

Abstract
Receptor Interacting Protein Kinase 1 (RIPK1) and RIPK3 are key adaptors that play critical roles in inflammatory and cell death signaling. Work in recent years have shown that their activities are tightly regulated by ubiquitination, phosphorylation and proteolysis. In addition to these post-translational modifications, the expression and activities of these kinases can further be tuned by environmental changes in pH and oxygen content. Proper control of these regulatory processes is crucial for the RIP kinases to execute their functions in immune responses and tissue homeostasis. In this review, we discuss recent advance in our understanding of the molecular mechanisms that regulate the activities of the RIP kinases. We will also discuss how the different regulatory mechanisms contribute to the functions of RIPK1 and RIPK3 in different pathophysiological settings.
Keywords: Ubiquitination, phosphorylation, Caspase, Inflammation, Apoptosis, Necroptosis
1. Introduction
Cell death is an important strategy employed by multicellular organisms to maintain tissue homeostasis and to defend against pathogen challenge. Members of the Receptor Interacting Protein Kinases (RIPKs) family are key regulators of cell death and inflammation. Recent studies showed that RIPK1 is a pleiotropic cell death adaptor that not only control apoptosis and necroptosis but also directly regulate inflammatory cytokine expression. In contrast to apoptosis, which is generally non-inflammatory and tolerogenic, lytic cell death such as necroptosis promotes inflammation. Necroptosis and other types of lytic cell death such as pyroptosis and ferroptosis are marked by cell swelling and rapid loss of plasma membrane integrity. Membrane rupture leads to the release of cellular contents such as IL1α, HMGB1, uric acid, ATP and DNA into the extracellular space. Because these cellular components are potent stimulants for inflammation, they have been referred to as “damage-associated molecular patterns” (DAMPs). Because of the prominent roles of RIPK1 and its associate kinase RIPK3 in cell death and inflammation, extensive work has been done in recent years to decipher the molecular mechanisms that regulate their activities. These regulatory processes are diverse in nature (e.g. post-translational modifications versus protein-protein interactions). Collectively, they tune the functions of these kinases to maintain tissue homeostasis and to promote functional differentiation of immune cells. As such, disease ensues when these regulatory processes are disrupted. Here, we will review recent findings on the mechanisms that regulate RIPK1 activities.
2. The activity of RIPK1 is controlled by different post-translational mechanisms
2.1. A ubiquitin scaffold restricts RIPK1 death-inducing activity
The biochemical events that regulate RIPK1 activity is best studied in TNFR1 signaling. TNF binding to its cognate receptor TNFR1 induces formation of a membrane signaling complex termed complex I [1]. RIPK1 is recruited to this complex via death domain (DD)-DD interaction with the cytoplasmic tail of the receptor. RIPK1 is heavily ubiquitinated in this complex. In addition to RIPK1, another DD-containing adaptor TRADD also binds to TNFR1. TRADD recruitment to complex I promotes RIPK1 ubiquitination through recruitment of the adaptors TRAF2, TRAF5 and the E3 ubiquitin ligases cIAP1 and cIAP2, which ubiquitinates RIPK1 and other adaptors in the complex. Hence, in Tradd−/− cells, recruitment and ubiquitination of RIPK1 to TNFR1 was severely impaired [2, 3] (Fig. 1). This led to the popular view that the ubiquitin scaffold on RIPK1 stabilizes complex I.
Figure 1. Regulation of RIPK1 activity by ubiquitination and phosphorylation.
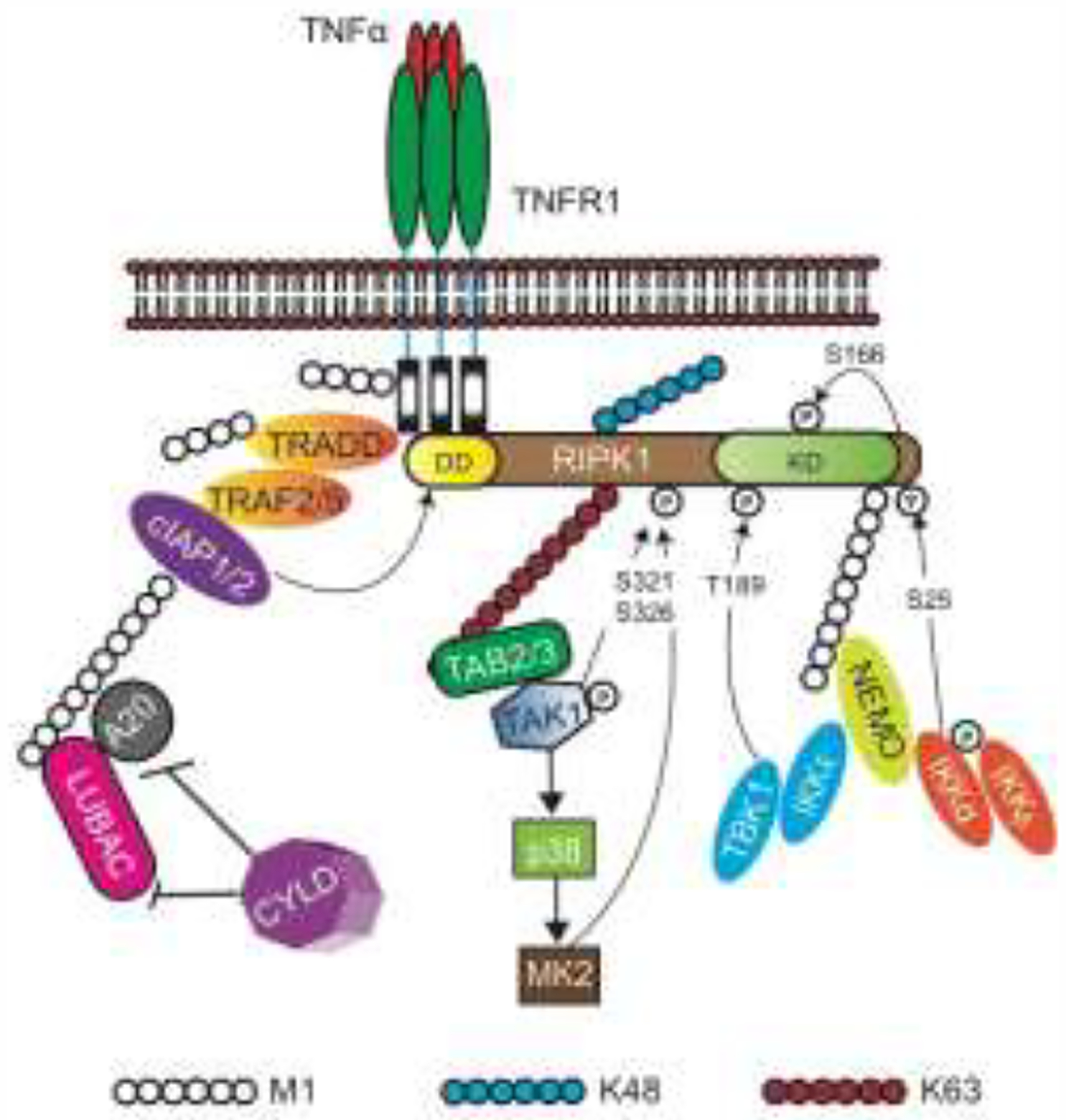
RIPK1 and TRADD bind to TNFR1 via DD interaction. TRADD subsequently recruits TRAF2/5 and cIAP1/2, which initiates the assembly of the ubiquitin scaffold of complex. Numerous components of complex I are modified by ubiquitination. In addition to cIAP1/2 and LUBAC, additional ubiquitinating and de-ubiquitinating enzymes such as cylindromatosis (CYLD) and A20 have also been implicated in regulating RIPK1 ubiquitination [66–71] (Table 1). Key kinases that phosphorylate RIPK1 at distinct sites are highlighted. cIAP, cellular inhibitor of apoptosis protein; IKK, inhibitor of NF-κB kinase; NEMO, NF-κB essential modulator NF-κB, nuclear factor kappa; RIPK, receptor-interacting protein kinase; TAB, TAK1-binding protein; TAK1, TGFβ activated kinase 1; TRADD, TNF-associated death domain; KD, Kinase domain.
The key E3 ligases that regulate RIPK1 ubiquitination are cIAP1, cIAP2 and the linear ubiquitin assembly complex (LUBAC). The IAPs were once thought to promote predominantly K63-linked RIPK1 ubiquitination. It is now clear that RIPK1 is modified by other types of ubiquitin linkages [4]. For example, using a cIAP1 mutant that is defective in the ubiquitin-associated (UBA) domain, it was shown that cIAP1 could stimulate K48-linked ubiquitination of RIPK1 [5]. Besides stabilizing the TNFR1 complex, early studies suggest that ubiquitinated RIPK1 also imposes steric hindrance to prevent transition of the membrane-bound complex I to the death-inducing complex II in the cytosol [6]. Consistent with the notion that cIAP-mediated RIPK1 ubiquitination inhibits cell death, combined deficiency of cIAP1 and cIAP2 or XIAP led to embryonic lethality that could be delayed by single allele loss of Ripk1 [7] (Table 1). Similar rescue of cell death and inflammation by Ripk1 heterozygous loss was also observed in mice with skin epidermal deletion of cIAP1 and cIAP2 [8].
Table 1:
E3 ligase and kinase deficiencies that affect RIPK1 activity
| Genetic deficiency | Viability | Phenotype | Biochemical Effect on RIPK1 | Genetic resuce by RIPK1 deficiency | Reference |
|---|---|---|---|---|---|
| Ciap1−/−Ciap2−/− | lethality at ~E10.5 | Cardiovascular defect | Inhibition of RIPK1 ubiquitination | single Ripkl-null allele extends survival till ~E14.5 | 7 |
| Ciap1−/−Xiap−/− | lethality at ~E10.5 | Cardiovascular defect | Reduction of RIPK1 ubiquitination | single Ripk1-null allele extends survival till weaning | 7 |
| Sharpin−/− | Viable | dermatitis, multi-organ inflammation | Impaired S25 phosphorylation and ubiquitination. Increased RIPK1 death-inducing activity. | full rescue by kinase inactive Ripk1K45A/K45A |
11, 14, 21 |
| Hoip−/− | lethality at ~E10.5 | Defective yolk sac vascularization, Excessive Endothelial Cell Death | Enhanced formation of RIPK1 death-inducing complex | survival extended to ~E14.5 by Rjpk1K45AJK45A | 11, 12 |
| Hoil1−/− | lethality at ~E10.5 | Disrupted vascular architecture and cell death in the yolk sac endothelium | Enhanced formation of RIPK1 death-inducing complex | survival extended to ~E14.5 by kinase inactive Ripk1K45A/K45A | 11 |
| Cyld−/− | Viable | Grossly normal | Increased length of RIPK1 ubiquitin chains. Inhibition of RIPK1 death-inducing activity. | N.D. | 66, 67, 71 |
| A20−/− | Die after 1 week | Runting, multi-organ inflammation | Enhanced RIPK1 activity and cell death | N.D. | 68, 69, 70 |
| Tbk1−/− | lethality at E13.5 - E14.5 | Severe liver degeneration | Loss of inhibitory phosphorylation at T189 (T190 for mouse RIPK1) | full rescue by kinase inactive Ripk1D138N/D138N |
18 |
| Mapkapk2 (MK2)−/− | Viable | No overt phenotype | Blockade of inhibitory phosphorylation at S321 and S336. Enhanced RIPK1 death-inducing activity | N.D. | 34, 35, 36 |
| Ikkα/β−/− | embryonic lethality | liver damage | Prevents inhibitory phosphorylation of RIPK1 at S25. Increased RIPK1 death-inducing activity. | N.D. | 16, 21 |
In contrast to the IAPs, LUBAC exclusively modify RIPK1 via M1-linked linear ubiquitination [9, 10]. LUBAC is composed of the catalytic subunit HOIP, HOIL-1 and the regulatory subunit SHARPIN. Disruption of any of the subunits of LUBAC causes hypersensitivity to TNF-induced cell death [11, 12]. Similar to the cIAP1/cIAP2 co-deletion model, inhibition of RIPK1 kinase activity modestly delayed embryonic lethality of Hoip−/− and Hoil1−/− mice [11, 13]. Inhibition of RIPK1 kinase activity was also insufficient to reverse skin inflammation in mice with skin epithelium specific deletion of Hoip or Hoil1 (HoipE-KO and Hoil1E-KO) [12]. Strikingly, RIPK1 kinase inhibition did significantly inhibit lethal skin inflammation in Hoil1E-KO mice when TNFR1 was also deleted [12]. These results indicate that LUBAC-mediated ubiquitination did regulate RIPK1 kinase activity, although this effect is likely mediated by receptors other than TNFR1.
In contrast to the Hoip−/− and Hoil1−/− mice, the chronic proliferative dermatitis (cpdm) mice, which contain a spontaneous mutation that leads to frame-shift and premature non-sense mutation of the Sharpin gene, are born alive but suffer from dermatitis and other immune inflammatory manifestations. Deletion of Tnf or genetic inactivation of RIPK1 kinase activity via the kinase inactive Ripk1K45A allele fully rescued the inflammatory disease [8, 9, 14] (Table 1). This is in stark contrast to mice lacking Hoip or Hoil1, whose inflammatory phenotypes were not restored by inhibition of RIPK1 kinase activity [11, 12]. Could the discrepant RIPK1 phenotypes in the different LUBAC mutant mice be caused by residual E3 ligase activity in cpdm mice? Regardless of the mechanism, these results do support the notion that the ubiquitin scaffold of complex I has a critical role in restricting the death-inducing activity of RIPK1 (Fig. 1). As we shall discuss later, a possible mechanism by which LUBAC inhibits the pro-death activity of RIPK1 is through the recruitment of kinases such as TBK1 and IKK [15–21].
Mass spectrometry revealed that many sites on RIPK1 were modified by ubiquitination [10]. Experimental evidence indicates that K377 of human RIPK1 (K376 in mouse RIPK1) is particularly important in controlling RIPK1 activity [22]. For example, reconstitution of K377RRIPK1 mutant in RIPK1-deficient cells led to strong induction of cell death [23]. Moreover, mice expressing the K376R-RIPK1 mutant were hypersensitive to TNF-induced cytotoxicity and died during embryogenesis around E13.5 [24, 25]. Consistent with an important role for the ubiquitin scaffold in recruiting inhibitory kinases to complex I, the K376R mutation appears to restrict inhibitory phosphorylation of RIPK1 by the kinase TAK1 [17, 25].
The concept that disruption of RIPK1 post-translational modification may be a pathogen sensing mechanism is illustrated in enteric bacterial infection. The LUBAC-generated ubiquitin scaffold has a critical function in “trapping” and restricting Salmonella Typhimurium growth [26]. This protective mechanism is disrupted by Shigella flexneri, which encodes two E3 ligases, IpaH1.4 and IpaH2.5, that directly binds to HOIL1 and triggers HOIP K48-linked ubiquitination and proteasomal degradation [27]. Disturbance of the LUBAC ubiquitin scaffold thus inhibits NF-κB activation. It is tempting to speculate that disruption of this ubiquitin scaffold will trigger RIPK1 activation and cell death (Fig. 2).
Figure 2. Pathogen targeting of RIPK1.
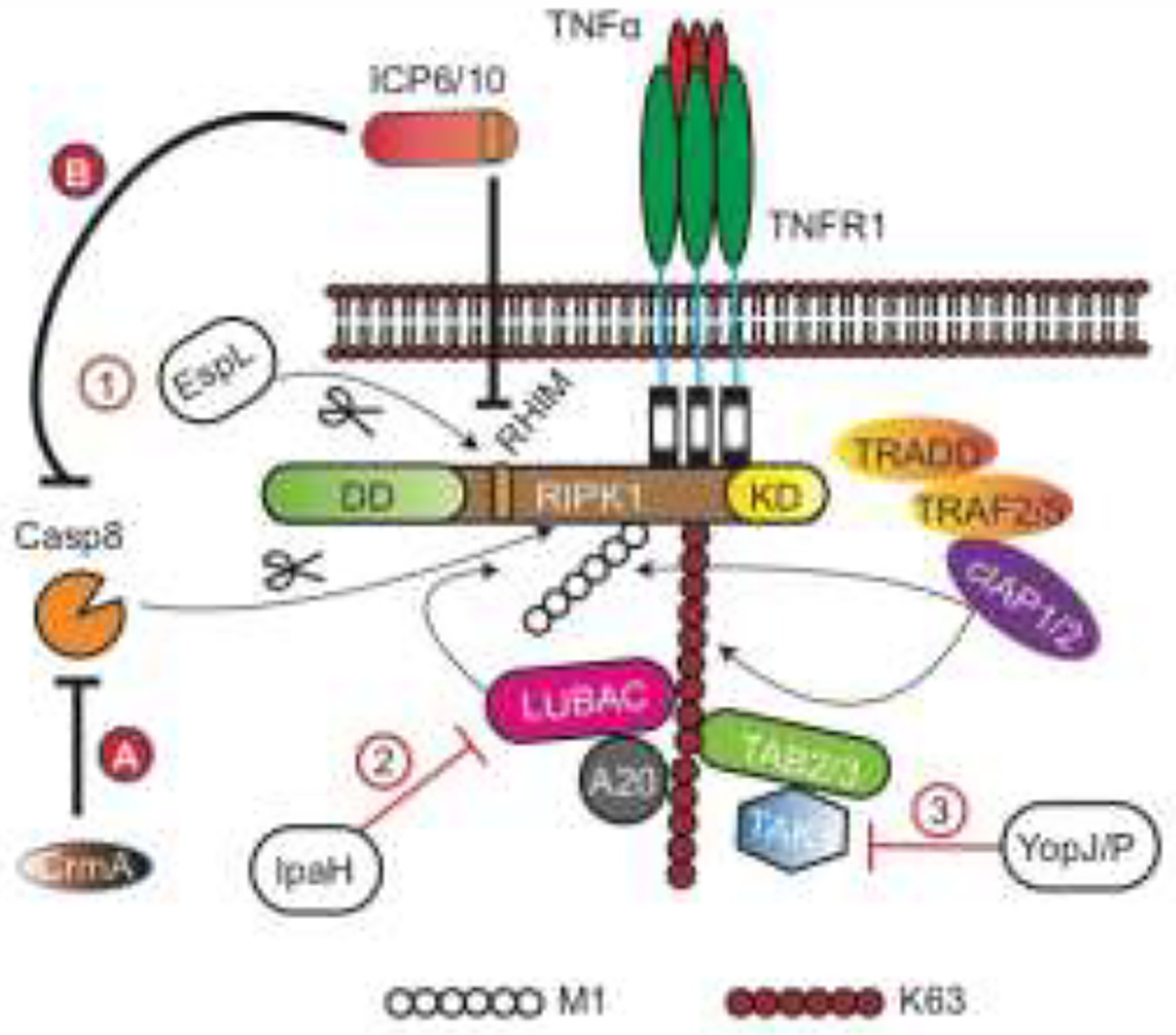
Several classes of bacterial effectors target different regulatory machineries that impinge on RIPK1 activity. For example, the T3SS effector EspL cleaves RIPK1 at the boundary of the RHIM, the Shigella flexneri E3 ligases IpaH1.4 and IpaH2.5 trigger proteasomal degradation of components of LUBAC, and the Yersinia effector YopJ/P inhibits TAK1. Bacterial interference of RIPK1 activity can either inhibit (e.g. EspL) or enhance (e.g. IpaH and YopJ/P) RIPK1-mediated inflammation and cell death. Orthopoxviruses encode CrmA or related inhibitors that block caspase 8, which indirectly inhibits RIPK1 cleavage to sensitize cells to necroptosis. On the other hand, the HSV1 and HSV2 inhibitors ICP6 and ICP10 exhibit dual activity against caspase 8 and the RHIM to block apoptosis and necroptosis. DD: Death domain, KD: kinase domain.
2.2. RIPK1 phosphorylation and pathogen sensing
Early mass spectrometry revealed multiple phosphorylation sites on RIPK1 [28]. The majority of these phosphorylation events do not impinge on RIPK1 activity [23, 29, 30]. However, several phospho-acceptor sites have been identified that either stimulate or inhibit RIPK1 activity. For example, phosphorylation of S166 in the kinase domain of RIPK1 has been widely used as a marker RIPK1 activation [31]. S166 phosphorylation was not detected in cells expressing kinase inactive RIPK1, indicating that this is likely an auto-phosphorylation event. S166 phosphorylation is not specific for cell death induction. In contrast, S161 phosphorylation has also been implicated to cause RIPK1 auto-activation and to promote interaction with RIPK3 during necroptosis [29]. However, the S161A-RIPK1 mutant had little effects on TNF-induced necroptosis [23]. Thus, further work is needed to accurately define the role of these phosphorylation events in regulating RIPK1 activity.
As we have alluded to in the previous section, RIPK1 ubiquitination and phosphorylation are intimately linked events. Many kinases that regulate RIPK1 activity are recruited to the ubiquitin scaffold of complex I. For example, NEMO and TAK1 binding to ubiquitinated RIPK1 brings the IKKα and IKKβ to the proximity of TAK1 to facilitate TAK1-mediated phosphorylation and activation of the IKKs. The IKKs phosphorylate RIPK1 at S25 in the kinase domain, leading to inhibition of its death-inducing activity (Fig. 1) [21]. Interestingly, this inhibitory switch appears to have a critical role in limiting bacterial pathogenesis. The Yersinia effector YopJ/P inhibits TAK1 and IKK activity to unleash RIPK1, which in turn stimulates caspase 8-dependent cleavage of the pore-forming gasdermin D (GSDMD) and cell death by pyroptosis [19, 32, 33] (Fig. 2). Mice that express the S25D-RIPK1 mutant were as defective as mice expressing the kinase inactive K45A-RIPK1 mutant in Yersinia-induced macrophage death and the control of bacterial replication [21] (Table 2). Hence, S25 phosphorylation is a key early event that restricts the death-inducing potential of RIPK1.
Table 2: Mutations of RIPK1 and the associated phenotypes.
(N/A = Not applicable
| Genetic deficiency | Viability | Phenotype | Effect on RIPK1 | Genetic Rescue |
Reference | |
|---|---|---|---|---|---|---|
| Mutations in Mice | Ripk1K45A/K45A | Viable | No overt defects | Loss of RIPK1 kinase activity | N/A | 15 |
| Ripk1D138N/D138N | Viable | No overt defects | Loss of RIPK1 kinase activity | N/A | 16 | |
| Ripk1K376R/K376R | lethality at ~E13.5 | Excessive cell death and severe inflammation | Disrupt RIPK1 ubiquitination | Co-deletion of Fadd and Ripk3 or Fadd and Mlkl | 23, 24 | |
| Ripk1S25D/S25D | Viable | No overt defects | Mimics IKK inhibitory phosphorylation | N/A | 20 | |
| Ripk1S321D/S321D | Not reported | Not reported | Mimics MK2 inhibitory phosphorylation | N/A | 33 | |
| Ripk1D325A/D325A | lethality at ~E10.5 | endocardial damage | Inhibit Caspase 8 cleavage of RIPK1 | Co-deletion of Fadd and Mlkl | 39, 41, 42 | |
| Ripk1RHIM/RHIM | P0 | Dermatitis, multi-organ inflammation | RIPK3(D161N), RHIM mutant RIPK3, RIPK3 deficiency, MLKL deficiency or Loss of ZBP1 | Deletion of Zbp1, Ripk3 or Mlkl | 30, 56 | |
| Human mutations | Heterozygous D324N, D324H and D324Y mutations in human | Viable | periodic fever, recurring lymph adenopathy | Inhibit Caspase 8 cleavage of RIPK1 | N/A | 39, 40 |
| Homozygous mutations of RIPK1 (I615T, T645M, Y426*, C601Y) | Viable | Primary immunodeficiency and/or Intestinal inflammation | Substantial reduction of RIPK1 protein expression | N/A | 43, 44 | |
| Biallelic mutations (frame-shift, deletion of exons or alternative splicing) | Viable | recurrent infections, intestinal inflammation and polyarthritis | Complete loss of RIPK1 protein expression | N/A | 43, 44, 45 |
In addition to TAK1 and the IKKs, the MAPKK MK2 also inhibits RIPK1 cytotoxic function via direct phosphorylation. However, unlike the IKKs, MK2 phosphorylates RIPK1 at S321/S336 away from the kinase domain [34–36]. This suggests that MK2 may indirectly regulate RIPK1 kinase and death-inducing activity via allosteric effects. TAK1 has also been shown to directly phosphorylate RIPK1 at S321 [17]. Similar to the IKKs, MK2 is also a target of inhibition by the Yersinia pestis effector YopJ/P (Fig. 2) [36]. Thus, RIPK1 may sit at the center of a regulatory hub that senses different perturbations by bacterial pathogens (Fig. 2).
TBK1 is another kinase that has recently joined the rank of RIPK1 inhibitory kinases. As in the case of the other inhibitory kinases, TBK1 recruitment to complex I was also facilitated by the LUBAC-generated ubiquitin scaffold via the adaptors TANK1 and NEMO (Fig. 1) [15]. Tbk1−/− mice suffer from embryonic lethality. Surprisingly, this lethality could be fully rescued by the RIPK1 kinase inactive allele D138N [18]. TBK1 and its associated kinase IKKε phosphorylate human RIPK1 at T189 (T190 in mouse RIPK1) [15, 18]. Loss of TBK1 function has been associated with neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS) or frontotemporal dementia (FTD) [37]. Interestingly, TAK1 expression is reduced in the aging brain [18]. When combined with heterozygous loss of Tbk1, this reduced TAK1 expression drives phenotypes that resemble human ALS/FTD. Remarkably, inhibition of RIPK1 kinase activity reversed these disease manifestations [18]. Thus, inhibitory RIPK1 phosphorylation events are not only relevant in pathogen defense, but also have critical roles in neuroinflammation.
2.3. Proteolytic processing – Lessons from human mutations
RIPK1 has long been known to be a substrate of caspase 8 [38, 39]. For the last decade, the dominant view has been that caspase 8 cleaves and inactivates RIPK1 to blunt necroptosis. However, this viewpoint has recently been challenged with the discovery of heterozygous human mutations that specifically inhibit RIPK1 cleavage. In these patients, the essential aspartate residue (D324) in the tetra-peptide caspase 8 cleavage site LQLD was substituted with another amino acid residue. Despite having a normal wild type Ripk1 allele, these patients suffer from early onset of periodic fever syndrome and lymphadenopathy marked by elevated inflammatory cytokine expression [40, 41]. This condition, which have been termed “cleavage-resistant RIPK1-induced autoinflammatory syndrome” (CRIA) [40], was recapitulated in mice that express the corresponding cleavage-resistant D325A-RIPK1 [40, 42]. Cells from the CRIA patients produced increased inflammatory cytokines in response to LPS and were hypersensitive to TNF-induced apoptosis and necroptosis. Unexpectedly, mice that are homozygous for the D325A mutation died during embryogenesis on E10.5, which was rescued by co-deletion of Ripk3 and Caspase8 [42, 43]. Collectively, these results indicate that RIPK1 cleavage by Caspase 8 not only inhibits necroptosis, but it also has critical roles in restraining the cell death and pro-inflammatory functions of RIPK1 (Fig. 3).
Figure 3. Cleavage and interaction with other RHIM adaptors prevent inadvertent RIPK1 activation.
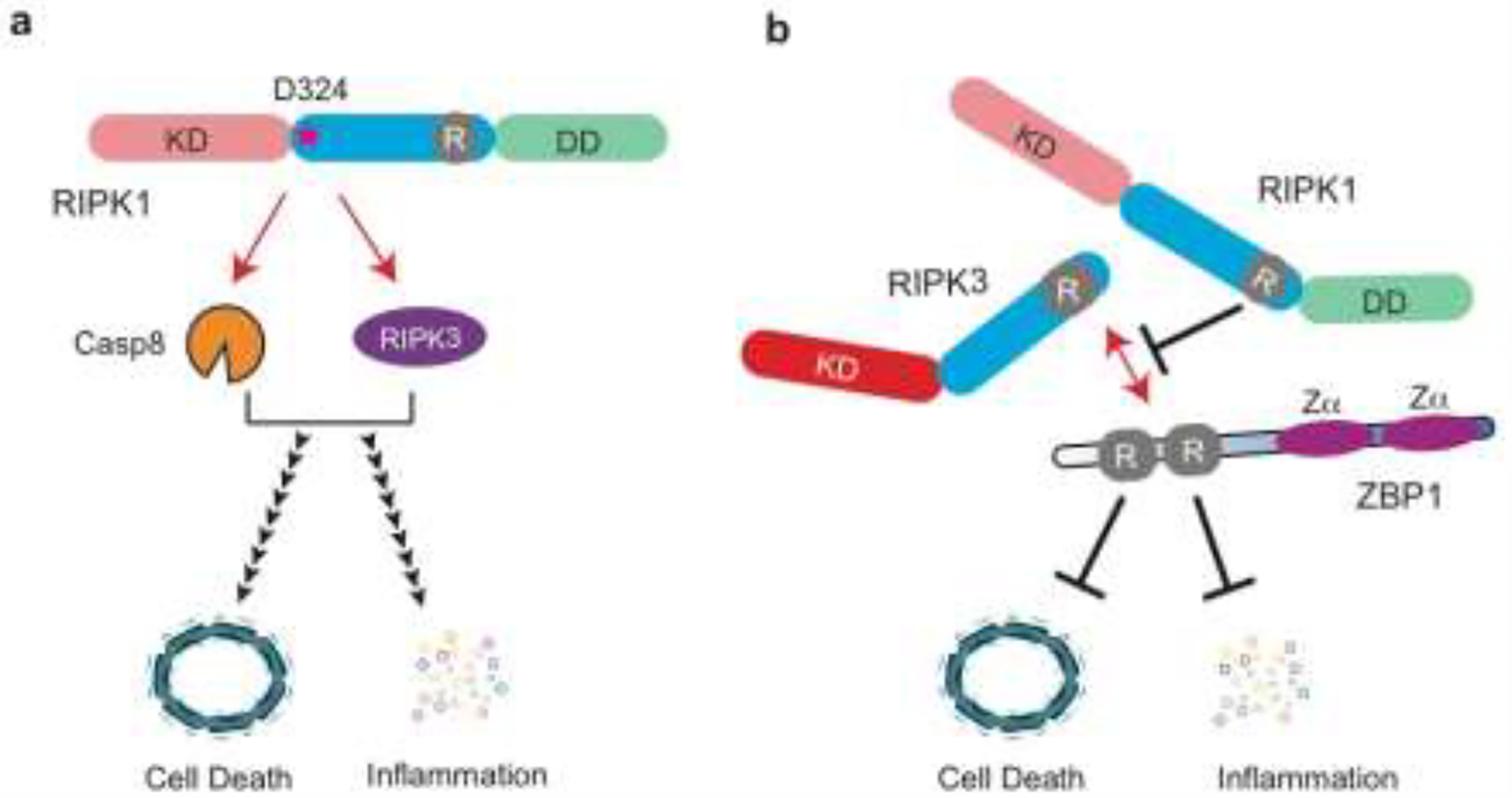
(a) Caspase 8 (Casp8) cleaves RIPK1 at D324. Heterozygous mutations at this cleavage site causes a spontaneous inflammatory disease in human. The non-cleavable RIPK1 unleashes Caspase 8 and RIPK3, which in turn stimulate cell death and elevated inflammatory cytokine production. The red stars represent mutation in D324. KD: kinase domain, R: RHIM, DD: death domain. (b) Genetic evidence indicates that RIPK1 sequesters ZBP1 and/or RIPK3 via the RHIM during perinatal development. This functional interaction is also critical for prevention of cell death and inflammation, and maintenance of homeostasis of the skin epithelium. When the RHIM of RIPK1 is disrupted by genetic manipulation, excessive ZBP1-RIPK3-mediated necroptosis occurs, leading to inflammation and lethality. Zα: zDNA-binding domain.
In addition to the cleavage-resistant mutations, homozygous mutations in Ripk1 that led to frameshift, alternative splicing, deletion of exons, or single amino acid substitutions have been identified. In these patients, RIPK1 protein expression was severely reduced or even completely lost [44–46]. Intriguingly, some of the mutations were located near the C-terminus of the RIPK1 protein, suggesting that reduced protein or mRNA stability might have contributed to the loss of RIPK1 protein expression. These patients are immunodeficient yet develop inflammatory bowel disease and polyarthritis [44–46] (Table 2). Thus, human RIPK1 mutations revealed the paradoxical nature of RIPK1 as both an inducer and an inhibitor of cell death and inflammation.
Many large DNA viruses encode caspase 8 inhibitors and thus indirectly restrict cleavage of RIPK1 (Fig. 2). The classic example being the Cytokine response modifier A (CrmA) from Cowpox virus, which inhibits caspase 1 and caspase 8 activity [47] (Fig. 2). The related orthopoxvirus vaccinia virus encode the CrmA ortholog B13R or Spi2, which inhibits death receptor-induced apoptosis but sensitizes cells to TNF-induced anti-viral necroptosis [48–52]. However, it is noteworthy that other large DNA viruses that encode caspase 8 inhibitors also encode inhibitors that block necroptosis. These include the herpes simplex virus 1 (HSV1) ICP6 and HSV2 ICP10 (Fig. 2). Interestingly, several recent studies revealed that enteropathogenic and enterohaemorrhagic bacteria Type 3 secretion system (T3SS) encodes EspL, an atypical cysteine protease that cleaves RIPK1 at the C-terminus of the RHIM [53]. EspL also cleaves other mammalian RHIM-containing adaptors in a similar fashion (Fig. 2). As a result, necroptosis and inflammatory cytokine signaling were blunted [53]. Since another T3SS effector NleB1 has been shown to covalently modify and inhibit the caspase 8 adaptor FADD [54, 55], EspL inhibition of the RHIM-containing adaptors ensures that there is no switch from apoptosis to necroptosis. This behavior is eerily similar to that of certain herpesviruses, which also simultaneously inhibit apoptosis and necroptosis [56]. The fact that bacterial and viral pathogens have both developed similar strategies to target the same cell death programs is a strong argument that RIPK1 and its related adaptors are key sentinels in host defense.
3. Non-PTM mechanisms that regulate RIPK1 activity
3.1. RHIM-mediated sequestration of RIPK1
The RHIM is defined by a highly conserved tetra-peptide core (e.g. IQIG or VQVG) flanked by mostly hydrophobic residues. The four mammalian RHIM-containing adaptors, RIPK1, RIPK3, the toll-like receptor 3 (TLR3)/TLR4 adaptor TRIF and the nucleic acid sensing adaptor ZBP1, are all critical signal adaptors for innate inflammation and cell death. In necroptosis, RIPK3 can be activated through RHIM-mediated binding with any of the other RHIM adaptors. This has led to the notion that RHIM-mediated interaction predominantly drives cell death (Fig. 3a). However, this dogma has recently been challenged. Mice that express a RIPK1 mutant with alanine substitutions in the RHIM core sequence (Ripk1mRHIM) died perinatally [31, 57] (Table 2). This was a surprising observation since mice expressing kinase inactive RIPK1 were viable [14, 49]. RIPK3 phosphorylation, which indicates activation of the kinase, was abundantly detected in different tissues of the Ripk1mRHIM mice. Moreover, a complex between RIPK3 and ZBP1 was detected in Ripk1mRHIM-expressing cells. Deletion of Zbp1, Ripk3 or Mlkl rescued the perinatal lethality of the Ripk1mRHIM mice [31, 57]. Thus, genetic evidence implicates that the RHIM of RIPK1 normally restricts ZBP1 and RIPK3 to prevent necroptosis during perinatal life. Although biochemical interaction between RIPK1 and ZBP1 or RIPK3 has not been detected, these results support the notion that RIPK1 has a kinase-independent but scaffold-dependent function in cell survival (Fig. 3b).
3.2. Other regulatory mechanisms of RIPK1 activity
HSP90 is a molecular chaperone for RIPK1 [58]. Inhibition of HSP90 with geldanamycin causes rapid degradation of RIPK1 and resistance to necroptosis [39]. In addition to RIPK1, recent results indicate that HSP90 also binds to the necroptosis adaptors RIPK3 and MLKL to promote their stability and necroptosis induction [59–62]. Hence, the effect of drugs that target HSP90 likely affect necroptosis at multiple levels. Besides regulation through protein-protein interaction and post-translational modifications, RIPK1 activity can also be regulated at a transcriptional level. For example, Ripk1 transcription was found to be inhibited under hypoxic conditions [63]. Furthermore, we recently found that RIPK1 activity is tunable in response to changes in the pH environment. At acidic pH, RIPK1 kinase activity and RIPK1-dependent apoptosis and necroptosis were both reduced [64]. Direct protonation of specific histidine residues is a common regulatory mechanism for pH-sensing channel proteins. As such, RIPK1 may be regulated by pH changes in a similar fashion. Environmental changes in pH can be a rapid and efficient way to tune RIPK1 activity during infection or in hypoxic tumor sites.
4. Concluding remarks
Our understanding of RIPK1 biology has undergone a major paradigm shift in recent years. It is now clear that RIPK1 is the fulcrum that balances cell survival and cell death signals. We argue that the different inhibitory mechanisms on RIPK1 have evolved to sense pathogen challenges. RIPK1 and the other RHIM signal adaptors share critical functions in innate immunity. Immune cells such as macrophages are often targeted by invading pathogens. Thus, interference of these sensor mechanisms will directly or indirectly impinge on RIPK1. Cell death in this scenario may be desirable as it will help to limit the bacterial or viral factory. However, these defense mechanisms do come with a price at times, such as that seen in the human CRIA patients. Time will tell if our increased understanding of these regulatory maneuvers will translate to successful pharmacologic targeting of RIPK1 in various inflammatory diseases [65].
Acknowledgement
We thank the Chan Lab members for discussion. This work is supported by NIH grant AI 119030.
Abbreviations:
- RHIM
RIP homotypic interaction motif
- LUBAC
linear ubiquitin assembly complex
- ZBP1
zDNA binding protein 1
- PTM
post-translation modification
- DD
death domain
- KD
kinase domain
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Micheau O and Tschopp J, Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell, 2003. 114(2): p. 181–90. [DOI] [PubMed] [Google Scholar]
- 2.Pobezinskaya YL, et al. , The function of TRADD in signaling through tumor necrosis factor receptor 1 and TRIF-dependent Toll-like receptors. Nat Immunol, 2008. 9(9): p. 1047–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ermolaeva MA, et al. , Function of TRADD in tumor necrosis factor receptor 1 signaling and in TRIF-dependent inflammatory responses. Nat Immunol, 2008. 9(9): p. 1037–46. [DOI] [PubMed] [Google Scholar]
- 4.Witt A and Vucic D, Diverse ubiquitin linkages regulate RIP kinases-mediated inflammatory and cell death signaling. Cell Death Differ, 2017. 24(7): p. 1160–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Annibaldi A, et al. , Ubiquitin-Mediated Regulation of RIPK1 Kinase Activity Independent of IKK and MK2. Mol Cell, 2018. 69(4): p. 566–580.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Donnell MA, et al. , NEMO inhibits programmed necrosis in an NFkappaB-independent manner by restraining RIP1. PLoS One, 2012. 7(7): p. e41238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moulin M, et al. , IAPs limit activation of RIP kinases by TNF receptor 1 during development. Embo j, 2012. 31(7): p. 1679–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anderton H, et al. , Inhibitor of Apoptosis Proteins (IAPs) Limit RIPK1-Mediated Skin Inflammation. J Invest Dermatol, 2017. 137(11): p. 2371–2379. [DOI] [PubMed] [Google Scholar]
- 9.Gerlach B, et al. , Linear ubiquitination prevents inflammation and regulates immune signalling. Nature, 2011. 471(7340): p. 591–6. [DOI] [PubMed] [Google Scholar]
- 10.de Almagro MC, et al. , Cellular IAP proteins and LUBAC differentially regulate necrosome-associated RIP1 ubiquitination. Cell Death Dis, 2015. 6: p. e1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peltzer N, et al. , LUBAC is essential for embryogenesis by preventing cell death and enabling haematopoiesis. Nature, 2018. 557(7703): p. 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taraborrelli L, et al. , LUBAC prevents lethal dermatitis by inhibiting cell death induced by TNF, TRAIL and CD95L. Nat Commun, 2018. 9(1): p. 3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peltzer N, et al. , HOIP deficiency causes embryonic lethality by aberrant TNFR1-mediated endothelial cell death. Cell Rep, 2014. 9(1): p. 153–165. [DOI] [PubMed] [Google Scholar]
- 14.Berger SB, et al. , Cutting Edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J Immunol, 2014. 192(12): p. 5476–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lafont E, et al. , TBK1 and IKKepsilon prevent TNF-induced cell death by RIPK1 phosphorylation. Nat Cell Biol, 2018. 20(12): p. 1389–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dondelinger Y, et al. , NF-kappaB-Independent Role of IKKalpha/IKKbeta in Preventing RIPK1 Kinase-Dependent Apoptotic and Necroptotic Cell Death during TNF Signaling. Mol Cell, 2015. 60(1): p. 63–76. [DOI] [PubMed] [Google Scholar]
- 17.Geng J, et al. , Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat Commun, 2017. 8(1): p. 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu D, et al. , TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging. Cell, 2018. 174(6): p. 1477–1491.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orning P, et al. , Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science, 2018. 362(6418): p. 1064–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Webb LV, et al. , Survival of Single Positive Thymocytes Depends upon Developmental Control of RIPK1 Kinase Signaling by the IKK Complex Independent of NF-kappaB. Immunity, 2019. 50(2): p. 348–361.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dondelinger Y, et al. , Serine 25 phosphorylation inhibits RIPK1 kinase-dependent cell death in models of infection and inflammation. Nat Commun, 2019. 10(1): p. 1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ea CK, et al. , Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell, 2006. 22(2): p. 245–57. [DOI] [PubMed] [Google Scholar]
- 23.McQuade T, Cho Y, and Chan FK, Positive and negative phosphorylation regulates RIP1-and RIP3-induced programmed necrosis. Biochem J, 2013. 456(3): p. 409–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang X, et al. , Ubiquitination of RIPK1 suppresses programmed cell death by regulating RIPK1 kinase activation during embryogenesis. Nat Commun, 2019. 10(1): p. 4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang Y, et al. , K63-linked ubiquitination regulates RIPK1 kinase activity to prevent cell death during embryogenesis and inflammation. Nat Commun, 2019. 10(1): p. 4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noad J, et al. , LUBAC-synthesized linear ubiquitin chains restrict cytosol-invading bacteria by activating autophagy and NF-kappaB. Nat Microbiol, 2017. 2: p. 17063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Jong MF, et al. , Shigella flexneri suppresses NF-κB activation by inhibiting linear ubiquitin chain ligation. Nature microbiology, 2016. 1(7): p. 16084–16084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Degterev A, et al. , Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol, 2008. 4(5): p. 313–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, et al. , RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun, 2017. 8: p. 14329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Delanghe T, Dondelinger Y, and Bertrand MJM, RIPK1 Kinase-Dependent Death: A Symphony of Phosphorylation Events. Trends Cell Biol, 2020. [DOI] [PubMed] [Google Scholar]
- 31.Newton K, et al. , RIPK1 inhibits ZBP1-driven necroptosis during development. Nature, 2016. 540(7631): p. 129–133. [DOI] [PubMed] [Google Scholar]
- 32.Peterson LW, et al. , RIPK1-dependent apoptosis bypasses pathogen blockade of innate signaling to promote immune defense. J Exp Med, 2017. 214(11): p. 3171–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sarhan J, et al. , Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc Natl Acad Sci U S A, 2018. 115(46): p. E10888–e10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jaco I, et al. , MK2 Phosphorylates RIPK1 to Prevent TNF-Induced Cell Death. Mol Cell, 2017. 66(5): p. 698–710.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dondelinger Y, et al. , MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death. Nat Cell Biol, 2017. 19(10): p. 1237–1247. [DOI] [PubMed] [Google Scholar]
- 36.Menon MB, et al. , p38(MAPK)/MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection. Nat Cell Biol, 2017. 19(10): p. 1248–1259. [DOI] [PubMed] [Google Scholar]
- 37.Abramzon YA, et al. , The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front Neurosci, 2020. 14: p. 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin Y, et al. , Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev, 1999. 13(19): p. 2514–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chan FK, et al. , A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem, 2003. 278(51): p. 51613–21. [DOI] [PubMed] [Google Scholar]
- 40.Lalaoui N, et al. , Mutations that prevent caspase cleavage of RIPK1 cause autoinflammatory disease. Nature, 2020. 577(7788): p. 103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tao P, et al. , A dominant autoinflammatory disease caused by non-cleavable variants of RIPK1. Nature, 2020. 577(7788): p. 109–114. [DOI] [PubMed] [Google Scholar]
- 42.Newton K, et al. , Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature, 2019. 574(7778): p. 428–431. [DOI] [PubMed] [Google Scholar]
- 43.Zhang X, Dowling JP, and Zhang J, RIPK1 can mediate apoptosis in addition to necroptosis during embryonic development. Cell Death Dis, 2019. 10(3): p. 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Y, et al. , Human RIPK1 deficiency causes combined immunodeficiency and inflammatory bowel diseases. Proc Natl Acad Sci U S A, 2019. 116(3): p. 970–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Uchiyama Y, et al. , Primary immunodeficiency with chronic enteropathy and developmental delay in a boy arising from a novel homozygous RIPK1 variant. J Hum Genet, 2019. 64(9): p. 955–960. [DOI] [PubMed] [Google Scholar]
- 46.Cuchet-Lourenço D, et al. , Biallelic RIPK1 mutations in humans cause severe immunodeficiency, arthritis, and intestinal inflammation. Science, 2018. 361(6404): p. 810–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou Q, et al. , Target protease specificity of the viral serpin CrmA. Analysis of five caspases. J Biol Chem, 1997. 272(12): p. 7797–800. [DOI] [PubMed] [Google Scholar]
- 48.Cho YS, et al. , Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell, 2009. 137(6): p. 1112–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Polykratis A, et al. , Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J Immunol, 2014. 193(4): p. 1539–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guo H, et al. , Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe, 2015. 17(2): p. 243–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang Z, et al. , RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe, 2015. 17(2): p. 229–42. [DOI] [PubMed] [Google Scholar]
- 52.Yu X, et al. , Herpes Simplex Virus 1 (HSV-1) and HSV-2 Mediate Species-Specific Modulations of Programmed Necrosis through the Viral Ribonucleotide Reductase Large Subunit R1. J Virol, 2016. 90(2): p. 1088–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pearson JS, et al. , EspL is a bacterial cysteine protease effector that cleaves RHIM proteins to block necroptosis and inflammation. Nat Microbiol, 2017. 2: p. 16258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li S, et al. , Pathogen blocks host death receptor signalling by arginine GlcNAcylation of death domains. Nature, 2013. 501(7466): p. 242–6. [DOI] [PubMed] [Google Scholar]
- 55.Pearson JS, et al. , A type III effector antagonizes death receptor signalling during bacterial gut infection. Nature, 2013. 501(7466): p. 247–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nailwal H and Chan FK, Necroptosis in anti-viral inflammation. Cell Death Differ, 2019. 26(1): p. 4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin J, et al. , RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature, 2016. 540(7631): p. 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lewis J, et al. , Disruption of hsp90 function results in degradation of the death domain kinase, receptor-interacting protein (RIP), and blockage of tumor necrosis factor-induced nuclear factor-kappaB activation. J Biol Chem, 2000. 275(14): p. 10519–26. [DOI] [PubMed] [Google Scholar]
- 59.Li D, et al. , A cytosolic heat shock protein 90 and cochaperone CDC37 complex is required for RIP3 activation during necroptosis. Proc Natl Acad Sci U S A, 2015. 112(16): p. 5017–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Park SY, et al. , Heat shock protein 90 inhibitor regulates necroptotic cell death via down-regulation of receptor interacting proteins. Pharmazie, 2015. 70(3): p. 193–8. [PubMed] [Google Scholar]
- 61.Zhao XM, et al. , Hsp90 modulates the stability of MLKL and is required for TNF-induced necroptosis. Cell Death Dis, 2016. 7: p. e2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jacobsen AV, et al. , HSP90 activity is required for MLKL oligomerisation and membrane translocation and the induction of necroptotic cell death. Cell Death Dis, 2016. 7: p. e2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moriwaki K, et al. , Differential roles of RIPK1 and RIPK3 in TNF-induced necroptosis and chemotherapeutic agent-induced cell death. Cell Death Dis, 2015. 6: p. e1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moriwaki K, Balaji S, and Ka-Ming Chan F, The death-inducing activity of RIPK1 is regulated by the pH environment. Sci Signal, 2020. 13(631). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weisel K, et al. , Randomized clinical study of safety, pharmacokinetics, and pharmacodynamics of RIPK1 inhibitor GSK2982772 in healthy volunteers. Pharmacol Res Perspect, 2017. 5(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Elliott PR, et al. , SPATA2 Links CYLD to LUBAC, Activates CYLD, and Controls LUBAC Signaling. Mol Cell, 2016. 63(6): p. 990–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wei R, et al. , SPATA2 regulates the activation of RIPK1 by modulating linear ubiquitination. Genes Dev, 2017. 31(11): p. 1162–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Priem D, et al. , A20 protects cells from TNF-induced apoptosis through linear ubiquitin-dependent and -independent mechanisms. Cell Death Dis, 2019. 10(10): p. 692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dziedzic SA, et al. , ABIN-1 regulates RIPK1 activation by linking Met1 ubiquitylation with Lys63 deubiquitylation in TNF-RSC. Nat Cell Biol, 2018. 20(1): p. 58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wertz IE, et al. , De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature, 2004. 430(7000): p. 694–9. [DOI] [PubMed] [Google Scholar]
- 71.Moquin DM, McQuade T, and Chan FK, CYLD deubiquitinates RIP1 in the TNFalpha-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS One, 2013. 8(10): p. e76841. [DOI] [PMC free article] [PubMed] [Google Scholar]