

Summary
Corticostriatal synaptic integration is partitioned among striosome (patch) and matrix compartments of the dorsal striatum, allowing compartmentalized control of discrete aspects of behavior. Despite the significance of such organization, it’s unclear how compartment-specific striatal output is dynamically achieved, particularly considering new evidence that overlap of afferents is substantial. We show that dopamine oppositely shapes responses to convergent excitatory inputs in mouse striosome and matrix striatal spiny projection neurons (SPNs). Activation of postsynaptic D1 dopamine receptors promoted the generation of long-lasting synaptically-evoked “up-states” in matrix SPNs but opposed it in striosomes, which were more excitable under basal conditions. Differences in dopaminergic modulation were mediated, in part, by dendritic voltage-gated calcium channels (VGCCs): pharmacological manipulation of L-type VGCCs reversed compartment-specific responses to D1 receptor activation. These results support a novel mechanism for the selection of striatal circuit components, where fluctuating levels of dopamine shift the balance of compartment-specific striatal output.
Graphical Abstract

eTOC
Prager et al. show that dopamine promotes the maintenance of dendritically-evoked “up-states” in mouse direct pathway matrix SPNs, but opposes it in striosomes. This requires postsynaptic D1 receptors, and involves differential engagement of L-type Ca2+ channels. These findings reveal a mechanism where fluctuations in dopamine may constrain compartment-specific striatal output.
Introduction
The dorsal striatum integrates convergent afferents from diverse cortical and subcortical brain regions to guide movement and motor-decision making. This involves the coordinated synaptic engagement of two complementary populations of striatal spiny projection neurons (SPNs), which ground the direct and indirect pathways of the basal ganglia (Cui et al., 2013; Gerfen and Surmeier, 2011). But there is a deeper and less understood organization embedded within the striatal network: SPNs are segregated into two anatomically- and chemically- defined compartments, known as striosomes (patches) and matrix (Gerfen, 1984; Graybiel and Ragsdale, 1978; Pert et al., 1976). Direct synaptic communication between striosome and matrix SPNs is minimal, and compartment-specific activity has been associated with unique behavioral tasks (Brimblecombe and Cragg, 2017; Canales and Graybiel, 2000; Crittenden and Graybiel, 2011; Friedman et al., 2017; Friedman et al., 2015; Jenrette et al., 2019; Nadel et al., 2020; Yoshizawa et al., 2018). Despite growing evidence that such compartmentalization is a crucial component of basal ganglia network function, our understanding of the mechanisms that regulate striosome vs matrix output is limited.
All striatal output is carried by SPNs. As striosomes account for only ~10–15% of striatal volume (Brimblecombe and Cragg, 2017; Crittenden and Graybiel, 2011), and few studies have attempted to make compartment-targeted recordings (Banghart et al., 2015; Crittenden et al., 2017; Kawaguchi et al., 1989; McGregor et al., 2019; Salinas et al., 2016; Smith et al., 2016), most of what we know about SPN function likely pertains to the matrix. SPNs are silent in the absence of synaptic drive, requiring coordinated glutamatergic innervation to overcome their hyperpolarized resting membrane potential and fire action potentials (Blackwell et al., 2003; Gerfen and Surmeier, 2011; Stern et al., 1998). In presumptive matrix SPNs these criteria can be fulfilled by the convergent activation of only about a dozen glutamatergic synapses, provided they are 1) spatially close to each other and 2) located on a distal dendrite (~100 μm or more from the soma) (Dorman et al., 2018; Du et al., 2017; Plotkin et al., 2011). Such synaptic synchronization induces a regenerative dendritic plateau potential that pushes the soma into a long-lasting depolarized “up-state” from which action potentials can fire (Du et al., 2017; Plotkin et al., 2011).
How are SPNs in striosomes and matrix differentially and task-dependently engaged? One mechanism involves preferential innervation by certain limbic-associated cortical regions (Friedman et al., 2017; Friedman et al., 2015; McGregor et al., 2019). But as a singular, generalizable explanation, this is problematic for two reasons: 1) recent work suggests that overlap of afferents to striosome and matrix SPNs is more substantial than previously appreciated (Smith et al., 2016) and 2) neuromodulation is likely a crucial determinant (Banghart et al., 2015; Brimblecombe and Cragg, 2015, 2017; Crittenden et al., 2017; Inoue et al., 2016; Miura et al., 2007; Prager and Plotkin, 2019; Salinas et al., 2016), which is highlighted by the fact that chronic intermittent administration of psychostimulants or dopamine agonists induces compartment-specific changes in immediate early gene expression (Canales and Graybiel, 2000). An unexplored possibility is that there are intrinsic differences in how striosome and matrix SPNs transform converging excitatory inputs into somatic state-transitions, and how this process is modulated by dopamine. Here we show that responses to convergent glutamatergic inputs are differentially modulated by dopamine: activation of D1 dopamine receptors (D1Rs) in D1R-expressing SPNs prolonged the duration of dendritically evoked up-states in the matrix but shortened the duration in striosomes. The opposing actions of D1R signaling were not due to differences in dopamine receptor activation and could be reversed by pharmacologically blocking or enhancing postsynaptic L-type voltage-gated Ca2+ channel (VGCC) activity.
Results
Since elevated striosome activity is often associated with hyperkinetic behavioral changes (Canales and Graybiel, 2000; Crittenden et al., 2017), and activity of D1R-expressing direct pathway SPNs (dSPNs, as opposed to D2R-expressing indirect pathway SPNs) is associated with action initiation (Gerfen and Surmeier, 2011; Kravitz and Kreitzer, 2012), we restricted our study to dSPNs residing in each compartment. To visualize dSPNs in striosome and matrix compartments, we crossed transgenic mice expressing eGFP under the control of the Nuclear receptor subfamily 4, group A, member 1 promoter (Nr4a1-eGFP; labeling striosomes) (Davis and Puhl, 2011) with drd1-tdTomato transgenic mice (labeling dSPNs) (Shuen et al., 2008) (Fig 1A, B). Consistent with previous reports using different transgenic mouse lines (Crittenden et al., 2017; McGregor et al., 2019; Smith et al., 2016) striosome dSPNs were more excitable than their matrix counterparts, as reflected by higher input resistance, lower rheobase current and larger miniature excitatory postsynaptic potential (mEPSP) amplitudes (Fig 1C, S1A–H). Though morphologically similar, total dendritic length was reduced in striosome dSPNs (S1I–L), a feature known to result in increased SPN somatic excitability (Gertler et al., 2008), and likely a reflection of the spatial constraints imposed by strict compartmental boundaries (Brimblecombe and Cragg, 2017; Fujiyama et al., 2011).
Figure 1. Matrix and striosome dSPNs support dendritically-evoked somatic up-states.

(A) Immunohistochemical localization of the striosome marker μ-opioid receptor (MOR; red) in a Nr4a1-eGFP (green) mouse (top). 2-photon maximum intensity projection (2P-MIP) image showing recording electrode placement in a parasagittal brain slice from a Nr4a1-eGFP × drd1-tdTomato mouse (bottom; scale bars = 500 μm).
(B) 2P-MIPs of recorded matrix and striosome dSPNs (scale bar = 30 μm).
(C) Corresponding current-voltage (IV) traces of matrix (top) and striosome (bottom) dSPNs.
(D-E) Representative somatic voltage traces in response to 2PLUG at a single distal dendritic spine (red) or multiple distal dendritic spines stimulated at threshold for up-state induction. 2P-MIPs of distal dendrites showing locations of 2PLUG (yellow circles) (right, scale bar = 2 μm).
(F) Somatic voltage traces of dendritically-evoked up-states aligned to the end of 2PLUG stimulation and normalized to peak amplitude.
(G) Box plots showing somatic up-state durations. m = matrix; s = striosome; ns = not significant.
(H) Box plots showing somatic membrane potentials of “down-” and “up-” states. *p<0.05, ***p<0.001. See also Figures S1–4 and Table S2.
Striatal output is ultimately dependent upon the capacity of SPNs to integrate coordinated synaptic events (Gerfen and Surmeier, 2011; Goldberg et al., 2003; Stern et al., 1998; Wilson and Kawaguchi, 1996). We therefore determined if dSPNs residing in striosomes vs matrix differ in their capacity to support dendritically-evoked up-states, a direct assay of SPN-intrinsic glutamatergic synaptic integration (Du et al., 2017; Plotkin et al., 2011). We evoked up-states in dSPNs by using 2-photon laser uncaging of glutamate (2PLUG; assisted by 2-photon laser scanning microscopy) to convergently activate 10–18 neighboring distal dendritic spines, independent of presynaptic identity (Fig 1D, E; S2). While both striosome and matrix dSPNs supported the generation of up-states, striosome dSPNs achieved a significantly more depolarized up-state membrane potential than their matrix counterparts (Fig 1F–H). Elevated up-state amplitude correlated with higher somatic input resistance (r = 0.3475; p = 0.0004), and was not due to differences in 1) synaptic strength (single spine 2PLUG-evoked EPSPs used to evoke up-states had similar amplitudes), 2) postsynaptic glutamate receptor composition (the amplitudes, kinetics and ratios of NMDA- and AMPA- receptor mediated 2PLUG-evoked current components, as well as miniature excitatory postsynaptic currents (mEPSCs), were similar) or 3) synaptic density (S3–4). Differences in up-state amplitudes did not translate to differences in the induction threshold or maintenance of up-states: no differences were observed in the number, density or dendritic location of spines required to evoke up-states, or the duration of up-states once evoked (Fig 1G, H; S2).
To predict how these intrinsic neuronal differences may impact compartment-specific striatal output we created data-derived computational models of striosome and matrix dSPNs. Detailed biophysical models were built from reconstructions of dSPNs (selected to capture the relative differences in dendritic length reported in S1), endowed with axospinous synaptic inputs and ion channels known to be integral to SPN function, and parametrically tuned using an iterative random selection process to match experimentally generated data (Fig 2A; S5) (Dorman et al., 2018; Jedrzejewski-Szmek et al., 2018). Corroborating our experimental results, simulated somatic up-state membrane potentials were significantly more depolarized in striosome vs matrix dSPNs (S5B, C). Further corroborating experimental results, a random forest regression revealed that the main determinants of up-state amplitude were voltage-gaged ion channels, not synaptic AMPA or NMDA receptors (S5E). Dynamic estimates of rheobase current demonstrated that membrane potential mapped directly onto excitability (Fig 2B, C), suggesting that ongoing excitatory synaptic drive will more readily evoke spiking during up-states in striosome vs matrix dSPNs. Indeed, applying randomly distributed synaptic inputs during simulated up-states showed this to be the case (Fig 2D, E). Moreover, these simulations suggest that striosome and matrix dSPNs respond differently to varying levels of ongoing afferent activity: lower frequencies of synaptic drive preferentially increased the firing rate of striosome dSPNs during up-states.
Figure 2. Biologically realistic computational models predict dSPN-intrinsic differences in compartment-specific output activity.

(A) Reconstructions of SPNs used to create striosome and matrix dSPN models.
(B) Box plots showing dynamic measures of rheobase currents during simulated down- and up- states, determined by 100 ms somatic current injection.
(C) Box plots showing firing rate induced by indicated current injections (100 ms) during simulated up-states.
(D) Simulations showing somatic responses to either dispersed excitatory synaptic inputs alone (at the indicated frequency) or dispersed excitatory synaptic inputs during a dendritically-evoked up-state.
(E) Box plots showing the change in firing rate caused by the addition of a dendritically-evoked up-state during dispersed excitatory synaptic inputs at indicated frequencies (timing same as in D). ns = not significant, *** p<0.001. See also Figure S5 and Tables S1 and S2.
While cell-intrinsic differences lay the groundwork for similar convergent synaptic stimulation to favor striosomal output, it cannot by itself explain bi-directional and task-dependent differences in compartment-specific activity. Both striatal circuit function and the behaviors it encodes are shaped by dopamine (Gerfen and Surmeier, 2011; Kravitz and Kreitzer, 2012; Surmeier et al., 2010). Furthermore, D1R activation enhances both neuronal excitability and the duration of dendritically-evoked up-states in uncategorized dSPNs (Lahiri and Bevan, 2020; Plotkin et al., 2011). Might elevations in dopamine bias the engagement of dSPNs in a compartment-specific manner? To test this, we pharmacologically activated D1Rs and determined the effect on dendritically-evoked up-states in striosome and matrix dSPNs. Strikingly, application of the selective D1R agonist dihydrexidine hydrochloride (DHX, 10 μM) had opposite effects on the maintenance of up-states, increasing up-state duration in matrix dSPNs (~30% median increase) and decreasing it in striosome dSPNs (~12% median decrease) (Fig 3A, B). This effect was blocked by pre-application of the selective D1R antagonist SCH 23390 hydrochloride (3μm) in both matrix and striosome dSPNs (S6A, B). Somatic up-state amplitudes were unaffected by DHX in matrix dSPNs (N=9, pre-drug median = −69.22 mV, post-drug median = −68.41 mV, W = −5, p = 0.8203) but were slightly reduced in striosome dSPNs (N=13, pre-drug median = −64.38 mV, post-drug median = −66.59 mV, W = −67, p = 0.0171). This alone cannot account for the DHX-induced shortening of up-states in striosome dSPNs, however, as there was not a significant correlation between changes in up-state amplitude and duration in these neurons (r=0.2207, p=0.4686).
Figure 3. D1R activation oppositely modulates dSPN up-state durations in matrix vs striosomes.
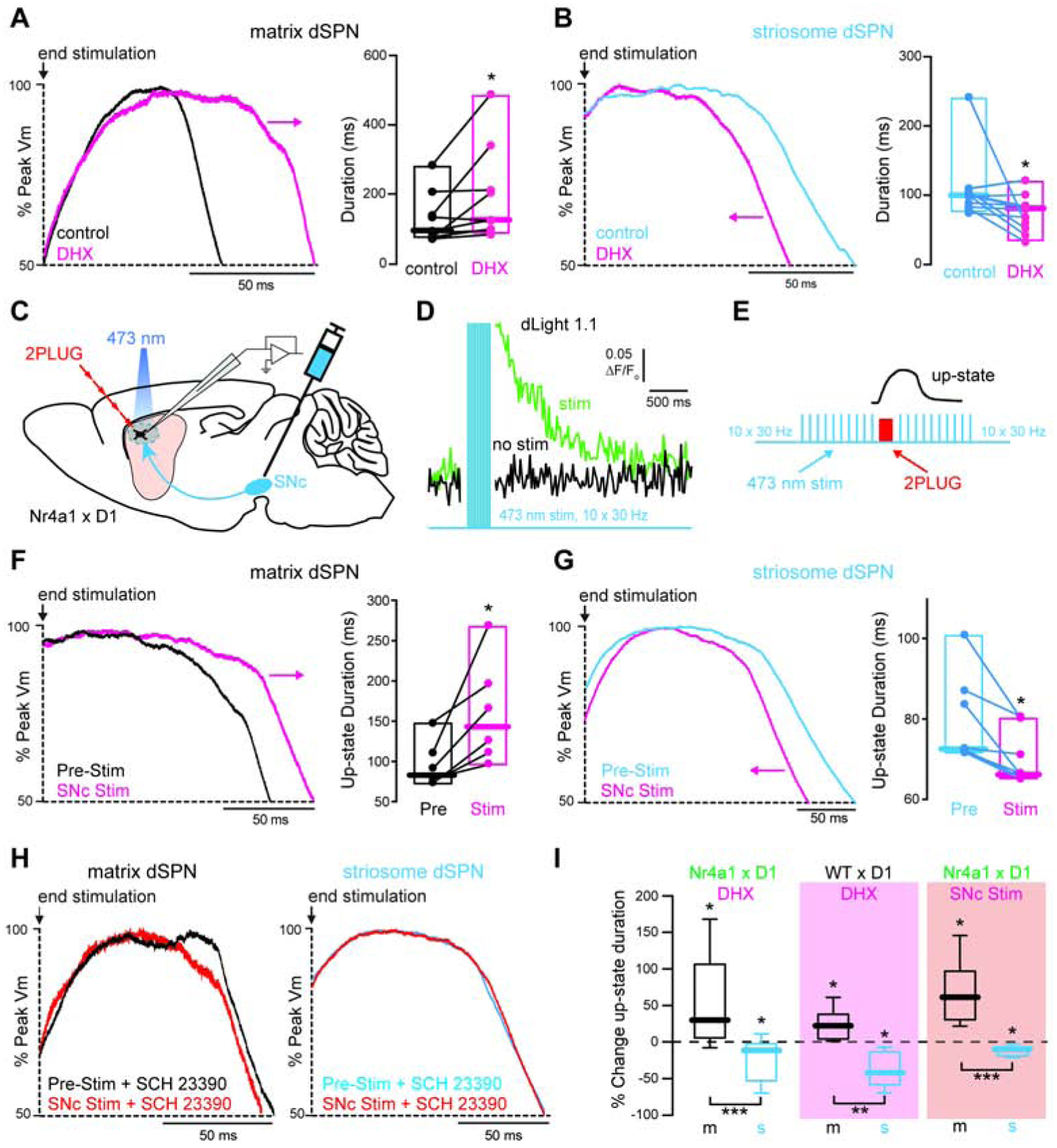
(A-B) Somatic up-state traces induced in (A) a matrix and (B) a striosome dSPN before and after application of the D1R agonist DHX. Arrows indicate direction of change in duration caused by DHX. Box plots show somatic up-state durations before and after DHX.
(C) Schematic of experimental design used in F-G.
(D) Line scan measuring changes in fluorescence of the genetically encoded dopamine sensor dLight 1.1 in dorsal striatum neuropil.
(E) Schematic of stimulation protocol used to coordinate 2PLUG-evoked up-states with endogenous dopamine release. Example somatic up-state from 1D shown above to illustrate timing.
(F-G) Somatic up-state traces induced in (F) a matrix and (G) a striosome dSPN before and after optogenetic stimulation of nigrostriatal dopaminergic (DAergic) fibers. Arrows indicate direction of change in duration caused by D1R activation. Box plots show somatic up-state durations before and in response to optogenetic stimulation.
(H) Somatic up-state traces in the presence of the D1R antagonist SCH 23390, before and in response to optogenetic simulation of nigrostriatal DAergic fibers.
(I) Box plots showing the percent change in somatic up-state duration caused by pharmacological activation of D1Rs in Nr4a1-eGFP × Drd1-tdTomato mice (from A-B) and WT × Drd1-tdTomato mice (from Fig S6D, E), or by transient optogenetic stimulation of DAergic fibers (from F-G). *p<0.05, **p<0.01, ***p<0.001. See also Figure S6 and Table S2.
Because transgenic expression of GFP has been linked to alterations in SPN electrical properties and responsiveness to dopamine (Chan et al., 2012; Cirnaru et al., 2019; Crittenden et al., 2014), we examined compartment-specific dopaminergic modulation in an alternate mouse model that does not rely on striosome- or matrix- specific transgenic markers. Compartments were visualized by unilaterally injecting an adeno-associated virus (AAV) expressing ChR2-EYFP into the left anterior cingulate cortex (ACC) of drd1-tdTomato mice. This produces robust axonal labeling in the ipsilateral striatum that largely avoids striosomes (Friedman et al., 2015). The location of each recording was later affirmed by post-hoc mu opioid receptor staining of recovered brain slices (S6C). Confirming our findings in Nr4a1-eGFP mice, we observed that DHX (10 μM) caused a ~22% median increase in up-state duration in matrix dSPNs and a ~41% median decrease in striosome dSPNs (S6D, E).
The data above show that prolonged pharmacological activation of D1Rs oppositely modulates the duration of synaptically-evoked up-states in striosome vs matrix dSPNs. The obvious question that arises is whether or not transient release of endogenous dopamine from substantia nigra pars compacta (SNc) neuron axons produces the same effect. To address this, we prepared acute brain slices from mice that received bilateral injections of an AAV expressing ChR2-mCherry into the lateral SNc. Local optogenetic stimulation of SNc axon fibers was optimized to evoke phasic elevations of dopamine that began just prior to and lasted during synaptically-evoked up-states (Fig 3C–E). Consistent with pharmacological experiments, phasic release of endogenous dopamine significantly increased the duration of synaptically-evoked up-states in matrix dSPNs (~61% median increase) and decreased the duration in striosome dSPNs (~9% median decrease) (Fig 3F, G). The effect of optogenetically evoked dopamine release on up-state duration was blocked by the D1R antagonist SCH 23390 in both neuron types (Fig 3H). Together these data demonstrate that D1R activation oppositely modulates state-transition durations in matrix and striosome dSPNs, and that this mechanism is 1) not due to the compartment labeling strategy used and 2) engaged by transient release of endogenous dopamine (Fig 3I)
How does activation of D1Rs produce opposite effects in dSPNs residing in different compartments? As these experiments were conducted in the presence of tetrodotoxin (1 μM) to prevent axonal propagation of presynaptic activity, the mechanism is likely postsynaptic and intrinsic to the SPNs themselves. SPN dendritic plateau potentials, and resulting up-states, are NMDA receptor-dependent (Plotkin et al., 2011). In presumptive matrix dSPNs, activation of D1Rs positively modulates NMDA receptor function (Cepeda et al., 1998), promoting an increase in up-state duration (Plotkin et al., 2011). D1R modulation of NMDA receptors in SPNs requires the activation of L-type voltage-gated Ca2+ channels (L-VGCCs) (Liu et al., 2004), which are enriched in SPN dendrites (Carter and Sabatini, 2004; Plotkin et al., 2013) but not required for the generation or maintenance of up-states under basal conditions (Plotkin et al., 2011). Could the failure of D1R activation to prolong up-state duration in striosome dSPNs be due to lower engagement of dendritic L-VGCCs? Two obvious mechanisms could lead to such a scenario: 1) basal engagement of L-VGCCs is lower or 2) D1R-enhancement of L-VGCC engagement is impaired. We tested the first hypothesis by comparing depolarization-evoked dendritic Ca2+ transients in the two compartments. Somatically-delivered depolarizing voltage steps evoked similar Ca2+ transients in distal dendritic spines of striosome and matrix dSPNs (Fig 4A, B). The contribution of L-VGCCs to these Ca2+ transients, however, was nearly 50% less in striosome dSPNs, as determined by application of the L-VGCC blocker isradipine (5 μM) (Fig 4A, C). Depolarization-evoked Ca2+ influx into SPN dendrites is mediated by a combination of L-, T- and R- type VGCCs (Carter and Sabatini, 2004). To determine if D1R modulation of L-VGCCs is impaired in striosome dSPNs we isolated the L-VGCC- mediated component of spinous Ca2+ transients by delivering a cocktail of ML 218 hydrochloride (1 μM) and SNX 482 (0.3 μM) to block T- and R- type VGCCs, respectively (Carter and Sabatini, 2004; Xiang et al., 2011). Confirming the results of the isradipine blockade experiments, the remaining L-VGCC-mediated component was significantly lower in striosome dSPNs (Fig 4D). Application of DHX enhanced L-VGCC- mediated Ca2+ transients similarly in both neuron populations (matrix median increase = 13.9%; striosome median increase = 12.5%), indicating that the signaling linkage between D1Rs and L-VGCCs is intact in striosome dSPNs (Fig 4E). Despite this, Ca2+ influx through L-VGCCs remained lower in striosome vs matrix dSPNs even in the presence of the D1R agonist. In fact, D1R-enhanced L-VGCC Ca2+ transients in striosome dSPNs were still smaller than unmodulated transients in matrix dSPNs (Fig 4E).
Figure 4. Bidirectional D1R-modulation of striosome and matrix dSPN somatic up-state duration is sensitive to L-VGCC engagement.

(A) Average distal dendritic spine Ca2+ transients (shaded areas are SEM) in the presence or absence of the L-VGCC channel antagonist isradipine. 2P-MIPs showing location of line scan (yellow) to the left (scale bars = 2 μm, 500 ms, 0.4 ΔG/Gsat; voltage step is to 0 mV).
(B) Boxplots showing depolarization-evoked Ca2+ transient amplitudes in distal dendritic spines.
(C) Boxplots showing the percent block of distal dendritic spine Ca2+ transients by isradipine.
(D) Boxplots showing distal dendritic spine Ca2+ transient amplitudes in the presence of the T- and R-type VGCC antagonists ML 218 and SNX 482.
(E) Boxplots showing distal dendritic spine Ca2+ transient amplitudes in the presence of ML 218 and SNX 482 before and after application of DHX. Even after application of DHX, Ca2 transients in striosomes remained lower than pre-DHX transients in the matrix.
(F-G) Somatic up-state traces induced in a matrix dSPN in the presence of isradipine (F) and a striosome dSPN in the presence of the L-VGCC agonist Bay K 8644 (G), before and after application of DHX. Box plots show somatic up-state durations.
(H-I) Somatic up-state traces induced in a matrix dSPN in the presence of Bay K 8644 (H) and a striosome dSPN in the presence of isradipine (I), before and after application of DHX. Box plots show somatic up-state durations.
(J) Box plots showing the percent change in somatic up-state duration induced by DHX in matrix and striosome dSPNs under control conditions (from Fig 3A-B), in the presence of isradipine and in the presence of Bay K 8644. *p < 0.05, **p < 0.01; ***p < 0.001. See also Table S2.
If the opposing effects of D1R activation on up-state duration are due to differences in dendritic L-VGCCs, then reversing compartmental differences in L-VGCC engagement should be able to reverse the compartment-specific responses to dopamine. Indeed, this was the case. Increasing L-VGCC activation with the L-VGCC agonist Bay K 8644 (1 μM) caused DHX to increase up-state duration in striosome dSPNs (14% median increase) and blocking L-VGCC activation with isradipine caused DHX to decrease up-state duration in matrix dSPNs (47.6% median decrease), essentially flipping the compartmental phenotypes (Fig 4F, G). Accentuating compartmental differences in L-VGCC engagement, on the other hand, further altered the responses to D1R activation: blocking L-VGCC activation with isradipine eliminated the effect of DHX on up-state duration in striosome dSPNs and increasing L-VGCC activation with Bay K 8644 caused DHX to decrease up-state duration in matrix dSPNs (16% median decrease) (Fig 4H, I). Together, these data suggest that not only is L-VGCC engagement an important determinant of compartment-specific responses to D1R activation, but graded engagement of L-VGCCs does not shape the response to dopamine in a purely linear way (Fig 4J). These data also demonstrate that while both populations of dSPNs possess the requisite machinery for dopamine to suppress up-state maintenance, the mechanism is likely compartment-specific, normally masked by L-VGCCs in matrix dSPNs, and only L-VGCC-dependent in striosome dSPNs.
Discussion
Our results describe a novel framework that resolves how regulation of compartment-specific striatal output may be achieved, independent of the identity or overlap of excitatory afferents. Our data suggest that under basal conditions dSPNs in striosome and matrix compartments integrate convergent excitatory synaptic inputs similarly, though ensuing somatic up-states reside closer to spike threshold in striosome dSPNs. Elevations in dopamine will reduce the time window during which direct pathway striosome SPNs can fire action potentials, while prolonging the time window in matrix dSPNs, biasing ongoing striatal output away from striosomes and towards the matrix. While the full complexity of the underlying mechanism must still be unraveled, the engagement of L-VGCCs plays a key role in determining the opposing responses to dopamine, offering a molecular target through which this process can be manipulated and tuned.
The present study demonstrates that the “associative capacity” (ability to generate and support dendritically-evoked up-states in response to coincident synaptic activation) of dSPNs is independent of the compartment in which they reside. This is consistent with our observations that the density and relative glutamate receptor contributions of dSPN dendritic spines are similar across compartments. Though the associative capacity of individual dendrites may be similar, our data imply that the frequency of dendritic plateaus and total resulting spikes per neuron may differ between compartments under physiological conditions. This is because 1) while proximal dendritic length and arborization are similar between dSPN types, the total real-estate of the dendritic tree that is capable of supporting synaptically-driven plateaus is approximately 55% less in striosomes (total measurable dendritic length >90 μm from soma: 143.4 μm, median, matrix; 79.7 μm, median, striosomes; see S1I–L), 2) while up-state durations are similar between dSPN types, up-state amplitudes reside closer to spike threshold in striosomes, increasing the likelihood of spike generation triggered by synaptic inputs arriving during the up-state (Plotkin et al., 2011; Stern et al., 1998) and 3) simulations in biologically realistic computational models revealed that lower frequencies of ongoing excitatory synaptic inputs are required to drive spiking during up-states in striosome vs matrix dSPNs. This sets a scenario where, under basal conditions, dSPNs in striosomes may display fewer dendritic plateaus (on a whole cell basis) but track ongoing cortical and thalamic activity with higher fidelity during each plateau.
Activation of D1Rs had opposite effects on up-state duration in striosome and matrix dSPNs. Since 1) D1R activation enhances dendritic NMDA receptor function and up-state duration in presumptive matrix dSPNs (Higley and Sabatini, 2010; Plotkin et al., 2011), 2) this enhancement requires Ca2+ entry through L-VGCCs (Cepeda et al., 1998; Liu et al., 2004) and 3) dendritic L-VGCC engagement is lower in striosome dSPNs, one would predict that D1R activation would simply have no effect on up-state duration when the requisite signaling elements for NMDA receptor enhancement are absent. So how does D1R activation decrease up-state duration in striosome dSPNs and in matrix dSPNs when L-VGCCs are blocked? Our data suggest that under permissive conditions D1R activation can decrease up-state duration in both neuron types, though the underlying mechanisms likely differ (requiring low engagement of L-VGCCs in striosome dSPNs and a L-VGCC-independent mechanism in matrix dSPNs). While dissecting the mechanisms is beyond the scope of this study, several possibilities may explain the reducing effect of D1Rs that is unmasked in matrix dSPNs when L-VGCCs are blocked. One potential mechanism involves modulation of other VGCCs. Inhibition of T-Type VGCCs reduces up-state duration in presumptive matrix dSPNs (Plotkin et al., 2011). Dopaminergic modulation of NMDA receptors in SPNs requires activation of PKA (Higley and Sabatini, 2010; Snyder et al., 1998). PKA activation can suppress T-type VGCC conductance in heterologous systems and isolated smooth muscle cells (Harraz and Welsh, 2013; Hu et al., 2009), though such modulation in SPNs remains to be determined. A second possibility is that D1R-induced PKA activation leads to the enhancement of a tonic GABA current (Janssen et al., 2009) that may attenuate the generation of dendritic plateau potentials (Du et al., 2017). Compartmental differences in GABAergic signaling have been reported (Banghart et al., 2015; Friedman et al., 2017), but as the MNI caging compound used in this study diminishes GABAergic signaling (Palma-Cerda et al., 2012), the impact that such a mechanism may have under our experimental conditions remains to be determined.
Activation of L-VGCCs was required for both the enhancing and reducing effects of D1R activation in striosome dSPNs, and the degree of L-VGCC activation determined the direction of the dopaminergic effect. Somewhat surprisingly, pharmacologically enhancing the already higher L-VGCC engagement in matrix dSPNs caused D1R activation to diminish up-state duration, rather than further augment it. Together this suggests that there is a nonlinear or “inverted U” relationship between L-VGCC engagement and response to D1R activation in both dSPN types. Such nonlinear effects of Ca2+ have been well described in numerous synaptic and behavioral phenomena and are typically attributed to Ca2+ concentration-dependent activation of phosphatases vs kinases (Dash et al., 2007; Lisman, 2001). But why does further enhancing L-VGCC engagement in matrix dSPNs reverse the effect of dopamine? Though the precise mechanism and physiological relevance of this remain to be determined, one possibility is that the boosted level of intracellular Ca2+ achieved in matrix dSPNs by Bay K 8644 application overwhelms spatial restrictions that normally confine the engagement of Ca2+-activated hyperpolarizing conductances (Bloodgood and Sabatini, 2007).
Manipulating L-VGCC engagement is sufficient to determine the directional effects of dopamine on dSPN up-state duration. That said, there are myriad targets of D1R signaling in SPNs that may also be involved (Surmeier et al., 2009). Taking advantage of our biologically realistic SPN models we used a random forest regression, which identifies what features are most important for predicting the dependent variable, to search for other possible candidates that may regulate up-state duration (S5F). Simulations revealed 3 potential targets: NMDA receptors, R-type VGCCs and Kv1.2 potassium channels. NMDA receptors and R-type VGCCs are already known to control SPN dendritic plateau duration (though modulation of R-type VGCCs by D1R signaling in SPNs is unclear) (Plotkin et al., 2011). Interestingly, D1R modulation of Kv1.2 has recently been shown to enhance the excitability of dSPNs during up-state transitions (Lahiri and Bevan, 2020) - our simulations suggest that dopaminergic modulation of this channel may also regulate the duration of up-states, though whether this contributes to compartmental differences remains to be determined.
Previous studies have shown that striosome dSPNs send direct axonal projections to dopaminergic neurons of the SNc, as well as other basal ganglia nuclei (Crittenden and Graybiel, 2011; Fujiyama et al., 2011). Though matrix dSPN axons may share a grossly similar trajectory (Smith et al., 2016), recent studies have demonstrated that striosome-originating SPN axons form tightly woven integrative units encapsulating dopaminergic neurons within the SNc, which in turn send dopaminergic projections throughout the dorsal striatum (Crittenden et al., 2016; Davis et al., 2018). As dSPN-SNc synapses are GABAergic (Brazhnik et al., 2008), it’s tempting to speculate that such projections may serve as a homeostat to keep striatal dopamine levels in check (Crittenden et al., 2016). If this is the case, our data suggest that elevations in striatal dopamine would decrease the duration of striosomal inhibition of SNc dopaminergic neurons, ultimately shunting such a feedback mechanism. This has fundamental implications for understanding how fluctuations in dopamine modulate basal ganglia network activity during goal-directed and habit learning (Darvas et al., 2014; Leventhal et al., 2014; Palmiter, 2008; Pennartz et al., 2009; Yin et al., 2004).
Overall, our data demonstrate that dopaminergic signaling through D1Rs can modulate the balance of compartment-specific striatal output. The opposing effects of D1R activation on synaptic integration in striosome and matrix dSPNs are 1) due, at least in part, to intrinsic differences in the compliment of dendritic VGCCs and 2) not dependent upon the presynaptic identity of converging axo-spinous inputs. As previous studies have suggested that up-state generation and action potential initiation are regulated by separate populations of synaptic inputs (Plotkin et al., 2011; Stern et al., 1998), dopamine may serve to act as a filter which temporally constrains the synaptic cues that can evoke dSPN spiking in striosomes following an associative event, while loosening this constraint in the matrix. These findings suggest that the gating of compartment-specific striatal output, a phenomenon now appreciated to be crucial for task-dependent behaviors (Bloem et al., 2017; Friedman et al., 2017; Friedman et al., 2015), represents an important parameter in the study of dopamine-dependent behavior and pathology involving the basal ganglia.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents may be directed to and will be fulfilled by Lead Contact, Dr. Joshua Plotkin, Joshua.plotkin@stonybrook.edu.
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Detailed datasets supporting the current study are available from the corresponding author on request. Source code is available in ModelDB at http://modeldb.yale.edu/266807.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All experimental procedures were performed in accordance with the United States Public Health Service Guide for Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, National Research Council), were approved by the Institutional Animal Care and Use Committee at Stony Brook University and conformed with the ARRIVE guidelines. Animals (n = 79; 33 males, 46 females) were housed 2–5 per cage (1–2 per cage if animals underwent surgical procedures) in an environmentally controlled room (20–23 °C, 30–70% humidity, 12 h light/dark cycles [350–400 lux]) with food and water available ad libitum. Cages were cleaned once a week and animal handling was minimized to reduce additional animal stress (Prager et al., 2011). No physical enrichment was provided. Experiments were conducted in 1.5–3-month-old male and female double-transgenic mice created by crossing Tg(Nr4a1-eGFP139GSAT) (Davis and Puhl, 2011) and Drd1a-tdTomato BAC (Ade et al., 2011; Shuen et al., 2008) transgenic mouse lines. Nr4a1-eGFP mice were visually genotyped at weaning (~postnatal day 21) with a blue LED and GFP filter-equipped goggles (Electron Microscopy Sciences) (Salinas et al., 2016) and Drd1a-tdTomato mice genotyped as previously described (Shuen et al., 2008). Randomization and blinding were not applicable to this study. Researchers attempted to experiment from both striosome and matrix dSPNs in each animal, when possible.
METHOD DETAILS
Surgical Procedures
Male and female mice were induced and maintained under anesthesia via inhalation of vaporized isoflurane (3% for induction and 1–2% for maintenance during surgery). After complete sedation was confirmed by tail pinch, animals were placed in a stereotaxic apparatus, the scalp was shaved and disinfected and eyes were covered with eye-protective gel (Puralube® Ophthalmic Ointment, Dechra Veterinary Products). Identical custom 1 μl Hamilton micro-volume syringes with a 50° bevel point (Hamilton, ref 65458–1) were used for viral injections targeting the Substantia nigra pars compacta (SNc; ±1.49 mm ML, −3.08 mm AP, and −4.11 mm DV), dorsal striatum (±2.40 mm ML, 0.50 mm AP, and −3.06 mm DV) or anterior cingulate cortex (ACC; +0.5 mm AP, +0.28 mm ML, −1.8 mm DV) and manipulated via a motorized stereotaxic injector (Quintessential Stereotaxic Injector QSI™, Stoelting Co., ref 53311) and Angle Two software (Leica Biosystems). 200 nL pAAV.CAG.hChR2(H134R)-mCherry.WPRE.SV40 (titer of 1×10−13 vg/ml) (sourced from Karl Deisseroth; Addgene viral prep # 100054-AAV5; http://n2t.net/addgene:100054; RRID: Addgene_100054) was slowly injected into the SNc bilaterally over 5 minutes; an additional 5 minutes were allowed for diffusion before slowly withdrawing the needle over 5 minutes. To qualitatively measure optogenetically-evoked dopamine release, wild type C57BL/6J mice received an additional injection of pAAV.CAG.dLight1.1 (200 nL at titer ≥7×1012 vg/ml) (sourced from Lin Tian; Addgene viral prep # 111067-AAV5; http://n2t.net/addgene:111067; RRID: Addgene_111067) into the dorsal striatum. To fluorescently label the matrix in WT × Drd1a-tdTomato BAC mice, 200nL of pAAV-hSyn-hChR2(H134R)-EYFP (titer: 1×1013 vg/mL) (Addgene, ref 26973-AAV9, RRID: Addgene_26973) was injected unilaterally into the left ACC. Throughout all surgical procedures and during recovery immediately after the procedure, animals were kept on a heating pad to stabilize body temperature at 37 °C. Animals were administered a postoperative analgesic, meloxicam (5mg/kg), via subcutaneous injection at the conclusion of the procedure and once daily thereafter as needed for 7 days. Animals receiving SNc viral injections were sacrificed 2–3 weeks following the surgical procedures; animals receiving ACC injections were sacrificed 10–14 days after surgical procedures. Cells from ACC injected animals were included in the final analysis only if tissue could be successfully processed for μ-opioid receptor staining post hoc to confirm electrode placement.
Electrophysiological Recordings
Ex vivo slice recordings.
Para-sagittal brain slices (275 μm) containing the dorsolateral striatum were obtained, as previously described (Plotkin et al., 2011). Briefly, mice were anesthetized with a mixture of ketamine (50mg/kg) and xylazine (4.5mg/kg), transcardially perfused with ice-cold artificial cerebral spinal fluid (ACSF) containing (in mM): 124 NaCl, 3 KCl, 1 CaCl2, 1.5 MgCl2 26 NaHCO3, 1 NaH2PO4 and 14 glucose and continuously bubbled with carbogen (95% O2 and 5% CO2). Slices were cut using a VT-1000s vibratome (Leica Microsystems, Buffalo Grove, IL) and transferred to a holding chamber where they were incubated at 32°C for 45 min in ACSF containing (in mM) 2 CaCl2 and 1 MgCl2, after which they were stored at room temperature (~21 °C) until recording (Plotkin et al., 2011).
Patch electrodes (3.5–5 MΩ) were pulled from thick-walled borosilicate glass using a Sutter P-1000 puller (Sutter Instruments, Novato, CA). Current clamp experiments were performed using an internal recording solution containing (in mM): 135 KMeSO4, 5 KCl, 10 HEPES, 2 Mg2+-ATP, 0.5 Na+-GTP, 5 Tris-phosphocreatine; 5 sodium phosphocreatine, 0.05 Alexa Fluor 568 hydrazide sodium salt (Invitrogen), and 0.2 Fluo-4 pentapotassium salt (Invitrogen), pH 7.25, 270–280 mOsm. Voltage clamp experiments were performed using an internal recording solution containing (in mM): 120 CsMeSO3, 5 NaCl, 10 TEA-Cl (tetraethylammonium-Cl), 10 HEPES, 5 Qx-314, 4 Mg2+-ATP, 0.3 Na+-GTP, 0.25 EGTA, and 0.05 Alexa Fluor 568 hydrazide sodium salt (Invitrogen), pH 7.25, 270–280 mOsm. For Ca2+ imaging experiments EGTA was omitted and replaced with 0.3 mM Fluo-5F pentapotassium salt (Invitrogen). Alexa 568 fluorescence was used to visualize cell bodies, dendrites and spines (Plotkin et al., 2011). Slices were transferred to a submersion-style recording chamber (Scientifica, Uckfield, UK), and electrophysiological recordings were made using a Multiclamp 700B amplifier (Molecular Devices, Jan Jose, CA), digitally sampled at 30 kHz and filtered at 1 KHz.
Following patch rupture, the internal solution was allowed to equilibrate for 10–15 min before imaging and the commencement of experimental procedures. In voltage-clamp mode (Vhold = −70mV), a single 20 ms, 5 mV depolarizing step was evoked to determine passive membrane properties. In current-clamp mode, rheobase was determined from 25 pA increments of depolarizing current injections (500 ms each, from −200 pA). All experiments except for those measuring membrane properties and optogentically evoked up-states were conducted in the presence of 1 μM tetrodotoxin citrate (TTX). mEPSCs were recorded for 120 s while voltage-clamped at −80 mV and mEPSPs were recorded for 360 s in current-clamp mode at Vrest. Access resistance was monitored regularly during recordings and cells were rejected if the resistance changed by more than 15% during the experiment. All recordings were performed at room temperature (21 ± 1°C) unless otherwise noted. Stimulation, acquisition and display were accomplished using Prairie View v5 (Bruker Nano, Inc., Middleton, WI).
Two-photon laser scanning microscopy.
Matrix/striosome compartments and dSPNs were identified by somatic enhanced-GFP and tdTomato two-photon-excited fluorescence using an Ultima Laser Scanning Microscope System (Bruker Nano, Inc., Middleton, WI). A Universal Illumination System with Transmitted Detector (Bruker Nano, Inc., Middleton, WI) was used to provide a bright-field transmission image in registration with the fluorescent images. Green and red signals were acquired using 810 nm pulsed light (90 MHz) excitation (Chameleon Ultra II, Coherent, Inc., Santa Clara, CA), unless otherwise noted. SPNs were patched under bright-field transmission using a high-sensitivity CMOS camera (Thor Labs, Newton, NJ) and a 60x/1.0 N.A. Olympus LUMPFL water-dipping lens.
Ca2+ imaging.
Dendritic Ca2+ transients were measured using Fluo-5F, as previously described (Shen et al., 2015). SPNs were voltage clamped, and dendritic VGCCs activated by a 300 ms depolarizing somatic voltage step from −80 to 0 mV. Green and red fluorescent line scan signals were acquired from distal dendritic spines at 5.342 ms and 512 pixels per line with 0.0776 μm2 pixels and 10 μs pixel dwell time. Ca2+ transients were expressed as ΔG/GSat to allow inter-cell comparisons (Bloodgood and Sabatini, 2007). Line scans started 500 ms before the stimulation protocol and continued ~1.5 s after the stimulation pulse ended, to obtain the baseline fluorescence and the decay of the optical signal after stimulation.
Two-photon laser uncaging.
2-photon laser uncaging of MNI-glutamate (2PLUG) was achieved using a second Chameleon-Ultra II laser tuned to 720nm, as previously described (Plotkin et al., 2011; Plotkin et al., 2013; Plotkin and Surmeier, 2014). 5 mM MNI-glutamate (Tocris Bioscience, Bristol, UK) dissolved in HEPES-based ACSF (containing (in mM): 124 NaCl, 3 KCl, 2 CaCl2, 1 MgCl2, 26 NaHCO3, 1 NaH2PO4, 26 HEPES and 14 glucose) was superfused over the slice using a perfusion syringe pump (World Precision Instruments, Sarasota, FL) and a multibarreled perfusion manifold (Cell MicroControls, Norfolk, VA) at a rate of 0.4ml/h. MNI-glutamate was uncaged using 1 ms pulses of 720 nm light (typically 10–20 mW at the sample plane, controlled by a dedicated Pockels cell modulator).
NMDA/AMPA receptor ratio measurements.
MNI-glutamate was uncaged adjacent to a single dendritic spine (1 ms duration) and uncaging laser power tuned to produce an EPSC of 10–20 pA from a holding potential of −80 mV. The holding potential was then changed to +40 mV and the same spine stimulated again. NMDA/AMPA ratios were calculated as previously described (Plotkin et al., 2014). Briefly, the AMPA receptor-mediated current component was measured as the peak inward current evoked at −80 mV (averaged from 3 trials), and the NMDA receptor-mediated component measured 50 ms after the peak outward current recorded at +40 mV (averaged from 3 trials). Unresponsive spines were excluded from analysis. A total of 3–4 distal spines (>100 μm from soma, measured by a straight line from the stimulation site to the origin of the dendrite at the soma) were measured per neuron.
Induction of dendritic plateau potentials (up-states).
Dendritic plateau potentials were evoked as previously described (Plotkin et al., 2011). Briefly, dSPNs were patch clamped in current clamp mode in the presence of 1 μM TTX (unless otherwise noted), and uncaging laser power was tuned to produce a somatic EPSP of 1–1.5 mV in response to 2PLUG adjacent to a single distal (>100 μm from soma) dendritic spine. Dendritic plateaus (and corresponding somatic up-states) were evoked by stimulating 10–18 neighboring distal dendritic spines in rapid succession (500 Hz), with this stimulation pattern repeated 1–4 times. Up-state duration was quantified as the time between the last uncaging stimulus and the return of the somatic membrane potential to 25% of the peak up-state amplitude. All cells included in the final analyses had calculated up-state durations of at least 70 ms, a duration above the empirically determined “break-point” between subthreshold and supralinear synaptic responses in both striosome and matrix dSPNs (Supplementary Fig. 2A–B). All pharmacological comparisons were performed before and after 10 min bath application.
Optogenetic release of endogenous dopamine.
Acute brain slices were prepared from Nr4a1-eGFP × Drd1a-tdTomato mice that received bilateral injections of AAV5-ChR2-mCherry into the SNc as described above. dSPNs were patch clamped and maintained at ~32 ± 1 °C, and a 473 nm solid state laser was used to locally stimulate nigrostriatal axons along the distal dendritic segment at which a 2PLUG-evoked plateau potential was previously induced. Optogenetic stimulus parameters were based on Yagishita et al. (Yagishita et al., 2014). Transient dopamine release was evoked immediately before and during a plateau potential: 10 sequential 473 nm laser stimuli (1 ms duration, 30 Hz, ~3 mW at sample plane) were delivered along the dendrite segment, the plateau-evoking 2PLUG stimuli initiated 33 ms after the last 473 laser stimulus, and the 10 sequential 473 nm laser stimuli delivered again beginning 33 ms after the final 2PLUG stimulus (Fig. 3E). Experiments were performed in the absence of TTX. Release of endogenous dopamine was confirmed using the genetically encoded dopamine sensor dLight1.1 (Patriarchi et al., 2018). Acute brain slices were prepared from WT mice receiving bilateral injections of AAV5-ChR2-mCherry into the SNc and AAV5-dLight1.1 into the dorsal striatum. Spiral line scans (920 nm, 21.2 ms with 0.0776 μm2 pixels and 10 μs pixel dwell) were used to measure changes in green fluorescence in dorsolateral striatum neuropil directly surrounding 10 sequential 473 nm laser stimuli (1 ms duration, 30 Hz, ~3 mW at sample plane). Line scans began 2000 ms before stimuli and continued for 5600 ms after the stimuli ended. A high-speed shutter was used to block signals to the green photomultiplier tube during optogenetic stimulation.
Image acquisition, reconstruction and analysis
At the conclusion of each experiment, whole cell z-series containing the soma and dendrites were acquired with 0.3662 μm2 pixels and 10-μs pixel dwell time; ~60–80 images were taken with 1 μm focal steps following each experiment. High magnification z-series of dendrites were acquired with 0.0776 μm2 pixels and 10 μs pixel dwell time; ~50–60 images were taken with 0.2 μm focal steps. Z-series were rendered as maximum intensity projections using PicViewer software (John Dempster, Strathclyde University). After recording, the location of the recorded cell was confirmed to be in a striosome or matrix compartment using a low-magnification (4X) objective.
Z-series images were deconvolved using a 60-iteration adaptive blind deconvolution algorithm with AutoQuant X software (Media Cybernetics, Rockville, MD). Neurons were semi-automatically reconstructed from whole cell z-series images using NeuronStudio (RRID:SCR_013798) (Rodriguez et al., 2008). All traces were manually inspected, including verifying that neurites trace back to the soma and reflect correct branching patterns. Sholl analysis was then performed to determine the number of dendritic crosses at concentric circles around the soma (intersections), cumulative dendritic length and branch points (Gertler et al., 2008). Data generated by NeuronStudio were confirmed by repeating Sholl analysis using ImageJ (RRID:SCR_003070). Spine density measurements were semi-automatically calculated from high magnification images of distal dendrites using NeuronStudio (Rodriguez et al., 2008). Dendrites were included in analysis only if blebbing was not present. Whole cell images were used only if cells could be fully visualized and the total dendritic length was greater than 650 μm.
µ-opioid receptor immunohistochemistry
Slices from ACC injected mice were washed once in 1x phosphate-buffered saline (PBS), transferred to 4% paraformaldehyde (PFA) for 4 hours, then to 30% sucrose overnight. Slices were re-sectioned from 275μm to 50μm on a cryostat (Leica Biosystems, ref # CM1950). Free-floating sections were washed in 1x PBS and incubated in blocking solution (3% Normal Goat Serum, 0.1% Triton-X100 in 1x PBS) for 2 hours at room temperature. Sections were then incubated in μ-opioid receptor (MOR) primary antibody (1:2000) (Abcam, ref ab10275, RRID: AB_2156356) overnight at 4°C, and goat anti-rabbit IgG H&L secondary antibody coupled to Alexa Fluor 647 (1:1000) (Abcam, ref 150083, RRID AB_2714032) for 2 hours at room temperature. Sections were counterstained with DAPI (Southern Biotech, ref 0100–20). Slide-mounted sections were imaged using a widefield fluorescence microscope (Olympus VS120 Virtual Slide Microscope, ref VS120-S6-W) and processed using ImageJ.
Computational Modeling
Modeled striosome and matrix dSPNs were based on a biophysically detailed SPN model that we previously published (Dorman et al., 2018). Morphologies were modified using two dSPNs from the Luebke repository on neuromorpho.org (ID: NMO_101636, NMO_101639) (Ascoli et al., 2007; Goodliffe et al., 2018). A neuron with shorter total dendritic length (but otherwise similar morphology) was used for the striosome dSPN as compared to the matrix dSPN, matching experimental observations (see Supplementary Fig. 1I–L). Spines were explicitly modeled as a cylindrical head (0.5 μm diameter, 0.5 μm length) and neck (0.12 μm diameter, 0.5 μm length) (Dorman et al., 2018) and were distributed on dendritic branches (greater than 25 μm from the soma) with a density of 1 spine/μm.
The models were endowed with voltage gated ion channels identified in SPNs (Supplementary Table 1), and Ca2+ dynamics were controlled by Ca2+ binding proteins, membrane pumps and radial diffusion in dendrites and spines (Dorman et al., 2018). Synaptic channels consisted of NMDAR and AMPAR channels with parameters consistent with Dorman et al. (2018) and conductances set to produce EPSPs close to 1 mV for a single synaptic input. The model was described using a declarative model specification (github.com/neurord/moose_nerp) and simulated using MOOSE with a timestep of 0.01 ms. Simulations were run on the Neuroscience Gateway Portal supercomputer (Sivagnanam et al., 2013).
Model Parameter Optimization.
Channel kinetic equations and parameters are similar to our previously reported model (Dorman et al., 2018), except the half activation voltages and time constants were allowed to be adjusted by the parameter optimization algorithm. Passive parameters (membrane resistivity, membrane capacitivity, and axial resistivity) and channel conductances were determined by fitting the model to hyperpolarizing and depolarizing current injection data from identified striosome and matrix dSPN electrophysiological traces using the automatic parameter optimization software ajustador (Jedrzejewski-Szmek et al., 2018) (http://github.com/neurord/ajustador).
Model parameter set variations.
To identify channel parameters that could modulate the amplitude or duration of up-states, a generic SPN model was used to generate 60,000 parameter sets by randomly varying channel conductance values for all voltage-gated and synaptic channels. Up-states were simulated for each parameter set as described below. Random forest regression was conducted in Python3.6 using the Scikit-Learn package to identify the channels that most strongly predicted either up-state amplitude or up-state duration (Supplementary Fig. 5E–F).
Populations of striosome and matrix dSPN specific models were randomly generated by using the optimized version of each compartment-specific model and randomly modulating channel conductances for the channels previously predicted to affect up-state amplitude and duration (A-type potassium channels, NMDA and AMPA synaptic channels, L-type, R-type, and T-type Ca2+ channels, and inward rectifying potassium channels). These parameter sets were each simulated with the set of simulations described below.
Model simulations.
Up-states were simulated by stimulating 14 neighboring distal spines twice each at 500 Hz. Rheobase simulations were conducted at rest or during up-states, using 25pA current steps and a 100 ms duration (the shortened duration was selected to be consistent with upstate durations). IV curves were generated for each parameter set to compute the model optimization fitness. For simulations with dispersed synaptic inputs, 100 inputs were randomly selected throughout the dendritic tree (except for the branch receiving clustered upstate-inducing inputs), and activation of dispersed inputs began 50ms after upstate stimulus onset and lasted for 300 ms. Dispersed input frequencies were varied from 0 to 500 Hz. Simulations for randomly varied parameter sets were included for statistical analysis strictly based on whether their optimization fitness value based on IV curves was sufficiently good (less than 2) and whether the parameter set actually produced an up-state. While the models captured experimental data and cell-type similarities and differences well, one parameter that was not fully captured was a lack of difference in plateau duration (Supplementary Fig. 5D). This likely reflects an unknown channel(s) not represented in the models, or randomly varying parameter values in the same ranges differentially affected plateau duration between the models. Comparisons that could be affected by this difference in duration were avoided; measures of spiking were therefore expressed as frequency or change in frequency.
QUANTIFICATION AND STATISTICAL ANALYSIS
Electrophysiological recordings and Ca2+ transients were analyzed using Igor Pro (WaveMetrics, Inc., Lake Oswego, OR; RRID:SCR_000325). Miniature EPSCs and EPSPs were analyzed (from 120 s or 360 s traces, rejected if holding current changed >20%) using the Mini Analysis program (Synaptosoft, Inc., Fort Lee, NJ; RRID:SCR_002184). Population data are reported as medians and presented as box and whisker plots, where boxes outline upper and lower quartiles and whiskers are upper and lower 10% values. Statistical differences were examined using 2-sided Mann-Whitney U, Wilcoxin signed-rank (for paired comparisons) or Kruskal Wallis (for multiple comparisons; followed by Mann-Whitney U) non-parametric tests of significance; normality was not assumed for comparisons. The Kolmogorov-Smirnov test was used to compare cumulative probabilities of mEPSCs. Because no obvious sex differences were observed (data available upon request), data from male and female mice were pooled. The ROUT method (Q = 1%) in GraphPad Prism was used to identify potential outliers (Motulsky and Brown, 2006). Data were analyzed prior to and again after exclusion criteria were enforced. Computational modeling analyses was performed in Python 3.6, using the Numpy, SciPy, Pandas, Matplotlib, and Scikit-Learn python packages. Random forest regression was performed using Scikit-Learn to identify channels associated with up-state duration and amplitude. Model simulation and analysis files are available in ModelDB (http://modeldb.yale.edu/266807). Statistical analysis of simulations from populations of striosome and matrix models followed the same statistical approach for experimental data unless otherwise described. Differences were considered statistically significant if p < 0.05. All statistical analyses were conducted using GraphPad Prism v7.0e (GraphPad Software, La Jolla, CA; RRID:SCR_002798) and for Supplementary Fig 4C Matlab R2020a (MathWorks, Natick, MA); figures were generated using Adobe Illustrator (San Jose, CA; RRID: SCR_010279).
Supplementary Material
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mu opioid receptor antibody | Abcam | RRID: AB_2156356 |
| Goat anti-rabbit IgG H&L (Alexa Fluor® 647) | Abcam | RRID: AB_2714032 |
| DAPI | Southern Biotech | 0100–20 |
| Bacterial and Virus Strains | ||
| pAAV5.CAG.hChR2(H134R)-mCherry.WPRE.SV40 | Gift from Karl Deisseroth | Addgene plasmid # 100054-AAV5; http://n2t.net/addgene:100054 RRID Addgene_100054 |
| pAAV5.CAG.dLight1.1 | Gift from Lin Tian Patriarchi et al., 2018 | Addgene plasmid #: 111067-AAV5; http://n2t.net/addgene:111067 RRID: Addgene_111067 |
| AAV9-hSyn-hChR2(H134R)-EYFP | Gift from Karl Deisseroth | Addgene plasmid # 26973-AAV9; Http://n2t.net/addgene:26973 RRID: Addgene_26973 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Tg(Drd1a-tdTomato)6Calak | Jackson Laboratories | RRID:IMSR_JAX:016204 |
| Mouse: Tg(Nr4a1-eGFP139Gsat) | GENSAT; Davis & Puhl, 2011 | Donated by Dr. Margaret Davis |
| Mouse: C57BL/6J | Jackson Laboratories | RRID:IMSR_JAX:000664 |
| Software and Algorithms | ||
| adjustador | Jȩdrzejewski-Szmek et al. 2018 | https://github.com/neurord/ajustador |
| Adobe Illustrator | Adobe | RRID: SCR_010279 |
| AutoQuant X | Media Cybernetics | RRID: SCR_002465 |
| GraphPad Prism V7.0e | Graphpad Software | RRID: SCR_002798 |
| Igor Pro | WaveMetrics, Inc. | RRID: SCR_000325 |
| ImageJ | National Institute of Health | RRID: SCR_003070 |
| Matlab R2020a | MathWorks | RRID: SCR_001622 |
| Mini Analysis | Synaptosoft | RRID: SCR_002184 |
| MOOSE | Upinder Bhalla, NCBS | https://moose.ncbs.res.in/ |
| Moose_nerp | Kim Blackwell, George Mason Univ | https://github.com/neurord/moose_nerp |
| NeuronStudio | Rodriguez et al., 2008 | RRID: SCR_013798 |
| Neuroscience Gateway Portal | Sivagnanam et al., 2013 | RRID:SCR_008915 |
| PicViewer V2.5.2 | John Dempster, Strathclyde University | http://spider.science.strath.ac.uk/sipbs/software_imaging.htm |
| Prairie View v5 | Bruker Nano, Inc | RRID: SCR_017142 |
| Python 3.6 | Python Software Foundation | RRID:SCR_008394 |
Highlights.
Both striosome and matrix dSPNs support dendritically-evoked somatic “up-states”
Dopamine oppositely modulates up-state length in striosome vs matrix dSPNs via D1Rs
Compartment-specific responses to D1R activation involve L-type Ca2+ channels
Changes in striatal dopamine may shift the balance of striosome vs matrix output
Acknowledgments
We thank Dr. Margaret Davis for providing Nr4a1-eGFP mice and advice, and Wendy Akmentin, Breanna Jones and Karthi Mayilvahanan for technical assistance. This work was supported by a NARSAD Young Investigator Award from the Brain & Behavior Research Foundation to J.L.P., R01 NS104089/NINDS to J.L.P., R01AA016022/NIAAA to K.T.B. and NIH F31DA047145–02 to D.B.D.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Ade KK, Wan Y, Chen M, Gloss B, and Calakos N (2011). An Improved BAC Transgenic Fluorescent Reporter Line for Sensitive and Specific Identification of Striatonigral Medium Spiny Neurons. Front Syst Neurosci 5, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascoli GA, Donohue DE, and Halavi M (2007). NeuroMorpho.Org: a central resource for neuronal morphologies. J Neurosci 27, 9247–9251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banghart MR, Neufeld SQ, Wong NC, and Sabatini BL (2015). Enkephalin Disinhibits Mu Opioid Receptor-Rich Striatal Patches via Delta Opioid Receptors. Neuron 88, 1227–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargas J, Howe A, Eberwine J, Cao Y, and Surmeier DJ (1994). Cellular and molecular characterization of Ca2+ currents in acutely isolated, adult rat neostriatal neurons. J Neurosci 14, 6667–6686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkefeld H, Sailer CA, Bildl W, Rohde V, Thumfart JO, Eble S, Klugbauer N, Reisinger E, Bischofberger J, Oliver D, et al. (2006). BKCa-Cav channel complexes mediate rapid and localized Ca2+-activated K+ signaling. Science 314, 615–620. [DOI] [PubMed] [Google Scholar]
- Blackwell KT, Czubayko U, and Plenz D (2003). Quantitative estimate of synaptic inputs to striatal neurons during up and down states in vitro. J Neurosci 23, 9123–9132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloem B, Huda R, Sur M, and Graybiel AM (2017). Two-photon imaging in mice shows striosomes and matrix have overlapping but differential reinforcement-related responses. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloodgood BL, and Sabatini BL (2007). Nonlinear regulation of unitary synaptic signals by CaV(2.3) voltage-sensitive calcium channels located in dendritic spines. Neuron 53, 249–260. [DOI] [PubMed] [Google Scholar]
- Brazhnik E, Shah F, and Tepper JM (2008). GABAergic afferents activate both GABAA and GABAB receptors in mouse substantia nigra dopaminergic neurons in vivo. J Neurosci 28, 10386–10398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brimblecombe KR, and Cragg SJ (2015). Substance P Weights Striatal Dopamine Transmission Differently within the Striosome-Matrix Axis. J Neurosci 35, 9017–9023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brimblecombe KR, and Cragg SJ (2017). The Striosome and Matrix Compartments of the Striatum: A Path through the Labyrinth from Neurochemistry toward Function. ACS Chem Neurosci 8, 235–242. [DOI] [PubMed] [Google Scholar]
- Canales JJ, and Graybiel AM (2000). A measure of striatal function predicts motor stereotypy. Nat Neurosci 3, 377–383. [DOI] [PubMed] [Google Scholar]
- Carter AG, and Sabatini BL (2004). State-dependent calcium signaling in dendritic spines of striatal medium spiny neurons. Neuron 44, 483–493. [DOI] [PubMed] [Google Scholar]
- Cazorla M, Shegda M, Ramesh B, Harrison NL, and Kellendonk C (2012). Striatal D2 receptors regulate dendritic morphology of medium spiny neurons via Kir2 channels. J Neurosci 32, 2398–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Colwell CS, Itri JN, Chandler SH, and Levine MS (1998). Dopaminergic modulation of NMDA-induced whole cell currents in neostriatal neurons in slices: contribution of calcium conductances. J Neurophysiol 79, 82–94. [DOI] [PubMed] [Google Scholar]
- Chan CS, Peterson JD, Gertler TS, Glajch KE, Quintana RE, Cui Q, Sebel LE, Plotkin JL, Shen W, Heiman M, et al. (2012). Strain-specific regulation of striatal phenotype in Drd2-eGFP BAC transgenic mice. J Neurosci 32, 9124–9132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirnaru MD, Melis C, Fanutza T, Naphade S, Tshilenge KT, Muntean BS, Martemyanov KA, Plotkin JL, Ellerby LM, and Ehrlich ME (2019). Nuclear Receptor Nr4a1 Regulates Striatal Striosome Development and Dopamine D1 Receptor Signaling. eNeuro 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crittenden JR, and Graybiel AM (2011). Basal Ganglia disorders associated with imbalances in the striatal striosome and matrix compartments. Front Neuroanat 5, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crittenden JR, Lacey CJ, Lee T, Bowden HA, and Graybiel AM (2014). Severe drug-induced repetitive behaviors and striatal overexpression of VAChT in ChAT-ChR2-EYFP BAC transgenic mice. Front Neural Circuits 8, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crittenden JR, Lacey CJ, Weng FJ, Garrison CE, Gibson DJ, Lin Y, and Graybiel AM (2017). Striatal Cholinergic Interneurons Modulate Spike-Timing in Striosomes and Matrix by an Amphetamine-Sensitive Mechanism. Front Neuroanat 11, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crittenden JR, Tillberg PW, Riad MH, Shima Y, Gerfen CR, Curry J, Housman DE, Nelson SB, Boyden ES, and Graybiel AM (2016). Striosome-dendron bouquets highlight a unique striatonigral circuit targeting dopamine-containing neurons. Proc Natl Acad Sci U S A 113, 11318–11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui G, Jun SB, Jin X, Pham MD, Vogel SS, Lovinger DM, and Costa RM (2013). Concurrent activation of striatal direct and indirect pathways during action initiation. Nature 494, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darvas M, Wunsch AM, Gibbs JT, and Palmiter RD (2014). Dopamine dependency for acquisition and performance of Pavlovian conditioned response. Proc Natl Acad Sci U S A 111, 2764–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash PK, Moore AN, Kobori N, and Runyan JD (2007). Molecular activity underlying working memory. Learn Mem 14, 554–563. [DOI] [PubMed] [Google Scholar]
- Davis MI, Crittenden JR, Feng AY, Kupferschmidt DA, Naydenov A, Stella N, Graybiel AM, and Lovinger DM (2018). The cannabinoid-1 receptor is abundantly expressed in striatal striosomes and striosome-dendron bouquets of the substantia nigra. PLoS One 13, e0191436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MI, and Puhl HL 3rd (2011). Nr4a1-eGFP is a marker of striosome-matrix architecture, development and activity in the extended striatum. PLoS One 6, e16619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorman DB, Jedrzejewska-Szmek J, and Blackwell KT (2018). Inhibition enhances spatially-specific calcium encoding of synaptic input patterns in a biologically constrained model. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K, Wu YW, Lindroos R, Liu Y, Rozsa B, Katona G, Ding JB, and Kotaleski JH (2017). Cell-type-specific inhibition of the dendritic plateau potential in striatal spiny projection neurons. Proc Natl Acad Sci U S A 114, E7612–E7621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman A, Homma D, Bloem B, Gibb LG, Amemori KI, Hu D, Delcasso S, Truong TF, Yang J, Hood AS, et al. (2017). Chronic Stress Alters Striosome-Circuit Dynamics, Leading to Aberrant Decision-Making. Cell 171, 1191–1205 e1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman A, Homma D, Gibb LG, Amemori K, Rubin SJ, Hood AS, Riad MH, and Graybiel AM (2015). A Corticostriatal Path Targeting Striosomes Controls Decision-Making under Conflict. Cell 161, 1320–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiyama F, Sohn J, Nakano T, Furuta T, Nakamura KC, Matsuda W, and Kaneko T (2011). Exclusive and common targets of neostriatofugal projections of rat striosome neurons: a single neuron-tracing study using a viral vector. Eur J Neurosci 33, 668–677. [DOI] [PubMed] [Google Scholar]
- Gerfen CR (1984). The neostriatal mosaic: compartmentalization of corticostriatal input and striatonigral output systems. Nature 311, 461–464. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, and Surmeier DJ (2011). Modulation of striatal projection systems by dopamine. Annu Rev Neurosci 34, 441–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertler TS, Chan CS, and Surmeier DJ (2008). Dichotomous anatomical properties of adult striatal medium spiny neurons. J Neurosci 28, 10814–10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JA, Kats SS, and Jaeger D (2003). Globus pallidus discharge is coincident with striatal activity during global slow wave activity in the rat. J Neurosci 23, 10058–10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodliffe JW, Song H, Rubakovic A, Chang W, Medalla M, Weaver CM, and Luebke JI (2018). Differential changes to D1 and D2 medium spiny neurons in the 12-month-old Q175+/− mouse model of Huntington’s Disease. PLoS One 13, e0200626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM, and Ragsdale CW Jr. (1978). Histochemically distinct compartments in the striatum of human, monkeys, and cat demonstrated by acetylthiocholinesterase staining. Proc Natl Acad Sci U S A 75, 5723–5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harraz OF, and Welsh DG (2013). Protein kinase A regulation of T-type Ca2+ channels in rat cerebral arterial smooth muscle. J Cell Sci 126, 2944–2954. [DOI] [PubMed] [Google Scholar]
- Higley MJ, and Sabatini BL (2010). Competitive regulation of synaptic Ca2+ influx by D2 dopamine and A2A adenosine receptors. Nat Neurosci 13, 958–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C, Depuy SD, Yao J, McIntire WE, and Barrett PQ (2009). Protein kinase A activity controls the regulation of T-type CaV3.2 channels by Gbetagamma dimers. J Biol Chem 284, 7465–7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue R, Suzuki T, Nishimura K, and Miura M (2016). Nicotinic acetylcholine receptor-mediated GABAergic inputs to cholinergic interneurons in the striosomes and the matrix compartments of the mouse striatum. Neuropharmacology 105, 318–328. [DOI] [PubMed] [Google Scholar]
- Janssen MJ, Ade KK, Fu Z, and Vicini S (2009). Dopamine modulation of GABA tonic conductance in striatal output neurons. J Neurosci 29, 5116–5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedrzejewski-Szmek Z, Abrahao KP, Jedrzejewska-Szmek J, Lovinger DM, and Blackwell KT (2018). Parameter Optimization Using Covariance Matrix Adaptation-Evolutionary Strategy (CMA-ES), an Approach to Investigate Differences in Channel Properties Between Neuron Subtypes. Front Neuroinform 12, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenrette TA, Logue JB, and Horner KA (2019). Lesions of the Patch Compartment of Dorsolateral Striatum Disrupt Stimulus-Response Learning. Neuroscience 415, 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai H, and Neher E (1992). Dihydropyridine-sensitive and omega-conotoxin-sensitive calcium channels in a mammalian neuroblastoma-glioma cell line. J Physiol 448, 161–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Wilson CJ, and Emson PC (1989). Intracellular recording of identified neostriatal patch and matrix spiny cells in a slice preparation preserving cortical inputs. J Neurophysiol 62, 1052–1068. [DOI] [PubMed] [Google Scholar]
- Kravitz AV, and Kreitzer AC (2012). Striatal mechanisms underlying movement, reinforcement, and punishment. Physiology (Bethesda) 27, 167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri AK, and Bevan MD (2020). Dopaminergic Transmission Rapidly and Persistently Enhances Excitability of D1 Receptor-Expressing Striatal Projection Neurons. Neuron 106, 277–290 e276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leventhal DK, Stoetzner C, Abraham R, Pettibone J, DeMarco K, and Berke JD (2014). Dissociable effects of dopamine on learning and performance within sensorimotor striatum. Basal Ganglia 4, 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE (2001). Three Ca2+ levels affect plasticity differently: the LTP zone, the LTD zone and no man’s land. J Physiol 532, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JC, DeFazio RA, Espinosa-Jeffrey A, Cepeda C, de Vellis J, and Levine MS (2004). Calcium modulates dopamine potentiation of N-methyl-D-aspartate responses: electrophysiological and imaging evidence. J Neurosci Res 76, 315–322. [DOI] [PubMed] [Google Scholar]
- Maylie J, Bond CT, Herson PS, Lee WS, and Adelman JP (2004). Small conductance Ca2+- activated K+ channels and calmodulin. J Physiol 554, 255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGregor MM, McKinsey GL, Girasole AE, Bair-Marshall CJ, Rubenstein JLR, and Nelson AB (2019). Functionally Distinct Connectivity of Developmentally Targeted Striosome Neurons. Cell Rep 29, 1419–1428 e1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNaughton NC, and Randall AD (1997). Electrophysiological properties of the human N-type Ca2+ channel: I. Channel gating in Ca2+, Ba2+ and Sr2+ containing solutions. Neuropharmacology 36, 895–915. [DOI] [PubMed] [Google Scholar]
- McRory JE, Santi CM, Hamming KS, Mezeyova J, Sutton KG, Baillie DL, Stea A, and Snutch TP (2001). Molecular and functional characterization of a family of rat brain T-type calcium channels. J Biol Chem 276, 3999–4011. [DOI] [PubMed] [Google Scholar]
- Miura M, Saino-Saito S, Masuda M, Kobayashi K, and Aosaki T (2007). Compartment-specific modulation of GABAergic synaptic transmission by mu-opioid receptor in the mouse striatum with green fluorescent protein-expressing dopamine islands. J Neurosci 27, 9721–9728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadel JA, Pawelko SS, Copes-Finke D, Neidhart M, and Howard CD (2020). Lesion of striatal patches disrupts habitual behaviors and increases behavioral variability. PLoS One 15, e0224715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisenbaum ES, and Wilson CJ (1995). Potassium currents responsible for inward and outward rectification in rat neostriatal spiny projection neurons. J Neurosci 15, 4449–4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata N, and Tatebayashi H (1990). Sodium current kinetics in freshly isolated neostriatal neurones of the adult guinea pig. Pflugers Arch 416, 594–603. [DOI] [PubMed] [Google Scholar]
- Palma-Cerda F, Auger C, Crawford DJ, Hodgson AC, Reynolds SJ, Cowell JK, Swift KA, Cais O, Vyklicky L, Corrie JE, and Ogden D (2012). New caged neurotransmitter analogs selective for glutamate receptor sub-types based on methoxynitroindoline and nitrophenylethoxycarbonyl caging groups. Neuropharmacology 63, 624–634. [DOI] [PubMed] [Google Scholar]
- Palmiter RD (2008). Dopamine signaling in the dorsal striatum is essential for motivated behaviors: lessons from dopamine-deficient mice. Ann N Y Acad Sci 1129, 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patriarchi T, Cho JR, Merten K, Howe MW, Marley A, Xiong WH, Folk RW, Broussard GJ, Liang R, Jang MJ, et al. (2018). Ultrafast neuronal imaging of dopamine dynamics with designed genetically encoded sensors. Science 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennartz CM, Berke JD, Graybiel AM, Ito R, Lansink CS, van der Meer M, Redish AD, Smith KS, and Voorn P (2009). Corticostriatal Interactions during Learning, Memory Processing, and Decision Making. J Neurosci 29, 12831–12838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pert CB, Kuhar MJ, and Snyder SH (1976). Opiate receptor: autoradiographic localization in rat brain. Proc Natl Acad Sci U S A 73, 3729–3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pifferi S, Dibattista M, and Menini A (2009). TMEM16B induces chloride currents activated by calcium in mammalian cells. Pflugers Arch 458, 1023–1038. [DOI] [PubMed] [Google Scholar]
- Plotkin JL, Day M, Peterson JD, Xie Z, Kress GJ, Rafalovich I, Kondapalli J, Gertler TS, Flajolet M, Greengard P, et al. (2014). Impaired TrkB receptor signaling underlies corticostriatal dysfunction in Huntington’s disease. Neuron 83, 178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin JL, Day M, and Surmeier DJ (2011). Synaptically driven state transitions in distal dendrites of striatal spiny neurons. Nat Neurosci 14, 881–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin JL, Shen W, Rafalovich I, Sebel LE, Day M, Chan CS, and Surmeier DJ (2013). Regulation of dendritic calcium release in striatal spiny projection neurons. J Neurophysiol 110, 2325–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin JL, and Surmeier DJ (2014). Multiphoton imaging approaches for studying striatal dendritic excitability. Methods Mol Biol 1183, 171–182. [DOI] [PubMed] [Google Scholar]
- Prager EM, Bergstrom HC, Grunberg NE, and Johnson LR (2011). The importance of reporting housing and husbandry in rat research. Front Behav Neurosci 5, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prager EM, and Plotkin JL (2019). Compartmental function and modulation of the striatum. J Neurosci Res 97, 1503–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Ehlenberger DB, Dickstein DL, Hof PR, and Wearne SL (2008). Automated three-dimensional detection and shape classification of dendritic spines from fluorescence microscopy images. PLoS One 3, e1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas AG, Davis MI, Lovinger DM, and Mateo Y (2016). Dopamine dynamics and cocaine sensitivity differ between striosome and matrix compartments of the striatum. Neuropharmacology 108, 275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Hernandez-Lopez S, Tkatch T, Held JE, and Surmeier DJ (2004). Kv1.2-containing K+ channels regulate subthreshold excitability of striatal medium spiny neurons. J Neurophysiol 91, 1337–1349. [DOI] [PubMed] [Google Scholar]
- Shen W, Plotkin JL, Francardo V, Ko WK, Xie Z, Li Q, Fieblinger T, Wess J, Neubig RR, Lindsley CW, et al. (2015). M4 Muscarinic Receptor Signaling Ameliorates Striatal Plasticity Deficits in Models of L-DOPA-Induced Dyskinesia. Neuron 88, 762–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuen JA, Chen M, Gloss B, and Calakos N (2008). Drd1a-tdTomato BAC transgenic mice for simultaneous visualization of medium spiny neurons in the direct and indirect pathways of the basal ganglia. J Neurosci 28, 2681–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivagnanam S, Majumdar A, Yoshimoto K, Astakhov V, Bandrowski A, Martone M, and Carnevale N (2013). Introducing the Neuroscience Gateway. In IWSG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JB, Klug JR, Ross DL, Howard CD, Hollon NG, Ko VI, Hoffman H, Callaway EM, Gerfen CR, and Jin X (2016). Genetic-Based Dissection Unveils the Inputs and Outputs of Striatal Patch and Matrix Compartments. Neuron 91, 1069–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder GL, Fienberg AA, Huganir RL, and Greengard P (1998). A dopamine/D1 receptor/protein kinase A/dopamine- and cAMP-regulated phosphoprotein (Mr 32 kDa)/protein phosphatase-1 pathway regulates dephosphorylation of the NMDA receptor. J Neurosci 18, 10297–10303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song SC, Beatty JA, and Wilson CJ (2016). The ionic mechanism of membrane potential oscillations and membrane resonance in striatal LTS interneurons. J Neurophysiol 116, 1752–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steephen JE, and Manchanda R (2009). Differences in biophysical properties of nucleus accumbens medium spiny neurons emerging from inactivation of inward rectifying potassium currents. J Comput Neurosci 27, 453–470. [DOI] [PubMed] [Google Scholar]
- Stern EA, Jaeger D, and Wilson CJ (1998). Membrane potential synchrony of simultaneously recorded striatal spiny neurons in vivo. Nature 394, 475–478. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Plotkin J, and Shen W (2009). Dopamine and synaptic plasticity in dorsal striatal circuits controlling action selection. Curr Opin Neurobiol 19, 621–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Shen W, Day M, Gertler T, Chan S, Tian X, and Plotkin JL (2010). The role of dopamine in modulating the structure and function of striatal circuits. Prog Brain Res 183, 149–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkatch T, Baranauskas G, and Surmeier DJ (2000). Kv4.2 mRNA abundance and A-type K(+) current amplitude are linearly related in basal ganglia and basal forebrain neurons. J Neurosci 20, 579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuckwell HC (2012). Quantitative aspects of L-type Ca2+ currents. Prog Neurobiol 96, 1–31. [DOI] [PubMed] [Google Scholar]
- Wilson CJ, and Kawaguchi Y (1996). The origins of two-state spontaneous membrane potential fluctuations of neostriatal spiny neurons. J Neurosci 16, 2397–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z, Thompson AD, Brogan JT, Schulte ML, Melancon BJ, Mi D, Lewis LM, Zou B, Yang L, Morrison R, et al. (2011). The Discovery and Characterization of ML218: A Novel, Centrally Active T-Type Calcium Channel Inhibitor with Robust Effects in STN Neurons and in a Rodent Model of Parkinson’s Disease. ACS Chem Neurosci 2, 730–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagishita S, Hayashi-Takagi A, Ellis-Davies GC, Urakubo H, Ishii S, and Kasai H (2014). A critical time window for dopamine actions on the structural plasticity of dendritic spines. Science 345, 1616–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HH, Knowlton BJ, and Balleine BW (2004). Lesions of dorsolateral striatum preserve outcome expectancy but disrupt habit formation in instrumental learning. Eur J Neurosci 19, 181–189. [DOI] [PubMed] [Google Scholar]
- Yoshizawa T, Ito M, and Doya K (2018). Reward-Predictive Neural Activities in Striatal Striosome Compartments. eNeuro 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Detailed datasets supporting the current study are available from the corresponding author on request. Source code is available in ModelDB at http://modeldb.yale.edu/266807.