

Abstract
The transforming growth factor-beta (TGFβ) pathway is essential during embryo development and in maintaining normal homeostasis. During malignancy, the TGFβ pathway is co-opted by the tumor to increase fibrotic stroma, to promote epithelial to mesenchymal transition increasing metastasis and producing an immune-suppressed microenvironment which protects the tumor from recognition by the immune system. Compelling preclinical data demonstrate the therapeutic potential of blocking TGFβ function in cancer. However, the TGFβ pathway cannot be described as a driver of malignant disease. Two small molecule kinase inhibitors which block the serine-threonine kinase activity of TGFβRI on TGFβRII, a pan-TGFβ neutralizing antibody, a TGFβ trap, a TGFβ antisense agent, an antibody which stabilizes the latent complex of TGFβ and a fusion protein which neutralizes TGFβ and binds PD-L1 are in clinical development. The challenge is how to most effectively incorporate blocking TGFβ activity alone and in combination with other therapeutics to improve treatment outcome.
Keywords: transforming growth factor-beta, TGFβ, glaunisertib, fresolimumab, bintrafusp alfa, decorin, vactosertib
Introduction to TGFβ
Transforming growth factor–β (TGFβ) pathway signaling activity is essential in embryo development and is frequently aberrantly active in the tumor microenvironment and tumor progression (1–5). The larger TGFβ family of secreted, homodimeric and hetero-dimeric proteins, including transforming growth factors, activins, NODAL, bone morphogenetic proteins and others, is among the critical regulators of tissue morphogenesis guiding early cell-fate decisions in embryo development, organogenesis, and in adults tissue homeostasis, inflammatory processes and wounding healing (6–9). In cancer, TGFβ signaling contributes to tumor progression, in part, contributing to an immunosuppressive microenvironment (10). The TGFβ protein family is encoded by 33 genes. These proteins are secreted in a pro-form and are activated in the extracellular matrix or are sequestered in an inactive form (1). There are over thirty TGFβ super-family members in humans including three TGFβ ligands, five activins, eight bone morphogenetic proteins (BMP) and fifteen growth and differentiation factors (GDF). TGFβ1, TGFβ2, and TGFβ3 are homodimeric polypeptides with a molecular weight of 25-kDa. The TGFβ2 crystal structure elucidated two monomers, each with two antiparallel pairs of β-strands that form a flat surface and a separate α–helix (10, 11). Two intrachain disulfide bonds form a ring that is threaded by a third intrachain disulfide bond forming a “cysteine knot”.
TGFβ family of ligands, receptors and signaling proteins
In the extracellular matrix, TGFβ exists as a latent propeptide. The latent TGFβ propeptide (LAP) is bound through disulfide bonds to latent TGFβ binding proteins (LTBPs). There are four latent TGFβ binding protein isoforms. Each isoform has specific roles in transport, maintaining TGFβ latency, TGFβ regulation and activation as well as microfibril organization and elastic fiber assembly (12). Latent TGFβ binding protein-1 (LTBP-1) binds with latent TGFβ in the extracellular matrix making latent TGFβ accessible to αvβ6 integrin which distorts the complex allowing release of active TGFβ.
Active TGFβ functions through specific high affinity receptors. Five TGFβ superfamily type I receptors and seven type II receptors have been identified in mammals (13, 14). The type I receptor family includes activin-like kinases (ALKs) 1 through 7. The type II receptors include TGFβ RII, BMP RII, activin RIIA and activin RIIB (15–18). The type I and type II receptors are structurally related transmembrane glycoproteins with an extracellular N-terminal ligand-binding domain with more than ten cysteine residues that regulate the dimeric structure, a transmembrane region, and a C-terminal serine/threonine kinase domain. The type I receptors have a highly conserved region GS domain that is rich in glycine and serine residues in the juxta-membrane domain next to the N-terminus of the kinase domain (19, 20).
The effects of the TGFβ family proteins are mediated by a set of receptor-activated mothers against decapentaplegic homologue (SMAD) transcription factors; however, resulting effects are highly context-dependent and differ depending on the cell type and the microenvironmental conditions. SMADs bind genome-wide with lineage-determined transcription factors and interact with the chromatin resulting in the specific cellular response (21, 22). SMAD2 and SMAD3 are transcription factors critical in TGFβ signaling. SMAD2 and SMAD3 form heterotrimeric complexes with SMAD4 in response to TGFβ. The trimeric SMAD complexes interact with varied cofactors to control TGFβ–dependent gene expression. Specificity is achieved when SMAD2 and SMAD3 form complexes with other transcription factors and cofactors. The crystal structure of SMAD3 complexed with transcription factor FOXH1 and the crystal structure SMAD2 in complex with transcriptional corepressor SKI provided a structural understanding of the protein binding. The structures showed that the MAD homology 2 (MH2) domains of SMAD2 and SMAD3 have multiple hydrophobic patches on their surfaces which interact in cooperative or competitive binding with transcriptional complex partners to transduce TGFβ signaling (23).
Epithelial-to-mesenchymal transitions
TGFβ signaling promotes cell motility by inducing epithelial-to-mesenchymal transitions (EMT) in normal physiology and development, and abnormally in cancer (24). Dis-regulated signaling by TGFβ superfamily members (activins, NODAL, bone morphogenetic proteins (BMPs), growth and differentiation factors (GDFs) and anti-Müllerian hormone), in addition to the TGFβ ligands 1, 2 and 3, is important to the development of tumors and metastasis and reflect the function of these proteins in normal embryonic development and tissue homeostasis. Malignant cells hijack the normally well-controlled functions of these secreted proteins to enable cells to grow at primary sites, and to disseminate and survive at distant sites (25–27). Loss or impairment of either TGF-β receptor make the signaling pathway non-functional (28).
In cells, TGFβ is involved in regulation of cell cycle, apoptosis, immune homeostasis and self-tolerance, EMT, a mark of cellular plasticity, and extracellular matrix composition regulation (7). TGFβ signaling alterations are involved in inflammatory and fibrotic diseases and tumor immune evasion (29). In the immune system, TGFβ is involved in B and T lymphocyte immunological response (1). TGFβ prevents the spontaneous activation of self-reactive T cells and sustains immune homeostasis in the presence of proinflammatory cytokines. TGFβ induces differentiation of effector T helper 17 (TH17) cells and inhibiting TGFβ receptor signaling blocks development of interleukin-17 secreting cells and TH17 cell–mediated autoimmunity. The differentiation of TH17 cells from CD4+ T cells can be inhibited by BMPs by blocking BMP receptor type 1 during CD4+ T cell activation resulting in generation of inflammatory effector cells expressing IL-17, interferon-β, and TNF family cytokines and TH17 cell lineage transcription factors (30).
High TGFβ concentrations in the tumor microenvironment and circulating blood are associated with aggressive cancer. TGF-β is an immunosuppressive cytokine that is frequently associated with mechanisms of tumor escape from immunosurveillance (31–34). TGFβ suppresses a key antitumor function of CD4+ T cells, interferon-β production through a process requiring the expression and phosphorylation of Smad proteins. Smad proteins can be detected in the mitochondria of CD4+ T cells and phosphorylated upon exposure to TGFβ. Phosphorylated Smad proteins can be detected in tumor-associated lymphocyte mitochondria (35). MAN1, an integral protein of the inner nuclear membrane encoded by the LEMD3 gene, inhibits TGFβ signaling by binding to Smad2 and Smad3. The three-dimensional structure of the MAN1-Smad2 complex has been determined from nuclear magnetic resonance and small-angle x-ray scattering studies. In cells, MAN1 can compete with the transcription factor FAST1 (fork-head activin signal transducer 1) for binding to Smad2 (36, 37). Progranulin, an autocrine growth factor with physiological and pathologic roles, increases dermal fibroblasts in wound healing and can activate cancer-associated fibroblasts. Progranulin mRNA and protein were elevated in a lesion from mice with bleomycin-induced dermal fibrosis. In bleomycin-treated mice, progranulin deficiency decreased dermal fibrosis and decreased myofibroblast differentiation. Decreased skin sclerosis in progranulin-deficient mice was accompanied by down-regulation of TGFβ receptor I and decreased Smad3 phosphorylation. Exogenous progranulin increased TGFβRI and phosphorylated Smad3 in cultured fibroblasts (38).
Malignant tumors are comprised of tumor cells and stromal cells including fibroblasts, endothelial cells, and inflammatory cells (39). Interactions between the tumor cells and the stroma influence cancer growth and metastasis (40–42).
Fibroblasts, in the vicinity of malignant cells, become activated and exhibit myofibroblastic characteristics that support invasive growth and metastasis. These cells are termed cancer-associated fibroblasts (CAFs) (43–45). Cancer cell exosomes containing microRNAs, proteins including TGFβ as well as DNA, are released and internalized by fibroblasts, thus promoting proliferation and expression of CAF markers and SMAD signaling. TGFβ inhibitors can block CAF marker expression (46). A human tumor xenograft model following long-term fractionated radiation, produced myofibroblasts which promoted tumor growth by enhancing angiogenesis (47). In another study, the tumor homing potential of mesenchymal stem cells (MSCs) was used to deliver a theranostic sodium iodide symporter transgene into a solid tumor. External beam radiotherapy increased the localization of the sodium iodide symporter transgene expressing MSCs to a HuH7 hepatocellular carcinoma xenograft then TGFβ was upregulated in the HuH7 tumors by exposure to external beam radiotherapy. The improved therapeutic effect was due to TGFβ produced, thus, leading to a focused amplification of MSC-based sodium iodide symporter transgene expression within the tumor milieu (48). TGFβ signaling controls expression of CDKN2A/B in some cancers. In cell culture, exogenous TGFβ induced ARF mRNA and protein, and ARF knockdown decreased TGFβ growth suppression. TGFβ stimulated ARF mRNA induction required SMAD2/3, p38MAPK, and SP1(49). In ovarian cancer, TGFβ has limits effects on cancer cells and contributes to ovarian tumor growth through tumor microenvironment effects especially through stimulation of CAFs (44, 45). In coculture experiments, TGFβ enhanced ovarian cancer cells aggressiveness by upregulating versican in CAFs. Versican expression was regulated through TGFβRII and SMAD signaling. Upregulated versican promoted the motility and invasion of ovarian cancer cells. A TGFβ–inducible gene signature specific to CAFs in high-grade serous ovarian tumors and showed that TGFβ stimulates ovarian cancer cell motility and invasion by upregulating the CAF-specific gene versican (50).
EMT Transitions
Several progenitor cell-types and cellular programs can produce myofibroblasts through EMT, a normal physiological process that regulates tissue development, remodeling and repair (51, 52). During the EMT process, epithelial cells lose polarity and undergo transition to a more mesenchymal phenotype (53, 55). Similarly, cancer cells invading adjacent tissues undergo a reversible EMT (55). TGFβ can induce myofibroblast differentiation of epithelial cells, endothelial progenitor cells, and quiescent fibroblasts in a process dependent on dimeric calpains which are non-lysosomal cysteine proteases. In cell culture, siRNA gene silencing demonstrated that TGFβ-induced EMT of an epithelial cell line depended on the activity of calpain 9 (56).
The differentiation of fibroblasts into highly activated, extracellular matrix-producing myofibroblasts when there is tissue injury is essential for normal tissue repair, however, myofibroblast persistence and accumulation can lead to pathological fibrosis reminiscent of the stromal response in malignancy. TGFβ1-induced myofibroblast differentiation increased ATF4 production leading to increased glycolysis and enhanced collagen production. ATF4 can promote the transcription of genes encoding enzymes of the serine-glycine biosynthetic pathway and glucose transporter 1 (GLUT1) (57). There is a SMAD-dependent crosstalk between ATF4 and TGFβ (58). In cell culture, blocking ATF4 expression decreased migration, invasion, proliferation, EMT and antiapoptotic and stemness markers. In xenograft models, silencing ATF4 decreased metastases, tumor growth, and prolonged drug response (59). A prospective genomic marker for radiation fibrosis study found that the C509T allele in TGFB1 is a key determinant of fibrosis (60). Using transcriptome profiling and epigenetic profiling TGFB2 was implicated in the inflammatory syndrome of systemic sclerosis (61, 62). TGFβ2 autocrine signaling induced a profibrotic state in cultured fibroblasts from systemic sclerosis patients. Inhibition of NF-kB or BRD4 blocked TGFB2 enhancer activity, decreased profibrotic gene expression, and decreased fibrosis in skin explants (63).
EMT can be regulated by SNAIL, ZEB and TWIST transcription factors as well as specific microRNAs (21, 6). The RAS-responsive element binding protein 1 (RREB1), a RAS transcriptional effector, is a TGFβ-activated SMAD transcription factor cofactor in EMT (4,22, 65). MAPK-activated RREB1 recruits TGFβ-activated SMAD factors to SNAIL. In carcinoma cells, TGFβ–SMAD and RREB1 increase expression of SNAIL and fibrogenic factors stimulating myofibroblasts, promoting intratumoral fibrosis and supporting tumor growth. RREB1 provides the link between RAS and TGFβ pathways for coordinated induction of developmental and fibrogenic EMT (4).
Cancer cells, with help of macrophages, migrate through extracellular matrix in the process of metastasis. In a 3D cell migration assay, macrophages enhanced the speed and persistence of cancer cell migration through the extracellular matrix depending upon matrix metalloproteinase activity. TNFα and TGFβ1, released by macrophages, stimulated migration through NF-kB–dependent MMP1 expression in malignant cells (66). The cadherin-11 promoted binding of macrophages to profibrotic myofibroblasts. Cadherin-11 was abundant at contact points between macrophages and myofibroblasts in fibrotic lungs. In culture, cadherin-11 junctions were found at adhesion sites between macrophages and myofibroblasts. When macrophage-myofibroblast pairs were in contact cadherin-11–mediated adhesion led to activation of latent TGFβ (67, 68). Epithelial carcinoma cells can undergo a partial or complete EMT in high TGFβ microenvironments and in low TGFβ microenvironments cells can revert to an epithelial phenotype. Prolonged TGFβ exposure can establish a more stable EMT phenotype and anticancer drug resistance in culture. Extended duration of TGFβ exposure increased mammalian target of rapamycin (mTOR) signaling and a mTOR inhibitor reduced cancer stem cell survival, chemoresistance and tumorigenesis in mice (69).
The TGFβ pathway can be activated by chemotherapeutics. A TGFβ protein trap, RER, can block TGFβ pathway activation by anticancer agents. Anticancer agents-induced gene expression changes associated with TGFβ pathway activation in cancer cells in culture. Due to anticancer induced TGFβ expression, TGFβ-induced EMT in tumor cells. TGFβ inhibitors including a TGFβ trap protein RER reversed this effect. Chemotherapeutics can stimulate TGFβ1 production and consequently enhance TGFβ signaling, EMT, and cancer stem cell features resulting in decreased chemo-sensitivity (70).
Transforming growth factor-beta in disease
Mutations in KRAS are frequent in lung adenocarcinoma. In a KRAS mutant mouse lung adenocarcinoma cell line, PPARγ activation inhibited cell growth in culture but promoted tumor progression when activated in microenvironmental myeloid cells. PPARγ activation in myeloid cells promoted TGFβ1 production, cancer cell migration and EMT. TGFβ1 induction of EMT in tumors is a critical process in cancer and TGFβ receptor inhibition could provide a useful treatment for KRAS-mutant epithelial tumors (71). The loss of TGFβ signaling in epithelial cells by TGFβRII deletion prevented bleomycin induced pulmonary fibrosis in mice. An epithelial cell response to injury involves integrins and TGFβ (72). Integrin adhesion receptors transmit force between extracellular ligands and the cellular actin cytoskeleton. In activation of precursor, pro-TGFβ1, integrins bind to the pro-domain, apply force, and release active TGFβ. Integrin αVβ6 binds pro-TGFβ1 in regions inside and outside the highly interdigitated interface to stabilize a specific integrin/pro-TGFβ orientation that defines the pathway which the actin-cytoskeleton-generated tensile force takes (73).
Ubiquitin-specific peptidase 11 (USP11) increased TGFβ-induced EMT and self-renewal in immortalized human mammary epithelial cells. Altering USP11 expression in human breast cancer cells affected migration in cell culture and metastasis in a xenograft. High USP11 expression in clinical breast cancer specimens correlated with poor outcome. Changing USP11 expression altered the TGFBR2 stability and TGFβ downstream signaling in human breast cancer cells (55). Deubiquitination of TGFBR2 by USP11 spares TGFBR2 from proteasomal degradation facilitating EMT and metastasis (74, 75). TGFBR2 knockout resulted in lung squamous cell carcinoma development in KRASG12D mice with a short latency, high penetrance, and extensive metastases. TGFBR2 knockout lung squamous cell carcinoma was similar to the human disease in histopathology, microenvironment and biomarker expression. Smad4 knockout did not produce lung squamous cell carcinoma; however, ERK1/2 inhibition in Smad4fl/fl;KRASG12D mice resulted in lung squamous cell carcinoma formation similar to TGFBR2 knockout tumors (76, 77). The consequence of SMAD4 deficiency/mutation in tumor response to radiotherapy was investigated using SMAD4 siRNA or SMAD4 shRNA to decrease SMAD4 expression in SMAD4 mutant cancer cells and clonogenic assay was applied to evaluate the effect on radiation response. SMAD4-depleted cancer cells were less responsive to radiotherapy based on clonogenic survival assay. Overexpression of wild-type SMAD4 in SMAD4-mutant cells restored radiosensitivity in an orthotopic xenograft model (76, 77).
Tregs and NK cells
A three-dimensional collagen-fibrin gel cell culture system was used to model the regulatory T cells (Treg) immunosuppressive effect. The model recapitulated the in vivo immune suppression rendering dissociated tumor cells resistant to killing by co-cultured activated, antigen-specific T cells and depended on cell-cell contact or cellular proximity. Immunosuppression was not observed when tumors excised from Treg-depleted mice were cultured in this system. TGFβ neutralizing antibodies prevented immunosuppression. Intravital microscopy allowed visualization of Tcells physically interacting with tumor resident Tregs. Tregs isolated from tumors suppressed CD8+ T cell–mediated killing, which depended on surface-bound TGFβ on the Tregs. Contact between Tregs and antitumor T cells in the tumor microenvironment inhibited tumor immunity in a TGFβ–dependent manner (78). The ability of natural killer (NK) cells to lyse allogeneic targets makes them an attractive platform for cell-based immunotherapy. NK cells are cytolytic, and their potent antitumor effects can be triggered by a lack of HLA expression on interacting target cells, as is the case for most solid tumors. The NK cell antitumor activity is limited by immunosuppressive TGFβ in the tumor microenvironment. NK cells were genetically altered to express variant TGFβ receptors, which couple a mutant TGFβ dominant-negative receptor to NK specific activating domains. These engineered receptors convert inhibitory TGFβ signals to activating signals. The altered NK cells were highly cytotoxic toward tumor cells in TGFβ supplemented media and prolonged survival of a xenograft tumor bearing mouse (79).
Cancers and other diseases
Melanoma which occurs in the epidermis then invades the dermis and metastasizes, stimulates production of microenvironmental TGFβ often secreted by skin adipocytes. TGFβ signaling produced a proliferative-to-invasive melanoma phenotype in cell culture. TGFβ inhibition blocked invasion (80). TGFβ signaling controls gene expression, in part, by targeting RNA binding proteins and non-coding RNAs. In cancer, TGFβ signaling alters RBP-RNA and RNA-RNA interactions resulting in cell growth and tumor cell dissemination (81). Pancreatic ductal adenocarcinoma that develop with an intact TGFβ pathway require ID1 proteins expressed in pancreatic ductal adenocarcinoma progenitor cells. Pancreatic ductal adenocarcinoma progression selects against TGFβ-mediated repression of ID1. AKT signaling and mechanisms linked to low-frequency genetic events converge on ID1 to preserve expression in pancreatic ductal adenocarcinoma, thus, ID1 may be a useful therapeutic target in pancreatic ductal adenocarcinoma (82, 83).
TGFβ is central to renal fibrosis in progressive chronic kidney diseases (72). An abundant long noncoding RNA present in TGFβ1–stimulated human tubular epithelial cells and fibrotic kidneys is TGFβ/Smad3-interacting long noncoding RNA (lnc-TSI) lnc-TSI is transcriptionally regulated by Smad3 binding with the MH2 domain of Smad3, and blocking the interaction of Smad3 with TGFβRI (77, 84). In liver, hepatic stellate cells that become myofibroblasts are a major source of extracellular matrix components during tissue fibrosis. TGFβ signaling enhances hepatocellular carcinoma progression by intrinsic activity as a growth factor and, by stimulating microenvironment changes supportive of tumor growth. Genetically altered mouse hepatic stellate cells were used to delete the gene encoding αv integrin, a mediator of fibrosis. In mice, these hepatic stellate cells protected from carbon tetrachloride–induced hepatic fibrosis, while deletion of other integrins did not. Pdgfrb-Cre targeted myofibroblasts in multiple organs showed that deletion of αv integrin was protective in multiple organ fibrosis (85). In head and neck squamous carcinoma TGFβ signaling loss increased response to radiation or cisplatin and inhibiting TGFβ signaling decreased homologous recombination repair and increased response to PARP inhibition. Thus, TGFβ has a role in DNA repair proficiency and supports use of TGFβ inhibitors in cancer (86). Liver injury may result in regeneration through hepatocyte proliferation; however, following acute severe injury regeneration may fail and senescence may proceed. In acute liver disease, p21-dependent hepatocellular senescence correlated with impaired regeneration and disease severity and depended on macrophage-derived TGFβ1. TGFβR1 inhibition reduced hepatic senescence and enhanced liver regeneration (87).
TGFβ signaling maintains joint homeostasis through IL-36. Human cartilage osteoarthritis was associated with decreased TGFBR2 and increased IL-36 (66). Mechanisms that govern the shift from joint homeostasis to osteoarthritis remain unknown. IL-36 is increased in osteoarthritis and IL-36R decreased in mice with tissue-specific TGFβR2 ablation. TGFβ signaling is prominent in osteogenesis imperfecta a genetic disorder of connective tissue characterized by brittle bones, fractures and extra-skeletal manifestations, with high expression of TGFβ target genes and high ratio of phosphorylated Smad2 to total Smad2 protein.
A TGFβ-induced EMT signature to score TGFβ-driven EMT was applied to pan-cancer data from The Cancer Genome Atlas to identify tumor types with evidence of TGFβ-induced EMT. Tumor types with high scores had lower survival rates than those with low scores and had a lower mutational burden in the TGFβ pathway (88). Dormant cancer cells are treatment resistant, breaking dormancy results in relapse following long periods of remission. The molecular mechanisms governing tumor cell dormancy have not been elucidated, however, data indicate that TGFβ family members are critical (89). IL1β and TGFβ1 trigger fibroblast recruitment to tumors and conversion into CAFs. Blocking this process with an IL1β neutralizing antibody and a TGFβR1 inhibitor can be countered by TGFβ activated kinase-mediated activation of the noncanonical TGFβ pathway. TGFβ activated kinase plus TGFβR1 inhibition blocks IL1β and TGFβ1-mediated fibroblast activation, decreasing the secretion of proinflammatory cytokines. The combination of a TGFβ activated kinase inhibitor plus TGFβR1 inhibitor reduced the metastatic capacity of tumor cells and the recruitment of fibroblasts.
Cancer and Other Diseases
Cancer develops through genetic injuries ending with malignant transformation. Numerous genes and pathways involved in progression to frank malignancy have been elucidated. Cancer develops following genetic aberrations in processes controlling proliferation, apoptosis, differentiation and genomic stability. Metastasis is a hallmark of malignancy (90). The process of metastasis is complex and involves dissemination of tumor cells from the primary tumor through the vascular and lymphatic systems and growth selectively in distant tissues and organs. TGFβ which is a growth suppressive cytokine in many normal situations becomes an active and important participant in malignant disease including angiogenesis, extracellular matrix deposition, immuno-suppression and metastasis growth promotion. TGFβ and its receptors are targets for protein therapeutics, small molecule kinase inhibitors and RNA-based therapeutics (91, 92). Neurofibromatosis type 2 syndrome is a very rare human genetic disease. NF2 loss results in reduced TGFβR2 expression, and increased expression of TGFβR1. TGFβR1 kinase inhibitors can restore cell differentiation and induce growth suppression in NF2-deficient Schwannoma cell line. The TGFβR1 kinase inhibitor, TEW7197, reduced tumor formation in a NF2-model mouse. From gene expression profiling, TEW7197 treatment increased lipid metabolism–related gene expression. Thus, reduction or deletion of TGFβR2 or NF2 induced TGFβR1 suggested that inhibition of TGFβ signaling may be beneficial in NF2-related cancers, mesothelioma and TGFβR2-mutated cancers (93). Non-small cell lung cancer (NSCLC) is largely incurable and is managed with chemotherapy, targeted agents, and immunotherapy. The mutations in NSCLC critical genes and the development of drugs targeted to those mutant proteins and immunotherapies have improved disease management for patients whose tumors have actionable mutations; however, cytotoxic chemotherapy remains an important treatment for many NSCLC patients. The efficacy of these therapies is limited by development of resistance. Aberrant high TGFβ expression in the tumor microenvironment promotes NSCLC progression and metastasis, and development of resistance to cytotoxic, targeted, and immunomodulatory therapeutic agents (95).
TGFβ downregulates the expression of ataxia telangiectasia–mutated (ATM) and mutS homolog 2 (MSH2) in cancer cells through a miRNA-mediated mechanism. DNA-repair genes expression was examined and BRCA1 was identified as downregulated by TGFβ through the miR181 family. Downregulating BRCA1, ATM, and MSH2, and high TGFβ produced the DNA damage response called a “BRCAness” phenotype, including poor DNA-repair and sensitivity to poly (ADP-ribose) polymerase (PARP) inhibitors. Breast cancer xenografts with active TGFβ signaling were resistant to doxorubicin and were sensitive to the PARP inhibitor veliparib. Doxorubicin with veliparib in combination were an effective treatment in TGFβ-high tumors (95).
Malignant cells often secrete high TGFβ that acts on normal cells in the tumor microenvironment as well as distant normal cells to suppress the antitumor immune response producing tumor immune tolerance, increasing fibrosis, augmenting angiogenesis, invasion and metastasis. Innate immune system dendritic cell subpopulations secrete TGFβ and contribute to the generation of regulatory T-cells that protect the tumor. TGFβ-directed therapy could reverse the immune suppression by this cytokine, decrease extracellular matrix formation, decrease angiogenesis, decrease osteolytic activity, and increase the sensitivity of malignant cells to cytotoxic therapies and immunotherapies. Normally, TGFβ acts as a tumor suppressor and is growth-inhibitory toward epithelial cells; however, in malignant disease, TGFβ supports tumor progression. In human patients, high TGFβ expression correlated with increased fibrosis. TGFβ supported pancreatic cancer progression through stromal and hematopoietic cell effects. TGFβR inhibition may be therapeutically useful pancreatic cancer especially in combination with immunotherapy, because malignant pancreatic cells frequently do not respond to TGFβ growth inhibition (96).
Therapeutic antibodies that block the programmed death-1 (PD-1)–programmed death-ligand 1 (PD-L1) pathway can produce durable responses in a subset of tumors (97). Analysis of tumors from patients with metastatic urothelial cancer treated with an anti-PD-L1, atezolizumab, indicated that treatment response depended upon CD8+ T-effector cell phenotype and high neoantigen or tumor mutation burden. Lack of response was associated with TGFβ signaling in CAFs and collagen-rich tumor stroma (98). In a mouse model, treatment with a TGFβ-blocking antibody and anti-PD-L1 antibody decreased TGFβ signaling and produced a robust anti-tumor immune response and tumor regression (99). TGFβ is an antitumor immune function repressor especially affecting the function of cytotoxic T lymphocytes. In preclinical models, adoptive transfer of TGFβ-insensitive CD8 T cells produced near complete regression of tumors which could not be reproduced by reduction in TGFβ signaling, through either genetic ablation or TGFβR1inhibition. In preclinical models, simultaneous inhibition of TGFβ and PD-L1 receptors resulted in increased survival. Combination regimens with TGFβ and PD-L1 pathway inhibitors may be therapeutically synergistic (100). Elevated plasma TGFβ can identify prognostically high-risk patients (101–104). In nude mice bearing subcutaneous human tumor xenografts, plasma levels of TGFβ were increased up to 37-fold compared with control mice. The controls had a TGFβ plasma level of 6.4 +/− 0.4 ng/ml while mice bearing the CaKi1 renal cell carcinoma had a TGFβ plasma level of 97.3 +/− 12.3 ng/ml. Fifteen tumor types were tested and the mean TGFβ plasma level in the xenograft bearing mice was 34.3 ng/ml, 5-times higher than in normal mice (105).
Therapeutic Approaches to TGFβ
TGFβ, the TGFβ signaling pathway and associated pathways have been under investigation as anticancer therapeutic targets for more than 30 years (105). Initial tool compounds including small molecules, antibodies and proteins tested in preclinical models demonstrated the therapeutic value of neutralizing/blocking the activity of TGFβ in cancer (106). Several investigational agents that target the TGFβ pathway have entered clinical trial (106). Clinical trials of an inhibitor of TGFβRI kinase activity, galunisertib, and a TGFβ neutralizing antibody, fresolimumab (GC1008), and other agents are underway (92).
Small Molecules
LY364947 is a small molecule identified in a high throughput screen designed to find compounds that could release the mink lung Mv1lu cell line from the growth inhibitory effects of TGFβ1 (108). Subsequent work showed that LY364947, a pyrazole-based compound, inhibited the serine-threonine kinase activity of TGFβR1. The TGFβ receptors are rare in the family of plasma member receptor kinases in being serine-threonine kinases and not receptor tyrosine kinases (RTKs). LY364947 is a potent TGFβR1/ALK5 inhibitor, IC50 = 59nM) (109). The kinase binding mode at the active site was elucidated by co-crystallization and X-ray analysis of potent inhibitors with the TGFβR-I receptor kinase domain (110). LY364947 inhibited Smad2 phosphorylation induced by TGFβ as well as fibronectin expression and MDA-MB-231 breast cancer cell invasion (111). Changes in tumor oxygenation and TGFβ expression was explored with single dose radiation therapy (20 Gy) or a single dose chemotherapy (cyclophosphamide, 250 mg/kg) in rats bearing 13762 mammary carcinoma growing subcutaneously in female Fischer 344 rat. Treatment with 20 Gy of radiation killed about two logs (99%) of 13762 tumor cells, and treatment with cyclophosphamide (250 mg/kg) killed about 1.5 logs (95%) of 13762 tumor cells. Hypoxia, measured by pO2 electrode, decreased in the tumors of treated animals until 6h posttreatment and then increased, so that 24h after administration of the radiation therapy or the chemotherapy the number of intra-tumoral blood vessels as determined by CD31 staining increased. TGFβ measured by radioimmunoassay, peaked in serum between 6h and 18h and again between 72h and 96h after radiation therapy. The TGFβ serum peak after cyclophosphamide was 3h after drug injection, with second peaks at 36h and 48h after drug administration. In the tumor, TGFβ peaked between 6h and 8h after drug administration (112, 113). LY364947 decreased the resistance of glioblastoma-initiating cells to radiation (113). Autocrine TGFβ signaling is important in cancer cell invasion and metastasis. Autocrine TGFβ supports cancer cell invasion by maintaining urokinase plasminogen activator (uPA) through protein secretion. Exposure to super-physiologic concentrations of paracrine/exogenous TGFβ further increased uPA expression and cell invasion. Both autocrine and paracrine TGFβ mediated uPA levels was controlled through the Smad4 protein. TGFβ maintains uPA expression through the Smad pathway, thus supporting tumor cell invasiveness (111). Glioblastoma (GBM) produce abundant TGFβ which, in response to radiation therapy, increases the DNA damage response. In murine (GL261) and human (U251, U87MG) glioma cells, response to radiation increased by approximately 25% when exposed to LY364947 before irradiation. GL261 neurosphere cultures were used to evaluate glioma initiating cells. LY364947 had no effect on neurosphere formation. Radiation decreased neurosphere formation by 28%. LY364947 treatment before irradiation decreased neurosphere formation by 75%. GL261 neurospheres produced 3.7-fold more TGFβ per cell than monolayer cultures, suggesting that TGFβ produced by glioma initiating cells promoted effective DNA damage response and self-renewal. Thus, LY364947 treatment of irradiated GL261 neurospheres decreased DNA damage responses, γH2AX and p53 phosphorylation, and induction of self-renewal signals, Notch1 and CXCR4 (114). Analysis of TCGA data and immunohistochemical phosphorylated SMAD2 staining in high grade gliomas indicated high but variable activation of the TGFβ pathway across glioma. Explant culture of seven high grade glioma specimen was used to assess whether LY364947 exposure could sensitize the explant cultures to radiation therapy. Immunofluorescence detection and image analysis of γH2AX foci, a DNA damage marker, and SOX2, a stem cell marker, increased after radiation. These responses were diminished by LY364947 in 5 of the 7 specimens (115).
Another small molecule TGFβ inhibitor tool compound, SB-431542 is an inhibitor of several TGFβ superfamily Type I activin receptor-like kinases, including ALK4, ALK5 and ALK7. The kinase domain of the TGFβ type I receptor, ALK5, and the substrate, Smad3 were expressed and screened for inhibitors. SB431542 emerged from the screen as an inhibitor of Smad3 phosphorylation with an IC50 of 94 nM. SB-431542 inhibited TGFβ1–induced nuclear Smad3 localization and inhibited TGFβ1–induced collagen I production. SB-431542 inhibited TGFβ-induced proliferation of human osteosarcoma cells. Although SB-431542 inhibits the kinase activity of TGFβ R1/ALK5 as well as the kinase activity of the activin type I receptor ALK4 and the nodal type I receptor ALK7, which are highly related to ALK5 in their kinase domains, SB-431542 had no effect on the ALK family members that recognize bone morphogenetic proteins (BMPs). Thus, SB-431542 was a selective inhibitor of endogenous activin and TGFβ signaling but did not alter BMP signaling. SB-431542 did not inhibit several other serine-threonine kinases such as components of the ERK, JNK, or p38 MAP kinase pathways (116, 117). SB-431542 suppressed TGFβ-induced growth stimulation of MG63 osteosarcoma cells. DNA microarray and quantitative real-time PCR analyses of RNAs from TGFβ-treated cells showed that several growth factors, including platelet-derived growth factorα, were induced by TGFβ in MG63 cells (118).
SB-431542 is often used in stem cell differentiation protocols. Embryonic vascular progenitor cells, also called endothelial precursor cells, can differentiate into mural cells/pericytes and endothelial cells (119). Members of the TGFβ superfamily are important during differentiation of vascular progenitor cells. TGFβ and activin inhibited proliferation and network formation by endothelial cells. SB-431542 exposure increased proliferation and network formation by endothelial cells, and increased the expression of claudin-5, an endothelial specific component of tight junctions (120). An effort was made to generate neurons from human pluripotent stem cells from cord blood-derived iPSCs (hiCBiPSCs). The differentiation of the cord blood-derived pluripotent stem cells was initiated by inhibiting TGFβ and BMP signaling by exposing the cells to dorsomorphin and SB-431542. A Sox1 and Pax6 positive neuronal progenitor cell population was induced using this protocol. The cells were further maturated into dopaminergic neurons (121).
The TGFβ inhibitor has found use in preclinical normal tissue and tumor cell organoid cultures. The cloning and propagation of human intestine and colon stem cells that sustain limited copy number and sequence variation despite extensive serial passaging and display cell-autonomous commitment to epithelial differentiation consistent with their origins along the intestinal tract required specialized culture media. The media was supplemented with a combination of growth factors and regulators of TGFβ, Wnt/β-catenin, EGF, IGF, and Notch pathways. Dissociated cells were seeded onto a feeder layer of lethally irradiated 3T3-J2 cells in media modified by the addition of R-spondin1, Jagged-1, Noggin, Rock inhibitor Y-27632, TGFβ inhibitor SB431542, and nicotinamide. The epigenetic commitment programs of these stem cells were stable, and replicative expansion was achieved (122). Culturing intestinal stem cells as 3D organoids resulted in heterogeneous cell populations, reflecting the in vivo cell type diversity. Intestinal stem cell organoid culture was conducted in a laminin-rich 3D matrix (Matrigel), within which cells develop a closed, cystic 3D structure. The growth factor additions for the monolayer and 3D cultures were very similar, both include Wnt agonist R-spondin, EGF, and BMP antagonist and TGF-β inhibitor SB-431542, the murine matrix and the focal production of Wnt3 by Paneth cells in 3D culture was the major difference between the systems (123).
The TGFβ inhibitor galunisertib (LY2157299) derived from the SAR initiated with LY364947, has entered clinical trial (124, 125). Galunisertib is an oral small molecule inhibitor of the TGFβRI kinase that downregulates the phosphorylation of SMAD2, abrogating activation of the TGFβ signaling pathway. Galunisertib has improved pharmaceutical properties and selectivity compared with LY364947. Mice bearing subcutaneously implanted Calu6 non-small cell lung carcinoma or MX-1 breast carcinoma xenografts were used to assess the relationship between galunisertib blood levels, pSMAD2,3 blood levels and response of the tumors to orally administered galunisertib. A model was developed that related tumor growth delay to pSMAD plasma concentrations (126). Galunisertib stimulated hematopoiesis from primary myelodysplastic syndrome bone marrow specimens via downregulation of SMAD2 phosphorylation. Myelodysplastic syndromes (MDS) are characterized by ineffective hematopoiesis, the molecular alterations that cause marrow failure continue to be investigated. The TGFβ pathway is constitutively activated in MDS progenitors. SMAD7, a negative regulator of TGFβ, was decreased in MDS marrow progenitors in a bone marrow tissue microarray by immunohistochemistry. Reduced SMAD7 in hematopoietic cells correlated with increased TGFβ–mediated gene transcription which could be inhibited by galunisertib. In vivo treatment with galunisertib decreased anemia in a TGFβ overexpressing transgenic mouse model of bone marrow failure. In culture, galunisertib stimulated hematopoiesis from primary MDS bone marrow specimens (127, 138).
Galunisertib has antitumor activity in breast, colon, lung cancers, and hepatocellular carcinoma xenograft tumors. Continuous long-term exposure to galunisertib caused cardiac toxicities in animals requiring adoption of a pharmacokinetic/pharmacodynamic-based dosing strategy. The use of the pharmacokinetic/pharmacodynamic model defined a therapeutic window with an appropriate safety profile for clinical investigation (125).
A galunisertib first-in-human dose (FHD) study was carried out in patients with cancer. Sixty-five patients (58 with glioma) with measurable, progressive malignancies were enrolled. Oral galunisertib was given morning and evening on an intermittent schedule of 14 days on and 14 days off (28-day cycle). Galunisertib monotherapy was studied in dose escalation (part A) first and then evaluated in combination with standard doses of lomustine (part B). Safety was assessed using Common Terminology Criteria for Adverse Events version 3.0. Antitumor activity was assessed by RECIST and Macdonald criteria. In part A, 16.6% (5/30) and in part B, 7.7% (2/26) of evaluable patients with glioma had either a complete (CR) or a partial response (PR). In both parts, 15 patients with glioma had stable disease (SD), 5 of whom had SD for 6 treatment cycles. Overall, clinical benefit was observed in 12 of 56 patients with glioma (21.4%). Galunisertib was safe, with no cardiac adverse events. The administration of galunisertib at 300 mg/day was deemed safe for future clinical investigation (128). In a subsequent trial, patients were randomized in a 2:1:1 ratio to galunisertib+lomustine, galunisertib monotherapy, or placebo+lomustine. The primary objective was overall survival (OS). One hundred fifty-eight patients (93.7% glioblastoma) were randomized: galunisertib+lomustine (n=79), galunisertib (n=39), and placebo+lomustine (n=40). Median OS in months for galunisertib+lomustine was 6.7 (5.3–8.5), 8.0 (5.7–11.7) for galunisertib alone, and 7.5 (5.6–10.3) for placebo+lomustine. There was no difference in OS for patients treated with galunisertib+lomustine compared with placebo+lomustine. Among 8 patients with IDH1 mutations, 7 patients were treated with galunisertib (monotherapy or with lomustine); OS ranged from 4 to 17 months. Galunisertib+lomustine failed to demonstrate improved OS relative to placebo+lomustine (NCT01582269) (129, 130).
TGF-β signaling is important in cardiovascular development and in cardiac repair. Because glaunisertib demonstrated cardiotoxicity in some preclinical models, patients treated with galunisertib underwent cardiovascular system monitoring in early trials. Patients (67 glioma and 12 solid tumor) enrolled in the first-in-human study received the TGFβ inhibitor galunisertib as monotherapy (n = 53) or in combination with lomustine (n=26). 2D echocardiography and Spectral Doppler (2D Echo with Doppler) were used to monitor patient cardiac performance every 2 months, in addition to electrocardiograms, thorax CT scans, and serum brain natriuretic peptide (BNP), troponin I, cystatin C, high-sensitivity C-reactive protein (hs-CRP). Treatment with galunisertib was not associated with cardiovascular toxicities. There were no increases of troponin I, BNP, or hs-CRP or reduction in cystatin C levels, which may indicate cardiovascular injury. Overall, cardiovascular monitoring for galunisertib did not detect medically relevant cardiac toxicity (131). Galunisertib is being investigated either as monotherapy or in combination with standard antitumor regimens (including nivolumab) in patients with cancer with high unmet medical needs such as glioblastoma, pancreatic cancer, and hepatocellular carcinoma (132).
Combination of galunisertib and dinutuximab, the anti-GD2 antibody, had increased efficacy with natural killer cells against neuroblastoma and preserved the function of in vitro expanded natural killer cells in AML and colon cancer models. Treatment of high-risk neuroblastoma using the anti-GD2 antibody dinutuximab induced antibody-dependent cell-mediated cytotoxicity. In cell culture, galunisertib suppressed SMAD activation in neuroblastoma cells induced by exogenous TGFβ1 or by patient blood and bone marrow plasma, and suppressed SMAD2 phosphorylation in human neuroblastoma cells growing in NSG mice. In NK cell cultures treated with exogenous TGFβ1, galunisertib suppressed SMAD2 phosphorylation and restored the expression of the TRAIL death ligand, the release of perforin and granzyme A, and the direct cytotoxicity and ADCC of activated NK cells against neuroblastoma cells (133). The TGFβR1 inhibitor, galunisertib, decreased TGFβ signaling in the high TGFβ immunosuppressive tumor microenvironment in a mouse liver metastases model of colon cancer. There was improved eradication of liver metastases in mice treated with adoptive NK cells combined with galunisertib compared with mice receiving NK cells only or TGFβ inhibition alone. The therapeutic efficacy of adoptive NK cell therapy was increased by approaches targeting TGFβ signaling (134). Galunisertib reversed TGFβ and regulatory T cell mediated suppression of human T cell proliferation. Combination with PD-L1 blockade and galunisertib increased tumor response in colon carcinoma models. Exposure to galunisertib in culture, reversed TGFβ and regulatory T cell mediated suppression of human T cell proliferation. Treatment of mice with 4T1-LP tumors resulted in dose-dependent antitumor activity with complete regressions in 50% of animals. This effect was CD8+ T cell dependent and increased T cell numbers in the tumors. Combination of galunisertib with PD-L1 blockade resulted in improved tumor response and complete regressions in colon carcinoma models, demonstrating the therapeutic potential of co-targeting TGFβ and PD-1/PD-L1 pathways (135). In metastatic colon cancers, tumor microenvironment features including little T-cell infiltration and/or increased TGFβ predict adverse outcomes. The mice carrying mutations found in colorectal cancers developed intestinal tumors with low mutational burden, T-cell exclusion and TGFβ-activated stroma. PD-1–PD-L1 immune checkpoint blockade had little effect in the tumors. TGFβ neutralization resulted in a robust cytotoxic T-cell and tumor response and made tumors susceptible to anti-PD-1–PD-L1 therapy (169).
Blocking TGF-β signaling in tumor cells in a tissue microenvironment was investigated in a panel of patient-derived xenografts were treated with TGFβR1 inhibitor galunisertib. In a cell culture clonogenic assay, galunisertib inhibited growth in some patient-derived xenografts. PDX bearing mice treated with galunisertib exhibited changes in TGFβ downstream signaling of genes in the TGF-β pathway. pSMAD2 protein expression and TGFβRI mRNA expression correlated with galunisertib effects in xenografts. Tumor cell TGFβ signaling does not fully account for the galunisertib antitumor activity indicating that xenograft models may be more representative than cell culture assays for assessing TGFβ inhibitor activity (136).
In advanced pancreatic cancer patients treated with galunisertib plus gemcitabine or gemcitabine plus placebo were examined for the effect of galunisertib on overall survival. Galunisertib (300 mg/day, orally) exposure metrics for each patient in the galunisertib plus gemcitabine arm (n=99) of this phase 2 study were calculated. The pharmacokinetics analysis included data from 297 patients/healthy subjects. Galunisertib was rapidly absorbed with peak concentrations attained within 0.5–2h and had an elimination half-life of 8h. Between-subject variance on clearance was estimated at 47%. A Weibull survival model with treatment effect (dose) estimated a hazard ratio of 0.796, after adjusting for patient baseline factors. There was a flat daily exposure–survival relationship within the observed exposure. Response covariates, such as reduction in CA19–9, time on treatment, and cumulative exposure over treatment cycles were identified as significant factors for survival for pancreatic cancer patients (137). The development of galunisertib is currently on hold.
Vactosertib (TEW-7197) is a selective small molecule inhibitor of TGFβR1 in clinical development. Vactosertib targets the adenosine-5-triphosphate binding site of TGFβR1, thereby inhibiting phosphorylation of the Smad2 and Smad3 proteins, the key mediators in TGFβ downstream signaling (138–141). TGFβ pathway activity produces tumor immune surveillance and resistance to immune checkpoint inhibitors. TGFβ responsive signatures in CAFs are associated with poor prognosis. Vactosertib safety, efficacy, and association with TGFβ response signatures were evaluated in patients with advanced solid tumors. Based on TCGA analysis, high fibroblast TGFβ response signatures were seen in pancreatic, lung, colorectal, and stomach cancers in association with poor prognosis (adjusted hazard ratio 1.27; P=1.06×10−8). In a phase I modified 3+3 dose-escalating study and patients (29) received vactosertib once daily over a dose range (30~340 mg) for 5 days with 2 days off. RNA sequencing of pretreatment tumor samples in 16 patients were evaluated for fibroblast TGFβ response signatures defined as the mean values of the expression of 171 genes. Vactosertib was safe and well tolerated, and maximum tolerated dose was not determined. The most common treatment-related adverse event was fatigue, while abdominal pain, AST elevation, and pulmonary edema occurred in one patient. Six of 17 patients who received ≥140 mg achieved stable disease (35.3%) and had higher fibroblast TGFβ response signatures than those with progressive disease. Vactosertib in combination with immune checkpoint inhibitors could be an effective therapy (NCT02160106; 142).
Protein Therapeutics
Decorin, a naturally occurring TGFβ1/2 antagonist, is a small leucine-rich proteoglycan which is involved in collagen fibrillogenesis, angiogenesis, and inflammation, and limits scaring in wound healing (143–150). The decorin core protein has leucine-rich tandem repeats with a glycoaminoglycan attached through the protein N-terminus. The major binding partners of decorin are TGFβ and collagen (151, 152).
Decorin acts as a tumor suppressor by blocking TGFβ immune-suppressive effects. Conversely, decorin has been described as a therapeutic target for osteosarcoma. Two osteosarcoma cell lines were established from the highly lung metastatic LM8 murine osteosarcoma cell line, the LM8-DCN which stably expressed human decorin and LM8-mock control. When the LM8 sublines were implanted subcutaneously in mice, fewer pulmonary metastases were observed in mice bearing LM8-DCN tumors compared to mice bearing LM8 and LM8-mock transfected tumors (153). In addition, the mice bearing LM8-DCN tumors survived longer than those with LM8 and LM8-mock tumors. There was no difference in the morphology or growth rates of the cells, but the motility and invasion of LM8 cells were inhibited by decorin suggesting that decorin prevented osteosarcoma lung metastasis.
TGFβ is involved in the in vivo resistance of the EMT-6/CTX and EMT-6/CDDP murine mammary tumors which are resistant to cyclophosphamide and cisplatin, respectively (106). Both of these tumors have higher intratumoral vessel counts than the EMT-6/parent tumor. Mice bearing the resistant tumors have higher plasma levels of TGFβ than mice bearing the parent tumors; however, upon treatment with cytotoxic therapies there was a greater rise in plasma TGFβ in mice bearing the parent tumor than in mice bearing the resistant tumors. In situ hybridization for TGFβ mRNA and immunohistochemical staining for TGFβ protein showed that the resistant tumor TGFβ was higher than in the parent tumor prior to treatment; however, after cytotoxic therapy the increase in TGFβ was greater in the parent tumor than in the resistant tumor. Treatment of tumor-bearing mice with the TGFβ inhibitor decorin did not alter the sensitivity of the parent tumor to cyclophosphamide or cisplatin as determined by tumor cell survival assay. However, administration of decorin increased the sensitivity of the EMT-6/CTX tumor to cyclophosphamide and of the EMT-6/CDDP tumor to cisplatin so that the tumor drug resistance was nearly ablated. A similar pattern was observed in the drug response of the bone marrow granulocyte-macrophage colony-stimulating factor of mice bearing each of the 3 tumors (154). The gene for TGFβ1 was transfected into the murine EMT-6/Parent mammary carcinoma tumor line to form the EMT6/PRK5 beta 1E tumor line. In monolayer culture, the EMT-6/PRK5 beta 1E tumor line secreted about 15-times more TGFβ1 into the medium than the EMT-6/Parent line. The response of the two cell lines to 4-hydroperoxycyclophosphamide, cisplatin, melphalan or thiotepa in monolayer culture was similar. When the EMT-6/PRK5 beta 1E cells were grown as a subcutaneous tumor in mice, plasma TGFβ1 was about 5-fold higher than in mice bearing the EMT-6/Parent tumor. In mice, the EMT-6/PRK5 beta 1E tumor was markedly resistant to a dose range of cyclophosphamide, cisplatin, melphalan and thiotepa compared with the EMT-6/Parent tumor. The bone marrow CFU-GM from the mice bearing the EMT-6/PRK5 beta 1E tumor was spared from the cytotoxicity of the drugs compared with the bone marrow CFU-GM from mice bearing the EMT-6/Parent tumor. Administration of decorin to mice bearing the EMT-6/PRK5 beta 1E tumor prior to drug treatment restored drug sensitivity to the tumor and to the bone marrow CFU-GM. Administration of decorin prior to drug treatment produced very little or no increase in the response of the EMT6/Parent tumor or the bone marrow CFU-GM from those animals. The EMT6/PRK5 beta 1E tumor model allowed the effect of secretion of TGFβ1 on therapeutic resistance to be assessed directly (155, 156).
Galacorin, human recombinant decorin, was investigated as a therapeutic for treatment of macular degeneration, diabetic retinopathy and diabetic macular edema (145). In rabbits, decorin decreased corneal scarring after eye surgery. In adult rats, treatment with decorin decreased spinal scarring and produced dissolution of mature scars by neutralizing TGFβ1/2 (146).
TGFβ neutralizing antibodies
The role of TGFβ in tumor therapeutic in vivo resistance was examined by administration of TGFβ-neutralizing antibodies, either 2G7 and 4A11 which supplied by Genentech, to animals bearing the EMT-6/Parent tumor or the antitumor alkylating resistant tumors, EMT-6/CTX or EMT-6/CDDP (156, 157). Treatment of tumor-bearing mice with a TGFβ neutralizing antibody by intraperitoneal injection daily on days 0–8 post-tumor cell implantation increased the sensitivity of the EMT6/Parent tumor to cyclophosphamide (CTX) and cisplatin (CDDP) and increased the sensitivity of the EMT-6/CTX tumor to CTX and the EMT6/CDDP tumor to CDDP. Bone marrow granulocyte-macrophage colony-forming unit survival was determined from the same mice. The increase in tumor sensitivity upon TGFβ neutralization was also observed in increased bone marrow CFU-GM sensitivity to CTX and CDDP. Mice bearing the EMT-6/CTX and EMT-6/CDDP tumors had higher serum lactate than normal or EMT-6/Parent tumor-bearing mice; lactate was decreased by the anti-TGFβ regimen. Treatment with TGFβ-neutralizing antibodies restored drug sensitivity in drug resistant tumors, altering both the tumor and host metabolic states (156, 157).
In cell culture, murine RenCa renal cell carcinoma cells do not response to TGFβ; however, in vivo the RenCa tumor does response to TGFβ blockade (158). When a pan-TGFβ neutralizing antibody 1D11 was administered to mice injected intravenously with RenCa cells, the number of lung metastases was decreased, if the antibody was administered at a high dose (50 mg/kg) and tumor progression was decreased (158). High TGFβ1 is produced by stromal cells which are anessential component of the leukemic microenvironment in the bone marrow niche and regulate cell proliferation, survival, and apoptosis. In cell culture, acute myeloid leukemia (AML) cells moved into a quiescent G0 state in the presence of TGFβ. The TGFβ-neutralizing antibody 1D11 abrogated TGFβ1 induced AML cell cycle arrest. Exposure to 1D11 enhanced cytarabine (Ara-C)– induced apoptosis of AML cells in hypoxic and normoxic conditions. Treatment with 1D11 along with Ara-C decreased leukemia burden and prolonged survival in an in vivo leukemia model (159). In the type I collagen of Crtap−/− mice treated with 1D11, the bone phenotype in osteogenesis imperfecta was corrected and the lung abnormalities improved (160).
Mice bearing GL261 subcutaneous tumors treated with 1D11 had increased tumor growth delay following radiation therapy compared with control mice. A local radiotherapy regimen in combination with anti-CD137 and anti-PD-1 produced an abscopal effect in a syngeneic murine tumor model. TGFβ neutralization with 1D11 enhanced treatment efficacy in syngeneic mouse tumors implanted in both flanks. Increases in CD8 T cells infiltrating the non-irradiated lesion were documented. Radiotherapy-induced TGFβ prevented abscopal benefits, however, TGFβ neutralization in combination with radio-immunotherapy resulted in improved efficacy (161).
Fresolimumab (GC1008) is a pan-TGFβ neutralizing antibody that is undergoing clinical trial in several fibrotic conditions and cancer. The crystal structure of the single-chain variable fragment of fresolimumab (GC1008 scFv) in complex with target TGFβ1 and in complex with TGFβ2 and with TGFβ3 has been described (162). Fresolimumab was radiolabeled with 89Zr for PET to analyze TGFβ expression, tumor uptake, and organ distribution at 3 doses and compared with 111In-IgG in a human TGFβ–transfected CHO xenograft, and in human breast cancer MDA-MB-231 tumor and metastasis model. 89Zr-fresolimumab distribution was similar to IgG in most organs and caused no toxicity in mice. It accumulated in primary tumors and metastases. High 89Zr-fresolimumab uptake was seen in sites of tumor ulceration and in scar tissue, processes in which TGFβ is known to be highly active (163). In three human phase 1 clinical trials, administration of fresolimumab was well tolerated (164).
Fresolimumab was evaluated in patients with advanced malignant melanoma and renal cell carcinoma. In a multi-center phase I trial, cohorts of patients with previously treated malignant melanoma or renal cell carcinoma received intravenous fresolimumab at escalating doses on days 0, 28, 42, and 56. Patients achieving at least stable disease received extended treatment consisting of 4 doses of fresolimumab every 2 weeks for up to 2 additional courses. Twenty-nine patients were treated, 22 in the dose-escalation and 7 in a safety cohort expansion. No dose-limiting toxicity was observed, and the maximum dose, 15 mg/kg, was determined to be safe. The development of reversible cutaneous keratoacanthomas/squamous-cell carcinomas (4 patients) and hyperkeratosis was the major adverse event observed. One malignant melanoma patient achieved a partial response, and six had stable disease with a median progression-free survival of 24 weeks (16.4–44.4 weeks) (165).
89Zr-fresolimumab PET was used to assess uptake of fresolimumab and to assess treatment outcome in patients with recurrent high-grade glioma. 89Zr-fresolimumab PET scans were acquired on day 2 or day 4 after injection. Patients were treated with fresolimumab (5 mg/kg, iv, every 3 wk). Twelve patients with recurrent high-grade glioma underwent 89Zr-fresolimumab PET 4 d after injection. All patients had clinical or radiologic progression after 1–3 infusions of fresolimumab. Median progression-free survival was 61d (25–80d), and median overall survival was 106d (37–417d). Although 89Zr-fresolimumab penetrated recurrent high-grade gliomas no clinical benefit was evident in this patient population (166).
Fresolimumab can cause cutaneous lesions which have been investigated for the clinical, histological, and immunohistochemical characteristics in skin biopsies from patients with treatment-emergent, cutaneous lesions during a phase 1 study of multiple fresolimumab doses with melanoma or renal cell carcinoma. Based on central review, four patients developed lesions with histological characteristics of keratoacanthomas and including a single case of well-differentiated squamous cell carcinoma. Following completion of fresolimumab, lesions spontaneously resolved (167).
Fresolimumab was tested in malignant pleura mesothelioma patients to assess clinical safety and response. Patients with progressive mesothelioma received fresolimumab (3mg/kg IV over 90 min every 21d) as part of an open-label Phase II trial. Although partial or complete radiographic responses were not observed, three patients showed stable disease at 3 mo. Serum from 5 of 13 patients had new or increased levels of antibodies against mesothelioma lysates as measured by immunoblotting. Patients who produced anti-tumor antibodies had increased median overall survival (15 vs 7.5mo, p < 0.03) compared with those who did not. The trial was terminated early for company business reasons (168).
A clinical trial in metastatic breast cancer assessed the feasibility, efficacy and immune enhancement of fresolimumab administration during radiotherapy. Metastatic breast cancer patients with at least three distinct metastatic sites were randomized to receive fresolimumab (1or 10 mg/kg). Patients receiving 10 mg/kg had a higher median overall survival than those receiving 1 mg/kg. The 10 mg/kg dose correlated with improved peripheral blood mononuclear cell counts and increased CD8 central memory T-cells; thus, these patients had a positive systemic immune response and longer median overall survival (NCT01401062; 169–173). Patients receiving the 10 mg/kg had improved peripheral blood mononuclear cell counts (172, 174). Additional analyses of residual peripheral blood mononuclear cells were performed using single cell network profiling and compared with data obtained from seven healthy female donors. Samples were analyzed for T-cell receptor modulated signaling via CD3 and CD28 crosslinking and measurement of evoked phosphorylation of AKT and ERK in CD4 and CD8 T-cell subsets defined by PD-1 expression. At baseline, a higher expression of PD-L1 was identified in patient monocytes compared to healthy donors. T cell receptor modulation revealed dysfunctional circulating T-cells in patient samples, and this was more pronounced in PD-1+ cells. Treatment with radiotherapy and fresolimumab did not resolve dysfunctional T-cell signaling. In culture PD-1 inhibition increased T-cell receptor signaling in PD-1+T-cells and not in PD-1 T-cells or in PD-1+T-cells from healthy donors. These findings support the notion of combining PD-1 blockade with TGFβ blockade and radiotherapy (NCT01401062; 167).
Bintrafusp alfa
Bifunctional molecules targeting two pathways simultaneously may provide an advantage compared with two single agents used in combination. Bintrafusp alfa (M7824, MSB0011359C) is a bifunctional fusion protein composed of an antibody directed to PD-L1 fused to the extracellular domain of human TGFβRII, a “trap” for the three TGFβ isoforms. Bintrafusp alfa efficiently, specifically, and simultaneously binds PD-L1 and neutralizes TGFβ. In mouse tumor models, Bintrafusp alfa suppressed tumor growth and metastasis more effectively than treatment with either an anti–PD-L1 antibody or TGFβ-trap. Bintrafusp alfa was effective in combination with radiotherapy or chemotherapy in mouse models (175). During the phase I dose-escalation study patients with advanced solid tumors received bintrafusp alfa (1, 3, 10 or 20 mg/kg once biweekly) until tumor progression, unacceptable toxicity or trial withdrawal (NCT02517398; 176). Nineteen patients were treated with bintrafusp alfa. Although 4 patients had grade 3 toxicities, an MTD was not reached. There was one confirmed complete response (cervical cancer), two durable confirmed partial responses (pancreatic cancer; anal cancer), one near-PR (cervical cancer), and two cases of prolonged stable disease in patients with growing disease at study entry (pancreatic cancer; carcinoid) (177).
Asian patients with recurrent gastric carcinoma who recurred after standard therapy were enrolled in the expansion cohort of a phase I trial and received bintrafusp alfa (1200 mg once every 2 weeks) until disease progression, unacceptable toxicity, or withdrawal. Thirty-one heavily pretreated patients received bintrafusp alfa for a median of 10.1 weeks. Six patients (19%) experienced grade 3 treatment-related adverse events. Ten patients (32%) had immune-related adverse events. The confirmed objective response rate per independent review committee was 16%; disease control rate was 26%. Median duration of response was 8.7 months (range 2.4–12.4). Responses occurred irrespective of PD-L1 expression or microsatellite instability status and appeared to correlate with high tumor TGFβ1 (178–181).
AVID200, a decoy receptor, is a rationally designed, highly potent inhibitor of TGFβ1/3. AVID200 has entered clinical trial (NCT03831438; 182).
Latent TGFβ1 Complex Stabilizer
Despite breakthroughs achieved with cancer checkpoint blockade therapy, many patients do not respond to PD-1 antibody (178). Human clinical and preclinical studies indicate that TGFβ signaling could be a point of intervention to increase response to checkpoint blockade therapy. Analysis of mRNA expression data from The Cancer Genome Atlas confirmed that TGFβ1 is the most prevalent TGFβ isoform expressed in many types of human tumors, suggesting that TGFβ1 may contribute to checkpoint blockade therapy resistance. A fully human antibody, SRK-181, that binds to TGFβ1-containing large latent complexes selectively through three regions but does not bind to active TGFβ or to the large latent complexes containing other TGFβ isoforms. SRK-181 prevented TGFβ1 activation in cell-based assays. Coadministration of SRK-181 and an anti–PD-1 antibody in mice harboring syngeneic tumors refractory to anti–PD-1 treatment resulted in tumor responses. Combined SRK-181 and anti–PD-1 treatment resulted in increased intra-tumoral CD8+ T-cells and decreased immunosuppressive myeloid cells (179).
Antisense RNA and siRNA
TGFβ is expressed at high levels by glioma cells and by glioblastoma tumors. In preclinical studies silencing TGFβ by siRNA by putting a triphosphate group at the end of siRNA, gene silencing was combined with immune activation via retinoic acid-inducible gene I (RIG-I). Systemic administration of this construct decreased tumor associated TGFβ in tumor-bearing mice and prolonged survival (170).
Phosphorothioate-locked nucleic acid (LNA)-modified antisense oligonucleotides, ISTH1047 and ISTH0047, which target TGFβ1/2 were assessed in cell culture and in tumor models. These antisense agents effectively targeted TGFβ1/2, decreased SMAD2 phosphorylation and reduced cellular migration and invasion. In vivo, ISTH1047 or ISTH0047 administration reduced SMAD2 phosphorylation in tumors and increased immune cell infiltration (183).
The pathophysiology of hypertrophic scars is characterized by prolonged inflammatory and proliferative wound healing. High expression TGFβ1 and COX-2 are key to the aberrant fibrogenic response. Silencing TGFβ1 and COX-2 expression with siRNA simultaneously in human fibroblasts resulted in an increase in apoptotic cells. Using human hypertrophic scar and skin graft implant models in mice, there were size reductions of the implanted tissues following intra-scar injection TGFβ1 and COX-2 specific siRNAs formulated in a Histidine Lysine Polymer (HKP). Silencing of the target genes resulted in down regulation of pro-fibrotic factors including α-SMA, hydroxyproline, collagen 1 and collagen 3 in the treated tissues. Intra-lesion injection of the TGF-β1 and COX-2 siRNAs reduced the size of hypertrophic scar implants by re-balancing of scar tissue deposition and degradation (184).
STP705, is an investigational siRNA therapeutic formulated in a polypeptide nanoparticle to target TGFβ1 and COX-2 gene expression. In preclinical animal models, STP705 administration increased T-cell penetration into tumors in the liver and increased the efficacy of an anti-PD-L1 antibody in a hepatocellular carcinoma model (185). In an ongoing clinical Phase II study evaluating STP705, administered by intralesional injection once per week for up to 6 weeks for treatment of non-melanoma skin cancer, interim analysis indicated that some patients had histological clearance of lesions. There were no adverse cutaneous skin reactions in the treated patients (NCT04293679; 186).
Conclusions and Directions
The critical involvement of the TGFβ family in malignant disease has been recognized for more than 30 years. TGFβ contributes to therapeutic resistance and to the immune-suppressive tumor microenvironment. Although small molecules, proteins and nucleic acid-based agents have been applied to neutralizing or blocking the tumor ‘protective’ effects of TGFβ, none of these agents has yet reached FDA approval. TGFβ is not a tumor-driver, in fact, malignant cells tend to be non-responsive to TGFβ, especially, if TGFβRII is not expressed. TGFβ acts primarily on the tumor microenvironment by silencing immune system activity against the tumor and increasing dense stroma production. The recent success of immune-therapeutics could be increased by combination with a TGFβ-activity inhibiting agent which could augment the effect of the vaccine, antibody, Car-T cells, or other immune-enhancer by removing a layer of tumor protection. The challenge going forward is to determine the optimal way to use inhibitors of the TGFβ pathway in the clinic to improve response to chemotherapy, radiation therapy and immunotherapy.
Figure 1.
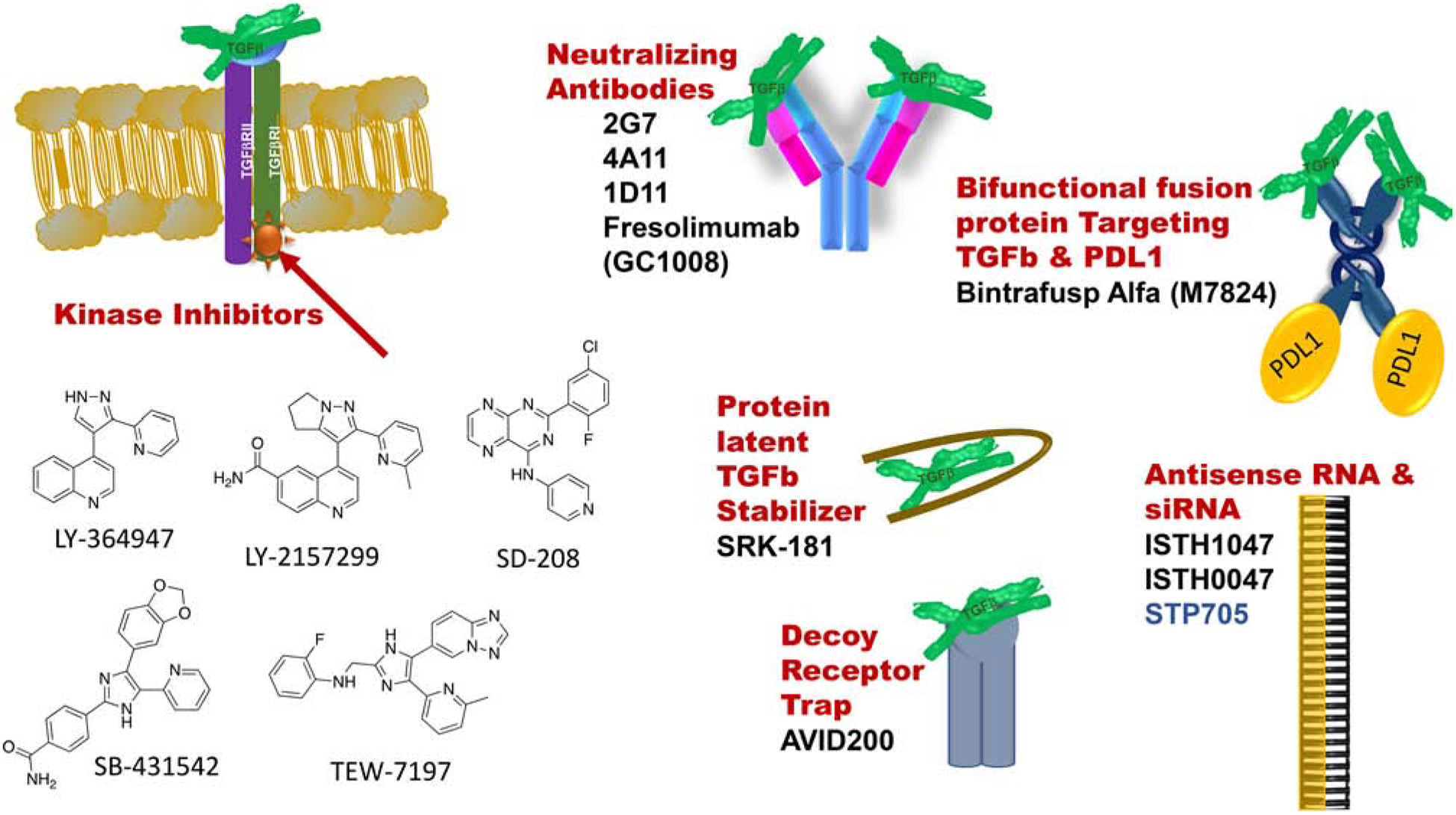
Schematic showing the variety of TGFβ pathway inhibitors and neutralizers being explored preclinically and clinically.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement
The author declares there are no conflicts of interest.
REFERENCES
- 1.Pickup M, Novitsky S, Moses HL. The roles of TGFβ in the tumor microenvironment. Nature Rev 2013; 13: 788–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yeh HW, Lee SS, Chang CY, Lang YD, Jou YS. A new switch for TGFβ in cancer. Cancer Res 2019; 79: 3797–805. [DOI] [PubMed] [Google Scholar]
- 3.Neuzillet C, Tijeras-Raballand A, Cohen R, Cros J, Faivre S, Raymond E, de Gramont A. Targeting the TGFβ pathway for cancer therapy. Pharmacol Therap 2015; 147: 22–31. [DOI] [PubMed] [Google Scholar]
- 4.Su J, Morgani SM, David CJ, Wang Q, Er EE, Huang YH, Basnet H, Zou Y, Shu W, Soni RK, Hendrickson RC, Hadjantonkis AK, Massague J. TGF-β orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1. Nature 2020; 577: 566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Gramont A, Faivre S, Raymond E. Novel TGF-β inhibitor ready for prime time in onco-immunology. OncoImmunol 2017; 6: e1257453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mullen AC, Wrana JL. TGF-b family signaling in embryonic and somatic stem-cell renewal and differentiation. Cold Spring Harbor Perspect Biol 2017; 9: a022186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akhurst RJ, Hata A. Targeting the TGFβ signaling pathway in disease. Nat Rev Drug Discov 2012; 11:790–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moses HL, Roberts AB, Derynck R. The discovery and early days of TGF-β: a historical perspective. Cold Spring Harbor Perspect Biol 2016; 8: a021865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roane BM, Arend RC, Birrer MJ. Review: targeting the transforming growth factor-beta pathway in ovarian cancer. Cancers 2019; 11: 668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Derynck R, Budi EH. Specificity, versatility and control of TGF-β family signaling. Sci Signal 2019; 12: eaav5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schlunegger MP, Grutter MG. An unusual feature revealed by the crystal structure at 2.2 Å resolution of human transforming growth factor-β2. Nature 1992; 358: 430–4. [DOI] [PubMed] [Google Scholar]
- 12.Robertson IB, Horiguchi M, Zilberberg L, Dabovic B, Hadjiolova K, Rifkin DB. Latent TGF-β-binding proteins. Matrix Biol 2015; 47: 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Derynck R, Akhurst RJ, Balmain A. TGF-β signaling in cancer: a double-edged sword. Trends Cell Biol 2001; 29:117–29. [DOI] [PubMed] [Google Scholar]
- 14.Massagué J How cells read TGF-β signals. Nat Rev Mol Cell Biol 2000; 1:169–78. [DOI] [PubMed] [Google Scholar]
- 15.Davod CJ, Massague J. Contextual determinants of TGFβ action in development, immunity and cancer. Nature Rev Molec Cell Biol 2018; 19: 419–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Attisano L, Wrana JL. Signal transduction by the TGF-β superfamily. Science 2002; 296:1646–7. [DOI] [PubMed] [Google Scholar]
- 17.Derynck R, Zhang YE. Smad-independent and smad-dependent pathways in TGF-β family signaling. Nature 2003; 425:577–84. [DOI] [PubMed] [Google Scholar]
- 18.Glick A TGF-β1, Back to the future. Cancer Biol Ther 2004; 3:276–83. [DOI] [PubMed] [Google Scholar]
- 19.Shi Y, Massagué J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 2003; 113:685–700. [DOI] [PubMed] [Google Scholar]
- 20.Heldin CH, Miyazono K, ten Dijke P. TGF-β signaling from cell membrane to nucleus through SMAD proteins. Nature 1997; 390:465–71. [DOI] [PubMed] [Google Scholar]
- 21.David CJ, Massagué J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol 2018; 19: 419–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.David CJ, Huang YH, Chen M, Su J, Zou Y, Bardeesy N, Iacobuzio-Donahue CA, Massagué J. TGF-β tumor suppression through a lethal EMT. Cell 2016; 164: 1015–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miyazono K, Suzuki H, Imamura T. Regulation of TGF-β signaling and its roles in progression of tumors. Cancer Sci 2003; 94: 230–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.D’Souza RCJ, Knittle AM, Nagaraj N, van Dinther M, Choudhary C, ten Dijke P, Mann M, Sharma K. Time-resolved dissection of early phosphoproteome and ensuing proteome changes in response to TGF-β. Sci Signal 2014; 7: issue 335 rs5. [DOI] [PubMed] [Google Scholar]
- 25.Bellomo C, Caja L, Moustakas A. Transforming growth factor-β as regulator of cancer stemness and metastasis. Brit J Cancer 2016; 115: 761–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moustakas A, Heldin CH. Dynamic control of TGF-β signaling and its links to the cytoskeleton. FEBS Letts 2008; 582: 2051–65. [DOI] [PubMed] [Google Scholar]
- 27.Wakefield LM, Hill CS. Beyond TGFβ: roles of other TGFβ superfamily members in cancer. Nature Rev 2013; 13: 328–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tian F, DaCosta Byfield S, Parks WT, Yoo S, Felici A, Tang B, Piek E, Wakefield LM, Roberts AB. Reduction in Smad2/3 signaling enhances tumorigenesis but suppresses metastasis of breast cancer cell lines. Cancer Res 2003; 63:8284–92. [PubMed] [Google Scholar]
- 29.Batlle E, Massague J. Transforming growth factor-β signaling in immunity and cancer. Immunity 2019; 50: 924–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Browning LM, Pietrzak M, Kuczma M, Simms CP, Kurczewska A, Refugia JM, Lowery DJ, Rempala G, Gutkin D, Ignatowicz L. TGF-β-mediated enhancement of Th17 cell generation is inhibited by bone morphogenetic protein receptor 1a signaling. Sci Signal 2018; 11: eaar2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Massagué J TGFbeta in Cancer. Cell 2008;134: 215–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leivonen SK, Kähäri YM. Transforming growth factor-β signaling in cancer invasion and metastasis. Int J Cancer 2007; 121:2119–24. [DOI] [PubMed] [Google Scholar]
- 33.Miyazono K, Moriwaki S, Ito T, Kurisaki A, Asashima M, Tanokura M. Hydrophobic patches on SMAD2 and SMAD3 determine selective binding cofactors. Sci Signal 2018; 11: eaao7227. [DOI] [PubMed] [Google Scholar]
- 34.Shah AH, Tabayoyong WB, Kundu SD, Kim SJ, Van Parijs L, Liu VC, Kwon E, Greenberg NM, Lee C. Suppression of tumor metastasis by blockade of transforming growth factor β signaling in bone marrow cells through a retroviral-mediated gene therapy in mice. Cancer Res 2002; 62:7135–8. [PubMed] [Google Scholar]
- 35.Dimeloe S, Gubser P, Loeliger J, Frick C, Develioglu L, Fischer M, Marquardsen F, Bantug GR, Thommen D, Lecoultre Y, Zippelius A, Langenkamp A, Hess C. Tumor-derived TGF-β inhibits mitochondrial respiration to suppress IFN-g production by human CD4+ T cells. Sci Signal 2019; 12: eeav3334. [DOI] [PubMed] [Google Scholar]
- 36.Bourgeois B, Gilquin B, Tellier-Lebegue C, Ostlund C, Wu W, Perez J, El Hage P, Lallemand F, Worman HJ, Zinn-Justin S. Inhibition of TGF-β signaling at the nuclear envelope: characterization of interactions between MAN1, Smad2 and Smad3 and PPM1A. Sci Signal 2013; 6: issue 280 ra49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bertero A, Brown S, Madrigal P, Osnato A, Ortmann D, Yiangou L, Kadiwala J, Hubner NC, de los Mozos IR, Sadee C, Lenaerts AS, Nakanoh S, Grandy R, Farnell E, Ule J, Stunnenberg HG, Mendjan S, Vallier L. The SMAD2/3 interactome reveals that TGFβ controls m6A mRNA methylation in pluripotency. Nature 2018; 555: 256–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang T, Zhang X, Chen A, Xiao Y, Sun S, Yan J, Cao Y, Chen J, Li F, Zhang Q, Huang K. Progranulin promotes bleomycin-induced skin sclerosis by enhancing transforming growth factor-β/Smad3 signaling through up-regulation of transforming growth factor-β type I receptor. Amer J Pathol 2019; 189: 1582–93. [DOI] [PubMed] [Google Scholar]
- 39.McSherry EA, Donatello S, Hopkins AM, McDonnell S. Molecular basis of invasion in breast cancer. Cell Mol Life Sci 2007; 64: 3201–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R, Weinberg RA. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007; 449: 557–63. [DOI] [PubMed] [Google Scholar]
- 41.Lin EY, Nguyen AV, Russell RG Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med 2001;193: 727–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res 1999; 59: 5002–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Padua D, Zhang X H-F, Wang Q et al. TGFβ primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 2008; 133: 66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Orimo A, Gupta PB, Sgroi DC, Arendzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF1/CXCL12 secretion. Cell 2005; 121: 335–48. [DOI] [PubMed] [Google Scholar]
- 45.Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, Fearon D, Greten F, Hingorani SR, Hunter T, Hynes RO, Jain RK, Janowitz T, Jorgensen C, Kimmelman AC, Kolonin MG, Maki RG, Powers RS, Pure E, Ramirez DC, Scherz-Shouval R, Sherman MH, Stewart S, Tlsty TD, Tuveson DA, Watt FM, Weaver V, Weeraratna AT, Werb Z. A framework for advancing our understanding of cancer-associated fibroblasts. Nature Rev Cancer 2020; 20: 174–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goulet CR, Bernard G, Tremblay S, Chaubaud S, Bolduc S, Pouliot F. Exosomes induce fibroblast differentiation into cancer-associated fibroblasts through TGFβ signaling. Molec Cancer Res 2018; 16: 1196–204. [DOI] [PubMed] [Google Scholar]
- 47.Shimura T, Sasatani M, Kawai H, Kamiya K, Kobayashi J, Komatsu K, Kunugita. Radiation-induced myofibroblasts promote tumor growth via mitochondrial ROS-activated TGFβ signaling. Molec Cancer Res 2018; 16: 1676–86. [DOI] [PubMed] [Google Scholar]
- 48.Schug C, Kitzberger C, Sievert W, Spellerberg R, Tutter M, Schmohl KA, Eberlein B, Biedermann T, Steiger K, Zach C, Schwaiger M, Multhoff G, Wagner E, Nelson PJ, Spitzweg C. Radiation-induced amplification of the TGFB1-induced mesenchymal stem cell-mediated sodium iodide symporter (NIS) gene 131I therapy. Clin Cancer Res 2019; August 22. [DOI] [PubMed] [Google Scholar]
- 49.Liu YT, Xu L, Bennett L, Hooks JC, Liu J, Zhou Q, Liem P, Zheng Y, Skapek SX. Identification of de novo enhancers activated by TGFβ to drive expression of CDKN2A and B in HeLa cells. Molec Cancer Res 2019; 17: 1854–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yeung TL, Leung CS, Wong KK, Samimi G, Thompson MS, Liu J, Zaid TM, Ghosh S, Birrer MJ, Mok SC. TGF-b modulates ovarian cancer invasion by upregulating CAF-derived versican in the tumor microenvironment. Cancer Res 2013; 73: 5016–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol 2006; 7: 131–42. [DOI] [PubMed] [Google Scholar]
- 52.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell 2008; 14: 818–29. [DOI] [PubMed] [Google Scholar]
- 53.Thiery JP. Epithelial-mesenchymal transition in development and pathologies. Curr Opin Cell Biol 2003; 15: 740–46. [DOI] [PubMed] [Google Scholar]
- 54.Wheelock MJ, Johnson KR. Cadherins as modulators of cellular phenotype. Annu Rev Cell Dev Biol 2003; 19: 207–35. [DOI] [PubMed] [Google Scholar]
- 55.Yang Y, Yang HH, Tang B, Wu AML, Flanders KC, Moshkovich N, Weinberg DS, Welsh MA, Weng J, Ochoa H, Hu TY, Herrmann M, Chen J, Edmondson EF, Simpson RM, Liu F, Liu H, Lee MP, Wakefield LM. The outcome of TGF-β antagonism in metastatic breast cancer models in vivo reflects a complex balance between tumor-suppressive and pro-progression activities of TGF-β. Clin Cancer Res 2020; 26: 643–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim DH, Beckett JD, Nagpal V, Seman-Senderos MA, Gould RA, Creamer TJ, MacFarlane EG, Chen Y, Bedja D, Butcher JT, Mitzner W, Rouf R, Hata S, Warren DS, Dietz HC. Calpain 9 as a therapeutic target in TGFβ-induced mesenchymal transition and fibrosis. Sci Transl Med 2019; 11: eaau2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Selvarajah B, Azuelos I, Plate M, Guillotin D, Forty EJ, Contento G, Woodcock HV, Redding M, Taylor A, Brunori G, Durrenberger PF, Ronzoni R, Blanchard AD, Mercer PF, Anastasiou D, Chambers RC. mTORC1 amplifies the ATF4-dependent de novo serine-glycine pathway to supply glycine during TGF-b1-induced collagen biosynthesis. Sci Signal 2019; 12: eeav3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ikemori R, Gabasa M, Duch P, Vizoso M, Bragado P, Arshakyan M, Luis IC, Marin A, Moran S, Castro M, Fuster G, Gea-Sorli S, Jauset T, Soucek L, Montuenga LM, Esteller M, Monso E, Peinado VI, Gascon P, Fillat C, Hilberg F, Reguart N, Alcaraz J. Epigenetic SMAD3 repression in tumor-associated fibroblasts impairs fibrosis and response to the antifibrotic drug nintedanib in lung squamous cell carcinoma. Cancer Res 2020; 80: 276–90. [DOI] [PubMed] [Google Scholar]
- 59.Gonzalez-Gonzalez A, Munoz-Muela E, Marchal JA, Cara FE, Molina MP, Cruz-Lozano M, Jimenez G, Verma A, Ramirez A, Qian W, Chen W, Kozielski AJ, Elemento O, Martin-Salvago MD, Luque RJ, Rosa-Garrido C, Landeira D, Quintana-Romero M, Rosato RR, Garcia MA, Ramirez-Tortosa CL, Kim H, Rodriguez-Aguayo C, Lopez-Berestein G, Sood AK, Lorente JA, Sanchez-Rovira P, Chang JC, Granados-Principal S. Activating transcription factor 4 modulates TGFβ-induced aggressiveness in triple-negative breast cancer via SMAD2/3/4 and mTORC2 signaling. Clin Cancer Res 2018; 24: 5697–709. [DOI] [PubMed] [Google Scholar]
- 60.Grossberg AJ, Lei X, Xu T, Shaitelman SF, Hoffman KE, Bloom ES, Stauder MC, Tereffe W, Schlembach PJ, Woodward WA, Buchholz TA, Smith BD. Association of transforming growth factor β polymorphism C-509T with radiation-induced fibrosis among patients with early stage breast cancer. JAMA Oncol 2018; 4: 1751–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao X, Kwan JYY, Yip K, Liu PP, Liu FF. Targeting metabolic dysregulation for fibrosis therapy. Nature Rev Drug Discovery 2020; 19: 57–75. [DOI] [PubMed] [Google Scholar]
- 62.Varga J, Pasche B. Anti-TGF-β therapy in fibrosis: recent progress and implication for systemic sclerosis. Curr Opin Rheumatol 2008; 20: 720–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shin JY, Beckett JD, Bagirzadeh R, Creamer TJ, Shah AA, McMahan Z, Paik JJ, Sampedro MM, MacFarlane EG, Beer MA, Warren D, Wigley FM, Dietz HC. Epigenetic activation and memory at a TGFB2 enhancer in systemic sclerosis. Science Transl Med 2019; 11: eeaw0790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Batlle E, Sancho E, Francí C, Domínguez D, Monfar M, Baulida J, García De Herreros A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2000; 2: 84–9. [DOI] [PubMed] [Google Scholar]
- 65.Oft M, Akhurst RJ, Balmain A. Metastasis is driven by sequential elevation of H-ras and Smad2 levels. Nat Cell Biol 2002; 4: 487–94. [DOI] [PubMed] [Google Scholar]
- 66.Li T, Chubinskaya S, Esposito A, jin X, Tagiafierro L, Loeser R, Hakimiyan AR, Longobardi L, Ozkan H, Spagnoli A. TGF-β type 2 receptor-mediated modulation of the IL-36 family can be therapeutically targeted in osteoarthritis. Sci Transl Med 2019; 11: eaan2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lodyga M, Cambridge E, Karvonen HM, Pakshir P, Wu B, Boo S, Kiebalo R, Kaarteenaho R, Glogauer M, Kapoor M. Cadherin-11-mediated adhesion of macrophages to myofibroblasts establishes a profibrotic niche of active TGF-β. Sci Signal 2019; 12: eaao3469. [DOI] [PubMed] [Google Scholar]
- 68.Hedrick E, Mohankumar K, Safe S. TGFβ-induced lung cancer cell migration is NR4A1-dependent. Molec Cancer Res 2018; 16: 1991–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Katsuno Y, Meyer DS, Zhang Z, Shokat KM, Akhurst RJ, Miyazono K, Derynck. Chronic TGF-β exposure drives stabilized EMT, tumor stemness and cancer drug resistance with vulnerability to bitopic mTOR inhibition. Sci Signal 2019; 12: eaau8544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhu H, Gu X, Xia L, Zhou Y, Bouamar H, Yang J, Ding X, Zwieb C, Zhang J, Hinck AP, Sun LZ, Zhu X. A novel TGF-β trap blocks chemotherapeutics-induced TGF-β1 signaling and enhances their anticancer activity in gynecological cancers. Clin Cancer Res 2018; 24: 2780–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sippel TR, Johnson AM, Li HY, Hanson D, Nguyen TT, Bullock BL, Poczobutt JM, Kwak JW, Kleczko E, Weiser-Evans MCM, Nemenoff RA. Activation of PPARγ in Myeloid cells promotes progression of epithelial lung tumors through TGF-β1. Molec Cancer Res 2019; 17: 1748–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Friedman SL, Sheppard D, Duffield JS, Violette S. Therapy for fibrotic diseases: nearing the starting line. Sci Transl Med 2013; 5: issue 167 167sr1. [DOI] [PubMed] [Google Scholar]
- 73.Dong X, Zhao B, Iacob RE, Zhu J, Koksal AC, Lu C, Engen JR, Springer TA. Force interacts with macromolecular structure in activation of TGF-β. Nature 2017; 542: 55–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Garcia DA, Baek C, Estrada MV, Tysl T, Bennett EJ, Yang J, Chang JT. USP11 enhances TGFβ-induced epithelial-mesenchymal plasticity and human breast cancer metastasis. Molec Cancer Res 2018; 16: 1172–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu Q, Ma L, Jones T, Palomero L, Pujana MA, Martinez-Ruiz H, Ha P, Murnane J, Cuartas I, Seoane J, Baumann M, Linge A, Barcellos-Hoff MH. Subjugation of TGFβ signaling by human papilloma virus in head and neck squamous cell carcinoma shifts DNA repair from homologous recombination to alternative end-joining. Clin Cancer Res 2018; 24: 6001–14. [DOI] [PubMed] [Google Scholar]
- 76.Wang Y, Tan X, Tang Y, Zhang C, Xu J, Zhou J, Cheng X, Hou N, Liu W, Yang G, Teng Y, Yang X. Dysregulated TGFBR2/ERK-Smad4/SOX2 signaling promotes lung squamous cell carcinoma formation. Cancer Res 2019; 79:4466–79. [DOI] [PubMed] [Google Scholar]
- 77.Wang F, Xia X, Yang C, Shen J, Mai J, Kim HC, Kirui D, Kang Y, Fleming JB, Koay EJ, Mitra S, Ferrari M. Shen H. SMAD4 gene mutation renders pancreatic cancer resistance to radiotherapy through promotion of autophagy. Clin Cancer Res 2018; 24: 3176–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Budhu S, Schaer DA, Li Y, Toledo-Crow R, Panageas K, Yang X, Zhong H, Houghton AN, Silverstein SC, Merghoub T, Wolchok JD. Blockade of surface-bound TGF-β on regulatory T cells abrogates suppression of effector T cell function in the tumor microenvironment. Sci Signal 2017; 10: eaak9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Burga RA, Yvon E, Chorvinsky E, Fernandes R, Cruz CRY, Bollard CM. Engineering the TGFβ receptor to enhance the therapeutic potential of natural killer cells as an immunotherapy for neuroblastoma. Clin Cancer Res 2019; 25: 4400–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Golan T, Parikh R, Jacob E, Vaknine H, Zemser-Werner V, Hershkovitz D, Malcov H, Leibou S, Reichman H, Sheinboim D, Percik R, Amar S, Brenner R, Greenberger S, Kung A, Khaled M, Levy C. Adipocytes sensitize melanoma cells to environmental TGF-β cues by repressing the expression of miR-211. Sci Signaling 2019; 12: eaav6847. [DOI] [PubMed] [Google Scholar]
- 81.Janakiraman H, House RP, Gangaraju VK, Diehl JA, Howe PH, Palanisamy V. The long (LNCRNA) and short (miRNA) of it: TGFβ-mediated control of RNA-binding proteins and noncoding RNAs. Molec Cancer Res 2018; 16: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huang YH, Hu J, Chen F, Lecomte N, Basnet H, David CJ, Witkin MD, Allen PJ, Leach SD, Hollmann TJ, Iacobuzio-Donahue CA, Massague J. ID1 mediates escapes from TGF-β tumor suppression in pancreatic cancer. Cancer Discovery 2020; 10: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Alvarez MA, Freitas JP, Mazher-Hussain S, Glazer ES. TGF-β inhibitors in metastatic pancreatic ductal adenocarcinoma. J Gastrointest Cancer 2019; 50: 207–13. [DOI] [PubMed] [Google Scholar]
- 84.Wang P, Luo ML, Song E, Zhou Z, Ma T, Wang T, Wang J, Jia N, Wang G, Nie S, Liu Y, Hou FF. Long noncoding RNA Inc-TSI inhibits renal fibrogenesis by negatively regulating the TGF-β/Smad3 pathway. Sci Transl Med 2018; 10: eaat2039. [DOI] [PubMed] [Google Scholar]
- 85.Henderson NC, Arnold TD, Katamura Y, Giacomini MM, Rodriguez JD, McCarty JH, Pellicoro A, Raschperger E, Betsholtz C, Ruminski PG, Griggs DW, Prinsen MJ, Maher JJ, Iredale JP, Lacy-Hulbert A, Adams RH, Sheppard D. Targeting of αv integrin identified a core molecular pathway that regulates fibrosis in several organs. Nature Med 2013; 19: 1617–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu S, Gonzalez-Prieto R, Zhang M, Geurink PP, Kooij R, Iyengar PV, van Dinther M, Bos E, Zhang X, Le Devedec SE, van der Water B, Koning RI, Zhu HJ, Mesker WE, Vertegaal ACO, Ovaa H, Zhang L, Martens JWM, ten Dijke P. Deubiquitinase activity profiling identifies UCHL1 as a candidate oncoprotein that promotes TGFβ-induced breast cancer metastasis. Clin Cancer Res 2020; 26: 1460–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bird TG, Muller M, Boulter L, Vincent DF, Ridgway RA, Lopez-Guadamillas E, Lu WY, Jamieson T, Govaere O, Campbell AD, Ferreira-Gonzalez S, Cole AM, Hay T, Simpson KJ, Clark W, Hedley A, Clarke M, Gentaz P, Nixon C, Bryce S, Kiourtis C, Sprangers J, Nibbs RB, Van Rooijen N, Bartholin L, McGreal SR, Apte U, Barry ST, Iredale JP, Clarke AR, Serrano M, Roskams TA, Sansom OJ, Forbes SJ. TGFβ inhibition restores a regenerative response in acute liver injury by suppressing paracrine senescence. Sci Transl Med 2018; 10: eaan1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Foroutan M, Cursons J, Hediyeh-Zadeh S, Thompson EW, Davis MJ. A transcriptional program for detecting TGFβ-induced EMT in cancer. Molec Cancer Res 2017; 15: 619–31. [DOI] [PubMed] [Google Scholar]
- 89.Prunier C, Baker D, ten Dijke P, Ritsma L. TGF-β family signaling pathways in cellular dormancy. Trends Cancer 2019; 5: 66–78. [DOI] [PubMed] [Google Scholar]
- 90.Meirson T, Hava Gil-Henn H, Samson AO. Invasion and metastasis: the elusive hallmark of cancer. Oncogene 2020; 39: 2024–6. [DOI] [PubMed] [Google Scholar]
- 91.Teicher BA. Transforming growth factor-β and the immune response to malignant disease. Clin Cancer Res 2007; 13: 6247–51. [DOI] [PubMed] [Google Scholar]
- 92.Pinkas J, Teicher BA. TGF-β in cancer and as a therapeutic target. Biochem Pharmacol 2006; 72: 523–9. [DOI] [PubMed] [Google Scholar]
- 93.Cho JH, Park S, Oh AY, Kang S, Yoon MH, Woo TG, Hong SD, Hwang J, Ha NC, Lee HY, Park BJ. Loss of NF2 induces TGF-β receptor I mediated non-canonical and oncogenic TGF-β signaling implication of therapeutic effect of TGF-β receptor I inhibitor on NF2 syndrome. Molec Cancer Therap 2018; 17: 2271–84. [DOI] [PubMed] [Google Scholar]
- 94.Eser PO, Janne PA. TGFβ in NSCLC and treatment options. Pharmacol Therap 2018; 184: 112–30. [DOI] [PubMed] [Google Scholar]
- 95.Liu L, Zhou W, Cheng CT Ren X, Somlo G, Fong MY, Chin AR, Li H, Yu Y, Xu Y, Timothy S, O’Connor F, O’Connor T, Ann DK, Stark JM, Wang SE. TGFβ induces “BRCAness” and sensitivity to PARP inhibition in breast cancer by regulating DNA-repair genes. Molec Cancer Res 2014; 12: 1597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Principe DR, Park A, Torres C, Dorman MJ, McKinney R, Munshi HG, Grippo PJ, Rana A. TGFβ blockade augments PD-1 inhibition to promote T-cell mediated regression of pancreatic cancer. Mol Cancer Ther 2019; 18: 613–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Demaria O, Cornen S, Daeron M, Morel Y, Medzhitov R, Vivier E. Harnessing innate immunity in cancer therapy. Nature 2019; 574: 45–56. [DOI] [PubMed] [Google Scholar]
- 98.Mani V, Bromley SK, Aijo T, Mora-Buch R, Carrizosa E, Warner RD, Hamze M, Sen DR, Chasse A, Lorant A, Griffith JW, Rahimi RA, McEntee CP, Jeffrey KL, Marangoni F, Travis MA, Lacey-Hulbert A, Luster AD, Mempel TR. Migratory DCs activate TGF-β to precondition naïve CD8+T cells for tissue-resident memory fate. Science 2019; 366: eaav5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mariathasan S, Turley SJ, Nickles D, Castigioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, Jhunjhunwala S, Banchereau R, Yang Y, Guan Y, Chalouni C, Ziai J, Senbabaoglu Y, Santoro S, Sheinson D, Hung J, et al. TGFβ attenuates tumor response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018; 554: 544–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Principe DR, DeCant B, Mascarrinas E, Wayne EA, Diaz AM, Akagi N, Hwang R, Pasche B, Dawson DW, Fang D, Bentrem DJ, Munshi HG, Jung B, Grippo PJ. TGFβ signaling in the pancreatic tumor microenvironment promotes fibrosis and immune evasion to facilitate tumorigenesis. Cancer Res 2016; 76: 2525–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tas F, Karabulut S, Yasasever CT, Duranyildiz D. Serum transforming growth factor-beta 1 (TGF-β1) levels have diagnostic, predictive, and possible prognostic roles in patients with melanoma. Tumour Biol 2014; 35: 7233–7. [DOI] [PubMed] [Google Scholar]
- 102.Kong FM, Anscher MS, Murase T, Abbott BD, Iglehart JD, Jirtle RL. Elevated plasma transforming growth factor-β1 levels in breast cancer patients decrease after surgical removal of the tumor. Ann Surg 1995; 222: 155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Anscher MS, Kong FM, Jirtle RL. The relevance of transforming growth factor β1 in pulmonary injury after radiation therapy. Lung Cancer 1998; 19: 109–20. [DOI] [PubMed] [Google Scholar]
- 104.Grainger DJ, Mosedale DE, Metcalfe JC. TGF-β in blood: a complex problem. Cytokine Growth Factor Rev 2000; 11: 133–45. [DOI] [PubMed] [Google Scholar]
- 105.Teicher BA. Malignant cells, directors of the malignant process: role of transforming growth factor-beta. Cancer & Metastasis Reviews 2001; 20: 133–43. [DOI] [PubMed] [Google Scholar]
- 106.Teicher BA, Herman TS, Holden SA, Wang Y, Pfeffer MR, Crawford JM, Frei E III. Tumor resistance to alkylating agents conferred by mechanisms operative only in vivo. Science 1990; 247:1457–61. [DOI] [PubMed] [Google Scholar]
- 107.Huynh LK, Hipolito CJ, ten Dijke P. A perspective on the development of TGF-β inhibitors for cancer treatment. Biomolecules 2019; 9: 742–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Howe PH, Cunningham MR, Leof EB. Inhibition of mink lung epithelial cell proliferation by transforming growth factor-β is coupled through a pertussis-toxin-sensitive substrate. Biochem J 1990; 266: 537–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sawyer JS, Anderson BD, Beight DW, Campbell RM, Jones ML, Herron DK, Lampe JW, McCowan JR, McMillen WT, Mort N, Parsons S, Smith EC, Vieth M, Weir LC, Yan L, Zhang F, Yingling JM. Synthesis and activity of new aryl- and heteroaryl-substituted pyrazole inhibitors of the transforming growth factor-beta type I receptor kinase domain. J Med Chem 2003; 46: 3953–6. [DOI] [PubMed] [Google Scholar]
- 110.Sawyer JS, Beight DW, Britt KS, Anderson BD, Campbell RM, Goodson T Jr, Herron DK, Li HY, McMillen WT, Mort N, Parsons S, Smith ECR, Wagner JR, Yan L, Zhang F, Yingling JM. Synthesis and activity of new aryl- and heteroaryl-substituted 5,6-dihydro-4H-pyrrolo[1,2-β] pyrazole inhibitors of the transforming growth factor-beta type I receptor kinase domain. Bioorg Med Chem Lett 2004; 14:3581–4. [DOI] [PubMed] [Google Scholar]
- 111.Shiou SR, Datta PH, Dhawan P, Law BK, Yingling JM, Dixon DA, Beauchamp RD. Smad4-dependent regulation of urokinase plasminogen activator secretion and RNA stability associated with invasiveness by autocrine and paracrine transforming growth factor-β. J Biol Chem 2006; 281: 33971–81. [DOI] [PubMed] [Google Scholar]
- 112.Kakeji Y, Maehara Y, Ikebe M, Teicher BA. Dynamics of tumor oxygenation, CD31staining, and transforming growth factor-beta levels after treatment with radiation or cyclophosphamide in the rat 13762 mammary carcinoma. Int. J Radiat Oncol Biol Phys 1997; 37: 1115–23. [DOI] [PubMed] [Google Scholar]
- 113.Teicher BA, Kakeji Y, Ara G, Herbst RS, Northey D. Prostate carcinoma response to therapy: in vivo resistance. In Vivo 1997; 11: 453–62. [PubMed] [Google Scholar]
- 114.Hardee ME, Marciscano AE, Medina-Ramirez CM, Zagzag D, Narayana A, Lonning SM, Barcellos-Hoff MH. Resistance of glioblastoma-initiating cells to radiation mediated by the tumor microenvironment can be abolished by Inhibiting Transforming Growth Factor-β. Cancer Res 2012; 72: 4119–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bayin NS, Ma L, Thomas C, Baitalmal R, Sure A, Fansiwala K, Bustoros M, Golfinos JS, Pacione D, Snuderl M, Zagzag D, Barcellos-Hoff MH, Placantonakis D. Patient-specific screening using high-grade glioma explants to determine potential radiosensitization by a TGFβ small molecule inhibitor. Neoplasia 2016; 18: 795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Laping NJ, Grygielko E, Mathur A, Butter S, Bomberger J, Tweed C, Martin W, Fornwald J, Lehr R, Harling J, Gaster L, Callahan JF, Olson BA. Inhibition of transforming growth factor (TGF)β 1–induced extracellular matrix with a novel inhibitor of the TGFβ type I receptor kinase activity: SB-431542. Mol Pharmacol 2002; 62: 58–64. [DOI] [PubMed] [Google Scholar]
- 117.Inman GJ, Nicolas FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, Laping NJ, Hill CS. SB-431542 is a potent and specific inhibitor of transforming growth factor-β superfamily type I activin receptor like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol 2002; 62: 65–74. [DOI] [PubMed] [Google Scholar]
- 118.Matsuyama S, Iwadate M, Kondo M, Saitoh M, Hanyu A, Shimizu K, Aburatani H, Mishima HK, Imamura T, Miyazono K, Miyazawa K. SB-431542 and Gleevec inhibit transforming growth factor-β-induced proliferation of human osteosarcoma cells. Cancer Res 2003; 63: 7791–8. [PubMed] [Google Scholar]
- 119.Bagley RG, Weber W, Rouleau C, Teicher BA. Pericytes and endothelial precursor cells: cellular interactions and contributions to malignancy. Cancer Res 2005; 65: 9741–50. [DOI] [PubMed] [Google Scholar]
- 120.Watabe T, Nishihara A, Mishima K, Yamashita J, Shimizu J, Miyazawa K, Nishikawa SI, Miyazono K. TGF-β receptor kinase inhibitor enhances growth and integrity of embryonic stem cell–derived endothelial cells. J Cell Biol 163: 1303–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Stanslowsky N, Haase A, Martin U, Naujock M, Leffler A, Dengler R, Wegner F. Functional differentiation of midbrain neurons from human cord blood-derived induces pluripotent stem cells. Stem Cell Res Ther 2014; 5: 35–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang X, Yamamoto Y, Wilson LH, Zhang T, Howitt B, Farrow MA, Kern F, Ning G, Hong Y, Khor CC, Chevalier B, Bertrand D, Wu L, Nagarajan N, Sylvester FA, Hyams JS, Devers T, Bronson R, Lacy DB, Ho KY, Crum CP, McKeon F, Xia W. Cloning and variation of ground state intestinal stem cells. Nature 2015; 522: 173–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Heo I, Clevers H. Expanding intestinal stem cells in culture. Cell Res 2015; 25:995–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Giannelli G, Villa E, Lahn M. Transforming growth factor-β as a therapeutic target in hepatocellular carcinoma. Cancer Res 2014; 74: 1890–4. [DOI] [PubMed] [Google Scholar]
- 125.Yingling JM, McMillen WT, Yan L, Huang H, Sawyer JS, Graff J, Clawson DK, Britt KS, Anderson BD, Beight DW, Desaiah D, Lahn MM, Benhadji KA, Lallena MJ, Holmgaard RB, Xu X, Zhang F, Manro JR, Oversen PW, Iyer CV, Brekken RA, Kalos MD, Driscoll KE. Preclinical assessment of galunisertib (LY2157299) monohydrate), a first-in-class transforming growth factor-β receptor type I inhibitor. Oncotarget 2018; 9: 6659–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bueno L, de Alwis DP, Pitou C, Yingling J, Lahn M, Glatt S, Tonciniz IF. Semi-mechanistic modeling of tumor growth inhibitory effects of LY2157299, a new type I receptor TGF-β kinase antagonist, in mice. Eur J Cancer 2008; 44: 142–50. [DOI] [PubMed] [Google Scholar]
- 127.Zhou L, McMahon C, Bhagat T, Alencar C, Yu Y, Fazzari M, Sohal D, Heuck C, Gundabolu K, Ng C, Mo Y, Shen W, Wickrema A, Kong G, Friedman E, Sokol L, Mantzaris G, Pellagatti A, Boultwood J, Platanias LC, Steidl U, Yan L, Yingling JM, Lahn MM, List A, Bitzer M, Verma A. Reduced SMAD7 leads to overactivation of TGF-β signaling in MDS that can be reversed by a specific inhibitor of TGF-β receptor I kinase. Cancer Res 2011; 71: 955–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rodon J, Carducci MA, Sepulveda-Sanchez JM, Azaro A, Calvo E, Seoane J, Brana I, Sicart E, Gueorguieva I, Cleverly AL, Pillay NS, Desaiah D, Estrem ST, Paz-Ares L, Holdhoff M, Blakeley J, Lahn MM, Baselga J. First-in-human dose study of the novel transforming growth factor-β receptor I kinase inhibitor LY2157299 monohydrate in patients with advanced cancer and glioma. Clin Cancer Res 2014; 21: 553–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. NCT01582269: A Study in Recurrent Glioblastoma (GB).
- 130.Brandes AA, Carpentier AF, Kesari S, Sepulveda-Sanchez JM, Wheeler HR, Chinot O, Cher L, Steinbach JP, Capper D, Specenier P, Rodon J, Cleverly A, Smith C, Gueorguieva I, Miles C, Guba SC, Desaiah D, Lahn MM, Wick W. A Phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro Oncol 2016; 18: 1146–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kovacs RJ, Maldonado G, Analia Azaro A, Fernandez MS, Romero FL, Sepulveda-Sanchez JM, Corretti M, Carducci M, Dolan M, Gueorguieva I, Cleverly AL, Pillay NS, Baselga J, Lahn MM. Cardiac safety of TGFβ receptor I kinase inhibitor LY2157299 monohydrate in cancer patients in a first-in-human dose study. Cardiovasc Toxicol 2015; 15:309–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Herbertz S, Sawyer JS, Stauber AJ, Gueorguieva I, Driscoll KE, Estrem ST, Cleverly AL, Desaiah D, Guba SC, Benhadji KA, Slapak CA, Lahn MM. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Design Develop Therap 2015; 9: 4479–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tran HC, Wan Z, Sheard MA, Sun J, Jackson JR, Malvar J, Xu Y, Wang L, Sposto R, Kim ES, Asgharzadeh S, Seeger RC. TGFβR1 blockade with galunisertib (LY2157299) enhances anti-neuroblastoma activity of anti-GD2 dinutuximab (ch14.18) with natural killer cells. Clin Cancer Res 2017; 23: 804–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Otegbeye F, Ojo E, Moreton S, Mackowski N, Lee DA, de Lima M, Wald DN. Inhibiting TGF-beta signaling preserves the function of highly activated, in vitro expanded natural killer cells in AML and colon cancer models. Plos One 2018; 13: e0197008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Holmgaard RB, Schaer DA, Li Y, Castaneda SP, Murphy MY, Xu X, Inigo I, Dobkin J, Manro JR, Iversen PW, Surguladze D, Hall GE, Novosiadly RD, Benhadji KA, Plowman GD, Kalos M, Driscoll KE. Targeting the TGFβ pathway with galunisertib, a TGFβRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J Immunother Cancer 2018; 6: 47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Maier A, Peille AL, Vuaroqueaux V, Lahn M. Anti-tumor activity of the TGF-β receptor kinase inhibitor galunisertib (LY2157299 monohydrate) in patient-derived tumor xenografts. Cell Oncol 2015; 38: 131–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gueorguieva I, Tabernero J, Melisi D, Macarulla T, Merz V, Waterhouse TH, Miles C, Lahn MM, Cleverly A, Benhadji KA. Population pharmacokinetics and exposure-overall survival analysis of the transforming growth factor-β inhibitor galunisertib in patients with pancreatic cancer. Cancer Chemotherap Pharmacol 2019; 84:1003–15. [DOI] [PubMed] [Google Scholar]
- 138.Santini V, Valcarcel D, Platzbecker U, Komrokji RS, Cleverly AL, Lahn MM, Janssen J, Zhao Y, Chiang A, Giagounidis A, Guba SC, Gueorguieva I, Girvan AC, da Silva Ferreira M, Bhagat TD, Pradhan K, Steidl U, Sridharan A, Will B, Verma A. Phase II study of the ALK5 inhibitor Galunisertib in very low-, low-, and intermediate-risk myelodysplastic syndromes. Clin Cancer Res 2019; 25:6976–85. [DOI] [PubMed] [Google Scholar]
- 139.Jung SY, Yug JS, Clarke JM, Bauer TM, Keedy VL, Hwang S, Kim SJ, Chung EK, Lee JI. Population pharmacokinetics of vactosertib, a new TGF-β receptor type I inhibitor, in patients with advanced solid tumors. Cancer Chemothera Pharmacol 2020; 85:173–83. [DOI] [PubMed] [Google Scholar]
- 140.Keedy VL, Bauer TM, Clarke JM, Hurwitz H, Baek I, Ha I, Ock CY, Nam SY, Kim M, Park N, Kim JY, Kim SJ. Association of TGF-β responsive signature with anti-tumor effect of vactosertib, a potent, oral TGF-β receptor type I (TGFBRI) inhibitor in patients with advanced solid tumors. J Clin Oncol 2018; 36: Abstr. 3031.30199311 [Google Scholar]
- 141.Ojo E, Otegbeye F, Moreton S, Wald D. Activity of TGF-b inhibition combined with NK adoptive cell therapy. J Clin Oncol 2016; 34: Abstr e14520 [Google Scholar]
- 142. NCT02160106: First in Human Dose Escalation Study of Vactosertib (TEW-7197) in Subjects with Advanced Stage Solid Tumors.
- 143.Danielson KG, Baribault H, Holmes DF, Graham H, Kadler KE, Iozzo RV. Targeted disruption of decorin leads to abnormal collagen fibril morphology and skin fragility. J Cell Biol. 1997; 136:729–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Järveläinen H, Sainio A, Wight TN. Pivotal role for decorin in angiogenesis. Matrix Biol 2015; 43:15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Nastase MV, Janicova A, Roedig H, Hsieh LT, Wygrecka M, Schaefer L. Small Leucine-Rich Proteoglycans in Renal Inflammation: Two Sides of the Coin. J Histochem Cytochem 2018; 66: 261–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ahmed Z, Bansal D, Tizzard K, Surey S, Esmaeili M, Gonzalez AM, Berry M, Logan A. Decorin blocks scarring and cystic cavitation in acute and induces scar dissolution in chronic spinal cord wounds. Neurobiol Dis 2014; 64:163–76. [DOI] [PubMed] [Google Scholar]
- 147.Grisanti S, Szurman P, Warga M, Kaczmarek R, Ziemssen F, Tatar O, Bartz-Schmidt KU. Decorin modulates wound healing in experimental glaucoma filtration surgery: a pilot study. Invest Ophthalmol Vis Sci 2005; 46:191–6. [DOI] [PubMed] [Google Scholar]
- 148.Sainio AO, Järveläinen HT. Decorin-mediated oncosuppression - a potential future adjuvant therapy for human epithelial cancers. Br J Pharmacol 2019; 176: 5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Pang X, Dong N, Zheng Z. Decorin as a multivalent therapeutic agent against cancer. Front Pharmacol 2020; 10: 1649.32063855 [Google Scholar]
- 150.Pang X, Dong N, Zheng Z. Small leucine-rich proteoglycans in skin wound healing. Front Pharmacol 2020; 10: 1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Neill T, Schaefer L, Iozzo RV. Small Leucine-Rich Proteoglycans in Skin Wound Healing. Adv Drug Deliv Rev 2016; 97:174–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Orgel JP, Eid A, Antipova O, Bella J, Scott JE. Decorin core protein (decoron) shape complements collagen fibril surface structure and mediates its binding. PLoS One 2009; 4(9):e7028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Shintani K, Matsumine A, Kusuzaki K, Morikawa J, Matsubara T, Wakabayashi T, Araki K, Satonaka H, Wakabayashi H, Iino T, Uchida A. Decorin suppresses lung metastases of murine osteosarcoma. Oncol Rep 2008; 19:1533–9. [PubMed] [Google Scholar]
- 154.Teicher BA Maehara Y, Kakeji Y, Ara G, Keyes SR, Wong J, Herbst R. Reversal of in vivo drug resistance by the transforming growth factor-β inhibitor decorin. Int J Cancer 1997; 71: 49–58. [DOI] [PubMed] [Google Scholar]
- 155.Teicher BA, Ikebe M, Ara G, Keyes SR, Herbst RS. Transforming growth factor-beta 1 overexpression produces drug resistance in vivo: reversal by decorin. In Vivo 1997; 11: 463–72. [PubMed] [Google Scholar]
- 156.Teicher BA, Holden SA, Ara G, Chen G. Transforming growth factor-β in in vivo resistance. Cancer Chemother. Pharmacol 1996; 37(6): 601–609. [DOI] [PubMed] [Google Scholar]
- 157.Anscher MS, Thrasher B, Rabbani Z, Teicher BA, Vujaskovic Z. Anti-TGFβ antibody 1D11 ameliorates normal tissue damage caused by high-dose radiation. Int J Radiat Oncol Biol Phys 2006; 65: 876–81. [DOI] [PubMed] [Google Scholar]
- 158.Perry K, Wong L, Liu V, Park I, Zhang Q, Rejen V, Huang X, Smith ND, Jovanovic B, Lonning S, Teicher BA, Lee C. Treatment of transforming growth factor-beta-insensitive mouse renca tumor by transforming growth factor-beta elimination. Urology 2008; 72: 225–9. [DOI] [PubMed] [Google Scholar]
- 159.Tabe Y, Shi YX, Zeng Z, Jin L, Shikami M, Hatanaka Y, Miida T, Hsu FJ, Andreeff M, Konopleva M. TGF-β-neutralizing antibody 1D11 enhances cytarabine induced apoptosis in AML cells in the bone marrow microenvironment. Plos One 2013; 8: e62785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Grafe I, Yang T, Alexander S, Homan EP, Lietman C, Jiang MM, Bertin T, Munivez E, Chen Y, Dawson B, Ishikawa Y, Weis MA, Sampath TK, Ambrose C, Eyre D, Bachinger HP, Lee B. Excessive transforming growth factor-β signaling is a common mechanism in osteogenesis imperfecta. Nature Med 2014; 20: 670–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Rodriguez-Ruiz ME, Rodriguez I, Mayorga L, Labiano T, Barbes B, Etxeberria I, Pinz-Sarvise M, Azpilikueeta A, Bolanos E, Sanamed MF, Berraondo P, Calvo FA, Barcelos-Hoff MH, Perez-Garcia JL, Melero I. TGFβ blockade enhances radiotherapy abscopal efficacy effects in combination with anti-PD1 and anti-CD137 immunostimulatory monoclonal antibodies. Molec Cancer Therap 2019; 18: 621–31. [DOI] [PubMed] [Google Scholar]
- 162.Moulin A, Mathieu M, Lawrence C, Bigelow R, Levine M, Hamel C, Marquette JP, Le Parc J, Loux C, Ferrari P, Capdevila C, Dumas J, Dumas B, Rak A, Bird J, Qiu H, Pan CQ, Edmunds T, Wei RR. Structures of a pan-specific antagonist antibody complex to different isoforms of TGFβ reveal structural plasticity of antibody-antigen interactions. Protein Science 2014; 23: 1698–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Munnink THO, Arjaans MEA, Timmer-Bosscha H, Schroder CP, Hesselink JW, Vedelaar SR, Walenkamp AME, Reiss M, Gregory RC, Lub-de Hooge MN, de Vries EGE. PET with the 89Zr-labeled transforming growth factor-β antibody fresolimumab in tumor models. J Nucl Med 2001; 52: 2001–8. [DOI] [PubMed] [Google Scholar]
- 164.Lonning S, Mannick J, McPherson JM. Antibody Targeting of TGF-β in Cancer Patients. Curr Pharmac Biotech 2011; 12: 2176–89. [DOI] [PubMed] [Google Scholar]
- 165.Morris JC, Tan AR, Olencki TE, Shapiro GI, Dezube BJ, Reiss M, Hsu FJ, Berzofsky JA, Lawrence DP. Phase I study of GC-1008 (fresolimumab): a human anti-transforming growth factor-beta (TGFβ) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. Plos One 2014; 9: e90353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Den Hollander MW, Bensch F, Glaudemans AWJM, Munnink THO, Enting RH, den Dunnen WFA, Heesters MAAM, Kruyt FAE, Lub-de Hooge MN, de Groot JC, Pearlberg J, Gietema JA, de Vries EGE, Walenkamp AME. TGF-β antibody uptake in recurrent high-grade glioma imaged with 89Zr-fresolimumab PET. J Nucl Med 2015; 56:1310–4. [DOI] [PubMed] [Google Scholar]
- 167.Lacouture ME, Morris JC, Lawrence DP, Tan AR, Olencki TE, Shapiro GI, Dezube BJ, Bzofsky JA, Hsu FJ, Guitart J. Cutaneous keratoacanthomas/squamous cell carcinomas associated with neutralization of transforming growth factor β by the monoclonal antibody fresolimumab (GC1008). Cancer Immunol Immunother 2015; 64: 437–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Stevenson JP, Kindler HL, Papasavvas E, Sun J, Jacobs-Small M, Hull J, Schwed D, Ranganathan A, Newick K, Heitjan DF, Langer CJ, McPherson JM, Montaner LJ, Albelda SM. Immunological effects of the TGFβ-blocking antibody GC1008 in malignant pleural mesothelioma patients. OncoImmunol 2013; 2: 8 e26218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, Sevillano M, Ibiza S, Canellas A, Hernando-Momblona X, Byrom D, Matarin JA, Calon A, Rivas EI, Nebreda AR, Riera A, Attolini CSO, Batle E. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018; 554: 538–43. [DOI] [PubMed] [Google Scholar]
- 170.Ellermeier J, Wei J, Duewell P, Hoves S, Stieg MR, Adunka T, Noerenberg D, Anders HJ, Mayr D, Poeck H, Hartmann G, Endres S, Schnurr M. Therapeutic efficacy of bifunctional siRNA combining TGF-β1 silencing with RIG-I activation in pancreatic cancer. Cancer Res 2013; 73: 1709–20. [DOI] [PubMed] [Google Scholar]
- 171. NCT01401062: Fresolimumab and radiotherapy in metastatic breast cancer.
- 172.Formenti SC, Lee P, Adams S, Goldberg JD, Li X, Xie MW, Ratikan JA, Felix C, Hwang L, Faull KF, Sayre JW, Hurvitz S, Glaspy JA, Comin-Anduix B, Demaria S, Schaue D, McBride WH. Focal irradiation and systemic transforming growth factor β blockade in metastatic breast cancer. Clin Cancer Res 2018; 24: 2493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Vanpouille-Box C, Diamond JM, Pilones KA, Zavadil J, Babb JS, Formenti SC, Barcellos-Hoff MH, Demaria S. TGFβ is a master regulator of radiation therapy-induced anti-tumor immunity. Cancer Res 2015; 75: 2232–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Formenti SC, Hawtin RE, Dixit N, Evensen E, Lee P, Goldberg JD, Li X, Vanpouille-Box C, Schaue D, McBride WH, Demaria S. Baseline T cell dysfunction by single cell network profiling in metastatic breast cancer patients. J ImmunoTherap Cancer 2019; 7: 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lan Y, Zhang D, Xu C, Hance KW, Marelli B, Qi J, Yu H, Qin G, Sircar A, Hernandez VM, Jenkins MH, Fontana RE, Deshpande A, Locke G, Sabzevari H, Radvanyi L, Lo KM. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGFβ. Sci Transl Med 2018; 10: eaan5488. [DOI] [PubMed] [Google Scholar]
- 176. NCT02517398: MSB0011359C (M7824) in Metastatic or Locally Advanced Solid Tumors.
- 177.Strauss J, Heery CR, Schlom J, Madan RA, Cao L, Kang Z, Lamping E, Marte JL, Dona hue RN, Grenga I, Cordes L, Christensen O, Mahnke L, Helwig C, Gulley JL. Phase I trial of M7824 (MSB0011359C), a bifunctional fusion protein targeting PD-L1 and TGFβ, in advanced solid tumors. Clin Cancer Res 2018; 24: 1287–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumors with combination immunotherapies. Nat Rev Drug Discov 2019; 18: 197–218. [DOI] [PubMed] [Google Scholar]
- 179.Martin CJ, Datta A, Littlefield C, Kalra A, Chapron C, Wawersik S, Dagbay KB, Brueckner CT, Nikiforov A, Danehy FT Jr., Streich FC Jr., Boston C, Simpson A, Jackson JW, Lin S, Danek N, Faucette RR, Raman P, Capili AD, Buckler A, Carven GJ, Schürpf T. Selective inhibition of TGFβ1 activation overcomes primary resistance to checkpoint blockade therapy by altering tumor immune landscape. Sci Transl Med 2020; 12: eaay8456. [DOI] [PubMed] [Google Scholar]
- 180.Kang YK, Bang YJ, Kondo S, Chung HC, Muro K, Dussault I, Helwig C, Osada M, Doi T. Safety and tolerability of bintrafusp alfa, a bifunctional fusion protein targeting TGF-β and PD-L1, in Asian patients with pretreated recurrent or refractory gastric cancer. Clin Cancer Res 2020; 26: 3202–10. [DOI] [PubMed] [Google Scholar]
- 181.Smalley Rumfield C, Pellom ST, Morillon II YM, Schlom J, Jochems C. Immunomodulation to enhance the efficacy of an HPV therapeutic vaccine. J ImmunoThera Cancer 2020; 8: e000612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. NCT03831438: Safety and tolerability study of AVID200 in patients with diffuse cutaneous systemic sclerosis.
- 183.Papachristodoulou A, Silginer M, Weller M, Schneider H, Hasenbach K, Janicot M, Roth P. Therapeutic targeting of TGF-β ligands in glioblastoma using novel antisense oligonucleotides reduces the growth of experimental gliomas. Clin Cancer Res 2019; 25: 7189–201. [DOI] [PubMed] [Google Scholar]
- 184.Zhou J, Zhao Y, Simonenko V, Xu JJ, Liu K, Wang D, Shi J, Zhong T, Zhang L, Zeng L, Huang B, Tang S, Lu AY, Mixson AJ, Sun Y, Patrick Y. Lu PY, Li Q. Simultaneous silencing of TGF-β1 and COX-2 reduces human skin hypertrophic scar through activation of fibroblast apoptosis. Oncotarget 2017; 8: 80651–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Molyneaux M, Xu J, Evans DM, Lu P. Effect on tumor growth by TGF-β1/COX-2 siRNA combination product (STP705) in a human cholangiocarcinoma (HuCCT-1) xenograft tumor model in nude mice. J Clin Oncol 2019; 37 suppl: Abstr 14652. [Google Scholar]
- 186. NCT04293679: Open Label, Dose Escalation Study for the Safety and Efficacy of STP705 in Adult Patients With isSCC.