

Abstract
OMA1 is a mitochondrial protease. Among its substrates are DELE1, a signaling peptide, which can elicit the integrated stress response, as well as the membrane-shaping dynamin-related GTPase OPA1, which can drive mitochondrial outer membrane permeabilization. OMA1 is dormant under physiological conditions but rapidly activated upon mitochondrial stress, such as loss of membrane potential or excessive reactive oxygen species. Accordingly, OMA1 was found to be activated in a number of disease conditions, including cancer and neurodegeneration. OMA1 has a predicted transmembrane domain and is believed to be tethered to the mitochondrial inner membrane. Yet, its structure has not been resolved and its context-dependent regulation remains obscure. Here, I review the literature with focus on OMA1’s biochemistry. I provide a good homology model of OMA1’s active site with a root-mean-square deviation of 0.9 Å and a DALI Z-score of 19.8. And I build a case for OMA1 actually being an integral membrane protease based on OMA1’s role in the generation of small signaling peptides, its functional overlap with PARL, and OMA1’s homology with ZMPSTE24. The refined understanding of this important enzyme can help with the design of tool compounds and development of chemical probes in the future.
Introduction
Eukaryotic cells over the course of evolution became compartmentalized thereby resembling an integrated system with distinct functional units that are in constant communication with one another. The nucleus for example holds the gene-regulatory dominance in this system and converses with other cell organelles, broadly speaking, by means of transcription and translation. Mitochondria on the other hand occupy a central position within eukaryotic cells in the regulation of energy-metabolic pathways. However, their means of communication with other cell compartments are less well understood. Recently, a new pathway comprising DELE1 was uncovered, which activated the integrated stress response upon mitochondrial malfunctioning (Fig. 1). DELE1 was found to be cleaved by the mitochondrial OMA1 protease in a context-dependent manner and released into the cytosol, where it activated the CHOP-signaling pathway [1, 2]. CHOP controls transcription of (integrated) stress-response genes upon accumulation of misfolded proteins in mitochondria and other compartments [3]. While this might point to proteolysis and discharge of signaling peptides as a more general manifestation of mitochondrial communications, I would like to focus here on the OMA1 protease and discuss its function and regulation in more details. OMA1 functionally overlaps with PARL and shows homology with ZMPSTE24, which suggests OMA1 is an integral membrane protease. A carboxy-terminal region in juxtaposition of the enzyme’s active center could serve as gatekeeper limiting substrate access or recognition. This model can help with the design of OMA1 inhibitors and—considering OMA1’s multiple disease implications—in due course guide the development of future drugs.
Figure 1:
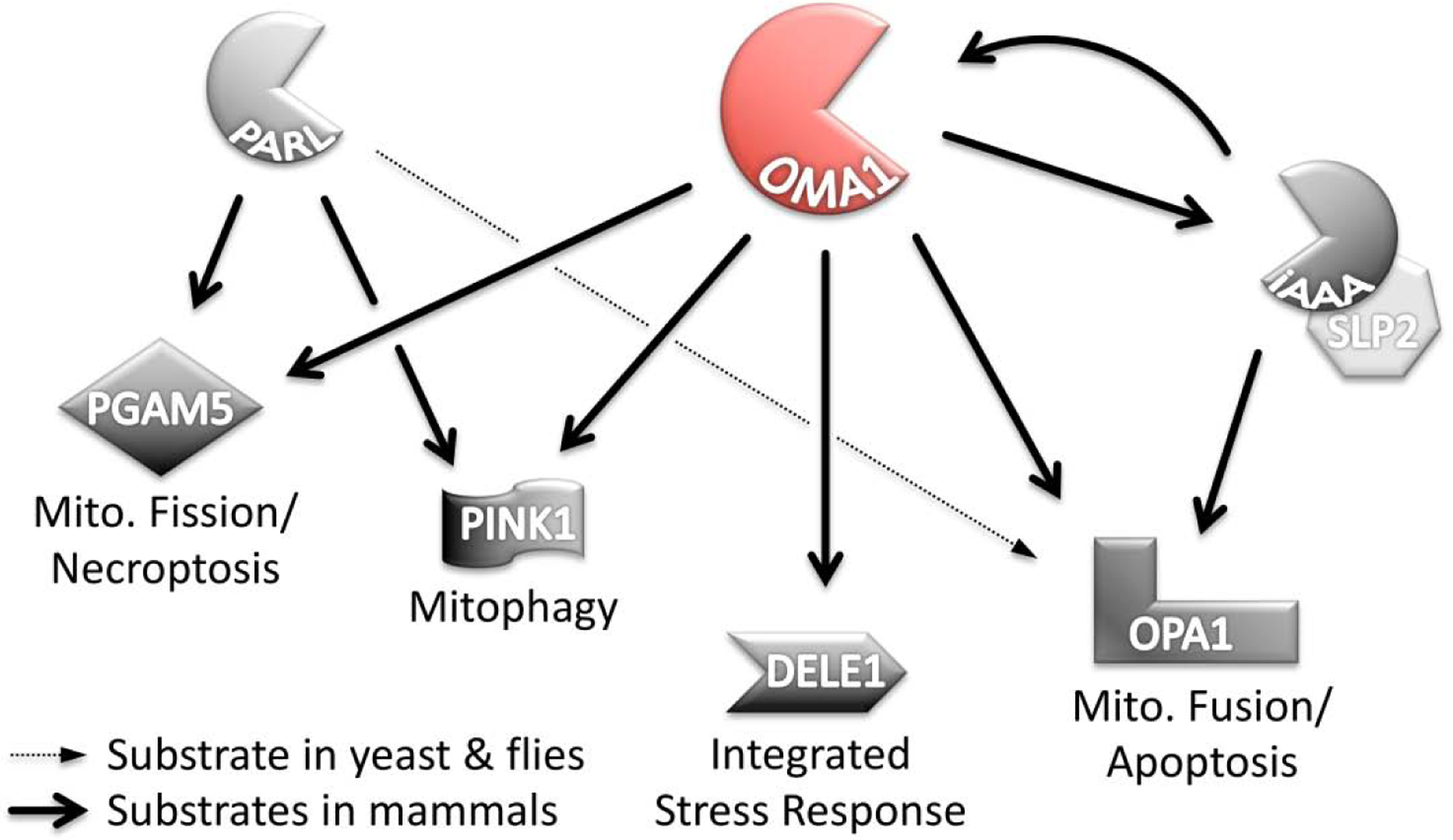
OMA1 shares substrates with PARL and the i-AAA protease. OMA1 can hydrolyze DELE1, OPA1, PGAM5 and PINK1, which leads to necroptosis, mitophagy, activation of the integrated stress response or apoptosis. OMA1 cleaves OPA1 at the S1 site, while the i-AAA protease cleaves OPA1 in close proximity at S2. Both proteases can digest each other in a context-dependent manner as well. PARL cleaves PGAM5 and PINK1, which both can be recognized by OMA1, too. PARL can hydrolyze OPA1 in yeast and flies but not in mammalian organisms.
Classification and Basic Function
OMA1 is distinct because the enzyme shows only little protease activity under physiological conditions, which raises questions of its context-dependent regulation. OMA1 is a conserved metalloendopeptidase in the mitochondrial inner membrane with proteolytic activity in the intermembrane space [4]. The enzyme has a HExxH Zn2+-binding motive and is classified according to the MEROPS database as M48 family member [5]. The OMA1 name is deduced from overlapping activity with m-AAA proteases. OMA1’s similarities and differences with the mitochondrial AAA proteases will be discussed below. For now, suffice it to say that OMA1—unlike AAA enzymes—works ATP-independently and has no ATP-binding domain. So how is OMA1 regulated then? The human OMA1 protein has 524 amino acids and a predicted molecular weight of 60 kDa (Uniprot: Q96E52). Yet, the mature OMA1 protein lacks its first 143 amino acids and migrates under denaturing gel-electrophoresis conditions just short of 40 kDa [6]. This implies OMA1 has either an amino-terminal import sequence or it is encoded as an apoprotein, or both. The short, mature form is functional (though not active), because it showed context-dependent activation when ectopically expressed in OMA1-null cells [6]. Interestingly, OMA1 starts to degrade upon activation as demonstrated by the complete disappearance of the OMA1 protein in Western-blots over time [6–8]. OMA1’s activation happens rapidly and the fading OMA1 signal can be noticed within minutes. OMA1’s degradation thus limits the time of the enzyme’s activity. This leaves us with the question: how is OMA1 activated?
Experimentally, OMA1 can be activated by a number of reagents that induce mitochondrial stress in one way or another (Table 1). The mitochondrial uncoupler CCCP is noteworthy because it led to the initial discovery of OMA1 [9, 10]. Oxidizing agents, such as hydrogen peroxide, rotenone or paraquat [6, 11, 12], cytotoxins, such as cisplatin [13], and antibiotics, such as actinonin, oligomycin, staurosporine or valinomycin can activate OMA1 in cell culture experiments as well [6, 9, 10, 14]. But not only pharmacological interventions trigger the enzyme, also genetic manipulations can result in OMA1 activation. For example, conditional expression of the BCL2 family proteins BID and BCL2L11 (BIM) in human osteosarcoma cells was found to activate OMA1 [15]. Loss of the complement C1q binding protein p32/C1QBP led to OMA1-dependent morphological changes of mitochondria [16]. And CHCHD2 mutations found in patients with Parkinson’s disease or CHCHD10 mutations found in patients with frontotemporal dementia or amyotrophic lateral sclerosis led to OMA1 activation when expressed in Hek293T or HeLa cells [17]. Embryonic fibroblasts from CHCHD2 knock-out mice showed enhanced OMA1 activity, too, which was further amplified by concomitant CHCHD10 knockout [17]. Genetic knockdown of the mitochondrial scaffolding proteins PHB2 and SLP2 appear to result in OMA1 activation as well [18, 19]. PHB2 and SLP2 are similar in their function in that they help organize lipid microdomains thereby controlling the AAA-proteases [20], which remove proteins from the inner membrane [21, 22]. The scaffolding proteins are believed to interact with distal parts of the AAA protease from the opposite side of the membrane. PHB2 binds in the intermembrane space to the m-AAA protease, which protrudes its active core into the matrix [23]. SLP2 binds at the same time on the matrix side to the i-AAA protease with its machinery extending into the intermembrane space [24]. But then SLP2 and PHB2 interact with one another suggesting a higher order organization of the AAA proteases all together [25]. Yet, it is peculiar that knockdown of the m-AAA protease in cell-culture studies resulted in OMA1 activation [9], but knockdown of the i-AAA protease did not [11].
Table 1:
Reagents triggering OMA1 activation.
| Reagent | Concentrations* | Referenece |
|---|---|---|
| Carbonyl cyanide m-chlorophenyl hydrazone (CCCP) | 10 µM or 20 µM for 30 min | Ehse et al. 2009 [9], Head et al. 2009 [10] |
| Oligomycin | 2 µM for 2 h | Ehse et al. 2009 [9] |
| Staurosporine | 2 µM for 3 h | Head et al. 2009 [10] |
| Actinonin | 150 µM for 6 h | Richter et al. 2013 [14] |
| Hydrogen peroxide (H2O2) | 200 µM for 3 h to 1 mM for 7 h | Anand et al. 2014 [11], Rainbolt et al. 2015 [12] |
| Paraquat | 5 mM for 8h | Baker et al. 2014 [6] |
| Rotenone | 50 µM for 10 h | Baker et al. 2014 [6] |
| Valinomycin | 1 µM for 1 h | Baker et al. 2014 [6] |
| Cisplatin | 5 µM for 24 h | Kong et al. 2014 [13] |
from the referenced studies, not necessarily indicative of potency.
OMA1 Interactions
Both i-AAA and m-AAA proteases are membrane-anchored, barrel-shaped oligomeric complexes, which are analogous to the proteasome in that they have a central pore-opening, an ATPase-powered protein-unfolding machinery, and a proteolytic cavity. The YME1L1 protein is the basic building block for the hexameric i-AAA protease, which shares the OPA1 protein as substrate with the OMA1 protease (Fig. 1). OPA1 is a membrane-shaping dynamin-related GTPase, which exists in a number of isoforms encoded by alternative splice-variants and/or generated by selective proteolysis of its amino-terminal membrane anchor [26, 27]. The ratios of the isoforms are critical determinants of OPA1’s function and are regulated by the i-AAA protease under physiological conditions [28–32]. OMA1 on the other hand can cleave all OPA1 species upon its activation, which is functionally connected to mitochondrial outer membrane permeabilization [33, 34]. OPA1 itself is not needed for OMA1 activation, because OMA1’s context-dependent DELE1 cleavage was not altered in OPA1 knockdown cells [1]. It is currently not known whether DELE1 is also an i-AAA protease substrate. But inhibition of another mitochondrial AAA protease, LONP1, did not result in OMA1-dependent DELE1 cleavage [1].
The aforementioned studies were all conducted in (mammalian) cell culture, which can help discern secondary effects and developmental defects prevalent in animal studies. To give an example, YME1L1 knockout has little impact on OMA1’s regulation in mouse embryonic fibroblasts [11]. Yet conditional YME1L1 knockout mice developed an OPA1-dependent cardiomyopathy ameliorated by simultaneous OMA1 knockout [35]. Conditional YME1L1 knockout in the mouse nervous system on the other hand resulted in developmental defects, which were aggravated by simultaneous OMA1 knockout [36]. It is conceivable that loss of YME1L1 in cardiomyocytes resulted in mitochondrial stress that triggered OMA1, while in the latter example OMA1 partially compensated for YME1L1 in the developing nervous system. Again, both OMA1 and YME1L1 can hydrolyze the OPA1 protein, which is essential for the development of flies, fish, mice and other models, including the differentiation of pluripotent stem cells [37–42]. But there is more to it. YME1L1 requires ATP for its activity [43], and YME1L1 became an OMA1 substrate itself in cells depleted of ATP and treated with hydrogen peroxide [12, 44]. To further complicate the matter, OMA1 was degraded by YME1L1 in the presence of ATP (or non-hydrolysable ATP analogs) upon hydrogen peroxide treatment [7, 8]. Mechanistically, hydrogen peroxide may impact intramolecular OMA1 and YME1L1 disulfide bonds [44, 45]. However, another study noted the accumulation of OMA1 degradation products in the absence of YME1L1 [46]. And from experiments with CCCP it was deduced that OMA1—once activated—degrades itself autocatalytically, because protease-dead mutant OMA1 was stable and did not vanish [6, 7]. Interestingly, this parallels experiments with protease-dead YME1L1, which was stable under hypoxic conditions indicating autocatalytic YME1L1 turn-over as well [47]. The current interpretation of all these results is that both OMA1 and i-AAA can degrade themselves and one another depending on the mitochondrial membrane potential (∆ψ), reactive oxygen species, and ATP levels thereby fine-tuning mitochondrial capacity and threshold levels for apoptosis [8, 48, 49]. The conditions under which all these experiments were conducted, however, are beyond physiological concentrations and illustrate some limitations of cellular studies.
What do we know about the eponymous m-AAA protease and its relationship with OMA1? AFG3L2 or AFG3L2 together with SPG7 (paraplegin) form homo- and hetero-hexameric m-AAA proteases with variable distributions across tissues and cell types [50, 51]. OMA1 is activated upon AFG3L2 knockdown in cellular assays, presumably in AFG3L2 knockout mice, and in patient-derived fibroblasts harboring pathogenic AFG3L2 mutations [9, 52, 53]. AFG3L2 is linked to a spectrum of disorders ranging from optic neuropathy to spinocerebellar ataxia (type 28, SCA28) to other neurodegenerative conditions depending on the type and inheritance of the mutation, and whether they impair the ATPase, or the protease domain [54, 55]. Paraplegin overexpression had some effects on OPA1 hydrolysis, which was slightly increased when both proteins were ectopically expressed (with a tag) in HeLa cells [26]. On the other hand, loss of paraplegin appears to have no impact on OMA1, because OPA1 cleavage (evoked by AFG3L2 knockdown) was consistent in mouse embryonic fibroblasts from wildtype and paraplegin knockout animals [9]. The authors of another study observed the accumulation of OMA1’s 60 kDa form in cells depleted of AFG3L2 while the 40 kDa mature enzyme diminished, which led them to conclude OMA1 could be an AFG3L2 substrate [56]. However, it is not fully understood if AFG3L2 knockdown in these experiments just disturbed mitochondrial function to a certain degree that impaired protein import. Comparable experiments with CCCP revealed that disruption of the membrane potential ∆ψ not only activated OMA1 but also stopped OMA1’s import, which resulted in accumulation of the 60 kDa form as well [10]. Nonetheless assuming OMA1 is indeed an AFG3L2 substrate, then OMA1’s lifecycle could present as follows: OMA1 is imported into mitochondria, where m-AAA proteases support its maturation and i-AAA proteases lead to OMA1’s degradation. Unless OMA1 becomes active, that is, because then OMA1 starts to self-degrade. The critical question though remains: how is OMA1 activated?
The AAA proteases interact with a number of proteins which can influence OMA1’s activity (Fig. 2). For example, the matrix protein DNAJC19 (TIM14) was fairly recently identified in a proteomics screen as PHB2 and AFG3L2 interaction partner [57]. Depletion of DNAJC19 in Hek293T cells led to OMA1-dependent OPA1 cleavage in this study, and the same remained true for PHB2 knock-down. Likewise, conditional neuronal PHB2 knockout mice showed increased OPA1 proteolysis in the brain along with a fatal tauopathy phenotype [58]. Yet, these mice lived significantly longer and had milder neurodegeneration on an OMA1 null genetic background, which places OMA1 downstream of PHB2 [59]. Interestingly, neuronal AFG3L2 knockout mice also developed tauopathy, which was comparable to the PHB2 phenotype [60]. So, for once the physical interactions aligned with the genetic interactions. PHB2 is one of two codependent prohibitins; PHB1 being the other one. Knockdown of either prohibitin results in the loss of the second one [c.f. 46, 61, 62, 63]. PHB1 silencing destructed the mitochondrial network [61], which implies increased OPA1 cleavage and elevated OMA1 activity. The cytotoxin aurilide was found to be a potent PHB1 inhibitor, and aurilide induced mitochondrial fragmentation and OPA1 cleavage as well [64]. Another study described an inverse correlation of prohibitin and OMA1 levels, whereby silencing of PHB1 resulted in accumulation of mature OMA1 (but not in hyperactivation). Prohibitin overexpression on the other hand diminished OMA1 protein levels, delayed BID-induced apoptosis, but did not prevent CCCP-induced OMA1 activation [46]. The aforementioned SLP2 is another component of the prohibitin complex, which upon deletion altered the OPA1 cleavage pattern [18]. PARL, a rhomboid protease in the inner membrane [65], formed a complex in a proteomics screen mainly with SLP2 and YME1L1, but also prohibitin and AFG3L2 among others [24]. Loss of PARL also altered OPA1 processing in vitro and in vivo—yet independent from CCCP-treatment [66, 67]. This is most likely an indirect interaction presumably mediated by OMA1 or YME1L1, because mammalian OPA1 is not recognized by PARL [26, 68]. The bottom line is that a conglomerate of proteases and structural proteins can directly or indirectly impact OMA1’s function, which on top of this appears to be functionally interrelated with cardiolipin [46, 47, 57], an abundant inner membrane phospholipid [69–71].
Figure 2:
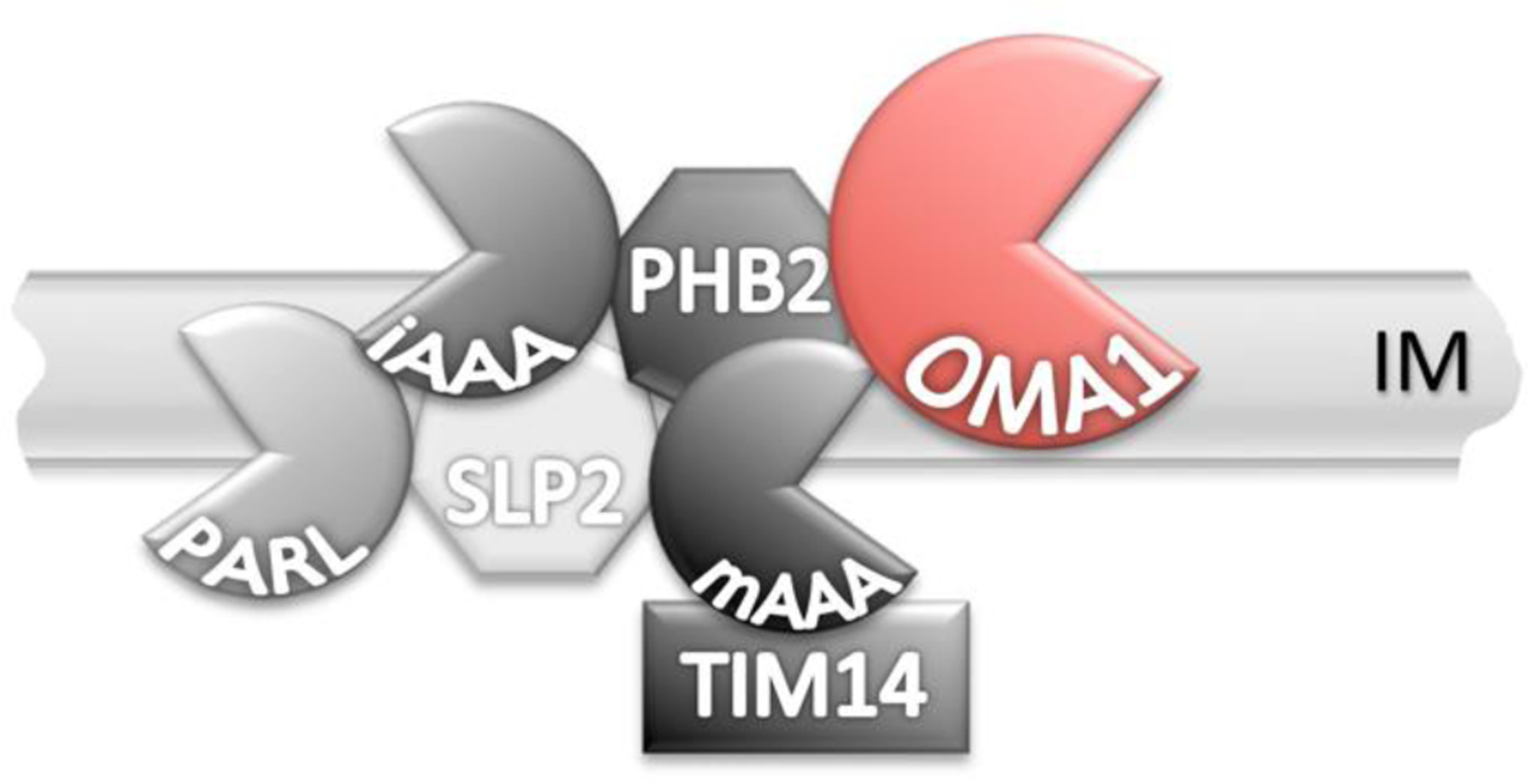
The AAA proteases are organized in a higher order complex. i-AAA faces the intramembrane space and can interact with SLP2 and PARL on the opposite site of the inner membrane (IM). m-AAA on the matrix side can interact with the prohibitins (PHB1 and PHB2) and presumably OMA1 on the intramembrane space-facing side of the IM. Genetic alterations of any of these members of the AAA protein complex impact OMA1 function.
OMA1—An Integral Membrane Protease?
PARL is worth a closer look despite the seemingly secondary effects on OMA1 and OPA1. PARL is mainly studied in the context of Parkinson’s disease for hydrolyzing PINK1 in the inner membrane [72–74]. PINK1 together with parkin and other Parkinson’s-associated proteins regulates mitochondrial autophagy—or mitophagy for short [75]. PARL sequentially proteolyzes PINK1’s amino-terminus just to release it into the cytosol where it is degraded by the N-end rule pathway [76, 77]. In malfunctioning mitochondria PINK1 is not digested by PARL anymore; PINK1 accumulates on the mitochondrial outer membrane, where it can initiate mitophagy [78–80]. Interestingly, mutations in PINK1’s import sequence resulted in incomplete mitochondrial translocation and subsequent degradation by OMA1 [81]. This means OMA1 can complement PARL’s function by hydrolyzing misrouted PINK1. PARL and OMA1 have one more substrate in common, PGAM5, which is a serine/threonine protein phosphatase implied in the regulation of the mitochondrial fission protein DRP1 among others [24, 82]. And as contemplated above, OMA1 can also complement YME1L1’s function processing OPA1 during development [36]. In this context it is remarkable that OPA1 in yeast (called MGM1) is processed by PARL (PCP1) and not OMA1 [83, 84]. OPA1 in flies—for OMA1’s absence—is also processed by PARL (rhomboid-7) [85]. So OMA1 appears to have taken on some of PARL’s duties over the course of evolution. PARL is an intramembrane cleaving protease—a mitochondrial inner membrane i-CLiP [65, 86, 87]. Is it plausible then that OMA1 is also an intramembrane cleaving protease? OMA1 cleaves OPA1 at the S1 cleavage site after arginine 194 (NP_056375). The i-AAA protease cleaves at the S2 cleavage site anywhere between 14 and 53 amino acids further downstream of S1 [26, 27]. OPA1 has a predicted carboxy-terminal transmembrane anchor from amino acid 97 to 113. For this reason, there is a compelling case to be made that OMA1 acts closer to the membrane than YME1L1.
PARL recognizes substrates from the matrix side [88] and interacts with the structural SLP2 protein and the i-AAA protease [24]. Could OMA1 on the other side of the bilayer interact in a similar manner with the structural prohibitin(s) and the m-AAA protease (see Fig. 2)? OMA1 was detected in yeast and mammalian cell extracts in higher molecular weight complexes [4, 6, 89, 90]. Pull-down experiments with two differently tagged OMA1 proteins further suggested OMA1 can form oligomers [6]. But then OMA1 and OPA1 also comigrated in higher order complexes [6]. A proteomics screen with PHB2 as bait found AFG3L2, YME1L1 and SLP2 among other proteins, but did not explicitly specify OMA1 as a significant PHB2-interacting partner [57]. Still, OMA1 comigrated with PHB1 and OPA1 under native gel-electrophoresis conditions in another study, which also identified a physical interaction with cardiolipin mediated through a hydrophobic region at the enzyme’s amino-terminal end [46]. Because membrane proteins are hydrophobic and have an inherent affinity for lipids, it is quite challenging to experimentally discern direct from indirect protein-protein interactions. Some interactions may also be transient in nature or occur only under certain conditions.
A predicted transmembrane domain at the amino-terminal end of the mature enzyme is believed to fix OMA1 to the inner membrane with the amino-terminal end in the matrix and its active site oriented towards the intermembrane space [4, 91, 92]. Yet, OMA1 belongs to the M48 family of metalloendopeptidases together with the zinc metallopeptidase STE24 (ZMPSTE24, also known as farnesylated-protein converting enzyme 1, FACE1). ZMPSTE24 is an integral membrane protein which cleaves the nuclear envelope protein prelaminin A [93]. ZMPSTE24 has seven α-helices that create a barrel-shaped cavity within the membrane [94]. While the barrel is buried in the bilayer, ZMPSTE24’s active site is situated right at its surface. The active site is formed by two helices, which are protruding from the membrane, and an enclosing β-strand sheet, which seals the barrel from the cytosol [95, 96]. (So technically ZMPSTE24 is not an i-CLiP because the active center is not in the membrane.) The architecture of ZMPSTE24’s active site assorts well with the Glu-zincin family. The catalytic Zn2+ is coordinated by the two histidines of the HExxH motif on transmembrane domain VI and a glutamate of α-helix VII. The β-sheets are formed by the loop connecting transmembrane domains V and VI. OMA1’s structure has not yet been resolved. Nonetheless homology models of the active site are fairly good with 18 % sequence identity, a DALI Z-score of 19.8 [97], and a root-mean-square deviation of 0.9 Å (Fig 3). Overall, the mature OMA1 protein could have six or seven α-helix that traverse the membrane (Fig 4), depending on whether α-helix VI crosses the membrane or is broken within the plane. The presence of proline residues suggests the latter. On the other hand, S2P is another intramembrane cleaving peptidase, which has a split α-helix that traverses the membrane [98]. Computer algorithms predict two to three transmembrane domains [99]. In silico studies further suggest OMA1’s α-helices have evenly spaced hydrophobic amino acids, which could indicate the formation of a barrel-shaped cavity in the membrane (Fig 4). In such a model OMA1’s active site is formed by the (extended) α-helical transmembrane domains IV and V and the β-sheet blades that connect α-helices III and IV (see Fig. 3). Therefore, OMA1 could indeed represent a novel integral membrane protease with a reaction chamber comparable to ZMPSTE24. Alternatively, OMA1 could form a multipass membrane protein similar to the rhomboid protease PARL, with which it shares functional homology.
Figure 3:
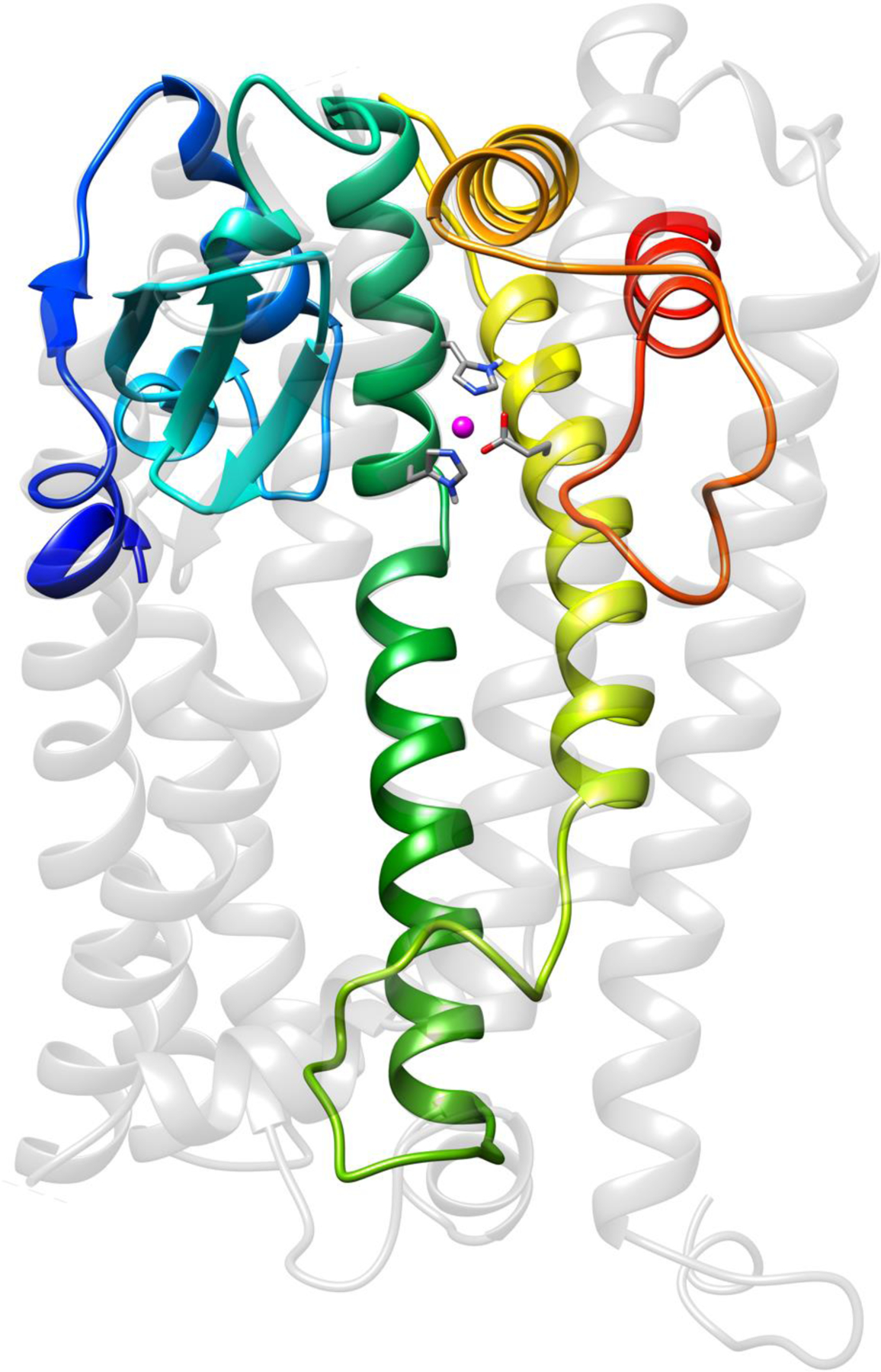
Homology model of OMA1’s active site based on ZMPSTE24 (PDB ID: 4AW6; shown greyed out.) The model is oriented with the amino-terminal end to the left (putative transmembrane domain III is depicted in blue) and the outset of the carboxyl end on the right (red). The catalytic zinc (pink) is coordinated by two histidines and a glutamate on transmembrane domains IV and V (green and yellow, respectively).
Figure 4:
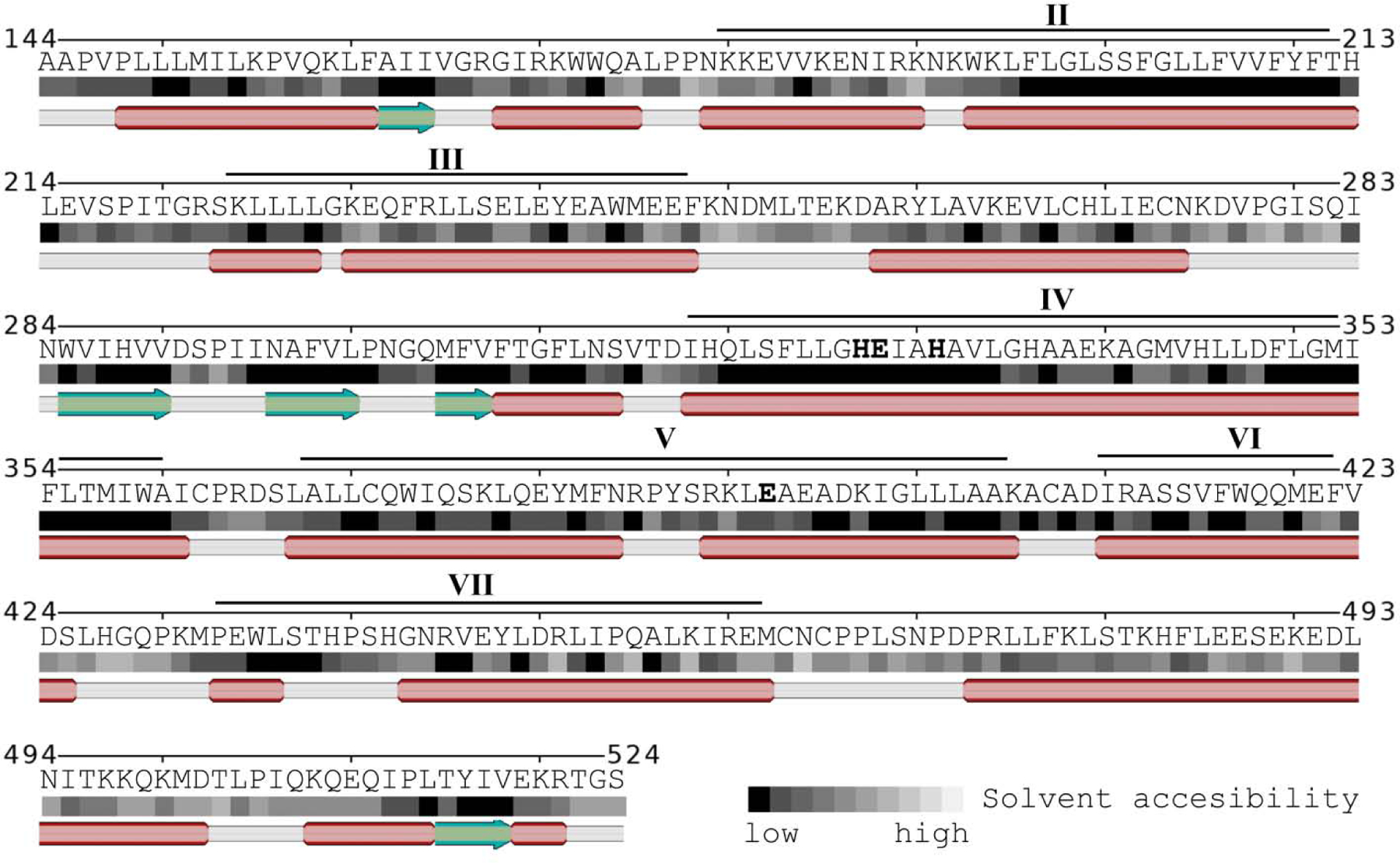
The mature OMA1 protein has a number of α-helices (red) with evenly spaced hydrophobic amino acids with low solvent accessibility (dark squares), which could form a protein with overall six or seven transmembrane domains (indicated by roman numbers). The β-sheets (green) connecting transmembrane III and IV form part of the active site stabilized by a short α-helix. The zinc-binding histidines and glutamate on transmembrane domains IV and V are printed in bold.
Implications
Concluding, it is quite possible that OMA1 is an autonomous integral membrane protease, though unlikely an i-CLiP. Three independent observations support this notion. First, OMA1’s participation in regulated proteolysis of short signaling peptides, which is a hallmark of integral membrane proteases. The discovery that OMA1 is involved in the generation of DELE1 fragments that can trigger the integrated stress response strongly supports this concept. The cleavage product S-OPA1 could be seen as a signaling peptide in the context of programmed cell death, too [33]. And it is tempting to speculate that OMA1’s fine-tuning of the energy metabolism could be mediated by signaling peptides as well. Homozygous OMA1 knock-out mice are viable but do show metabolic impairments [100]. Second, OMA1’s functional overlap with the integral membrane protease PARL suggests that OMA1 could be an integral membrane protease. PARL’s active site is rather promiscuous [101] and specificity is established through substrate recognition domains apart from the active site [88, 102], which seems a common feature of integral membrane proteases. For example, ZMPSTE24 has potentially broader substrate specificity as well, as it can also clear proteins from the clogged translocon [103, 104]. And γ-secretase, which is critical for Alzheimer’s disease, is an intramembrane cleaving protease complex safeguarding its membrane-exposed active center by extramembrane portions of the complex [105, 106]. Parts of OMA1’s α-helix VI are in juxtaposition to the active cleft and could function as gate-keeper or be involved in substrate recognition. Short 5 or 10 amino acid deletions in this region prevented self-cleavage while still preserving OMA1’s proteolytic activity towards OPA1 [7]. This establishes α-helix VI’s proximity to the active center. This region of the protein could therefore present an OMA1 substrate in itself or be involved in OMA1’s regulation. And last but not least, OMA1’s homology with the integral membrane protease ZMPSTE24 is a strong indicator that OMA1 is an integral membrane protease. Membrane proteases are inherently challenging to drug and to date no specific OMA1 inhibitors have been disclosed. Epigallocatechin gallate (EGCG), a polyphenol from green tea, was found to reduce hydrogen peroxide-dependent OMA1 activation [107]. The authors of this study speculate EGCG could bind to OMA1. However, they did not thoroughly address whether EGCG functioned as reactive oxygen species scavenger in their experiments. Two other studies showed that chloramphenicol could prevent OMA1 activation upon actinonin-treatment and AFG3L2-knock-down, thereby adding proteotoxicity to the list of alleged OMA1 activators [14, 108]. Chloramphenicol is an inhibitor of protein synthesis on mitochondrial ribosomes [109]. Then again ZMPSTE24 is a known off-target for a number of HIV1 protease inhibitors [110–112] and it would be interesting to see whether any of these can inhibit OMA1.
OMA1’s context-dependent regulation is still not fully understood and clearly needs more investigation. At this stage, it also cannot be ruled out that OMA1’s carboxy-terminal region is involved in protein-protein interactions, for example forming dimers or oligomers like the AAA proteases. OMA1’s physical interactions with lipids and functional interactions with the enzymes mediating their synthesis clearly needs more research as well. Potent and specific tool compounds that can modulate OMA1’s function in a precise and defined way would certainly help answer many of the open questions around this important enzyme. Considering OMA1 an integral membrane protease may help with the identification and design of the first OMA1 inhibitors.
Acknowledgements
Thank you for the generous support of my research by the National Institute on Aging (NIA) of the National Institutes of Health (NIH) under Award Number R43AG063642 and by the Department of Energy’s Lawrence Berkeley National Laboratory in Berkeley, CA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
Dr. Marcel V. Alavi is shareholder of 712 North Inc., a California-based pharmaceutical company.
References
- [1].Fessler E, Eckl EM, Schmitt S, Mancilla IA, Meyer-Bender MF, Hanf M, Philippou-Massier J, Krebs S, Zischka H, Jae LT, A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol, Nature 579 (2020) 433–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Guo X, Aviles G, Liu Y, Tian R, Unger BA, Lin YT, Wiita AP, Xu K, Correia MA, Kampmann M, Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway, Nature 579 (2020) 427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D, An integrated stress response regulates amino acid metabolism and resistance to oxidative stress, Mol Cell 11 (2003) 619–633. [DOI] [PubMed] [Google Scholar]
- [4].Kaser M, Kambacheld M, Kisters-Woike B, Langer T, Oma1, a novel membrane-bound metallopeptidase in mitochondria with activities overlapping with the m-AAA protease, J Biol Chem 278 (2003) 46414–46423. [DOI] [PubMed] [Google Scholar]
- [5].Rawlings ND, Waller M, Barrett AJ, Bateman A, MEROPS: the database of proteolytic enzymes, their substrates and inhibitors, Nucleic Acids Res 42 (2014) D503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Baker MJ, Lampe PA, Stojanovski D, Korwitz A, Anand R, Tatsuta T, Langer T, Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics, EMBO J 33 (2014) 578–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhang K, Li H, Song Z, Membrane depolarization activates the mitochondrial protease OMA1 by stimulating self-cleavage, EMBO Rep 15 (2014) 576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Rainbolt TK, Lebeau J, Puchades C, Wiseman RL, Reciprocal Degradation of YME1L and OMA1 Adapts Mitochondrial Proteolytic Activity during Stress, Cell reports 14 (2016) 2041–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ehses S, Raschke I, Mancuso G, Bernacchia A, Geimer S, Tondera D, Martinou JC, Westermann B, Rugarli EI, Langer T, Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1, J Cell Biol 187 (2009) 1023–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Head B, Griparic L, Amiri M, Gandre-Babbe S, van der Bliek AM, Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells, J Cell Biol 187 (2009) 959–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, Langer T, The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission, J Cell Biol 204 (2014) 919–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rainbolt TK, Saunders JM, Wiseman RL, YME1L degradation reduces mitochondrial proteolytic capacity during oxidative stress, EMBO Rep 16 (2015) 97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kong B, Wang Q, Fung E, Xue K, Tsang BK, p53 is required for cisplatin-induced processing of the mitochondrial fusion protein L-Opa1 that is mediated by the mitochondrial metallopeptidase Oma1 in gynecologic cancers, J Biol Chem 289 (2014) 27134–27145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Richter U, Lahtinen T, Marttinen P, Myohanen M, Greco D, Cannino G, Jacobs HT, Lietzen N, Nyman TA, Battersby BJ, A mitochondrial ribosomal and RNA decay pathway blocks cell proliferation, Curr Biol 23 (2013) 535–541. [DOI] [PubMed] [Google Scholar]
- [15].Jiang X, Jiang H, Shen Z, Wang X, Activation of mitochondrial protease OMA1 by Bax and Bak promotes cytochrome c release during apoptosis, Proc Natl Acad Sci U S A. 2014. October 14;111(41):14782–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Noh S, Phorl S, Naskar R, Oeum K, Seo Y, Kim E, Kweon HS, Lee JY, p32/C1QBP regulates OMA1-dependent proteolytic processing of OPA1 to maintain mitochondrial connectivity related to mitochondrial dysfunction and apoptosis, Sci Rep 10 (2020) 10618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Liu YT, Huang X, Nguyen D, Shammas M, Wu B, Dombi E, Springer DA, Poulton J, Sekine S, Narendra DP, Loss of CHCHD2 and CHCHD10 activates OMA1 peptidase to disrupt mitochondrial cristae phenocopying patient mutations, Hum Mol Genet. 2020. June 3;29(9):1547–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tondera D, Grandemange S, Jourdain A, Karbowski M, Mattenberger Y, Herzig S, Da Cruz S, Clerc P, Raschke I, Merkwirth C, Ehses S, Krause F, Chan DC, Alexander C, Bauer C, Youle R, Langer T, Martinou JC, SLP-2 is required for stress-induced mitochondrial hyperfusion, EMBO J 28 (2009) 1589–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Merkwirth C, Dargazanli S, Tatsuta T, Geimer S, Lower B, Wunderlich FT, von Kleist-Retzow JC, Waisman A, Westermann B, Langer T, Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria, Genes Dev 22 (2008) 476–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Deshwal S, Fiedler KU, Langer T, Mitochondrial Proteases: Multifaceted Regulators of Mitochondrial Plasticity, Annu Rev Biochem. 2020. June 20;89:501–528. [DOI] [PubMed] [Google Scholar]
- [21].Glynn SE, Multifunctional Mitochondrial AAA Proteases, Front Mol Biosci 4 (2017) 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Leonhard K, Herrmann JM, Stuart RA, Mannhaupt G, Neupert W, Langer T, AAA proteases with catalytic sites on opposite membrane surfaces comprise a proteolytic system for the ATP-dependent degradation of inner membrane proteins in mitochondria, EMBO J 15 (1996) 4218–4229. [PMC free article] [PubMed] [Google Scholar]
- [23].Tatsuta T, Model K, Langer T, Formation of membrane-bound ring complexes by prohibitins in mitochondria, Mol Biol Cell 16 (2005) 248–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wai T, Saita S, Nolte H, Muller S, Konig T, Richter-Dennerlein R, Sprenger HG, Madrenas J, Muhlmeister M, Brandt U, Kruger M, Langer T, The membrane scaffold SLP2 anchors a proteolytic hub in mitochondria containing PARL and the i-AAA protease YME1L, EMBO Rep 17 (2016) 1844–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Da Cruz S, Parone PA, Gonzalo P, Bienvenut WV, Tondera D, Jourdain A, Quadroni M, Martinou JC, SLP-2 interacts with prohibitins in the mitochondrial inner membrane and contributes to their stability, Biochim Biophys Acta 1783 (2008) 904–911. [DOI] [PubMed] [Google Scholar]
- [26].Ishihara N, Fujita Y, Oka T, Mihara K, Regulation of mitochondrial morphology through proteolytic cleavage of OPA1, EMBO J 25 (2006) 2966–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Song Z, Chen H, Fiket M, Alexander C, Chan DC, OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L, J Cell Biol 178 (2007) 749–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dietz JV, Bohovych I, Viana MP, Khalimonchuk O, Proteolytic regulation of mitochondrial dynamics, Mitochondrion 49 (2019) 289–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].MacVicar T, Langer T, OPA1 processing in cell death and disease - the long and short of it, J Cell Sci 129 (2016) 2297–2306. [DOI] [PubMed] [Google Scholar]
- [30].Ohba Y, MacVicar T, Langer T, Regulation of mitochondrial plasticity by the i-AAA protease YME1L, Biol Chem. 2020. May 26; 401(6–7): 877–890. [DOI] [PubMed] [Google Scholar]
- [31].Del Dotto V, Fogazza M, Carelli V, Rugolo M, Zanna C, Eight human OPA1 isoforms, long and short: What are they for?, Biochim Biophys Acta Bioenerg 1859 (2018) 263–269. [DOI] [PubMed] [Google Scholar]
- [32].McBride H, Soubannier V, Mitochondrial function: OMA1 and OPA1, the grandmasters of mitochondrial health, Curr Biol 20 (2010) R274–276. [DOI] [PubMed] [Google Scholar]
- [33].Alavi MV, Targeted OMA1 therapies for cancer, Int J Cancer 145 (2019) 2330–2341. [DOI] [PubMed] [Google Scholar]
- [34].Alavi MV, Fuhrmann N, Dominant optic atrophy, OPA1, and mitochondrial quality control: understanding mitochondrial network dynamics, Mol Neurodegener 8 (2013) 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wai T, Garcia-Prieto J, Baker MJ, Merkwirth C, Benit P, Rustin P, Ruperez FJ, Barbas C, Ibanez B, Langer T, Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice, Science 350 (2015) aad0116. [DOI] [PubMed] [Google Scholar]
- [36].Sprenger HG, Wani G, Hesseling A, Konig T, Patron M, MacVicar T, Ahola S, Wai T, Barth E, Rugarli EI, Bergami M, Langer T, Loss of the mitochondrial i-AAA protease YME1L leads to ocular dysfunction and spinal axonopathy, EMBO Mol Med 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Caglayan S, Hashim A, Cieslar-Pobuda A, Jensen V, Behringer S, Talug B, Chu DT, Pecquet C, Rogne M, Brech A, Brorson SH, Nagelhus EA, Hannibal L, Boschi A, Tasken K, Staerk J, Optic Atrophy 1 Controls Human Neuronal Development by Preventing Aberrant Nuclear DNA Methylation, iScience 23 (2020) 101154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Shahrestani P, Leung HT, Le PK, Pak WL, Tse S, Ocorr K, Huang T, Heterozygous mutation of Drosophila Opa1 causes the development of multiple organ abnormalities in an age-dependent and organ-specific manner, PLoS One 4 (2009) e6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Alavi MV, Bette S, Schimpf S, Schuettauf F, Schraermeyer U, Wehrl HF, Ruttiger L, Beck SC, Tonagel F, Pichler BJ, Knipper M, Peters T, Laufs J, Wissinger B, A splice site mutation in the murine Opa1 gene features pathology of autosomal dominant optic atrophy, Brain 130 (2007) 1029–1042. [DOI] [PubMed] [Google Scholar]
- [40].Davies VJ, Hollins AJ, Piechota MJ, Yip W, Davies JR, White KE, Nicols PP, Boulton ME, Votruba M, Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function, Human molecular genetics 16 (2007) 1307–1318. [DOI] [PubMed] [Google Scholar]
- [41].Rahn JJ, Stackley KD, Chan SS, Opa1 is required for proper mitochondrial metabolism in early development, PLoS One 8 (2013) e59218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Herkenne S, Ek O, Zamberlan M, Pellattiero A, Chergova M, Chivite I, Novotna E, Rigoni G, Fonseca TB, Samardzic D, Agnellini A, Bean C, Di Benedetto G, Tiso N, Argenton F, Viola A, Soriano ME, Giacomello M, Ziviani E, Sales G, Claret M, Graupera M, Scorrano L, Developmental and Tumor Angiogenesis Requires the Mitochondria-Shaping Protein Opa1, Cell Metab 31 (2020) 987–1003 e1008. [DOI] [PubMed] [Google Scholar]
- [43].Coppola M, Pizzigoni A, Banfi S, Bassi MT, Casari G, Incerti B, Identification and characterization of YME1L1, a novel paraplegin-related gene, Genomics 66 (2000) 48–54. [DOI] [PubMed] [Google Scholar]
- [44].Brambley CA, Marsee JD, Halper N, Miller JM, Characterization of Mitochondrial YME1L Protease Oxidative Stress-Induced Conformational State, J Mol Biol 431 (2019) 1250–1266. [DOI] [PubMed] [Google Scholar]
- [45].Bohovych I, Dietz JV, Swenson S, Zahayko N, Khalimonchuk O, Redox Regulation of the Mitochondrial Quality Control Protease Oma1, Antioxid Redox Signal 31 (2019) 429–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Anderson CJ, Kahl A, Fruitman H, Qian L, Zhou P, Manfredi G, Iadecola C, Prohibitin levels regulate OMA1 activity and turnover in neurons, Cell Death Differ. 2020. June; 27(6):1896–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].MacVicar T, Ohba Y, Nolte H, Mayer FC, Tatsuta T, Sprenger HG, Lindner B, Zhao Y, Li J, Bruns C, Kruger M, Habich M, Riemer J, Schwarzer R, Pasparakis M, Henschke S, Bruning JC, Zamboni N, Langer T, Lipid signalling drives proteolytic rewiring of mitochondria by YME1L, Nature 575 (2019) 361–365. [DOI] [PubMed] [Google Scholar]
- [48].Jones E, Gaytan N, Garcia I, Herrera A, Ramos M, Agarwala D, Rana M, Innis-Whitehouse W, Schuenzel E, Gilkerson R, A threshold of transmembrane potential is required for mitochondrial dynamic balance mediated by DRP1 and OMA1, Cell Mol Life Sci 74 (2017) 1347–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bohovych I, Fernandez MR, Rahn JJ, Stackley KD, Bestman JE, Anandhan A, Franco R, Claypool SM, Lewis RE, Chan SS, Khalimonchuk O, Metalloprotease OMA1 Fine-tunes Mitochondrial Bioenergetic Function and Respiratory Supercomplex Stability, Sci Rep 5 (2015) 13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Koppen M, Metodiev MD, Casari G, Rugarli EI, Langer T, Variable and tissue-specific subunit composition of mitochondrial m-AAA protease complexes linked to hereditary spastic paraplegia, Molecular and cellular biology 27 (2007) 758–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Sacco T, Boda E, Hoxha E, Pizzo R, Cagnoli C, Brusco A, Tempia F, Mouse brain expression patterns of Spg7, Afg3l1, and Afg3l2 transcripts, encoding for the mitochondrial m-AAA protease, BMC Neurosci 11 (2010) 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Tulli S, Del Bondio A, Baderna V, Mazza D, Codazzi F, Pierson TM, Ambrosi A, Nolte D, Goizet C, Toro C, Baets J, Deconinck T, DeJonghe P, Mandich P, Casari G, Maltecca F, Pathogenic variants in the AFG3L2 proteolytic domain cause SCA28 through haploinsufficiency and proteostatic stress-driven OMA1 activation, J Med Genet 56 (2019) 499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Maltecca F, De Stefani D, Cassina L, Consolato F, Wasilewski M, Scorrano L, Rizzuto R, Casari G, Respiratory dysfunction by AFG3L2 deficiency causes decreased mitochondrial calcium uptake via organellar network fragmentation, Human molecular genetics 21 (2012) 3858–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Puchades C, Ding B, Song A, Wiseman RL, Lander GC, Glynn SE, Unique Structural Features of the Mitochondrial AAA+ Protease AFG3L2 Reveal the Molecular Basis for Activity in Health and Disease, Mol Cell 75 (2019) 1073–1085 e1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Caporali L, Magri S, Legati A, Del Dotto V, Tagliavini F, Balistreri F, Nasca A, La Morgia C, Carbonelli M, Valentino ML, Lamantea E, Baratta S, Schols L, Schule R, Barboni P, Cascavilla ML, Maresca A, Capristo M, Ardissone A, Pareyson D, Cammarata G, Melzi L, Zeviani M, Peverelli L, Lamperti C, Marzoli SB, Fang M, Synofzik M, Ghezzi D, Carelli V, Taroni F, ATPase Domain AFG3L2 Mutations Alter OPA1 Processing and Cause Optic Neuropathy, Ann Neurol. 2020. July;88(1):18–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Consolato F, Maltecca F, Tulli S, Sambri I, Casari G, m-AAA and i-AAA complexes coordinate to regulate OMA1, the stress-activated supervisor of mitochondrial dynamics, J Cell Sci 131 (2018). [DOI] [PubMed] [Google Scholar]
- [57].Richter-Dennerlein R, Korwitz A, Haag M, Tatsuta T, Dargazanli S, Baker M, Decker T, Lamkemeyer T, Rugarli EI, Langer T, DNAJC19, a mitochondrial cochaperone associated with cardiomyopathy, forms a complex with prohibitins to regulate cardiolipin remodeling, Cell Metab 20 (2014) 158–171. [DOI] [PubMed] [Google Scholar]
- [58].Merkwirth C, Martinelli P, Korwitz A, Morbin M, Bronneke HS, Jordan SD, Rugarli EI, Langer T, Loss of prohibitin membrane scaffolds impairs mitochondrial architecture and leads to tau hyperphosphorylation and neurodegeneration, PLoS Genet 8 (2012) e1003021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Korwitz A, Merkwirth C, Richter-Dennerlein R, Troder SE, Sprenger HG, Quiros PM, Lopez-Otin C, Rugarli EI, Langer T, Loss of OMA1 delays neurodegeneration by preventing stress-induced OPA1 processing in mitochondria, J Cell Biol 212 (2016) 157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kondadi AK, Wang S, Montagner S, Kladt N, Korwitz A, Martinelli P, Herholz D, Baker MJ, Schauss AC, Langer T, Rugarli EI, Loss of the m-AAA protease subunit AFG(3)L(2) causes mitochondrial transport defects and tau hyperphosphorylation, EMBO J 33 (2014) 1011–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kasashima K, Ohta E, Kagawa Y, Endo H, Mitochondrial functions and estrogen receptor-dependent nuclear translocation of pleiotropic human prohibitin 2, J Biol Chem 281 (2006) 36401–36410. [DOI] [PubMed] [Google Scholar]
- [62].Jian C, Xu F, Hou T, Sun T, Li J, Cheng H, Wang X, Deficiency of PHB complex impairs respiratory supercomplex formation and activates mitochondrial flashes, J Cell Sci 130 (2017) 2620–2630. [DOI] [PubMed] [Google Scholar]
- [63].Steglich G, Neupert W, Langer T, Prohibitins regulate membrane protein degradation by the m-AAA protease in mitochondria, Molecular and cellular biology 19 (1999) 3435–3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Sato S, Murata A, Orihara T, Shirakawa T, Suenaga K, Kigoshi H, Uesugi M, Marine natural product aurilide activates the OPA1-mediated apoptosis by binding to prohibitin, Chemistry & biology 18 (2011) 131–139. [DOI] [PubMed] [Google Scholar]
- [65].Spinazzi M, De Strooper B, PARL: The mitochondrial rhomboid protease, Semin Cell Dev Biol 60 (2016) 19–28. [DOI] [PubMed] [Google Scholar]
- [66].Cipolat S, Rudka T, Hartmann D, Costa V, Serneels L, Craessaerts K, Metzger K, Frezza C, Annaert W, D’Adamio L, Derks C, Dejaegere T, Pellegrini L, D’Hooge R, Scorrano L, De Strooper B, Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling, Cell 126 (2006) 163–175. [DOI] [PubMed] [Google Scholar]
- [67].Griparic L, Kanazawa T, van der Bliek AM, Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage, J Cell Biol 178 (2007) 757–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Duvezin-Caubet S, Jagasia R, Wagener J, Hofmann S, Trifunovic A, Hansson A, Chomyn A, Bauer MF, Attardi G, Larsson NG, Neupert W, Reichert AS, Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology, J Biol Chem 281 (2006) 37972–37979. [DOI] [PubMed] [Google Scholar]
- [69].Horvath SE, Daum G, Lipids of mitochondria, Prog Lipid Res 52 (2013) 590–614. [DOI] [PubMed] [Google Scholar]
- [70].Mejia EM, Hatch GM, Mitochondrial phospholipids: role in mitochondrial function, J Bioenerg Biomembr 48 (2016) 99–112. [DOI] [PubMed] [Google Scholar]
- [71].Sam PN, Avery E, Claypool SM, Proteolytic Control of Lipid Metabolism, ACS Chem Biol 14 (2019) 2406–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Deas E, Plun-Favreau H, Gandhi S, Desmond H, Kjaer S, Loh SH, Renton AE, Harvey RJ, Whitworth AJ, Martins LM, Abramov AY, Wood NW, PINK1 cleavage at position A103 by the mitochondrial protease PARL, Human molecular genetics 20 (2011) 867–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ, Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL, J Cell Biol 191 (2010) 933–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Meissner C, Lorenz H, Weihofen A, Selkoe DJ, Lemberg MK, The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking, Journal of neurochemistry 117 (2011) 856–867. [DOI] [PubMed] [Google Scholar]
- [75].Lemasters JJ, Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging, Rejuvenation Res 8 (2005) 3–5. [DOI] [PubMed] [Google Scholar]
- [76].Varshavsky A, N-degron and C-degron pathways of protein degradation, Proceedings of the National Academy of Sciences of the United States of America 116 (2019) 358–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Yamano K, Youle RJ, PINK1 is degraded through the N-end rule pathway, Autophagy 9 (2013) 1758–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Rub C, Wilkening A, Voos W, Mitochondrial quality control by the Pink1/Parkin system, Cell Tissue Res 367 (2017) 111–123. [DOI] [PubMed] [Google Scholar]
- [79].Bader V, Winklhofer KF, PINK1 and Parkin: team players in stress-induced mitophagy, Biol Chem. 2020. May 26;401(6–7):891–899. [DOI] [PubMed] [Google Scholar]
- [80].Sekine S, Youle RJ, PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol, BMC Biol 16 (2018) 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Sekine S, Wang C, Sideris DP, Bunker E, Zhang Z, Youle RJ, Reciprocal Roles of Tom7 and OMA1 during Mitochondrial Import and Activation of PINK1, Mol Cell 73 (2019) 1028–1043 e1025. [DOI] [PubMed] [Google Scholar]
- [82].Wang Z, Jiang H, Chen S, Du F, Wang X, The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways, Cell 148 (2012) 228–243. [DOI] [PubMed] [Google Scholar]
- [83].Sesaki H, Southard SM, Hobbs AE, Jensen RE, Cells lacking Pcp1p/Ugo2p, a rhomboid-like protease required for Mgm1p processing, lose mtDNA and mitochondrial structure in a Dnm1p-dependent manner, but remain competent for mitochondrial fusion, Biochem Biophys Res Commun 308 (2003) 276–283. [DOI] [PubMed] [Google Scholar]
- [84].McQuibban GA, Saurya S, Freeman M, Mitochondrial membrane remodelling regulated by a conserved rhomboid protease, Nature 423 (2003) 537–541. [DOI] [PubMed] [Google Scholar]
- [85].McQuibban GA, Lee JR, Zheng L, Juusola M, Freeman M, Normal mitochondrial dynamics requires rhomboid-7 and affects Drosophila lifespan and neuronal function, Curr Biol 16 (2006) 982–989. [DOI] [PubMed] [Google Scholar]
- [86].Lysyk L, Brassard R, Touret N, Lemieux MJ, PARL Protease: A Glimpse at Intramembrane Proteolysis in the Inner Mitochondrial Membrane, Curr Biol. 2006. May 23;16(10):982–9. [DOI] [PubMed] [Google Scholar]
- [87].Jeyaraju DV, Sood A, Laforce-Lavoie A, Pellegrini L, Rhomboid proteases in mitochondria and plastids: keeping organelles in shape, Biochim Biophys Acta 1833 (2013) 371–380. [DOI] [PubMed] [Google Scholar]
- [88].Cho S, Dickey SW, Urban S, Crystal Structures and Inhibition Kinetics Reveal a Two-Stage Catalytic Mechanism with Drug Design Implications for Rhomboid Proteolysis, Mol Cell 61 (2016) 329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Khalimonchuk O, Jeong MY, Watts T, Ferris E, Winge DR, Selective Oma1 protease-mediated proteolysis of Cox1 subunit of cytochrome oxidase in assembly mutants, J Biol Chem 287 (2012) 7289–7300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Bohovych I, Donaldson G, Christianson S, Zahayko N, Khalimonchuk O, Stress-triggered activation of the metalloprotease Oma1 involves its C-terminal region and is important for mitochondrial stress protection in yeast, J Biol Chem 289 (2014) 13259–13272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Levytskyy RM, Bohovych I, Khalimonchuk O, Metalloproteases of the Inner Mitochondrial Membrane, Biochemistry 56 (2017) 4737–4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Lopez-Pelegrin M, Cerda-Costa N, Martinez-Jimenez F, Cintas-Pedrola A, Canals A, Peinado JR, Marti-Renom MA, Lopez-Otin C, Arolas JL, Gomis-Ruth FX, A novel family of soluble minimal scaffolds provides structural insight into the catalytic domains of integral membrane metallopeptidases, J Biol Chem 288 (2013) 21279–21294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Davies BS, Fong LG, Yang SH, Coffinier C, Young SG, The posttranslational processing of prelamin A and disease, Annu Rev Genomics Hum Genet 10 (2009) 153–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Goblirsch BR, Wiener MC, Ste24: An Integral Membrane Protein Zinc Metalloprotease with Provocative Structure and Emergent Biology, J Mol Biol. 2020. August 21;432(18):5079–5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Quigley A, Dong YY, Pike AC, Dong L, Shrestha L, Berridge G, Stansfeld PJ, Sansom MS, Edwards AM, Bountra C, von Delft F, Bullock AN, Burgess-Brown NA, Carpenter EP, The structural basis of ZMPSTE24-dependent laminopathies, Science 339 (2013) 1604–1607. [DOI] [PubMed] [Google Scholar]
- [96].Pryor EE Jr., Horanyi PS, Clark KM, Fedoriw N, Connelly SM, Koszelak-Rosenblum M, Zhu G, Malkowski MG, Wiener MC, Dumont ME, Structure of the integral membrane protein CAAX protease Ste24p, Science 339 (2013) 1600–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Holm L, Benchmarking fold detection by DaliLite v.5, Bioinformatics 35 (2019) 5326–5327. [DOI] [PubMed] [Google Scholar]
- [98].Feng L, Yan H, Wu Z, Yan N, Wang Z, Jeffrey PD, Shi Y, Structure of a site-2 protease family intramembrane metalloprotease, Science 318 (2007) 1608–1612. [DOI] [PubMed] [Google Scholar]
- [99].Adamczak R, Porollo A, Meller J, Accurate prediction of solvent accessibility using neural networks-based regression, Proteins 56 (2004) 753–767. [DOI] [PubMed] [Google Scholar]
- [100].Quiros PM, Ramsay AJ, Sala D, Fernandez-Vizarra E, Rodriguez F, Peinado JR, Fernandez-Garcia MS, Vega JA, Enriquez JA, Zorzano A, Lopez-Otin C, Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice, EMBO J 31 (2012) 2117–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Schafer A, Zick M, Kief J, Steger M, Heide H, Duvezin-Caubet S, Neupert W, Reichert AS, Intramembrane proteolysis of Mgm1 by the mitochondrial rhomboid protease is highly promiscuous regarding the sequence of the cleaved hydrophobic segment, J Mol Biol 401 (2010) 182–193. [DOI] [PubMed] [Google Scholar]
- [102].Dickey SW, Baker RP, Cho S, Urban S, Proteolysis inside the membrane is a rate-governed reaction not driven by substrate affinity, Cell 155 (2013) 1270–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Ast T, Michaelis S, Schuldiner M, The Protease Ste24 Clears Clogged Translocons, Cell 164 (2016) 103–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Kayatekin C, Amasino A, Gaglia G, Flannick J, Bonner JM, Fanning S, Narayan P, Barrasa MI, Pincus D, Landgraf D, Nelson J, Hesse WR, Costanzo M, Myers CL, Boone C, Florez JC, Lindquist S, Translocon Declogger Ste24 Protects against IAPP Oligomer-Induced Proteotoxicity, Cell 173 (2018) 62–73 e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Wolfe MS, Substrate recognition and processing by gamma-secretase, Biochim Biophys Acta Biomembr 1862 (2020) 183016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Wolfe MS, Structure and Function of the gamma-Secretase Complex, Biochemistry 58 (2019) 2953–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Nan J, Nan C, Ye J, Qian L, Geng Y, Xing D, Rahman MSU, Huang M, EGCG protects cardiomyocytes against hypoxia-reperfusion injury through inhibition of OMA1 activation, J Cell Sci 132 (2019). [DOI] [PubMed] [Google Scholar]
- [108].Richter U, Ng KY, Suomi F, Marttinen P, Turunen T, Jackson C, Suomalainen A, Vihinen H, Jokitalo E, Nyman TA, Isokallio MA, Stewart JB, Mancini C, Brusco A, Seneca S, Lombes A, Taylor RW, Battersby BJ, Mitochondrial stress response triggered by defects in protein synthesis quality control, Life Sci Alliance 2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Grivell LA, Reijnders L, de Vries H, Altered mitochondrial ribosomes in a cytoplasmic mutant of yeast, FEBS Lett 16 (1971) 159–163. [DOI] [PubMed] [Google Scholar]
- [110].Clark KM, Jenkins JL, Fedoriw N, Dumont ME, Human CaaX protease ZMPSTE24 expressed in yeast: Structure and inhibition by HIV protease inhibitors, Protein Sci 26 (2017) 242–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Coffinier C, Hudon SE, Farber EA, Chang SY, Hrycyna CA, Young SG, Fong LG, HIV protease inhibitors block the zinc metalloproteinase ZMPSTE24 and lead to an accumulation of prelamin A in cells, Proceedings of the National Academy of Sciences of the United States of America 104 (2007) 13432–13437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Hudon SE, Coffinier C, Michaelis S, Fong LG, Young SG, Hrycyna CA, HIV-protease inhibitors block the enzymatic activity of purified Ste24p, Biochem Biophys Res Commun 374 (2008) 365–368. [DOI] [PMC free article] [PubMed] [Google Scholar]