

Abstract
Introduction:
Primary progressive apraxia of speech (PPAOS) is a neurodegenerative syndrome in which patients present with an isolated motor speech disorder. Some PPAOS patients develop parkinsonism and other features of progressive supranuclear palsy (PSP) and/or corticobasal syndrome (CBS) over time. We aimed to assess the evolution of parkinsonian characteristics in PPAOS patients who had been followed yearly for at least six years.
Methods:
From a large cohort of 46 PPAOS patients, eight were followed yearly for > 6-years in multiple NIH-funded grants. Parkinsonian and other features, including bradykinesia, tremor, rigidity, postural instability, apraxia, ocular motor function and cognition were assessed at each visit, and research criteria applied for PSP and CBS diagnosis. Neurological, speech-language test scores, and [18F]fluorodeoxyglucose PET (FDG-PET) and MRI midbrain volumes were assessed.
Results:
A Parkinson’s plus syndrome developed in all eight patients (100%). Bradykinesia was the earliest feature, followed by rigidity and postural instability. Tremor was not a significant feature. Parkinsonism, limb apraxia and ocular motor impairment tended to develop four-to-five years after onset with some patients having slight asymmetric parkinsonism. Six patients (75%) met research criteria for probable PSP, although only one for PSP-Richardson’s syndrome; three patients met criteria for possible CBS. Slightly asymmetric, left-sided, hypometabolism was observed on FDG-PET, not matching asymmetry of Parkinsonism. Midbrain hypometabolism was absent-minimal. Three patients had progressive midbrain volumes in the PSP-Richardson’s syndrome range.
Conclusions:
A Parkinson’s plus syndrome may inevitably develop in PPAOS supporting PPAOS as an early presentation of a Parkinson’s plus disorder.
Keywords: Apraxia of speech, Parkinsonism, PSP, corticobasal syndrome, PET
INTRODUCTION
Apraxia of speech is a motor speech disorder affecting the planning and/or programming of speech [1]. It can occur acutely after stroke or be progressive and resultant from a neurodegenerative disease [2]. In the context of neurodegeneration, agrammatic aphasia may accompany the disorder but patients may have isolated apraxia of speech and be diagnosed with primary progressive apraxia of speech (PPAOS) [3]. The prevalence of PPAOS has been estimated to be approximately 4.4 per 100,000 people [4] although it is likely higher as patients with PPAOS are often misdiagnosed. Audio files demonstrating PPAOS characteristics have been published [5] and PPAOS is associated with hypometabolism on [18F]fluorodeoxyglucose (FDG) PET and atrophy on MRI, predominantly in the superior premotor cortex and supplementary motor area [3, 6].
We previously assessed general progression in PPAOS patients over two visits, approximately two years apart, and found that most of them developed some Parkinsonism over the years, particularly bradykinesia with some showing rigidity [7]. Interestingly, a subset of patients also developed clinical features that are typically observed in progressive supranuclear palsy (PSP) and/or corticobasal syndrome (CBS), including limb apraxia, gait and balance problems, vertical gaze slowing or palsy and ocular motor impersistence which emerged approximately 5 years after onset [7]. PPAOS has been associated with corticobasal degeneration (CBD) at autopsy [8, 9] in a patient that had developed subtle parkinsonism relatively early in the disease, with frank CBS symptoms observed a couple of years before death [8]. The development of parkinsonism has also been observed in other individual cases, presumed to have had PPAOS [5, 10].
Little is known about the temporal evolution of specific parkinsonian and other features, whether parkinsonian features are asymmetric, whether PPAOS patients evolve to meet recent research criteria for PSP [11] or CBS [12] and whether over time PPAOS evolves into a an entity that resembles a Parkinson plus disorder. To address these unknowns, we evaluated the development and evolution of parkinsonism in eight PPAOS patients who underwent at least six yearly serial clinical evaluations.
METHODS
Patient recruitment:
The Neurodegenerative Research Group (NRG) at Mayo Clinic has been recruiting patients with progressive AOS into NIH-funded grants since July 2010. All patients undergo yearly longitudinal assessments including speech/language/neurological/neuropsychological evaluations, volumetric MRI and FDG-PET scans. To best capture the evolution of parkinsonism over a long time period we selected patients who had at least six yearly visits and who met published PPAOS criteria at onset [3]. Any patient with even mild (but unequivocal) evidence of aphasia or more than one PSP or CBS feature at onset was excluded. A total of eight patients met inclusion and exclusion criteria.
Speech-language evaluation:
Judgments concerning the presence/absence/severity of AOS and aphasia at each visit were made by consensus between at least two speech-language pathologists (JRD, EAS, HMC, RLU) based on reviewing video recordings of speech and language examinations, as described [3]. Speech/language evaluations included the motor speech disorder (MSD) severity rating scale [13] which rates motor speech disorder (AOS or dysarthria) severity and the Western Aphasia Battery (WAB)- Revised [14] to assess aphasia presence and severity [WAB-Aphasia Quotient (AQ)]. For the MSD, a score below 10 was considered impaired on motor speech [2], and for the WAB-AQ, below 93.8 [14] was considered supportive of aphasia.
Neurological evaluation:
Neurologic examinations were performed by a Movement Disorders expert (KAJ). Parkinsonism was assessed with the MDS Sponsored revision of the Unified Parkinson’s Disorder Rating Scale part-III (MDS-UPDRS-III) [15], limb apraxia with the WAB praxis subtest [16], and ocular motor impairment with the PSP Saccadic Impairment Scale (PSIS)[17] and PSP Rating Scale (PSPRS)-Ocular motor sub-scale [18]. Testing also included the Montreal Cognitive Assessment battery (MoCA) [19] for general cognition, Frontal Assessment Battery (FAB) [20] for executive function, and Neuropsychiatric Inventory Questionnaire (NPI-Q) [21] for neuropsychiatric features. Neurological diagnoses were determined at a consensus meeting involving three movement disorders and/or behavioral neurologists with expertise in degenerative parkinsonian disorders (KAJ, HB, FA). Cut-points to define abnormal were based on published data for: MoCA<26 [19]; FAB<16 [22] and NPI-Q>4 [23]. For the MDS-UPDRS-III, we used a cut-point of >7.95 and for the limb apraxia subset of the WAB, a cut-point of < 60 suggested the presence of limb apraxia. Cut-points were calculated based on the 95th percentile of scores from a cohort of 21 motorically normal control subjects (median age=67; range=[45, 85]; male/female =10/11). That is, 95% of normal subjects scored ≤ 7.95 on the MDS-UPDRS-III and 60 on the WAB praxis test. For ocular motor dysfunction, we used two different scales. For the PSIS, a score of 2 depicts slowing in vertical eye saccades and 3 or higher vertical supranuclear gaze palsy. For the ocular motor sub-score of the PSPRS, the total score was 12; a score of ≥2 was considered as evidence of eye movement abnormalities.
The Movement Disorder Society clinical criteria for PSP (MDS-PSP) [11] were applied to the final visit for all patients to determine whether they met criteria for PSP. Each patient was assigned an ocular motor (01, 02 or 03), akinesia (A1, A2, A3), postural instability (P1, P2, P3), and corticobasal (C3) level, as defined in the criteria [11]. All patients met criteria for a speech-language designation (C1) given that all had a speech disorder. The Multiple Allocation Extinction (MAX) rules [24] designed to reduce the number of multiple diagnoses from the MDS-PSP criteria were also applied. Research criteria for CBS [12] were applied to the final visits.
FDG-PET:
FDG-PET scans were acquired with a PET/CT scanner (GE Healthcare) while operating in 3D mode. Participants were injected with 18F-FDG of approximately 459 MBq (range 367–576 MBq), and after a 30-minute uptake period an 8-minute 18F-FDG scan was performed. Individual-level patterns of hypometabolism were assessed using 3D stereotactic surface projections [25] using CortexID (GE Healthcare) whereby activity at each voxel is Z-scored to an age-segmented normative database. We compare FDG-PET at baseline and last visit.
MRI:
Volumetric MRI scans were obtained at 3T, as previously described [3]. Midbrain volume was calculated at the baseline and last visit, and volumes were divided by total intracranial volume to correct for head size. Midbrain volumes were compared to previously published healthy control and PSP-Richardson’s syndrome cohorts [26].
The study was approved by the Mayo Clinic IRB.
RESULTS
Baseline characteristics:
Among the eight PPAOS patients, 5 (63%) were male (Supplementary Table 1). The median age at onset was 66 years [Interquartile range (IQR) of 54–74]. The median duration (time from onset to first visit) was 3.5 (IQR: 2–5.5) years and median disease duration (onset to last visit) was 9 (IQR: 8–11) years.
Evolution of specific parkinsonian features:
All eight patients developed parkinsonian features over time (Figure 1). Bradykinesia was one of the earliest findings, with abnormalities observed within the first four years of onset in six patients, and it steadily worsened over time. Four patients had asymmetric bradykinesia at one time point. Patient 1 had subtle asymmetric, left greater than right-side, bradykinesia at first visit and continued to be asymmetric on subsequent visits. Patients 5 and 7 developed asymmetric bradykinesia after the second visit. For patient 5, it was right side greater than left, and for patient 7, left greater than right. Patient 2 became right-side asymmetric at the last visit. The remaining for patients (patients 3, 4, 6 and 8) had relatively symmetric yet progressive bradykinesia. Rigidity developed later in time for all patients but one who did not develop any rigidity (patient 2). Except for patient 5, who showed right-sided asymmetry as he did for bradykinesia, the rest of the six patients showed relatively symmetric rigidity. When assessing axial and limb rigidity patterns separately, half of our patients showed axial greater than appendicular rigidity. In three of those, it was within the first five years of disease onset. The other four patients showed the opposite pattern, with half showing this pattern within four years of onset. Progressive postural instability was observed in all but one patient (patient 7). It emerged as an early finding in patients 1, 3 and 4, but a late finding in patients 2, 5 and 8. Tremor was not a prominent feature in any patient.
Figure 1: The evolution of bradykinesia, rigidity, tremor and postural instability.
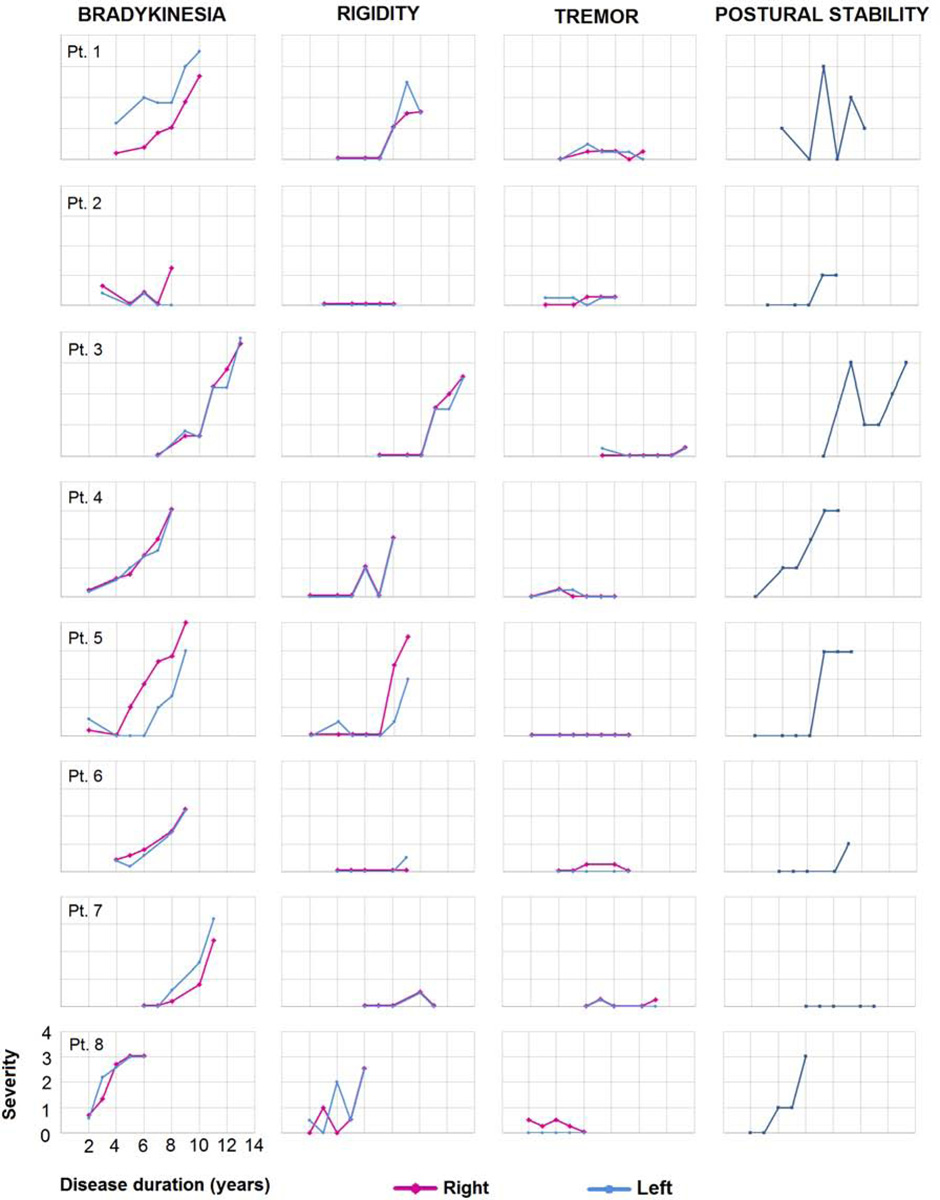
- Subtest scores of bradykinesia on finger tapping, hand movement, pronation-supination, toe-tapping and leg agility were summed up for each side and graphs for ‘bradykinesia’ variable were drawn accordingly.
- Subtest scores of the upper and lower extremity were summed up for each side and graphs for ‘rigidity’ variable were drawn accordingly.
- Subtest scores of postural and kinetic tremors of the hand, resting tremors of the upper and lower extremity were summed up and graphs for the ‘tremor’ variable were drawn accordingly.
- For the ‘postural instability’ variable, the subtest score for postural stability was used to create the graphs.
Evolution of neurological test results:
The MDS-UPDRS-III scores increased over time for all eight patients (Figure 2), with abnormalities (>7.95) developing after approximately four years in all but one patient (patient 8). The PSPRS-Ocular motor scale scores became abnormal (above 2) over time for all but two patients (patients 2 and 6). Three patients (1, 5 and 7) developed vertical supranuclear palsy (i.e. a score of 3+ on PSIS), three patients (4, 7 and 8) developed slowing in vertical eye movements (i.e. score of 2 on PSIS), and two patients (2 and 6) showed no abnormalities on the PSIS. Abnormalities on the PSIS tended to occur after five years from onset. This was similar for the PSPRS-Ocular motor scale, with the exception of patient 8 who showed abnormalities between two and three years after disease onset. Except for two patients (patients 1 and 6), all showed more than subtle evidence of limb apraxia on the WAB limb apraxia test, with prominent impairment developing late in the disease course.
Figure 2: Longitudinal performance of all patients on select clinical measures.
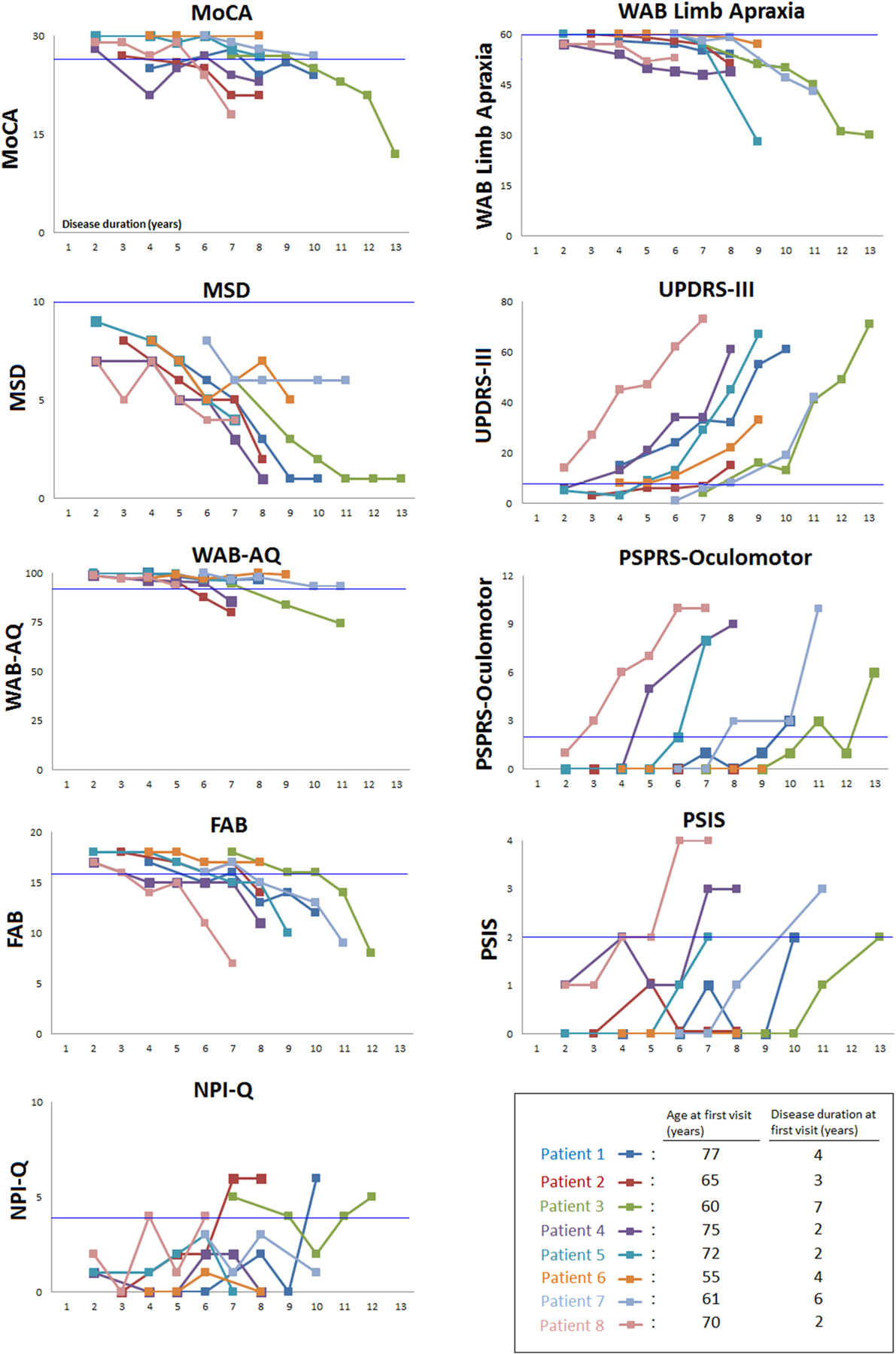
Normal cutpoints are noted with the bright blue line. MoCA = Montreal Cognitive Assessment; MSD= Motor Speech Disorder; WAB-AQ= Western Aphasia Battery- Revised aphasia quotient; FAB = Frontal Assessment Battery; NPI-Q = Neuropsychiatric Inventory Questionnaire; WAB limb apraxia= Western Aphasia Battery limb apraxia subtest; UPDRS-III= Unified Parkinson’s Disorder Rating Scale III; PSPRS-Ocular motor= Progressive Supranuclear Palsy (PSP) Rating Scale Ocular motor subtest; PSIS= PSP Saccadic Impairment Scale
All patients except one (patient 6) showed decreasing performance on the MoCA over time, with all but three (patients 5, 6 and 7) eventually scoring below the impairment cut-point (Figure 2). Cognition was relatively preserved within the first 3.5 years, with declines starting on average six years after onset. As expected, motor speech impairment on the MSD was abnormal for all patients from the first visit and it continued to decline over time. Three patients (2, 3 and 4) developed aphasia six years after onset. All but one (patient 6) showed decreased frontal lobe executive function on the FAB approximately seven years after disease onset. Three patients (1, 2 and 3) showed impaired NPI-Q in their last visits, after six years of disease duration. For the other five patients, we did not observe many behavioral abnormalities given their NPI-Q scores showed fluctuations within the normal range.
Application of PSP/CBS diagnostic criteria:
When MDS-PSP criteria were applied at the last visit (Supplementary Table 1), all but two of our patients fulfilled criteria for ocular motor impairment (01 or 02) and akinesia (A2 or A3), and hence would meet criteria for possible PSP-SL and probable PSP-P. Of these, three (patients 3, 4 and 5) also met C3 and hence would also meet criteria for possible PSP-CBS. One (patient 8) met criteria for postural instability (P1) since unprovoked falls were observed within three years of onset and hence would also meet criteria for probable PSP-RS. When MAX criteria were applied to these six patients, patient 8 was diagnosed with probable PSP-RS and the other five with probable PSP-P. Two patients (patients 2 and 6) had an isolated speech disorder at last visit (i.e. C1) and, hence, met criteria for suggestive of PSP-SL. When research criteria for CBS were applied, three patients (patients 3, 4 and 5) met possible CBS criteria (Supplementary Table 1). No patient developed limb dystonia, postural/action limb myoclonus or an alien limb phenomenon.
Imaging findings:
Progression was observed on FDG-PET in all eight patients (Figure 3). Hypometabolism was predominantly observed in premotor cortex, always worse on the left, although asymmetry was variable. Decline in striatal hypometabolism was observed and was relatively symmetric. In four patients (patients 2, 3, 4 and 5) greater change occurred in the left striatum compared to the right over time (change = 0.14 vs 0.05; p=0.039) [(median baseline left striatum: 1.54 (IQR: 1.44–1.6), right striatum: 1.52 (IQR: 1.44–1.6; median at last visit: left striatum 1.40 (IQR: 1.33– 1.44), right striatum: 1.47 (IQR: 1.36–1.48)]. Patterns of FDG-PET cortical asymmetry at baseline and last visit did not match well with clinical parkinsonian asymmetry from UPDRS-III. Absent-minimal midbrain hypometabolism was observed across patients. Baseline midbrain volumes were similar to controls in all but one patient (patient 1) (Figure 4). Progression was observed by the time of the last visit, with three patients (patients 1, 7 and 8) having volumes similar to PSP-Richardson’s syndrome.
Figure 3: Superior views of FDG-PET scans of first and last visits for each patient showing hypometabolism as age-corrected Z-scores.
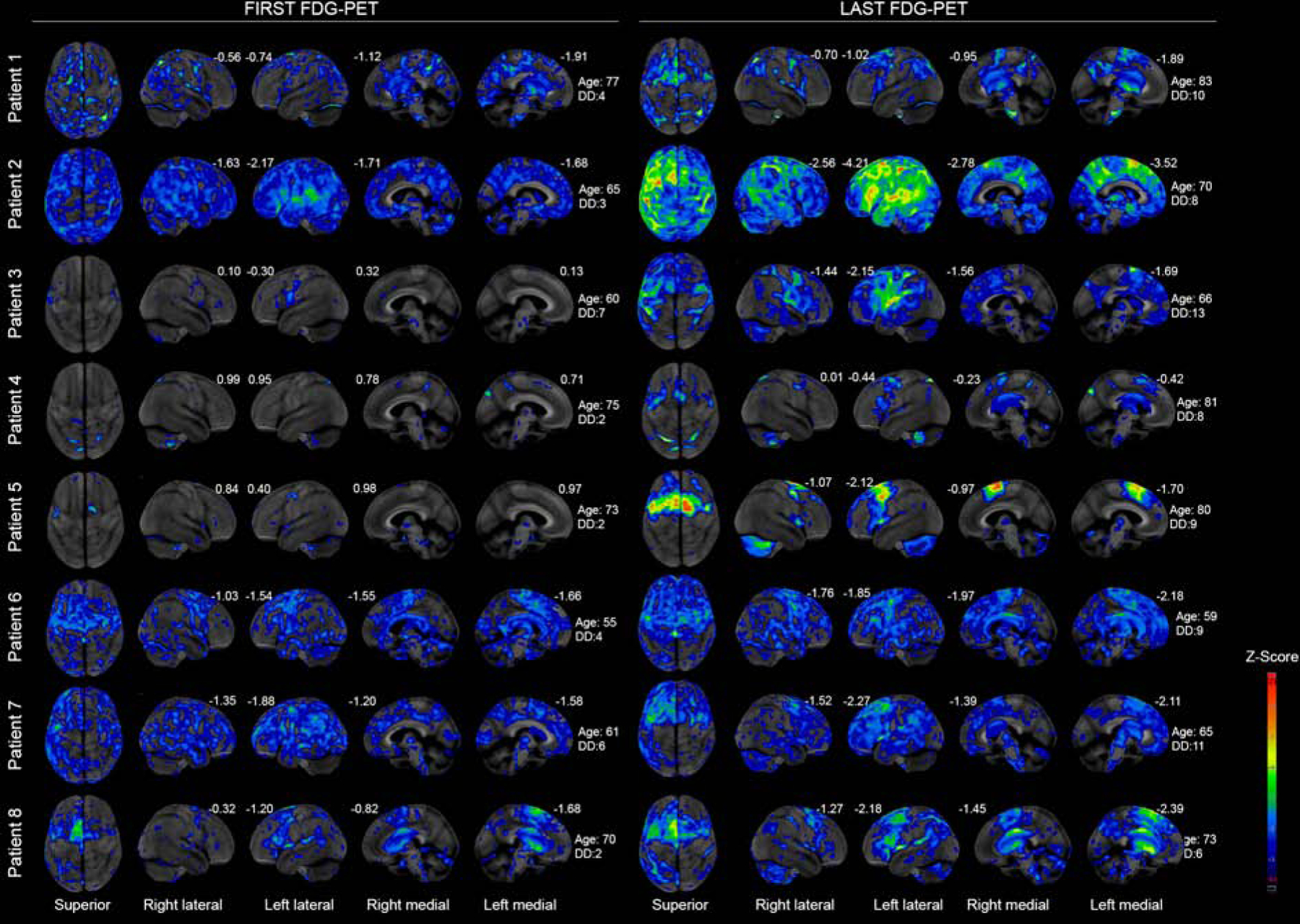
DD=Disease duration. On the left-hand side, first visit images and corresponding age at scanning and PPAOS duration are demonstrated. On the right-hand side, last visit images and corresponding age at scanning and disease duration are demonstrated. Numbers are Z-scores compared to metabolism of the same region in an age match normal control population.
Figure 4: Midbrain volumes at first and last visits for each PPAOS patient compared to healthy controls and PSP-Richardson syndrome.
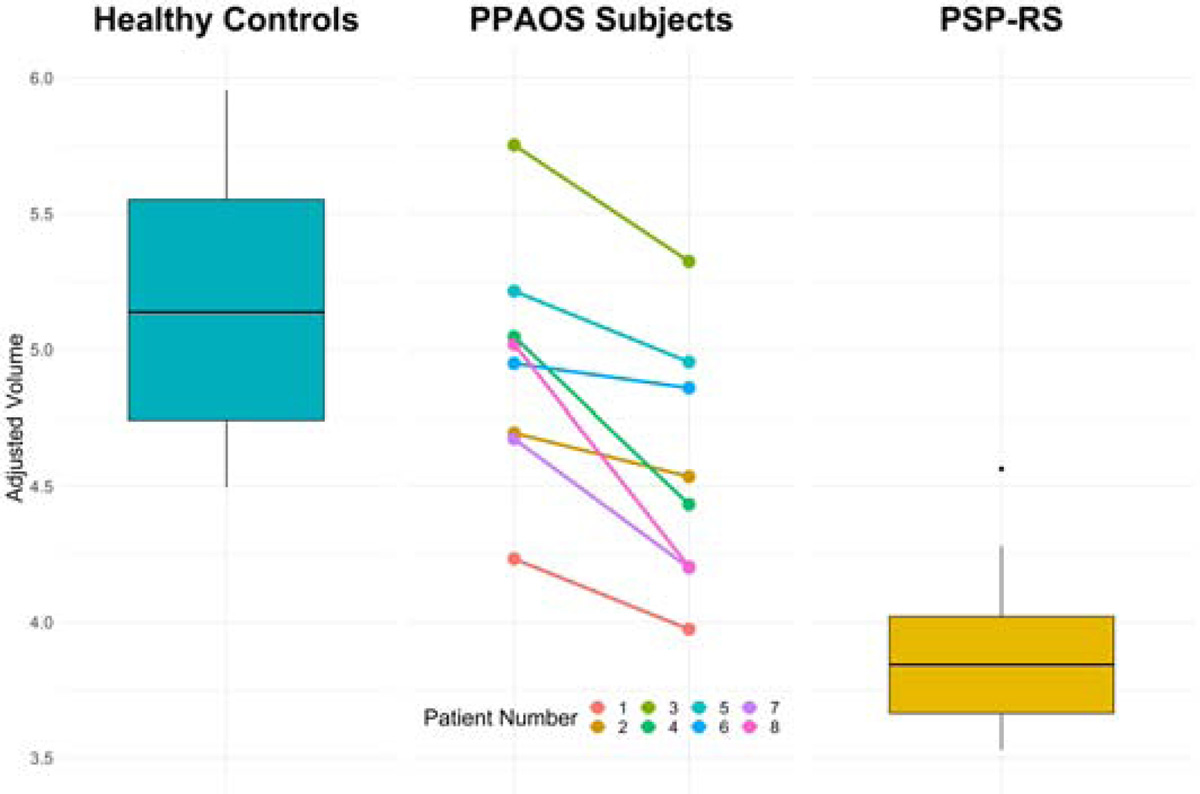
Volumes are adjusted for total intracranial volume. Boxes show median, 25th and 75th percentiles with whiskers extending to 95th percentiles.
DISCUSSION
In this longitudinal study we characterize the development and progression of parkinsonism in patients with PPAOS and long term follow-up. We show that bradykinesia is one of the earliest developing features, followed by rigidity and postural instability, and that ocular motor impairment and limb apraxia also commonly develops. The majority of PPAOS patients eventually met diagnostic criteria for probable PSP-P with a subset also meeting criteria for possible CBS.
All patients developed some parkinsonian features, with scores on the MDS-UPDRS III tending to increase around four-to-five years after disease onset. Bradykinesia emerged earlier than the other characteristic signs and was slightly asymmetric for some patients. Bradykinesia/akinesia is a commonly seen extrapyramidal finding and used as a diagnostic criterion for both Parkinson’s disease (PD) and other parkinsonian disorders, including PSP [11] and CBS [12]. For CBS, asymmetry is used to determine diagnosis, with more asymmetric motor functioning observed in CBS compared to PSP. We suspected that asymmetry in bradykinesia in our patients may be emerging from asymmetric basal ganglia involvement as noted on FDG-PET over time although we acknowledge that FDG-PET is not as sensitive to basal ganglia dysfunction as it is for cortical dysfunction. In all eight patients, the emergence of rigidity and postural instability followed bradykinesia. Rigidity is an important component of the clinical presentation of PSP and CBS [11, 12]. However, its characteristics can differ. Rigidity in PSP is almost always more prominent in axial muscles, neck, and upper trunk, than appendicular. On the contrary, in CBS it usually begins in one limb and ultimately affects all four limbs with less axial rigidity [27]. Four of our patients (50%) showed axial rigidity greater than appendicular rigidity and the other four showed the opposite pattern. Postural instability is one of the core features of PSP and while it is not a core feature of CBS, it can develop later in the disease course [27]. Unlike bradykinesia, rigidity, and postural instability, we did not observe a significant role of tremor in PPAOS.
We also observed the emergence of limb apraxia and ocular motor impairment in some of our patients. Limb apraxia occurs in 45–81% of CBS patients, as a part of cortical dysfunction [12] and can result from damage to the supplementary motor area [28]. Two patients with the lowest limb apraxia scores on their last visit did indeed fulfill criteria for a possible CBS. The majority of our PPAOS patients (75%) had a vertical gaze abnormality and was impaired on the PSIS (≥2) four to five years into the disease. They also performed in the abnormal range on the PSPRS-Ocular motor scale, however, given the larger range, the PSPRS-Ocular motor scale revealed less clear progression of eye movement abnormality over time. Therefore, it appears that the majority of PPAOS patients will develop some features of both PSP and CBS over time. Of our 46 PPAOS patients all but 2 have developed parkinsonian features to date and 44 (96%) have evolved into a Parkinson plus syndrome with some features typically observed in PSP and CBS, although here they tend to more often overlap (like a hybrid syndrome) [29]. With-that-said, other classic features of these two syndromes, such as early falls, axial and appendicular dystonia, stimulus sensitive myoclonus and alien-limb phenomenon, are not observed.
We also observed progression on other neurological and language measures. We observed a consistent decline on the MoCA, around six years after onset, slightly later than the development of parkinsonism and a similar development of aphasia, also about six years after onset; AOS always remained the most prominent speech or language feature. In fact, three patients scored near zero on the MSD scale at their last visits, indicating mutism. Executive dysfunction progressed in all but one patient approximately seven years into the disease. We did not observe much evidence for psychiatric features, with progression on the NPI-Q being difficult to assess due to fluctuations in scores, possibly related to treatment effects on depression, anxiety and agitation that can occur in PPAOS.
Anatomically, PPAOS is associated with involvement of the supplementary motor area and lateral premotor cortex [3, 6, 30]. It does appear that over time there is greater involvement of these areas, as well as spread into motor cortex and prefrontal cortex [7, 26]. Midbrain atrophy has also been observed in some patients but is typically mild compared to PSP-Richardson’s syndrome [26], as was observed in three patients in this study.
The evolution of PPAOS into a Parkinson-plus syndrome has clinical relevance as such patients typically will not be evaluated by movement disorder experts early in their illness, limiting recognition of this syndrome in the field. Most patients with PPAOS first present to their primary care physician and later on are referred to a range of different specialists. In our experience, only 20% of PPAOS patients are correctly diagnosed early in the disease course, with an equal number receiving a functional (psychogenic) or psychiatric diagnosis. Recognition of this syndrome will require knowledge about the existence of the syndrome, recognizing its characteristic features [5, 30] and utilizing tests such as FDG-PET which is sensitive to showing subtle hypometabolism in the supplementary motor area and premotor cortex.
According to the new MDS-PSP diagnostic criteria [11], the majority of patients (6/8) showed characteristics of more than one PSP variant. When we applied the MAX criteria [24], five of eight patients met criteria for ‘probable PSP-P’, one for ‘probable PSP-RS’, and two for ‘suggestive of PSP-SL’ mirroring our experience when applying the MDS-PSP criteria to a large cohort of patients with speech and language disorders, whereby a large number were diagnosed as PSP-P by MAX criteria [26].
The main strength of our study is the 6 years of detailed yearly clinical examinations by leading experts in movement disorders and speech and language pathology. On-the-other hand, the main limitation is the lack of pathological confirmation.
CONCLUSION
PPAOS patients appear to inevitably develop a Parkinson’s plus syndrome and hence should be recognized as an early presentation of such.
Supplementary Material
Highlights.
Primary progressive apraxia of speech (PPAOS) is a neurodegenerative syndrome in which patients present with an isolated motor speech disorder.
PPAOS patients develop parkinsonism and evolve into a Parkinson-plus disorder over the disease course.
The evolved parkinsonian syndrome share clinical features with both progressive supranuclear palsy (PSP) and corticobasal syndrome (CBS).
Acknowledgements
This study was funded by National Institute of Health grants R01-DC12519, R01-DC010367, R01-DC14942 and R01-NS89757.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest: None
REFERENCES:
- [[1].McNeil MR, Robin DA, Schmidt RA, Apraxia of speech: Definition and differential diagnosis, Clinical management of sensorimotor speech disorders 2 (2009) 249–267. [Google Scholar]
- [2].Duffy JR, Motor speech disorders-e-book: Substrates, differential diagnosis, and management, Elsevier Health Sciences; 2013. [Google Scholar]
- [3].Josephs KA, Duffy JR, Strand EA, Machulda MM, Senjem ML, Master AV, Lowe VJ, Jack CR Jr., Whitwell JL, Characterizing a neurodegenerative syndrome: primary progressive apraxia of speech, Brain 135(Pt 5) (2012) 1522–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Whitwell JL, Duffy JR, Strand EA, Machulda MM, Tosakulwong N, Weigand SD, Senjem ML, Spychalla AJ, Gunter JL, Petersen RC, Jack CR Jr., Josephs KA, Sample size calculations for clinical trials targeting tauopathies: a new potential disease target, J Neurol 262(9) (2015) 2064–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Utianski RL, Duffy JR, Clark HM, Strand EA, Boland SM, Machulda MM, Whitwell JL, Josephs KA, Clinical Progression in Four Cases of Primary Progressive Apraxia of Speech, Am J Speech Lang Pathol 27(4) (2018) 1303–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Whitwell JL, Duffy JR, Strand EA, Machulda MM, Senjem ML, Gunter JL, Kantarci K, Eggers SD, Jack CR Jr., Josephs KA, Neuroimaging comparison of primary progressive apraxia of speech and progressive supranuclear palsy, Eur J Neurol 20(4) (2013) 629–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Josephs KA, Duffy JR, Strand EA, Machulda MM, Senjem ML, Gunter JL, Schwarz CG, Reid RI, Spychalla AJ, Lowe VJ, Jack CR Jr., Whitwell JL, The evolution of primary progressive apraxia of speech, Brain 137(Pt 10) (2014) 2783–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tetzloff KA, Duffy JR, Strand EA, Machulda MM, Boland SM, Utianski RL, Botha H, Senjem ML, Schwarz CG, Josephs KA, Whitwell JL, Clinical and imaging progression over 10 years in a patient with primary progressive apraxia of speech and autopsy-confirmed corticobasal degeneration, Neurocase 24(2) (2018) 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Josephs KA, Duffy JR, Strand EA, Whitwell JL, Layton KF, Parisi JE, Hauser MF, Witte RJ, Boeve BF, Knopman DS, Dickson DW, Jack CR Jr., Petersen RC, Clinicopathological and imaging correlates of progressive aphasia and apraxia of speech, Brain 129(Pt 6) (2006) 1385–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Broussolle E, Tommasi M, Mauguière F, Chazot G, Progressive anarthria with secondary parkinsonism: a clinico-pathological case report, J Neurol Neurosurg Psychiatry 55(7) (1992) 577–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hoglinger GU, Respondek G, Stamelou M, Kurz C, Josephs KA, Lang AE, Mollenhauer B, Muller U, Nilsson C, Whitwell JL, Arzberger T, Englund E, Gelpi E, Giese A, Irwin DJ, Meissner WG, Pantelyat A, Rajput A, van Swieten JC, Troakes C, Antonini A, Bhatia KP, Bordelon Y, Compta Y, Corvol JC, Colosimo C, Dickson DW, Dodel R, Ferguson L, Grossman M, Kassubek J, Krismer F, Levin J, Lorenzl S, Morris HR, Nestor P, Oertel WH, Poewe W, Rabinovici G, Rowe JB, Schellenberg GD, Seppi K, van Eimeren T, Wenning GK, Boxer AL, Golbe LI, Litvan I, P.S.P.S.G. Movement Disorder Society-endorsed, Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria, Mov Disord 32(6) (2017) 853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Armstrong MJ, Litvan I, Lang AE, Bak TH, Bhatia KP, Borroni B, Boxer AL, Dickson DW, Grossman M, Hallett M, Josephs KA, Kertesz A, Lee SE, Miller BL, Reich SG, Riley DE, Tolosa E, Troster AI, Vidailhet M, Weiner WJ, Criteria for the diagnosis of corticobasal degeneration, Neurology 80(5) (2013) 496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yorkston KM, Strand E, Miller R, Hillel A & Smith K, Speech deterioration in amyotrophic lateral sclerosis: Implications for the timing of intervention, Journal of Medical Speech-Language Pathology 1 (1993) 35–46. [Google Scholar]
- [14].Kertesz A, Western aphasia battery test manual, Psychological Corp 1982.
- [15].Goetz CG, Tilley BC, Shaftman SR, Stebbins GT, Fahn S, Martinez-Martin P, Poewe W, Sampaio C, Stern MB, Dodel R, Dubois B, Holloway R, Jankovic J, Kulisevsky J, Lang AE, Lees A, Leurgans S, LeWitt PA, Nyenhuis D, Olanow CW, Rascol O, Schrag A, Teresi JA, van Hilten JJ, LaPelle N, Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): scale presentation and clinimetric testing results, Mov Disord 23(15) (2008) 2129–70. [DOI] [PubMed] [Google Scholar]
- [16].Kertesz A, Western aphasia battery (Revised), San Antonio, TX; PsychCorp; (2007). [Google Scholar]
- [17].Whitwell JL, Master AV, Avula R, Kantarci K, Eggers SD, Edmonson HA, Jack CR Jr., Josephs KA, Clinical correlates of white matter tract degeneration in progressive supranuclear palsy, Arch Neurol 68(6) (2011) 753–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Golbe LI, Ohman-Strickland PA, A clinical rating scale for progressive supranuclear palsy, Brain 130(Pt 6) (2007) 1552–65. [DOI] [PubMed] [Google Scholar]
- [19].Nasreddine ZS, Phillips NA, Bedirian V, Charbonneau S, Whitehead V, Collin I, Cummings JL, Chertkow H, The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment, J Am Geriatr Soc 53(4) (2005) 695–9. [DOI] [PubMed] [Google Scholar]
- [20].Dubois B, Slachevsky A, Litvan I, Pillon B, The FAB: a Frontal Assessment Battery at bedside, Neurology 55(11) (2000) 1621–6. [DOI] [PubMed] [Google Scholar]
- [21].Kaufer DI, Cummings JL, Ketchel P, Smith V, MacMillan A, Shelley T, Lopez OL, DeKosky ST, Validation of the NPI-Q, a brief clinical form of the Neuropsychiatric Inventory, J Neuropsychiatry Clin Neurosci 12(2) (2000) 233–9. [DOI] [PubMed] [Google Scholar]
- [22].Bezdicek O, Ruzicka F, Fendrych Mazancova A, Roth J, Dusek P, Mueller K, Ruzicka E, Jech R, Frontal Assessment Battery in Parkinson’s Disease: Validity and Morphological Correlates, J Int Neuropsychol Soc 23(8) (2017) 675–684. [DOI] [PubMed] [Google Scholar]
- [23].Tetzloff KA, Duffy JR, Clark HM, Utianski RL, Strand EA, Machulda MM, Botha H, Martin PR, Schwarz CG, Senjem ML, Reid RI, Gunter JL, Spychalla AJ, Knopman DS, Petersen RC, Jack CR, Lowe VJ, Josephs KA, Whitwell JL, Progressive agrammatic aphasia without apraxia of speech as a distinct syndrome, Brain 142(8) (2019) 2466–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Grimm MJ, Respondek G, Stamelou M, Arzberger T, Ferguson L, Gelpi E, Giese A, Grossman M, Irwin DJ, Pantelyat A, Rajput A, Roeber S, van Swieten JC, Troakes C, Antonini A, Bhatia KP, Colosimo C, van Eimeren T, Kassubek J, Levin J, Meissner WG, Nilsson C, Oertel WH, Piot I, Poewe W, Wenning GK, Boxer A, Golbe LI, Josephs KA, Litvan I, Morris HR, Whitwell JL, Compta Y, Corvol JC, Lang AE, Rowe JB, Hoglinger GU, How to apply the movement disorder society criteria for diagnosis of progressive supranuclear palsy, Mov Disord 34(8) (2019) 1228–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Minoshima S, Frey KA, Koeppe RA, Foster NL, Kuhl DE, A diagnostic approach in Alzheimer’s disease using three-dimensional stereotactic surface projections of fluorine-18-FDG PET, Journal of Nuclear Medicine 36(7) (1995) 1238. [PubMed] [Google Scholar]
- [26].Whitwell JL, Stevens CA, Duffy JR, Clark HM, Machulda MM, Strand EA, Martin PR, Utianski RL, Botha H, Spychalla AJ, Senjem ML, Schwarz CG, Jack CR Jr., Ali F, Hassan A, Josephs KA, An Evaluation of the Progressive Supranuclear Palsy Speech/Language Variant, Mov Disord Clin Pract 6(6) (2019) 452–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wenning GK, Litvan I, Jankovic J, Granata R, Mangone CA, McKee A, Poewe W, Jellinger K, Ray Chaudhuri K, D’Olhaberriague L, Pearce RK, Natural history and survival of 14 patients with corticobasal degeneration confirmed at postmortem examination, J Neurol Neurosurg Psychiatry 64(2) (1998) 184–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Leiguarda R, Merello M, Balej J, Apraxia in corticobasal degeneration, Adv Neurol 82 (2000) 103–21. [PubMed] [Google Scholar]
- [29].Josephs KA, Eggers SD, Jack CR Jr., Whitwell JL, Neuroanatomical correlates of the progressive supranuclear palsy corticobasal syndrome hybrid, Eur J Neurol 19(11) (2012) 1440–6. [DOI] [PubMed] [Google Scholar]
- [30].Josephs KA, Duffy JR, Strand EA, Machulda MM, Senjem ML, Lowe VJ, Jack CR Jr., Whitwell JL, Syndromes dominated by apraxia of speech show distinct characteristics from agrammatic PPA, Neurology 81(4) (2013) 337–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.