

Graphical abstract
Keywords: SARS-CoV-2, COVID-19, Polymerase, 3CLpro, Therapeutic targets, α-Glucosidase inhibitor
Abstract
COVID-19 pandemic, caused by SARS-CoV-2, has drastically affected human health all over the world. After the emergence of the pandemic the major focus of efforts to attenuate the infection has been on repurposing the already approved drugs to treat COVID-19 adopting a fast-track strategy. However, to date a specific regimen to treat COVID-19 is not available. Over the last few months a substantial amount of data about the structures of various key proteins and their recognition partners involved in the SARS-CoV-2 pathogenesis has emerged. These studies have not only provided the molecular level descriptions of the viral pathogenesis but also laid the foundation for rational drug design and discovery. In this review, we have recapitulated the structural details of four key viral enzymes, RNA-dependent RNA polymerase, 3-chymotrypsin like protease, papain-like protease and helicase, and two host factors including angiotensin-converting enzyme 2 and transmembrane serine protease involved in the SARS-CoV-2 pathogenesis, and described the potential hotspots present on these structures which could be explored for therapeutic intervention. We have also discussed the significance of endoplasmic reticulum α-glucosidases as potential targets for anti-SARS-CoV-2 drug discovery.
1. Introduction
The current coronavirus infectious disease 19 (COVID-19) pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has shown unprecedented impact on healthcare, economies and social life all over the world (Bayham and Fenichel, 2020, Burkle, 2020). As of September 21st, 2020 over 30.9 million people have been confirmed with COVID-19 with over 959,000 deaths since the emergence of the infection (WHO, 2020). An effective vaccine to prevent the infection yet to be available despite a significant number of vaccine candidates have been reported which are in pre-clinical development or under clinical trials (Al-Kassmy et al., 2020, Checcucci et al., 2020, Wang et al., 2020a). Any specific treatment regimen against COVID-19 is also not available to date.
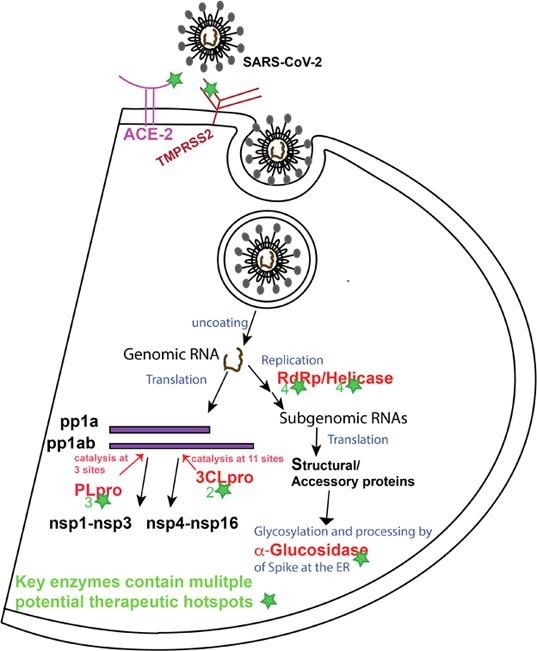
SARS-CoV-2 belongs to the Coronavirinae subfamily in the Coronaviridae family of the Nidovirales order (Lu et al., 2020). Angiotensin-converting enzyme 2 (ACE2) present at the cellular surface of various types of tissues acts as a primary receptor of SARS-CoV-2. The receptor-binding domain (RBD) in the S1 subunit of the viral spike (S) protein mediates the binding of the virus to ACE2. Subsequently, priming of the S protein by a host protease enzyme, transmembrane serine protease 2 (TMPRSS2) enables the fusion of membranes and viral entry (Hoffmann et al., 2020, Wan et al., 2020, Wrapp and Wang, 2020b).
SARS-CoV-2 is a positive sense RNA virus with genome size of 29.9 kb which contains 11 open reading frames (ORF). Orf1a and orf1ab encode the pp1a and pp1ab replicase polyproteins (Fig. 1 ) (Chan and Kok, 2020, Licastro et al., 2020). The replicase polyproteins are cleaved by papain like protease (PLpro) at 3 different N-terminus sites and by 3-chymotrypsin-like protease (3CLpro) at 11 different sites to release non-structural proteins. Overall, the SARS-CoV-2 genome encodes 16 non-structural proteins (nsp1-nsp16), 4 structural proteins including spike, the membrane protein, envelope and nucleocapsid, and accessory proteins 3, 6, 7a, 7b, 8, and 10. Non-structural proteins are translated from viral genomic RNA in the form of replicase polyproteins while structural and accessory proteins are translated from sub-genomic RNAs. Six of the non-structural proteins are enzymes in nature, which include the PLpro domain in nsp3, nsp5 (3CLpro), nsp12 (RNA-dependent RNA polymerase), nsp13 (helicase), nsp14 (N7-methyltransferase) and nsp16 (2′-O-methyltransferase), while nsp7-nsp10 play regulatory roles in the replication cycle of the virus (Chellapandi and Saranya, 2020, Lei et al., 2020, Shereen et al., 2020).
Fig. 1.

Genome structure of SARS-CoV-2.
After the emergence of the COVID-19 pandemic, early efforts to discover treatments were focused on evaluating the efficacy of already known drugs particularly antiviral agents against SARS-CoV-2. Several clinically approved drugs including RdRp inhibitor remdesivir that was initially approved to treat Ebola virus, a well know antimalarial drug chloroquine and its derivative hydroxychloroquine and a combination therapy consisting of HIV protease inhibitors, ritonavir and lopinavir were approved by the World Health Organization (WHO) in March 2020 for the SOLIDARITY worldwide clinical trials, to evaluate the clinical efficacy of these drugs against COVID-19 (Cao et al., 2020, Gadebusch Bondio and Marloth, 2020, Hung et al., 2020, Sterling and Irwin, 2015, Wang et al., 2020b, Wang et al., 2020d). Drug repurposing and various clinical trials of already approved drugs to evaluate their efficacy against COVID-19 have been reviewed in detail by several authors (Lima et al., 2020, Pandey et al., 2020, Rahman et al., 2020, Santos et al., 2020, Tu et al., 2020).
Considering the global impact of the COVID-19 pandemic and possibility of re-emergence of coronavirus infections in future, there is an urgent need of developing new antiviral agents particularly with broad-spectrum efficacy against different coronaviruses. In this regard, different viral proteins involved in the viral replication process that are highly conserved or contain conserved motifs among coronaviruses, represent potential targets for broad-spectrum anti-coronavirus therapeutic development. Several recently published reviews have provided an overview of the coronavirus therapeutic targets and details of different in vitro and/or in vivo characterized anti-coronavirus active agents (McKee et al., 2020, Su et al., 2020a, Tiwari et al., 2020). Targeting specific structural features of the proteins crucial for viral replication cycle to interfere with their functions is an important strategy to identify new antiviral drugs.
Advancement in biotechnology to efficiently produce viral proteins and structure determination techniques particularly cryogenic electron microscopy (cryo-EM) along with high commitment of the scientific community during the pandemic have led the emergence of structures of various key proteins of SARS-CoV-2 also including novel scaffolds (Chen et al., 2020, Gao et al., 2020, Hillen and Kokic, 2020, Jin et al., 2020, Lan et al., 2020, Melero et al., 2020, Shang et al., 2020, Shin and Mukherjee, 2020, te Velthuis et al., 2012, Wrapp and Wang, 2020a, Yin et al., 2020). In this review we recapitulated the structural features of the key catalytic proteins involved in the SARS-CoV-2 pathogenesis, their main catalytic sites and other structural regions involved in interactions with partner proteins resulting in functional outcomes crucial for the viral pathogenesis and propagation. We have also highlighted the known inhibitors of these proteins in the context of describing their interactions with the target proteins according to the available structural data. Plenty of studies on different aspects of SARS-CoV-2 are emerging, however, here we focused on four viral enzymes, PLpro, 3CLpro, helicase and RdRp, and two host factors including ACE2, TMPRSS2 to discuss potential structural hotspots that could be explored as target sites for therapeutic intervention. Moreover, we also discussed α-glucosidases of human endoplasmic reticulum (ER) as potential therapeutic targets against SARS-CoV-2.
2. Key enzymes of SARS-CoV-2 replication cycle
2.1. PLpro of SARS-CoV-2
The PLpro enzyme of coronaviruses is a catalytic domain of the large multidomain nsp3 protein (Lei et al., 2018). It cleaves the pp1a/ab polyprotein at the junctions of nsp1/nsp2, nsp2/nsp3 and nsp3/nsp4 releasing nsp1, nsp2 and nsp3 thereby playing important role in the viral replication cycle. PLpro recognizes a conserved sequence consisting of Leu-X-Gly-Gly and cleaves at the carboxyl-terminal of the last glycine residue. SARS-CoV-2 PLpro shows 83% sequence identity with its counterpart of SARS-CoV.
To date several crystal structures of SAR-CoV-2 PLpro and its complexes with different ligands have been reported in protein data banks. Structurally, the enzyme consists of a canonical right-handed thumb, palm and fingers architecture also containing a forth N-terminus ubiquitin-like sub-domain (Fig. 2 a). The catalytic site of PLpro is conserved among all coronaviruses and consists of a catalytic triad of Cys-111, His-272 and Asp-286. During catalysis, Cys-111 acts as a nucleophile, His-272 plays a role of general acid/base and Asp-286 promotes the deprotonation of Cys-111. The catalytic site is present in a ridge between the palm and the thumb domains (Shin and Mukherjee, 2020).
Fig. 2.

SARS-CoV-2 PLpro exhibits multiple potential therapeutic hotspots. (A) X-ray crystal structure of SARS-CoV-2 PLpro shown in cartoon, with the illustration of fingers, palm, thumb and ubiquitin-like (UL) domains (PDB ID 6wuu). The catalytic residues are displayed with green sticks and the two ubiquitin-binding sites (UBS1 and UBS2) are colored blue and purple, respectively. (B) The structure of SARS-CoV-2 PLpro overlaid by its complex with the dual action inhibitor, GRL0617 showing the interactions of the inhibitor with Tyr-268 that has adopted different conformation in the complex structure (PDBID 7jir). (C) Chemical structures of different SARS-CoV-2 PLpro inhibitors.
Recently, Rut et al investigated the substrate specificity of PLpro by using a combinatorial library of tetrapeptides where systematic variations of natural and unnatural amino acids were incorporated at positions P2, P3 and P4 by keeping Gly at the P1 position. The study suggested very high specificity of the enzyme for Gly at the P2 site, preference for positive and hydrophobic residues at the P3 site and a high preference for large bulky hydrophobic residues at the P4 position. X-ray structures of SARS-CoV-2 PLpro show that the P2 binding sub-pocket is very narrow and can accommodate only a very small side chain such as H atom of glycine. Whereas the P4 binding pocket is wide and surrounded by several hydrophobic residues suitable for bulky hydrophobic side chains. Such findings led to the development of peptide inhibitors consisting of unnatural amino acids at the P3 and P4 positions and vinyl ester at the N-terminus as a warhead for the formation of covalent bond. These inhibitors covalently link to Cys-111 through thioether bond and perfectly fit in the substrate binding pocket as has been depicted by their X-ray crystal structures in complex with SARs-CoV-2 PLpro (Rut et al., 2020). These inhibitors, VIR250 and VIR251 (Fig. 2c), inhibit the catalytic activity of the enzyme and binding of ubiquitin and interferon stimulated gene 15 (ISG15) to SARS-CoV-2 PLpro and do not bind to any other protease, thereby showing high specificity for SARS-CoV-2 PLpro (Rut et al., 2020). Structural description of the binding interactions of these inhibitors provides essential information for the design of new pharmacophores and potent inhibitors of this enzyme. Recently, an FDA approved drug against leukemia, 6-thioguanine was reported to inhibit SARS-CoV-2 PLpro with IC50 of 0.1 to 0.5 µM and viral replication with EC50 of 0.647 ± 0.374 μM. However, the structural basis of this inhibition by 6-thioguanine remains to be characterized (Swaim et al., 2020).
In addition to processing the pp1a to produce functional nsp1, nsp2 and nsp3, SARS-CoV-2 PLpro can bind and hydrolyze ubiquitin and ubiquitin like protein ISG15. These two cellular proteins contain the Leu-X-Gly-Gly cleavage motif of PLpro at their C-terminus. Ubiquitin and ISG15 are cellular regulatory proteins that can be post-translationally incorporated to a target protein through the formation of isopeptide bond between the C-terminus of these proteins and ɛ-amino group of lysine side chains of the target protein. PLpro can hydrolyze the isopeptide bonds removing ubiquitin and ISG15 from the host proteins therefore the enzyme is also referred to as isopeptidase. ISG15 and ubiquitin are signaling elements that play an important role in the innate immune response against viruses. Deubiquitinating and deISGylating activities of SARS-Cov-2 PLpro thus antagonize ubiquitin-dependent cellular response against the virus. In this regard, PLpro has been demonstrated to inhibit the production of cytokines involved in the activation of host innate immune response. It has been shown to interfere with the activation of transcriptional factor interferon regulatory factor-3 (IRF3) and NFkB signaling pathway thereby mediating an antagonistic effect against antiviral host immune response.
Interestingly, the catalytic activity of PLpro leading to deubiquitination and deISGylation of the host protein or just it’s binding to cellular protein-bound ubiquitin and ISG15 has been shown to mediate the antagonistic effect of viral PLpro. Therefore, inhibiting the catalytic activity of PLpro as well as intervening it’s binding to cellular protein-bound ubiquitin and ISG15 can restore cellular antiviral innate immune response against the virus. Inhibition of coronavirus PLpro thus, can lead to inhibit the viral replication and overcome the viral antagonistic effect on the host innate immune response against the virus. A small organic molecule GRL-0617 (snyder457) and its three derivatives (Fig. 2c) have been reported to bind to PLpro at the entrance of its catalytic groove and block the binding of its substrate. GRL0617 inhibits the viral replication and promotes the antiviral immunity (Shin and Mukherjee, 2020). These compounds fits into a binding pocket formed by the movement of Tyr-268 containing loop (Fig. 2b) and the binding is mediated primarily by hydrophobic interactions of naphthyl ring of these inhibitors and hydrophobic pocket formed by Tyr-268 and the two conserved proline residues, Pro-247 and Pro-248. In addition to the main catalytic pocket the pocket formed between Tyr-268 and Pro-247/248 represents an important potential hotspot for therapeutic intervention.
Structurally, PLpro exhibits two ubiquitin binding sites namely UBS1 and UBS2 that can accommodate Lys-48 linked diubiquitin and ISG15 in a similar manner involving both the sites or monoubiquitin at one of the sites. UBS1 encompasses the finger and palm sub-domains while UBS2 is primarily present in the thumb subdomain of PLpro. The X-ray crystal structure of the complex of ISG15 and SARS-CoV-2 PLpro shows the N-terminus domain of ISG15 occupies the UBS2 site while its C-terminus domain occupies UBS1 of PLpro (Fig. 2) (Shin and Mukherjee, 2020). Interrupting the binding of ISG15 and/or ubiquitin at the UBS1 and UBS2 sites of viral PLpro would potentially overcome the viral antagonizing effect against the host innate immune response. The UBS1 and UBS2 sites thus represent potential target sites to block such binding. However, no such inhibitor binding at any of the UBS1 and UBS2 sites of PLpro has been reported to date. Taking together, SARS-CoV-2 PLpro represents an important target for dual effect, inhibition of viral replication and propagation, and restoring the antiviral immunity.
2.2. 3CLpro of SARS-CoV-2
3CLpro is one of the most important components of viral replication as it cleaves the replicase polyprotein after its translation at 11 different sites releasing most of the functional protein components of replicases, which is the reason this protease is also referred to as main protease of coronaviruses. 3CLpro is highly conserved among all coronaviruses with 96% sequence conservation. Two amino acid residues His-41 and Cys-145 involved in catalysis are highly conserved. In addition to these catalytic residues His-163 and Phe-140 facilitate the binding of glutamine at the P1 position of the peptide substrate through two H bonds between NΣ2 atom of His-163 and backbone carbonyl oxygen of Phe-140 with OΣ1 and NΣ2 atoms of substrate glutamine, respectively (Xue et al., 2008). A typical oxyanion whole is formed between Cys-145 and Gly-143 to derive the catalysis (Fig. 3 a). The substrate specificity of 3CLpro is primarily defined by the residues at the P1, P1′ and P2 positions of the peptide substrate. These positions are highly conserved in all coronaviruses in particular the presence of glutamine at the P1 position (N-terminus of the scissile bond) of the substrate is strictly required for 3CLpro binding across all coronaviruses (Fig. 3c) (Ullrich and Nitsche, 2020).
Fig. 3.

Potential therapeutic hotspots on SARS-CoV-2 3CLpro. (A) Cartoon presentation of SARS-CoV-2 3CLpro single protamer, three domains I, II and III are labeled, and two catalytic residues are displayed with green sticks. The structural region involved in making the dimer interface is colored purple. Ala-285 that mediates inter-protamer contacts, and Glu-290 and its salt bridge-forming counterpart from the second protamer, Arg-4 are displayed with sticks. (B) Molecular surface display of the substrate-binding pocket of the enzyme. Locations of key residues and individual sub-pockets of the substrate-binding site are labeled. (C) Tetrapeptide sequence of the cleavage motif in the substrate showing conservation among different coronaviruses particularly for the P1 site. (D) Chemical structures of different inhibitors of 3CLpro.
Most of the SARS-CoV-2 3CLpro inhibitors reported to date are covalent inhibitors where different warheads have been used to form covalent linkage with the thiol side chain of the key catalytic residue, Cys-145. In all these designed inhibitors, the lactam ring has been used as a surrogate of the P1 glutamine residue. Another common feature in the inhibitors reported by three different research groups is the presence of hydrophobic groups at the P2 site. Zhang et al., have reported the design of α-ketoamide derivatives by incorporating the ketoamide group as a warhead for covalent linkage of the inhibitor. These inhibitors form covalent bond with the side chain of Cys-145, resulting from a nucleophilic attack by the thiol group of Cys-145 on the α-keto group of the inhibitor to form thiohemiketal. One of the potent inhibitors of this class (Fig. 3b) shows IC50 of 0.67 µM against purified SARS-CoV-2 3CLpro and EC50 of ~4 µM in viral replication assay. The structure of this inhibitor in complex with 3CLpro shows that the lactam ring at the P1 position fits into the S1 pocket, cyclopropyl methyl side chain at the P2 position occupies position in the S2 sub-pocket and tert-butyloxycarbonyl (Boc), a protecting group on the P3 residue stabilizes the binding through hydrophobic interactions with proline-168. Boc has also been associated with its role in mediating transport of the inhibitor across the cell membrane (Zhang and Lin, 2020).
Dai et al., have reported a group of covalent inhibitors incorporating the aldehyde group as a warhead for covalent linkage. These compounds (11a and 11b) (Fig. 3d) also contain the lactam group at the P1 site that fits into the S1 subsite. The cyclohexyl ring or the 3-fluorophenyl group at the P2 site inserts in the S2 cavity and is stabilized primarily through hydrophobic interactions. The indole group at the P3 position is primarily stabilized by hydrophobic interactions. These compounds inhibit SARS-CoV-2 infection with EC50 of 0.04 to 0.05 µM and show promising safety profile and pharmacokinetic properties (Dai and Zhang, 2020).
Another covalent inhibitor, N3 was reported by Jin et al., (Jin et al., 2020). This inhibitor forms a covalent bond with Cys-145 presumably through Michael addition reaction between the Cys-145 thiol side chain and Cβ of the vinyl group of the inhibitor. This inhibitor also contains the lactam ring at the P1 position that inserts in the S1 sub-pocket. The side chain of leucine at the P2 site is inserted in the S2 sub-pocket while at the P3 and P4 positions hydrophobic residues are present. This compound inhibits the SARS-CoV-2 replication with EC50 of 16.8 µM. The same group also identified a few small organic molecules through in silico screening of drug libraries followed by in vitro validation. The most potent compound among these was ebselen (Fig. 3d) that inhibited SARS-CoV-2 3CLpro with IC50 of 0.67 µM and viral replication with EC50 of 4.7 µM. The mass spectrometry analysis showed that these compounds were also covalently linked to the Cys-145 of 3CL-pro (Jin et al., 2020).
The only non-covalent inhibitor of SARS-CoV-2 3CLpro reported to date with sub-micromolar IC50 is baicalein (Fig. 3d), a flavonoid of plant origin. Baicalein has been reported to inhibit SARS-CoV-2 3CLpro with IC50 of 0.94 ± 0.2 µM and SARS-CoV-2 infection in the Vero cells with EC50 of 2.94 ± 1.19 µM. X-ray crystal structure of the complex of this compound and 3CLpro shows it’s binding at the catalytic site of the enzyme. The binding is stabilized by several H bonds between three hydroxyl groups of the inhibitor and the backbone atoms of key residues of catalytic pocket including Gly-143, Ser-144, Cys-145, and hydrophobic interactions between it’s phenyl ring and the S2 sub-pocket (Su et al., 2020b). All these inhibitors and structural description of their interactions with the enzyme provide insight into rational design of more potent inhibitors of 3CLpro as potential candidates to treat SAR-CoV-2 infections.
The 3CLpro enzyme forms a homodimer and it is catalytically active only in the dimer form (Pillaiyar et al., 2016, Zhang and Lin, 2020). Therefore, from inhibitor design perspective, in addition to the main substrate binding site the interprotamer contact surface represents a potential target site to interrupt the dimer formation leading to inhibit the catalytic activity of the enzyme (Fig. 3a). However, no example of such inhibitor of 3CLpro exists to date.
2.3. RdRp of SARS-CoV-2
RdRp of positive sense RNA viruses plays a central role in the viral replication cycle. RdRp catalyzes the synthesis of negative-strand RNA (−RNA), new genomic RNA and sub-genomic messenger RNAs during viral replication (Kim and Chang, 2013, Shereen et al., 2020, te Velthuis et al., 2012). Moreover, the N-terminal domain of the coronavirus RdRp exhibits nucleotidyltransferase enzymatic activity that also plays an important role in viral replication (Lehmann et al., 2015). RdRp shares 98% sequence identity between SARS-CoV-2 and SARS-CoV. The RdRp domain is present in the NSP12 subunit of the viral genome and is translated as a part of the pp1ab polyprotein (Fig. 1), and successively cleaved from the polyprotein by proteolytic activity of the viral 3CLpro enzyme. Subsequently, RdRp is incorporated into a membrane-associated complex for its functions. The viral proteins that interact with RdRp to form the complex include NSP7, NSP8 and NSP13 (Chen et al., 2020, Subissi et al., 2014). Recently, several cryo-EM structures of SARS-CoV-2 RdRp have been reported. These structures include complex of RdRp with its cofactors nsp7 and nsp8 (Gao et al., 2020), RdRp-nsp7-nsp8 complex containing bound RNA (Hillen and Kokic, 2020), an inhibitor remdesivir (Yin et al., 2020) and nsp13 (Chen et al., 2020). The structure of RdRp-nsp7-nsp8 complex without bound RNA indicates that binding of nsp7 and nsp8 to RdRp is not primarily dependent on the bound RNA.
RdRp consists of 920 amino acid residues. Its structure adopts a typical canonical right-hand architecture containing the palm, the thumb and the fingers sub-domains, an interface domain, an N-terminus nidovirus RdRp-associated nucleotidyl tranferase (NiRAN) and a β hairpin. Two highly conserved amino acid residues Asp-760 and Asp-761 are the main catalytic residues that are present in the palm subdomain of the protein and the catalytic groove is surrounded by the palm, the thumb and the fingers sub-domains. In the RNA bound holo-RdRp structure, two nsp8 binds to RdRp opposite to the catalytic groove and their helical extensions primarily consisting of positively charged residues protrude in parallel to the exiting RNA while binding to it (Hillen and Kokic, 2020). Nsp13-bound RdRp replication machinery contains two nsp13 molecules. The zinc-binding N-terminal domain of each of the nsp13 molecules interacts with the protruding helical extension of each copy of nsp8 while the ATpase domain of each helicase is positioned in front of holo-RdRp (Chen et al., 2020). The NiRAN domain contains Mg+2 and nucleotide-binding sites. The NiRAN domain might be involved in the activation of nucleoside triphosphate to be incorporated into the growing chain of RNA where Mg+2 plays a crucial role in the transferase activity of the domain. However, such role of the NiRAN domain remains to be experimentally confirmed.
Nucleotide analogues can mimic natural nucleotide substrates of the enzyme to be incorporated into the new RNA chain and inhibit further elongation of the RNA. Generally, three types of nucleotide analogue RdRp inhibitors have been reported in positive sense RNA viruses. The analogues that lack 3′ –OH group, halt the RNA strand elongation after being incorporated (Mitsuya et al., 1990). Some nucleotide analogues do not halt the RNA strand synthesis but incorporate a permanent mutation in the RNA due to modified base, such compounds are called mutagens (Crotty et al., 2000). Some analogues contain natural base and 3′ –OH of the ribose but the ribose ring is modified. These inhibitors after incorporation interrupt translocation as the termination of the RNA strand occurs after the incorporation of three more bases; such termination is referred to as non-obligate chain termination. Every type of nucleotide analogue inhibitors of RdRp needs to be in the form of nucleoside triphosphate to mimic the natural nucleotide substrates of the enzyme (Warren et al., 2014). Nucleoside triphosphates are less stable and have limited permeability across the cell membrane. Therefore, such drugs are developed in the form of pro-drugs that contain hydrophobic groups capping the polar phosphate to facilitate passage across the cell membrane. Inside the cell the hydrophobic groups are removed and subsequently triphosphate form representing the active drug is produced. Remdesivir that has been approved for a widespread phase-3 clinical trial to treat COVID-19 is an adenosine analogue that is incorporated into the RNA strand by RdRp resulting in non-obligate chain termination during RNA synthesis (Jorgensen et al., 2020, Sisay, 2020). The triphosphate active form of remdesivir has been shown to be more reactive to RdRp than its natural substrate ATP (Yin et al., 2020). Ribavirin, a previously approved antiviral drug, is also a nucleotide analogue RdRp inhibitor, and is under clinical trial in combination therapy against COVID-19 (Hung et al., 2020). Favipiravir, a broad-spectrum antiviral drug is a structural analogue of the guanine base and is the inhibitor of viral RdRp. This drug has also shown limited efficacy against SARS-CoV-2 in clinical use (Jomah et al., 2020). Sofosbuvir another nucleotide analogues has been demonstrated in silico to inhibit SARS-CoV-2 RNA replication (Jácome et al., 2020).
Multiple nucleotide analogue RdRp inhibitor drugs have shown efficacy against SARS-CoV-2 in clinical trials to varying extent and are at the early phases of development (Gadebusch Bondio and Marloth, 2020), however, no non-nucleoside SARS-CoV-2 RdRp inhibitor has been reported to date. Recent structures of RdRp in complex with RNA and different cofactor proteins provide essential insight into the rational design of RdRp inhibitors. In SARS-CoV-2 RdRp, besides the main catalytic groove containing catalytic sub-site around the Asp-760 and Asp761 residues, there is a well defined ADP and Mg+2 binding site in the NiRAN domain. Although the precise role of the NiRAN domain in RNA transcription and replication remains to be elucidated yet this site represents a potential hotspot to be targeted for therapeutic intervention (Fig. 4 ). RdRp requires it’s binding to the nsp7 and nsp8 cofactor proteins for RNA replication process. SARS-CoV-2 RdRp exhibits two distinct nsp8 and one nsp7 binding sites (Fig. 4). These sites can be explored to identify pockets for inhibitors that could interrupt the binding of RdRp to the essential cofactor proteins. Similarly, complementary sites on nsp8 and nsp7 could also be exploited to interrupt their binding to RdRp as a potential strategy to discover new inhibitors of SARS-CoV-2 replication.
Fig. 4.

SARS-CoV2 RdRp contains multiple potential therapeutic hotspots. Molecular surface presentation of SARS-CoV-2 RdRp, with catalytic residues, nsp8-intearcting region and nsp7-binding region of RdRp colored red, cyan and purple, respectively. Remdesivir bound in the catalytic pocket and ADP bound in the NiRAN domain, are depicted as yellow and green sticks, respectively.
2.4. Helicase of SARS-CoV-2
Coronavirus helicase (NSP13) catalyzes the unwinding of duplex RNA into single strand nucleic acid chains during RNA transcription and replication (Snijder et al., 2016), the process is indispensable for viral replication and propagation. The process of nucleic acid unwinding is driven by the energy from hydrolysis of NTP, which is also catalyzed by helicase. SARS-CoV-2 helicase contains distinct RNA and NTP binding sites. In addition to these two important binding sites there is a β19-β20 loop encompassing residues 331–357 that plays a crucial role during the nucleic acid unwinding process (Jia et al., 2019). However, a detailed mechanism of how the β19-β20 loop plays a role in RNA duplex unwinding yet to be delineated. Helicase exhibits high sequence conservation among all coronaviruses (Jia et al., 2019, Mirza and Froeyen, 2020), in particular it shows 99.8% sequence identity between SARS-CoV-2 and SARS-CoV (Iftikhar et al., 2020). A typical CoV helicase consists of a zinc-binding domain, which is bisected by domains 1B and stalk from the main catalytic domains 1A and 2A.
Recently, cryo-EM structures of SARS-CoV-2 helicase and its complex with RNA-bound RdRp-nsp7-nsp8 transcription/replication machinery (holo RdRp) have been reported. In these structures, two nsp13 have been shown to bind to holo RdRp. In the structure, the N-terminal zinc-binding domain (ZBD) of each nsp13 interacts with the N-terminal helical protrusion of each copy of nsp8. ZBD and the catalytic domain 1A of one of the nsp13 molecules also make contacts with the thumb domain of RdRp and nsp8 head region, respectively. The amino acid residues involved in all these interactions are universally conserved among coronaviruses (Chen et al., 2020). Binding of nsp13 to RdRp complex has been shown to significantly enhance the NTPase and unwinding activity of the helicase enzyme in SARS-CoV(Jia et al., 2019). SARS-CoV-2 helicase structure incorporates five different potential hotspots that can be explored to interfere with its regular function and for therapeutic intervention. These hotspots include, distinct NTP and RNA binding sites, the β19-β20 loop, the nsp8/RdRp binding site on ZBD and a site on the 1A catalytic domain that mediates interaction of nsp13 with the head region of nsp8 (Fig. 5 ).
Fig. 5.

Cartoon presentation of SARS-CoV-2 helicase. Individual domains including zinc-binding domain, stalk, and domains 1A, 1B and 2A are labeled. Residues involved in ATP and nucleic acid binding are depicted as yellow and brown sticks, respectively. The β19-β20 loop is colored dark blue while residues involved in mediating binding of helicase to the RdRp-nsp7-nsp8 replication complex are colored cyan.
In addition to the essential role in RNA transcription and replication, SARS-CoV-2 nsp13 has also been demonstrated to potently antagonize the interferon pathway and suppress the production of primary interferon. Interferon plays an essential role in the host defense against viruses. Nsp13 inhibits the nuclear localization of interferon regulatory factor-3 (IRF3) that acts as transcriptional factor of interferon β (Yuen et al., 2020). Nsp13 therefore, inhibits the production of primary interferon and interferon pathways. However, structural description of nsp13 interactions with any of the factors of interferon pathway yet to be described. Intervening the interferon antagonizing effect of nsp13 could potentially restore the antiviral immune response and suppress the viral propagation.
Although the coronavirus helicase enzyme incorporates several potential therapeutic hotspots but only a few inhibitors of this enzyme have been reported. Shum et al have reported the synthesis of different DNA aptamers, which inhibit DNA unwinding activity of SARS-CoV helicase in vitro with IC50 ranging from 87 to 120 nM (Shum and Tanner, 2008). Purine derivatives have been shown to inhibit ATPase as well as RNA-duplex unwinding activity of SARS-CoV helicase with IC50 of 8.6 and 41.6 μM (Cho et al., 2015). Mirza et al., recently performed extensive in silico screening followed by molecular dynamics simulation to identify three aromatic compounds as novel potential inhibitors of SARS-CoV-2 helicase (Mirza and Froeyen, 2020). White et al., recently identified two previously known drugs lumacaftor and cepharanthine to inhibit SARS-CoV-2 Nsp13 ATPase activity with in vitro IC50 values of 0.3 and 0.4 mM (White et al., 2020). The structural description of all these inhibitors and their interactions with SARS-CoV-2 helicase will provide basis for the rational design of new potent inhibitors of helicase as potential antiviral agents.
3. Host factors
Targeting host factors involved in key steps of viral pathogenesis has advantage of less chances of the emergence of drug-resistant viral variants, particularly in genetically variable viruses (Edinger et al., 2014). However, on the contrary, targeting a host factor may result in significant modulation in the associated physiological function leading to side effects. Nevertheless, to contain a life threatening viral infection by targeting a host factor the associated side effects could be compromised.
3.1. ACE2 a potential target for therapeutic intervention against SARS-CoV-2
ACE2 is a key component of the renin angiotensin system (RAS) that contributes to maintain the blood pressure and balance of electrolytes thereby playing an important role in cardiovascular and kidney functions. Renin cleaves angiotensinogen to produce angiotensin I, which is further cleaved by angiotensin converting enzyme (ACE) into angiotensin II. Angiotensin I and II are further processed by ACE2 to produce angiotensin 1–9 and angiotensin 1–7, respectively. Angiotensin 1–7 stimulates vasodilation while angiotensin II acts as a vasoconstrictor. Therefore, a balance between angiotensin II and angiotensin 1–7 is important to maintain the homeostasis (Imai et al., 2010, Kuba et al., 2013). ACE2 is a transmembrane glycoprotein with 120 kDa molecular mass with an extracellular catalytic domain. ACE2 is expressed in several types of tissue including lungs, heart, kidneys, testes and colon.
Enzymatically, ACE2 acts as a carboxypeptidase and exhibits the conserved active site HEXXH motif for the enzymatic activity. ACE2 was identified as the primary receptor of SARS-CoV in 2003 (Li et al., 2003) and further studies suggested that binding of viral spike protein to ACE2 did not block its catalytic activity (Kuba et al., 2013). SARS-CoV-2 binds to ACE2 with roughly 10 fold higher affinity as compared to SARS-CoV explaining the basis of much higher transmissibility of SARS-CoV-2 (Tai et al., 2020). Recently, the crystal structure of the complex of ACE2 and the receptor-binding domain (RBD) of SARS-CoV-2 spike has been solved by Shang et al., that delineates the key interactions mediating the viral recognition of its ACE2 receptor (Shang et al., 2020). Interference with these interactions by targeting either of the interacting proteins could inhibit the viral entry to the target cell. Viral spike RBD-binding site on ACE2 is rather shallow and wide without a very well defined pocket (Fig. 6 a). However, small pockets within the wide binding site could be identified and targeted to interfere with the interactions between the viral spike and ACE2.
Fig. 6.

The ACE2 and TMPRSS2 host factors. (A) ACE2 in complex with RBD of the SARS-CoV-2 S protein (PDB ID 6m0j). The ACE2 structure is displayed in grey surface whereas its S protein interacting region and its catalytic site are colored green and purple, respectively. ACE2 bound SARS-CoV-2 S protein RBD is shown as a cyan cartoon. (B) Cartoon presentation of the ACE2 RBD interface with interacting residues of both proteins shown as sticks. (C) Cartoon presentation of the TMPRSS2 homology modeled structure. The catalytic triad is shown as cyan sticks and the two domains of the outer membrane region of TMPRSS2 are labeled. (D) Chemical structures of the two known inhibitors of TMPRSS2.
Recent molecular dynamics study has shown that peptides surrogate of the spike-binding motif of ACE2 inhibits the interaction of the S protein to ACE2 by binding to the S protein (Baig et al., 2020). Monteil et al., have shown that a clinical grade recombinant soluble ACE2 protein can inhibit entry of SARS-CoV-2 to engineered human blood vessel and kidney organoids (Monteil et al., 2020), suggesting blocking the interactions between ACE2 and the viral S protein as a promising strategy for therapeutic intervention against SARS-CoV2.
Similarly, spike RBD peptide mimetics consisting of natural and unnatural amino acids could be designed that can bind to ACE2 and block the binding of the viral spike protein. Targeting viral spike-binding site on ACE2 may not significantly affect the normal physiological function of ACE2 as blocking this site may not affect the enzymatic activity of ACE2 which is required for its normal biological functions. Moreover, several inhibitors of ACE2 carboxypeptidase activity are known that inhibit ACE2 catalyzed proteolysis (Takahashi et al., 2015). Such inhibitors are less likely to intervene the binding of SARS-CoV-2 S protein to the ACE2 receptor as the ACE2 catalytic site is in a distinct structural region than the S-protein binding site (Fig. 6a).
3.2. TMPRSS2 as a potential target for therapeutic intervention against SARS-CoV-2
SARS-CoV-2 cellular entry is mediated by the binding of its S protein to the ACE2 receptor and subsequent priming of the S protein by cellular protease that involves the cleavage of the S protein at two different sites namely S1/S2 and S2′. Such priming enables the fusion of viral membrane to the host cellular membrane. Transmembrane serine protease 2 (TMPRSS2) catalyzes this proteolytic cleavage of the S protein (Hoffmann et al., 2020). In the S1/S2 cleavage site of SARS-CoV-2 there is an insertion of four residues (PRRA) as compared to SARS-CoV – the S1/S2 cleavage motifs of SARS-CoV and SARS-CoV-2 consist of SLLR|S and RRAR|S, respectively while the S2′ cleavage site in the two viruses is identical. SARS-CoV requires cysteine protease, cathepsin for the cleavage of S1/S2 site and TMPRSS2 for the cleavage of S2′ site, while in SARS-CoV-2 cleavage at both sites occurs by TMPRSS2 (Hoffmann et al., 2020). This difference has been suggested as the basis of higher transmissibility of SARS-CoV-2 as compared to SARS-CoV(Wang et al., 2020c). TMPRSS2 is a member of transmembrane serine proteases. Different members of this family are expressed in a variety of tissues in a tissue dependent manner. TMPRSS2 has also been reported to play an important role in the cellular entry of avian influenza viruses through priming of hemagglutinin of these viruses (Shin and Seong, 2017). Therefore, TMPRSS2 could be targeted for the development of broad-spectrum antiviral agents.
TMPRSS2 consists of an N-terminal cytoplasmic domain, a hydrophobic transmembrane domain and an extracellular scavenger receptor domain followed by the C-terminal serine protease domain. The serine protease domain contains the Ser-His-Asp catalytic triad where the serine residue acts as a nucleophile during catalysis. Recently, Hoffmann et al., described the inhibition of SARS-CoV-2 and SARS-CoV in lungs cell line Calu-3 and in primary human lungs cell by camostat mesylate, a synthetic serine protease inhibitor previously approved for the treatment of chronic pancreatitis and oral squamous cell carcinoma (Hoffmann et al., 2020). Similarly another serine protease inhibitor nafamostat mesilate (Fig. 6d) which was approved in Japan to treat acute pancreatitis, has been shown to inhibit SARS-CoV-2 entry to the Vero cells with EC50 of 22.5 μM (Wang et al., 2020b). Although these drugs have shown viral entry inhibition in vitro, their efficacy to treat COVID-19 remains to be studied. As the structure of TMPRSS2 is not available yet, to gain an understanding of its structural features we homology modeled the structure of extracellular domain of TMPRSS2 using serine protease hepsin (ID: 5ce1.1.A) as template that showed 34% sequence identity. The structure of extracellular portion incorporates a scavenger receptor domain and a typical serine protease domain. The putative substrate-binding site with catalytic triad has been depicted in Fig. 6c. The TMPRSS2 modeled structure could be used to rationally design its inhibitors until its cryo-EM or X-ray structure is available.
3.3. Endoplasmic reticulum (ER) glucosidases as potential targets for therapeutic intervention against SARS-CoV-2
The S protein of SARS-CoV-2 is highly glycosylated as each protamer of the S protein trimer incorporates 22 N-glycosylation sites. Watanabe et al., have recently reported the mass spectrometry based glycan profiling of the recombinant S protein of SARS-CoV-2.According to this profiling, SARS-CoV-2 exhibits different glycoforms including high-mannose, complex and hybrid type glycans covering the whole surface of the S protein(Watanabe et al., 2020). Sanda et al., have reported the presence of 9O-linked glycans on the recombinantly expressed S protein, these glycosylation sites also include Thr-678 that is present in the vicinity of TMPRSS2 cleavage site on the S protein and is unique to SAR-CoV-2(Sanda et al., 2020). Co– and post-translationally incorporated glycans on viral proteins play diverse roles in viral pathobiology. Glycans mediate protein folding and stability, and play important roles in defining viral tropism and immune evasion by shielding the protein epitopes from the immune system(Dalziel et al., 2014, Watanabe et al., 2019). In the SARS-CoV-2 S protein no mutation of any N-glycosylation site has been reported since the emergence of this infection suggesting essential roles of glycans in the SARS-CoV-2 pathogenesis and survival(Watanabe et al., 2020).
Intervening the glycosylation process of viral proteins also represents an important strategy for therapeutic intervention in particular for highly glycosylated viruses such as coronaviruses. N-glycosylation represents the most common type of glycosylation in eukaryotes that occurs in the lumen of endoplasmic reticulum (ER) by the incorporation of a preformed glycan precursor Glc3Man9GlcNAc2 to the asparagine residue present in the NXS/T sequon where “X” represents any residue except proline. The incorporated glycan is processed by a series of enzymes present in the ER and the Golgi apparatus. Initial steps of this processing involve stepwise trimming of the three glucose saccharides from one of the terminal branches of Man9- GlcNAc2 by the α-glucosidase I and α-glucosidas II enzymes, in succession (Fig. 7 ). The trimmed Man9-GlcNAc2 or Man8-GlcNAc2 is transported to the Golgi apparatus for further glycan processing.
Fig. 7.

Glycosylation processing of glycoproteins in the ER and chemical structures of two well-known glucosidase inhibitors.
Inhibition of ER α-glucosidases represents a potential approach to discover new antiviral agents. Celgosivir, the precursor of a known ER α-glucosidase I inhibitor castanospermine, has shown efficacy against hepatitis C virus (HCV) infections and this potential drug underwent phase II clinical trials in combination therapy with pegylated interferon-α and ribavirin to treat hepatitis C (Durantel, 2009) – HCV envelope protein E2 exhibits 11 N-glycosylation sites(Iacob et al., 2008). Chapel et al., has reported the effect of ER glucosidase inhibitors on HCV morphogenesis. HCV virus like particles (VLPs) produced in the presence of ER α-glucosidase inhibitors contained primarily unprocessed glycans and partially miss-folded envelope proteins(Chapel et al., 2006). Ma et al., recently reported the inhibition of different hemorrhagic viruses including the yellow fever and ebola viruses by an ER α-glucosidase I inhibitor, IHVR-19029 in vitro and in animal models when used in combination with favipiravir (Ma et al., 2018). In 2014 two siblings were reported with a rare genetic disorder of glycosylation type IIb resulting from mutations in the gene encoding ER glucosidases. These siblings were resistant to viral infections indicating key roles of ER glucosidases in viral morphology and pathogenesis (Sadat et al., 2014). Effect of different immunosugar inhibitors of different glycan processing enzymes of the ER and the Golgi apparatus on the infectivity of different influenza viruses in vitro and in mouse models have been described (Tyrrell et al., 2017).
ER α-glucosidase inhibitors not only can alter the glycosylation processing of the viral glycoproteins leading to affect viral fitness but also can alter the morphology of viral receptor proteins if those are glycosylated. Zhao et al., have shown that α-glucosidase I inhibitor, IHVR-17028 inhibits the entry of lentiviral particles pseudo-typed with envelopes of SARS-CoV and human coronavirus NL-63 due to the altered glycosylation of ACE2 which is a glycoprotein (Zhao et al., 2015). Therefore, inhibition of ER glucosidases can potentially inhibit the SARS-CoV-2 infections through dual mechanisms; by altering the glycosylation of the viral S protein leading to impact its morphogenesis and by altering the glycosylation of the SARS-CoV-2 receptor ACE2 leading to impair its proper structure resulting in an effect on viral-receptor recognition and subsequent entry. Most of the known inhibitors with antiviral effects are the derivative of well-known glucosidase inhibitors 1-deoxynojirimycin (DNJ) and castanospermine (CAST) (Fig. 7). These derivatives have shown inhibition of different enveloped viruses. However, studies in animal models show rather moderate inhibition of viral infections by these drugs suggesting a need of designing more potent inhibitors. Chang J et al., has provided a comprehensive review on different glucosidase inhibitors and their antiviral effects in detail (Chang et al., 2013).
The human ER glucosidases structures are not yet available, however, structures of glucosidase I from yeast and glucosidase II from different organisms have been reported (Barker and Rose, 2013, Caputo et al., 2016). Solving the structure of human ER glucosidases through cryo-EM or X-ray crystallography will provide detailed insight into the rational design of more potent and specific ER glucosidase inhibitors.
Although ER α-glucosidase inhibition is a potential approach for antiviral drug discovery, however, such inhibition is expected to alter the glycosylation of every cellular glycoprotein leading to impart adverse physiological effects. Therefore, tissue specific cellular marker targeted drug delivery can minimize the off target effects of host targeted drugs, in general (Zhao et al., 2020). Recently, Bouhaddou et al., reported a mass spectrometry based phosphoproteomics and scanning electron microscopy based morphological analyses of the SARS-CoV-2 infected cells and showed an altered morphology of the infected cells with filopodial protrusions on the cellular surface (Bouhaddou et al., 2020). Such morphological exclusiveness of SARS-CoV-2 infected cells and associated cell surface markers can be exploited for an infected cell specific drug delivery. Warfield et al., very recently reported that single dose of a derivative of DNJ, N-(9′-methoxynonyl)-1-deoxynojirimycin, an ER α-glucosidase inhibitor, prevented the death of mice infected with lethal doses of dengue and H1N1 influenza viruses. The single dose of the drug showed extended pharmacological effect in animal models and safety in human subjects at a dose as high as 1 g(Warfield et al., 2020). Such findings highlight a novel paradigm for the development of treatments against acute viral infections.
4. Conclusions
SARS-CoV-2 infections have emerged as one of the most formidable health risks and are adversely affecting economies, healthcare, educational system, social and personal lives all over the world. The COVID-19 cases continue to increase in many parts of the world despite vigorous and collective global efforts of its containment. Since the emergence of this pandemic substantial multidisciplinary and cohesive research efforts have resulted in the creation of unprecedented knowledge about the virus and its cross talk with the host. Molecular structures of various SARS-CoV-2 proteins and their complexes with interacting partners explaining the structural basis of viral pathogenesis and its replication cycle have become available as a result of striking progress in technology and extraordinary efforts of the scientific community. In this review we recapitulated the structural information of four viral factors, RdRp, 3CLpro, PLpro and helicase and two host factors including ACE2 and TMPRSS2 in the context of explaining potential hotspots to target for drug development against SARS-CoV-2 in addition to discussing ER glucosidases as potential drug targets. Future efforts should be made to identify new potential inhibitors of the SARS-CoV-2 infections by exploring different potential target sites on proteins in addition to their main catalytic sites. Moreover, solving the structures of human ER glucosidases would also be an important future perspective for rational drug design. In brief, by combining the emerging structural information, computer-aided drug design and medicinal chemistry techniques, rational drug design efforts should result in the development of a number of potential drugs against SARS-CoV-2.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgements
We highly acknowledge the Department of Biology, SBA School of Science and Engineering at Lahore University of Management Sciences for a continues support of research in the form of resources.
Authors contributions
MS wrote a major part of the original draft, SS provided guidance and supervision in the manuscript preparation, formal analysis, and reviewing & editing.
References
- Al-Kassmy J., Pedersen J., Kobinger G. Vaccine Candidates against Coronavirus Infections. Where Does COVID-19 Stand? Viruses. 2020;12:861–879. doi: 10.3390/v12080861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baig M.S., Alagumuthu M., Rajpoot S., Saqib U. Identification of a Potential Peptide Inhibitor of SARS-CoV-2 Targeting its Entry into the Host Cells. Drugs in R&D. 2020;20:161–169. doi: 10.1007/s40268-020-00312-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker M.K., Rose D.R. Specificity of Processing α-glucosidase I is guided by the substrate conformation: crystallographic and in silico studies. J. Biol. Chem. 2013;288:13563–13574. doi: 10.1074/jbc.M113.460436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayham J., Fenichel E.P. Impact of school closures for COVID-19 on the US health-care workforce and net mortality: a modelling study. The Lancet. Public health. 2020;5:e271–e278. doi: 10.1016/S2468-2667(20)30082-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhaddou M., Memon D., Meyer B., White K.M., Rezelj V.V., Correa Marrero M., Polacco B.J., Melnyk J.E., Ulferts S., Kaake R.M., Batra J., Richards A.L., Stevenson E., Gordon D.E., Rojc A., Obernier K., Fabius J.M., Soucheray M., Miorin L., Moreno E., Koh C., Tran Q.D., Hardy A., Robinot R., Vallet T., Nilsson-Payant B.E., Hernandez-Armenta C., Dunham A., Weigang S., Knerr J., Modak M., Quintero D., Zhou Y., Dugourd A., Valdeolivas A., Patil T., Li Q., Hüttenhain R., Cakir M., Muralidharan M., Kim M., Jang G., Tutuncuoglu B., Hiatt J., Guo J.Z., Xu J., Bouhaddou S., Mathy C.J.P., Gaulton A., Manners E.J., Félix E., Shi Y., Goff M., Lim J.K., McBride T., O'Neal M.C., Cai Y., Chang J.C.J., Broadhurst D.J., Klippsten S., De Wit E., Leach A.R., Kortemme T., Shoichet B., Ott M., Saez-Rodriguez J., tenOever B.R., Mullins R.D., Fischer E.R., Kochs G., Grosse R., García-Sastre A., Vignuzzi M., Johnson J.R., Shokat K.M., Swaney D.L., Beltrao P., Krogan N.J. The Global Phosphorylation Landscape of SARS-CoV-2 Infection. Cell. 2020;182:685–712.e619. doi: 10.1016/j.cell.2020.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkle F.M. Declining Public Health Protections within Autocratic Regimes: Impact on Global Public Health Security, Infectious Disease Outbreaks, Epidemics, and Pandemics. Prehosp. Disaster Med. 2020;35:237–246. doi: 10.1017/S1049023X20000424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao B., Wang C., Antinori S., Cossu M.V., Ridolfo A.L., Rech R., Bonazzetti C., Pagani G., Gubertini G., Coen M., Magni C., Castelli A., Borghi B., Colombo R., Giorgi R., Angeli E., Mileto D., Milazzo L., Vimercati S., Pellicciotta M., Corbellino M., Torre A., Rusconi S., Oreni L., Gismondo M.R., Giacomelli A., Meroni L., Rizzardini G., Galli M. Compassionate remdesivir treatment of severe Covid-19 pneumonia in intensive care unit (ICU) and Non-ICU patients: Clinical outcome and differences in post-treatment hospitalisation status. Trials. 2020;158 doi: 10.1016/j.phrs.2020.104899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputo, A.T., Alonzi, D.S., Marti, L., Reca, I.B., Kiappes, J.L., Struwe, W.B., Cross, A., Basu, S., Lowe, E.D., Darlot, B., Santino, A., Roversi, P., 2016. Structures of mammalian ER α-glucosidase II capture the binding modes of broad-spectrum iminosugar antivirals 113, E4630-4638. [DOI] [PMC free article] [PubMed]
- Chan, J.F., Kok, K.H., 2020. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan 9, 221-236. [DOI] [PMC free article] [PubMed]
- Chang J., Block T.M., Guo J.-T. Antiviral therapies targeting host ER alpha-glucosidases: current status and future directions. Antiviral Res. 2013;99:251–260. doi: 10.1016/j.antiviral.2013.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapel C., Garcia C., Roingeard P., Zitzmann N., Dubuisson J., Dwek R.A., Trepo C., Zoulim F., Durantel D. Antiviral effect of α-glucosidase inhibitors on viral morphogenesis and binding properties of hepatitis C virus-like particles. J. Gen. Virol. 2006;87:861–871. doi: 10.1099/vir.0.81503-0. [DOI] [PubMed] [Google Scholar]
- Checcucci E., Piramide F., Pecoraro A., Amparore D., Campi R., Fiori C., Elhage O., Kotecha P., Vyakarnam A., Serni S., Dasgupta P., Porpiglia F. The vaccine journey for COVID-19: a comprehensive systematic review of current clinical trials in humans. Panminerva Med. 2020 doi: 10.23736/S0031-0808.20.03958-0. [DOI] [PubMed] [Google Scholar]
- Chellapandi P., Saranya S. Genomics insights of SARS-CoV-2 (COVID-19) into target-based drug discovery. Med. Chem. Res. 2020:1–15. doi: 10.1007/s00044-020-02610-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Malone B., Llewellyn E., Grasso M., Shelton P.M.M., Olinares P.D.B., Maruthi K., Eng E.T., Vatandaslar H., Chait B.T., Kapoor T.M., Darst S.A., Campbell E.A. Structural Basis for Helicase-Polymerase Coupling in the SARS-CoV-2 Replication-Transcription Complex. Cell. 2020;182:1560–1573.e1513. doi: 10.1016/j.cell.2020.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho J.B., Lee J.M., Ahn H.C., Jeong Y.J. Identification of a Novel Small Molecule Inhibitor Against SARS Coronavirus Helicase. J. Microbiol. Biotechnol. 2015;25:2007–2010. doi: 10.4014/jmb.1507.07078. [DOI] [PubMed] [Google Scholar]
- Crotty S., Maag D., Arnold J.J., Zhong W., Lau J.Y., Hong Z., Andino R., Cameron C.E. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat. Med. 2000;6:1375–1379. doi: 10.1038/82191. [DOI] [PubMed] [Google Scholar]
- Dai W., Zhang B. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science (New York N.Y.) 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalziel M., Crispin M., Scanlan C.N., Zitzmann N., Dwek R.A. Emerging principles for the therapeutic exploitation of glycosylation. Science (New York N.Y.) 2014;343:1235681. doi: 10.1126/science.1235681. [DOI] [PubMed] [Google Scholar]
- Durantel, D., 2009. Celgosivir, an alpha-glucosidase I inhibitor for the potential treatment of HCV infection. Current opinion in investigational drugs (London, England: 2000) 10, 860-870. [PubMed]
- Edinger T.O., Pohl M.O., Stertz S. Entry of influenza A virus: host factors and antiviral targets. J. Gen. Virol. 2014;95:263–277. doi: 10.1099/vir.0.059477-0. [DOI] [PubMed] [Google Scholar]
- Gadebusch Bondio M., Marloth M. The “Historic Study” SOLIDARITY-Research's Answer to the Sars-CoV-2 Pandemic. Ntm. 2020;28:219–225. doi: 10.1007/s00048-020-00257-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y., Yan L., Huang Y., Liu F., Zhao Y., Cao L., Wang T., Sun Q., Ming Z., Zhang L. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science (New York N.Y.) 2020;368:779–782. doi: 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillen H.S., Kokic G. Structure of replicating SARS-CoV-2 polymerase. Nature. 2020;584:154–156. doi: 10.1038/s41586-020-2368-8. [DOI] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Kruger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A., Muller M.A., Drosten C., Pohlmann S. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181:271–280.e278. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung I.F., Lung K.C., Tso E.Y., Liu R., Chung T.W., Chu M.Y., Ng Y.Y., Lo J., Chan J., Tam A.R., Shum H.P., Chan V., Wu A.K., Sin K.M., Leung W.S., Law W.L., Lung D.C., Sin S., Yeung P., Yip C.C., Zhang R.R., Fung A.Y., Yan E.Y., Leung K.H., Ip J.D., Chu A.W., Chan W.M., Ng A.C., Lee R., Fung K., Yeung A., Wu T.C., Chan J.W., Yan W.W., Chan W.M., Chan J.F., Lie A.K., Tsang O.T., Cheng V.C., Que T.L., Lau C.S., Chan K.H., To K.K., Yuen K.Y. Triple combination of interferon beta-1b, lopinavir-ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: an open-label, randomised, phase 2 trial. Lancet (London, England) 2020;395:1695–1704. doi: 10.1016/S0140-6736(20)31042-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacob R.E., Perdivara I., Przybylski M., Tomer K.B. Mass spectrometric characterization of glycosylation of hepatitis C virus E2 envelope glycoprotein reveals extended microheterogeneity of N-glycans. J. Am. Soc. Mass Spectrom. 2008;19:428–444. doi: 10.1016/j.jasms.2007.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iftikhar H., Ali H.N., Farooq S., Naveed H., Shahzad-ul-Hussan S. Identification of potential inhibitors of three key enzymes of SARS-CoV2 using computational approach. Comput. Biol. Med. 2020;122 doi: 10.1016/j.compbiomed.2020.103848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y., Kuba K., Ohto-Nakanishi T., Penninger J.M. Angiotensin-converting enzyme 2 (ACE2) in disease pathogenesis. Circul. J. Off. J. Jap. Circul. Soc. 2010;74:405–410. doi: 10.1253/circj.cj-10-0045. [DOI] [PubMed] [Google Scholar]
- Jácome R., Campillo-Balderas J.A., Ponce de León S., Becerra A., Lazcano A. Sofosbuvir as a potential alternative to treat the SARS-CoV-2 epidemic. Sci. Rep. 2020;10:9294. doi: 10.1038/s41598-020-66440-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Z., Yan L., Ren Z., Wu L., Wang J., Guo J., Zheng L., Ming Z., Zhang L., Lou Z. Delicate structural coordination of the Severe Acute Respiratory Syndrome coronavirus Nsp13 upon ATP hydrolysis. Nucleic Acids Res. 2019;47:6538–6550. doi: 10.1093/nar/gkz409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z., Du X., Xu Y. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Jomah S., Asdaq S.M.B., Al-Yamani M.J. Clinical efficacy of antivirals against novel coronavirus (COVID-19): A review. J. Infect. Public Health. 2020;13:1187–1195. doi: 10.1016/j.jiph.2020.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen S.C.J., Kebriaei R., Dresser L.D. Remdesivir: Review of Pharmacology, Pre-clinical Data, and Emerging Clinical Experience for COVID-19. Pharmacotherapy. 2020;40:659–671. doi: 10.1002/phar.2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C.W., Chang K.M. Hepatitis C virus: virology and life cycle. Clin. Mol. Hepatol. 2013;19:17–25. doi: 10.3350/cmh.2013.19.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuba K., Imai Y., Penninger J.M. Multiple functions of angiotensin-converting enzyme 2 and its relevance in cardiovascular diseases. Circul. J. Off. J. Japan. Circul. Soc. 2013;77:301–308. doi: 10.1253/circj.cj-12-1544. [DOI] [PubMed] [Google Scholar]
- Lan J., Ge J., Yu J. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- Lehmann K.C., Gulyaeva A., Zevenhoven-Dobbe J.C., Janssen G.M., Ruben M., Overkleeft H.S., van Veelen P.A., Samborskiy D.V., Kravchenko A.A., Leontovich A.M., Sidorov I.A., Snijder E.J., Posthuma C.C., Gorbalenya A.E. Discovery of an essential nucleotidylating activity associated with a newly delineated conserved domain in the RNA polymerase-containing protein of all nidoviruses. Nucleic Acids Res. 2015;43:8416–8434. doi: 10.1093/nar/gkv838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei J., Kusov Y., Hilgenfeld R. Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antiviral Res. 2018;149:58–74. doi: 10.1016/j.antiviral.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei X., Dong X., Ma R., Wang W., Xiao X., Tian Z., Wang C., Wang Y., Li L., Ren L., Guo F., Zhao Z., Zhou Z., Xiang Z., Wang J. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 2020;11:3810. doi: 10.1038/s41467-020-17665-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., Choe H., Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licastro D., Rajasekharan S., Dal Monego S., Segat L., D'Agaro P., Marcello A. Isolation and Full-Length Genome Characterization of SARS-CoV-2 from COVID-19 Cases in Northern Italy. J. Virol. 2020;94 doi: 10.1128/JVI.00543-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima W.G., Brito J.C.M., Overhage J., Nizer W. The potential of drug repositioning as a short-term strategy for the control and treatment of COVID-19 (SARS-CoV-2): a systematic review. Arch. Virol. 2020;165:1729–1737. doi: 10.1007/s00705-020-04693-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., Wang W., Song H., Huang B., Zhu N., Bi Y., Ma X., Zhan F., Wang L., Hu T., Zhou H., Hu Z., Zhou W., Zhao L., Chen J., Meng Y., Wang J., Lin Y., Yuan J., Xie Z., Ma J., Liu W.J., Wang D., Xu W., Holmes E.C., Gao G.F., Wu G., Chen W., Shi W., Tan W. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet (London, England) 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J., Zhang X., Soloveva V., Warren T., Guo F., Wu S., Lu H., Guo J., Su Q., Shen H., Solon E., Comunale M.A., Mehta A., Guo J.T., Bavari S., Du Y., Block T.M., Chang J. Enhancing the antiviral potency of ER α-glucosidase inhibitor IHVR-19029 against hemorrhagic fever viruses in vitro and in vivo. Antiviral Res. 2018;150:112–122. doi: 10.1016/j.antiviral.2017.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee D.L., Sternberg A., Stange U., Laufer S., Naujokat C. Candidate drugs against SARS-CoV-2 and COVID-19. Nature. 2020;157 doi: 10.1016/j.phrs.2020.104859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melero R., Sorzano C.O.S., Foster B., Vilas J.L., Martínez M. Continuous flexibility analysis of SARS-CoV-2 spike prefusion structures. IUCrJ. 2020;7:1059–1069. doi: 10.1107/S2052252520012725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza M.U., Froeyen M. Structural elucidation of SARS-CoV-2 vital proteins: Computational methods reveal potential drug candidates against main protease, Nsp12 polymerase and Nsp13 helicase. J Pharm Anal. 2020;10:320–328. doi: 10.1016/j.jpha.2020.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsuya H., Yarchoan R., Broder S. Molecular targets for AIDS therapy. Science (New York N.Y.) 1990;249:1533–1544. doi: 10.1126/science.1699273. [DOI] [PubMed] [Google Scholar]
- Monteil V., Kwon H., Prado P., Hagelkrüys A., Wimmer R.A., Stahl M., Leopoldi A., Garreta E., Hurtado Del Pozo C., Prosper F., Romero J.P., Wirnsberger G., Zhang H., Slutsky A.S., Conder R., Montserrat N., Mirazimi A., Penninger J.M. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell. 2020;181:905–913.e907. doi: 10.1016/j.cell.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey A., Nikam A.N., Shreya A.B., Mutalik S.P., Gopalan D., Kulkarni S., Padya B.S., Fernandes G., Mutalik S., Prassl R. Potential therapeutic targets for combating SARS-CoV-2: Drug repurposing, clinical trials and recent advancements. Life Sci. 2020;256 doi: 10.1016/j.lfs.2020.117883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillaiyar T., Manickam M., Namasivayam V., Hayashi Y., Jung S.H. An Overview of Severe Acute Respiratory Syndrome-Coronavirus (SARS-CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy. J. Med. Chem. 2016;59:6595–6628. doi: 10.1021/acs.jmedchem.5b01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman S., Hossain M., Hasan M., Khan M.K., Khatun A., Dash R., Uddin M.J., Serafin M.B., Bottega A., Foletto V.S., da Rosa T.F., Hörner A., Hörner R. Drug repositioning is an alternative for the treatment of coronavirus COVID-19. Drug Dev. Res. 2020;55 doi: 10.1016/j.ijantimicag.2020.105969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rut, W., Lv, Z., Zmudzinski, M., 2020. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: A framework for anti-COVID-19 drug design. Sci. Adv. 6. [DOI] [PMC free article] [PubMed]
- Sadat M.A., Moir S., Chun T.W., Lusso P., Kaplan G., Wolfe L., Memoli M.J., He M., Vega H., Kim L.J.Y., Huang Y., Hussein N., Nievas E., Mitchell R., Garofalo M., Louie A., Ireland D.C., Grunes C., Cimbro R., Patel V., Holzapfel G., Salahuddin D., Bristol T., Adams D., Marciano B.E., Hegde M., Li Y., Calvo K.R., Stoddard J., Justement J.S., Jacques J., Priel D.A.L., Murray D., Sun P., Kuhns D.B., Boerkoel C.F., Chiorini J.A., Di Pasquale G., Verthelyi D., Rosenzweig S.D. Glycosylation, hypogammaglobulinemia, and resistance to viral infections. New Engl. J. Med. 2014;370:1615–1625. doi: 10.1056/NEJMoa1302846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanda, M., Morrison, L., Goldman, R., 2020. N and O glycosylation of the SARS-CoV-2 spike protein. bioRxiv. [DOI] [PMC free article] [PubMed]
- Santos J., Brierley S., Gandhi M.J. Repurposing Therapeutics for Potential Treatment of SARS-CoV-2: A Review. Viruses. 2020;12:705. doi: 10.3390/v12070705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J., Ye G., Shi K., Wan Y., Luo C., Aihara H. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;581:221–224. doi: 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shereen M.A., Khan S., Kazmi A., Bashir N., Siddique R. COVID-19 infection: Origin, transmission, and characteristics of human coronaviruses. J. Adv. Res. 2020;24:91–98. doi: 10.1016/j.jare.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin D., Mukherjee R. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. 2020;587:657–662. doi: 10.1038/s41586-020-2601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin W.-J., Seong B.L. Type II transmembrane serine proteases as potential target for anti-influenza drug discovery. Expert Opin. Drug Discov. 2017;12:1139–1152. doi: 10.1080/17460441.2017.1372417. [DOI] [PubMed] [Google Scholar]
- Shum K.T., Tanner J.A. Differential inhibitory activities and stabilisation of DNA aptamers against the SARS coronavirus helicase. Chembiochem: Eur. J. Chem. Biol. 2008;9:3037–3045. doi: 10.1002/cbic.200800491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisay M. Available Evidence and Ongoing Clinical Trials of Remdesivir: Could It Be a Promising Therapeutic Option for COVID-19? Front. Pharmacol. 2020;11:791. doi: 10.3389/fphar.2020.00791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., Decroly E., Ziebuhr J. The Nonstructural Proteins Directing Coronavirus RNA Synthesis and Processing. Adv. Virus Res. 2016;96:59–126. doi: 10.1016/bs.aivir.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterling T., Irwin J.J. ZINC 15–Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015;55:2324–2337. doi: 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, H., Su, H., Zhou, F., Huang, Z., Ma, X., Natarajan, K., Zhang, M., Huang, Y., 2020a. Molecular Insights into Small Molecule Drug Discovery for SARS-CoV-2. Angewandte Chemie (International ed. in English). [DOI] [PubMed]
- Su H.X., Yao S., Zhao W.F., Li M.J., Liu J., Shang W.J., Xie H., Ke C.Q., Hu H.C., Gao M.N., Yu K.Q., Liu H., Shen J.S., Tang W., Zhang L.K., Xiao G.F., Ni L., Wang D.W., Zuo J.P., Jiang H.L., Bai F., Wu Y., Ye Y., Xu Y.C. Anti-SARS-CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol. Sin. 2020:1–11. doi: 10.1038/s41401-020-0483-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subissi L., Posthuma C.C., Collet A., Zevenhoven-Dobbe J.C., Gorbalenya A.E., Decroly E., Snijder E.J., Canard B., Imbert I. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. PNAS. 2014;111:E3900–3909. doi: 10.1073/pnas.1323705111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaim, C.D., Perng, Y.C., Zhao, X., Canadeo, L.A., Harastani, H.H., Darling, T.L., Boon, A.C.M., Lenschow, D.J., Huibregtse, J.M., 2020. 6-Thioguanine blocks SARS-CoV-2 replication by inhibition of PLpro protease activities. bioRxiv. [DOI] [PMC free article] [PubMed]
- Tai, W., He, L., Zhang, X., Pu, J., Voronin, D., 2020. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine 17, 613-620. [DOI] [PMC free article] [PubMed]
- Takahashi S., Yoshiya T., Yoshizawa-Kumagaye K., Sugiyama T. Nicotianamine is a novel angiotensin-converting enzyme 2 inhibitor in soybean. Biomed. Res. (Tokyo, Japan) 2015;36:219–224. doi: 10.2220/biomedres.36.219. [DOI] [PubMed] [Google Scholar]
- te Velthuis A.J., van den Worm S.H., Snijder E.J. The SARS-coronavirus nsp7+nsp8 complex is a unique multimeric RNA polymerase capable of both de novo initiation and primer extension. Nucleic Acids Res. 2012;40:1737–1747. doi: 10.1093/nar/gkr893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari V., Beer J.C., Sankaranarayanan N.V., Swanson-Mungerson M., Desai U.R. Discovering small-molecule therapeutics against SARS-CoV-2. Drug Discovery Today. 2020;25:1535–1544. doi: 10.1016/j.drudis.2020.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Y.F., Chien C.S., Yarmishyn A.A., Lin Y.Y., Luo Y.H., Lin Y.T., Lai W.Y., Yang D.M., Chou S.J., Yang Y.P., Wang M.L., Chiou S.H. A Review of SARS-CoV-2 and the Ongoing Clinical Trials. Viruses. 2020;21 doi: 10.3390/ijms21072657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrrell, B.E., Sayce, A.C., Warfield, K.L., Miller, J.L., 2017. Iminosugars: Promising therapeutics for influenza infection 43, 521-545. [DOI] [PMC free article] [PubMed]
- Ullrich S., Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 2020;30 doi: 10.1016/j.bmcl.2020.127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y., Shang J., Graham R., Baric R.S., Li F. Receptor Recognition by the Novel Coronavirus from Wuhan: an Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020;94 doi: 10.1128/JVI.00127-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J., Peng, Y., Xu, H., Cui, Z., Williams, R.O., 3rd, 2020a. The COVID-19 Vaccine Race: Challenges and Opportunities in Vaccine Formulation 21, 225. [DOI] [PMC free article] [PubMed]
- Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., Shi Z., Hu Z., Zhong W., Xiao G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Qiu Y., Li J.-Y., Zhou Z.-J., Liao C.-H., Ge X.-Y. A unique protease cleavage site predicted in the spike protein of the novel pneumonia coronavirus (2019-nCoV) potentially related to viral transmissibility. Virol. Sin. 2020:1–3. doi: 10.1007/s12250-020-00212-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y., Zhou, F., Zhang, D., Zhao, J., Du, R., Hu, Y., Cheng, Z., Gao, L., Jin, Y., Luo, G., Fu, S., Lu, Q., Du, G., Wang, K., Lu, Y., Fan, G., Zhang, Y., Liu, Y., Ruan, S., Liu, W., Jaki, T., Hayden, F.G., Horby, P.W., 2020d. Evaluation of the efficacy and safety of intravenous remdesivir in adult patients with severe COVID-19: study protocol for a phase 3 randomized, double-blind, placebo-controlled, Multicentre Trial 21, 422. [DOI] [PMC free article] [PubMed]
- Warfield, K.L., Alonzi, D.S., Hill, J.C., Caputo, A.T., Roversi, P., Kiappes, J.L., 2020. Targeting Endoplasmic Reticulum α-Glucosidase I with a Single-Dose Iminosugar Treatment Protects against Lethal Influenza and Dengue Virus Infections 63, 4205-4214. [DOI] [PubMed]
- Warren T.K., Wells J., Panchal R.G., Stuthman K.S., Garza N.L., Van Tongeren S.A., Dong L., Retterer C.J., Eaton B.P., Pegoraro G., Honnold S., Bantia S., Kotian P., Chen X., Taubenheim B.R., Welch L.S., Minning D.M., Babu Y.S., Sheridan W.P., Bavari S. Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430. Nature. 2014;508:402–405. doi: 10.1038/nature13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y., Bowden T.A., Wilson I.A., Crispin M. Exploitation of glycosylation in enveloped virus pathobiology. Biochim. Biophys. Acta, Gen. Subj. 2019;1863:1480–1497. doi: 10.1016/j.bbagen.2019.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y., Allen J.D., Wrapp D., McLellan J.S., Crispin M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science (New York N.Y.) 2020;369:330–333. doi: 10.1126/science.abb9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White M.A., Lin W., Cheng X. Discovery of COVID-19 Inhibitors Targeting the SARS-CoV-2 Nsp13 Helicase. J. Phys. Chem. Lett. 2020;11:9144–9151. doi: 10.1021/acs.jpclett.0c02421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. 2020. Corona virus disease, COVID-19 situation report, 21 September 2020, 21 September 2020 ed. World Health Organization.
- Wrapp, D., Wang, N., 2020a. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation 367, 1260-1263. [DOI] [PMC free article] [PubMed]
- Wrapp D., Wang N. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science (New York N.Y.) 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X., Yu H., Yang H., Xue F., Wu Z., Shen W., Li J., Zhou Z., Ding Y., Zhao Q., Zhang X.C., Liao M., Bartlam M., Rao Z. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J. Virol. 2008;82:2515–2527. doi: 10.1128/JVI.02114-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, W., Mao, C., Luan, X., 2020. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir 368, 1499-1504. [DOI] [PMC free article] [PubMed]
- Yuen, C.K., Lam, J.Y., Wong, W.M., Mak, L.F., Wang, X., Chu, H., Cai, J.P., Jin, D.Y., 2020. SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists 9, 1418-1428. [DOI] [PMC free article] [PubMed]
- Zhang L., Lin D. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science (New York N.Y.) 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X., Guo F., Comunale M.A., Mehta A., Sehgal M., Jain P., Cuconati A., Lin H., Block T.M., Chang J., Guo J.T. Inhibition of endoplasmic reticulum-resident glucosidases impairs severe acute respiratory syndrome coronavirus and human coronavirus NL63 spike protein-mediated entry by altering the glycan processing of angiotensin I-converting enzyme 2. Antimicrob. Agents Chemother. 2015;59:206–216. doi: 10.1128/AAC.03999-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z., Ukidve A., Kim J., Mitragotri S. Targeting Strategies for Tissue-Specific Drug Delivery. Cell. 2020;181:151–167. doi: 10.1016/j.cell.2020.02.001. [DOI] [PubMed] [Google Scholar]