

Abstract
N6-methyladenosine (m6A) plays an important role in many cancers. However, few studies have examined the role of m6A in colorectal CRC. To examine the effect of m6A on CRC, we studied the genome of 591 CRC cases from The Cancer Genome Atlas (TCGA). The relationship between the messenger RNA (mRNA) expression, copy number variation (CNVs), and mutations of m6A “Writers,” “Readers,” and “Erasers,” prognosis, immune cell infiltration, and genetic mutations in CRC cases were analyzed. CNVs and mutations were found in thirteen m6A regulators. As expected, gain and amplification of m6A regulators increased the mRNA expression of these regulators, while deletion led to reduction in the mRNA expression. Moreover, CNVs and mutation of these regulators were significantly associated with APC, TP53, and microsatellite instability (MSI) status (p < 0.001, p < 0.001, and p = 0.029, respectively). CNVs of m6A regulators also correlated with inferred immune cell infiltration in CRC tissues, especially in colon tissues. Additionally, alterations of RBM15, YTHDF2, YTHDC1, YTHDC2, and METTL14 genes were related to the worse overall survival and disease-free survival (DFS) of CRC patients. Specifically, the deletion status of “Writers” was also correlated to the DFS of CRC patients (p = 0.02). Gene set enrichment analysis found that FTO was involved in mRNA 3′ end processing, polyubiquitin binding, and RNA polymerase promoter elongation, while YTHDC1 was related to interferon-alpha and gamma response. In conclusion, a novel relationship was identified between CNVs and mutations of m6A regulators with prognosis and inferred immune function of CRC. These findings will improve the understanding of the relationship of m6A in CRC.
1. Introduction
Colorectal cancer (CRC) is one of the most lethal malignant diseases worldwide. It is the third leading cause of cancer-related death and third most commonly diagnosed cancer in both men and women in the United States [1]. Despite many studies clarifying the tumor biology of CRC, the incidence and mortality of patients with CRC are still high. Therefore, effective solutions are urgently needed for earlier diagnosis and to predict prognosis.
The concept of epigenetics was first discussed in 1942; since then, epigenetics has been proven to play vital roles in carcinogenesis and cancer progression [2]. RNA modifications are considered as a kind of epigenetics [3] and currently, more than 170 RNA modifications are recognized [4]. Cellular RNAs (rRNAs, transfer RNAs (tRNAs), mRNAs, small nuclear RNAs (snRNAs), lncRNAs and miRNAs, and others) contain over a hundred structurally distinct posttranscriptional modifications at thousands of sites [3]. It has been discovered that RNA modifications regulate most steps of the gene expression, from DNA transcription to mRNA translation [5, 6]. It was not until recently that expansive enthusiasm for RNA modification resurged, provoked by identification of internal mRNA modification, most conspicuously N6-methyladenosine (m6A), methylated at the N6 position of adenosine.
M6A has been the most studied RNA modification to date. It was first reported as the main pattern of eukaryotic mRNA methylation [7]. M6A modification on RNA is abundant near the stop codon and 3′-untranslated region (3′-UTR) [8, 9] and translated near 5′-UTR in a cap-independent manner [10], thereby regulating RNA transcription, translation, and metabolism. The effectors in m6A pathways include “writers” and “erasers” that install and remove the methylation and “readers” that recognize it, respectively. “Writers” include methyltransferase-like 3 (METTL3) [11], METTL14 [12], Wilms tumor 1-associated protein (WTAP) [13], RBM15/15B [14], and KIAA1429 (VIRMA) [15], which introduce the methyl code to target RNAs; “Erasers” mainly include fat mass and obesity-associated protein (FTO) [16] and alkB homologue 5 (ALKBH5) [17], which both selectively delete the methyl code from target RNAs; ‘Readers' such as YT521-B homology (YTH) domain containing 1 (YTHDC1), YTHDC2 [18], YTH N6-methyl-adenosine RNA binding protein 1 (YTHDF1), YTHDF2 [19], eukaryotic initiation factor (eIF) 3 [14], IGF2 mRNA binding proteins (IGF2BP) families [20], heterogeneous nuclear ribonucleoprotein (HNRNP) protein families [21], and zinc finger CCCH domain-containing protein 13 (ZC3H13) [22] can decipher the m6A methylation code.
Growing evidence suggests that m6A modification has been playing an important role in various cancers. This modification is closely linked to increased tumor proliferation, carcinogenesis, migration, and metastasis [23, 24]. In CRC, it has been reported that METTL3 could facilitate tumor progression [25], and YTHDF1 regulates tumorigenicity and cancer stem cell-like activity [26]. Also, m6A modification is related to the lncRNA RP11 activity to trigger the dissemination of CRC cells via upregulation of Zeb1 [27]. RNA modification genes have also been shown to modify T cell activation in cancers. However, there are relatively few studies on the extensive role of m6A RNA modification regulators in CRC prognosis, immune status, and other clinicopathological features. We hypothesize that m6A modification is widely involved in CRC.
This study is aimed at improving the understanding of m6A in CRC and providing some evidence for future examination of the role of RNA m6A methylation in CRC.
2. Materials and Methods
2.1. Datasets
The clinical information, copy number variation (CNV) data, somatic mutation data, and RNA expression data were obtained from The Cancer Genome Atlas (TCGA) cBioportal platform (https://www.cbioportal.org/). The data about immune cell infiltration in tissues was obtained from TIMER (https://cistrome.shinyapps.io/timer/) [28].
2.2. Data Grouping and Analysis
CRC cases with CNV, mutation, and clinicopathological information were retrieved from the TCGA database (Colorectal Adenocarcinoma Project). CNV was identified using segmentation analysis and GISTIC algorithm in the cBioportal platform. The relationship between clinicopathological characteristics was analyzed according to the status of CNV and/or mutation: “patients with mutation and/or CNV of m6A regulators” and “patients without CNV or mutation.” For the RNA-seq data, mRNA expression Z-scores, RSEM (Batch normalized from Illumina HiSeq_RNASeqV2), were obtained from the cBioportal platform. Then, the mRNA expression level was analyzed according to the CNV status of m6A genes. Gene set enrichment analysis (GSEA) was performed using GSEA 3.0 (http://software.broadinstitute.org/gsea/index.jsp) in which the hallmark gene set “h.all.v6.0.symbols.gmt” was adopted. In this study, cases were divided into two groups according to the median expression of m6A regulators' mRNA. Gene sets with a nominal p value <0.05 and the false discovery rate (FDR) < 0.25 were considered to be significantly enriched.
2.3. M6A Regulatory Gene Selection
A comprehensive method was adopted to identify m6A regulatory genes. First, a list of m6A regulators was retrieved from published literature, and then the list was mapped into the TCGA database to exclude the genes of which the data are not available in the database. In total, thirteen m6A RNA modification genes were identified. “Writer”: METTL3, METTL14, WTAP, VIRMA, and RBM15; “Eraser”: FTO and ALKBH5; “Reader”: ZC3H13, YTHDC1, YTHDC2, YTHDF1, YTHDF2, and HNRNPC.
2.4. Statistical Analysis
Data and figures were analyzed using R programming version 3.6.1. The chi-square test or Mann–Whitney U test was used to analyze the correlation between m6A regulators and clinicopathological characteristics. The Kaplan-Meier curve and log-rank test were used to detect the effect of m6A regulatory genes on prognosis. Cox proportional hazard regression models were performed. The overall survival (OS) and disease-free survival (DFS) were defined as the number of months from the initial diagnosis until death or recurrence or the last follow-up, respectively. The infiltration level of immune cells in cancer tissues for each CNV category was compared with those in the normal tissues using a two-sided Wilcoxon rank-sum test in the immune cells' infiltration analysis.
3. Results
3.1. M6A Regulatory Gene CNV Status
591 cases with CNV and mutation data were included in our study among which CNVs were observed in all of the m6A regulator genes. Mutations were observed in all of the thirteen m6A regulators, however, only in a small number of the cases (Figure 1(a)). Moreover, the number of deletion CNV events (1797) was almost equal to the number of gain events (1573) (Figure 1(b)). Notably, 76.1% of the cases harbored YTHDF1 CNVs which was the most commonly observed among all the m6A regulators, while only 28.26% of cases acquired WTAP CNVs (Figure 1(c)).
Figure 1.
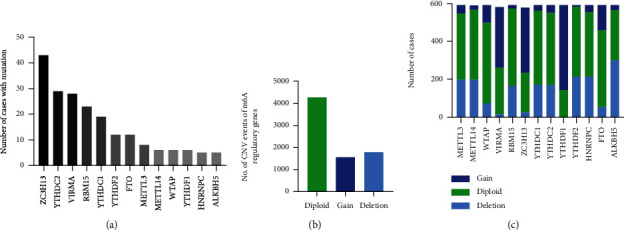
CNVs and mutation events of m6A regulators. (a) Mutation events of m6A regulators in the cohort. (b) Total CNV events of m6A regulators in the cohort. (c) CNV distribution of each m6A regulator.
3.2. CNVs of m6A Regulators with Clinicopathological and Molecular Features
To fully explore the significance of CNVs/mutation of m6A regulators in CRC, we next questioned whether CNVs are associated with clinicopathological characteristics. Age, sex, tumor stage, vascular invasion, lymphovascular invasion, perineural invasion, and sidedness were evaluated. The data revealed that tumor staging was related with CNVs/mutation of m6A regulatory genes, and there was a borderline significance (p = 0.058) between M staging and CNV/mutation status (Table 1). To further understand the mechanisms underlying CNVs/mutation of the m6A regulatory genes, we analyzed the relationship between CNVs/mutation and common gene mutation in CRC, such as KRAS, NRAS, APC (Adenomatous polyposis coli), and TP53. The data showed that APC and TP53 mutations were significantly associated with m6A regulator CNVs/mutation which is consistent with previous data [29], indicating that cases with APC or TP53 mutation were prone to have the m6A regulatory gene CNVs/mutation (Table 2). We also analyzed the relationship between the MSI status and CNVs/mutation, and data showed that MSS cases were prone to have the m6A modification genes' CNVs/mutation (Table 2).
Table 1.
CNVs/Mutation of m6A regulators with clinicopathological characteristics.
| Without mutation/CNV | With mutation/CNV | p value | |
|---|---|---|---|
| Age | 0.583 | ||
| ≤ 65 | 16 | 284 | |
| > 65 | 11 | 263 | |
| Gender | 0.761 | ||
| Male | 12 | 300 | |
| Female | 13 | 264 | |
| Stage | 0.045 | ||
| I | 4 | 98 | |
| II | 15 | 202 | |
| III | 6 | 167 | |
| IV | 0 | 82 | |
| T stage | 0.938 | ||
| Tis | 0 | 1 | |
| T1 | 0 | 19 | |
| T2 | 5 | 98 | |
| T3 | 17 | 384 | |
| T4 | 3 | 62 | |
| N stage | 0.222 | ||
| N0 | 19 | 319 | |
| N1 | 3 | 141 | |
| N2 | 3 | 103 | |
| M stage | 0.058 | ||
| M0 | 21 | 421 | |
| M1 | 0 | 79 | |
| Primary site∗ | 0.307 | ||
| RCC | 10 | 175 | |
| LCC | 5 | 177 | |
| Vascular invasion | 1 | ||
| Without | 13 | 378 | |
| With | 4 | 118 | |
| Lymphovascular invasion | 0.821 | ||
| Without | 13 | 306 | |
| With | 7 | 205 | |
| Perineural invasion | 0.680 | ||
| Without | 6 | 157 | |
| With | 1 | 56 |
∗RCC: right-sided colon cancer; LCC: left-sided colon cancer.
Table 2.
Correlation of m6A regulatory genes' CNVs/Mut and common genes' mutation.
| Without MUT/CNV | With MUT/CNV | p | ||
|---|---|---|---|---|
| APC | WT | 14 | 126 | <0.001 |
| MUT | 11 | 440 | ||
| TP53 | WT | 21 | 203 | <0.001 |
| MUT | 4 | 363 | ||
| KRAS | WT | 17 | 321 | 0.363 |
| MUT | 8 | 245 | ||
| NRAS | WT | 21 | 530 | 0.141 |
| MUT | 4 | 36 | ||
| MSI status | MSI | 7 | 68 | 0.029 |
| MSS | 4 | 174 |
WT: wild type; MUT: mutation; MSI: microsatellite instability; MSS: microsatellite stable; CNV: copy number variation.
To investigate whether CNVs/mutations affected the mRNA expression of m6A regulators, we then examined the relationship between the CNV status and individual m6A regulator mRNA expression in cases with the mRNA expression data. As expected, copy number loss of m6A regulators was associated with lower expression of mRNAs, while copy number gains were related with the higher expression of these mRNAs (Figures 2(a)–2(m)). We also analyzed the correlation of the mRNA expression among all of the thirteen m6A regulators. It suggested that YTHDC1 was correlated with several other genes including VIRMA, METTL14, ZC3H13, and FTO, indicating a central role of YTHDC1 in the m6A modification. In addition, three pairs of genes, WTAP/HNRNPC, METTL14/YTHDC2, and FTO/ZC3H13, were also closely correlated with each other (Figure 2(n)).
Figure 2.
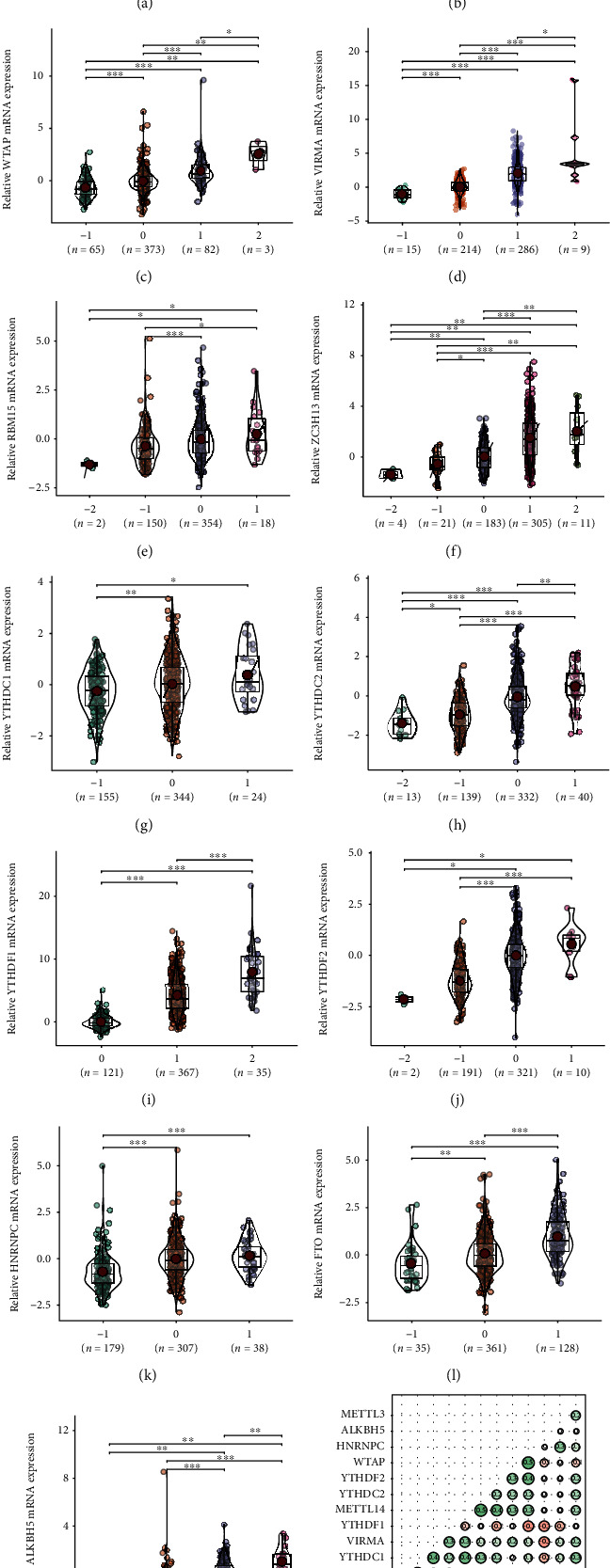
(a)–(m) Effect of CNVs on the mRNA expression of each m6A regulator (-2: deep deletion; -1: shallow deletion; 0: diploid; 1: gain; 2: amplification). (n) mRNA correlation among thirteen m6A regulators. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
3.3. Immune Cell Infiltration and CNVs of m6A Regulators
The immune system plays an important role in carcinogenesis, especially in therapeutic efficacy of immunotherapy. The tumor microenvironment (TME) often contains large numbers of infiltrating myeloid cells including monocytes, macrophages, dendritic cells, and granulocytes. These cells exert various functions in the TME, ranging from regulating the immune process to drug sensitivity. Therefore, we examined whether CNVs of m6A regulators were linked with changes to inferred immune cells infiltration. As shown in Figure 3, CNVs of m6A regulators were closely correlated with immune cell infiltration in the COAD (colon adenocarcinoma) cohort, while in the READ (rectal adenocarcinoma) cohort, the effect of CNVs of m6A genes was reduced. The published literature on the m6A modification also reveals that the m6A modification is related to immune cell activation [30], homeostasis [28], and immune response [31].
Figure 3.

CNVs affect immune cell infiltration in CRC tissues. Blue grid represents that CNV is statistically involved in immune cell infiltration in cancer tissues compared with normal tissues. Grey grid represents that data is not available while white grid represents that p > 0.05. COAD: colon adenocarcinoma; READ: rectum adenocarcinoma.
3.4. CNVs of m6A Regulators with the Prognosis of CRC Patients
We next addressed the relationship between prognosis and m6A regulatory genes to better interpret the role of m6A regulators. The data indicated that RBM15 played an important role in the CRC outcome. Cases with nondiploid (loss/gain) CNVs of RBM15 had a worse OS (HR = 1.89, p = 0.001) and DFS (HR = 1.78, p = 0.005) rates compared with patients who had diploid CNV in the three genes (Figures 4(a) and 4(c)). Moreover, when patients were divided into three groups according to loss, gain, and diploid CNV status, it showed that patients with gain of CNVs in RBM15 had the worse OS compared with patients that had loss or diploid CNV status (gain: HR = 2.57, p = 0.018; loss: HR = 1.91, p = 0.003) (Figure 4(b)). In addition, nondiploid YTHDF2 yielded poorer OS compared to diploid YTHDF2 (HR = 1.43, p = 0.064) (Figure 4(d)). However, there was only a borderline significance for patients that had gain of YTHDF2 mRNA that led to a worse OS when compared with loss and diploid CNVs of YTHDF2 (Figure 4(e)). Moreover, alterations of YTHDC1, YTHDC2, and METTL14 were linked to poorer DFS as shown in Figures 4(f)–4(h). As “Writer” loss was related with poor DFS as shown in Table 3, we further stratified the patients according to the CNV status of “Writer.” The data suggested that patients with gain of “Writer” had better DFS than those without indicating that the upregulation of “Writer” is related to good survival outcome of CRC (Figure 4(i)).
Figure 4.
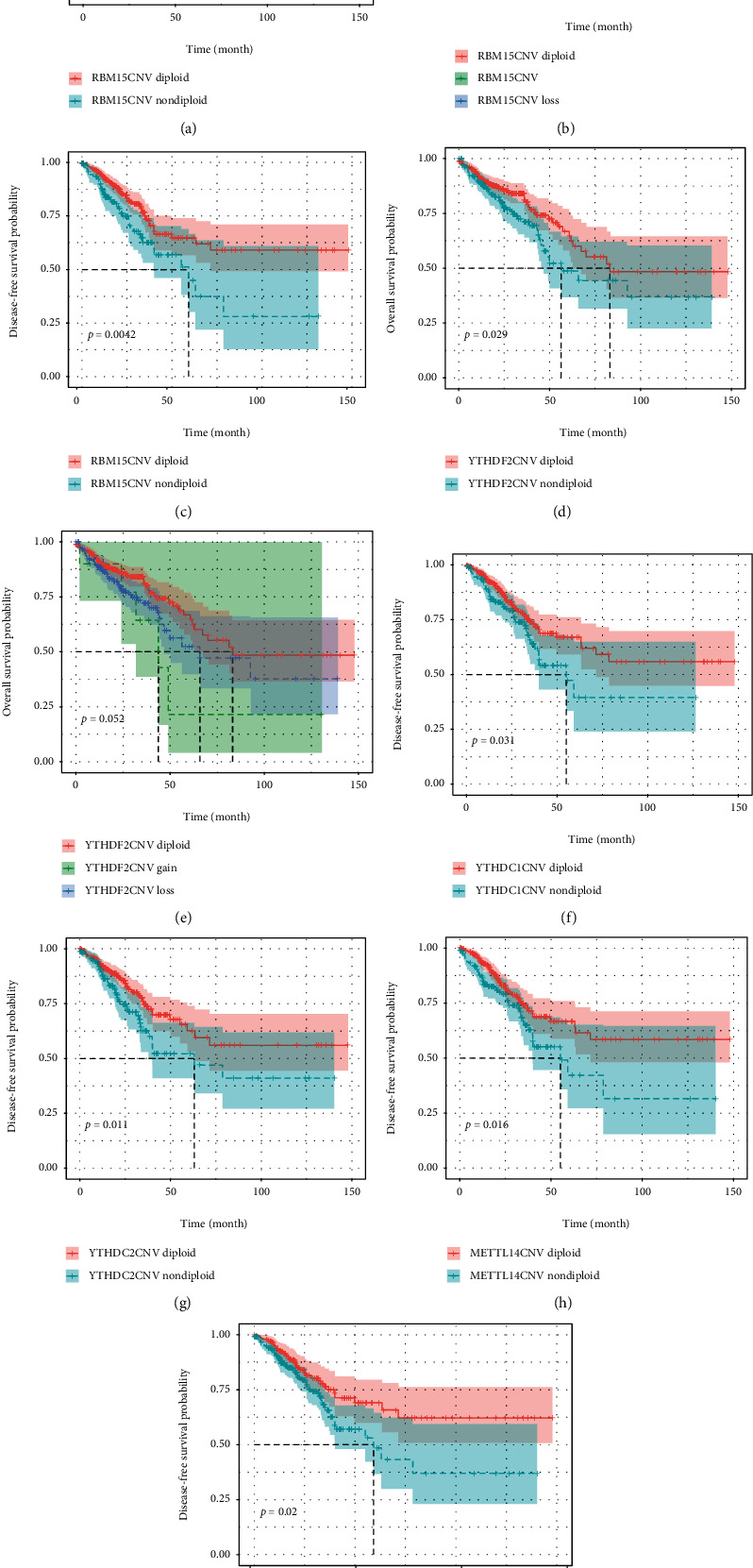
Relationship between CNVs of m6A regulators and OS and DFS. OS: overall survival; DFS: disease-free survival.
Table 3.
Univariate and multivariate COX regression analysis of m6A regulatory genes for CRC patients' OS and DFS.
| Characteristics | OS | DFS | ||||||
|---|---|---|---|---|---|---|---|---|
| Univariate | Multivariate | Univariate | Multivariate | |||||
| HR (95% CI) | p | HR (95% CI) | p | HR (95% CI) | p | HR (95% CI) | p | |
| Age (>65 vs ≤65) | 2.5 (1.68-3.72) | 0.000 | 2.61 (1.08-6.28) | 0.033 | 1.05 (0.72-1.53) | 0.811 | 1.09 (0.70-1.71) | 0.700 |
| Sex (male vs female) | 1.07 (0.75-1.53) | 0.707 | 1.35 (0.91-1.98) | 0.132 | ||||
| Tumor stage | ||||||||
| I | 1 | 1 | 1 | |||||
| II | 1.62 (0.71-3.68) | 0.249 | NA (0-inf) | 0.998 | 2.22 (0.99-4.99) | 0.054 | 2.16 (0.38-12.47) | 0.388 |
| III | 3.07 (1.37-6.88) | 0.006 | NA (0-inf) | 0.997 | 3.62 (1.61-8.12) | 0.002 | 3.17 (0.45-22.27) | 0.246 |
| IV | 7.96 (3.55-17.86) | 0.000 | NA (0-inf) | 0.997 | 8.45 (3.66-19.48) | <0.001 | 1.08 (0.07-16.48) | 0.959 |
| T stage | ||||||||
| T1 | 1 | 1 | 1 | |||||
| T2 | 0.83 (0.18-3.92) | 0.816 | NA (0-inf) | 0.999 | 0.92 (0.2-4.25) | 0.914 | 1.16 (0.14-9.88) | 0.890 |
| T3 | 1.92 (0.47-7.8) | 0.363 | NA (0-inf) | 0.999 | 2.05 (0.5-8.36) | 0.315 | 1.03 (0.09-11.36) | 0.980 |
| T4 | 5.92 (1.4-25.06) | 0.016 | NA (0-inf) | 0.999 | 6.19 (1.45-26.54) | 0.014 | 3.01 (0.27-34.02) | 0.370 |
| N stage | ||||||||
| N0 | 1 | 1 | 1 | |||||
| N1 | 1.77 (1.13-2.78) | 0.013 | 0.11 (0.02-0.63) | 0.013 | 1.8 (1.14-2.84) | 0.011 | 0.86 (0.26-2.86) | 0.805 |
| N2 | 4 (2.63-6.08) | <0.001 | 0.45 (0.09-2.32) | 0.338 | 3.58 (2.27-5.64) | <0.001 | 1.81 (0.54-6.05) | 0.334 |
| M stage (M1 vs M0) | 4.28 (2.85-6.42) | <0.001 | 3.59 (2.26-5.68) | <0.001 | 6.97 (0.78-62.58) | 0.083 | ||
| Site | ||||||||
| Abdomen | 1 | 1 | ||||||
| Colon | 0.42 (0.06-3.02) | 0.389 | 0.35 (0.05-2.54) | 0.301 | ||||
| Rectum | 0.32 (0.04-2.42) | 0.267 | 0.36 (0.05-2.72) | 0.323 | ||||
| TP53 (wild type vs mutant) | 0.97 (0.67-1.4) | 0.860 | 0.91 (0.61-1.35) | 0.630 | ||||
| APC (wild type vs mutant) | 1.08 (0.7-1.66) | 0.719 | 1.27 (0.82-1.97) | 0.276 | ||||
| KRAS (wild type vs mutant) | 1.17 (0.81-1.67) | 0.408 | 0.7 (0.48-1.03) | 0.070 | 0.755 (0.48-1.19) | 0.223 | ||
| NRAS (wild type vs mutant) | 0.86 (0.44-1.7) | 0.663 | 1.55 (0.63-3.81) | 0.339 | ||||
| BRAF (wild type vs mutant) | 0.76 (0.46-1.23) | 0.264 | 1.12 (0.6-2.08) | 0.729 | ||||
| MSI status (MSS vs MSI) | 0.52 (0.29-0.93) | 0.027 | 0.31 (0.13-0.76) | 0.01 | 0.79 (0.39-1.62) | 0.526 | ||
| Vascular invasion (with vs without) | 2.28 (1.52-3.41) | 0.000 | 0.74 (0.31-1.77) | 0.494 | 1.77 (1.14-2.75) | 0.012 | 1.18 (0.64-2.17) | 0.598 |
| Lymphovascular invasion (with vs without) | 2 (1.36-2.95) | 0.000 | 1.86 (0.68-5.11) | 0.228 | 1.66 (1.12-2.47) | 0.012 | 0.78 (0.42-1.44) | 0.420 |
| Perineural invasion (with vs without) | 1.56 (0.8 - 3.03) | 0.188 | 1.61 (0.83-3.12) | 0.157 | ||||
| Lymph node count | ||||||||
| 0-11 | 1 | 1 | 1 | 1 | ||||
| 12-23 | 0.55 (0.34-0.89) | 0.016 | 0.67 (0.10-4.70) | 0.688 | 0.68 (0.38-1.2) | 0.181 | 2.32 (1.13-4.74) | 0.020 |
| > 24 | 0.46 (0.27-0.77) | 0.003 | 0.18 (0.02-1.51) | 0.114 | 0.58 (0.32-1.06) | 0.076 | 1.44 (0.88-2.36) | 0.140 |
| CNV/MUT (with vs without) | 0.66 (0.31-1.43) | 0.295 | 1.02 (0.38-2.78) | 0.964 | ||||
| WTAPCNV | ||||||||
| Deep deletion | 1 | 1 | ||||||
| Shallow deletion | 0.13 (0.02-1) | 0.050 | 1.92 (0.15-24.62) | 0.615 | 1 | |||
| Diploid | 0.08 (0.01-0.62) | 0.015 | 0.43 (0.03-5.85) | 0.528 | 0.7 (0.41-1.2) | 0.189 | ||
| Gain | 0.11 (0.01-0.82) | 0.032 | 0.47 (0.04-6.24) | 0.57 | 0.9 (0.45-1.78) | 0.764 | ||
| Amplification | 0.19 (0.01-3.11) | 0.247 | NA (0-inf) | 0.999 | 1.43 (0.19-10.83) | 0.727 | ||
| Writer loss eraser gain status | ||||||||
| LNGN | 1 | 1 | 1 | 1 | ||||
| LNGP | 0.64 (0.29-1.42) | 0.273 | 0.51 (0.10-2.48) | 0.4 | 1.04 (0.48-2.27) | 0.917 | 1.19 (0.46-3.07) | 0.730 |
| LPGN | 1.43 (0.95-2.13) | 0.083 | 0.59 (0.21-1.67) | 0.316 | 1.54 (0.97-2.43) | 0.066 | 1.58 (0.90-2.76) | 0.110 |
| LPGP | 0.87 (0.48-1.57) | 0.637 | 0.22 (0.04-1.09) | 0.064 | 1.77 (1.03-3.03) | 0.038 | 1.64 (0.85-3.14) | 0.140 |
CNV: copy number variation; LN(P)GN(P): loss negative (positive) gain negative (positive). OS: overall survival; DFS: disease-free survival.
To test whether CNVs of m6A regulatory genes were independent prognostic factors, cox regression analysis was performed. The results suggested that “Writer” loss positive/“Eraser” gain positive was associated with poorer DFS; however, it was not an independent factor according to the multivariate analysis. In addition, age, tumor stage, tumor stage, T/N/M stage, MSS (microsatellite stable), vascular invasion, lymph-vascular invasion, and lymph node count were associated with OS. In addition, age, tumor stage, and lymph node count were independent factors of OS indicated by multivariate analysis. Tumor stage, T/N/M stage, vascular invasion, lymph-vascular invasion, and “Writer” loss status/“Eraser” gain status were correlated with DFS, and only the lymph-node count was an independent factor after multivariate analysis (Table 3).
3.5. Gene Set Enrichment Assay (GSEA)
Due to the importance of CNVs in m6A regulators in carcinogenesis, we next questioned whether specific pathways are changed by these m6A regulatory genes. We explored the enriched gene sets in samples with low or high m6A regulatory gene mRNA expression levels in 524 cases. The GSEA results implied that the FTO low expression was associated with polyubiquitin binding, mRNA 3′ end processing, and transcription elongation from RNA polymerase II (Figures 5(a)–5(c)). These biological processes are widely involved in tumors' malignancy [32–34]. In addition, the YTHDC1 low expression was related with the interferon-gamma response and interferon-alpha (Figures 5(d) and 5(e)).
Figure 5.
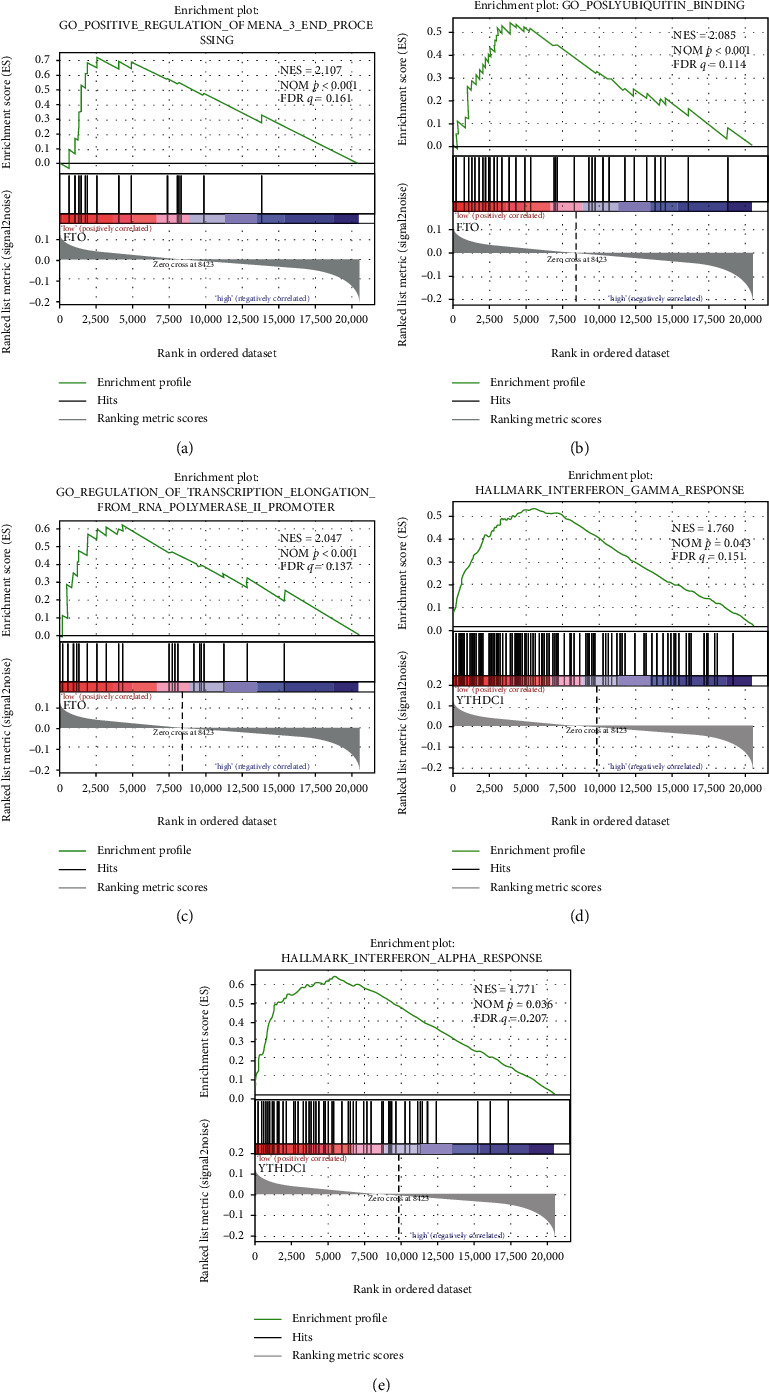
GSEA results of the mRNA level of FTO and YTHDC1. Gene set enrichment plots of (a) mRNA 3′ end processing, (b) polyubiquitin binding, (c) transcription from RNA polymerase promoter, (d) interferon-gamma, and (e) interferon-alpha response.
4. Discussion
Previous TCGA studies have assessed genomic correlation [35], immune cells infiltration, and molecular characterization [36] of CRC. In our study, we comprehensively explored the effect of CNVs and mutations in m6A regulators on the mRNA expression, immune cell infiltration, metabolic pathways, and CRC patient survival. We observed that CNVs affected the mRNA expression of m6A regulators, immune cell infiltration in CRC tissues, and patient prognosis. Furthermore, the dysregulated mRNA expression correlated with immune cell regulatory pathways.
Our data shows that the majority of CRC patients had acquired CNV or mutations to m6A modification genes, with 76%, 64%, and 58% cases harboring YTHDF1, ZC3H13, and VIRMA CNV, respectively. The frequency of alterations is much higher than other cancers, such as acute myelocytic leukemia [29] and renal cancer [37]. It suggests that the m6A modification plays a more important role in CRC. It has been shown that various m6A correlated genes are involved in the regulation of carcinogenesis, proliferation, migration, and stem cell-like activity [25–27]. Interestingly, “Reader” ZC3H13 and “Writer” VIRMA are both predisposed to mutation and copy number gain, as positively correlated with each other as shown in Figure 2(n).
An important discovery was the significant association between APC and TP53 mutations and CNV and mutations to m6A modification genes. APC and TP53 mutations are common in CRC. APC is a tumor suppressor gene frequently mutated in CRC. Mutation and inactivation of this gene are a key, and early event almost uniquely observed in colorectal tumorigenesis. The correlation between m6A regulators and APC mutation suggests the role of the m6A modification in the early stage of CRC. Consistently, in AML patients, it has been reported that mutations and CNVs of m6A regulators are related to TP53 mutations [29]. As epigenetic regulations unlikely lead to genomic alterations, it is reasonable that alterations of m6A genes are induced by other functional events, probably through upregulation of cancer promoters or downregulation of tumor inhibitors. Interestingly, the MSI status was also correlated to CNV and mutations in m6A regulators.
Different consensus molecular subtype (CMS) classifications have been developed to facilitate clinical translation [38], among which CMS1 (MSI Immune) is featured with hypermutated, microsatellite instable, and strong immune activation [38]. MSI is a crucial biomarker in the prognosis of CRC, and MSI-high (MSI-H) status is associated with a better prognosis compared to MSS CRC [39]. The analysis of the MSI status and m6A genes' CNVs and mutation showed that cases with MSS are prone to CNVs and mutations to m6A regulators. Additionally, cases with nondiploid m6A genes have a poorer prognosis as shown in Figure 4, which is consistent with that patients harboring MSS that have a worse survival compared with those with MSI-H [40]. These data warrant further study about the biological and clinical roles of m6A genes alteration in CRC.
Right-sided and left-sided colon cancer exhibit different molecular features. Left-sided colon cancer (LCC) harbors more chromosomal instability pathway-related mutations, including APC, KRAS, and P53 mutations, while MSI and DNA mismatch repair pathways are commonly observed in the right-sided tumors (RCC) [41]. We found m6A regulator CNVs/mutation is statically related with MSS, APC mutation, and p53 mutation which are features of left-sided cancer. However, there is no significant correlation between the regulators CNVs/mutation and sidedness of colon cancer. This could be due to the limited sample size in our study or too much missing information regarding the MSI status.
CRC is dominated by diverse and plastic immune cell infiltration. These immune cells have an important effect on tumor development. Some populations of cells are strongly correlated with DFS and OS [38]. Also, genomic correlates of immune cell infiltrate in colorectal carcinoma [35]. In our study, we analyzed immune cell infiltration and the CNV status of m6A regulators. Nondiploid CNV status (deletion and gain) has a significant effect on CRC immune cell infiltration. B cells, CD8+ T cells, macrophages, neutrophils, and dendritic cells are affected by gene deletion or gain. It is consistent with previous studies that show macrophages, dendritic cells, neutrophils, B cells, and T cells which are involved in the outcomes of CRC [42]. It is reported that METTL3 could facilitate macrophage polarization through the methylation of STAT1 mRNA [43], and mettl3-mediated m6A methylation promotes dendritic cell activation [30]. Moreover, YTHDF1 has been proved to be related with the antigen-specific CD8+ T cell antitumor response [44]. It is not surprising that patients from the COAD cohort have higher susceptibility to CNVs than those in the READ when it comes to immune cell infiltration. Colon and rectal cancers differ from each other in terms of anatomic location of the tumor, prognosis, recurrence rate, and treatment strategy. Additionally, the immune profile of colon is different from that of rectum. It is reported that CD3+ T lymphocytes, CD8+ T lymphocytes, and the effector molecule granzyme B infiltration are correlated with OS of colon cancer but not rectal cancer [45], while lymphocytic infiltration is associated with relapse and distant metastasis of rectal cancer but not colon cancer according to the study of Nagtegaal et al. [46]. These data illustrate the role of immune systems in CRC and CNVs of m6A regulators that could be used as a promising target to treat CRC.
In conclusion, we systematically demonstrated the effect of CNVs and mutations to m6A regulators on the mRNA expression, immune cell infiltration, prognosis, and metabolic pathway switch of CRC. Our study provides evidence for future study on the role of RNA m6A methylation in CRC and therapeutic target to treat CRC.
Acknowledgments
The study was funded by the China Scholarship Council (201808230083).
Data Availability
The data used to support the findings of this study are available in the Cancer Genome Atlas database (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga).
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- 1.Siegel R. L., Miller K. D., Jemal A. Cancer statistics, 2019. CA: a Cancer Journal for Clinicians. 2018;69(1):7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.Wainwright E. N., Scaffidi P. Epigenetics and cancer stem cells: unleashing, hijacking, and restricting cellular plasticity. Trends Cancer. 2017;3(5):372–386. doi: 10.1016/j.trecan.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He C. Grand challenge commentary: RNA epigenetics? Nature Chemical Biology. 2010;6(12):863–865. doi: 10.1038/nchembio.482. [DOI] [PubMed] [Google Scholar]
- 4.Boccaletto P., Machnicka M. A., Purta E., et al. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Research. 2018;46(D1):D303–D307. doi: 10.1093/nar/gkx1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Helm M., Motorin Y. Detecting RNA modifications in the epitranscriptome: predict and validate. Nature Reviews. Genetics. 2017;18(5):275–291. doi: 10.1038/nrg.2016.169. [DOI] [PubMed] [Google Scholar]
- 6.Delaunay S., Frye M. RNA modifications regulating cell fate in cancer. Nature Cell Biology. 2019;21(5):552–559. doi: 10.1038/s41556-019-0319-0. [DOI] [PubMed] [Google Scholar]
- 7.Desrosiers R., Friderici K., Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proceedings of the National Academy of Sciences of the United States of America. 1974;71(10):3971–3975. doi: 10.1073/pnas.71.10.3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meyer K. D., Saletore Y., Zumbo P., Elemento O., Mason C. E., Jaffrey S. R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149(7):1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ke S., Alemu E. A., Mertens C., et al. A majority of m6A residues are in the last exons, allowing the potential for 3' UTR regulation. Genes & Development. 2015;29(19):2037–2053. doi: 10.1101/gad.269415.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meyer K. D., Patil D. P., Zhou J., et al. 5′ UTR m6A promotes cap-independent translation. Cell. 2015;163(4):999–1010. doi: 10.1016/j.cell.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schumann U., Shafik A., Preiss T. METTL3 gains R/W access to the epitranscriptome. Molecular Cell. 2016;62(3):323–324. doi: 10.1016/j.molcel.2016.04.024. [DOI] [PubMed] [Google Scholar]
- 12.Liu J., Yue Y., Han D., et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA _N_ 6-adenosine methylation. Nature Chemical Biology. 2014;10(2):93–95. doi: 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ping X. L., Sun B. F., Wang L., et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Research. 2014;24(2):177–189. doi: 10.1038/cr.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meyer K. D., Jaffrey S. R. Rethinking m6A readers, writers, and erasers. Annual Review of Cell and Developmental Biology. 2017;33(1):319–342. doi: 10.1146/annurev-cellbio-100616-060758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwartz S., Mumbach M. R., Jovanovic M., et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Reports. 2014;8(1):284–296. doi: 10.1016/j.celrep.2014.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jia G., Fu Y., Zhao X., et al. _N_ 6-Methyladenosine in nuclear RNA is a major substrate of the obesity- associated FTO. Nature Chemical Biology. 2011;7(12):885–887. doi: 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng G., Dahl J. A., Niu Y., et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Molecular Cell. 2013;49(1):18–29. doi: 10.1016/j.molcel.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haussmann I. U., Bodi Z., Sanchez-Moran E., et al. m6A potentiates _Sxl_ alternative pre-mRNA splicing for robust _Drosophila_ sex determination. Nature. 2016;540(7632):301–304. doi: 10.1038/nature20577. [DOI] [PubMed] [Google Scholar]
- 19.Wu R., Yao Y., Jiang Q., et al. Epigallocatechin gallate targets FTO and inhibits adipogenesis in an mRNA m6A-YTHDF2-dependent manner. International Journal of Obesity. 2018;42(7):1378–1388. doi: 10.1038/s41366-018-0082-5. [DOI] [PubMed] [Google Scholar]
- 20.Muller S., Glass M., Singh A. K., et al. IGF2BP1 promotes SRF-dependent transcription in cancer in a m6A- and miRNA-dependent manner. Nucleic Acids Research. 2019;47(1):375–390. doi: 10.1093/nar/gky1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao B. S., Roundtree I. A., He C. Post-transcriptional gene regulation by mRNA modifications. Nature Reviews. Molecular Cell Biology. 2017;18(1):31–42. doi: 10.1038/nrm.2016.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wen J., Lv R., Ma H., et al. Zc3h13 regulates nuclear RNA m6A methylation and mouse embryonic stem cell self-renewal. Molecular Cell. 2018;69(6):1028–1038.e6. doi: 10.1016/j.molcel.2018.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma J. Z., Yang F., Zhou C. C., et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6) -methyladenosine-dependent primary MicroRNA processing. Hepatology. 2017;65(2):529–543. doi: 10.1002/hep.28885. [DOI] [PubMed] [Google Scholar]
- 24.Chen X. Y., Zhang J., Zhu J. S. The role of m6A RNA methylation in human cancer. Molecular Cancer. 2019;18(1):p. 103. doi: 10.1186/s12943-019-1033-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li T., Hu P. S., Zuo Z., et al. METTL3 facilitates tumor progression via an m6A-IGF2BP2-dependent mechanism in colorectal carcinoma. Molecular Cancer. 2019;18(1):p. 112. doi: 10.1186/s12943-019-1038-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bai Y., Yang C., Wu R., et al. YTHDF1 regulates tumorigenicity and cancer stem cell-like activity in human colorectal carcinoma. Frontiers in Oncology. 2019;9:p. 332. doi: 10.3389/fonc.2019.00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu Y., Yang X., Chen Z., et al. m(6)A-induced lncRNA RP11 triggers the dissemination of colorectal cancer cells via upregulation of Zeb1. Mol Cancer. 2019;18(1):p. 87. doi: 10.1186/s12943-019-1014-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H. B., Tong J., Zhu S., et al. m6A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature. 2017;548(7667):338–342. doi: 10.1038/nature23450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kwok C. T., Marshall A. D., Rasko J. E., Wong J. J. Genetic alterations of m6A regulators predict poorer survival in acute myeloid leukemia. Journal of Hematology & Oncology. 2017;10(1):p. 39. doi: 10.1186/s13045-017-0410-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang H., Hu X., Huang M., et al. Mettl3-mediated mRNA m6A methylation promotes dendritic cell activation. Nature Communications. 2019;10(1):p. 1898. doi: 10.1038/s41467-019-09903-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li L. J., Fan Y. G., Leng R. X., Pan H. F., Ye D. Q. Potential link between m6A modification and systemic lupus erythematosus. Molecular Immunology. 2018;93:55–63. doi: 10.1016/j.molimm.2017.11.009. [DOI] [PubMed] [Google Scholar]
- 32.Hollerer I., Grund K., Hentze M. W., Kulozik A. E. mRNA 3'end processing: a tale of the tail reaches the clinic. EMBO Molecular Medicine. 2013;6(1):16–26. doi: 10.1002/emmm.201303300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song L., Luo Z. Q. Post-translational regulation of ubiquitin signaling. The Journal of Cell Biology. 2019;218(6):1776–1786. doi: 10.1083/jcb.201902074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saldi T., Cortazar M. A., Sheridan R. M., Bentley D. L. Coupling of RNA polymerase II transcription elongation with pre-mRNA splicing. Journal of Molecular Biology. 2016;428(12):2623–2635. doi: 10.1016/j.jmb.2016.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giannakis M., Mu X. J., Shukla S. A., et al. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Reports. 2016;15(4):857–865. doi: 10.1016/j.celrep.2016.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou J., Wang J., Hong B., et al. Gene signatures and prognostic values of m6A regulators in clear cell renal cell carcinoma - a retrospective study using TCGA database. Aging. 2019;11(6):1633–1647. doi: 10.18632/aging.101856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guinney J., Dienstmann R., Wang X., et al. The consensus molecular subtypes of colorectal cancer. Nature Medicine. 2015;21(11):1350–1356. doi: 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Battaglin F., Naseem M., Lenz H. J., Salem M. E. Microsatellite instability in colorectal cancer: overview of its clinical significance and novel perspectives. Clinical Advances in Hematology & Oncology. 2018;16(11):735–745. [PMC free article] [PubMed] [Google Scholar]
- 40.Petrelli F., Ghidini M., Cabiddu M., et al. Microsatellite instability and survival in stage II colorectal cancer: a systematic review and meta-analysis. Anticancer Research. 2019;39(12):6431–6441. doi: 10.21873/anticanres.13857. [DOI] [PubMed] [Google Scholar]
- 41.Baran B., Mert Ozupek N., Yerli Tetik N., Acar E., Bekcioglu O., Baskin Y. Difference between left-sided and right-sided colorectal cancer: a focused review of literature. Gastroenterology Research. 2018;11(4):264–273. doi: 10.14740/gr1062w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fridman W. H., Pages F., Sautes-Fridman C., Galon J. The immune contexture in human tumours: impact on clinical outcome. Nature Reviews. Cancer. 2012;12(4):298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 43.Liu Y., Liu Z., Tang H., et al. TheN6-methyladenosine (m6A)-forming enzyme METTL3 facilitates M1 macrophage polarization through the methylation ofSTAT1mRNA. American Journal of Physiology. Cell Physiology. 2019;317(4):C762–C775. doi: 10.1152/ajpcell.00212.2019. [DOI] [PubMed] [Google Scholar]
- 44.Han D., Liu J., Chen C., et al. Anti-tumour immunity controlled through mRNA m6A methylation and YTHDF1 in dendritic cells. Nature. 2019;566(7743):270–274. doi: 10.1038/s41586-019-0916-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deschoolmeester V., Baay M., Van Marck E., et al. Tumor infiltrating lymphocytes: an intriguing player in the survival of colorectal cancer patients. BMC Immunology. 2010;11(1):p. 19. doi: 10.1186/1471-2172-11-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nagtegaal I. D., Marijnen C. A., Kranenbarg E. K., et al. Local and distant recurrences in rectal cancer patients are predicted by the nonspecific immune response; specific immune response has only a systemic effect--a histopathological and immunohistochemical study. BMC Cancer. 2001;1(1):p. 7. doi: 10.1186/1471-2407-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are available in the Cancer Genome Atlas database (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga).