

Abstract
The bone marrow microenvironment (BMM) provides input via production of cytokines, chemokines, extracellular matrixes in the context of lower oxygen levels that influences self-renewal, survival, differentiation, progression and therapeutic resistance of multiple myeloma and leukemic cells. Within the context of the BMM, tumor cells are supported by osteoblasts, bone marrow stromal cells (BMSCs), fibroblasts, myeloid cells, endothelial cells and blood vessels as well as extracellular matrix (ECM) that contribute to tumor progression. Environmental mediated-drug resistance (EM-DR) contains cell adhesion mediated drug resistance (CAM-DR) and soluble factor mediated drug resistance (SM-DR) that contributes to de-novo drug resistance. In this review, we focus on the crosstalk between the BMM and tumor cells as well as mechanisms underlying the BMM contributing to drug resistance in hematologic malignancies.
Keywords: bone marrow microenvironment (BMM), cell adhesion mediated drug resistance (CAM-DR), soluble mediated drug resistance (SM-DR)
Introduction
Tumor cells although driven by the expression of oncogenes and deletions of tumor suppressors genes efficiently utilize cues from the tumor microenvironment that are favorable for survival and disease progression. The bone marrow microenvironment (BMM) is known as a tumor sanctuary and has been recognized to play a critical role in tumor development, progression, metastasis and therapeutic resistance. The bone marrow (BM) is the major location for hematopoiesis and as such contains gradients of chemokines, cytokines and oxygen content that supports the pluripotent hematopoietic stem cells (HSCs) as well as provides the necessary cues for mobilization and differentiation of HSCs toward the myeloid or lymphoid progenitor cell [1]. In the adult BM, HSCs are located near the peri-sinusoidal and peri-arteriolar regions of the BM (Figure 1). In vivo studies indicate that the O2 content of the BM is 1–4% and is significantly lower than what is found in the peripheral blood which ranges from 10–13% O2 content [2]. The hypoxic environment contributes to preserving normal HSC quiescence and self-renewal capacity in part due to HIF dependent alterations in gene expression [3]. The BM is an environment that a cancer stem cell could potentially highjack as quiescence is known to protect from cytotoxic agents as well as targeted therapy. N-cadherin expression of HSCs is also considered to sustain HSCs in a quiescent state [4, 5]. Experimental evidence indicates that depletion of N-cadherin on HSCs decreased the ability of HSCs to bind to the endosteal surface [6, 7]. The quiescent state is considered to protect HSCs from stress [8]. The vascular niche serves as a microenvironment that regulates hematopoiesis, stem cell mobilization and homing, proliferation and differentiation of actively cycling, and short-term HSCs [9]. The vascular niche produces factors such as SDF-1 which is critical for mobilization, homing and engraftment in HSCs [10]. Experimental evidence derived from both in vitro and in vivo models demonstrate that hematopoietic tumors will highjack the micro-domains contained within the bone marrow niche for survival and immune invasion. Clinical evidence indicates that minimal residual disease is found in the bone marrow microenvironment following standard of care treatment of leukemia’s as well multiple myeloma. Together, these data suggest that the BM may contribute to failure to eliminate minimal residual disease in patients and limits the success of current chemotherapeutic strategies. Survival pathways emanating from the niche include activation of signaling cascades that in turn activate classical survival signals including but not limited to the PI3K/AKT, MAPK, and JAK/STAT pathways. In addition, tumor cells can highjack the niche to promote quiescence a cell state which is also known to confer resistance to standard cytotoxic drugs. This review will discuss the impact of bone marrow stroma cells on survival and drug resistance of hematopoietic tumors as well as potential opportunities for targeting these interactions to improve patient outcomes.
Figure 1: Bone marrow microenvironment (BMM) is a dynamic and complex network.
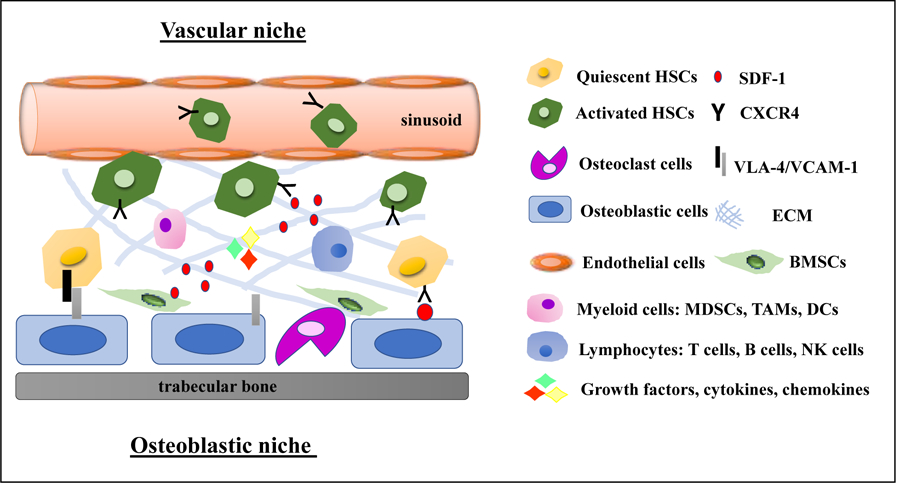
The BMM consists of several cells including quiescent/activated hematopoietic stem cells (HSCs), osteoclast cells, osteoblastic cells, endothelial cells, myeloid cells (myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and dendritic cells (DC)), lymphocytes (T cells, B cells and nature killer (NK) cells), bone marrow stromal cells (BMSCs) extracellular matrix (ECM), vasculature, and soluble factors such as growth factors, cytokines, chemokines.
Interactions between hematologic malignancies and bone marrow microenvironment (BMM)
CXCR4/SDF-1 axis is critical for the cross-talk between tumor cells and BMM [11]. Stromal cell-derived factor-1 (SDF-1, also known as CXCL12) is a homeostatic chemokine that signals through its receptor, CXCR4, for hematopoiesis, development and organization of the immune system. BM stromal cells (BMSCs) are a major source for SDF-1 secretion. Upon stimulation by its agonist SDF-1, CXCR4 is phosphorylated on serine/threonine residues leading to the activation of the JAK/STAT pathway, Wnt/β-catenin, JNK/PI3K pathway, NF-kB signaling, ERK1/2 and Ras/Raf pathways [12]. Of note many of the signaling pathways that can be transiently regulated by the BMM mimic oncogene dependency such as activation of PI3K, JAK/STAT and ERK1/2 pathways. BM niche maintains various function of HSCs supporting stem cell adhesion, self-renewal, apoptosis, mobilization and homing [13*–15]. A process whereby HSCs leaves the BM and migrates between blood circulation and the BM niche is commonly referred to as mobilization and homing [15]. Integrins, selectins and chemokines are known to affect the homing of HSCs. CXCR4/SDF-1 related with stem cell motility, tissue repair and regeneration [15, 16*]. Considering the BM niche is critical for supporting the survival and self-renewal of HSCs it is perhaps not surprising that minimal residual disease following standard of care treatment of leukemias and multiple myeloma (MM) is found in the BM niche [17*, 18]. These clinical observations support the translational need to understand the mechanism(s) that spares tumor cells from chemotherapy while residing in the BM niche. Hematopoietic tumor cells highjack mechanisms used by stem cells to home and survive in the BM niche [11]. Homing is a multistep process, including signaling by the SDF-1, activation of lymphocyte function-associated antigen 1(LFA-1), α4β1 integrin, and cytoskeleton rearrangement [19*, 20]. Once tumor cells reside in the niche exposure to BMSCs is sufficient to cause resistance to mechanistically and structurally diverse chemotherapeutic agents. In addition, the BM niche is known to confer resistance to FAS and trail ligand mediated cell death and thus may also contribute to immune evasion [21*]. The resistant phenotype is typically reversible as removal from exposure to components of the BM niche leads to a rapid reversal of drug resistance [22].
Soluble factors-mediated drug resistance (SM-DR)
A variety of soluble factors such as growth factors (vascular endothelial growth factor (VEGF), FGF, G-CSF, GM-CSF and TGFs), cytokines (IL-3, IL-6, IL-11, TNFα, IFNs) and chemokine factors (SCF and SDF-1) contribute to survival, growth and drug resistance in hematologic malignancies [23, 24]. For example, cross talk between tumor cells and BMSCs are the major source of IL-6 (Figure 2). IL-6 up-regulates the antiapoptotic protein Mcl-1 and Bcl-XL through inducing JAK/STAT3, PI3K/AKT and MEK/MAPK signaling pathways that are known to augment survival and drug resistance in MM [25–27]. Autocrine IL-6 derived from MM patients exhibited resistance to dexamethasone-induced apoptosis, whereas MM cells that do not produce IL-6 cells were sensitive to dexamethasone [28]. Mesenchymal stem cells (MSCs) from MM patient produced abnormally high amounts of IL-6 as compared to MSCs derived from healthy donor [24]. A monoclonal antibody directed against IL-6 (Siltuximab) has been designed to neutralize IL-6 has entered into clinical trials [29, 30]. VEGF can be secreted by both MM cells and BMSCs and is associated with angiogenesis, but can also directly support proliferation, migration and drug resistance in MM cells [31, 32]. Moreover, VEGF enhances BMSCs secreted IL-6, indicating VEGF is not only an autocrine growth factor but can also trigger of IL-6-mediated paracrine MM mediated growth [25]. VEGF also increased microvessel density (MVD) in the BM that correlated with disease progression and poor prognosis. A small peptide CXCR4 antagonist, LY2510924, can inhibit BMSCs-mediated resistance to chemotherapy-induced apoptosis in primary acute myelogenous leukemia (AML) [33*, 34]. Cytokine signaling from BMM provides a protective sanctuary niche for chronic myelogenous leukemia (CML) cell survival in the presence of BCR-ABL kinase inhibitor [35]. Experimental evidence supports that activation of STAT3 is a causative mediator of BCR-ABL-independent resistance in CML [34]. Inhibition of pSTAT3 using siRNA strategies or treatment with a Jak inhibitor sensitize CML to tyrosine kinase inhibitor (TKI) in the context of conditioned media derived from BMSCs. Cell lines used in this study included the BCR-ABL driven K562 and KU-812 CML cell lines and was further confirmed using primary CML patient derived samples [37–39**]. Soluble factors released by bone marrow-derived mesenchymal stomal cells (HS-5) lead to activate STAT3 in CML (K562) that was sufficient to caused resistance to imatinib mesylate, nilotinib and dastinib-mediated cell death [38]. Specifically, treatment with ruxolitinib, a JAK2 inhibitor which targets the JAK-STAT pathway, enhances TKI-induced apoptosis in CML cell lines (K562 and KU-812) and primary patient derived cell lines [40]. A phase I clinical trial (NCT01702064) of ruxolitinib in combination with nilotinib in CML patients showed to be safe, and well-tolerated and encouraging responses were reported [37].
Figure 2. SM-DR and CAM-DR contribute to drug resistance.
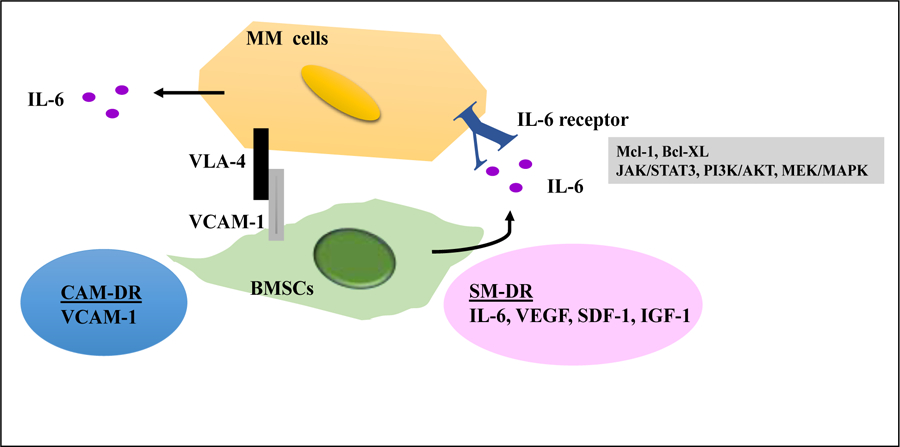
BMSCs are the major source of IL-6. IL-6 up-regulates the Mcl-1 and Bcl-XL through JAK/STAT3, PI3K/Akt and MEK/MAPK that are known to augment survival and drug resistance. CAM-DR induces a multidrug resistant phenotype as cell adhesion can protect tumors cells from chemotoxic agents in MM. CAM-DR can also cause to express of soluble factor in conferring tumor cell drug resistance.
Cell adhesion-mediated drug resistance (CAM-DR)
The interactions between tumor cells and the BMM can occur via direct contact with ECM components, including microfibrillar collagen type-VI, fibronectin (FN), collagen, vitronectin, osteopontin, laminin, or stromal cells. These interactions regulate cell proliferation, differentiation, survival and migration in normal hematopoietic, epithelial cells and various tumor cells [41]. It has been established by multiple studies that cancer cell adhesion to various matrixes is sufficient to confer de novo drug resistance a phenomenon which has been referred to as CAM-DR [42–48]. Cell adhesion is mediated by adhesion molecules which include but not limited to integrins, immuno-globulin superfamily and selectins. The vascular cell adhesion molecule-1 (VCAM-1) and Fibronectin (FN) are two known ligands for α4β1 integrin commonly referred to as very late antigen-4, (VLA-4) [49–51]. Inside-out activation of VLA-4 is required for this integrin to be competent to bind matrix [52]. Moreover, α4β1 is highly expressed on MM patients suggesting that VLA-4 is an important adhesive molecule for disease progression [53]. Bortezomib decreases α4β1 expression and results in inhibition of CAM-DR in MM a finding that may contribute to the clinical success of bortezomib [49]. The selection of doxorubicin-resistant(8226/DOX6) and melphalan-resistant (8226/LR5) MM cells correlated with overexpression of VLA-4. Conversely, 8226/DOX6 and 8226/LR5 cells removed from drug reverted to drug sensitive state which correlated with lower VLA-4 expression. These data suggest that drug exposure of cytotoxic drugs selects for increased expression of VLA-4. Moreover, the drug sensitive 8226 MM cells (8226/S) adhered to FN as a substrate conferred resistance to the apoptotic effects of doxorubicin and melphalan treatment. This adhesion mediated drug resistance referred to as CAM-DR was not caused by reduced drug accumulation or upregulation of anti-apoptotic Bcl-2 family members [42, 54]. CAM-DR induces a multidrug resistant phenotype as cell adhesion can protect tumors cells from multiple structurally and mechanistically diverse chemotoxic agents in MM [42, 54–57] and leukemia model systems [58–60]. Furthermore, CAM-DR can also augment signaling pathways that cause increased expression of soluble factors in conferring tumor cell drug resistance. For example, interaction between MM and BMSCs enhances BMSCs to secrete IL6 and GM-CSF and MM cells to release IL1B, TGFB, IL6 and VEGF [43, 60, 61] the supportive bi-directional signaling between tumor and stroma cells which culminate to support survival following cytotoxic insult. VLA-4 is a useful indicator for drug resistance and as a therapeutic target in MM therapy. Biomedical imaging techniques such as skeletal survey and 18F-fluorodeoxyglucose (FDG)/positron emission tomography (PET) are tools for diagnosing and stage MM patients [62]. However, both of these approaches have limitations. Experimental results indicated that VLA-4-targeted PET 64Cu-CB-TE1A1P-LLP2A showed high uptake in the 5TGM1 mouse tumors and high binding to the human MM cell lines RPMI-8226, indicating that VLA-4-targeted imaging with 64Cu-CB-TE1A1P-LLP2A is another tool to image MM tumors [63, 64]. A murine anti-integrin-α4 antibody (PS/2) inhibited tumor burden in BM using both in vitro and in vivo models. Natalizumab, a recombinant humanized IgG4 monoclonal antibody blocked tumor growth tumor growth and chemosensitized MM cells to bortezomib, suggesting combination with VLA-4 and bortezomib enhanced MM cytotoxicity and could be a strategy to improve patient outcome [64, 65]. A clinical trial (NTC00675428) to study the safety profile and the anti-tumor activity of natalizumab in patients of relapsed/refractory MM was used to evaluate possible correlations with clinical activity. Unfortunately, this trial was noted to be terminated due to low enrollment [66].
Epigenetic alterations
In contrast to the acquired drug resistance largely driven by genetic mutations, accumulating evidence suggest that epigenetic regulation contributes to the EM-DR induced by environment cues. Recent literature support that chromatin regulators play a critical role in BMM-induced drug resistance and constitute promising targets of therapeutic epi-drug development for various cancers. Due to space limitation, we will summarize BMM-mediated drug resistance regulated by epigenetic regulators in MM [67, 68].
Inhibitors of histone deacetylases (HDACs) are promising accessory therapeutic agents for MM treatment [69]. The impact of HDAC inhibitors on BM-mediated drug resistance in MM are topics of several recent studies. RPMI8226 and U266 cells pretreated by romidepsin a class I histone deacetylase inhibitor substantially reverses drug resistance to bortezomib, melphalan, and dexamethasone as induced by stromal UBE6T-7 cells [70]. Remarkably, romidepsin treatment decreases the expression of CD49d, a key component of the VAL-4 integrin that mediates the MM adhesion to stromal cells [71]. Another type I HDAC inhibitor BG45 triggers apoptosis of MM in the presence of BMSCs. This inhibitor reduces phosphorylation of STAT3 [72]. It is likely that BG45 compromises the translocation of SATA3 into the nucleus, where it functions as a transcription factor involved in chromatin remodeling for transcriptional regulation [73].
Histone methylations are another major group of histone post-translational modifications receiving extensive investigations on epigenetic transcriptional regulation [74]. Adhesion of MM to BMSCs activates the PI3K/AKT pathway which induces EZH2 deactivation through its phosphorylation at Ser21 [75, 76]. The phosphorylation of EZH2 results in hypo-methylation of H3K27 at promoters of anti-apoptosis genes and supports their sustained expression to counteract drug-induced apoptosis. Remarkably, pharmacological inhibition of the PI3K/AKT pathway compromises cell adhesion-mediated drug resistance in refractory myeloma mouse models [76]. In contrast to EZH2, KDM6B is a H3K27me3 demethylase. TNF-alpha or BMSCs conditioned medium upregulates expression of KDM6B through the NF-κB signaling pathway. Deletion of KDM6B systematically downregulates the expression of genes related to the growth-promoting MAPK pathway in a demethylase-independent manner in MM.1S cells [77]. Therefore, targeting KDM6B protein itself other than its enzymatic activity is necessary to mitigate soluble factor-induced drug resistance of MM. All these advances highlight the therapeutic potential of targeting chromatin regulators for improved treatment of MM.
Additional contributors to drug resistance in the BMM
Exosomes serves as a role in cell-cell communication contribute to immune suppression, angiogenesis, metastasis and drug resistance [78]. Exosomes are activated under cellular activation or stress and released from cancer cells and other cells from BMM such as fibroblasts and immunocytes to promote angiogenesis, modulate signaling pathways of stromal cells and regulates immune response [79, 80*]. For example, exosomes released from hypoxic MM promote angiogenesis through the HIF-FIH signaling pathway [81]. Exosomes released by AML alter proliferative, angiogenesis, migration of BMSCs and can reprogram the niche of the tumor microenvironment [82]. Fibroblast-derived exosomes transfer mtDNA that was associated with promoting cancer stem-like cells, leading to endocrine therapy resistance in breast cancer cells [83].
Acidosis of the tumor microenvironment can affect the cytotoxicity of anticancer drugs, tumor cell invasion and immune dysfunctions to maintain tumor survival [84]. The pH of the extracellular in tumor cells is significantly more acidic than normal tissues and while the pH of intracellular contents of both is similar. The efficacy of uptake of weakly basic drugs such as doxorubicin, mitoxantrone, vincristine and vinblastine are impaired in acidic conditions due to charge of the molecule and decreases the effect of those anti-cancer agents [85].
Adhesion of tumor cells to BMSCs is more complex than adhesion of integrin to a single matrix as BMSCs secrete multiple matrixes and present multiple cell-adhesion molecules (CAM) allowing for cell-cell adhesions. BMSCs are often referred to as mesenchymal stem cells due to their plasticity and contribute to cancer progression through secreting cytokines and growth factors [86]. MM BM MSCs support MM.1S and RPMI.8226 cell growth by releasing exosome. Conversely, the exosome from normal BM-MSCs inhibited these cell growth [87]. BMSCs can differentiate into cancer-associated fibroblasts (CAFs) by releasing TGF-β [88]. CXCL-8 derived from patients of acute myeloid leukemia (AML) BM-MSCs support leukemogenesis through PI3K/AKT signaling pathway in AML HL60 and THP1 cell lines. [89*]. In summary, BM-MSCs are main component of hematopoietic niche to control self-renewal and differentiation of hematopoietic stem/progenitor cells (HSPCs) [88, 90, 91] that modulate the pathogenesis of a variety of hematologic malignancy such as acute lymphoblastic leukemia (ALL), AML, MM, lymphomas, chronic myeloid leukemia (CML), and myelodysplastic syndromes (MDS) [92].
Although immune checkpoint inhibitors to date have failed as a therapeutic strategy for treatment of MM. Myeloid lineages in the BMM include myeloid-derived suppressor cells (MDSCs) that directly influence tumor growth [93]. Infiltrated MDSCs suppress the cytotoxic activities of NK and the adaptive immune response to promote tumor vascularization and contributes to the escape of immune-surveillance and expansion of various cancer types, including melanoma, multiple myeloma, hepatocellular carcinoma, [94]. Thus targeting MDSC’s maybe a tractable approach for immunotherapy for the treatment of MM.
Perspectives
The effectiveness of chemotherapy is often pursued in the context of unicellular models. This model will likely provide targets critical for survival of a tumor cell but may not accurately reflect the tumor cell signaling pathways in the context of the tumor microenvironment. To decrease minimal residual disease this may require combination strategies that target the BMM in conjunction with ongogenic drivers such as the Jak inhibitor and Nilotinob ongoing clinical trial in CML [37]. Clinical trials designed to target the BMM with corresponding correlative data derived from tissue will provide insight whether targeting the BMM is a tractable strategy for reducing minimal residual disease and relapse. Resistance to chemotherapy remains a persistent challenge in the clinic. Targeting BMM compartments is one interesting strategy aimed to inhibit BMM-mediated drug resistance to improve drug response. One of the hurdles for targeting the BMM is that single agent activity may not be achieved, and efficacy is not realized until combination studies are pursued making the clinical development pathway more challenging. Additional challenges will be an understanding of the evolution of resistance as part of a personalized approach to the treatment of cancer as the BMM provides the potential for multiple signaling pathways that contribute to resistance which likely evolves during disease progression. To date it is not clear whether targeting one specific receptor-cytokine or adhesion-matrix interaction will be sufficient to reverse resistance associated with the tumor microenvironment. Strategies designed to target downstream of cell adhesion molecules such as FAK or integrin linked kinase may eliminate concerns of redundancy of adhesion mediated signaling. Similarly, inhibiting the JAK/STAT pathway for cytokine signaling maybe more effective compared to blocking a single cytokine receptor. However, due to the complexity of the bone marrow microenvironment and the diversity of the niche delineating diagnostic biomarkers of response signatures for targets associated with SM-DR, CAM-DR and EM-DR will be required to guide combination strategies with standard of care agents to improve individualized patient outcomes.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The author declare no conflict of interest.
Reference
- [1].Sugiyama T, Kohara H, Noda M, Nagasawa: Maintenance of the hematopoietic stem cell pool by CXCl2-CXCR4 chemokinw signaling in bone marrow stromal cell niches. Immunity 2006, 25:977–988. [DOI] [PubMed] [Google Scholar]
- [2].Spencer JA, Ferraro F, Roussakis E, Klein A, Wu J, Runnels JM, et al. ,: Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508:269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wielockx B, Grinenko T, Mirtschink P, Chavakis T: Hypoxia pathway proteins in normal and malignant hematopoiesis. Cells 2019, 8:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Guerrouahen BS, AI-Hijji I, Tabrizi AR: Osteoblastic and vascular endothelial niches, their control on normal hematopoietic stem cells, and their consequences on the development of leukemia. Stem Cells Int 2011, 2011:375857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kondo M: Lymphoid and myeloid lineage commitment in multipotent hematopoietic progenitors. Immunol Rev 2010, 238:37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Arai F, Suda T: Maintenance of quiescent hematopoietic stem cells in the osteoblastic niche. Ann N Y Acad Sci 2007, 1106:41–53. [DOI] [PubMed] [Google Scholar]
- [7].Kiel MJ, Radice GL, Morrison SJ. Lack of evidence that hematopoietic stem cells depend on N-cadherin-mediated adhesion to osteoblasts for their maintenance. Cell Stem Cell 2007, 1:204–217. [DOI] [PubMed] [Google Scholar]
- [8].Hosokawa K, Arai F, Yoshihara H, et al. Knockdown of N-cadherin suppresses the long-term engraftment of hematopoietic stem cells. Blood 2010, 116:554–563. [DOI] [PubMed] [Google Scholar]
- [9].Li J: Quiescence regulators for hematopoietic stem cell. Expe Hema 2011, 39:511–520. [DOI] [PubMed] [Google Scholar]
- [10].Avecilla ST, Hattori K, Heissig B, Tejada R, Liao F, Shido K, et al. : Chemokine-mediated interaction of hematopoietic progenitors with the bone marrow vascular niche is required for thrombopoiesis. Nat Met 2014, 10:64–71. [DOI] [PubMed] [Google Scholar]
- [11].Burger JA, Kipps TJ: their microenvironment. CXCR4: a key receptor in the cross talk between tumor cells and Blood 2006, 107:1761–1767. [DOI] [PubMed] [Google Scholar]
- [12].Ullah TR: The role of CXCR4 in multiple myeloma: cells’ journey from bone marrow to beyond. J Bone Oncol 2019, 17:100253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13]*.Himburg HA, Termini CM, Schlussel L, Kan J, Li M, Zhao L, Fang T, et al. : Distinct bone marrow source of pleiotrophin control hematopoietic stem cell maintenance and regeneration. Cell Stem Cell 2018, 23:370–381.Pleiotrophin secretion by BMSCs is essential for HSC maintenance and HSC regeneration following irradiation.
- [14].Jung Y, Wang J, Schneider A, Sun YX, Koh-Paige AJ, Osman NI, McCauley LK, et al. ,: Regulation of SDF-1(CXCL12) production by osteoblasts; a possible mechanism for stem cell homing. Bone 2006, 38:497–508. [DOI] [PubMed] [Google Scholar]
- [15].Wright DE, Wagers AJ, Gulati AP, Johnson FL, Weissman IL: Physiological migration of hematopoietic stem and progenitor cells. Science 2001, 294:1933–1936. [DOI] [PubMed] [Google Scholar]
- [16]*.Huang X, Guo B, Liu S, Wan J, Broxmeyer HE.: Neutralizing negative epigenetic regulation by HDAC5 enhances human hematopoietic stem cell homing and engraftment. Nat Commun 2018, 9:2741.HDAC5 inhibition increases human HSCs homing and engraftment.
- [17]*.Romano A, Palumbo GA, Parrinello NL, Conticello C, Martello M, Terraqna C: Minimal residual disease assessment within the bone marrow of multiple myeloma: a review of caveats, clinical significance and future perspectives. Front Oncol 2019, 9:699.Minimal residual disease (MRD) has been updated to include in criteria of responses of patient with MM by the International Myeloma Working Group (IMWG). This paper highlights the more recent techniques developed to assess MRD in MM, their limits in sensitivity, applicability and application in clinical contexts.
- [18].Cartson KS, Guzman ML: Is Minimal Residual Disease Monitoring Clinically Relevant in Adults with Acute Myelogenous Leukemia? Curr Hematol Malig Rep 2013, 8:109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19]*.Buffone AJ, Anderson NR, Hammer DA.: Migration against the direction of flow is LFA-1-dependent in human hematopoietic stem and progenitor cells. J Cell Sci 2018, 131:jcs205575.LFA-1 is a key receptor during the trafficking of HSPCs to the BM.
- [20].Parmo-Cabanas M, Bartolome RA, Wright N, Hidalgo A, Drager AM, Teixido J: Integrin α4β1 involvement in stromal cell-derived factor-1α-promoted myeloma cell transendothelial migration and adhesion: role of cAMP and the actin cytoskeleton in adhesion. Exp Cell Res 2004, 294:571–580. [DOI] [PubMed] [Google Scholar]
- [21]*.de Loff M, de Jong S, Kruyt FAE: Multiple interactions between cancer cells and the tumor microenvironment modulate TRAIL signaling: implications for TRAIL receptor targeted therapy. Front Immunol 2019, 10:1530.Tumor necrosis factor (TNF) related apoptosis-inducing ligand (TRAIL) signaling is considered therapeutic target. However, the efficacy of TRAIL-receptor agonists is disappointed. This paper highlights the impact of the tumor microenvironment on the outcome of TRAIL signaling in tumors cells and discusses the efficacy of TRAIL-based therapy.
- [22].Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS: Cell adhesion mediated drug resistance (CAM-DR): Role of integrins and resistance to apoptosis in human myeloma cell lines. Blood 1999, 93:1658–1667. [PMC free article] [PubMed] [Google Scholar]
- [23].Zhro L, Liu W, Xiao J, Cao B: The role of exosomes and “exosomal shuttle microRNA” in tumorigenesis and drug resistance. Cancer Lett 2015, 356:339–346. [DOI] [PubMed] [Google Scholar]
- [24].Jiang X, Lopez A, Holyoake T, Evaes A, Eaves C: Autocrine production and action of IL-3 and granulocyte colony-stimulating factor in chronic myeloid leukemia. Proc Natl Acad Sci USA 1999, 96:12804–12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Catlett-Falcone R, Landowski TH, Oshiro MM, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 1999, 10:105–115. [DOI] [PubMed] [Google Scholar]
- [26].Cheung WC,Van Ness B: Distinct IL-6 signal transductionleads to growth arrest and deathin B cells or growth promotion and cell survival in myeloma cells. Leukemia 2002, 16:1182–1188. [DOI] [PubMed] [Google Scholar]
- [27].Meads MB, Hazlehurst LA, Dalton WS: The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin Cancer Res 2008, 14:2519–2526. [DOI] [PubMed] [Google Scholar]
- [28].Rosean TR, Tompkins VS, Tricot G, Holman CJ, Olivier AK, Zhan F, Janz S: Preclinical validation of interleukin 6 as a therapeutic target in multiple myeloma. Immuno Res 2014, 59:188–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Voorhees PM, Manges RF, Sonneveld P, Jagannath S, Somlo G, Krishnan A, et al. : A phase 2 multicentre study of siltuximab, an anti-interleukin-6 monoclonal antibody, in patients with relapsed or refractory multiple myeloma. Br J Haematol 2013, 16:357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Orlowski RZ, Gercheva L, Williams C, Sutherland H, Robak T, Masszi T, et al. : A Phase II, Randomized, Double-Blind, Placebo-Controlled Study of Siltuximab (Anti-IL-6 mAb) and Bortezomib versus Bortezomib Alone in Patients with Relapsed or Refractory Multiple Myeloma. Am J Hematol 2015, 90:42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Podar K, Tai YT, Lin BK, Narsimhan RP, Sattler M, Kijima T, et al. : Vascular endothelial growth factor-induced migration of multiple myeloma cells is associated with beta 1 integrin-and phosphatidylinositol3-kinase-dependent PKC alpha activation. J Biol Chem 2002, 8:277. [DOI] [PubMed] [Google Scholar]
- [32].Podar K, Tonon G, Sattler M, Tai Yt, Legouill S, Yasui H, et al. The small-molecule VEGF receptor inhibitor pazopanib (GW786034B) targets both tumor and endothelial cells in multiple myeloma. Proc Natl Acad Sci USA 2006, 103:19478–19483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33]*.Boddu P, Borthakur G, Koneru M, Huang Xuelin, Naqvi K, Wierda W, et al. : Initial report of a phase I study of LY2510924, idarubicin and cytarabine in relapsed/refractory acute myeloid leukemia. Front Oncol 2018, 8:369.This initial report of LY2510924 with idarubicin and cytarabine is safe in relapsed/refractory AML.
- [34].Cho BS, Zeng Z, Mu H, Wang Z, Konoplev S, McQueen T, et al. : Antileukemia activity of the novel peptidic CXCR4 antagonist LY2510924 as monotherapy and in combination with chemotherapy. Blood 2015, 126:222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Williams RT, den Besten W, Sherr CJ. Cytokine-dependent imatinib resistance in mouse BCRABL + Arf-null lymphoblastic leukemia. Genes Dev 2007; 21:2283–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ering AM, Kraft IL, Page BD, O’Hare T, Gunning PT, Deininger MW: STAT3 as a mediator of BCR-ABL1-independnet resistance in chronic myeloid leukemia. Leuk Suppl 2014, 3:S5–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sweet K, Hazlehurst L, Sahakian E, Powers J, Nodzon L, Kayali F, et al. : A phase I clinical trial of ruxolitinib in combination with nilotinib in chronic myeloid leukemia patients with molecular evidence of disease. Leuk Res 2018, 74:89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bewry NN, Nair RR, Emmons MF, Boulware D, Pinilla-Ibarz J, Hazlehurst LA: Stat3 contributes to resistance toward BCR-ABL inhibitors in a bone marrow microenvironment model of drug resistance. Mol Cancer Ther 2008, 7:3169–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39]**.Yagi K, Shimada A, Sendo T.: Pharmacological inhibition of JAK3 enhances the antitumor activity of imatinib in human chronic myeloid leukemia. Eur J Pharmacol 2018, 825:28–33.Jak3 is important in CML cell survival and Jak3 inhibitor synergistically enhanced the antitumor effects of IMA in CML and reduced the expression of stem cell marker.
- [40].Nair RR, Tolentino JH, Argilagos RF, Zhang L, Pinilla-lbarz J, Hazlehurst LA: Potentiation of Nilotinib-mediated cell death in the context of the bone marrow microenvironment requires a promiscuous JAK inhibitor in CML. Leuk Res 2012, 36:756–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Dalton WS, Hazlehurst L, Shain K, Landowski T, Alsina M: Targeting the bone marrow microenvironment in hematologic malignancies. Semin Hematol 2004, 41:1–5. [DOI] [PubMed] [Google Scholar]
- [42].Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS: Cell adhesion mediated drug resistance (CAM-DR): Role of integrins and resistance to apoptosis in human myeloma cell lines. Blood 1999, 93:1658–1667. [PMC free article] [PubMed] [Google Scholar]
- [43].Shain KH, Landowski TH, Dalton WS: Adhesion-mediated intracellular redistribution of c-Fas-associated death domain-like IL-1-coverting enzyme-like inhibitory protein-long confers resistance to CD95-induced apoptosis in hematopoietic cancer cell lines. J Immunol 2002, 168:2544–2553. [DOI] [PubMed] [Google Scholar]
- [44].Li ZW, Dalton WS: Tumor microenvironment and drug resistance in hematologic malignancies. Blood Rev 2006, 20:333–342. [DOI] [PubMed] [Google Scholar]
- [45].Anreddy N, Hazlehurst LA: Targeting intrinsic and extrinsic vulnerabilities for the treatment of multiple myeloma. J Cell Biochem 2017, 118:15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Nair RR, Gebhard AW, Emmons MF, Hazlehurst LA: Emerging strategies for targeting cell adhesion in multiple myeloma. Adv Pharmacol 2012, 65:143–189. [DOI] [PubMed] [Google Scholar]
- [47].Nair RR, Tolentino J, Hazlehurst LA: The bone marrow microenvironment as a sanctuary for minimal residual disease in CML. Biochem Pharmacol 2010, 80:602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bellamy WT, Dalton WS, Gleason MC, Grogan TM, Trent JM: Development and Characterization of a Melphalan-resistant Human Multiple Myeloma Cell Line. Cancer Res 1991, 51:995–1002. [PubMed] [Google Scholar]
- [49].Noborio-Hatano K, Kikuchi J, Takatoku M, Shimizu R, Wada T, Ueda M, et al. : Bortezomib overcomes cell-adhesion-mediated drug resistance through downregulation of VLA-4 expression in multiple myeloma. Oncogene 2009, 28:231–242. [DOI] [PubMed] [Google Scholar]
- [50].Peled A, Kollet O, Ponomaryov T, Petit I, Franitza S, Grabovsky V, et al. : The Chemokine SDF-1 activates the integrins LFA-1, VLA-4, and VLA-5 on immature human CD34(+) cells: role in transendothelial/stromal migration and engraftment of NOD/SCID mice. Blood 2000, 95:3289–3296. [PubMed] [Google Scholar]
- [51].Li ZW, Dalton WS: Tumor microenvironment and drug resistance in hematologic malignancies. Blood Rev 2006, 20:333–342. [DOI] [PubMed] [Google Scholar]
- [52].Luo BH, Carman CV, Springer TA: Structural basis of integrin regulation and signaling. Annu Rev Immunol 2007, 25:619–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Petty JM, Lenox CC, Weiss DJ, Poynter ME, Suratt BT: Crosstalk between CXCR4/SDF-1 and VLA-4/VCAM-1 pathways regulates neutrophil retention in the bone marrow. J Immunol 2009, 182:604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Damiano JS, Dalton WS: Integrin-mediated drug resistance in multiple myeloma. Leuk Lymphoma 2000, 38:71–81. [DOI] [PubMed] [Google Scholar]
- [55].Hazlehurst LA, Enkemann SA, Beam CA, Argilagos RF, Painter J, Shain KH, et al. : Genotypic and phenotypic comparisons of de novo and acquired melphalan resistance in an isogenic multiple myeloma cell line model. Cancer Res 2003, 63:7900–7906. [PubMed] [Google Scholar]
- [56].Hazlehurst LA, Damiano JS, Buyuksal I, Pledger WJ, Dalton WS: Adhesion to fibronectin via β1 integrins regulates p27kip1 levels and contributes to cell adhesion mediated drug resistance (CAM-DR). Oncogene 2000, 19:4319–4327. [DOI] [PubMed] [Google Scholar]
- [57].Meads MB, Hazlehurst LA, Dalton WS: The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin Cancer Res 2008, 14:2519–2526. [DOI] [PubMed] [Google Scholar]
- [58].Hazlehurst LA, Argilagos RF, Emmons M, Boulware D, Beam CA, Sullivan DM, et al. ,: Cell adhesion to fibronectin (CAM-DR) influences acquired mitoxantrone resistance in U937 cells. Cancer Res 2006, 66:2338–2345. [DOI] [PubMed] [Google Scholar]
- [59].Dafmiano JS, Hazlehurst LA, Dalton WS: Cell adhesion-mediated drug resistance (CAM-DR) protects the K562 chronic myelogenous leukemia cell line from apoptosis induced by BCR/ABL inhibition, cytotoxic drugs, and gamma -irradiation. Leukemia 2001, 15:1232–1239. [DOI] [PubMed] [Google Scholar]
- [60].Hazlehurst LA, Valkov N, Wisner L, Storey JA, Boulware D, Sullivan DM, et al. ,: Reduction in drug-induced DNAdouble-strand breaks associated with b1 integrin–mediated adhesion correlates with drug resistance in U937 cells. Blood 2001, 98:1897–1903. [DOI] [PubMed] [Google Scholar]
- [61].Shain KH, Landowski TH, Dalton WS: Adhesion-mediated intracellular redistribution of c-Fas-associated death domain-like IL-1-coverting enzyme-like inhibitory protein-long confers resistance to CD95-induced apoptosis in hematopoietic cancer cell lines. J Immunol 2002, 168:2544–2553. [DOI] [PubMed] [Google Scholar]
- [62].Soodgupta D, Hurchla MA, Jiang M, Zheleznyak A, Weibaecher KN, Anderson CJ, et al. : Very late antigen-4 (α4β1integrin) targeted PET imaging for multiple myeloma. PLoS One 2013, 8:e55841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Beaino W, Anderson CJ: PET imaging of very late antigen-4 in melanoma: comparison of 68Ga and 64Cu-labeled NODAGA and CB-TE1A1P-LLP2-A conjugates. J Nucl Med 2014, 55:1856–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Podar K, Zimmerhackl A, Fulciniti M, Tonon G, Hainz U, Tai YT, et al. : The selective adhesion molecule inhibitor natalizumab decreases multiple myeloma cell growth in the bone marrow microenvironment: therapeutic implications. Br J Haematol 2011, 155:438–448. [DOI] [PubMed] [Google Scholar]
- [65].Olson DL, Burkly LC, Leone DR, Dolinski BM, Lobb RR: Anti-alpha4 integrin monoclonal antibody inhibits multiple myeloma growth in a murine model. Mol Cancer Ther 2005, 4:91–99. [PubMed] [Google Scholar]
- [66].Touzeau C, Moreau P, Dumontet C: Monoclonal antibody therapy in multiple myeloma. Leukemia 2017, 31:1039–1047. [DOI] [PubMed] [Google Scholar]
- [67].Garcia-Gomez A, Rodriguez-Ubreva J, Ballestar E: Epigenetic interplay between immune, stromal and cancer cells in the tumor microenvironment. Clin Immunol 2018, 196:64–71. [DOI] [PubMed] [Google Scholar]
- [68].Sylvestre M, Tarte K, Roulois D: Epigenetic mechanisms driving tumor supportive microenvironment differentiation and function: A role in cancer therapy? Epigenomics 2020, 12:157–169. [DOI] [PubMed] [Google Scholar]
- [69].Harada T, Hideshima T, Anderson KC: Histone deacetylase inhibitors in multiple myeloma: From bench to bedside. Int J Hematol 2016, 104:300–309. [DOI] [PubMed] [Google Scholar]
- [70].Sripayap P, Nagai T, Hatano K, Kikuchi J, Furukawa Y, Ozawa K: Romidepsin overcomes cell adhesion-mediated drug resistance in multiple myeloma cells. Acta Haematol 2014, 132:14. [DOI] [PubMed] [Google Scholar]
- [71].Minami J, Suzuki R, Mazitschek R, Gorgun G, Ghosh B, Cirstea D, Hu Y, Mimura N, Ohguchi H, Cottini F, Jakubikova J et al. : Histone deacetylase 3 as a novel therapeutic target in multiple myeloma. Leukemia 2014, 28:680–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Wingelhofer B, Neubauer HA, Valent P, Han X, Constantinescu SN, Gunning PT, Muller M, Moriggl R: Implications of stat3 and stat5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer. Leukemia 2018, 32:1713–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Harada T, Oda A, Ohguchi H, Grondin Y, Tenshin H, Hiasa M, Teramachi J, Oura M, Sogabe K, Fujii S, Nakamura S et al. : Novel therapeutic rationale for targeting hdac1 and pim2 in multiple myeloma. Blood 2019, 134 (Supplement_1)3111. [Google Scholar]
- [74].Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K: High-resolution profiling of histone methylations in the human genome. Cell 2007, 129:823–837. [DOI] [PubMed] [Google Scholar]
- [75].Kikuchi J, Koyama D, Wada T, Izumi T, Hofgaard PO, Bogen B, Furukawa Y: Phosphorylation-mediated ezh2 inactivation promotes drug resistance in multiple myeloma. J Clin Invest 2015, 125:4375–4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Cha TL, Zhou bP, Xia W, Wu Yadi, Yang CC, Chen CT, et al. : Akt-mediated phosphorylation of EZH2 suppresses methylation of lysing 27 in histone H3. Science 2005, 310:306–310. [DOI] [PubMed] [Google Scholar]
- [77].Ohguchi H, Harada T, Sagawa M, Kikuchi S, Tai YT, Richardson PG, Hideshima T, Anderson KC: Kdm6b modulates mapk pathway mediating multiple myeloma cell growth and survival. Leukemia 2017, 31:2661–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Azmi AS, Bao B, Sarkar FH: Exosomes in cancer development, metastasis and drug resistance: a comprehensive review. Cancer Metastasis Rev 2013, 32:623–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Wu T, Dai Y: Tumor microenvironment and therapeutic response. Cancer Lett 2017, 387:61–68. [DOI] [PubMed] [Google Scholar]
- [80]*.Kao CY, Papoutsakis ET: Extracellular vesicles: exosomes, microparticles, their parts, and their targets to enable their biomanufacturing and clinical applications. Current Opin in Biote 2019, 69:89–98.Extracellular vesicles (EVs) are generated from mammalian cells under activation, stress, carry RNAs, protein and lipids form their parent cells and are considered candidates for gene therapies.
- [81].Huan J, Homick NI, Shurtleff MJ, Skinner AM, Goloviznina NA, Roberts CT Jr, et al. : RNA trafficking by acute myelogenous leukemia exosomes. Cancer Resc 2012, [DOI] [PubMed]
- [82].Umezu T, Tadokoro H, Azuma K, Yoshizawa S, Ohyashiki K, Ohyashiki JH: Exosomal miR-135b shed from hypoxic multiple myeloma cells enhances angiogenesis by targeting factor-inhibiting HIF-1. Blood 2014, 123:3748–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Sansone P, Savini C, Kurelac I, Change Q, Amato LB, Strillacci A, et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc Natl Acad Sci USA 2017, 114:E9066–E9075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Huber V, Camisaschi C, Berzi A, Ferro S, Lugini L, Triulzi T, et al. : Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semi in Canc Biol 2017, 43:74–89. [DOI] [PubMed] [Google Scholar]
- [85].Tredan O, Galmarini CM, Patel K, Tannock IF: Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst 2007, 99:1441–1454. [DOI] [PubMed] [Google Scholar]
- [86].Senthebane DA, Rowe A, Thomford NE, Shipanqa H, Munro D, Mazeedi MAMA et al. : The role of tumor microenvironment in chemoresistance: to survive, keep your enemies closer. Int J Mol Sci 2017, 18:1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Roccaro AM, Sacco A, Maiso P, Azab AK, Tai YT, Reagan M et al. : BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J Clin Invest 2013, 123:1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Phinney DG: Functional heterogeneity of mesenchymal stem cells: implications for cell therapy. J Cell Biochem 2012, 113:2806–12. [DOI] [PubMed] [Google Scholar]
- [89]*.Cheng J, Li Y, Liu S, Jiang Y, Ma J, Wan L et al. : CXCL8 derived from mesenchymal stromal cells supports survival and proliferation of acute myeloid leukemia cells through the PI3K/AKT pathway. FASEB J 2019, 33:4755–4764The role of proinflammatory cytokines secreted by the BMSCs in the progression of AML is unclear. This study shows that the role of CXCL8 derived form AMK BMSCs on the growth of AML by directed contact coculture system. Inhibition of CXCL8-CXCR2 axis showed therapeutic potential in various solid tumors.
- [90].Chen X, Armstrong MA, Li G: Mesenchymal stem cells in immunoregulation. Immunol Cell Biol 2006, 84:413–21. [DOI] [PubMed] [Google Scholar]
- [91].Klopp AH, Gupta A, Spaeth E, Andreeff M, Marini F: Concise review: Dissecting a discrepancy in the literature: do mesenchymal stem cells support or suppress tumor growth? Stem Cells 2011, 29:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Krampera M: Mesenchymal stromal cells: more than inhibitory cells. Leukemia 2011, 25:565–6. [DOI] [PubMed] [Google Scholar]
- [93].Pitt JM, Marabelle A, Eggermont A, Soria JC, Kroemer G, Zitogel L: Targeting the tumor microenvironment: removing obstruction to anticancer immune responses and immunotherapy. Ann Oncol 2016, 27:1482–1492. [DOI] [PubMed] [Google Scholar]
- [94].Munn DH, Bronte V: Immune suppressive mechanisms in the tumor microenvironment. Curr Opin Immunol 2016, 39:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]