

Abstract
Objective:
Lipoprotein(a) (Lp(a]) is an independent risk factor for cardiovascular diseases and plasma levels are primarily determined by variation at the LPA locus. We performed a genome-wide association study (GWAS) in the UK Biobank to determine whether additional loci influence Lp(a) levels.
Approach and Results:
We included 293,274 White British individuals in the discovery analysis. Approximately 93,095,623 variants were tested for association with natural log-transformed Lp(a) levels using linear regression models adjusted for age, sex, genotype batch, and 20 principal components of genetic ancestry. After quality control, 131 independent variants were associated at genome-wide significance (P ≤ 5 × 10−8). In addition to validating previous associations at LPA, APOE, and CETP, we identified a novel variant at the APOH locus, encoding beta2-glycoprotein I (β2GPI). The APOH variant rs8178824 was associated with increased Lp(a) levels (β [95% CI] (ln nmol/L), 0.064 [0.047, 0.081]; P = 2.8 × 10−13) and demonstrated a stronger effect after adjustment for variation at the LPA locus (β [95% CI] (ln nmol/L), 0.089 [0.076, 0.10]; P = 3.8 × 10−42). This association was replicated in a meta-analysis of 5,465 European-ancestry individuals from the Framingham Offspring Study and Multi-Ethnic Study of Atherosclerosis (β [95% CI] (ln mg/dL), 0.16 [0.044, 0.28]; P = 0.0071).
Conclusions:
In a large-scale GWAS of Lp(a) levels, we identified APOH as a novel locus for Lp(a) in individuals of European ancestry. Additional studies are needed to determine the precise role of β2GPI in influencing Lp(a) levels as well as its potential as a therapeutic target.
Keywords: Lipoprotein(a), Lipids, Lipoproteins, GWAS
Subject codes: Lipids and Cholesterol, Genetics, Genetic Association Studies
Graphical Abstract
Introduction
Lipoprotein(a) (Lp[a]) is an independent risk factor for both coronary artery disease and aortic valve stenosis1,2. Lp(a) consists of a low-density lipoprotein (LDL)-like particle covalently bound to the glycoprotein apolipoprotein(a) (apo[a]). Levels of Lp(a) are primarily controlled by the size of the apo(a) protein, with smaller apo(a) isoforms leading to higher concentrations of plasma Lp(a). This size polymorphism is caused by a variable number of kringle IV type 2 repeats in the LPA gene. Together with other sequence variation in LPA, these kringle IV type 2 repeats are estimated to explain more than 90% of variability in Lp(a) concentration in individuals of European ancestry3,4.
Lp(a) plasma concentrations vary widely between populations, with African-ancestry individuals having 2–3 fold higher levels than European-ancestry individuals5,6. The distribution of Lp(a) is right-skewed across populations, with most individuals having very low levels7. While the precise physiological functions of Lp(a) are still unclear, there is evidence that it has proatherogenic and proinflammatory properties8. In pathophysiological studies, Lp(a) or apo(a) have been detected in both the lesioned intima of human arteries9–12 and in aortic valve lesions13,14.
Several genome-wide association studies (GWAS) of Lp(a) have been performed, highlighting LPA as the major genetic determinant of Lp(a) levels4,7,15–20. However, these studies have been limited by small sample sizes (N<15,000), sparse genotyping arrays, or a focus on founder populations. In this study, we aimed to identify novel loci for Lp(a) by performing a GWAS in nearly 300,000 individuals from the UK Biobank. The findings could provide further insights into the regulation and clearance of Lp(a) particles and highlight novel targets for Lp(a)-lowering therapies.
Materials and Methods
The data that were analyzed for this study are available from the UK Biobank or from dbGAP in the case of the Multi-Ethnic Study of Atherosclerosis and the Framingham Offspring Study. For details, please see the Major Resources Table in the Supplemental Material.
Study Population
The UK Biobank study recruited over 500,000 individuals aged 40–69 years from 22 recruitment centers across the United Kingdom between 2006 and 2010. Participants provided blood samples for DNA extraction and biomarker analysis and completed a series of questionnaires, as previously described21. UK Biobank received ethical approval from the North West Multi-Centre Research Ethics Committee and all participants provided written informed consent. All relevant internal review boards approved this study. Only White British individuals were included in the discovery analysis to reduce confounding by ancestry, where White British ancestry was determined using a combination of self-reported ethnicity and results from a principal component analysis21.
Phenotyping
Lp(a) (nmol/L) was measured using an immunoturbidimetric analysis on a Randox AU5800. Measurements were taken at the initial assessment visit (2006–2010) or the first repeat assessment visit (2012–2013). Measurements that returned an error from the analyzer or were outside of the reportable range (3.80–189 nmol/L) were excluded (n=91,426). Additional phenotypes are described in the Supplemental Material.
Genotyping
Genotyping was performed using the Affymetrix UK BiLEVE Axiom array on an initial 50,000 participants, while the remaining 450,000 participants were genotyped using the Affymetrix UK Biobank Axiom array. Quality control and imputation were performed centrally by the UK Biobank, as described previously21. Briefly, genetic markers were tested for batch effects, plate effects, departure from Hardy-Weinberg equilibrium, sex effects, array effects, and discordance across control replicates; markers that failed at least one test in a given batch had their genotype calls set to missing. Imputation was performed using only markers present on both the UK BiLEVE UK Biobank Axiom arrays, and markers that failed quality control in more than one batch, had a >5% missing rate, or had a minor allele frequency <0.0001 were removed. Samples with unusually high heterozygosity or >5% missing rate were excluded from analysis.
GWAS for Lp(a)
Associations of 93,095,623 genetic variants with natural log-transformed Lp(a) were tested in linear regression models assuming additive genetic effects in PLINK version 2.022,23. All models were adjusted for age, sex, genotype batch, and 20 principal components of ancestry. Variants with minor allele frequency < 0.01 or imputation quality score < 0.3 were removed (n=83,266,620). Clumping was performed on variants reaching genome-wide significance (P ≤ 5 × 10−8) with PLINK version 1.922,24; index variants were chosen greedily starting with the lowest p-value, and variants less than 1Mb away from an index variant with an r2 > 0.01 were assigned to that index variant’s clump. The most significant independent variants in each locus (lead variants) were queried for previous associations using PhenoScanner (accessed 02/04/2020)25,26. In a conditional analysis, all lead variants were tested for association with Lp(a) after additional adjustment for assessment center.
Conditional analysis of the LPA locus
A weighted LPA-region genetic risk score was created using all independent genome-wide significant variants in the LPA region. Lead variants outside the LPA region were tested for association with Lp(a) after adjusting for age, sex, genotype batch, 20 principal components, and the LPA-region genetic risk score.
Replication in Other Populations
The lead variant in each locus was tested for association with Lp(a) in other ethnic groups from the UK Biobank containing at least 1,000 unrelated individuals (South Asians, Black Africans, and Black Caribbeans). Lp(a) (nmol/L) was natural log-transformed and models were adjusted for age, sex, genotype batch, and 20 principal components. Lead variants were also assessed in a fixed-effects meta-analysis of self-reported White individuals from the Multi-Ethnic Study of Atherosclerosis (MESA) and the Framingham Offspring Study. The variant rs1065853 was not available in these cohorts so rs7412 was used as a proxy (linkage disequilbrium r2=0.99 in the UK Biobank). Cohort descriptions and model details are provided in the Supplemental Material.
Statistical Analyses
Two-sided p-values ≤ 5 × 10−8 were considered significant in the GWAS and two-sided p-values ≤ 0.05 were considered significant in all other analyses. Proportion of variance explained was calculated for independent significant variants in the LPA region independently, when modeled together, and when combined as a weighted genetic risk score.
Results
A total of 293,274 individuals with Lp(a) measurements were included in the study. Demographic characteristics of the UK Biobank Lp(a) subset are presented in Table I in the Supplemental Material, with individuals stratified by Lp(a) levels. Individuals with Lp(a) levels greater than or equal to the median were more likely to have coronary artery disease and aortic stenosis but were less likely to be female or diabetic (all P<0.001). These individuals also had higher levels of LDL cholesterol (corrected and uncorrected for Lp[a]) and high-density lipoprotein cholesterol (all P<0.001).
Following quality control, 9,829,003 variants with a minor allele frequency > 0.01 remained for further analysis. The association of these variants with Lp(a) showed no substantial inflation in the test statistics (genomic inflation factor [λ] = 1.03, Figure 1). After clumping, we identified 131 independent variants associated with Lp(a) at the genome-wide significance level of P ≤ 5 × 10−8. The most significant variant, rs10455872 in LPA, explained 29% of variance in Lp(a) levels (Table II in Supplemental Material). There were 126 other independent variants in the LPA region, explaining an additional 20% of variance.
Figure 1:
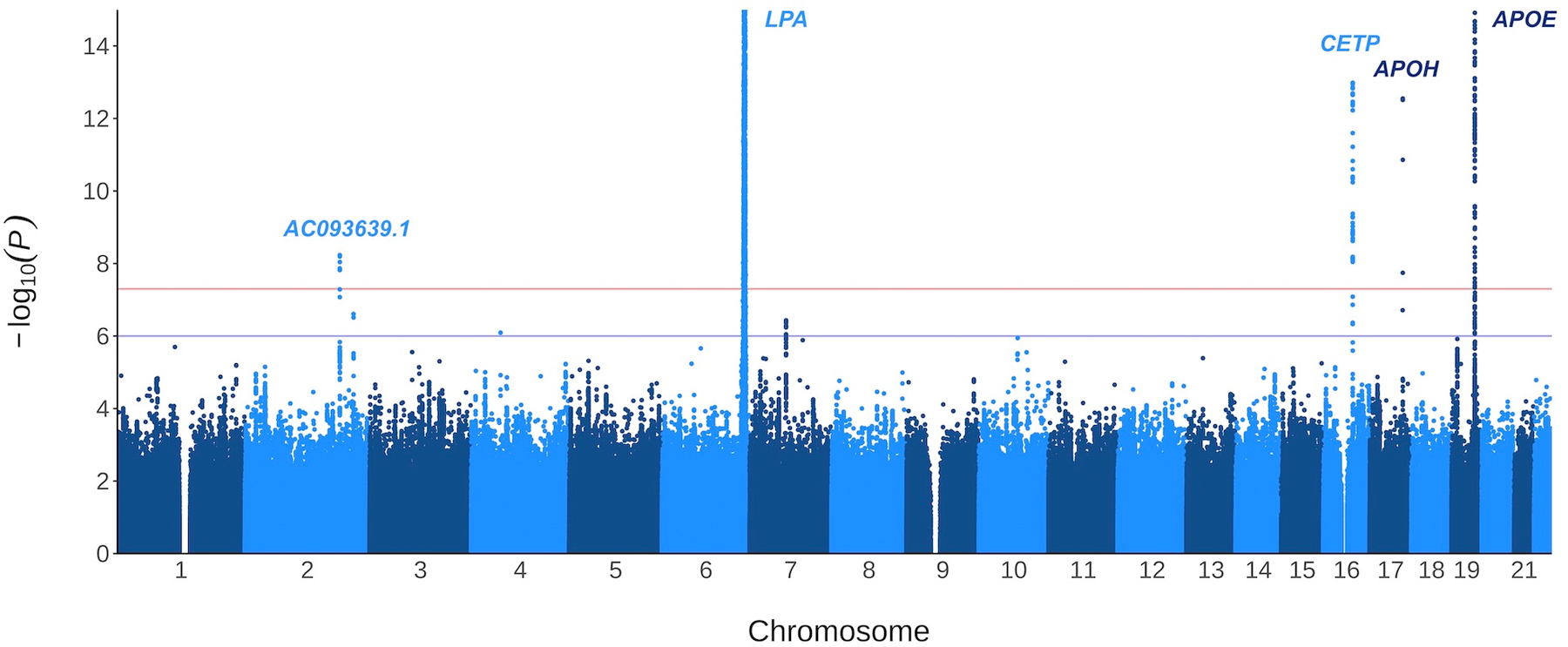
Log-transformed p-values for the association of 9,829,003 variants with natural log-transformed Lp(a). The plot has been cropped for better resolution, truncating only the signals at chromosome 6 (LPA region) and chromosome 19 (APOE region).
Outside the LPA region, we identified variants in four loci (Table 1). Variant rs1065853 on chromosome 19, located downstream of APOE, was associated with decreased Lp(a) levels (β [95% CI] (ln nmol/L), −0.11 [−0.12, −0.10]; P = 2.8 × 10−96), as was variant rs247617 on chromosome 16, located upstream of CETP (β [95% CI] (ln nmol/L), −0.023 [−0.030, −0.017]; P = 1.0 × 10−13). On chromosome 17, rs8178824 in APOH was associated with an increase in Lp(a) (β [95% CI] (ln nmol/L), 0.064 [0.047, 0.081]; P = 2.8 × 10−13). Finally, variant rs826128 on chromosome 2, located in the long non-coding RNA AC093639.1, was associated with decreased Lp(a) levels (β [95% CI] (ln nmol/L), −0.039 [−0.053, −0.026]; P = 5.9 × 10−9). The median Lp(a) level by genotype is shown for all lead variants in Table III of the Supplemental Material. Additional adjustment for assessment center did not materially change the results (Table IV in Supplemental Material).
Table 1:
Association of lead variants with Lp(a).
| Variant | CHR | Position | Genes in Locus | Minor Allele (Freq) | β [95% CI] ((ln nmol/L) | P | Variants in Locus* |
|---|---|---|---|---|---|---|---|
| rs10455872 | 6 | 161010118 | LPA, ZDHHC14, SNX9† | G (0.076) | 1.7 [1.7,1.7] | 6.2 × 10−22,136 | 127 |
| rs1065853 | 19 | 45413233 | APOE | T (0.080) | −0.11 [−0.12, −0.10] | 2.8 × 10−96 | 1 |
| rs247617 | 16 | 56990716 | CETP | A (0.32) | −0.023 [−0.030, −0.017] | 1.0 × 10−13 | 1 |
| rs8178824 | 17 | 64224775 | APOH | T (0.030) | 0.064 [0.047,0.081] | 2.8 × 10−13 | 1 |
| rs826128 | 2 | 184797074 | AC093639.1 | A (0.054) | −0.039 [−0.053, −0.026] | 5.9 × 10−9 | 1 |
Number of independent (r2 < 0.01), genome-wide significant variants.
Additional genes (+/− 3.5 MB from LPA): TULP4, SYTL3, EZR, RP1-155D22.1, RSPH3, RP1-111C20.4, FNDC1, RP11-125D12.1, RP11-125D12.2, RP3-393E18.1, SOD2, ACAT2, PNLDC1, RP1-249F5.3, IGF2R, SLC22A1, SLC22A2, SLC22A3, LPAL2, PLG, RP11-235G24.1, RP11-235G24.2, RP11-235G24.3, RP3-428L16.1, MAP3K4, AGPAT4, PARK2.
The lead variant in each locus was evaluated for association with Lp(a) in the following populations from the UK Biobank: South Asian (n=6,101), Black African (n=2,510), and Black Caribbean (n=3,207). Consistent with results in White British individuals, rs10455872 was associated with increased Lp(a) in South Asians (β [95% CI] (ln nmol/L), 1.01 [0.82,1.19]; P = 8.5 × 10−26) and Black Caribbeans (β [95% CI] (ln nmol/L), 0.81 [0.59, 1.03]; P = 1.4 × 10−12). This variant showed no association in Black Africans, likely owing to its low frequency in this population (minor allele frequency = 6.3 × 10−4). The variant rs1065853 near APOE was significantly associated with Lp(a) across ethnic groups, with Black African individuals demonstrating the largest decrease in levels per minor allele (T) (β [95% CI] (ln nmol/L), −0.28 [−0.34, −0.21]; P = 9.4 × 10−16). The lead variants in CETP, APOH, and AC093639.1 were not significantly associated in South Asians, Black Africans, or Black Caribbeans (Table V in Supplemental Material).
The lead variants were also tested for association with Lp(a) in a meta-analysis of 5,465 European-ancestry individuals from the Multi-Ethnic Study of Atherosclerosis and the Framingham Offspring Study. Both rs10455872 in LPA and rs8178824 in APOH were significantly associated with increased Lp(a) levels in the meta-analysis (rs10455872: β [95% CI] (ln mg/dL), 2.1 [2.0, 2.2]; P = 6.0 × 10−534, and rs8178824: β [95% CI] (ln mg/dL), 0.16 [0.044, 0.28]; P = 0.0071). The lead variants in APOE, CETP, and AC093639.1 showed no significant effects (Table VI in Supplemental Material).
The weighted LPA-region genetic risk score contained 127 variants and explained 44% of the variance in Lp(a) levels. After adjusting for this score, variants in APOE, CETP, and APOH showed stronger effects on Lp(a) (Table VII in Supplemental Material). Conversely, the variant rs826128 on chromosome 2 showed a decreased effect and no longer reached genome-wide significance (β [95% CI] (ln nmol/L), −0.016 [−0.026, −0.0058]; P = 0.0019).
Discussion
We performed a GWAS for plasma Lp(a) levels in 293,274 White British individuals from the UK Biobank. We confirmed the association of loci in the LPA region with Lp(a) levels, as well as APOE and CETP. In addition, we identified APOH as a novel risk locus and replicated this association in a meta-analysis of two independent cohorts.
As expected, our association study identified many significant variants in the LPA gene and the surrounding region. Despite imposing a stringent r2 threshold (≤ 0.01), 127 variants were independently associated with Lp(a). Together, the top 4 variants explained 40% of the variance in Lp(a) levels, while the remaining 123 explained an additional 9%. Consistent with previous work1,20,27, the variant rs10455872 was the most strongly associated with Lp(a), explaining 29% of variation in Lp(a) levels alone.
Outside of the LPA region, we identified variants at four loci, two of which have been previously associated with Lp(a) or Lp(a)-cholesterol: APOE and CETP. Our lead variant near the APOE locus, rs1065853, is in high linkage disequilibrium with the apoE2-defining variant rs7412 (r2=0.99), which has been previously associated with decreased Lp(a)4,19,28,29. Relative to the apoE3 and apoE4 isoforms, apoE2 has a lower affinity for LDL receptors and LDL receptor-related protein I, potentially leading to less competition for Lp(a) binding and greater clearance of Lp(a)30. Upstream of the CETP locus, the variant rs247617 was also associated with decreased Lp(a) levels. This finding is consistent with clinical studies showing that inhibition of cholesteryl ester transfer protein, the product of CETP, decreases Lp(a) levels31–33. This variant is also in high linkage disequilibrium with rs247616 (r2=0.99), which has been previously associated with Lp(a)-cholesterol28.
Apart from APOE and CETP, no other loci outside the LPA region have been associated with Lp(a) levels at the genome-wide significance level. Here, we identify rs8178824 in APOH as significantly associated with increased Lp(a) and provide independent replication. Relative to rs10455872, the effect size of rs8178824 is small, with individuals homozygous for the minor allele having a median Lp(a) level only 4.7 nmol/L higher than individuals with two major alleles. However, this effect is comparable to those seen for APOE and CETP, where the difference in homozygous genotype classes is 10.5 nmol/L and 1.9 nmol/L, respectively. As demonstrated previously with treatments targeting CETP, which produced reductions in Lp(a) of more than 30%31–33, therapeutic targeting of the APOH locus could have a more substantial effect on Lp(a) than the effects of a single variant.
The APOH locus encodes beta2-glycoprotein I (β2GPI), a single chain plasma protein with a high affinity for negatively charged surfaces34. Recently, β2GPI has been shown to interact with proprotein convertase subtilisin/kexin-9 (PCSK9)35, whose inhibition leads to reductions in LDL cholesterol36. This evidence is supported by previous studies demonstrating that genetic variation in APOH is associated with decreased levels of LDL cholesterol37–40 and peak particle diameter41. In vitro, β2GPI has also been shown to bind to Lp(a), both through the phospholipids of the LDL component and through the kringle IV-domain of apo(a)42. Given that apo(a) is a major site for the accumulation of negatively charged oxidized phospholipids43, the interaction of β2GPI and Lp(a) may be primarily mediated through binding of β2GPI to these phospholipids.
The variant we identified in APOH is in perfect linkage disequilibrium with rs1801689 (r2=1.0). Interestingly, the amino acid change caused by rs1801689 (Cys325Gly, also known as Cys306Gly) has been shown to disrupt the ability of β2GPI to bind to phospholipids44. This change may reduce β2GPI’s affinity for oxidized phospholipids on apo(a), thereby allowing more free molecules of β2GPI and Lp(a) to circulate in the plasma. Indeed, rs1801689 has also been previously associated with increased levels of plasma β2GPI45,46. The potential role of β2GPI in lipid metabolism is further supported by the observation that it can accelerate triglyceride clearance in rats47. Future studies should investigate whether the presence of β2GPI similarly affects Lp(a) clearance or affects its pathogenicity through other mechanisms.
This study has several strengths and limitations. The UK Biobank discovery sample was larger than any previous Lp(a) GWAS, and thus had more power to detect novel associations. In addition, we were able to replicate our novel finding in APOH in a meta-analysis of two other European-ancestry cohorts. However, the APOH variant showed no association with Lp(a) in other ethnicities from the UK Biobank. The lack of replication observed for this variant and others may reflect reduced power due to smaller sample sizes, different allele frequencies, or different patterns of linkage disequilibrium48; nonetheless, additional studies in larger non-European cohorts are warranted. Another limitation of our study is the high percentage of individuals missing Lp(a) measurements (>20%) in the UK Biobank due to the assay’s limited reportable range (3.80–189 nmol/L); our results may therefore not apply to individuals with very high levels of Lp(a).
In summary, we have performed a large-scale GWAS of Lp(a) levels, validating previous loci and identifying APOH as a novel locus. Our findings provide further insight into the regulation of Lp(a) levels and highlight β2GPI as a potential therapeutic target in individuals with elevated Lp(a).
Supplementary Material
Highlights.
We have performed a large-scale genome-wide association study of Lp(a) levels.
We confirmed the association of variants in LPA, APOE, and CETP with Lp(a).
We identified APOH as a novel risk locus for Lp(a), highlighting β2GPI as a determinant of Lp(a) levels and a potential therapeutic target.
Acknowledgements
This research has been conducted using the UK Biobank Resource under application number 41025, with supporting data from the Framingham Heart Study of the National Heart Lung and Blood Institute (NHLBI) of the National Institutes of Health and Boston University School of Medicine, as well as the Multi-Ethnic Study of Atherosclerosis (MESA). We thank the investigators, staff, and participants of these cohorts for their valuable contributions. A full list of participating MESA institutions can be found at http://www.mesa-nhlbi.org.
Sources of Funding
Ms Hoekstra was supported by an award from the Canadian Institutes of Health Research (CIHR) for the duration of the study. Ms Chen has received grants from the McGill University Faculty of Medicine and McGill University Health Centre Foundation. Dr Thanassoulis has received research support from the CIHR, the Heart and Stroke Foundation of Canada, the NHLBI (grant R01 HL128550), the Doggone Foundation, the Courtois Foundation, and the Satoko Shibata and Richard Ingram Foundation. Dr Thanassoulis receives salary support from the Fonds de Recherche Santé–Québec.
MESA and the MESA SHARe project are conducted and supported by the NHLBI in collaboration with MESA investigators. Support for MESA is provided by contracts 75N92020D00001, HHSN268201500003I, N01-HC-95159, 75N92020D00005, N01-HC-95160, 75N92020D00002, N01-HC-95161, 75N92020D00003, N01-HC-95162, 75N92020D00006, N01-HC-95163, 75N92020D00004, N01-HC-95164, 75N92020D00007, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169, UL1-TR-000040, UL1-TR-001079, UL1-TR-001420. Also supported in part by the National Center for Advancing Translational Sciences, CTSI grant UL1TR001881, and the National Institute of Diabetes and Digestive and Kidney Disease Diabetes Research Center grant DK063491 to the Southern California Diabetes Endocrinology Research Center. The funding for SHARe genotyping was provided by NHLBI contract N02-HL-64278. Genotyping was performed at Affymetrix (Santa Clara, California, USA) and the Broad Institute of Harvard and MIT (Boston, Massachusetts, USA) using the Affymetrix Genome-Wide Human SNP Array 6.0. The Framingham Heart Study acknowledges the support of contracts NO1-HC-25195, HHSN268201500001I and 75N92019D00031 from the NHLBI and grant supplement R01 HL092577-06S1 for this research.
Abbreviations
- Lp(a)
Lipoprotein(a)
- GWAS
Genome-wide association study
- Apo(a)
Apolipoprotein(a)
- LDL
Low-density lipoprotein
- β2GPI
Beta2-glycoprotein I
- MESA
Multi-Ethnic Study of Atherosclerosis
Footnotes
Disclosures
Dr Thanassoulis has participated in advisory boards for Amgen, Sanofi/Regeneron, Ionis, HLS Therapeutics, and Servier Canada and has received research grants from Ionis and Servier for research outside the scope of this work. Dr Tsimikas is a co-inventor and receives royalties from patents owned by UCSD on oxidation-specific antibodies and of biomarkers related to oxidized lipoproteins and is a co-founder and has an equity interest in Oxitope, Inc and its affiliates (“Oxitope”) as well as in Kleanthi Diagnostics, LLC (“Kleanthi”). Although these relationships have been identified for conflict of interest management based on the overall scope of the project and its potential benefit to Oxitope and Kleanthi, the research findings included in this particular publication may not necessarily relate to the interests of Oxitope and Kleanthi. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies.
References
- 1.Clarke R, Peden JF, Hopewell JC, et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med 2009;361:2518–2528. [DOI] [PubMed] [Google Scholar]
- 2.Thanassoulis G, Campbell CY, Owens DS, et al. Genetic Associations with Valvular Calcification and Aortic Stenosis. N Engl J Med 2013;368:503–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boerwinkle E, Leffert CC, Lin J, Lackner C, Chiesa G, Hobbs HH. Apolipoprotein(a) gene accounts for greater than 90% of the variation in plasma lipoprotein(a) concentrations. J Clin Invest 1992;90:52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mack S, Coassin S, Rueedi R, et al. A genome-wide association meta-analysis on lipoprotein (a) concentrations adjusted for apolipoprotein (a) isoforms. J Lipid Res 2017;58:1834–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kraft HG. Frequency distributions of apolipoprotein(a) Kringle IV repeat alleles and their effects on lipoprotein(a) levels in Caucasian, Asian, and African populations: The distribution of null alleles is non-random. Eur J Hum Genet 1996;4:74–87. [DOI] [PubMed] [Google Scholar]
- 6.Cobbaert C, Kesteloot H. Serum lipoprotein(a) levels in racially different populations. Am J Epidemiol 1992;136:441–449. [DOI] [PubMed] [Google Scholar]
- 7.Kronenberg F, Utermann G. Lipoprotein(a): Resurrected by genetics. J Intern Med 2013;273:6–30. [DOI] [PubMed] [Google Scholar]
- 8.Orsó E, Schmitz G. Lipoprotein(a) and its role in inflammation, atherosclerosis and malignancies. Clin Res Cardiol Suppl 2017;12:31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rath M, Niendorf A, Reblin T, Dietel M, Krebber HJ, Beisiegel U. Detection and quantification of lipoprotein(a) in the arterial wall of 107 coronary bypass patients. Arteriosclerosis 1989;9:579–592. [DOI] [PubMed] [Google Scholar]
- 10.Niendorf A, Rath M, Wolf K, Peters S, Arps H, Beisiegel U, Dietel M. Morphological detection and quantification of lipoprotein(a) deposition in atheromatous lesions of human aorta and coronary arteries. Virchows Arch A Pathol Anat Histopathol 1990;417:105–111. [DOI] [PubMed] [Google Scholar]
- 11.Hoff HF, Neil JO, Yashiro A. Partial characterization of Iipoproteins containing apo [a] in human atherosclerotic lesions. J Lipid Res 1993;34:789–798. [PubMed] [Google Scholar]
- 12.Reblin T, Meyer N, Labeur C, Henne-Bruns D, Beisiegel U. Extraction of lipoprotein(a), apo B, and apo E from fresh human arterial wall and atherosclerotic plaques. Atherosclerosis 1995;113:179–188. [DOI] [PubMed] [Google Scholar]
- 13.O’Brien KD, Reichenbach DD, Marcovina SM, Kuusisto J, Alpers CE, Otto CM. Apolipoproteins B, (a), and E accumulate in the morphologically early lesion of “degenerative” valvular aortic stenosis. Arterioscler Thromb Vasc Biol 1996;16:523–532. [DOI] [PubMed] [Google Scholar]
- 14.Torzewski M, Ravandi A, Yeang C, et al. Lipoprotein(a)-Associated Molecules Are Prominent Components in Plasma and Valve Leaflets in Calcific Aortic Valve Stenosis. JACC Basic to Transl Sci 2017;2:229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Y, Ma H, Zhu Q, et al. A genome-wide association study on lipoprotein (a) levels and coronary artery disease severity in a Chinese population. J Lipid Res 2019;60:1440–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu W, Cheng YC, Chen K, et al. Evidence for several independent genetic variants affecting lipoprotein (a) cholesterol levels. Hum Mol Genet 2015;24:2390–2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ober C, Nord AS, Thompson EE, et al. Genome-wide association study of plasma lipoprotein(a) levels identifies multiple genes on chromosome 6q. J Lipid Res 2009;50:798–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qi Q, Workalemahu T, Zhang C, Hu FB, Qi L. Genetic variants, plasma lipoprotein(a) levels, and risk of cardiovascular morbidity and mortality among two prospective cohorts of type 2 diabetes. Eur Heart J 2012;33:325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li J, Lange LA, Sabourin J, Duan Q, Valdar W, Willis MS, Li Y, Wilson JG, Lange EM. Genome- and exome-wide association study of serum lipoprotein (a) in the Jackson Heart Study. J Hum Genet 2015;60:755–761. [DOI] [PubMed] [Google Scholar]
- 20.Zabaneh D, Kumari M, Sandhu M, et al. Meta analysis of candidate gene variants outside the LPA locus with Lp(a) plasma levels in 14,500 participants of six White European cohorts. Atherosclerosis 2011;217:447–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bycroft C, Freeman C, Petkova D, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018;562:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang CC, Chow CC, Tellier LCAM, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Purcell S, Chang C. PLINK 2.0 www.cog-genomics.org/plink/2.0/
- 24.Purcell S, Chang C. PLINK 1.9 www.cog-genomics.org/plink/1.9/
- 25.Staley JR, Blackshaw J, Kamat MA, et al. PhenoScanner: A database of human genotype-phenotype associations. Bioinformatics 2016;32:3207–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kamat MA, Blackshaw JA, Young R, Surendran P, Burgess S, Danesh J, Butterworth AS, Staley JR. PhenoScanner V2: an expanded tool for searching human genotype-phenotype associations. Bioinformatics 2019;35:4851–4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ronald J, Rajagopalan R, Cerrato F, Nord AS, Hatsukami T, Kohler T, Marcovina S, Heagerty P, Jarvik GP. Genetic variation in LPAL2, LPA, and PLG predicts plasma lipoprotein(a) level and carotid artery disease risk. Stroke 2011;42:2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zekavat SM, Ruotsalainen S, Handsaker RE, et al. Deep coverage whole genome sequences and plasma lipoprotein(a) in individuals of European and African ancestries. Nat Commun 2018;9:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moriarty PM, Varvel SA, Gordts PLSM, McConnell JP, Tsimikas S. Lipoprotein(a) Mass Levels Increase Significantly According to APOE Genotype: An Analysis of 431 239 Patients. Arterioscler Thromb Vasc Biol 2017;37:580–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Phillips MC. Apolipoprotein e isoforms and lipoprotein metabolism. IUBMB Life 2014;66:616–623. [DOI] [PubMed] [Google Scholar]
- 31.Bloomfield D, Carlson GL, Sapre A, Tribble D, McKenney JM, Littlejohn TW, Sisk CMC, Mitchel Y, Pasternak RC. Efficacy and safety of the cholesteryl ester transfer protein inhibitor anacetrapib as monotherapy and coadministered with atorvastatin in dyslipidemic patients. Am Heart J 2009;157:352–360. [DOI] [PubMed] [Google Scholar]
- 32.Cannon CP, Shah S, Dansky HM, et al. Safety of anacetrapib in patients with or at high risk for coronary heart disease. N Engl J Med 2010;363:2406–2415. [DOI] [PubMed] [Google Scholar]
- 33.Thomas T, Zhou H, Karmally W, et al. CETP (Cholesteryl Ester Transfer Protein) Inhibition with Anacetrapib Decreases Production of Lipoprotein(a) in Mildly Hypercholesterolemic Subjects. Arterioscler Thromb Vasc Biol 2017;37:1770–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miyakis S, Giannakopoulos B, Krilis SA. Beta 2 glycoprotein I-function in health and disease. Thromb Res 2004;114:335–346. [DOI] [PubMed] [Google Scholar]
- 35.Melendez QM, Wooten CJ, Krishnaji ST, Knagge K, Kirchner D, Lopez D. Identification of Novel Proteins Interacting with Proprotein Convertase Subtilisin/Kexin 9 2020;3:1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Horton JD, Cohen JC, Hobbs HH. Molecular biology of PCSK9: its role in LDL metabolism. Trends Biochem Sci 2007;32:71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leduc MS, Shimmin LC, Klos KLE, Hanis C, Boerwinkle E, Hixson JE. Comprehensive evaluation of apolipoprotein H gene (APOH) variation identifies novel associations with measures of lipid metabolism in GENOA. J Lipid Res 2008;49:2648–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Asselbergs FW, Guo Y, Van Iperen EPA, et al. Large-scale gene-centric meta-analysis across 32 studies identifies multiple lipid loci. Am J Hum Genet 2012;91:823–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elbers CC, Guo Y, Tragante V, et al. Gene-Centric Meta-Analysis of Lipid Traits in African, East Asian and Hispanic Populations. PLoS One 2012;7:e50198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Willer CJ, Schmidt EM, Sengupta S, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet 2013;45:1274–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bossé Y, Feitosa MF, Després JP, Lamarche B, Rice T, Rao DC, Bouchard C, Pérusse L, Vohl MC. Detection of a major gene effect for LDL peak particle diameter and association with apolipoprotein H gene haplotype. Atherosclerosis 2005;182:231–239. [DOI] [PubMed] [Google Scholar]
- 42.Köchl S, Fresser F, Lobentanz E, Baier G, Utermann G. Novel interaction of apolipoprotein(a) with β−2 glycoprotein I mediated by the kringle IV domain. Blood 1997;90:1482–1489. [PubMed] [Google Scholar]
- 43.Leibundgut G, Scipione C, Yin H, et al. Determinants of binding of oxidized phospholipids on apolipoprotein (a) and lipoprotein (a). J Lipid Res 2013;54:2815–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sanghera DK, Wagenknecht DR, McIntyre JA, Kamboh MI. Identification of structural mutations in the fifth domain of apolipoprotein H (β2-glycoprotein I) which affect phospholipid binding. Hum Mol Genet 1997;6:311–316. [DOI] [PubMed] [Google Scholar]
- 45.Kamboh MI, Manzi S, Mehdi H, Fitzgerald S, Sanghera DK, Kuller LH, Atson CE. Genetic variation in apolipoprotein H (β2-glycoprotein I) affects the occurrence of antiphospholipid antibodies and apolipoprotein H concentrations in systemic lupus erythematosus. Lupus 1999;8:742–750. [DOI] [PubMed] [Google Scholar]
- 46.Mather KA, Thalamuthu A, Oldmeadow C, et al. Genome-wide significant results identified for plasma apolipoprotein H levels in middle-aged and older adults. Sci Rep 2016;6:23675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wurm H, Beubler E, Polz E, Holasek A, Kostner G. Studies on the possible function of β2-glycoprotein-I: Influence in the triglyceride metabolism in the rat. Metabolism 1982;31:484–486. [DOI] [PubMed] [Google Scholar]
- 48.Gurdasani D, Barroso I, Zeggini E, Sandhu MS. Genomics of disease risk in globally diverse populations. Nat Rev Genet 2019;20:520–535. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.