

Abstract
Serotonin neurotransmission is largely governed by the regulation of the serotonin transporter (SERT). SERT is modulated in part by cholesterol, but the role of cholesterol and lipid signaling intermediates in regulating SERT are unknown. Serotonergic neurons were treated with statins to decrease cholesterol and lipid signaling intermediates. Contrary to reported decreases in 5-HT uptake after cholesterol depletion, biochemical and imaging methods both showed that statins increased 5-HT uptake in a fluoxetine-dependent manner. Simvastatin lowered the Km without changing Vmax for 5-HT or SERT distribution to the plasma membrane. Cholesterol repletion did not block enhanced 5-HT uptake by simvastatin but the enhanced uptake was blocked by lipid isoprenylation intermediates farnesyl pyrophosphate and geranylgeranyl pyrophosphate. Blockade of geranylgeranylation alone without statins also enhanced 5-HT uptake. Overall, this study revealed a specific neuronal effect of statin drugs and identified lipid signaling through geranylgeranylation within the isoprenylation pathway regulates SERT in a cholesterol-independent manner.
Keywords: Serotonin transporter, serotonin uptake, Isoprenylation, Cholesterol, Simvastatin, Lipid Signaling
Graphical Abstract
1. Introduction
Serotonin (5-HT) in the brain is important for sleep, cognition, memory, and mood. Given that the synaptic regulation of 5-HT is mediated primarily by the serotonin transporter (SERT), a dysregulation of SERT can have marked physiological consequences (Amsterdam et al., 2013; Frick et al., 2015). Investigations into the regulation of SERT have traditionally focused on kinase-mediated phosphorylation but few studies have examined the role of lipid signaling in the regulation of SERT despite the brain being one of the most lipid-rich organs in the body (Sastry, 1985).
Lipids such as cholesterol are localized in organelle and plasma membranes of the neuron and enriched in segments of the plasma membrane where SERT and other monoamine transporters are distributed (Chang et al., 2012; Laursen et al., 2018; Magnani et al., 2004). Consequently, cholesterol is positioned to interact with SERT in lipid regions to affect the localization and function of SERT (Ferraro et al., 2016). While cholesterol itself has been implicated in regulating 5-HT transport, it is unknown if SERT is regulated through biosynthetic lipid intermediates of cholesterol.
The lipid precursors of cholesterol are synthesized in the endoplasmic reticulum beginning with the rate-limiting enzyme HMG-CoA reductase and the sequential production of intermediates mevalonate, farnesyl pyrophosphate (FPP), squalene, and finally cholesterol. Moreover, the intermediate FPP diverges from the cholesterol biosynthetic pathway before the production of cholesterol to yield geranylgeranyl pyrophosphate (GGPP). Both FPP and GGPP comprise the isoprenylation pathway, which canonically lipidates proteins such as small GTPases by adding hydrophobic phospholipids that tether proteins to the plasma membrane. Whereas lipidated proteins modulate multiple signaling processes at the plasma membrane, the role of isoprenylation in SERT regulation is not known.
The current study addressed the roles of cholesterol biosynthesis and isoprenylation in the regulation of SERT. The statin class of cholesterol lowering drugs was used based on their property to inhibit both cholesterol and isoprenylation. Immortalized serotonergic cells derived from the rat raphe nucleus were treated with statin drugs in combination with interventions of cholesterol synthesis and isoprenylation lipid intermediates (squalene, mevalonate, FPP, GGPP) to differentially determine the roles of cholesterol and isoprenylation in mediating the transport of 5-HT by SERT. The findings reveal a previously undisclosed effect of statins and a heretofore unidentified mechanism that regulates SERT.
2. Materials and Methods
2.1. Reagents and Antibodies
Hygromycin (30–240-CR), Zeocin (R25001), and Fetal Bovine Serum (FBS) (MT35010CV) were purchased from Fisher Scientific (Carlsbad, CA). Mevalonate (41288), Squalene (S3626), SyntheChol (S5442), Methyl-β-cyclodextrin (mβCD) (C4555), and DAPI (D9542) were purchased from Sigma Aldrich (St. Louis, MO). Atorvastatin (10493), Fluvastatin (10010337), Simvastatin (10010344), Farnesyl pyrophosphate (FPP) (63250), Geranylgeranyl pyrophosphate (GGPP) (63330), and Fluoxetine (14418) were purchased from Cayman Chemical (Ann Arbor, MI). Geranylgeranyl transferase type I inhibitor GGTI-298 (361184), G418 (29065A), anti-mouse secondary antibody (SC-516102), and anti-rabbit secondary antibody (SC2004) were purchased from Santa Cruz (Dallas, TX). DMEM (11330–057), sulfo-NHS-S-S-Biotin (21331), Alexa Fluor 488 secondary antibody (A1100), and Rhodamine Phalloidin (R415) were purchased from ThermoFisher Scientific (Cincinnati, OH). Farnesyl transferase inhibitor I (344510) was purchased from Millipore Sigma (Cincinnati, OH), anti-pan cadherin primary antibody (6528) was purchased from Abcam (Branford, CT), Lamin B1 (12987–1-AP) was purchased from Proteintech (Rosemont, IL), anti-SERT primary antibody (RA24330) was purchased from Neuromics (Minneapolis, MN), and 5-HT primary antibody (20080) was purchased from Immunostar (Hudson, WI).
2.2. Preparation of RN46A-B14 cells with Overexpressed SERT-Myc
Serotonergic RN46A-B14 cells with a temperature-sensitive SV40 large T-antigen (Eaton et al., 1995a; Eaton et al., 1995b) were provided by Scott Whittemore (Louisville, KY). RN46A-B14 cells exhibit a neuronal phenotype, express vesicular monoamine transporter (VMAT), aromatic amino acid decarboxylase (AADC) and SERT, and constitutively release 5-HT via vesicular docking at the plasma membrane (Eaton et al., 1995b; Stansley and Yamamoto, 2013). Thus, these cells have the ability to synthesize 5-HT, express SERT, package 5-HT into vesicles, as well as release and reuptake 5-HT (Eaton et al., 1995a; White et al., 1994; Bethea et al., 2003,). The RN46A-B14 cell line has been authenticated by the European Collection of Authenticated Cell Cultures (ECACC) under the Public Health England. Cells were stably transfected with c-myc-tagged SERT to provide ample SERT expression and to allow for identification of overexpressed SERT using antibodies for c-myc. For stable SERT-Myc expression, rat SERT cDNA was amplified in a pCMV6 vector (Origene, 0084884) with a c-myc tag. The insert was digested and cloned into the Xba1 and Kpn1 sites in the pcDNA3.1/Zeo (−) vector (Invitrogen, V86520) and contained the Zeocin resistance gene that permitted selection of stable cell lines with expression driven by the CMV promoter. The pcDNA3.1-SERTMyc construct was transfected into the RN46A-B14 cells and colonies were selected with 200 μg/ml Zeocin. Colonies 3 and 12 out of 22 colonies were selected for the experiments in this study based on neuronal-like morphology, comparable SERT expression between both colonies verified by western blot, and comparable SERT function verified by 5-HT uptake.
2.3. SERT-Myc Cell Culture
RN46A-B14 cell stocks were incubated in T75 flasks at 33°C in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS, 250μg/ml G418, 100μg/ml Hygromycin and 200μg/ml Zeocin. G418 was required to select for the large T antigen and hygromycin was used to select for brain-derived neurotrophic factor production, both of which are important for the immortalized and serotonergic phenotype of the cells. Zeocin selection maintained stable SERT overexpression. T75 cell stocks were kept at a 33C° permissive temperature which allows for continued division in cell culture. Cells were switched to 37C° when plated on 6 well plates prior to uptake to slow growth and limit confluency prior to experimental assays. This switch to 37C°, combined with reducing serum helped to slow cell growth so cells did not overgrow during the 3 day pre-drug incubation. Stocks were split onto 6 well plates (100,000 cells/well) for 72hr prior to addition of any reagents. After 72hr, cells were washed with serum- and antibiotic-free DMEM. The following reagents were used independently and applied for a 20min incubation in serum-free DMEM followed by simvastatin for 24hr: mevalonate, squalene, SyntheChol, FPP, and GGPP. Fluoxetine was applied 1hr prior to the addition of 5-HT for uptake and was not included in the incubation period during the 24hr incubation with 0.5μM simvastatin. In other experiments, geranylgeranyl transferase type I inhibitor GGTI-298, farnesyl transferase inhibitor I, atorvastatin, and fluvastatin were also applied to cells in DMEM for 24hr. mβCD was applied for 30min in serum-free DMEM prior to 5-HT uptake.
2.4. 5-HT Uptake
Measurement of 5-HT uptake in SERT-Myc cells was modified from that described by Zhu et al (2006). Briefly, SERT-Myc cells were incubated in 10μM MAO inhibitor pargyline for 30min to prevent the degradation of 5-HT. 5-HT uptake was initiated by addition of 0.1μM 5-HT (or a range of 1nM-20μM 5-HT for Michaelis Menten kinetics) to the media and incubated at 37°C for 10min. Uptake was terminated by the transfer of media and nonadherent cells to ice-cold HBSS followed by an ice cold HBSS wash over ice. All cells were collected in ice cold HBSS and centrifuged at 600xg. The supernatant was discarded and pellets were stored at −20°C until assayed for 5-HT by HPLC.
2.5. 5-HT Measurement by High Pressure Liquid Chromatography
Cell pellets were sonicated in 0.25M perchloric acid and centrifuged at 14,000xg for 20min at 4°C to precipitate protein. Supernatant (20μl) was injected into a HPLC and analyzed with EZChrom software (Scientific Software Inc.). Samples were quantified by comparison of sample peak height to the peak height of a 400pg 5-HT standard. Cell pellets from initial spin down were dissolved in 1N NaOH and protein was quantified by a Bradford assay. 5-HT was normalized to total protein measured in the cell pellet (pg 5-HT/μg protein). Data from the fluoxetine inhibition kinetics (Figure 2B, D) were further transformed to calculate the serotonin Ki for SERT in the presence of increasing fluoxetine concentrations. Calculations were based on the Cheng and Prusoff equation for a competitive inhibitor IC50= Ki (1+([5-HT]/km) (Cheng and Prusoff 1973).
2.6. 5-HT Measurement by Immunocytochemical Detection
Cells were plated on poly-D-lysine coated cover slips in 6 well plates (50,000 cells/well) and simvastatin with fluoxetine, GGPP, or GGTI-298 treatments were completed as described above. Following treatment with 5-HT for 10min, cells were washed twice with ice-cold phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde for 10min at room temperature, and washed twice with PBS for 5min. Cells were permeabilized with 0.05% saponin in 0.5% PBS-Triton (PBS-T) solution for 2min, washed twice with PBS for 5min, and then blocked with 10% norma goat serum (NGS) in PBS-T for 1hr at room temperature. Cells were incubated with 5-HT primary antibody (1:40,000) in 5% NGS and PBS-T overnight at 4°C. The 5-HT polyclonal antibody is raised against 5-HT conjugated to bovine serum albumin by paraformaldehyde. Subsequently, cells were washed three times with PBS-T and incubated with Alexa Fluor 488 secondary antibody (1:1000) in 5% NGS in PBS-T for 1hr at room temperature. Cells were washed three times with PBS-T, then incubated with a solution containing 300nM DAPI and 165nM Rhodamine Phalloidin for 5min at room temperature. Cells were washed again with PBS-T and mounted on slides with Vectashield and kept at 4°C until evaluation. Images were collected at 6 × 2μm z axis steps using a Zeiss LSM800 laser scanning confocal microscope equipped with a 20x objective at excitation wavelengths of 405nm (DAPI, Em: 400–510nm), 488nm (Alexa Fluor 488, Em: 510–575 nm) and 561nm (Rhodamine Phalloidin, Em: 575–700nm). The microscope was equipped with Zeiss Zen 2.6 (blue edition) software (Carl Zeiss, Inc.). Maximum projection images of the exported Z-stack files were captured and visualized using ImageJ image software ver.1.51n (Abramoff et al., 2004). All treatment conditions within an experimental cohort were stained and imaged simultaneously, and cells within each experimental cohort were imaged using identical microscope settings. No modifications of brightness or contrast were imposed by ImageJ.
2.7. Biotinylation and Separation of Plasma Membrane SERT and Intracellular SERT
Separation of biotinylated plasma membrane SERT was modified from Loweth et al., 2014. In brief, cells were treated for 24hr with vehicle or 0.5μM simvastatin and a 5-HT uptake assay was performed. Cell pellets were centrifuged at 600xg for 5min. Pellets were resuspended and incubated for 30min in sodium-citrate buffer activated sulfo-NHS-S-S-Biotin on a 360° end-over-end rotator at 4°C. The reaction was quenched with 100mM Glycine for 20min and samples were centrifuged at 20,000xg for 2min at 4°C. Biotinylated cell pellets were sonicated in lysis buffer (25mM HEPES, 500mM NaCl, 1mM PMSF, 20mM NaF, 0.1% IGEPAL, 2mM EDTA, 1X HALT protease inhibitor), centrifuged at 20,000xg for 2min at 4°C, and supernatant was stored at −20°C until the avidin-biotin pull-down. Bradford protein analysis was performed on the stored and lysed biotinylated cell samples and 100μg of protein was incubated with equal amounts of PBS pre-washed NeutrAvidin Agarose Resin (Thermo Scientific, 29204), followed by overnight incubation. The following day, incubated sample was removed from the resin and the overnight incubation was repeated with freshly-washed NeutrAvidin Agarose Resin. Sample-bound resin from the first incubation (membrane fraction) was stored in 2xLDS and 200mM DTT at −20°C. After the second overnight incubation in resin, non-biotinylated sample that did not bind to the resin was removed and placed in fresh Eppendorf tubes (intracellular fraction) and stored in 2xLDS and 200mM DTT at −20°C. 2xLDS and 200mM DTT was also added to the remaining second fraction of bound resin, and then the first and second bound fractions were pooled together, spun on a centrifugal filter unit with a 0.45μm filter (Millipore, UFC30HV00) at 11,000xg for 5min at 4°C. Samples were stored at −20°C until prepared for western analysis. Equivalent volumes of protein in LDS were loaded for each sample.
2.8. Cholesterol Measurement in SERT-Myc Cells
The Amplex Red Assay (ThermoFisher, A12216) was used to measure total cholesterol in RN46-B14 cells with overexpressed SERT treated with and without simvastatin and SyntheChol. Cells were collected over ice and washed with HBSS, centrifuged at 600xg for 5min, and the pellets were washed in ice cold HBSS. Pellets were then sonicated in 300μl of the provided reaction buffer and plated onto a 96 well plate for a 30min incubation with amplex red working solution containing HRP, cholesterol oxidase, cholesterol esterase, and reaction buffer. Total cell cholesterol content was quantified using a cholesterol standard curve based on increasing concentrations of cholesterol incubated in a 96 well plate with working solution prior to measurement of the fluorescent signal. A fluorescent plate reader was used to excite (520nM) and measure emitted fluorescence (590nM) from the samples.
2.9. Western Blot Analysis
Cell pellets were prepared in LDS and DTT prior to loading onto a NuPAGE4–12% bis tris polyacrylamide gel (Invitrogen, NP0335BOX) for gel electrophoresis using the XCell SureLock™ Mini gel electrophoresis system in MOPS running buffer. Following SDS-PAGE, gels were transferred onto PVDF membranes and blocked for 1hr in 0.5% nonfat dry milk solubilized in 0.5% TBS-T. Membranes were then incubated overnight with primary antibody in 4°C on an orbital shaker. The following morning, membranes were removed from 4°C, washed three times for 5min in 0.5% TBS-T on a rocking shaker at room temperature, and incubated in secondary antibody at a 1:2500 dilution for 1hr, followed by three 5min washes in 0.5% TBS-T. Membranes were developed in chemiluminescent reagent (VWR PI34578) and imaged in a Fujifilm imager and analyzed with Multi Gauge V3.0 software. Primary and secondary antibody combinations tested include: anti-pan cadherin primary (1:1000) with anti-mouse secondary (1:2500), anti-SERT primary (1:1000) with anti-rabbit secondary (1:2500), and anti-lamin B1 primary (1:1000) with anti-rabbit secondary (1:2500) all in 0.5% nonfat dry milk solubilized in 0.5% TBS-T.
2.10. Statistical Analysis
Experiments were replicated with at least three independent experiments and each independent experiment had 2–3 technical replicates. Statistical analyses were performed on the mean of each independent experiment and data were represented as the mean ± SEM. Under conditions in which samples were pooled from more than one well (Figure 3C), the pooled data were treated as the mean of an independent experiment. Statistical analyses were performed using one- and two-way ANOVA where applicable, followed by the appropriate post-hoc test to determine statistical comparison between different treatments. p-values less than 0.05 were considered significant.
3.0. Results
3.1. Statins Enhance 5-HT Uptake in a Dose- and SERT-dependent Manner.
Atorvastatin, fluvastatin, and simvastatin treatment enhanced 5-HT uptake at concentrations of 1μM and 10μM, but did not significantly affect uptake at 0.1μM (Figure 1). Additional and lower concentrations of simvastatin ranging between 0.1–1μM were tested to recapitulate concentrations measured in the brain after oral simvastatin dosing (Johnson-Anuna et al., 2005).
Figure 1-. Statins enhance 5-HT uptake in a dose-dependent manner.

A) Atorvastatin, Fluvastatin, and Simvastatin were applied to SERTMyc cells for 24hr at 0.1, 1, and 10μM concentrations. Each statin significantly enhanced 5-HT uptake at 1 and 10μM (n=3/grp, 1-Way ANOVA significant drug effect (p<0.0001), *p<0.01 Tukey Post-Hoc comparison to Vehicle).
Simvastatin also enhanced 5-HT uptake at 0.1μM and 0.5μM (Figure 2A). To assess that simvastatin-enhanced uptake was mediated by SERT, the SERT inhibitor fluoxetine was used (Zhu et al., 2006). Fluoxetine (1μM) blocked 5-HT uptake in both vehicle and simvastatin-treated cells, indicating a SERT-specific mechanism of 5-HT uptake and its enhancement by simvastatin (Figure 2B, C), which was not a result of changes in endogenous 5-HT (Figure 2D).
Figure 2-. Simvastatin enhances 5-HT uptake in a SERT-dependent manner.

A) A lower concentration range of simvastatin was applied to SERTMyc cells for 24hr (0.001, 0.01, 0.1, 0.5, and 1μM), resulting in significantly enhanced 5-HT uptake at 0.1, 0.5, and 1μM (n=4–5/grp, 1-Way ANOVA significant simvastatin effect (p<0.001), *p<0.01 Tukey Post-Hoc comparison to Vehicle). B) Enhanced 5-HT uptake by 0.5μM simvastatin applied for 24hr was blocked by 1μM of the SERT inhibitor fluoxetine, applied 30min prior to 5-HT uptake (n=4/grp, except Veh/Fluox n=2, 2-Way ANOVA significant Fluoxetine effect (p<0.01), #p<0.05 Tukey Post-Hoc comparison to Veh/Veh, *p<0.001 Tukey Post-Hoc comparison to 0.5μM Sim/Veh). C) SERTMyc cells post-fixed after 5-HT uptake assay and stained for 5-HT. Prior to 5-HT uptake assay and fixation, SERTMyc cells were treated with or without 0.5μM simvastatin for 24hr followed by 1μM fluoxetine applied 30min prior to 5-HT uptake. D.) SERTMyc cells treated with 0.5μM simvastatin for 24hr does not change endogenous basal 5-HT content (no added 5-HT) within SERTMyc cells (n=3/grp, t-test between Veh and Sim not significant). Cells were collected as described in the 5-HT uptake methods, but exogenous 5-HT was not added for 10min. Data are represented as mean ± SEM. Veh=Vehicle, Sim=simvastatin, Fluox=fluoxetine.
3.2. Simvastatin decreases the Km without changing Vmax for 5-HT uptake, and decreases the Ki of fluoxetine.
To determine how SERT activity was altered by simvastatin, Michaelis-Menten kinetics were experimentally determined by exposing cells to 0.5μM simvastatin or vehicle for 24hr and uptake was measured in the presence of increasing concentrations of 5-HT. The 5-HT concentration range selected was based on the maximal plateau in 5-HT uptake for Vmax calculation in the RN46-B14 expression system. Simvastatin reduced Km values from a Km of 2.213μM in vehicle-treated cells to 0.679μM (Figure 3A, D). The Vmax for 5-HT uptake was not significantly different between simvastatin (232.2μM) compared to vehicle-treated (246.1μM) cells, suggesting that the amount of SERT at the plasma membrane remained constant with simvastatin treatment. Values for vehicle-treated cells correspond with previously published Km and Vmax values from raphe rat brain slices from which these cells were derived and brain synaptosomal preparations (Bunin et al., 1998; Butler et al., 1988; Montañez et al., 2003; Shaskan and Snyder, 1970). The Ki was determined at a fixed concentration of 100nM 5-HT and increasing concentrations of the SERT inhibitor, fluoxetine. The fluoxetine inhibitor concentration range was selected based on a plateau of inhibition at higher concentrations that represented maximal SERT saturation by fluoxetine. Simvastatin reduced the Ki for fluoxetine from 5.98nM to 1.18nM at a 5-HT concentration of 100nM (Figure 3B, D). Similar to the Km and Vmax, the Ki transformed from the IC50 and Km in vehicle-treated samples corresponded with previously published transformed Ki values for SERT (Barker et al., 1998).
Figure 3-. Simvastatin decreases the Michaelis-Menten Km for 5-HT uptake without changing Vmax or membrane SERT, and decreases the Ki of fluoxetine.

A) SERTMyc cells treated with 0.5μM simvastatin for 24hr significantly decreases the Michaelis-Menten Km, but does not change SERT uptake Vmax (n=5/grp, *p<0.0001 t-test comparing vehicle vs. simvastatin Km and Vmax). B) SERTMyc cells treated with 0.5μM simvastatin for 24hr shifts SERT Ki at 100nM 5-HT, with increasing concentrations of SERT inhibitor fluoxetine (n=4/grp, *p<0.05 t-test comparing vehicle vs. simvastatin Ki). C) Simvastatin does not change densitometry measures of SERT in total, membrane separated, and intracellular fractions after 0.5μM, 24hr simvastatin treatment. Representative blots demonstrate enrichment of membrane localized pan-cadherin in membrane and total fractions that is not present in the intracellular fraction, and intracellular fractions with lamin B1. D) Mean values and SEM for SERT uptake Km and Vmax (from graph A) and IC50 and Ki (from graph B) in vehicle and simvastatin-treated cells. Data are represented as mean ± SEM. T=Total SERT, M and Mem=Membrane SERT, I and Intra=Intracellular SERT, V=Vehicle, S=Simvastatin.
3.3. Simvastatin does not alter total SERT or SERT localization.
Total SERT, intracellular SERT, and plasma membrane SERT were measured by biotinylation of SERT at the membrane and subsequent streptavidin separation (Figure 3C). Densitometry measures of SERT immunoreactivity by Western blot revealed no changes in SERT in total cell homogenate, or the corresponding membrane and intracellular fractions of these samples. Representative densitometry images of pan-cadherin and SERT on the same western blot indicate membrane enrichment in the total and membrane fractions. Densitometry images for lamin B1 on the same western blot reveal intracellular protein within the lanes loaded with intracellular fractions.
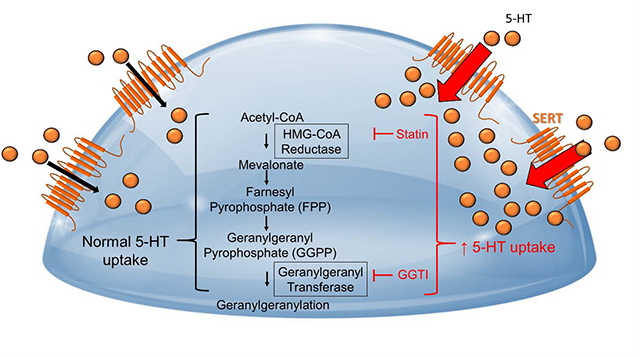
3.4. Simvastatin-enhanced 5-HT Uptake is Dependent on the Cholesterol Synthesis Pathway, but Independent of Cholesterol
The cholesterol biosynthetic path, the effect of simvastatin, and its relationship to the isoprenylation pathway is depicted in Figure 4. To test the role of cholesterol biosynthetic intermediates in simvastatin-enhanced uptake, intermediates mevalonate and squalene (Figure 5A) were included in the media during simvastatin treatment. Mevalonate blocked simvastatin-enhanced 5-HT uptake in a dose-dependent manner (Figure 5A). In contrast, squalene did not block simvastatin-enhanced 5-HT uptake. To further test the contribution of cholesterol, cholesterol content of the cells was measured to determine the effect of simvastatin within the 24hr timeframe of exposure. Total cell cholesterol was decreased by simvastatin (Figure 5B) and was repleted with addition of the cholesterol delivery reagent SyntheChol (Smith et al., 2017; Whiteley et al., 2017). Regardless, SyntheChol during simvastatin treatment did not block the enhanced 5-HT uptake by simvastatin (Figure 5B). In contrast, treatment with the cholesterol depleting reagent mβCD at concentrations of 10 and 20mg/ml for 30min decreased 5-HT uptake (Figure 5C) similar to previous studies (Magnani et al., 2004; Scanlon et al., 2001).
Figure 4-. Cholesterol synthesis and mechanism of action of inhibitors.

Simvastatin blocks the rate limiting enzyme in cholesterol synthesis HMG-CoA reductase. Simvastatin also blocks the generation of downstream biosynthetic intermediates including mevalonate, farnesyl pyrophosphate (FPP), geranylgeranyl pyrophosphate (GGPP), squalene, and cholesterol. FPP and GGPP isoprenylation and downstream protein regulation are also inhibited by simvastatin or can be directly inhibited by blocking FPP-mediated farnesylation with Farnesyl transferase inhibitors (FT inhibitors) or GGPP-mediated geranylgeranyl transferase type 1 inhibitors (GGT-1 inhibitors).
Figure 5-. Simvastatin-enhanced 5-HT uptake is dependent on the cholesterol synthesis pathway, but independent of cholesterol.

A.) Mevalonate or squalene were applied to SERTMyc cells during treatment with 1μM simvastatin for 24hr. Repletion of mevalonate blocked simvastatin enhanced 5-HT uptake at 10μM and 50μM (n=3/grp, 1-Way ANOVA significant drug effect (p<0.0001) within Simvastatin-treated groups, *p<0.01 Tukey Post-Hoc comparison to Simvastatin/ 0μM Mevalonate)( 2-Way ANOVA significant simvastatin mevalonate interaction (p<0.05) within Veh, Veh+50μM Mevalonate, Simvastatin, Simvastatin+50μM Mevalonate, #p<0.01 Tukey Post-Hoc comparison between Veh and Simvastatin/0μM Mevalonate). Squalene repletion did not block enhanced 5-HT uptake (n=3/grp, 1-Way ANOVA not significant within simvastatin-treated groups) (#p<0.01 t-test comparing Veh and Simvastatin). B) SERTMyc cells treated with 0.5μM simvastatin for 24hr decreased total cell cholesterol (results were quantified as nM Cholesterol/μg protein and normalized to % Veh), which was repleted with SyntheChol. Repletion of total cell cholesterol with SyntheChol during treatment with simvastatin for 24hr blocked the simvastatin-induced cholesterol depletion (n=8–12/grp, 2-Way ANOVA significant interaction (p<0.001), *p<0.01 Tukey Post-Hoc comparison between Veh and Sim, #p<0.01 Tukey Post-Hoc comparison between Sim and Sim+Chol). However, SyntheChol did not block enhanced 5-HT uptake by simvastatin treatment (n=6–7/grp, 2-Way ANOVA significant simvastatin effect (*p<0.001), no significant Tukey Post-Hoc Comparison between Sim, and Sim+Chol). C) SERTMyc cells treated with 10 and 20mg/ml mβCD for 30min significantly decreased 5-HT uptake (n=4, 1-Way ANOVA significant mβCD effect (p<0.001), *p<0.01 Tukey Post-Hoc comparison between 10 or 20mg/ml and 0mg/ml). Data are represented as mean ± SEM. Veh=Vehicle, Sim=Simvastatin, Chol=SyntheChol, mβCD= methyl-β-Cyclodextrin.
3.5. Simvastatin-enhanced 5-HT Uptake is Dependent on Isoprenylation
In addition to the inhibition of cholesterol synthesis, simvastatin inhibits the synthesis of the isoprenylation pathway intermediates FPP and GGPP prior to the production of cholesterol (see Figure 4). The addition of FPP and GGPP blocked simvastatin-enhanced uptake in a concentration-dependent manner (Figure 6). To determine if GGPP specifically regulates SERT independent of simvastatin, the enzyme for geranylgeranylation (geranylgeranyl transferase type 1 (GGTase)), (Figure 7) was blocked by GGTI-298 at concentrations up to the IC70 (10μM) of the inhibitor (Chu et al., 2015; McGuire et al., 1996). Similar to simvastatin, inhibition of GGTase enhanced 5-HT uptake (Figure 7A, C) and was blocked in a SERT-dependent manner (Figure 7E, F). Both HMG-CoA reductase and GGTase inhibition did not change endogenous 5-HT, but 10μM of GGTI-298 produced a larger fold increase in 5-HT uptake than 0.5μM simvastatin (Figure 7D). Farnesyl transferase (FTase) was also blocked by the Farnesyl transferase I inhibitor at concentrations up to the IC70 (500nM) of the inhibitor, but had no effect on 5-HT uptake (Figure 7B).
Figure 6-. Simvastatin-enhanced 5-HT uptake is dependent on isoprenylation.

A) FPP was applied to SERTMyc cells during treatment with 0.5μM simvastatin for 24hr. FPP repletion blocks simvastatin-enhanced 5-HT uptake dose dependently at 2μM and 4μM FPP, but not at 0.5μM FPP (n=4–6/grp, 1-Way ANOVA significant drug effect (p<0.0001) within Simvastatin-treated groups, *p<0.05 Tukey Post-Hoc comparison between Simvastatin/0μM FPP)( 2-Way ANOVA significant simvastatin FPP interaction (p<0.05) within Veh, Veh+4μM FPP, Simvastatin, Simvastatin+4μM FPP, #p<0.001 Tukey Post-Hoc comparison between Veh and Simvastatin/0μM FPP). B) GGPP repletion also blocks enhanced 5-HT uptake dose dependently at 4μM GGPP, but not at 2 and 0.5μM GGPP (n=3–5/grp, 1-Way ANOVA significant drug effect (p<0.0001) within simvastatin-treated groups, *p<0.05 Tukey Post-Hoc comparison between simvastatin and 0μM GGPP)(2-Way ANOVA significant simvastatin GGPP interaction (p<0.01) within Veh, Veh+4μM GGPP, Simvastatin, Simvastatin+4μM GGPP, #p<0.01 Tukey Post-Hoc comparison between Veh and Simvastatin). Data are represented as mean ± SEM. Veh=Vehicle. C) SERTMyc cells post-fixed after 5-HT uptake assay and stained for 5-HT. Prior to 5-HT uptake assay and fixation, SERTMyc cells were treated with or without 0.5μM simvastatin and 4μM GGPP.
Figure 7-. Geranylgeranylation inhibition enhances 5-HT uptake independent of simvastatin, is SERT-dependent.

A.) Geranylgeranyl transferase Type 1 (GGTase) inhibitor GGTI-298 dose-dependently increases 5-HT uptake at concentrations up to IC70 inhibitor concentrations (10μM GGTI-298). Results were quantified as pg 5-HT/μg protein and normalized to % Veh. (n=4/grp, 1-Way ANOVA significant GGTI-298 effect (p<0.001), *p<0.01 Tukey’s Post-Hoc comparison to Veh. B.) Farnesyl Transferase (FTase) inhibitor I up to the IC70 inhibitor concentration (500nM Farnesyl Transferase Inhibitor I) does not enhance 5-HT uptake (n=3/grp, One-Way ANOVA not significant). C) SERTMyc cells post-fixed after 5-HT uptake assay and stained for 5-HT. Prior to 5-HT uptake assay and fixation, SERTMyc cells were treated with Vehicle, 1, 2, 4, and 10μM GGTI-298 for 24hr. D) 10μM GGTI increases 5-HT uptake more than 0.5μM simvastatin, without changing basal 5-HT (n=4/grp, 2-Way ANOVA with Tukey’s Post-Hoc comparison to Veh *p<0.05, or comparing Sim and GGTI #p<0.001). E.) Enhanced 5-HT uptake by 10μM GGTI-298 applied for 24hr was blocked by 1μM of the SERT inhibitor fluoxetine, applied 30min prior to 5-HT uptake (n=3/grp, 2-Way ANOVA significant Fluoxetine effect (p<0.01), #p<0.05 Tukey Post-Hoc comparison to Veh,Veh, *p<0.001 Tukey Post-Hoc comparison to 10μM GGTI-298). F.) SERTMyc cells post-fixed after 5-HT uptake assay and stained for 5-HT. Prior to 5-HT uptake assay and fixation, SERTMyc cells were treated with or without 10μM GGTI-298 for 24hr followed by 1μM fluoxetine applied 30min prior to 5-HT uptake Data are represented as mean ± SEM. Veh= Vehicle, Fluox= Fluoxetine, GGTI=Geranylgeranyl transferase type 1 inhibitor GGTI-298, GGTase=Geranylgeranyl Transferase 1, FTase=Farnesyl Transferase.
4.0. Discussion
The effects of the biosynthetic pathway for cholesterol on the uptake of 5-HT by 5-HTergic neurons were examined. Contrary to initial expectations, the statin class of cholesterol lipid lowering drugs did not decrease, but enhanced the uptake of 5-HT in a manner independent of cholesterol per se. Additional experiments revealed that simvastatin altered the kinetics of 5-HT uptake by SERT and enhanced uptake through decreases in the isoprenylation pathway that diverges from the cholesterol synthesis pathway.
The dose-dependent increase in 5-HT uptake by lipid-lowering statin drugs (Figure 1) suggests a broad range of concentrations at which statins enhance SERT uptake. The concentrations of statins that increase 5-HT uptake are also within the range of concentrations in the brain reported after acute systemic treatment (Johnson-Anuna et al., 2005). Additionally, chronic treatments with simvastatin decreased brain cholesterol, FPP, and GGPP in rodent models (Eckert et al., 2009) and reduced other cholesterol biosynthetic intermediates in humans (Serrano-Pozo et al., 2010). As both simvastatin and atorvastatin penetrate the blood-brain barrier (Kandiah and Feldman 2009; Saheki et al., 1994), the current in vitro findings using 5-HT neurons are suggestive of a pronounced inhibition on the isoprenylation pathway by simvastatin that likely have effects on SERT activity in the brain.
The findings provide 3 lines of supportive evidence that statin-enhanced 5-HT uptake is independent of cholesterol per se. First, repletion of cholesterol with SyntheChol or the introduction of the immediate cholesterol precursor squalene did not alter the effect of simvastatin on enhanced 5-HT uptake (Figure 5). Second, the depletion of cholesterol (Figure 5C) with mβCD reduced 5-HT uptake similar to that observed by others in which mβCD or Nystatin dose-dependently decreased SERT-dependent 5-HT uptake through removal of cholesterol or the binding and sequestering of plasma membrane cholesterol (Magnani et al., 2004; Scanlon et al., 2001). Lastly, increases in 5-HT uptake were recapitulated with the geranylgeranylation transferase inhibitor GGTI-298 in the absence of simvastatin (Figure 7) and support both the cholesterol-independent effects of simvastatin and the role of geranylgeranylation on 5-HT uptake.
The cholesterol independent effect of statins on 5-HT uptake is also supported by the differences in the magnitude of cholesterol depletions produced by statins compared to the direct sequestration and removal of cholesterol. Simvastatin modestly decreased total cholesterol by 20% in the RN46A-B14 cells transfected with SERT (Figure 5B). Although the total cell cholesterol content in these cells is similar to HEK cells (Magnani et al., 2004), mβCD depletes free cholesterol by 40–50% in HEK cells (Magnani et al., 2004) and up to 85% in the plasma membrane (Scanlon et al., 2001). The low but pharmacologically relevant concentrations of simvastatin used over 24hr may have been insufficient to significantly deplete plasma membrane cholesterol yet was sufficient to alter 5-HT uptake through the divergent isoprenylation path. Thus, the interactions of cholesterol with SERT at the plasma membrane likely remain intact with statin treatment (Ferraro et al., 2016; Laursen et al., 2018; Ikonen, 2008) and permit increases in the activity of SERT.
The addition of the isoprenylation intermediates FPP and GGPP also blocked simvastatin-enhanced uptake and further illustrates that simvastatin enhances 5-HT uptake alternatively through a decrease in isoprenylation (Figure 6). Inhibition of the enzyme geranylgeranyl transferase with GGTI-298 reproduced the effects of simvastatin and further supports the role of decreased isoprenylation and the GGPP modification of proteins in mediating increased 5-HT uptake through SERT (Figure 7). However, inhibition of the enzyme farnesyl transferase with farnesyl transferase inhibitor I (Figure 7B) did not reproduce the effects of simvastatin and indicates more specifically that inhibition of geranylgeranylation and not farnesylation increases 5-HT uptake by SERT. The concentrations of GGTI-298 produced a more amplified effect on 5-HT uptake by SERT compared to simvastatin (Figure 7D). Although GGTase inhibition is more proximal to changes in SERT compared to HMG-CoA reductase inhibition that could produce a greater increase in 5-HT uptake, it is also likely that the concentrations of simvastatin compared to GGTI-298 differ in the magnitude of inhibition on isoprenylation and thus differences in 5-HT uptake by SERT.
These findings are suggestive of changes in SERT activity without changes in the density/amount of SERT with simvastatin treatment. Michaelis-Menten kinetics revealed significant decreases in the Km of SERT for 5-HT without changes in Vmax (Figure 3A, D). The kinetic results using RN46A-B14 cells transfected with SERT are similar to literature reports of Km values ranging from 1–4μM using brain slices or synaptosomes (Butler et al., 1988; Codd and Walker 1987; Daws et al., 2005) and transformed Ki values of 6.6nM (Barker et al., 1998). Based on these kinetics changes, alterations in 5-HT uptake by simvastatin are due to activity-dependent effects of SERT since simvastatin did not change the amount of plasma membrane SERT, intracellular SERT, and total SERT (myc-tagged and non myc-tagged SERT) (Figure 3C) and did not alter endogenous 5-HT in the absence of added 5-HT (Figure 2D, 7D).
5-HT uptake blockers are known to block uptake of 5-HT by RN46A cells (Zhu et al., 2006). Our findings illustrate that fluoxetine also blocks the effects of simvastatin on 5-HT uptake measured by biochemical and immunocytochemical methods (Figs 2B, C) and support the role of SERT in mediating 5-HT uptake by these cells. Further kinetic analyses with fluoxetine show decreases in the Ki of fluoxetine for SERT with simvastatin (Figure 3B, D). Fluoxetine binds SERT in an intermediate outward, extracellular-facing conformation (Tavoulari et al., 2009) whereas 5-HT serves as a substrate to shift SERT to an inward, intracellular-facing conformation for uptake into neurons (Zhang and Rudnick 2006). Therefore, the current findings of a decreased Ki for fluoxetine and enhanced 5-HT uptake do not suggest changes in the orientation of SERT at the membrane but rather a change in the rate of 5-HT uptake by SERT. Further details of any specific conformational changes in SERT induced by simvastatin remain to be determined.
The immunocytochemical evidence of 5-HT uptake provides robust visualization of the enhanced intracellular accumulation of 5-HT (Figures 2C, 6C, 7C, 7F) that is blocked with fluoxetine (Figure 2C, 7F) and the addition of GGPP (Figure 6C). The specificity of the selected 5-HT antibody is supported by the inhibition of intracellular 5-HT signal with fluoxetine (Figure 2C). Furthermore, the distribution of 5-HT throughout the cell and perinuclear space are likely due to the broad distribution of SERT throughout the cell processes and soma.
While only a single cell model was used in the current experiments, the SERTMyc cells are comparable to the cholesterol content of HEK293 and HeLa cell lines, SERT in brain synaptosomes, and platelets (Magnani et al., 2004), SERT Vmax, Ki, and Km for 5-HT uptake (Barker al., 1998; Butler et al., 1988; Codd and Walker 1987) and the magnitude of decrease in 5-HT uptake after mβCD treatment of HEK293 cells (Magnani et al., 2004; Scanlon et al., 2001). Moreover, the increase in 5-HT uptake produced by simvastatin on RN46A-B14 cells with transfected SERT is similar to the increase in 5-HT uptake by platelets of humans that have received statins (Vevera et al., 2005). Thus, the current findings can be extended to other cell types but it remains to be determined if the changes in 5-HT uptake by platelets after statin treatment are mediated through SERT and isoprenylation.
GGPP geranylgeranylates small GTPases (Hoffman et al., 2000; Joyce and Cox, 2003; Park et al., 2002) and inhibition of GGPP can increase the activity of GTPases (Dunford et al., 2006; Khan et al, 2011) that in turn, could increase the synthesis of the phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) (Weernink et al., 2003) known to bind and regulate SERT (Anderluh et al., 2017). Alternatively, inhibition of GGPP also activates kinase signaling pathways that regulate SERT (reviewed in Bermingham and Blakely 2016). Regardless, it is unknown how geranylgeranylation affects kinase signaling, but increases in small GTPase activity (Dunford et al., 2006; Kahn et al., 2011) through extended GGPP inhibition (24hr) could be involved. It also remains to be determined if statins or isoprenylation inhibition can affect changes in SERT phosphorylation, a primary modification by which SERT is regulated (Ramamoorthy et al., 1998; Ramamoorthy et al., 2007; Samuvel et al., 2005; Sørensen et al., 2014; Zarpellon et al., 2008).
These findings demonstrate the first evidence of a specific lipid-signaling pathway in monoamine transporter regulation. The results support a unique role for the inhibition of geranylgeranylation by statins and GGTI-298 in promoting uptake of 5-HT by SERT. The reduced availability of extracellular 5-HT by enhanced 5-HT uptake through SERT could explain the adverse neurophysiological effects of statin treatment (Thompson et al., 2016) that include decrements in attention, psychomotor speed, and cognition (Kotti et al., 2006; Muldoon et al., 2000; Muldoon et al., 2004; Suraweera et al., 2016). These results could pertain to a large percentage of the population given that 1 in every 4 adults over 40 years old is prescribed a statin (Gu et al., 2014). Regardless of statin side effects, our findings have broader implications and contribute to a growing interest in lipid signaling in the brain as it relates to the role of 5-HT in major depression (Walther et al., 2018), schizophrenia (Mueller and Meador-Woodruff, 2020) and Alzheimer’s disease (Jeong et al., 2018).
Acknowledgments
Funding
This work was supported by NIH DA42737, the PhRMA Predoctoral Fellowship, and the Indiana University Cagiantas Scholarship.
Abbreviations:
- CMV
cytomegalovirus
- DAPI
4′,6-diamidino-2-phenylindole
- DMEM
dulbecco’s modified eagle medium
- DTT
dithiothreitol
- FPP
farnesyl pyrophosphate
- GGPP
geranygeranyl pyrophosphate
- GGTI-298
geranylgeranyl transferase inhibitor-298
- HBSS,
Hank’s Balanced Salt Solution
- HEK 293
Human embryonic kidney 293 cells
- HMG-CoA
3-hydroxy-3-methylglutaryl-CoA
- HRP
horseradish peroxidase
- IGEPAL
octylphenoxypolyethoxyethanol
- LDS
lithium dodecyl sulfate
- mβCD
methyl-β-cyclodextrin
- MAO
monoamine oxidase
- MOPS
3-(N-morpholino)-propane sulfonate
- NGS
normal goat serum
- PMSF
phenylmethylsulfonyl fluoride
- PVDF
polyvinylidene fluoride or polyvinylidene difluoride
- 5-HT
5-hydroxytryptophan, serotonin
- SERT
serotonin transporter
Footnotes
Declaration of Interests: The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abramoff MD, Magalhaes PJ, Ram SJ. “Image Processing with ImageJ”. Biophotonics International, volume 11, issue 7, pp. 36–42, 2004. [Google Scholar]
- Amsterdam JD, Newberg AB, Newman CF, Shults J, Wintering N, and Soeller I. (2013). Change over time in brain serotonin transporter binding in major depression: effects of therapy measured with [123I]-ADAM SPECT. Journal of Neuroimaging, 23(4), 469–476. doi: 10.1111/jon.12035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderluh A, Hofmaier T, Klotzsch E, Kudlacek O, Stockner T, Sitte HH, and Schütz GJ. (2017). Direct PIP2 binding mediates stable oligomer formation of the serotonin transporter. Nature Communications, 8(1). doi: 10.1038/ncomms14089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker EL, Perlman MA, Adkins EM, Houlihan WJ, Pristupa ZB, Niznik HB, Blakely RD. (1998). High affinity recognition of serotonin transporter antagonists defined by species-scanning mutagenesis. The Journal of Biological Chemistry, 273, 19459–19468. doi: 10.1074/jbc.273.31.19459 [DOI] [PubMed] [Google Scholar]
- Bermingham DP and Blakely RD. (2016). Kinase-dependent regulation of monoamine neurotransmitter transporters. Pharmacological Reviews, 68(4):888–953. doi: 10.1124/pr.115.012260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bethea CL, Lu NZ, Reddy A, Shlaes T, Streicher JM, Whittemore SR. (2003). Characterization of reproductive steroid receptors and response to estrogen in a rat serotonergic cell line. Journal of Neuroscience Methods, 127(1):31–41. doi: 10.1016/s0165-0270(03)00095-5 [DOI] [PubMed] [Google Scholar]
- Bunin MA, Prioleau C, Mailman RB, and Wightman RM. (1998). Release and uptake rates of 5-hydroxytryptamine in the dorsal raphe and substantia nigra reticulata of the rat brain. Journal of Neurochemistry, 70(3):1077–1087. doi: 10.1046/j.1471-4159.1998.70031077.x [DOI] [PubMed] [Google Scholar]
- Butler J, Tannian M, Leonard BE. (1988). The chronic effects of desipramine and sertraline on platelet and synaptosomal 5HT uptake in olfactory bulbectomised rats. Progress in Neuropsychopharmacology & Biological Psychiatry, 12(5):585–594. doi: 10.1016/0278-5846(88)90004-8 [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. (1973). Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochemical Pharmacology, 22 (23): 3099–108. doi: 10.1016/0006-2952(73)90196-2 [DOI] [PubMed] [Google Scholar]
- Chu UB, Duellman T, Weaver SJ, Tao Y, and Yang J. (2015). Endothelial protective genes induced by statin are mimicked by ERK5 activation as triggered by a drug combination of FTI-277 and GGTI-298. Biochimica et Biophysica Acta, 1850(7):1415–1425. doi: 10.1016/j.bbagen.2015.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codd EE, Walker RF. (1987). Platelet serotonin uptake: methods and strain differences. Psychiatry Research, 44:169–178. doi: 10.1016/0165-1781(87)90051-5 [DOI] [PubMed] [Google Scholar]
- Daws LC, Montañez S, Owens WA, Gould GG, Frazer A, Toney GM, and Gerhardt GA. (2005) Transport mechanisms governing serotonin clearance in vivo revealed by high-speed chronoamperometry. Journal of Neuroscience Methods, 143:49–62. doi: 10.1016/j.jneumeth.2004.09.011 [DOI] [PubMed] [Google Scholar]
- Dunford JE, Rogers MJ, Ebetino FH, Phipps RJ, and Coxon FP. (2006). Inhibition of protein prenylation by bisphosphonates causes sustained activation of Rac, Cdc42, and Rho GTPases. Journal of Bone and Mineral Research, 21(5):684–694. doi: 10.1359/jbmr.060118 [DOI] [PubMed] [Google Scholar]
- Eaton MJ, Staley JK, Globus MY, Whittemore SR. (1995a). Developmental regulation of early serotonergic neuronal differentiation: the role of brain-derived neurotrophic factor and membrane depolarization. Developmental Biology; 170(1):169–182. doi: 10.1006/dbio.1995.1205 [DOI] [PubMed] [Google Scholar]
- Eaton MJ, Whittemore SR. (1995b). Adrenocorticotropic hormone activation of adenylate cyclase in raphe neurons: multiple regulatory pathways control serotonergic neuronal differentiation. Journal of Neurobiology; 28(4):465–481. doi: 10.1002/neu.480280407 [DOI] [PubMed] [Google Scholar]
- Eckert GP, Hooff GP, Strandjord DM, Igbavboa U, Volmer DA, Muller WE, and Wood WG. (2009). Regulation of the brain isoprenoids farnesyl- and geranylgeranylpyrophosphate is altered in male Alzheimer patients. Neurobiological Disorders, 35(2):251–257. doi: 10.1016/j.nbd.2009.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro M, Masetti M, Recanatini M, Cavalli A, and Bottegoni G (2016). Mapping cholesterol interaction sites on serotonin transporter through coarse-grained molecular dynamics. PLOS ONE, 11(12), e0166196. doi: 10.1371/journal.pone.0166196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frick A, Åhs F, Engman J, Jonasson M, Alaie I, Björkstrand J, Frans Ö, Faria V, Linnman C, Appel L, et al. (2015). Serotonin synthesis and reuptake in social anxiety disorder:a positron emission tomography study.JAMA Psychiatry, 72(8):794–802. doi: 10.1001/jamapsychiatry.2015.0125. [DOI] [PubMed] [Google Scholar]
- Hoffman GR, Nassar N, and Cerione RA. (2000). Structure of the Rho family GTP-binding protein dc42 in complex with the multifunctional regulator RhoGDI. Cell, 100(3):345–356. doi: 10.1016/s0092-8674(00)80670-4 [DOI] [PubMed] [Google Scholar]
- Ikonen E (2008). Cellular cholesterol trafficking and compartmentalization. Nature Reviews Molecular Cell Biology, 9(2), 125–138. doi: 10.1038/nrm2336 [DOI] [PubMed] [Google Scholar]
- Jeong A, Suazo KF, Wood WG, Distefano MD, and Li L. (2018). Isoprenoids and protein prenylation: implications in the pathogenesis and therapeutic intervention of Alzheimer’s disease. Critical reviews in biochemistry and molecular biology, 53(3), 279–310. doi: 10.1080/10409238.2018.1458070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson-Anuna LN, Eckert GP, Keller JH, Igbavboa U, Franke C, Fechner T, Schubert-Zsilavecz M, Karas M, Muller WE, and Wood WG. (2005). Chronic administration of statins alters multiple gene expression patterns in mouse cerebral cortex. Journal of Pharmacology and Experimental Therapeutics, 312:786–793. doi: 10.1124/jpet.104.075028 [DOI] [PubMed] [Google Scholar]
- Joyce PL and Cox AD. (2003). Rac1 and Rac3 are targets for geranylgeranyltransferase I inhibitor-mediated inhibition of signaling, transformation, and membrane ruffling. Cancer Research, 63(22):7959–7967. [PubMed] [Google Scholar]
- Khan OM, Ibrahim MX, Jonsson IM, Karlsson C, Liu M, Sjogren AK, Olofsson FJ, Brisslert M, Andersson S, Ohlsson C, et al. (2011). Geranylgeranyltransferase type I (GGTase-I) deficiency hyperactivates macrophages and induces erosive arthritis in mice. The Journal of Clinical Investigation, 121(2): 628–39. doi: 10.1172/JCI43758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandiah N and Feldman HH. (2009). Therapeutic potential of statins in Alzheimer’s disease. Journal of the Neurological Sciences, 283(1–2):230–234. doi: 10.1016/j.jns.2009.02.352 [DOI] [PubMed] [Google Scholar]
- Kotti TJ, Ramirez DM, Pfeiffer BE, Huber KM, and Russell DW. (2006). Brain cholesterol turnover required for geranylgeraniol production and learning in mice. Proceedings of the National Academy of Sciences of the United States of America, 103(10): 3869–3874. doi: 10.1073/pnas.0600316103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laursen L, Severinsen K, Kristensen KB, Periole X, Overby M, Müller HK, Schiøtt B, and Sinning S. (2018). Cholesterol binding to a conserved site modulates the conformation, pharmacology, and transport kinetics of the human serotonin transporter. Journal of Biological Chemistry, 293(10), 3510–3523. doi: 10.1074/jbc.m117.809046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loweth JA, Scheyer AF, Milovanovic M, LaCrosse AL, Flores-Barrera E, Werner CT, Li X, Ford KA, Le T, Olive MF, et al. (2014). Synaptic depression via mGluR1 positive allosteric modulation suppresses cue-induced cocaine craving. Nature Neuroscience,17(1):73–80. doi: 10.1038/nn.3590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnani F, Tate CG, Wynne S, Williams C, and Haase J. (2004). Partitioning of the serotonin transporter into lipid microdomains modulates transport of serotonin. Journal of Biological Chemistry, 279(37):38770–38778. doi: 10.1074/jbc.M400831200 [DOI] [PubMed] [Google Scholar]
- McGuire TF, Qian Y, Vogt A, Hamilton AD, Sebti SM. (1996). Platelet-derived growth factor receptor tyrosine phosphorylation requires protein geranylgeranylation but not farnesylation. Journal of Biological Chemistry, 271(44):27402–27407. doi: 10.1074/jbc.271.44.27402 [DOI] [PubMed] [Google Scholar]
- Montañez S, Owens WA, Gould GG, Murphy DL, and Daws LC. (2003). Exaggerated effects of fluvoxamine in heterozygote serotonin transporter knockout mice. Journal of Neurochemistry, 86:210–219. doi: 10.1046/j.1471-4159.2003.01836.x [DOI] [PubMed] [Google Scholar]
- Mueller TM and Meador-Woodruff JH (2020). Post-translational protein modifications in schizophrenia. NPJ Schizophrenia, 6(1):5. doi: 10.1038/s41537-020-0093-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muldoon MF, Barger SD, Ryan CM, Flory JD, Lehoczky JP, Matthews KA, and Manuck SB. (2000). Effects of lovastatin on cognitive function and psychological well-being. American Journal of Medicine, 108(7): 538–546. doi: 10.1016/s0002-9343(00)00353-3 [DOI] [PubMed] [Google Scholar]
- Muldoon MF, Ryan CM, Sereika SM, Flory JD, and Manuck SB. (2004). Randomized trial of the effects of simvastatin on cognitive functioning in hypercholesterolemic adults. American Journal of Medicine, 117(11): 823–829. doi: 10.1016/j.amjmed.2004.07.041 [DOI] [PubMed] [Google Scholar]
- Park HJ, Kong D, Iruela-Arispe L, Begley U, Tang D, and Galper JB. (2002). 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors interfere with angiogenesis by inhibiting the geranylgeranylation of RhoA. Circulation Research, 91(2):143–150. doi: 10.1161/01.res.0000028149.15986.4c [DOI] [PubMed] [Google Scholar]
- Gu Q, Paulose-Ram R, Burt VL, and Kit BK. (2014). Prescription Cholesterol-lowering medication use in adults aged 40 and over: United States, 2003–2012. NCHS Data Brief. (177):1–8. [PubMed] [Google Scholar]
- Ramamoorthy S, Giovanetti E, Qian Y, and Blakely RD. (1998). Phosphorylation and regulation of antidepressant-sensitive serotonin transporters. Journal of Biological Chemistry, 273, 2458–2466. doi: 10.1074/jbc.273.4.2458 [DOI] [PubMed] [Google Scholar]
- Ramamoorthy S, Samuvel DJ, Buck ER, Rudnick G, and Jayanthi LD. (2007). Phosphorylation of threonine residue 276 is required for acute regulation of serotonin transporter by cyclic GMP. Journal of Biological Chemistry, 282, 11639–11647. doi: 10.1074/jbc.M611353200 [DOI] [PubMed] [Google Scholar]
- Saheki A, Terasaki T, Tamai I, and Tsuji A. (1994). In vivo and in vitro blood-brain barrier transport of 3-hydroxy-3-methylglutaryl coenzyme a (hmg-coa) reductase inhibitors. Pharmaceutical Research, 11(2), 305–311. doi: 10.1023/a:1018975928974 [DOI] [PubMed] [Google Scholar]
- Samuvel DJ, Jayanthi LD, Bhat NR, and Ramamoorthy S. (2005). A role for p38 mitogen-activated protein kinase in the regulation of the serotonin transporter: Evidence for distinct cellular mechanisms involved in transporter surface expression. Journal of Neuroscience, 25, 29–41. doi: 10.1523/JNEUROSCI.3754-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry PS. (1985). Lipids of nervous tissue: composition and metabolism. Progress in Lipid Research, 24 69–176. doi: 10.1016/0163-7827(85)90011-6 [DOI] [PubMed] [Google Scholar]
- Suraweera C, de Silva V, Hanwella R. (2016). Simvastatin-induced cognitive dysfunction: two case reports. Journal of Medical Case Reports, 10:83. doi: 10.1186/s13256-016-0877-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanlon SM, Williams DC, and Schloss P. (2001). Membrane cholesterol modulates serotonin transporter activity. Biochemistry, 40(35), 10507–10513. doi: 10.1021/bi010730z [DOI] [PubMed] [Google Scholar]
- Serrano-Pozo A, Vega GL, Lütjohann D, Locascio JJ, Tennis MK, Deng A, Atri A, Hyman BT, Irizarry MC, and Growdon JH. (2010). Effects of Simvastatin on cholesterol metabolism and Alzheimer disease biomarkers . Alzheimer Disease & Associated Disorders, 24(3):220–6. doi: 10.1097/wad.0b013e3181d61fea [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaskan EG and Snyder SH. (1970). Kinetics of serotonin accumulation into slices from rat brain: relationship to catecholamine uptake. Journal of Pharmacology and Experimental Therapeutics, 175(2):404–418. [PubMed] [Google Scholar]
- Smith WS, Baker EJ, Holmes SE, Koster G, Hunt AN, Johnston DA, Flavell SU, & Flavell DJ. (2017). Membrane cholesterol is essential for triterpenoid saponin augmentation of a saporin-based immunotoxin directed against CD19 on human lymphoma cells. Biochimica et Biophysica Acta (BBA) - Biomembranes, 1859(5), 993–1007. 10.1016/j.bbamem.2017.02.013 [DOI] [PubMed] [Google Scholar]
- Sørensen L, Strømgaard K, and Kristensen AS. (2014). Characterization of intracellular regions in the human serotonin transporter for phosphorylation sites. ACS Chemical Biology, 9(4):935–944. doi: 10.1021/cb4007198 [DOI] [PubMed] [Google Scholar]
- Stansley BJ, Yamamoto BK. (2013). L-dopa-induced dopamine synthesis and oxidative stress in serotonergic cells. Neuropharmacology; 67:243–251. doi: 10.1016/j.neuropharm.2012.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavoulari S, Forrest LR, and Rudnick G. (2009). Fluoxetine (Prozac) binding to serotonin transporter is modulated by chloride and conformational changes. Journal of Neuroscience, 29(30), 9635–9643. doi: 10.1523/jneurosci.0440-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson PD, Panza G, Zaleski A, and Taylor B. (2016). Statin-associated side effects. Journal of the American College of Cardiology, 67(20): 2395–2410. doi: 10.1016/j.jacc.2016.02.071 [DOI] [PubMed] [Google Scholar]
- Vevera J, Fisar Z, Kvasnicka T, Zdenek H, Stárková L, Ceska R, Papezová H. (2005). Cholesterol-lowering therapy evokes time-limited changes in serotonergic transmission. Psychiatry Research, 133(2–3):197–203. doi: 10.1016/j.psychres.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Walther A, Cannistraci CV, Simons K, Durán C, Gerl MJ, Wehrli S, & Kirschbaum C. (2018). Lipidomics in major depressive disorder. Frontiers in psychiatry, 9, 459. doi: 10.3389/fpsyt.2018.00459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weernink PAO, Meletiadis K, Hommeltenberg S, Hinz M, Ishihara H, Schmidt M, & Jakobs KH. (2003). Activation of type i phosphatidylinositol 4-phosphate 5-kinase isoforms by the Rho GTPases, RhoA, Rac1, and Cdc42. Journal of Biological Chemistry, 279(9), 7840–7849. doi: 10.1074/jbc.m312737200 [DOI] [PubMed] [Google Scholar]
- White LA, Eaton MJ, Castro MC, et al. (1994). Distinct regulatory pathways control neurofilament expression and neurotransmitter synthesis in immortalized serotonergic neurons. Journal of Neuroscience; 14(11 Pt 1):6744–6753. doi: 10.1523/JNEUROSCI.14-11-06744.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteley L, Haug M, Klein K, Willmann M, Bohn E, Chiantia S, Schwarz S. (2017). Cholesterol and host cell surface proteins contribute to cell-cell fusion induced by the Burkholderia type VI secretion system 5. PLoS One;12(10):e0185715. doi: 10.1371/journal.pone.0185715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarpellon A, Donella-Deana A, Folda A, Turetta L, Pavanetto M, and Deana R. (2008) Serotonin (5-HT) transport in human platelets is modulated by Src-catalysed Tyr-phosphorylation of the plasma membrane transporter SERT. Cellular Physiology and Biochemistry, 21, 87–94. doi: 10.1159/000113750 [DOI] [PubMed] [Google Scholar]
- Zhang YW and Rudnick G. (2006). The cytoplasmic substrate permeation pathway of serotonin transporter. Journal of Biological Chemistry, 281(47), 36213–36220. doi: 10.1074/jbc.m605468200 [DOI] [PubMed] [Google Scholar]
- Zhao ZS, Manser E, Chen XQ, Chong C, Leung T, Lim L. (1998). A conserved negative regulatory region in alphaPAK: inhibition of PAK kinases reveals their morphological roles downstream of Cdc42 and Rac1. Molecular and Cellular Biology, 18(4):2153–2163. doi: 10.1128/mcb.18.4.2153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu CB, Blakely RD, and Hewlett WA. (2006). The proinflammatory cytokines interleukin-1beta and tumor necrosis factor-alpha activate serotonin transporters. Neuropsychopharmacology, 31(10):2121–2131. doi: 10.1038/sj.npp.1301029 [DOI] [PubMed] [Google Scholar]