

Abstract
Heart failure is a leading cause of death in the United States. Diabetes, also known as diabetes mellitus (DM), exponentially increases the risk of heart failure. The increase in oxidative stress and metabolic dysfunction caused by DM can lead to DNA damage and the development of diabetic cardiomyopathy. Ataxia telangiectasia mutated kinase (ATM) is a DNA damage response protein with a primary nuclear function to regulate cell cycle progression in response to double-strand DNA breaks, acts as a redox sensor, and facilitates DNA repair. ATM deficiency associates with the development of insulin resistance and DM. Consequently, patients with Ataxia telangiectasia, a rare autosomal recessive disorder, have an increased risk of developing heart failure. The main objective of this review is to summarize the shared metabolic and cardiac abnormalities associated with DM and ATM deficiency, with a focus on the development of heart failure.
Keywords: ATM, Diabetes, Diabetic Cardiomyopathy, Heart Failure
Introduction
Patients with diabetes mellitus (DM) have an increased risk of developing heart failure. An astonishing fact is that 30 million Americans are diabetic and 90 million are prediabetic[1]. As average life expectancy rate increases, the prevalence of patients with DM and heart failure has progressively accelerated in western and developing countries[2]. DM is a metabolic disorder characterized by the inability to properly use, transport and store nutritional energy, resulting in hyperglycemia[3]. There are two main types of DM, insulin-dependent type 1 DM and non-insulin-dependent type 2 DM. This review is focused on type 2 DM which accounts for ~90-95% diabetic diagnosis[1,4]. In addition to hyperglycemia, DM associates with insulin resistance, inflammation, mitochondrial dysfunction, endothelial dysfunction and increased oxidative stress[5,6].
Ataxia telangiectasia mutated kinase (ATM) is a 370kDa serine/threonine kinase that belongs to the PI3-kinase (PI3K) protein family. ATM is a regulator of the G1/S checkpoint, and is present in the nucleus, cytoplasm and mitochondria[7]. Activation of ATM occurs in response to double-strand DNA (dsDNA) breaks, increased reactive oxygen species (ROS) production, oxidative damage, and other genotoxic stressors[8] . In response to dsDNA breaks, ATM and accessory proteins (MDC1, 53PB1, and BRACA1) are recruited to the site of dsDNA breaks where ATM becomes activated via auto-phosphorylation of Ser1981[9,10]. Upon activation, ATM terminates cell cycle progression until DNA repair has been completed[10]. ATM is also a redox sensor and is activated in response to oxidative stress. While dsDNA breaks cause ATM to become activated in monomer form, oxidation of ATM results in a covalently bound dimer form[11]. The ATM gene is located on chromosome 11 q22-23[11 ]. While many different types of mutations have been identified on the ATM gene, the majority are nonsense mutations which cause premature stop codons resulting in truncation, and dysfunctional ATM protein[12,13]. Mutations in the ATM gene result in a multisystem disorder called Ataxia telangiectasia (AT) which causes cardiac, neurological, immunological, and endocrinological abnormalities[14–17]. Approximately 2% of the population has a heterozygous mutation in the ATM gene. Individuals with a heterozygous mutation exhibit a less severe AT phenotype. However, they are at a higher risk of developing DM, heart failure and cancer [7,8,10,18].
DM and ATM deficiency share many risk factors for the development of heart disease such as insulin resistance, increased oxidative stress, endothelial dysfunction and mitochondrial dysfunction[6,11]. DM and ATM deficiency independently associate with the development and acceleration of heart failure[6,7,19,20]. Since ATM contributes to both nuclear and cytoplasmic signaling, disruption in ATM function can directly contribute to metabolic complications such as DM and the development of heart disease[11]. Not much is known about the role of ATM in the development of diabetic cardiomyopathy (DCM). This review article summarizes the role of ATM in myocardial remodeling using myocardial infarction (MI) as a model, and discusses the shared metabolic abnormalities during DM and ATM deficiency, and how this may exacerbate the progression of heart failure.
DM and Heart Failure
DCM is a complex, multifactorial heart failure syndrome that is characterized by cardiac structural and functional abnormalities independent of hypertension, coronary artery disease and dyslipidemia[3,6]. The risk of developing heart failure is doubled in DM males and four times as likely in DM females as compared to nondiabetic patients[4]. Approximately 20-30% of diabetic patients have heart failure which greatly increases the risk of morbidity and mortality[6,21]. Modifications in cardiac energy metabolism and insulin signaling are the key metabolic abnormalities associated with hyperglycemia and the development and progression of DCM[6]. Hyperglycemia also causes altered calcium homeostasis, increased glycation end products, ROS production, mitochondrial dysfunction and cardiac metabolic dysfunction[4,6], which can contribute to the development of the structural and functional abnormalities that are hallmarks of DCM[6,19]. The early stages of DCM are generally asymptomatic. However, diastolic dysfunction, left ventricular hypertrophy, fibrosis and cardiac remodeling have been noted[3,6]. The later stages of DCM associate with systolic dysfunction, heart failure and increased morbidity and mortality[3,6]. When cardiomyopathy and DM occur concomitantly, the development of heart failure is greatly accelerated[4]. While DCM occurs independent of hypertension, 80% of diabetic patients are hypertensive, which further exacerbates the risk of heart disease[6,22,23].
ATM, DCM and Heart Failure
ROS-mediated DNA oxidation and fragmentation have been observed in the myocardium during DM[24]. Since ATM is activated in response to dsDNA breaks and oxidation, ATM may play a role in the development and progression of DCM. The DNA damage incurred during DM directly contributes to the cardiac structural and functional alterations, which are the hallmarks of DCM[24,25]. Decreased LV compliance, increased atrial filling, lengthened isovolumetric relaxation, variations in left ventricular end diastolic volume (LVEDV), decreased early diastolic filling, cardiomyocyte stiffness, and diastolic dysfunction are often observed in early and advanced stages of DCM[6,26]. During late stages, an increase in left ventricular diameter, decreased percent ejection fraction (%EF), increased filling pressures and advanced systolic dysfunction are observed[6,27]. Complications from advanced systolic dysfunction in DCM often lead to the acceleration of heart failure. Previous work from our laboratory has shown that stimulation of β-adrenergic receptor (β-AR) increases ATM expression in the heart and cardiac myocytes[28]. ATM expression was also found to be higher in the infarct left ventricular region of mice 1 and 3 days post-MI[29]. Similar to DCM, ATM deficiency associates with cardiac structural and functional abnormalities. Mice lacking ATM (KO) exhibited decreased LV mass, LV end diastolic diameter (LVEDD), LV end systolic diameter (LVESD), LVEDV, and LV end systolic volume (LVESV) versus wild-type (WT) mice. In addition, ATM KO mice displayed an increased E/A wave ratio and decreased deceleration time for the E-wave[30]. Using heterozygous KO (ATM deficient) mice, it was demonstrated that ATM plays an important role in β-AR-stimulated myocardial remodeling with respect to heart function, apoptosis and fibrosis[28]. ATM deficiency differentially affected myocardial remodeling in response to early (1 and 7 days post-MI) versus late (14 and 28 days post-MI) phase. During early MI phase, MI-mediated decrease in %EF and percent fractional shortening (%FS) was significantly lower in ATM heterozygous KO mice versus their wild-type (WT) counterparts. Additionally, LVESD and LVEDD were lower in ATM deficient hearts 4 hrs, 1,3 and 7 days post-MI[29,31,32]. In contrast, exacerbated LV dysfunction was observed in ATM deficient mice as evidenced by a greater decrease in %EF and FS, and increase in LVESV 14 and 28 days post-MI. Mortality post-MI was also higher in ATM deficient hearts post-MI[33,34].
Increased cardiac fibrosis is a common feature in the diabetic and ATM deficient heart[6,8]. Fibrosis and cardiac remodeling are initial indicators of DCM and lead to the development of cardiac dysfunction and heart failure[3,35]. Cardiac fibrosis is caused by extracellular matrix (ECM) remodeling in response to cardiac damage. While ECM deposition is protective in early cardiac injury, excessive ECM deposition causes myocardial stiffening and cardiac dysfunction[36]. Consequently, myocardial fibrosis is a major cause of heart failure[8,36]. Cardiac interstitial, perivascular and replacement fibrosis has been observed in diabetic patients independent of hypertension and coronary artery disease[37]. Further, DM associates with left and right ventricle interstitial fibrosis with increased type I and III collagen deposition[37]. Additionally, increased fibrosis in the heart has been observed in DM mice and rat models. Of note, severity of cardiac fibrosis may depend on comorbidities, species, age, and sex [37]. ATM deficiency also associates with augmented cardiac fibrosis[8]. ATM deficient mice exhibit increased cardiac fibrosis at basal levels, and following β-AR stimulation[28]. Similarly, ATM deficient mice display increased cardiac fibrosis 3, 7 and 28 days post-MI compared to WT[29,31,34].
Cardiac hypertrophy is another major contributor to the development of heart failure and occurs during ATM deficiency and DM[7,11,27,30,37]. Myocyte hypertrophy occurs to compensate for the increased hemodynamic load caused by the progressive decrease in cardiomyocytes during cardiac injury[8,38]. Similar to fibrosis, hypertrophy is beneficial during early cardiac injury, but progressively leads to cardiac dysfunction and heart failure[27]. Left ventricular hypertrophy is noted during the early stages of DCM[27]. Interestingly, hypertrophy as indicated by an increase in myocyte cross sectional area and increased expression of atrial natriuretic peptide was observed in the myocardium of ATM KO mice [29,30]. ATM deficient mice also exhibit increased myocyte cross-sectional area prior to and 28 days post-MI[34].
Angiogenesis promotes myocyte survival during ischemia through the mitigation of hypoxia. DM associates with two aberrant angiogenic responses. The kidneys and retina display excessive angiogenesis, while decreased angiogenesis occurs in the heart and peripheral limbs[39]. Impaired angiogenesis is suggested to be a major contributor of DM-induced ischemic heart disease (IHD). Cardiomyocytes may exert anti-angiogenic effects in the heart via the exosomal transfer of miR-320 into endothelial cells[40]. ATM deficiency also associates with decreased cardiac angiogenesis post-MI[33]. Since angiogenesis is an important protective mechanism post-MI, ATM deficiency may negatively affect this protective mechanism during development of DCM.
The impairment of insulin metabolic signaling observed in DM and ATM deficiency is known to accelerate the development and progression of heart failure[8,20,27,41]. Insulin signaling promotes the activation of endothelial nitric oxide synthase (eNOS) and nitric oxide (NO) production, which are critical for optimal cardiac function[27]. Consequently, decreased myocardial eNOS activation and NO production is observed during DM, which can lead to cardiac macro and microvascular disease. This may further promote cardiac fibrosis through the upregulation of collagen cross linking enzymes[27]. Moreover, insulin resistance and ATM deficiency associate with downregulation of the PI3K/Akt signaling in cardiovascular tissues which may result in decreased NO production, a critical vasodilator[6,11,27,42]. Though future studies are needed, the altered PI3K/Akt pathway, insulin resistance and decreased NO bioavailability may also explain why DM and AT patients exhibit vascular complications such as macro-vascular and microvascular disease[5,9,11]. The role of ATM specifically in DCM using ATM deficient mice remains to be investigated. However, changes in structural and functional parameters of the heart, fibrosis, angiogenesis and hypertrophy in ATM deficient hearts post-MI clearly indicate that ATM has the potential to play a critical role in the development of DCM.
Cardiac Energy Metabolism during DM and ATM deficiency
Myocardial tissue has the highest metabolic requirement in the body. The primary metabolic pathway utilized for myocardial ATP production is oxidative phosphorylation using long chain fatty acids and glucose[43]. Given that fatty acid oxidation accounts for 50-70% of cardiac energy production[3], cardiomyocytes possess multiple proteins for fatty acid transport such as fatty acid transport protein (FATP), fatty acid binding protein (FABP), and cluster of differentiation 36 (CD36) also known as fatty acid translocase (FAT)[3]. Glucose oxidation provides approximately 10% of myocardial energy production[43]. Cardiac basal glucose uptake is provided by glucose transporter 1 (GLUT1), which is constitutively expressed on the sarcolemma membrane[43], whereas the majority of cardiac glucose uptake is facilitated through insulin-dependent glucose transporter 4 (GLUT4), which translocates to the plasma membrane in response to insulin and contraction[11,43]. The natural competition/balance of glucose or free fatty acid substrate utilization was first described in 1963 and is known as the Randle cycle, where increased blood glucose inhibits free fatty acid oxidation[44]. Therefore, the utilization of fatty acid or glucose is dependent on the substrate availability[45]. Studies suggest that cardiac metabolic flexibility is essential in maintaining adequate ATP synthesis under normal and hypoxic conditions[43]. Unfortunately, insulin resistance, DM and heart failure significantly affect metabolic adaption during starvation and cardiac injury[46].
Hyperglycemia and hyperlipidemia observed in DM increase the storage and use of free fatty acid substrates in the myocardium, thereby inhibiting glucose utilization and decreasing metabolic flexibility[3,46]. DM further associates with impaired metabolic flexibility through attenuation in signaling that regulates the translocation of GLUT4 to the cardiomyocyte cell membrane, impairing glucose uptake, NO production and calcium homeostasis in the DM myocardium [20,35,45]. Moreover, a downregulation of AMP-activated protein kinase (AMPK), a master regulator of energy homeostasis, is noted in DCM resulting in inhibited glucose uptake and upregulation of FAT and FATP1, shifting metabolism towards fatty acid oxidation[4,6]. However, excessive utilization of free fatty acids can lead to an accumulation of toxic byproducts and cause cardiac lipotoxicity[4,45,47] In addition, this rise in fatty acid uptake and oxidation potentially contributes to the mitochondrial dysfunction, increased ROS production and development of DCM[3,43]. This may lead to decreased ATP production, decreased cardiac contractility, altered calcium handling, inflammation and exacerbation of myocyte death[4,45]. Interestingly, an increase in glucose and free fatty acids in the cytosol of myocytes have been observed during heart failure[46]. However, substrate entry into mitochondria is decreased, resulting in gluco- and lipo-toxicity and an increase in ketone substrate utilization which further decreases metabolic flexibility[46] contributing to worsening of DCM.
Although, the role of ATM in the development of DCM needs further investigation, evidence from non-cardiac cells and tissue suggest that ATM deficiency may share several similar pathways to DM when it comes to shifting cardiac energy metabolism. In L6 muscle cells transfected with ATM, insulin caused a dramatic increase in GLUT4 translocation[18]. Increase in cell surface GLUT4 may help shift the balance toward glucose metabolism[48]. Since DM and ATM deficiency associate with decreased AMPK activity, this suggests that AMPK activation may serve as a potential target to mitigate some of the metabolic changes that occur in the diabetic heart. Interestingly, some anti-diabetic drugs such as metformin are shown to decrease hepatic gluconeogenesis, increase insulin sensitivity and stimulate AMPK activation, which in-turn, decreases lipid and cholesterol synthesis[46]. Although the mechanism isn’t well understood, metformin treatment also activates AMPK, which reduces insulin resistance and increases glucose uptake in cardiac tissue, skeletal muscle and insulin resistant cardiomyocytes[27,46]. Interestingly, a genome-wide association study identified an association for a SNP (single-nucleotide polymorphism) at a locus containing ATM gene for the treatment success of metformin[13]. Further, inhibition of ATM using KU-55933 inhibited metformin-induced AMPK activation in hepatic cells, suggesting that ATM plays a role in the glucose lowering effects of metformin upstream of AMPK[49]. Collectively, these studies provide a role for ATM in the regulation of glucose homeostasis and metformin response. Of note, ATM heterozygous knockout mice (hKO) with ApoE null mutation (ATM +/−/ApoE−/−) display abnormal lipid metabolism and hypercholesterolemia as compared to the WT control[50,51]. Increased ROS derived from mitochondrial dysfunction may serve as the driving force behind the development of metabolic abnormalities observed during ATM deficiency. Treatment with mitochondrial-targeted antioxidant MitoQ successfully reversed multiple features of metabolic abnormalities detected in ATM +/−/ApoE−/− mice[52]. Oxidative activation of ATM causes a glucose metabolic shift from glycolysis to the pentose phosphate pathway, thereby causing anti-oxidative response which increases the survival and proliferation of cardiac progenitor cells in diabetic hearts[11,53]. Together, these studies provide evidence that ATM activation may play a protective role in the development of metabolic syndrome associated with DM. However, future investigations are needed to clarify the role of ATM in the development of DCM.
Insulin Resistance during DM and ATM Deficiency
Insulin signaling plays an important role in cardiac cellular homeostasis[4]. During normal cardiac insulin metabolic signaling, insulin binds to the insulin receptor, activating insulin receptor substrates, PI3K and Akt (Fig 2). This signaling cascade stimulates the activation of IRS1/2 transcription, inactivates Akt substrate 160 and translocates GLUT4 to the plasma membrane, stimulating glucose uptake and preventing lipolysis[43,45]. Insulin resistance or insufficient insulin production are two primary causes of diabetic hyperglycemia[54]. In DM, insulin resistance results in overproduction and release of insulin by the pancreatic β-cells, which causes hyperinsulinemia. With time, the β-cells become dysfunctional and cannot produce the quantity of insulin necessary to compensate for the insulin resistance[3,45], further complicating therapy. As a result, a decrease of insulin-mediated glucose uptake and increased free fatty acid oxidation is observed (Fig 2) [3,45].
Fig 2. Schematic depiction of insulin signaling during Diabetes and ATM deficiency.
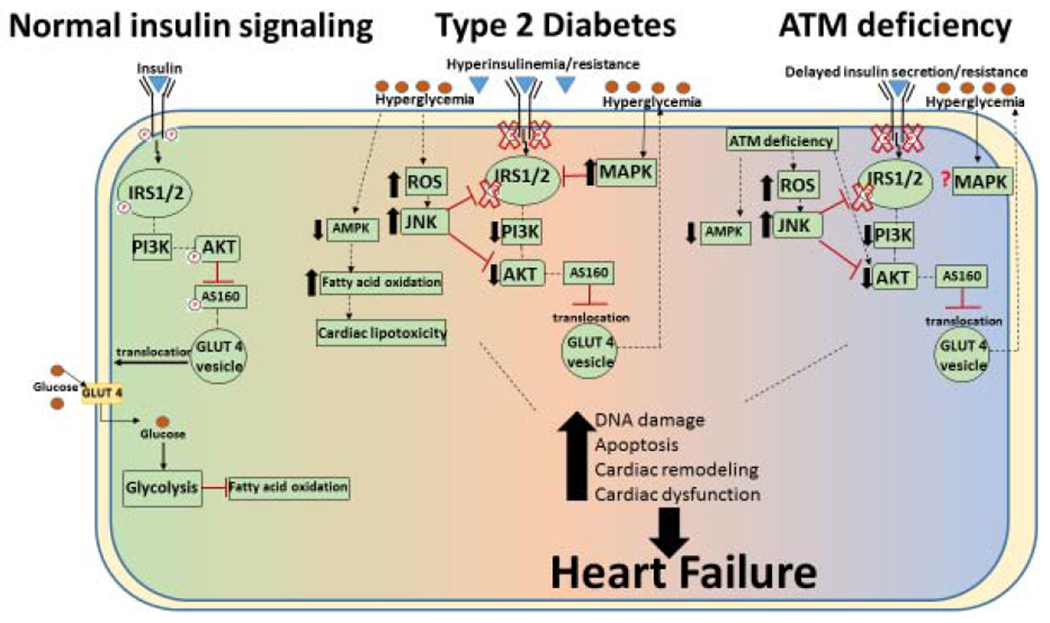
Left: Insulin responsive signaling: The intracellular signaling cascade begins with the binding of insulin to the insulin receptor which activates IRS1/2 (insulin receptor substrate proteins 1 and 2) and downstream targets such as Akt. Active Akt may phosphorylate Akt substrate 160 (AS160), and stimulate GLUT4 translocation to the plasma membrane and allowing glucose uptake. Center: Type 2 diabetic insulin signaling: Overproduction/release of insulin causes hyperinsulinemia. As insulin resistance increases, decreased insulin-mediated glucose uptake occurs resulting in decreased activation of IRS1/2 and Akt, phosphorylation of AS160, GLUT 4 translocation and glucose uptake. Persistent hyperglycemia then causes an increase in reactive oxygen species (ROS), and may activate JNK and decrease AMPK activation. These changes may shift the metabolism toward fatty acid oxidation and cardiac lipotoxicity. Together, these alterations can cause DNA damage, apoptosis, cardiac remodeling and cardiac dysfunction, leading to heart failure. Right: Insulin resistance during ATM deficiency: Delayed insulin secretion and insulin resistance is observed during ATM deficiency with decreased activation of IRS1/2 and Akt. This may decrease AS160 phosphorylation and glucose uptake. ATM deficiency independently associates with mechanisms suggested to cause insulin resistance such as decreased AMPK activation, increased JNK activation and higher oxidative stress. ATM deficiency also associates with increased DNA damage, apoptosis, cardiac remodeling and cardiac dysfunction.
ATM is established as an insulin responsive protein that plays a significant role in the glucose metabolic pathway[9,18]. Metabolic abnormalities such as hyperglycemia, insulin resistance and diminished/delayed insulin secretion have been observed in AT patients and ATM deficient mouse model[9,41,55]. Altered mitochondrial function and associated oxidative stress observed in cardiomyocytes during ATM deficiency are implicated (but not fully elucidated) in the development of metabolic dysfunction and insulin resistance[7,9,11].
Alteration in PI3K/Akt signaling is a major contributor to the development of insulin resistance and type 2 DM. Akt stimulates the transcription and translocation of GLUT4, which mediates the majority of cardiac glucose transport. Decreased PI3K/Akt signaling and associated decrease in GLUT4 expression has been observed in diabetic hearts and is linked to the acceleration of heart failure[4,6]. ATM KO hearts exhibit lower Akt activation at basal levels and in response to β-AR stimulation. Akt activation was also lower in the myocardium of ATM deficient mice 4hrs post-MI[30,32]. Therefore, the attenuation of PI3K/Akt signaling during ATM deficiency may contribute to the decrease in GLUT4 translocation, glucose uptake and insulin resistance observed in DM[6,18,20]. While ATM plays a large role in glucose metabolism, the effect of ATM deficiency on GLUT1 expression are still being elucidated. In skeletal muscle, ATM deficiency upregulated GLUT1 transcription with resultant increase in basal glucose uptake and insulin resistance[56]. Conversely, inhibition of ATM decreased cell surface GLUT1 with concomitant reduction of glucose transport in L6 myoblasts[57].
Increased JNK activation may also play a role in the development of insulin resistance by directly phosphorylating IRS 1 and 2, and reducing Akt activation and GLUT4 translocation[6,58]. JNK activation induces rat myocyte apoptosis, and is suggested to promote the development of the cardiovascular abnormalities seen in DM and DCM[6,58]. Enhanced JNK activation is also common during ATM deficiency, and is known to cause insulin resistance through the disruption of insulin signaling[11,59–61]. Chloroquine (an ATM activator) reduced metabolic abnormalities, JNK activation and the development of atherosclerosis while improving glucose tolerance in ATM +/−/ApoE−/− mice[60].
Activation of mitogen activated protein kinases (MAPK) pathway is suggested to contribute to cardiac hypertrophy, fibrosis, functional alterations, cardiomyocyte insulin resistance, acceleration of DCM and heart failure[6,62,63]. Extracellular signal-regulated kinase 1 and 2 (ERK1/2) belong to the MAPK family and play a role in transmitting extracellular signals to the intracellular molecular targets. ERK1/2 signaling contributes to cell survival, growth, differentiation, and proliferation [64,65]. Activation of ERK1/2 has been observed in response to hyperglycemia-induced stress and insulin signaling[64,65]. Interestingly, selective inhibition of ERK pathway is suggested to ameliorate insulin resistance [64] and may be a potential therapeutic target for DM. Lack of ATM associates with decreased ERK1/2 activation in neurosphere cells [66]. Mouse embryonic fibroblasts exhibit decreased ERK1/2 activation and defective differentiation into adipocytes[67]. DNA damage is suggested to activate ERK1/2 in an ATM-dependent manner in fibroblasts[68]. ERK1/2 activation was higher in the myocardium of WT mice 4 hr post-MI. However, ERK1/2 activation remained unchanged in mice with ATM deficiency[32]. Future investigations are warranted to investigate the role of ERK1/2 signaling in the development of insulin resistance and DM during ATM deficiency.
Conclusion
ATM is a versatile kinase involved in regulation of cell cycle progression, coordination of DNA damage response, vesicle/protein transport mechanisms, glucose metabolism, oxidative stress and mitochondrial function[7,8]. Consequently, ATM deficient individuals have an enhanced susceptibility to developing metabolic disorders, IHD and cancer[8]. Additionally, disruption of cytoplasmic signaling initiated by ATM contributes to metabolic complications, insulin resistance, DM, heart disease and heart failure[8,11]. Cardiovascular alterations observed during ATM deficiency further demonstrate the importance and versatility of ATM. ATM deficiency associates with increased cardiac fibrosis, hypertrophy, and an array of structural and functional abnormalities[8,29,31,34]. Further, ATM deficiency and DM independently associate with insulin resistance, metabolic impairment, hyperglycemia, hyperinsulinemia, dyslipidemia, hypertension, increased atherosclerosis, and cardiac structural and functional abnormalities (Fig 1). Additionally, DM and ATM deficiency exhibit alterations in metabolic associated proteins, including decreased AMPK, PI3K/Akt and ERK1/2 activation, and increased oxidative stress and JNK activation (Fig 2). Taken together, this information suggests that activation of ATM and ATM-dependent mechanisms may serve as potential therapeutic targets to protect against the metabolically-induced cardiovascular dysfunction observed during DM, and potentially reduce the development of heart failure. This is consistent with the observation of decreased ATM activation in muscle cells during insulin resistance[18]. Since DM and ATM deficiency share many metabolic and cardiac abnormalities (Fig. 1), it can be speculated that DM may cause a decrease in the expression of ATM, disturbing ATM-dependent metabolic mechanisms and causing cardiac structural and functional abnormalities leading to DCM and heart failure. Therefore, future studies are crucial to the understanding of the role of ATM and other signaling pathways involved in DM-mediated development and progression of DCM, and to the development of therapeutic approaches to decrease the risk of heart failure in diabetic patients.
Fig 1. Shared metabolic and cardiovascular abnormalities of diabetes mellitus (DM) and Ataxia telangiectasia mutated kinase (ATM) deficiency.
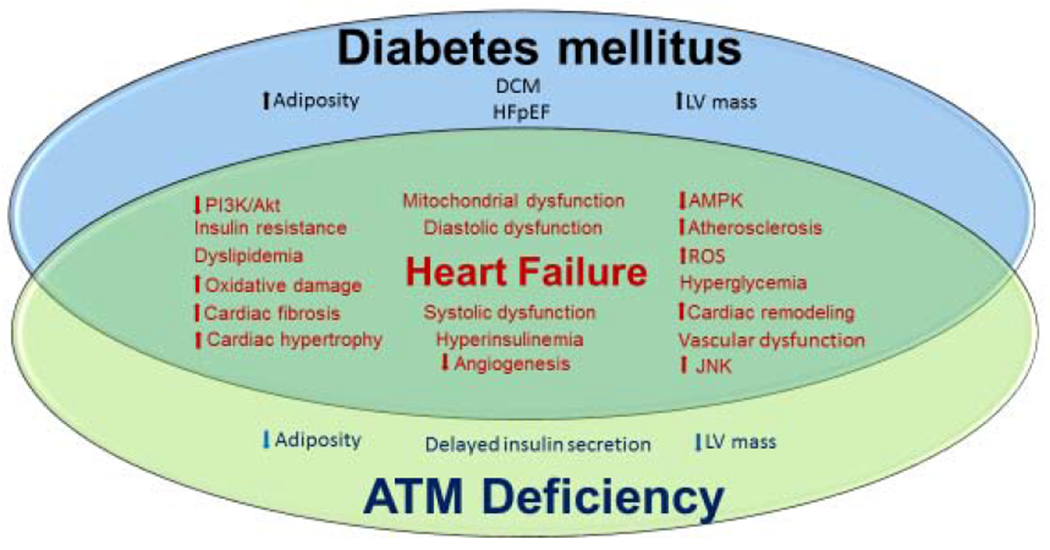
Alterations of AMPK, PI3K/Akt, and JNK independently associate with DM and ATM deficiency, and have been implicated in the inhibited glucose uptake and insulin resistance observed in both conditions. Further, aberrant metabolic conditions such as dyslipidemia, hyperinsulinemia, hyperglycemia and insulin resistance are features of DM and ATM deficiency, and contribute to vascular dysfunction. Mitochondrial dysfunction, increased ROS production and associated oxidative damage are also present in DM and during ATM deficiency. Lastly, DM and ATM deficiency independently associate with cardiac structural and functional abnormalities including increased cardiac fibrosis, hypertrophy, systolic and diastolic dysfunction. Collectively, all of the aforementioned conditions contributes to the development and progression of diabetic cardiomyopathy and heart failure.
Acknowledgments
Funding
This work is supported by Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development Merit Review Award BX004045, NIH NHLBI Grant R15HL141947 and funds from the Institutional Research and Improvement account (to K. Singh).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
No conflicts of interest, financial or otherwise, are declared by the authors.
Conflict of Interest
None
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
● of special interest
●● of outstanding interest
- 1.Centers for Disease Control and Prevention USD of H and HS: National diabetes datistics report. Centers Dis Control Prev 2017, [Google Scholar]
- 2.Ofstad AP, Atar D, Gullestad L, Langslet G, Odd &, Johansen E: The heart failure burden of type 2 diabetes mellitus-a review of pathophysiology and interventions. Hear Fail Rev 2018, 23:303–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Isfort M, Stevens SCW, Schaffer S, Jong CJ, Wold LE: Metabolic dysfunction in diabetic cardiomyopathy. Hear Fail Rev 2014, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kenny HC, Abel ED: Heart failure in type 2 diabetes mellitus. Circ Res 2019, 124:121–141. [DOI] [PMC free article] [PubMed] [Google Scholar]; ●● This paper explores pathophysiological risk factors beyond hyperglycemia, which contribute to heart failure in diabetic patients. Additionally, pharmacological therapies for patients with concomitant diabetes and heart failure were discussed.
- 5.Wu Y, Ding Y, Tanaka Y, Zhang W: Risk factors contributing to type 2 diabetes and recent advances in the treatment and prevention. Int J Med Sci 2014, 11:1185–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jia G, Hill MA, Sowers JR: Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res 2018, 122:624–638. [DOI] [PMC free article] [PubMed] [Google Scholar]; ● This paper highlights pathophysiological mechanisms that contribute to the metabolic and cardiovascular dysfunction present during diabetic cardiomyopathy.
- 7.Blignaut M, Loos B, Botchway SW, Parker AW, Huisamen B: Ataxia-telangiectasia mutated is located in cardiac mitochondria and impacts oxidative phosphorylation. Sci Rep 2019, 9:4782. [DOI] [PMC free article] [PubMed] [Google Scholar]; ●● This is the first paper to show that ATM localizes within the inner mitochondrial membrane of cardiomyocytes during normoxic conditions, interacts with the electron transport chain and may play a role in oxidative phosphorylation.
- 8.Thrasher P, Singh M, Singh K: Ataxia-telangiectasia mutated kinase: role in myocardial remodeling. J rare Dis Res Treat 2017, 2:32–37. [PMC free article] [PubMed] [Google Scholar]
- 9.Amirifar P, Ranjouri MR, Yazdani R, Abolhassani H, Aghamohammadi A: Ataxia-telangiectasia: a review of clinical features and molecular pathology. Pediatr Allergy Immunol 2019, 30:277–288. [DOI] [PubMed] [Google Scholar]; ● This paper provides an extensive assessment of nuclear and cytoplasmic functions of ATM, clinical manifestations, and associated complications of ATM deficiency.
- 10.Ambrose M, Gatti RA: Pathogenesis of ataxia-telangiectasia: the next generation of ATM functions. Blood 2013, 121:4036–4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Espach Y, Lochner A, Strijdom H, Huisamen B: ATM protein kinase signaling, type 2 diabetes and cardiovascular disease. Cardiovasc Drugs Ther 2015, 29:51–58. [DOI] [PubMed] [Google Scholar]
- 12.Hassin-Baer S, Bar-Shira A, Gilad S, Galanty Y, Khosravi R, Lossos A, Giladi N, Weitz R, Ben-Zeev B, Goldhammer Y, et al. : Absence of mutations in ATM, the gene responsible for ataxia telangiectasia in patients with cerebellar ataxia. J Neurol 1999, 246:716–9. [DOI] [PubMed] [Google Scholar]
- 13.Ditch S, Pauli TT: The ATM protein kinase and cellular redox signaling: beyond the DNA damage response. Trends Biochem Sci 2012, 37:15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rothblum-Oviatt C, Wright J, Lefton-Greif MA, McGrath-Morrow SA, Crawford TO, Lederman HM: Ataxia telangiectasia: A review. Orphanet J Rare Dis 2016, 11:159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shiloh Y, Ziv Y: The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 2013, 14:197–210. [PubMed] [Google Scholar]
- 16.Nowak-Wegrzyn A, Crawford TO, Winkelstein JA, Carson KA, Lederman HM: Immunodeficiency and infections in ataxia-telangiectasia. Natl Institutes Heal 2004, 144:505–516. [DOI] [PubMed] [Google Scholar]
- 17.van Os NJH, Roeleveld N, Weemaes CMR, Jongmans MCJ, Janssens GO, Taylor AMR, Hoogerbrugge N, Willemsen MAAP: Health risks for ataxia-telangiectasia mutated heterozygotes: a systematic review, meta-analysis and evidence-based guideline. Clin Genet 2016, 90. [DOI] [PubMed] [Google Scholar]
- 18.Halaby M-J, Hibma JC, He J, Yang D-Q: ATM protein kinase mediates full activation of Akt and regulates glucose transporter 4 translocation by insulin in muscle cells. Cell Signal 2008, 20:1555–1563. [DOI] [PubMed] [Google Scholar]
- 19.Thomas MC: Type 2 diabetes and heart failure: challenges and solutions. Curr Cardiol Rev 2016, 12:249–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Riehle C, Abel ED: Insulin signaling and heart failure. Clrc Res 2016, 118:1151–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dunlay SM, Michael Givertz C-CM, David Aguilar C-C, Larry Allen FA, Michael Chan F, Akshay Desai FS, Anita Deswal M, Victoria Vaughan Dickson F, Mikhail Kosiborod FN, Carolyn Lekavich FL, et al. : On behalf of the american heart association heart failure and transplantation committee of the council on clinical cardiology; council on cardiovascular and stroke nursing; and the heart failure society of america type 2 diabetes mellitus and heart failur. Circulation 2019, 140:294–324. [Google Scholar]
- 22.Zhou M-S, Wang A, Yu H: Link between insulin resistance and hypertension: what is the evidence from evolutionary biology? Dlabetol Metab Syndr 2014, 6:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soares Felício J, Cavalcante Koury C, Tavares Carvalho C, Felício Abrahão Neto J, Barbosa Mileo K, Pontes Arbage T, Dias Silva D, Ferreira de Oliveira A, Soares Peixoto A, Bentes Figueiredo Junior A, et al. : Present insights on cardiomyopathy in diabetic patients. Curr Diabetes Rev 2016, 12:384–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Al Flroob AM, Abukhalil MH, Hussein OE, Mahmoud AM: Pathophysiological mechanisms of diabetic cardiomyopathy and the therapeutic potential of epigallocatechin-3-gallate. Biomed Pharmacother 2019, 109:2155–2172. [DOI] [PubMed] [Google Scholar]
- 25.Volpe CMO, Villar-Delfino PH, Dos Anjos PMF, Nogueira-Machado JA: Cellular death, reactive oxygen species (ROS) and diabetic complications review-Article. Cell Death Dis 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zoppini G, Bergamini C, Bonapace S, Trombetta M, Mantovani A, Toffalini A, Lanzoni L, Bertolini L, Zenari L, Bonora E, et al. : Left ventricular chamber dilation and filling pressure may help to categorise patients with type 2 diabetes. BMJ Open Diab Res Care 2018, 6:529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jia G, Demarco VG, Sowers JR: Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol 2016, 12:144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Foster CR, Singh M, Subramanian V, Singh K: Ataxia telangiectasia mutated kinase plays a protective role in β-adrenergic receptor-stimulated cardiac myocyte apoptosis and myocardial remodeling. Mol Cell Biochem 2011, 353:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Daniel LL, Daniels CR, Harirforoosh S, Foster CR, Singh M, Singh K: Deficiency of ataxia telangiectasia mutated kinase delays inflammatory response in the heart following myocardial infarction. J Am Heart Assoc 2014, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Foster CR, Zha Q, Daniel LL, Singh M, Singh K: Lack of ATM induces structural and functional changes in the heart: role in β-adrenergic receptor-stimulated apoptosis. Exp Physiol 2012, 94:506–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Foster CR, Daniel LL, Daniels CR, Dalai S, Singh M, Singh K: Deficiency of ataxia telangiectasia mutated kinase modulates cardiac remodeling following myocardial infarction: involvement in fibrosis and apoptosis. PLoS One 2013, 8:e83513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thrasher PR, Scofield SLC, Dalai S, Crawford CC, Singh M, Singh K: Ataxia telangiectasia mutated kinase deficiency impairs the autophagic response early during myocardial infarction. Am J Physiol Hear Circ Physiol 2018, 315:48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]; ● ATM deficiency was found to decrease AMPK and Akt following myocardial infarction and impaired the autophagic response after cardiac injury
- 33.Jia L, Zhang W, Ma Y, Chen B, Liu Y, Piao C, Wang Y, Yang M, Liu T, Zhang J, et al. : Haplodeficiency of ataxia telangiectasia mutated accelerates heart failure after myocardial infarction. J Am Heart Assoc 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]; ●● This study explored the role of ATM deficiency on heart failure following myocardial infarction (MI). ATM deficiency was found to impair angiogenesis post-MI, which may contribute to accelerated heart failure.
- 34.Daniel LL, Scofield SLC, Thrasher P, Dalai S, Daniels CR, Foster CR, Singh M, Singh K: Ataxia telangiectasia-mutated kinase deficiency exacerbates left ventricular dysfunction and remodeling late after myocardial infarction. Am J Physiol - Hear Circ Physiol 2016, 311: H445–H452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jia G, Whaley-Connell A, Sowers JR: Diabetic cardiomyopathy: a hyperglycaemia-and insulin-resistance-induced heart disease. Diabetologia 2018, 61:21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]; ●● This paper discusses the evolution of diabetic cardiomyopathy and pathophysiological mechanisms, and demonstrates the complexity of concomitant diabetic cardiomyopathy and heart failure, preventative strategies and treatment options.
- 36.Hinderer S, Schenke-Layland K: Cardiac fibrosis - a short review of causes and therapeutic strategies. Adv Drug Deliv Rev 2019, 146:77–82. [DOI] [PubMed] [Google Scholar]
- 37.Russo I, Frangogiannis NG: Diabetes-associated cardiac fibrosis: cellular effectors, molecular mechanisms and therapeutic opportunities. J Mol Cell Cardiol 2016, 90:84–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.St John Sutton MG, Sharpe N: Left Ventricular Remodeling After Myocardial Infarction Pathophysiology and Therapy Clinical Cardiology: New Frontiers. 2000. [DOI] [PubMed] [Google Scholar]
- 39.Costa PZ, Soares R: Neovascularization in diabetes and its complications. Unraveling the angiogenic paradox. Life Sci 2013, 92:1037–1045. [DOI] [PubMed] [Google Scholar]
- 40.Wang X, Huang W, Liu G, Cai W, Millard RW, Wang Y, Chang J, Peng T, Fan GC: Cardiomyocytes mediate anti-angiogenesis in type 2 diabetic rats through the exosomal transfer of miR-320 into endothelial cells. J Mol Cell Cardiol 2014, 74:139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miles PD, Treuner K, Latronica M, Olefsky JM, Barlow C: Impaired insulin secretion in a mouse model of ataxia telangiectasia. Am J Physiol Metab 2007, 293:E70–E74. [DOI] [PubMed] [Google Scholar]
- 42.Janus A, Szahidewicz-Krupska E, Mazur G, Doroszko A: Insulin resistance and endothelial dysfunction constitute a common therapeutic target in cardiometabolic disorders. Mediators Inflamm 2016, 2016:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chanda D, Luiken JJFP, Glatz JFC: Signaling pathways involved in cardiac energy metabolism. FEBS Lett 2016, 590:2364–2374. [DOI] [PubMed] [Google Scholar]
- 44.Randle PJ, Garland PB, Hales CN, Newsholme EA: The glucose fatty acid cycle its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 281:785–789. [DOI] [PubMed] [Google Scholar]
- 45.Ormazabal V, Nair S, Elfeky O, Aguayo C, Salomon C, Zuniga FA: Association between insulin resistance and the development of cardiovascular disease. Cardiovasc Diabetol 2018, 17:122. [DOI] [PMC free article] [PubMed] [Google Scholar]; ● This paper provided a mechanistic description of insulin resistance and its effects on the development of cardiovascular disease and atherosclerosis.
- 46.Maack C, Lehrke M, Backs J, Heinzel FR, Hulot JS, Marx N, Paulus WJ, Rossignol P, Taegtmeyer H, Bauersachs J, et al. : Heart failure and diabetes: metabolic alterations and therapeutic interventions: a state-of-the-art review from the translational research committee of the heart failure Aasociation-european society of cardiology. Eur Heart J 2018, 39:4243–4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stanley WC, Recchia FA, Lopaschuk GD: Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 2005, 85:1093–1129. [DOI] [PubMed] [Google Scholar]
- 48.Shao D, Tian R, Compr P: Glucose transporters in cardiac metabolism and hypertrophy overview of glucose transporter. Compr Physiol 2015, 6:331–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou K, Bellenguez C, Spencer CCA, Bennett AJ, Coleman RL, Tavendale R, Hawley SA, Donnelly LA, Schofield C, Groves CJ, et al. : Common variants near ATM are associated with glycemic response to metformin in type 2 diabetes. Nat Genet 2011, 43:117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mercer JR, Cheng KK, Figg N, Gorenne I, Mahmoudi M, Griffin J, Vidal-Puig A, Logan A, Murphy MP, Bennett M: DNA damage links mitochondrial dysfunction to atherosclerosis and the metabolic syndrome. Circ Res 2010, 107:1021–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu DF, Yang H, Xiang W, Zhou LC, Shi MJ, Julies G, LaPlante JM, Ballard BR, Guo ZM: Heterozygous mutation of ataxia-telangiectasia mutated gene aggravates hypercholesterolemia in apoE-deficient mice. J Lipid Res 2005, 46:1380–1387. [DOI] [PubMed] [Google Scholar]
- 52.Mercer JR, Yu E, Figg N, Cheng KK, Prime TA, Griffin JL, Masoodi M, Vidal-Puig A, Murphy MP, Bennett MR: The mitochondria-targeted antioxidant mitoQ decreases features of the metabolic syndrome in ATM +/−/ApoE−/− mice. Free Radio Biol Med 2012, 52:841–849. [DOI] [PubMed] [Google Scholar]
- 53.Katare R, Oikawa A, Cesselli D, Beltrami AP, Avolio E, Muthukrishnan D, Munasinghe PE, Angelini G, Emanueli C, Madeddu P: Boosting the pentose phosphate pathway restores cardiac progenitor cell availability in diabetes. Cardiovasc Res 2013, 97:55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jun HS, Bae HY, Lee BR, Koh KS, Kim YS, Lee KW, Kim H man, Yoon JW: Pathogenesis of non-insulin-dependent (type II) diabetes mellitus (niddm) - genetic predisposition and metabolic abnormalities. Adv Drug Deliv Rev 1999, 35:157–177. [DOI] [PubMed] [Google Scholar]
- 55.Connelly PJ, Smith N, Chadwick R, Exley AR, Shneerson JM, Pearson ER: Recessive mutations in the cancer gene ataxia telangiectasia mutated ( ATM ), at a locus previously associated with metformin response, cause dysglycaemia and insulin resistance. Diabet Med 2016, 33:371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ousset M, Bouquet F, Fallone F, Biard D, Dray C, Valet P, Salles B, Muller C: Cell cycle loss of ATM positively regulates the expression of hypoxia inducible factor 1 (HIF-1) through oxidative stress: role in the physiopathology of the disease. Cell Cycle 2010, 9:2814–2822. [DOI] [PubMed] [Google Scholar]
- 57.Andrisse S, Patel GD, Chen JE, Webber AM, Spears na LD, Koehler RM, Robinson-Hill RM, Ching JK, Jeong I, Fisher JS: ATM and glut1-S490 phosphorylation regulate glutl mediated transport in skeletal muscle. PLoS One 2013, 8:e66027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Solinas G, Becattini B: JNK at the crossroad of obesity, insulin resistance, and cell stress response. Mol Metab Metab 2017, 6:174–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McGill JB, Johnson M, Hurst S, Cade WT, Yarasheski KE, Ostlund RE, Schechtman KB, Razani B, Kastan MB, McClain DA, et al. : Low dose chloroquine decreases insulin resistance in human metabolic syndrome but does not reduce carotid intima-media thickness. Diabetol Metab Syndr 2019, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schneider JG, Finck BN, Ren J, Standley KN, Takagi M, Maclean KH, Bernal-Mizrachi C, Muslin AJ, Kastan MB, Semenkovich CF: ATM-dependent suppression of stress signaling reduces vascular disease in metabolic syndrome. Cell Metab 2006, 4:377–389. [DOI] [PubMed] [Google Scholar]
- 61.Guleria A, Chandna S: ATM kinase: much more than a DNA damage responsive protein. DNA Repair (Amst) 2016, 39:1–20. [DOI] [PubMed] [Google Scholar]
- 62.Xu Z, Sun J, Tong Q, Lin Q, Qian L, Park Y, Zheng Y, Struijker-Boudier HAJ: The role of erk1/2 in the development of diabetic cardiomyopathy. Int J Mol Sci 2016, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lakshmanan AP, Harima M, Sukumaran V, Soetikno V, Thandavarayan RA, Suzuki K, Kodama M, Nagata M, Takagi R, Watanabe K: Modulation of AT-1R/AMPK-MAPK cascade plays crucial role for the pathogenesis of diabetic cardiomyopathy in transgenic type 2 diabetic (spontaneous diabetic torii) rats. Biochem Pharmacol 2012, 83:653–660. [DOI] [PubMed] [Google Scholar]
- 64.Ozaki K, Awazu M, Tamiya M, Iwasaki Y, Harada A, Kugisaki S, Tanimura S, Kohno M: Targeting the ERK signaling pathway as a potential treatment for insulin resistance and type 2 diabetes. Am J Physiol Endocrinol Metab 2016, 310:643–651. [DOI] [PubMed] [Google Scholar]
- 65.Xu Z, Sun J, Tong Q, Lin Q, Qian L, Park Y, Zheng Y: The role of ERK1/2 in the development of diabetic cardiomyopathy. Int J Mol Sci 2016, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim J, Wong PKY: Loss of ATM impairs proliferation of neural stem cells through oxidative stress-mediated p38 MAPK signaling. Stem Cells 2009, 27:1987–1998. [DOI] [PubMed] [Google Scholar]
- 67.Takagi M, Ogawa Y, Correspondence SM: ATM regulates adipocyte differentiation and contributes to glucose homeostasis. CellReports 2015, 10:957–967. [DOI] [PubMed] [Google Scholar]
- 68.Tang D, Wu D, Hirao A, Lahti JM, Liu L, Mazza B, Kidd VJ, Mak TW, Ingram AJ: ERK activation mediates cell cycle arrest and apoptosis after DNA damage independently of p53. J Biol Chem 2002, 277:12710–12717. [DOI] [PubMed] [Google Scholar]