, article 6590609:1–17. doi: 10.1155/2017/6590609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu H., Gao X., Lin J., et al. MicroRNA-20b suppresses the expression of ZFP-148 in viral myocarditis. Molecular and Cellular Biochemistry. 2017;429(1-2):199–210. doi: 10.1007/s11010-017-2947-7. [DOI] [PubMed] [Google Scholar]
- 3.Bang A., Xuefei L., Ge L., Haitao Y. Interleukin-37 ameliorates coxsackievirus B3-induced viral myocarditis by modulating the Th17/regulatory T cell immune response. Journal of Cardiovascular Pharmacology. 2017;69:305–313. doi: 10.1097/FJC.0000000000000476. [DOI] [PubMed] [Google Scholar]
- 4.Weihui G., Zhen Z., Yang C., Yumei L. MiR-223/Pknox1 axis protects mice from CVB3-induced viral myocarditis by modulating macrophage polarization. Experimental Cell Research. 2018;366:41–48. doi: 10.1016/j.yexcr.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 5.Zhao G., Zhang H. M., Qiu Y., Ye X., Yang D. Cleavage of desmosomal cadherins promotes γ-catenin degradation and benefits Wnt signaling in coxsackievirus B3-induced destruction of cardiomyocytes. Frontiers in Microbiology. 2020;11:p. 767. doi: 10.3389/fmicb.2020.00767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu H., Chuang L., Ping L. Interleukin-27 ameliorates coxsackievirus-B3-induced viral myocarditis by inhibiting Th17 cells. Virology Journal. 2015;12(1):189–189. doi: 10.1186/s12985-015-0418-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weidong L., Mao C., Xu L., et al. Morroniside alleviates coxsackievirus B3-induced myocardial damage apoptosis via restraining NLRP3 inflammasome activation. RSC Advances. 2019;9:1222–1229. doi: 10.1039/c8ra08662a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tianlong L., Mingjie Z., Haiyan N., et al. Astragalus polysaccharide from Astragalus Melittin ameliorates inflammation via suppressing the activation of TLR-4/NF-κB p65 signal pathway and protects mice from CVB3-induced virus myocarditis. International Journal of Biological Macromolecules. 2018;126 doi: 10.1016/j.ijbiomac.2018.12.207. [DOI] [PubMed] [Google Scholar]
- 9.Farhan R., Ramail S., Alessandra D., et al. Simvastatin reduces TGF-β1-induced SMAD2/3-dependent human ventricular fibroblasts differentiation: role of protein phosphatase activation. International Journal of Cardiology. 2018;270:228–236. doi: 10.1016/j.ijcard.2018.06.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhuowang G., Youming C., Wang B., et al. MFGE8 attenuates Ang-II-induced atrial fibrosis and vulnerability to atrial fibrillation through inhibition of TGF-β1/Smad2/3 pathway. Journal of Molecular and Cellular Cardiology. 2020;139:164–175. doi: 10.1016/j.yjmcc.2020.01.001. [DOI] [PubMed] [Google Scholar]
- 11.Sun X. H., Fu J., Sun D. Q. Halofuginone alleviates acute viral myocarditis in suckling BALB/c mice by inhibiting TGF-β1. Biochemical and Biophysical Research Communications. 2016;473(2):558–564. doi: 10.1016/j.bbrc.2016.03.118. [DOI] [PubMed] [Google Scholar]
- 12.Chen P., Xie Y., Shen E., et al. Astragaloside IV attenuates myocardial fibrosis by inhibiting TGF-β1 signaling in coxsackievirus B3-induced cardiomyopathy. European Journal of Pharmacology. 2011;658(2-3):168–174. doi: 10.1016/j.ejphar.2011.02.040. [DOI] [PubMed] [Google Scholar]
- 13.Zhang H., Yu J., Sun H., et al. Effects of ubiquitin-proteasome inhibitor on the expression levels of TNF-α and TGF-β1 in mice with viral myocarditis. Experimental and Therapeutic Medicine. 2019;18(4):2799–2804. doi: 10.3892/etm.2019.7895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liang L., Gao Y. Effect of lipid factor CTRP9 on myocardial remodeling induced by isoproterenol in mice. Zhonghua Yi Xue Za Zhi. 2018;98:3025–3031. doi: 10.3760/cma.j.issn.0376-2491.2018.37.016. [DOI] [PubMed] [Google Scholar]
- 15.Dajun Z., Pan F., Yang S., et al. Cardiac-derived CTRP9 protects against myocardial ischemia/reperfusion injury via calreticulin-dependent inhibition of apoptosis. Cell Death and Disease. 2018;9:p. 723. doi: 10.1038/s41419-018-0726-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nariman M., Reza F., Solaleh E., et al. Association of circulating CTRP9 with soluble adhesion molecules and inflammatory markers in patients with type 2 diabetes mellitus and coronary artery disease. PLoS One. 2018;13 doi: 10.1371/journal.pone.0192159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takahiro K., Koji O., Rei S., et al. CTRP9 protein protects against myocardial injury following ischemia-reperfusion through AMP-activated protein kinase (AMPK)-dependent mechanism. Journal of Biological Chemistry. 2012;287:18965–18973. doi: 10.1074/jbc.M112.357939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hui S., Yuexing Y., Ming W. X., Bond L. W., Liang M. X. Inhibition of CTRP9, a novel and cardiac-abundantly expressed cell survival molecule, by TNFα-initiated oxidative signaling contributes to exacerbated cardiac injury in diabetic mice. Basic Research in Cardiology. 2013;108:p. 315. doi: 10.1007/s00395-012-0315-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mingyang X., Shuzhen L., Hengliang S., Xie Y., Wan D. Cardiology Department Of. Effects of overexpression of CTRP9 mediated by AAV9 on myocardial fibrosis in diabetic rats. Journal of Zhengzhou University. 2017;52:570–574. [Google Scholar]
- 20.Takahiro K., Rei S., Koji O., et al. C1q/tumor necrosis factor-related protein 9 protects against acute myocardial injury through an adiponectin receptor I-AMPK-dependent mechanism. Molecular and Cellular Biology. 2015;35:2173–2185. doi: 10.1128/MCB.01518-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tomas Nicola M., Beck Laurence H., Catherine M., et al. Thrombospondin Type-1 domain-containing 7A in idiopathic membranous nephropathy. The New England Journal of Medicine. 2014;371(24):2277–2287. doi: 10.1056/NEJMoa1409354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roberts David D., Miller Thomas W., Rogers Natasha M., Mingyi Y., Isenberg Jeffrey S. The matricellular protein thrombospondin-1 globally regulates cardiovascular function and responses to stress via CD47. Matrix Biology. 2012;31(3):162–169. doi: 10.1016/j.matbio.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lingyun C., Prasadh R. D., Silverstein Roy L. Thrombospondin-1 modulates VEGF signaling via CD36 by recruiting SHP-1 to VEGFR2 complex in microvascular endothelial cells. Blood. 2013;122:1822–1832. doi: 10.1182/blood-2013-01-482315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ying X., Marcin D., Carlos G., et al. Endogenous thrombospondin 1 protects the pressure-overloaded myocardium by modulating fibroblast phenotype and matrix metabolism. Hypertension. 2011;58:902–911. doi: 10.1161/HYPERTENSIONAHA.111.175323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Almen G. C., Wouter V., Rick V. L., et al. MicroRNA-18 and microRNA-19 regulate CTGF and TSP-1 expression in age-related heart failure. Aging Cell. 2011;10(5):769–779. doi: 10.1111/j.1474-9726.2011.00714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y., Xu L. Y., Lam K. S., Lu G., Cooper G. J., Xu A. Proteomic characterization of human serum proteins associated with the fat-derived hormone adiponectin. Proteomics. 2006;6(13):3862–3870. doi: 10.1002/pmic.200500840. [DOI] [PubMed] [Google Scholar]
- 27.Jefferson B., Ali M., Grant S., et al. Thrombospondin-1 exacerbates acute liver failure and hepatic encephalopathy pathology in mice by activating transforming growth factor β1. The American Journal of Pathology. 2020;190(2):347–357. doi: 10.1016/j.ajpath.2019.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tian X., Wang Y., Wenjie D., et al. Thrombospondin-1 production regulates the inflammatory cytokine secretion in THP-1 cells through NF-κB signaling pathway. Inflammation. 2017;40:1606–1621. doi: 10.1007/s10753-017-0601-x. [DOI] [PubMed] [Google Scholar]
- 29.Ruicheng Z., Liangbao Z., Jufeng Z., Youming P. Complement-C1q TNF-related protein 3 alleviates mesangial cell activation and inflammatory response stimulated by secretory IgA. American Journal of Nephrology. 2016;43:460–468. doi: 10.1159/000446353. [DOI] [PubMed] [Google Scholar]
- 30.Tan B. K., Adya R., Chen J., et al. Metformin decreases angiogenesis via NF-κB and Erk1/2/Erk5 pathways by increasing the antiangiogenic thrombospondin-1. Cardiovascular Research. 2009;83 doi: 10.1093/cvr/cvp131. [DOI] [PubMed] [Google Scholar]
- 31.Wang T., Shuang C., Wang W. X., et al. Aberrant PD-1 ligand expression contributes to the myocardial inflammatory injury caused by coxsackievirus B infection. Antiviral Research. 2019;166:1–10. doi: 10.1016/j.antiviral.2019.03.007. [DOI] [PubMed] [Google Scholar]
- 32.Xingmei Q., Sidong X. Intein-mediated backbone cyclization of VP1 protein enhanced protection of CVB3-induced viral myocarditis. Scientific Reports. 2017;7:p. 41485. doi: 10.1038/srep41485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jinyu C., Yu S., Chunnan L., et al. Nox4-dependent ROS production is involved in CVB3-induced myocardial apoptosis. Biochemical and Biophysical Research Communications. 2018;503:1641–1644. doi: 10.1016/j.bbrc.2018.07.093. [DOI] [PubMed] [Google Scholar]
- 34.Fangqiang S., Kong F., Hongqing Z., Yongqin Z., Ming L. Ulinastatin protects against CVB3-induced acute viral myocarditis through Nrf2 activation. Inflammation. 2018;41:803–810. doi: 10.1007/s10753-018-0734-6. [DOI] [PubMed] [Google Scholar]
- 35.Yang S., Wei Y., Yuexing Y., et al. C1q/tumor necrosis factor–related protein-9, a novel adipocyte-derived cytokine, attenuates adverse remodeling in the ischemic mouse heart via protein kinase A activation. Circulation. 2013;128 doi: 10.1161/CIRCULATIONAHA.112.000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mingxin L., Lin Y., Wei L., et al. C1q/TNF-related protein-9 promotes macrophage polarization and improves cardiac dysfunction after myocardial infarction. Journal of Cellular Physiology. 2019;234:18731–18747. doi: 10.1002/jcp.28513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanxing B., Cheng L., Yang Y., et al. C1q/TNF-related protein 9 protects diabetic rat heart against ischemia reperfusion injury: role of endoplasmic reticulum stress. Oxidative Medicine and Cellular Longevity. 2016;2016:14. doi: 10.1155/2016/1902025.1902025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bao Q., Bangying Z., Ya S., et al. Intermittent hypoxia mediated by TSP1 dependent on STAT3 induces cardiac fibroblast activation and cardiac fibrosis. eLife. 2020;9 doi: 10.7554/eLife.49923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krishna Smriti M., Jonathan G. The role of thrombospondin-1 in cardiovascular health and pathology. International Journal of Cardiology. 2013;168(2):692–706. doi: 10.1016/j.ijcard.2013.04.139. [DOI] [PubMed] [Google Scholar]
- 40.Lopezdee Zenaida P., Kenneth P., Gutierrez Linda S. Thrombospondin-1: multiple paths to inflammation. Mediators of Inflammation. 2011;2011:10. doi: 10.1155/2011/296069.296069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhenjie L., Stephanie M., Jun R., et al. Thrombospondin-1 (TSP1) contributes to the development of vascular inflammation by regulating monocytic cell motility in mouse models of abdominal aortic aneurysm. Circulation Research. 2015;117:p. 129. doi: 10.1161/CIRCRESAHA.117.305262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nor Jacques E., Mitra Raj S., Sutorik Michelle M., Mooney David J., Castle Valerie P., Polverini P. J. Thrombospondin-1 induces endothelial cell apoptosis and inhibits angiogenesis by activating the caspase death pathway. Journal of Vascular Research. 2000;37:209–218. doi: 10.1159/000025733. [DOI] [PubMed] [Google Scholar]
- 43.Lan Z. H. A. N. G., Jing L. I. U., Tian-long L. I. U., et al. Astragalus triterpenoid saponins attenuated CVB3-induced cardiac fibrosis via suppressed TGF-β1/Smad2,p38MAPK signal pathway. Molecular Cardiology of China. 2018;18:2631–2634. [Google Scholar]
- 44.Wen Y., Hailian B., Lixin L., et al. Knockout of immunoproteasome subunit β2i ameliorates cardiac fibrosis and inflammation in DOCA/salt hypertensive mice. Biochemical and Biophysical Research Communications. 2017;490:84–90. doi: 10.1016/j.bbrc.2017.05.011. [DOI] [PubMed] [Google Scholar]
- 45.Liang S., Xianqun M., Wang Z., Jiali L., Kuang H., Wang Q. Polysaccharide from Ephedra sinica Stapf inhibits inflammation expression by regulating factor-β1/Smad2 signaling. International Journal of Biological Macromolecules. 2018;106:947–954. doi: 10.1016/j.ijbiomac.2017.08.096. [DOI] [PubMed] [Google Scholar]
- 46.Masoomeh B., Farhad Z., Mehrdad H., Majid K., Mansooreh S. TGF- β1–mediated apoptosis associated with SMAD-dependent mitochondrial Bcl-2 expression. Clinical Lymphoma, Myeloma & Leukemia. 2012;12:138–143. doi: 10.1016/j.clml.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 47.Hui S., Yong Z., Xiuping B., Shaohua L., Guohai S. Valsartan blocks thrombospondin/transforming growth factor/Smads to inhibit aortic remodeling in diabetic rats. Diagnostic Pathology. 2015;10 doi: 10.1186/s13000-015-0246-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang R., Dan L., Jing O., et al. Leonurine alleviates LPS-induced myocarditis through suppressing the NF-кB signaling pathway. Toxicology. 2019;422:1–13. doi: 10.1016/j.tox.2019.04.011. [DOI] [PubMed] [Google Scholar]
- 49.Remels A. H. V., Derks Wouter J. A., Cilleropastor B., et al. NF-κB-mediated metabolic remodelling in the inflamed heart in acute viral myocarditis. Biochimica et Biophysica Acta. 2018;1864:2579–2589. doi: 10.1016/j.bbadis.2018.04.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The CTRP9 expression in different age groups of VMC and the VP1 expression in different treated groups of H9c2 cells.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.