

Abstract
Jab1, also known as Csn5/Cops5, is a key subunit of the COP9 Signalosome, a highly conserved macromolecular complex. We previously reported that the conditional knockout of Jab1 in mouse limb buds and chondrocytes results in severely shortened limbs and neonatal lethal chondrodysplasia, respectively. In this study, we further investigated the specific role of Jab1 in osteoblast differentiation and postnatal bone growth by characterizing a novel mouse model, the Osx-cre; Jab1flox/flox conditional knockout (Jab1 cKO) mouse, in which Jab1 is deleted in osteoblast precursor cells. Jab1 cKO mutant mice appeared normal at birth, but developed progressive dwarfism. Inevitably, all mutant mice died prior to weaning age. The histological and micro-computed tomography analysis of mutant long bones revealed severely altered bone microarchitecture, with a significant reduction in trabecular thickness. Moreover, Jab1 cKO mouse tibiae had a drastic decrease in mineralization near the epiphyseal growth plates, and Jab1 cKO mice also developed spontaneous fractures near the tibiofibular junction. Additionally, our cell culture studies demonstrated that Jab1 deletion in osteoblast precursors led to decreased mineralization and a reduced response to TGFβ and BMP signaling. Moreover, an unbiased reporter screen also identified decreased TGFβ activity in Jab1-knockdown osteoblasts. Thus, Jab1 is necessary for proper osteoblast differentiation and postnatal bone growth, likely in part through its positive regulation of the TGFβ and BMP signaling pathways in osteoblast progenitor cells.
Keywords: Jab1/Csn5/Cops5, COP9 Signalosome, Osteoblast Precursors, TGFβ/BMP Signaling, Bone Microarchitecture
1. Introduction
Bone homeostasis is a lifelong process in which bone is remodeled by the concerted actions of bone forming osteoblasts and bone resorbing osteoclasts (1). Moreover, osteoblast derived osteocytes are mechanosensors that coordinate the activity of osteoblasts and osteoclasts (1). Osteoblasts also play essential roles in regulating hematopoietic cells in the bone marrow (2). Many signaling pathways are crucial for proper bone development and homeostasis, especially the Transforming Growth Factor β (TGFβ) and Bone Morphogenetic Protein (BMP) signaling pathways (3, 4). Canonical TGFβ/BMP signaling is transduced through downstream effector R-Smads, Smad2/3 (TGFβ) and Smad1/5/8 (BMP). The activation of R-Smads is mediated by a ligand binding induced dimerization of Type I and Type II TGFβ or BMP receptors, resulting in phosphorylation and activation of the R-Smads. Activated R-Smads then bind with Smad4, and translocate to the nucleus to promote transcription of TGFβ/BMP target genes (3, 4). The TGFβ/BMP signaling pathway is evolutionarily conserved in metazoans and plays many crucial roles in bone and cartilage development and homeostasis. TGFβ/BMP dysregulation results in various osteochondrodysplasias and other diseases, including cancer (4).
Jab1, also known as Cops5/Csn5, is an essential component of the evolutionarily conserved COP9 Signalosome (CSN) complex (5). Jab1 is the only CSN subunit that harbors a JAMM (Jab1/MPN/Mov34 metalloenzyme) motif necessary for the enzymatic removal of NEDD8 (deNEDDylation), a small, ubiquitin-like protein, from the Cullin subunit of the Cullin-RING Ligases (CRLs), largest family of E3 ubiquitin ligases (5). The CSN complex is required for animal survival, which is highlighted by the fact that the knockout of any individual CSN subunit in mice so far all result in early embryonic lethality (6, 7). Interestingly, Jab1 was also independently identified as a co-activator of c-Jun and JunD (8). Outside of the CSN complex, Jab1 also plays a versatile role in regulating the localization, stability, and activity of many transcription factors, including p53, HIF-1α, AP-1 family members, and various Smads (8-13). Thus, Jab1 is essential for regulating many cellular functions, including differentiation, apoptosis, proliferation, DNA damage response, and numerous signal transduction pathways (7). Our lab previously demonstrated that Jab1 is necessary for the successive stages of mouse skeletogenesis during embryogenesis, likely through regulating the TFGβ/BMP signaling pathways in a context-specific manner (14, 15). Prx1-cre-mediated deletion of Jab1 specifically in osteochondral progenitor cells of the limb buds resulted in severely shortened limbs at birth with a drastically disorganized epiphyseal growth plate, impaired osteoblastogenesis, decreased chondrogenesis, and a reduced response to BMP signaling (15). On the other hand, the chondrocyte-specific deletion of Jab1 using a Col2a1-cre driver resulted in severe chondrodysplasia and neonatal lethality, with a severe growth plate defect (14). Col2a1-cre; Jab1flox/flox chondrocytes displayed increased apoptosis, impaired cell cycle progression, and increased expression of BMP signaling effectors (14). Thus, Jab1 is necessary for proper skeletal development during mouse embryogenesis, at least in part through its regulation of BMP signaling activity, in a stage-specific manner. However, the specific role of Jab1 in osteoblast progenitor cells and postnatal bone growth was unknown until this study.
Thus, to investigate the role of Jab1 in bone homeostasis, we deleted Jab1 in osteoblast precursor cells using the Osx1-GFP::Cre (hereafter referred to as Osx-cre) deleter (16). Interestingly, while the Osx-cre; Jab1flox/flox conditional knockout (cKO) mice appeared normal at birth, they then all developed progressive dwarfism. Surprisingly, Jab1 cKO mice died with 100% penetrance prior to weaning age. Histological and micro-computed tomography (μCT) analyses revealed that these mice have significantly decreased postnatal bone growth, a delayed secondary ossification center formation, and severely altered bone microarchitecture. Furthermore, there is impaired osteoblast differentiation and mineralization, concomitant with a reduced response to TGFβ/BMP signaling in Jab1 cKO osteoblast precursor cells.
2. Materials and Methods
2.1. Mouse Studies
All animal protocols were approved by the IACUC of Case Western Reserve University. All mice were maintained and housed at Case Western Reserve University’s animal facility under standard conditions, as in previous studies (14, 15). The Jab1flox/flox mice were obtained from Dr. Ruggero Pardi (17). Osx1-GFP::Cre mice (16) and Gt(ROSA)26Sortm1Sor (18) mice were purchased from The Jackson Laboratory (Stock No. 006361 and 003309, respectively).
2.2. Cell Culture
The osteoblast precursor cell line MC3T3-E1 (CRL-2593) and mouse bone marrow stromal cell (mBMSC) (CRL-12424) lines were purchased from ATCC (Manassas, VA, USA) and were cultured according to the manufacturer’s instructions. For lentiviral infection, MC3T3-E1 and mBMSCs cells were infected with Mission shRNAs (Sigma-Aldrich, St. Louis, MO, USA) specifically targeting Jab1 (shRNAs 1, 2, and 3 correspond to TRCN0000304598, TRCN0000311065, and TRCN0000302049, respectively) or a Non-Mammalian shRNA control (SHC002V) at MOI = 3 (MC3T3-E1 cells) or MOI = 5 (mBMSCs cells) per the manufacturer’s protocol (19). 48 hours after infections, cells were selected using puromycin at concentrations of 3.5μg/mL or 6μg/mL for MC3T3-E1 and mBMSCs, respectively. The Cignal Finder Reporter Array #CCA-901L-12 (Qiagen) was used to measure signal transduction pathway reporter activities according to the manufacturer’s protocol as previously described (19). Briefly, the control and Jab1-knockdown MC3T3-E1 cells were reverse transfected by plating into 96-well plates, and signal pathway reporter activities were measured using the dual luciferase assay kit (Promega, Madison, WI, USA) according to the manufacturer’s protocol.
Primary mouse calvarial osteoblasts were isolated from P5 Jab1flox/flox mice by serial Collagenase P digestions, and expanded for 48hrs, as previously described (20). Cells were then infected with adenoviruses expressing either Cre Recombinase (Ad-Cre) or Green Fluorescent Protein (Ad- GFP) (Gene Transfer Vector Core) at MOI = 500, and analyzed as previously described (14, 15). Osteoblast differentiation was induced by culturing cells in α-MEM supplemented with 5 mM β-Glycerophosphate and 100μg/mL ascorbic acid for 21 days, as previously described (20).
2.3. Western Blotting
Total proteins were extracted from MC3T3-E1 cells and mBMSCs in RIPA buffer (Thermo Fisher Scientific, Waltham, MA, USA), or from mouse primary calvarial osteoblasts in 5% SDS, and supplemented with Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). Subsequent western blotting was performed as previously described (14, 15, 19). The antibodies used in this study are listed in Supplementary Table S1.
2.3. RNA Isolation, cDNA Synthesis, and Real-Time Quantitative RT-PCR
Total RNAs were isolated from MC3T3-E1 cells and primary calvarial osteoblasts using TRIzol reagent (Invitrogen) and the PureLink RNA Mini kit (Ambion, Foster City, CA, USA) as previously described (14, 15, 19). The primers in this study are listed in Supplementary Table S2.
2.4. Histology
The entire P18 mouse hindlimbs were separated from the pelvis, placed into a cassette with the lateral side down, and fixed for 16 hrs in 10% neutral buffered formalin. After 3 washes of PBS, the hindlimbs were decalcified in 0.5M EDTA for 21 days. Tissues were then embedded in paraffin, sectioned at 5μm thickness, and stained with H&E as previously described (14, 15, 19, 21). Von Kossa staining (22), Oil Red O Staining, (23), and β-galactosidase staining (24) was performed as previously described.
2.5. Micro-Computed Tomography (μCT)
Whole tibiae were excised from P18-P20 mice and fixed in 10% neutral buffered formalin for 16 hrs. After 3 washes in PBS, the samples were stored in 70% ethanol at 4°C. Prior to μCT analysis, tibiae were first rehydrated in PBS containing calcium and magnesium to minimize mineral dissolution (25). Scans were conducted in a Bruker SkyScan1172 (Bruker MicroCT, Kontich, Belgium) with a 11 MPix camera at an isotropic voxel size of 8 μm employing a 0.5mm thick aluminum filter. An applied x-ray tube voltage of 50kV with an x-ray intensity of 100μA was applied over 180 degrees of rotation with acquisition every 0.45 degrees. Reconstruction was carried out with a modified Feldkamp algorithm (26) using the SkyScan NRecon software accelerated by GPU (27). Tibial reconstruction alignments were adjusted with SkyScan DataView software to a consistent orientation. The selection of the Region/volume of interest (ROI/VOI), the segmentation to binary, and the morphometric analysis were all performed using SkyScan CT-Analyser (“CTAn”) software. Morphometry measurements for both cortical and trabecular bone were based on previously reported guidelines (28). Tibial lengths were measured from proximal to distal epiphyses. The cortical VOI was defined as 0.5mm (62 slices) beginning 10 slices proximally offset (0.08mm) from 58% of the metaphysis-metaphysis length, which is the mean position of the tibia-fibula junction (TFJ) in wild type mice. At this age, the proximal metaphysis does not have a clear cortical-trabecular separation (29). Therefore, the entire proximal metaphysis was analyzed for 3D trabecular morphometry. Due to significant size differences among genotypes, the analysis of trabecular VOI was defined as 25% of the metaphysis-metaphysis length.
2.6. Statistical Analysis
All experiments were conducted using at least 3 biological replicates. Data are expressed as mean ± standard deviation. With the exception of μCT data, all statistical analysis was performed using a 2-tailed Student’s t-test on normally distributed data, with p < 0.05 considered significant. Statistical tests on μCT data were performed with GraphPad Prism version 8.4.2 software (GraphPad Software, La Jolla, CA, USA). Data were assessed using D’Agostino & Pearson normality testing and Brown-Forsythe test for Equal Variance. Normally distributed data exhibiting equal variance were analyzed by one-way ANOVA with post-hoc analysis by Bonferroni’s multiple comparisons test. Data that were not normally distributed or displaying equal variance were tested by Kruskal-Wallis one-way ANOVA with post-hoc analysis by Dunn’s multiple comparisons test.
3. Results
3.1. The loss of Jab1 in osteoblast progenitor cells results in a progressive postnatal dwarfism phenotype and early lethality.
To investigate the specific role of Jab1 in postnatal bone growth, we deleted Jab1 specifically in osteoblast precursor cells with a commonly used Osx-cre deleter to generate Osx-cre; Jab1flox/flox (Jab1 cKO) mice (16). To confirm the cells targeted by the Osx-cre transgene, we first crossed Osx-cre only mice with Rosa26-LacZ mice. The β-galactosidase staining of long bones from Osx-cre; Rosa26-LacZ mice revealed the expression of Osx-cre in osteoblasts lining the diaphyseal periosteum, the trabecular bone, the secondary ossification center, and also in the hypertrophic chondrocytes and osteocytes, as previously reported (30, 31) (Figure S1). It was previously reported that the expression of the Osx-cre transgene alone might result in a minor bone phenotype in mice (30, 32, 33). Thus, in this study, we performed all mouse experiments using Osx-cre only mice as controls, in addition to wild type controls.
Both Jab1 cKO and Osx-cre only mice appeared grossly normal at birth and through 6 days of age. By 7 days of age, both Osx-cre only and Jab1 cKO mice begin to develop dwarfism. Our results are in agreement with previous studies showing a decrease in the length and weight of Osx-cre only mice at around 1 week of age (30, 33). However, whereas the Osx-cre only mice have a normal lifespan/healthspan, and their length stabilizes thereafter, the dwarfism in Jab1 cKO mice progressively worsened and they also began to appear lethargic around postnatal day 12 (Figure 1A). Unexpectedly, all Jab1 cKO mice die by postnatal day 21, around their weaning age. Indeed, Jab1 cKO mice had significantly decreased body length and weight by postnatal day 18 compared with wild type mice and Osx-cre only controls (Figure 1B). These results demonstrate that the expression of Jab1 in osteoblast precursor cells is essential for survival and for maintaining normal postnatal growth in mice.
Figure 1. Loss of Jab1 in osteoblast precursor cells resulted in progressive dwarfism and early lethality.

(A) Representative pictures of wild type and Jab1 cKO mice at postnatal days 6, 12, and 18, displaying the progressive dwarfism. (B) The quantification of the length and weight of wild type, Osx-cre only, and Jab1 cKO mice at postnatal day 18. Error bars represent means ± SD. ** p < 0.01, *** p < 0.001, ***** p < 0.00005 when Osx-cre only and Jab1 cKO are compared with wild type. # p < 0.05 when Jab1 cKO is compared with Osx-cre only. Wild type, n = 12; Osx-cre only, n = 8; Jab1 cKO, n = 9.
3.2. The loss of Jab1 in osteoblast precursors drastically alters trabecular and cortical bone structure and delays the formation of the secondary ossification center.
Next, we collected tibiae from 18-21-day old wild type, Osx-cre only, and Jab1 cKO mice to identify the overall differences in bone structure of early postnatal bones. Histological analysis revealed that the primary trabeculae in Jab1 cKO mice are thinner compared with wild type and Osx-cre only mice (Figure 2A). Proper secondary ossification center formation is critical for growth plate formation and long bone growth. Interestingly, our histological analysis also revealed that the secondary ossification center in Jab1 cKO mice was much smaller than both the wild type and Osx-cre only mice (Figure 2B). This might be due to a delay in the initial formation of the SOC, as the histology at postnatal day 9 shows a drastically decreased size of the secondary ossification center in Jab1 cKO, which persists through postnatal day 16 (Figure S2). The histology also uncovered that the cortical bone is more porous in Jab1 cKO mice compared with Osx-cre only and wild type mice (Figure 2C). Thus, Jab1 expression in osteoblast precursor cells is critical for proper secondary ossification center formation and proper bone growth.
Figure 2. Jab1 cKO mice exhibited severely altered bone morphology and delayed secondary ossification center formation.

Hematoxylin and Eosin staining in the proximal tibiae of wild type, Osx-cre only, and Jab1 cKO mice at 18 days of age. (A) Representative images of primary trabeculae. Scale bars, 100μm. (B) Representative images of the secondary ossification centers (SOC). Scale bars, 100μm. (C) Representative images of the cortical bone (CB). Scale bars, 50μ.
3.3. Jab1 deficiency in osteoblast precursor cells severely impairs bone microarchitecture.
To further characterize the in vivo effect of Jab1 loss on postnatal bone growth, we performed detailed micro-computed tomography (μCT) analysis on the tibiae of 18-21-day-old wild type, Osx-cre only, and Jab1 cKO mice. Representative reconstructions of tibiae from each genotype are presented in Figure 3A. First of all, we observed some heterogeneity among individual samples of Jab1 cKO tibiae (Figure S3). Nonetheless, in agreement with the length measurements of the entire mouse (Figure 1B), μCT reconstructions and quantification of the tibial length confirmed that Jab1 cKO mice had significantly shorter tibiae compared with wild type and Osx-cre only mice (Figures 3A and 3B). Next, μCT quantification revealed that both the periosteal and endosteal perimeter were significantly decreased in Jab1 cKO mutants, indicating decreased bone size (Figure S4). Moreover, similarly to our histological analysis, the μCT analysis revealed that the primary trabecular bone in Jab1 cKO and Osx-cre only mice appeared to be decreased when compared with wild type mice (Figure 3A, third row, and Figure S5A). Indeed, uCT quantification of trabecular bone confirmed that there were significant decreases in the trabecular thickness of Jab1 cKO and Osx-cre only tibiae compared with wild type tibiae. There was also a trend towards increased trabecular separation in Jab1 cKO bones compared with Osx-cre only and wild type bones (Figure 4A). Additionally, uCT quantification of trabecular bone volume (BV) and total volume (TV) revealed a significant decrease in Jab1 cKO tibiae compared with wild type tibiae, as expected based on the smaller size of Jab1 cKO mice (Figures 1A and 4B). We also observed a trend towards decreased BV and TV in the tibiae of Jab1 cKO mice when compared with Osx-cre only mice (Figure 4B). However, due to the proportional decrease in both bone volume and tissue volume within genotypes, the bone volume fraction (BV/TV) was not significantly changed among Jab1 cKO, Osx-cre only, and wild type controls (Figure 4B). Furthermore, uCT analysis demonstrates that the cortical bone was thicker in both Jab1 cKO and Osx-cre only tibiae compared with wild type (Figures 3A, 5A, and S5B). Furthermore, when compared with wild type tibiae, the cortical bone area (BA) was either slightly decreased in Jab1 cKO or was similar in Osx-cre only tibiae. There were also decreases in the total cross sectional area (TA) in Jab1 cKO and Osx-cre only tibiae compared with wild type tibiae. Thus, when compared with wild type, there was a significantly increased cortical bone area fraction (BA/TA) in Osx- cre only mice, which was exacerbated in Jab1 cKO mice (Figure 5B). Interestingly, cross sections of cortical bone appeared to be more porous in Jab1 cKO and Osx-cre only bones compared with wild type bones, similarly to our histological analysis (Figures 2C and 3A, bottom row).
Figure 3. Jab1 cKO mice exhibited severely altered bone microarchitecture and compromised mineralization.

(A) The μCT reconstructions of tibiae from Jab1 cKO, wild type, and Osx-cre only littermates at 18 days of age. Top row: entire tibiae. White arrow, callus-like structure. Second row: the bone present in the first 25% of the metaphysis from the proximal tibial growth plate. Third row: cross section taken from box in the second row showing trabecular bone inside the metaphysis. Bottom row: cross sections taken at 58% of the diaphyseal length from the proximal tibial growth plate. (B) The μCT quantification of the whole tibial length. Error bars represent means ± SD. ** p < 0.01 when Osx-cre only and Jab1 cKO are compared with wild type. † p < 0.001 when Jab1 cKO is compared with Osx-cre only. Wild type, n = 12; Osx-cre only, n = 8; Jab1 cKO n = 9. (C) The μCT reconstructions of three individual Jab1 cKO tibiae showing the apparent lack of mineralization at the proximal and distal ends (white arrows). (D) The μCT reconstruction of the callus-like structure in a Jab1 cKO tibia. Left, a rotated view of the Jab1 cKO tibia from 3A with the callus-like structure indicated by a white arrow. Right, newly formed bone inside the callus-like structure.
Figure 4. Loss of Jab1 in osteoblast precursors led to reduced trabecular thickness.
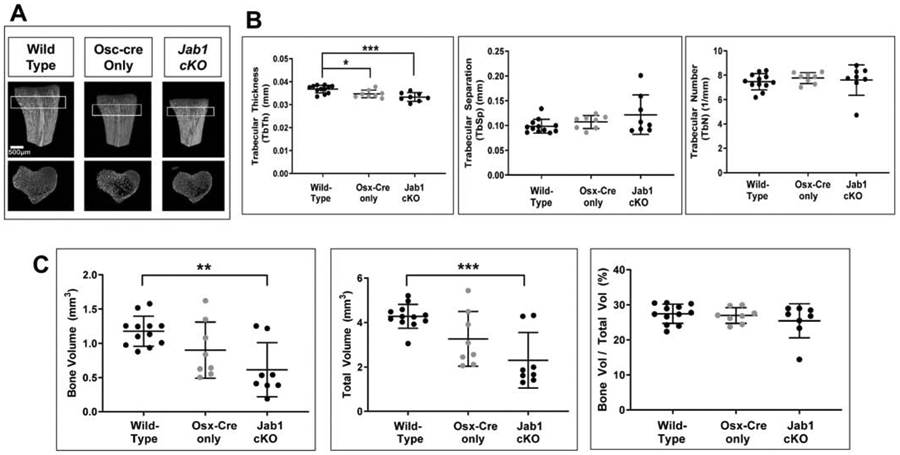
(A) The μCT quantification of trabecular thickness, trabecular separation, and trabecular number in wild type, Osx-cre only, and Jab1 cKO mice. (B) The μCT quantification of bone volume (BV), total volume (TV), and trabecular bone volume fraction (BV/TV) in wild type, Osx-cre only, and Jab1 cKO mice. Wild type, n = 12; Osx-cre only, n = 8; Jab1 cKO, n = 9. * p < 0.05, ** p < 0.01, *** p < 0.001 by one-way ANOVA with post-hoc analysis by Bonferroni’s multiple comparisons test.
Figure 5. Loss of Jab1 in osteoblast precursors led to increased cortical thickness and cortical bone fraction.
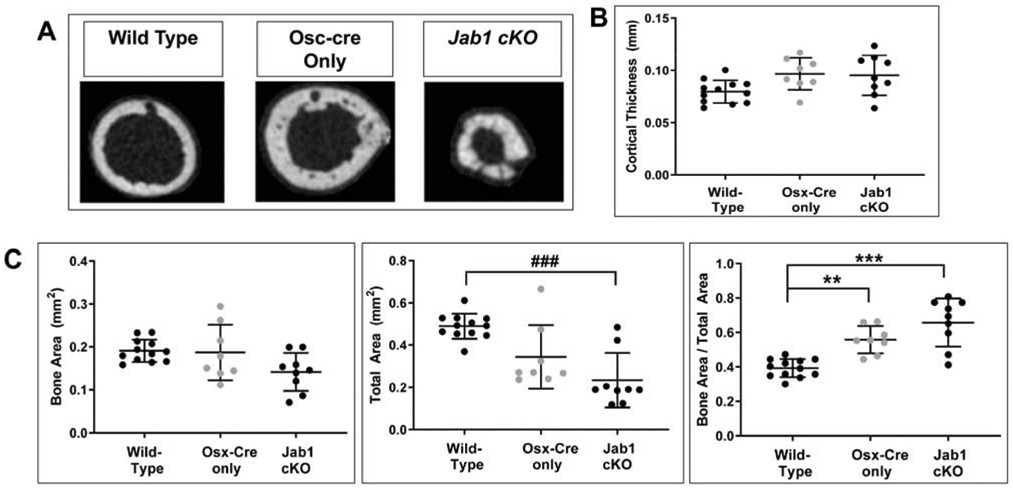
(A) The μCT quantification of cortical thickness. (B) The μCT quantification of cortical bone area (BA), total cross sectional area (TA), and cortical bone fraction (BA/TA). Wild type, n = 12; Osx-cre only, n = 8; Jab1 cKO, n = 9. For parametric analyses, * p < 0.05, ** p < 0.01, *** p < 0.001 by one-way ANOVA with post-hoc analysis by Bonferroni’s multiple comparisons test. For non-parametric analyses, ### p < 0.001 by Kruskal-Wallis one-way ANOVA with post-hoc analysis by Dunn’s multiple comparisons test.
Intriguingly, in some Jab1 cKO bones, a large portion of their proximal and distal ends appeared to be poorly mineralized (Figure 3C), even though gross examinations showed that there were tissues present in these areas. We also observed that multiple Jab1 cKO mice had callus-like structures present near their distal tibiofibular junction (Figures 3A, 3D, and S3). In contrast, no similar callus-like structures were ever identified in any of the tibiae isolated from wild type or Osx-cre only mice (Figure S3). A closer examination of this callus-like structure in Jab1 cKO tibiae revealed that the area inside this callus-like structure was likely to be newly formed bone (Figure 3D). This finding suggests that Jab1 cKO bones might be more susceptible to incurring spontaneous fractures compared with wild type and Osx-cre only bones. Therefore, the loss of Jab1 in osteoblast precursors significantly alters long bone microarchitecture, impairs proper mineralization, and might lead to spontaneous fractures.
3.4. The loss of Jab1 in osteoblast precursors inhibits osteoblast differentiation and mineralization, likely in part through the positive regulation of TGFβ/BMP signaling.
Our previous study in Prx1-cre; Jab1flox/flox mice showed that Jab1 positively regulates BMP signaling in osteochondral progenitor cells (15). Thus, we hypothesized that the loss of Jab1 specifically in osteoblast precursor cells also results in impaired BMP-mediated osteoblast differentiation and mineralization. To this end, we first knocked down Jab1 expression in primary calvarial osteoblasts isolated from Jab1flox/flox mice, using adenoviruses expressing either Cre Recombinase (Ad-Cre) or, as a control, a GFP reporter (Ad-GFP) (14, 15). We first confirmed the Jab1 knockdown efficiency at the RNA and protein levels (Figure 6A). Next, we cultured the primary osteoblasts in osteoblast differentiation media for 21 days and examined mineralization status by performing Von Kossa staining (Figure 6A). Von Kossa staining revealed fewer black nodules in Jab1-knockdown versus control primary osteoblasts, indicating that Jab1 is required for robust osteoblast differentiation and mineralization. To confirm this finding, we also performed Jab1-knockdown experiments in a widely used mouse preosteoblastic cell line, MC3T3-E1 (34), using lentiviral shRNAs specifically targeting Jab1. Again, von Kossa staining revealed that the depletion of Jab1 expression nearly abolished nodule formations in MC3T3-E1 cells as well (Figure 6B). Together, these results confirm that Jab1 deficiency drastically reduced osteogenic potential in osteoblast precursor cells, which may account for the impaired mineralization and altered bone structure observed in our histological and μCT analyses of Jab1 cKO mice (Figures 2 and 3).
Figure 6. Jab1 deficiency inhibited osteoblast differentiation and mineralization ex vivo.

(A) Left and middle panels: Jab1 RNA and protein expression levels in control and Jab1-knockdown mouse primary calvarial osteoblasts. Right panel: von Kossa staining in control and Jab1-knockdown mouse primary calvarial osteoblasts after 21 days of culture in osteoblast differentiation media. (B) Left and middle panels: Jab1 RNA and protein expression levels in control and Jab1-knockdown MC3T3-E1 cells. Right panel: von Kossa staining in Jab1-knockdown MC3T3-E1 cells after 21 days of culture in osteoblast differentiation media. (C) Left and middle panel: Jab1 RNA and protein expression levels in Jab1-knockdown mBMSCs. Right panel: oil red O staining of mBMSCs cultures.
Mesenchymal stem cells (MSCs) are multipotent cells with the ability to differentiate into multiple mesenchymal cell lineages, including osteoblasts and adipocytes (35). It is also well-established that the loss of osteogenic differentiation is often associated with an increased adipogenic potential (35). Thus, to determine the effect of Jab1 loss on MSC adipogenesis, we performed a lentiviral Jab1-knockdown in a mouse bone marrow stromal cell (mBMSC) line, and then induced them to differentiate into adipocytes (Figure 6C). Indeed, we found that there were increased lipid droplets in mBMSCs by Oil Red O staining in Jab1-knockdown mBMSCs. Thus, the loss of Jab1 might lead to an increase in adipogenic potential and decreased osteoblast differentiation and mineralization.
To further explore the underlying mechanism of Jab1-mediated osteoblast differentiation, we next examined the expression of a master osteoblast differentiation transcription factor Runx2 upon Jab1-knockdown in osteoblast precursor cells (36). Interestingly, we found that the loss of Jab1 in MC3T3-E1 cells resulted in no significant change in the Runx2 expression at either the RNA or protein level (Figure 7A and 7B). However, we detected a significant decrease in the mRNA expression of Osteocalcin, the most specific late-stage marker of osteoblast differentiation, in Jab1-knockdown MC3T3-E1 cells (Figure 7A). This finding is consistent with the mineralization defect we observed in Jab1 cKO mice (Figures 3C), indicating that the loss of Jab1 in osteoblast precursors might lead to a late-stage differentiation defect. Next, to determine the effect of Jab1 loss on major signaling pathways, we performed an unbiased reporter screen in MC3T3-E1 cells (Figure 7C). Impressively, we found that of 45 major signaling pathway reporters, 31 (68%) of them were clearly altered in MC3T3-E1 cells upon Jab1-knockdown (Figure 7C). Furthermore, of those 31 reporters, 26 (83%) were downregulated, including a Smad2/3 reporter for the TGFβ pathway (Figure 7C). All together, these data are consistent with the established function of Jab1 as a transcriptional co-factor and with others’ previous reports demonstrating a link between Jab1 and the TGFβ/BMP signaling pathway (8, 11, 12, 14, 15, 37-39).
Figure 7. Jab1-knockdown inhibited osteoblast differentiation likely in part through the TGFβ/BMP signaling pathways.

(A) RNA expression levels of Runx2 and Osteocalcin in Jab1-knockdown MC3T3-E1 cells. (B) Protein expression levels of Runx2 and Jab1 in Jab1-knockdown MC3T3-E1 cells. (C) An unbiased reporter screen in Jab1-knockdown MC3T3-E1 cells. The black arrow indicates a canonical Smad2/3 TGFβ reporter. A detailed list of the pathways in this screen can be found in Supplementary Table 3. (D) Western blot analysis of Jab1 and phospho-Smad1/5/8 in primary calvarial osteoblasts cultured in osteoblast differentiation media for 0 and 21 days, respectively. (E) Western blot analysis of Jab1 and phospho-Smad1/5/8 in control and Jab1-knockdown primary calvarial osteoblasts after 21 days of culture in osteoblast differentiation media. (F) Western blot analysis of phospho-Smad1/5/8 (top) and phospho-Smad2/3 (bottom) in control and Jab1-knockdown MC3T3-E1 cells treated with 300ng/mL BMP7 or 2ng/mL TGFβ-3 for 1 hour, respectively.
TGFβ is crucial for many aspects of bone biology and osteoblast differentiation, and is tightly regulated at many levels. Therefore, we decided to further investigate the effect of Jab1 loss on osteoblast’s response to TGFβ/BMP stimulation. When primary mouse calvarial osteoblasts were cultured in osteoblast differentiation media for 21 days, Jab1 was expressed at both an early (Day 0) and a late stage (Day 21) of osteoblast differentiation (Figure 7D), hinting that Jab1 is required throughout osteoblast differentiation. We also observed a higher level of phospho-Smad1/5/8, a key downstream BMP effector, at Day 21 versus Day 0 (Figure 7D). Next, we infected primary calvarial osteoblasts isolated from Jab1flox/flox mice with adenoviruses expressing either Cre Recombinase (Ad-cre) or a GFP control (Ad-GFP), induced them to differentiate into osteoblasts, and performed western blotting for phospho-Smad1/5/8 (Figure 7E). Interestingly, we found that a decrease in the levels of Jab1 resulted in a major decrease in the levels of phospho-Smad1/5/8 in primary osteoblasts (Figure 7E). Furthermore, we treated control and Jab1-knockdown MC3T3-E1 preosteoblasts with BMP7 (300ng/mL) or TGFβ-3 (2ng/mL), and determined the expressions of phospho-Smad1/5/8 and phospho-Smad2/3, a key TGFβ downstream effector, respectively (Figure 7F). Indeed, there was a decreased response to both TGFβ and BMP signaling upon Jab1-knockdown in MC3T3-E1 cells. These results suggest that Jab1 promotes osteoblast differentiation and mineralization at least in part through stimulating TGFβ/BMP signaling.
4. Discussion
Jab1 is an essential subunit of the COP9 Signalosome, and regulates many vital cellular functions, including cell proliferation, differentiation, and apoptosis. Indeed, the conditional knockout of Jab1 in any given tissue studied to date all results in severe defects to progenitor cell survival and differentiation (5). Our previous work has demonstrated that, as a master developmental regulator, Jab1 is necessary for the successive stages of skeletogenesis by regulating the TGFβ/BMP signaling pathways in a context-specific manner (14, 15). Interestingly, master developmental regulators are often reactivated during tumorigenesis to promote malignancy via their specific effects on cell proliferation, differentiation, and survival. Indeed, as a potential oncogene, JAB1 has been shown to be overexpressed in many different forms of cancer (7). To that end, we have recently demonstrated that JAB1 overexpression specifically in osteoblasts can increase the incidence of p53-dependent osteosarcoma formation in mice (19). Moreover, a recently developed small molecular inhibitor of JAB1, CSN5i-3, has shown promising results in reducing cell viability, including in osteosarcoma (19). Therefore, a better understanding of the physiological role of Jab1 in osteoblasts will facilitate the development of better Jab1-targeted therapeutic options for osteosaercoma and other cancers.
In the present study, we provide strong evidence that the expression of Jab1 in osteoblast precursor cells is essential for proper postnatal bone growth and osteoblast differentiation in mice, likely at least in part through the regulation of TGFβ/BMP signaling. To our knowledge, whereas dwarfism and impaired bone structure is relatively common in mice in which a gene of interest is deleted using the the same Osx-cre driver, early postnatal lethality is extremely rare among them. Very strikingly, Jab1 cKO mutant mice all die around weaning age. While the Osx-cre transgene mainly targets cells at the earliest stages of osteoblast differentiation, it is also known to target some other cell types, including hypertrophic chondrocytes, osteocytes, stromal cells, and perivascular cells (30, 31). Indeed, our β-galactosidase staining in Osx-cre; Rosa26 LacZ tibiae revealed strong LacZ expressions in osteoblasts, as well as osteocytes and hypertrophic chondrocytes (Figure S1). Because osteoblasts constitute a major component of the hematopoietic stem cell niche (2), we speculate that the early lethality in Jab1 cKO mice might result from defective hematopoiesis. Interestingly, a recent study demonstrated that Osx-cre-mediated deletion of Tgfbr2 resulted in early lethality at 4-weeks of age, dwarfism, reduced osteoblast maturation, and perturbed hematopoiesis (40). Additionally, Jab1 has been linked to hematopoietic defects in mice (41, 42). Thus, the early lethality of Jab1 cKO mice might be the result of defective hematopoiesis, through the regulation of the HSC niche and/or through TGFβ/BMP signaling, which warrants further investigation.
Osx-cre transgenic mice by themselves were reported to have a transient dwarfism phenotype (30, 33). However, despite our study corroborating the finding that Osx-cre only mice are smaller beginning at P7, the Jab1 cKO mice were still significantly smaller than Osx-cre only mice at postnatal day 18 (Figures 1 and 3A). It is also important to note that the expression of the Osx-cre transgene by itself did not affect osteoblast differentiation (33), thus the bone defects observed in the Jab1 cKO mice were most likely due to the effect of Jab1 deletion. Indeed, the bone defects observed in some Jab1 cKO mice were so severe that the mineralization could not even be detected by μCT, and spontaneous fractures occurred (Figure S3). Thus, based on the early lethality, significantly shorter stature, multiple bone defects, and changes in most μCT parameters, we conclude that Jab1 plays an essential role in maintaining proper bone microarchitecture in mice.
In contrast to what we previously reported in mice with Jab1 deleted specifically in chondrocytes, where we found an enhanced response to exogenous BMP treatment (14), the loss of Jab1 in osteoblast progenitor cells resulted in a reduced response to BMP treatment, highlighting the context-specific role of Jab1 in the regulation of key developmental signaling pathways. Indeed, Jab1 has been reported to positively and negatively regulate TGFβ/BMP signaling through controlling the stability and degradation of Smad7 and Smad4, respectively (11, 12). Moreover, several studies have shown that Jab1 inhibits BMP signaling by negatively regulating Smad1/5 (14, 37, 43). As the TGFβ/BMP signaling pathway is tightly regulated at multiple levels (4, 44), the role of Jab1 in regulating TGFβ/BMP signaling during skeletal development is likely to be highly context-dependent, and remains to be further investigated.
Our reporter assay indicates that Jab1 activates many signal transduction pathways in osteoblasts. Notably, a canonical Smad2/3 TGFβ reporter activity decreased upon Jab1 deletion, and the response to exogenous TGFβ/BMP stimulation was also reduced in osteoblasts. Moreover, in both primary calvarial osteoblasts and preosteoblastic MC3T3-E1 cells, the mineralization was greatly reduced upon loss of Jab1 (Figure 6). Additionally, we provide evidence that the loss of Jab1 resulted in a shift from osteoblastogenesis to adipogenesis in mouse bone marrow stromal cells. It is well-established that low bone mass can result from a lineage shift in mesenchymal stem cells from osteoblasts to adipocytes, which can contribute to osteoporosis (45). Thus, a better understanding of the role of Jab1 in promoting osteoblast differentiation and postnatal bone growth in osteoblast precursor cells might help promote better therapeutic treatments of bone diseases such as osteoporosis.
Supplementary Material
Highlights.
A novel Osx-cre; Jab1flox/flox conditional knockout mouse model exhibits early lethality around weaning age.
Osx-cre; Jab1flox/flox tibiae display severely impaired bone microarchitecture and mineralization.
Jab1 in osteoblasts promotes osteoblast differentiation, at least in part through TGFβ and BMP signaling.
Acknowledgements
The authors thank Dr. Ruggero Pardi for the generous gift of the Jab1 conditional allele Jab1flox/flox mice. The authors also thank Teresa Pizzuto for her expert histology work. This study was supported in part by the NIAMS of the National Institutes of Health R01 AR068361 to G.Z., the NIDCR of the National Institutes of Health R03 DE019190 and DE019190A1S1 to G.Z., and the NIAMS T32 AR007505 to W.E.S. and L.A.B.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
The authors declare that there are no potential conflicts of interest.
REFERENCES
- 1.Kenkre JS, Bassett J. The bone remodelling cycle. Ann Clin Biochem. 2018;55(3):308–27. [DOI] [PubMed] [Google Scholar]
- 2.Frisch BJ. The hematopoietic stem cell niche: What's so special about bone? Bone. 2019;119:8–12. [DOI] [PubMed] [Google Scholar]
- 3.Salazar VS, Gamer LW, Rosen V. BMP signalling in skeletal development, disease and repair. Nat Rev Endocrinol. 2016;12(4):203–21. [DOI] [PubMed] [Google Scholar]
- 4.Derynck R, Budi EH. Specificity, versatility, and control of TGF-beta family signaling. Sci Signal. 2019;12(570). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li P, Xie L, Gu Y, Li J, Xie J. Roles of Multifunctional COP9 Signalosome Complex in Cell Fate and Implications for Drug Discovery. J Cell Physiol. 2017;232(6):1246–53. [DOI] [PubMed] [Google Scholar]
- 6.Kato JY, Yoneda-Kato N. Mammalian COP9 signalosome. Genes Cells. 2009;14(11):1209–25. [DOI] [PubMed] [Google Scholar]
- 7.Liu G, Claret FX, Zhou F, Pan Y. Jab1/COPS5 as a Novel Biomarker for Diagnosis, Prognosis, Therapy Prediction and Therapeutic Tools for Human Cancer. Front Pharmacol. 2018;9:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Claret FX, Hibi M, Dhut S, Toda T, Karin M. A new group of conserved coactivators that increase the specificity of AP-1 transcription factors. Nature. 1996;383(6599):453–7. [DOI] [PubMed] [Google Scholar]
- 9.Oh W, Lee EW, Sung YH, Yang MR, Ghim J, Lee HW, et al. Jab1 induces the cytoplasmic localization and degradation of p53 in coordination with Hdm2. J Biol Chem. 2006;281(25):17457–65. [DOI] [PubMed] [Google Scholar]
- 10.Bae MK, Ahn MY, Jeong JW, Bae MH, Lee YM, Bae SK, et al. Jab1 interacts directly with HIF-1alpha and regulates its stability. J Biol Chem. 2002;277(1):9–12. [DOI] [PubMed] [Google Scholar]
- 11.Wan M, Cao X, Wu Y, Bai S, Wu L, Shi X, et al. Jab1 antagonizes TGF-beta signaling by inducing Smad4 degradation. EMBO Rep. 2002;3(2):171–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim BC, Lee HJ, Park SH, Lee SR, Karpova TS, McNally JG, et al. Jab1/CSN5, a component of the COP9 signalosome, regulates transforming growth factor beta signaling by binding to Smad7 and promoting its degradation. Mol Cell Biol. 2004;24(6):2251–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Samsa WE, Zhou X, Zhou G. Signaling pathways regulating cartilage growth plate formation and activity. Semin Cell Dev Biol. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen D, Bashur LA, Liang B, Panattoni M, Tamai K, Pardi R, et al. The transcriptional co-regulator Jab1 is crucial for chondrocyte differentiation in vivo. J Cell Sci. 2013;126(Pt 1):234–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bashur LA, Chen D, Chen Z, Liang B, Pardi R, Murakami S, et al. Loss of jab1 in osteochondral progenitor cells severely impairs embryonic limb development in mice. J Cell Physiol. 2014;229(11):1607–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodda SJ, McMahon AP. Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development. 2006;133(16):3231–44. [DOI] [PubMed] [Google Scholar]
- 17.Panattoni M, Sanvito F, Basso V, Doglioni C, Casorati G, Montini E, et al. Targeted inactivation of the COP9 signalosome impairs multiple stages of T cell development. J Exp Med. 2008;205(2):465–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21(1):70–1. [DOI] [PubMed] [Google Scholar]
- 19.Samsa WE, Mamidi MK, Bashur LA, Elliott R, Miron A, Chen Y, et al. The crucial p53-dependent oncogenic role of JAB1 in osteosarcoma in vivo. Oncogene 2020. DOI: 10.1038/s41388-020-1320-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou G, Zheng Q, Engin F, Munivez E, Chen Y, Sebald E, et al. Dominance of SOX9 function over RUNX2 during skeletogenesis. Proc Natl Acad Sci U S A. 2006;103(50):19004–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang B, Mamidi MK, Samsa WE, Chen Y, Lee B, Zheng Q, et al. Targeted and sustained Sox9 expression in mouse hypertrophic chondrocytes causes severe and spontaneous osteoarthritis by perturbing cartilage homeostasis. Am J Transl Res. 2020;12(3):1056–69. [PMC free article] [PubMed] [Google Scholar]
- 22.Samsa WE, Vasanji A, Midura RJ, Kondratov RV. Deficiency of circadian clock protein BMAL1 in mice results in a low bone mass phenotype. Bone. 2016;84:194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang B, Cotter MM, Chen D, Hernandez CJ, Zhou G. Ectopic expression of SOX9 in osteoblasts alters bone mechanical properties. Calcif Tissue Int. 2012;90(2):76–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou G, Garofalo S, Mukhopadhyay K, Lefebvre V, Smith CN, Eberspaecher H, et al. A 182 bp fragment of the mouse pro alpha 1(II) collagen gene is sufficient to direct chondrocyte expression in transgenic mice. J Cell Sci. 1995; 108 ( Pt 12):3677–84. [DOI] [PubMed] [Google Scholar]
- 25.Gustafson MB, Martin RB, Gibson V, Storms DH, Stover SM, Gibeling J, et al. Calcium buffering is required to maintain bone stiffness in saline solution. J Biomech. 1996;29(9):1191–4. [DOI] [PubMed] [Google Scholar]
- 26.Feldkamp LA, Davis LC, Kress JW. Practical cone-beam algorithm. Journal of the Optical Society of America A. 1984, 10.1364/josaa.1.000612;1. [DOI] [Google Scholar]
- 27.Yan G, Tian J, Zhu S, Dai Y, Qin C. Fast cone-beam CT image reconstruction using GPU hardware. Journal of X-Ray Science and Technology. 2008;16(4):225–34. [Google Scholar]
- 28.Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Muller R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res. 2010;25(7):1468–86. [DOI] [PubMed] [Google Scholar]
- 29.Bortel EL, Duda GN, Mundlos S, Willie BM, Fratzl P, Zaslansky P. Long bone maturation is driven by pore closing: A quantitative tomography investigation of structural formation in young C57BL/6 mice. Acta Biomater. 2015;22:92–102. [DOI] [PubMed] [Google Scholar]
- 30.Huang W, Olsen BR. Skeletal defects in Osterix-Cre transgenic mice. Transgenic Res. 2015;24(1):167–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen J, Shi Y, Regan J, Karuppaiah K, Ornitz DM, Long F. Osx-Cre targets multiple cell types besides osteoblast lineage in postnatal mice. PLoS One. 2014;9(1):e85161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davey RA, Clarke MV, Sastra S, Skinner JP, Chiang C, Anderson PH, et al. Decreased body weight in young Osterix-Cre transgenic mice results in delayed cortical bone expansion and accrual. Transgenic Res. 2012;21(4):885–93. [DOI] [PubMed] [Google Scholar]
- 33.Wang L, Mishina Y, Liu F. Osterix-Cre transgene causes craniofacial bone development defect. Calcif Tissue Int. 2015;96(2):129–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sudo H, Kodama HA, Amagai Y, Yamamoto S, Kasai S. In vitro differentiation and calcification in a new clonal osteogenic cell line derived from newborn mouse calvaria. J Cell Biol. 1983;96(1):191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Q, Shou P, Zheng C, Jiang M, Cao G, Yang Q, et al. Fate decision of mesenchymal stem cells: adipocytes or osteoblasts? Cell Death Differ. 2016;23(7):1128–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Komori T Molecular Mechanism of Runx2-Dependent Bone Development. Mol Cells. 2020;43(2):168–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sangadala S, Yoshioka K, Enyo Y, Liu Y, Titus L, Boden SD. Characterization of a unique motif in LIM mineralization protein-1 that interacts with jun activation-domain-binding protein 1. Mol Cell Biochem. 2014;385(1-2):145–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.He F, Lu D, Jiang B, Wang Y, Liu Q, Liu Q, et al. X-linked intellectual disability gene CUL4B targets Jab1/CSN5 for degradation and regulates bone morphogenetic protein signaling. Biochim Biophys Acta. 2013;1832(5):595–605. [DOI] [PubMed] [Google Scholar]
- 39.Li J, Gu Z, Li S, Xiao Z, Sun K. Reverse correlation of Jab1 and Smad4 in PANC-1 cells involved in the pathogenesis of pancreatic cancer. Int J Clin Exp Pathol. 2015;8(8):9279–85. [PMC free article] [PubMed] [Google Scholar]
- 40.Abou-Ezzi G, Supakorndej T, Zhang J, Anthony B, Krambs J, Celik H, et al. TGF-beta Signaling Plays an Essential Role in the Lineage Specification of Mesenchymal Stem/Progenitor Cells in Fetal Bone Marrow. Stem Cell Reports. 2019;13(1):48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mori M, Yoneda-Kato N, Yoshida A, Kato JY. Stable form of JAB1 enhances proliferation and maintenance of hematopoietic progenitors. J Biol Chem. 2008;283(43):29011–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sinha S, Dwivedi TR, Yengkhom R, Bheemsetty VA, Abe T, Kiyonari H, et al. Asrij/OCIAD1 suppresses CSN5-mediated p53 degradation and maintains mouse hematopoietic stem cell quiescence. Blood. 2019;133(22):2385–400. [DOI] [PubMed] [Google Scholar]
- 43.Haag J, Aigner T. Jun activation domain-binding protein 1 binds Smad5 and inhibits bone morphogenetic protein signaling. Arthritis Rheum. 2006;54(12):3878–84. [DOI] [PubMed] [Google Scholar]
- 44.Thielen NGM, van der Kraan PM, van Caam APM. TGFbeta/BMP Signaling Pathway in Cartilage Homeostasis. Cells. 2019;8(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abdallah BM, Kassem M. New factors controlling the balance between osteoblastogenesis and adipogenesis. Bone. 2012;50(2):540–5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.