

1 |. CASE PRESENTATION
A 34-year-old primigravida woman, a communication coordinator for a state farm bureau, was at 13 weeks’ gestation of an uncomplicated pregnancy (in vitro fertilization) when she awoke with left eye vision loss. Her blood pressure was 180/125. She was afebrile and did not appear ill. She reported no other symptoms, no previous health problems or hospitalizations. Her only medication was a prenatal vitamin. Physical examination was normal except for hypertension and serous retinal detachments in her left eye, related to her severe hypertension. Her hemoglobin was 132 g/L, platelet count 55 × 109L (she had no previous platelet counts), creatinine 103.4 μmol/L, total bilirubin 12 μmol/L, haptoglobin 630 mg/L, lac-tate dehydrogenase (LDH) 5.54 μkat/L (normal range, 1.67–4.18 μkat/L), urine protein > 3000 mg/L. Very few schistocytes were seen on her peripheral blood smear. Brain magnetic resonance imaging (MRI) demonstrated a recent small infarction in her right parietal lobe. The fetus was normal (fetal heart rate and ultrasound). Antihypertensive treatment was begun.
Her presentation had all of the characteristics of preeclampsia with severe features, an obstetric emergency. However, preeclampsia very rarely occurs before 20 weeks’ gestation. When symptoms of preeclampsia with severe features occur before 20 weeks’ gestation, alternative diagnoses, such as thrombotic thrombocytopenic purpura (TTP), must be considered.1
The hematology consultant considered TTP and requested ADAMTS13 activity measurement. However TTP did not explain her sudden severe hypertension. Also, she did not have the characteristic laboratory features of TTP: she was not anemic, she had no evidence of hemolysis, and her thrombocytopenia was not severe. The PLASMIC score was five, indicating only a 5%−24% probability of TTP.2 Plasma exchange treatment for TTP was begun because there was no apparent alternative diagnosis.
The hematology consultant had focused on acquired, autoimmune TTP, which is much more common than hereditary TTP in adults. The incidence of acute episodes of acquired TTP is approximately 3/106 population/year.3 The estimated prevalence of hereditary is only 1/106 population.4 However, when TTP initially presents during pregnancy, hereditary TTP may be almost as frequent as acquired TTP5 because severe complications of pregnancy occur in almost all women with hereditary TTP.4 Also, hereditary TTP may not have the characteristic clinical features of acquired TTP. For example, stroke may occur in patients with hereditary TTP who have neither anemia nor thrombocytopenia.4
Her blood pressure was controlled, her vision improved and her platelet count increased. On day 4, ADAMTS13 activity was reported as 6% (normal, ≥61%) with no functional inhibitor. From additional history, we learned that our patient had no previous signs of hereditary TTP. She had never been told of problems at the time of her birth that required transfusion or a prolonged hospital stay. She had not had symptoms of thrombocytopenia. There was no was no family history of thrombocytopenia, TTP, or kidney disease. Her father takes medicine for hypertension. Her mother and two sisters are healthy. Neither sister has been pregnant.
These data together with her presenting features suggested a diagnosis of hereditary TTP. However, our experience is that a functional ADAMTS13 inhibitor was not identified in 17% of patients with their initial episode of acquired TTP.4 Therefore confirmation of the diagnosis of hereditary TTP requires documentation of ADAMTS13 mutations.
After 3 days, plasma exchange was replaced by plasma infusion, the effective treatment for hereditary TTP. This change was considered to be safe because her platelet count had increased to 97 × 109/L and her LDH had returned to normal ( 2.42 ukat/L). She continued to improve and was discharged on day 7; her platelet count was 201 × 109/L. We subsequently learned that her blood pressure had been 130/90 at her routine prenatal appointment 2 weeks before admission, and that high blood pressures (138–182/90–119) had been documented four times during the previous 2 years. However, there had been no diagnosis of hypertension and she had received no treatment; high blood pressures were assumed to be caused by anxiety and apprehension.
It was fortunate that we did not know of her previous severe hypertension at the time of her hospitalization. If we had known, we may have assumed that this was an exacerbation of chronic hypertension and that severe hypertension alone could have caused her presenting clinical features.6
During the next 9 weeks she continued plasma prophylaxis, 750 mL (3 units, 12.5 mL/kg body weight) every 2 weeks. Her blood pressures (on labetalol) and platelet counts remained normal. At 17 weeks’ gestation, she had a normal fetal ultrasound. At 22 weeks’ gestation, fetal demise was discovered on a routine obstetric evaluation, 4 days following a plasma infusion. The following day, at a routine hematology evaluation, she described fatigue. Blood pressure was 178/100, hemoglobin 51 g/L, platelet count 20 × 109 L, creatinine 129 umol/L, LDH 24.65 ukat/L. She was readmitted to the hospital. Plasma exchange was begun. Her dead fetus was delivered on day 3. She was discharged 4 days later.
These data were consistent with an exacerbation of her hereditary TTP. Plasma exchange (rather than plasma infusion) was begun, because of the severity of her exacerbation and because ADAMTS13 sequencing to confirm the diagnosis of hereditary TTP had not yet been done.
Examination of her placenta with routine hematoxylin & eosin staining demonstrated the diagnostic features of severe maternal vascular malperfusion, including placental hypoplasia (weight, 75 g, <third percentile for 22 weeks’ gestation), decidual arteriopathy with intravascular thrombi and fibrinoid necrosis, multiple infarctions, diffuse distal villous hypoplasia, and increased perivillous fibrin deposition (Figure 1A). These features are characteristic of preeclampsia with severe features; they also occur in women with chronic hypertension.7 Because the thrombi had no hyalinization, they were recent (within several days), caused by her TTP exacerbation. Confocal microscopy images of immunostained placental sections demonstrated that the thrombi were primarily composed of von Willebrand factor (VWF) (Figure 1B).
FIGURE 1.
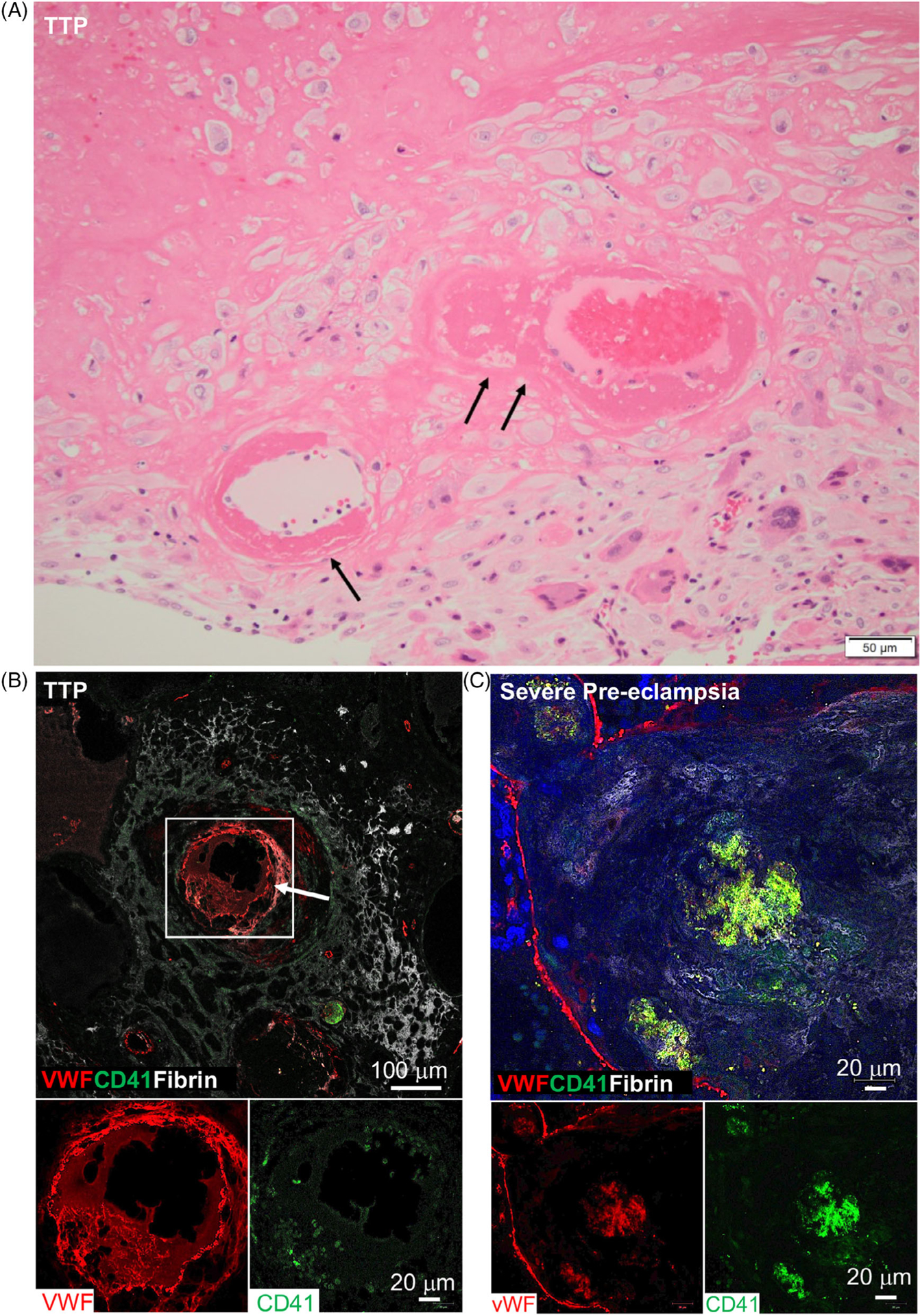
A, Routine (hematoxylin & eosin) images of the patient’s placenta (first pregnancy). The vessels in the basal plate illustrate the spectrum of decidual arterioles with fibrinoid necrosis (single arrow) and mural thrombi (double arrows). These are characteristic histologic features of severe maternal vascular malperfusion. B, Confocal microscopy images of immunostained sections of the patient’s placenta (first pregnancy). A decidual arteriole (white square, arrow) is partially occluded by a thrombus primarily composed of VWF (red stain) with surrounding perivascular fibrin (white stain). The lower two images illustrate VWF (red stain) but few platelets (CD41, green stain) in the thrombus. C, Confocal microscopy images of immunostained sections of the placenta from a patient with preeclampsia with severe features, without TTP, at 31 weeks’ gestation. A decidual arteriole is partially occluded by a thrombus composed of fibrin (white stain), platelets (CD 41, green stain), and VWF (red stain) When the stains are combined, the combination of green and red appears as yellow. The lower two images illustrate the individual stain for VWF (red) and platelets (green)
These data document that thrombosis caused by hereditary TTP and chronic hypertension can cause placental injury described as maternal vascular malperfusion, which is characteristic of preeclampsia with severe features. Thrombi composed of VWF are consistent with the pathogenesis of TTP but we could find only three previous reports describing the composition of thrombi in patients with TTP.8–10
At follow-up evaluations, her blood pressures (on labetalol) and platelet counts were normal without plasma prophylaxis. The diagnosis of hereditary TTP was confirmed by identification of pathogenic sequence variants of both ADAMTS13 genes: p.R1060W, p. W910*. These sequence variants have been previously reported in multiple unrelated families with hereditary TTP.
Six months after her hospital discharge, she began her second pregnancy (also with in vitro fertilization). We managed her with more intensive plasma prophylaxis to prevent an exacerbation of her TTP. We began at gestational week 4 with 750 mL every 2 weeks. At gestational week 11, we increased the frequency to weekly. At gestational week 31, when her platelet count decreased to 187 × 109/L and her blood pressure increased to 134/90, we increased the frequency to twice weekly. The following week she was admitted for preeclampsia with severe features (blood pressure 152/116, hemoglobin 107 g/L, platelets 149 × 109/L). Her hypertension was controlled, and on day 5 she had an uncomplicated caesarean delivery. Three days later she was discharged. Although her infant remained in the hospital for 7 weeks, he remains healthy. Currently, her blood pressures (on labetalol) and platelet counts are normal without plasma prophylaxis.
More intensive plasma prophylaxis was succesful for prevention of TTP exacerbation. Her chronic hypertension, and likely also her hereditary TTP, increased her risk for preeclampsia.
Her placenta was not collected for evaluation. Therefore, we examined the placenta of a 20-year-old woman who had preeclampsia with severe features at 31 weeks’ gestation, without clinical features of TTP (platelets 154 −190 × 109/L, total bilirubin 5.13–6.84 umol/L, LDH 3.33–4.45 umol/L). Routine staining demonstrated the diagnostic features of severe maternal vascular malperfusion (not shown). Confocal microscopy images of immunostained placental sections documented that the thrombi contained VWF in addition to fibrin and platelets (Figure 1C).
The unexpected discovery of VWF as a component of thrombi in the absence of clinical features of TTP may suggest that VWF participates in the pathogenesis of thrombosis in all women who have preeclampsia with severe features. The patient’s course is summarized in Table 1.
TABLE 1.
Summary of our patient’s clinical course
| Gestational age weeks | |
|---|---|
| Clinical features and outcomes | |
| First pregnancy | |
| 13 | Presentation with hypertension, stroke and moderate thrombocytopenia. Hereditary TTP suspected. Begin plasma prophylaxisa, every 2 weeks. |
| 17 | Normal fetal ultrasound. |
| 22 | TTP exacerbation. Intrauterine fetal demise. Subsequently, biallelic ADAMTS13 mutations confirmed diagnosis of hereditary TTP. |
| Second pregnancy | |
| 4 | Begin plasma prophylaxis, every 2 weeks. |
| 11 | Increase plasma to weekly (approaching the gestational age when complications occurred in her first pregnancy). |
| 31 | Increase plasma to twice weekly (blood pressure ↑, platelet count ↓). |
| 32 | Preeclampsia with severe features. C-section, healthy infant. |
Each plasma infusion was 750 mL (12.5 mL/kg).
2 |. DISCUSSION
Pregnancy has been recognized as a time of great risk for women with hereditary TTP since the disorder was first described.4,11 Hereditary TTP may be asymptomatic until thrombotic complications occur during pregnancy.4,5,12–15 However, recognition of hereditary TTP during pregnancy may be difficult because it may mimic the clinical characteristics of preeclampsia with severe features1 and it may not have the clinical characteristics of the more common acquired TTP.4
Treatment of hereditary TTP is simply the replacement of ADAMTS13 by plasma infusion. Prophylactic plasma can provide effective protection from TTP exacerbation throughout pregnancy for women with previously diagnosed hereditary TTP. The amount of plasma and the frequency of infusions is adjusted to maintain a safe level of ADAMTS13 activity (considered to be >10%)16 or simply to maintain the woman’s unique normal platelet count.17 The requirement for plasma increases during pregnancy as the risk for thrombosis increases. We believe that our patient’s exacerbation of TTP at 22 weeks’ gestation of her first pregnancy (Table 1) could have been prevented with more frequent plasma infusions. In her subsequent pregnancy, weekly then twice-weekly plasma prophylaxis was effective for preventing TTP exacerbation. However, plasma prophylaxis could not prevent the occurrence of preeclampsia with severe features. Our patient’s chronic hypertension increased her risk for preeclampsia.6 Hereditary TTP may also increase risk for preeclampsia. If a patient has adverse reactions to plasma, an alternative is beroctocog alfa, the plasma-derived factor VIII concentrate used for hemophilia A.18 Beroctocog alfa contains sufficient ADAMTS13 activity for weekly prophylactic treatment. Current clinical trials with recombinant ADAMTS13 suggest that it will become the standard for prophylaxis when it is approved for treatment of patients with hereditary TTP.19
The unique placental circulation may contribute to exacerbations of hereditary TTP. Normal placental development involves remodeling of the maternal decidual spiral arterioles, increasing their lumen diameter five to 10-fold.20 This allows increased maternal blood flow to the placenta with decreased resistance. When remodeling fails to occur, blood flow to the placenta is decreased. The blood flows with higher velocity and greater turbulence, increasing risk for thrombosis of decidual arterioles and causing villous hypoplasia. The placental pathology that results from the failure of normal decidual spiral arteriole remodeling is described as maternal vascular malperfusion.7 The classic clinical association with maternal vascular malperfusion is preeclampsia. Maternal vascular malperfusion is also associated with chronic hypertension and multiple other disorders.7 Hereditary TTP could contribute to the thrombosis of decidual arterioles and therefore also contribute to the histologic lesions of maternal vascular malperfusion and the clinical features of preeclampsia.
Our patient’s initial crisis at 13 weeks’ gestation of her first pregnancy could have been caused by synergistic effects of her chronic hypertension and hereditary TTP. Chronic hypertension could have impaired the remodeling of the decidual spiral arterioles and initiated the development of maternal vascular malperfusion.7 The resulting high velocity and turbulent arteriolar blood flow could have contributed to the exacerbation of her TTP. Turbulent blood flow at high velocity creates shear stress that facilitates the assembly of VWF multimers into thicker bundles and longer strands that can obstruct blood flow.21 The emergence of the VWF multimers from the decidual arterioles into the intervillous spaces brings them to a region of lower flow velocity that has a high concentration of platelets.22 Since the ultra-large VWF multimers of TTP have their A-1 platelet-binding domains continually exposed, the milieu of the intervillous spaces would facilitate platelet-VWF binding. Increased platelet binding to the ultra-large VWF multimers in patients with TTP increases risk for thrombosis. This is suggested by the ability of caplacizumab, which blocks platelet binding to VWF, to promptly stop symptoms of TTP.23
Our patient’s TTP exacerbation at 22 weeks’ gestation of her first pregnancy caused placental maternal vascular malperfusion resulting in fetal loss. Immunostaining of her placenta demonstrated that the decidual arteriolar thrombi were composed primarily of VWF, consistent with the pathogenesis of TTP. This observation suggests that hereditary TTP can cause the pathologic features of maternal vascular malperfusion. Although we were unable to examine our patient’s placenta from her second pregnancy, when she had preeclampsia with severe features at 32 weeks’ gestation, we suspect that VWF could have been a component of her decidual arteriolar thrombi, because of the turbulent circulation caused by maternal vascular malperfusion. To determine the composition of decidual arteriolar thrombi in patients without TTP, we examined the placenta of a patient who had preeclampsia with severe features, but no evidence for TTP. Her decidual arteriolar thrombi also contained VWF. This was unexpected. However, it is consistent with the experimental observations that turbulent, high velocity blood flow can create large VWF multimers which can contribute to vascular obstruction.21
Our patient’s experience suggests that chronic hypertension can substantially increase the risk for severe complications during pregnancy in women with hereditary TTP. We suspect that patients with hereditary TTP may have increased frequency of chronic hypertension, because we have previously documented that patients in remission following episodes of acquired TTP have increased prevalence of hypertension.24 However, hypertension was not mentioned in two recent reports of large registries of hereditary TTP14,15 and in a com prehensive review of hereditary TTP.4 Blood pressure was also infrequently reported in our systematic review of 180 case reports of patients with hereditary TTP, 2001–2020. However, hypertension was described in only 34 patients, commonly in children.25 We anticipate that future reports by the hereditary TTP Registries14,15 will include data on the prevalence of hypertension.
The key point from our experience with this patient is that hereditary TTP can mimic the clinical and pathologic abnormalities of preeclampsia with severe features early in the second trimester. Therefore, when manifestations of preeclampsia with severe features occur before 20 weeks’ gestation, hereditary TTP should be suspected. Our experience also suggests that the unique placental circulation may contribute to the high risk for exacerbation of hereditary TTP during pregnancy.
Footnotes
CONFLICT OF INTEREST
This project had no outside support. The authors have no conflict with this topic or these data.
REFERENCES
- 1.Gestational Hypertension and Preeclampsia. ACOG Practice Bulletin No. 202. American College of Obstetricians and Gynecologists. Obstet Gynecol. 2019;133:e1–e25. [Google Scholar]
- 2.Bendapudi PK, Hurwitz S, Fry A, et al. Derivation and extrenal validation of the PLASMIC score for rapid assessment of adults with thrombotic microangiopathies: a cohort study. Lancet Haematol. 2017;3026:1–8. [DOI] [PubMed] [Google Scholar]
- 3.Page EE, Kremer Hovinga JA, Terrell DR, Vesely SK, George JN. Thrombotic thrombocytopenic purpura: diagnostic criteria, clinical features, and long-term outcomes from 1995 through 2015. Blood Adv. 2017;1:590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kremer Hovinga JA, George JN. Hereditary thrombotic thrombocytopenic purpura. N Eng J Med. 2019;381:1653–1662. [DOI] [PubMed] [Google Scholar]
- 5.Moatti-Cohen M, Garrec C, Wolf M, et al. Unexpected frequency of Upshaw-Schulman syndrome in pregnancy-onset thrombotic thrombocytopenic purpura. Blood. 2012;119:5888–5897. [DOI] [PubMed] [Google Scholar]
- 6.Seely EW, Ecker J. Chronic hypertension in pregnancy. Circulation. 2014;128:1254–1261. [DOI] [PubMed] [Google Scholar]
- 7.Ernst LM. Maternal vascular malperfusion of the placental bed. APMIS. 2018;126:551–560. [DOI] [PubMed] [Google Scholar]
- 8.Asada Y, Sumiyoshi A, Hayashi T, Suzumiya J, Kaketani K. Immunohistochemistry of vascular lesions in thrombotic thrombocytopenic purpura, with special reference to factor VIII related antigen. Thromb Res. 1985;38:469–479. [DOI] [PubMed] [Google Scholar]
- 9.Tsai HM, Chandler WL, Sarode R, et al. Von Willebrand factor and von Willebrand factor-cleaving metalloprotease activity in Escherichia coli O157:H7-associated hemolytic uremic syndrome. Pediatr Res. 2001;49:653–659. [DOI] [PubMed] [Google Scholar]
- 10.Hosler GA, Cusumano AM, Hutchins GM. Thrombotic thrombocytopenic purpura and hemolytic uremic syndrome are distinct pathologic entities. A review of 56 autopsy cases. Arch Pathol Lab Med. 2003;127:834–839. [DOI] [PubMed] [Google Scholar]
- 11.Fuchs WE, George JN, Dotin LN, Sears DA. Thrombotic thrombocytopenic purpura. Occurrence two years apart during late pregnancy in two sisters. JAMA. 1976;235:2126. [DOI] [PubMed] [Google Scholar]
- 12.Fujimura Y, Matsumoto M, Isonishi A, et al. Natural history of Upshaw-Schulman syndrome based on ADAMTS13 gene analysis in Japan. J Thromb Haemost. 2011;9(Suppl 1):283–301. [DOI] [PubMed] [Google Scholar]
- 13.Von Krogh AS, Kremer Hovinga JA, Tjonnfjord GE, et al. The impact of congenital thrombotic thrombocytopenic purpura on pregnancy complications. Thromb Haemost. 2014;111:1180–1183. [DOI] [PubMed] [Google Scholar]
- 14.Alwan F, Vendramin C, Liesner R, et al. Characterization and treatment of congenital thrombotic thrombocytopenic purpura. Blood. 2019;133:1644–1651. [DOI] [PubMed] [Google Scholar]
- 15.van Dorland HA, Mansouri Taleghani M, Sakai K, et al. The International Hereditary Thrombotic Thrombocytopenic Purpura (TTP) Registry: key findings at enrollment until 2017. Haematologica. 2019;104:2107–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Epperla N, Hemauer K, Friedman KD, George JN, Foy P. Congenital thrombotic thrombocytopenic purpura related to a novel mutation in ADAMTS13 gene and management during pregnancy. Am J Hematol. 2016;91:644–646. [DOI] [PubMed] [Google Scholar]
- 17.Buckley MF, James JW, Brown DE, et al. A novel approach to the assessment of variations in the human platelet count. Thromb Haemost. 2000;83:480–484. [PubMed] [Google Scholar]
- 18.Aledort LM, Singleton TC, Ulsh PJ. Treatment of congenital thrombotic thrombocytopenic purpura: a new paradigm. J Pediatr Hematol Oncol. 2017;39:524–527. [DOI] [PubMed] [Google Scholar]
- 19.Scully M, Knobl P, Kentouche K, et al. A recombinant human ADAMTS-13: first-in-human study evaluating pharmacokinetics, safety and tolerability in cTTP patients. Blood. 2017;130:2055–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moser G, Guettler J, Forstner D, Gauster M. Maternal platelets - friend or foe of the human placenta? Int J Mol Sci. 2019;20:5639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng Y, Chen J, Lopez JA. Flow-driven assembly of VWF fibres and webs in in vitro microvessels. Nat Commun. 2015;6:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reese JA, Peck JD, Yu Z, et al. Platelet sequestration and consumption in the placental intervillous space contribute to lower platelet counts during pregnancy. Am J Hematol. 2019;94:E8–E11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chander DP, Loch MM, Cataland SR, George JN. Caplacizumab therapy without plasma exchange for acquired thrombotic thrombocytopenic purpura. N Engl J Med. 2019;381:92–94. [DOI] [PubMed] [Google Scholar]
- 24.Deford CC, Reese JA, Schwartz LH, et al. Multiple major morbidities and increased mortality during long-term follow-up after recovery from thrombotic thrombocytopenic purpura. Blood. 2013;122:2023–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Borogovac A, Tarasco E, Kremer Hovinga JK, George JN. Hypertension in patients with hereditary thrombotic thrombocytopenic purpura. eJHaem. 2020. 10.1002/jha2.29. [DOI] [PMC free article] [PubMed]