

Abstract
Despite successful therapeutic strategies in the prevention and treatment of arteriosclerosis, the cardiovascular complications remain a major clinical and societal issue worldwide. Increased vascular calcification promotes arterial stiffness and accelerates cardiovascular morbidity and mortality. Upregulation of the Runt-related transcription factor 2 (Runx2), an essential osteogenic transcription factor for bone formation, in the cardiovascular system has emerged as an important regulator for adverse cellular events that drive cardiovascular pathology. This review discusses the regulatory mechanisms that are critical for Runx2 expression and function, and highlights the dynamic and complex cross-talks of a wide variety of posttranslational modifications (PTMs), including phosphorylation, acetylation, ubiquitination, and O-GlcNAcylation, in regulating Runx2 stability, cellular localization and osteogenic transcriptional activity. How the activation of an array of signaling cascades by circulating and local microenvironmental factors upregulates Runx2 in vascular cells and promotes Runx2-mediated osteogenic trans-differentiation of vascular smooth muscle cells (VSMC) and expression of inflammatory cytokines that accelerate macrophage infiltration and vascular osteoclast formation is summarized. Furthermore, the increasing appreciation of a new role of Runx2 upregulation in promoting VSMC phenotypic switch, and protein modulation by O-GlcNAcylation and Runx2-dependent repression of SMC-specific gene expression are discussed. Further exploring the regulation of this key osteogenic transcription factor and its new perspectives in the vasculature will provide novel insights into the transcriptional regulation of VSMC phenotype switch, reprograming and vascular inflammation that promote the pathogenesis of arteriosclerosis.
Keywords: Runx2, arteriosclerosis, smooth muscle phenotypic switch, vascular calcification, inflammation, posttranslational modification
Graphical Abstract
INTRODUCTION
The flexibility and elasticity of healthy arteries enable proper distribution of oxygen and nutrients to all organs in the body. Vascular homeostasis and normal vascular tones are attributed largely to the contractile feature of vascular smooth muscle cells (VSMC), the major cells in the tunic media of the elastic arteries. Dysregulation of VSMC proliferation, migration, senescence, differentiation and trans-differentiation promotes medial and intimal thickening and arterial stiffening1. Thickening and stiffening of the arterial walls are characteristics of arteriosclerosis, which is often associated with atherosclerosis and represents an independent risk factor that predicts adverse cardiovascular outcomes in aging populations and patients with diabetes mellitus, hypertension, chronic kidney disease (CKD) and atherosclerosis1. On the other hand, metabolic disorders and cardiovascular complications further accelerate arteriosclerosis2. Increased vascular calcification has been demonstrated in aging arteries as well as in patients with various cardiovascular and metabolic complications, leading to arterial stiffening and increased risk of cardiovascular morbidity and mortality 3–7.
Emerging studies in the last two decades have provided molecular, cellular and genetic evidence uncovering vascular calcification as an active cell-driven process featuring osteogenic differentiation of vascular cells. In particular, mineralization of VSMC contributes significantly to the development of vascular calcification in the tunica media as well as atherosclerotic intima, which reduces arterial elasticity and increases arterial stiffening8, 9. The high plasticity of VSMC allows the phenotypic transition from the contractile to a synthetic phenotype, featuring down regulation of the SMC-specific contractile proteins, such as smooth muscle α-actin (SMA, Acta2), SM22α (Tagln) and smooth muscle cell myosin heavy chain (SMMHC, Myh11). VSMC share a similar mesenchymal origin to the bone-forming osteoblasts. During the osteogenic trans-differentiation of VSMC, downregulation of SMC marker genes is associated with upregulation of the key osteogenic transcription factors for osteoblast formation, resulting in increased expression of “bone markers” in VSMC, such as osteocalcin (OC), collagen type I alpha 1 (Col1α1), bone sialoprotein (osteopontin, OPN) and alkaline phosphatase (ALP) 10–12, which promotes the transformation of VSMC into bone-like cells.
Among the key regulators for osteogenic differentiation and calcification of VSMC, ample evidence has demonstrated that the Runt-related transcription factor 2 (Runx2/CBFα1/AML3) is an essential and sufficient regulator for osteogenic differentiation and calcification of vascular cells. A causal role of the Runx2 in promoting calcification and matrix protein production has been demonstrated in vascular cells 10, 12, 13. Importantly, the expression of Runx2 is low in healthy vasculature, but markedly increased Runx2 in calcified arteries in animal models and human subjects with atherosclerosis, diabetes mellitus and CKD14–17. From the studies of two independently generated SMC-specific Runx2 deletion mouse models, Giachelli’s group and our group have demonstrated that SMC-specific Runx2 deficiency inhibits intimal calcification in atherosclerosis-prone mice as well as medial calcification in CKD mice16, 18. Of note, SMC-specific Runx2 deficiency did not affect basal VSMC phenotype and normal vascular development 16, 18, which is consistent with the observations of normal cardiovascular systems in the Runx2 global knockout mice19, 20. The potent inhibitory effects of Runx2 deficiency on VSMC calcification in vitro as well as medial and intimal vascular calcification in vivo have supported a definitive role of VSMC and SMC-specific expression of Runx2 in the development of vascular calcification12, 16, 18.
This review discusses the important structure and function relationship of Runx2, its regulation of VSMC reprograming and inflammation that contributes to the pathogenesis of arteriosclerosis, highlighting the crucial function determined by both the post-translational modification mechanisms and the interaction of Runx2 with key regulatory signals.
Runx2 expression and structure: lessons learned from bone biology
The Runx2 transcription factor is indispensable for both osteoblast differentiation and chondrocyte maturation, thus it is essential for the development of bone and cartilage19–21. Ablation of Runx2 or Runx2 osteogenic activity in mice results in defects in skeletal development 19–21. In humans, mutations of RUNX2 cause cleidocranial dysplasia (CCD), an autosomal dominant disorder characterized by hypoplastic or absent clavicles, large fontanelles, dental anomalies, delayed skeletal development or other skeletal abnormalities22, 23. The defects in bone and tooth development resulting from the Runx2 deficiency in mouse and human subjects; and the transcriptional regulation of Runx2 on an array of osteoblast-specific matrix proteins, including OC, Col1α1, osteopontin, and bone matrix metalloproteinase 13 (MMP-13), have established the essential role of Runx2 as the master osteogenic transcription factor and marker for osteoblast differentiation24–26.
• Runx2 expression and functional domains
The human RUNX2 gene is located at the short arm of chromosome 6 at position 21.1 (6p21.1)27; and the murine Runx2 gene is located on chromosome 1728. Expression runx2 is transcriptionally regulated by two promoters, the distal P1 promoter and the proximal P2 promoter (Figure 1A). The P1 promoter drives expression of type II runx2 transcript encoded by the exon 1–8 that is mainly expressed in bone progenitor cells and upregulated during bone formation29; while the P2 promoter drives expression of type I runx2 transcript encoded by the exon 2–8 that is expressed in both osteogenic and non-osteogenic mesenchymal tissues30. Consistently, mice defective of the runx2-P1 promoter exhibit CCD-like symptoms with severe defects in skeletal and tooth development 29. A conserved syntenic linkage has been identified between Runx2 and the suppressor of Ty3 homolog (Supt3h) gene, a ubiquitously expressed gene that is constitutively transcribed in the opposite direction using a promoter in the first intron of Runx231, 32. Studies by Stein and colleagues have demonstrated a bone-differentiation-specific regulation of the Supt3h promoter on the Runx2-P1 promoter via direct association, thus driving Runx2 transcription. The binding sites for several transcriptional co-factors have been identified in the Runx2 promoters, which regulate the runx2 transcription and thus promote osteogenesis33–35. For instance, binding of a homeodomain transcription factor Dlx5 to the Runx2-P1 promoter increases Runx2 transcription35. In contrast, binding of the homeodomain containing transcription factors Msx2 to the Dlx5 binding sites on the Runx2-P1 promoter inhibits Runx2 transcription35, suggesting differential regulation of runx2 expression via different transcriptional factors. Binding of other transcriptional factors, such as estrogen receptor alpha and TWIST1, a basic helix-loop-helix transcription factor to the Runx2-P2 promoter has also been found to increase Runx2 transcription36, 37. In addition, Runx2 binds to its own promoter via the OSE2, a cis-acting element originally identified as a Runx2-binding site on the osteocalcin promoter, and regulates its own expression25. Therefore, the Runx2 regulation is complex, dynamic and context-dependent.
Figure 1. RUNX2 gene and protein structure.

A) RUNX2 gene structure. Two major RUNX2 isoforms are transcribed from the P1 and P2 promoters respectively, indicating by the ATG start codons, which are encoded by exon 1–8 (type II) or exons 2–8 (type I). The DNA binding Runt homology domain (RHD) is encoded by exons 2–5. B) RUNX2 protein structure. In addition to the RHD domain, RUNX2 proteins contain a glutamine/alanine (QA) rich region, a nuclear-localization signal (NLS), a proline/serine/threonine (PST) rich region, a nuclear matrix targeting signal (NMTS), and a C-terminal VWRPY domain for TLE/Groucho interactions.
All the three Runx family members, Runx1, Runx2, and Runx3, share a 128-amino acids long evolutionarily conserved “Runt-homology domain” (RHD, Figure 1B). RHD is responsible for Runx DNA binding and heterodimerization with the core binding factor β (PEBP2/CBFβ). The non-DNA binding β subunit enhances the DNA binding affinity of Runx proteins and prevents Runx2 proteins from proteolytic degradation by the ubiquitin-proteasome system38–40. In addition, Runx2 proteins contain a glutamine-alanine (QA) domain, consisting of 23 glutamine repeats and 17 alanine repeats, and the proline-serine-threonine (PST) rich domain located in the C-terminus of the RHD domain. Both QA and PST function as transactivation domains28, 41. Runx2 binding to a number of other transcription factors has been shown to regulate the transcriptional activity of Runx2, including TWIST42, sex-determining region Y-box 9 (Sox9)43, Yes-associated protein (YAP)44 and Smads 45, 46 (Figure 2). TWIST interacts with the Runx2 DNA-binding domain via the Twist box that inhibits Runx2 osteogenic function during skeletal development in mice42. The highly conserved last five amino acids at the Runx2 C-terminus, the VWRPY motif, serve as a transcriptional repression motif via mediating the interaction of Runx2 with the transcriptional repressor transducin-like Enhancer of split 2 (TLE2), a mammalian homologue of Groucho in Drosophila41, 47.
Figure 2. Runx2 interaction with various cytosolic and nuclear proteins that regulate its post-translational modification, cellular localization and stability, and transcriptional activity.

The binding proteins of the Runt-related transcription factor 2 (Runx2) regulate Runx2 posttranslational modifications (PTMs), including phosphorylation, ubiquitination, acetylation and O-GlcNAcylation are shown (top left); Runx2 nuclear translocation, DNA binding and transcriptional activities are highlighted (top right). The key regulators for Runx2 PTMs and Runx2 transactivity are shown in the respective box. In addition, binding of Runx2 to the CBF consensus sequence TGT/cGGT on the target genes, via its Runt-homology domain, regulates the transcription of the target genes (bottom).
A nuclear localization signal (NLS) consisting of a 9-amino-acid motif is identified to mediate nuclear translocation and function of Runx241. Mutations in the Runx2 NLS impairs Runx2 nuclear localization and function, resulting in CCD in patients48. Binding of Runx2 to non-activated signal transducer and activator of transcription 1(STAT1) inhibits nuclear translocation of Runx2, thus decreasing Runx2 in the nucleus and attenuating Runx2 osteogenic transactivity49. Accordingly, STAT1 deficiency increased Runx2 DNA-binding activities in osteoblasts, resulting in accelerated osteoblast differentiation and enhanced bone formation in mice49. Furthermore, a nuclear matrix targeting signal (NMTS) consisting of 38 amino acids in the PST domain is determined to direct Runx2 to the nuclear matrix. Deficiency in the Runx2 NMTS domain does not affect nuclear localization of Runx2 or Runx2-DNA binding, but impairs nuclear matrix localization of Runx2 and its osteogenic transcriptional activity50, 51. In BMP2-induced osteoblast formation, binding of Smad 1 and Smad 5 to the Runx2 NMTS domain facilitates nuclear matrix localization of Runx2 and enhances Runx2 transcriptional activity, while the disruption of the Runx2-Smad binding impairs BMP2-mediated osteoblast differentiation51–53.
The importance of the C-terminal Runx2 functional domains in the commitment and progression of the osteoblast differentiation is further supported by the impaired bone formation in the Runx2 mutant mice with the exon 8 ablation51. Although containing the intact Runx2 RHD DNA binding domain, the Runx2 exon 8 truncation in mice results in bone defects and embryonic lethality similarly to the Runx2 null mutant mice19–21. Accordingly, these studies have demonstrated the importance of the C-terminal regulatory domains in mediating the osteogenic transcriptional activity of Runx2, and Runx2-dependent osteogenesis.
• Post-translational modifications of Runx2
The osteogenic function of Runx2 is controlled by multifaceted regulatory mechanisms. In addition to the regulation of expression of Runx2, post-translational modifications (PTMs) of Runx2 interface with different factors to control Runx2 transcriptional activity spatiotemporally. The discussions below highlight several well-characterized PTMs, including phosphorylation, acetylation, ubiquitination and O-GlcNAcylation, and their impacts on Runx2 stability, nuclear localization, and its ability to bind to DNA and other co-factors that are essential for the Runx2 transcriptional activity (Figure 2).
- Runx2 Phosphorylation
Phosphorylation is the most common PTM on many proteins, which is regulated by a wide variety of kinases that add a phosphate group to an amino acid, and protein phosphatases that catalyze the cleavage of the phosphodiester bond and remove the phosphate group. Protein phosphorylation leads to conformational and functional changes of proteins, as well as their physical interactions with other molecules. Phosphorylation of Runx2 has been demonstrated to be an important regulatory mechanism for Runx2 osteogenic transactivity and osteoblast formation in vitro and skeleton development in mice54–56. Many important kinases positively regulate phosphorylation of Runx2 and its function, such as the extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK) and p38 MAPK-mediated phosphorylation of Runx2 increases Runx2 transcriptional activity that is critical for osteoblast formation in vitro and skeletal development in vivo54–56. In human bone marrow stromal cells, elevated Runx2 phosphorylation, but not Runx2 transcription or protein expression, is highly associated with increased Runx2 transcriptional activity during the in vitro osteoblast differentiation57. Multiple kinase-selective phosphorylation sites have been identified on the serine and threonine residues on Runx2. The serine 301 (Ser301) and Ser319 within the Runx2 PST domain as well as Ser43 and Ser510 are activated by the ERK/MAPK signaling pathway. Mutations of Runx2 Ser301 and Ser319 inhibit MAPK-induced Runx2 activation, osteoblast-specific gene expression and osteoblast differentiation58. The p38 MAPK induces Runx2 phosphorylation on Ser28, Ser244, Ser472 and Ser395 in addition to Ser301 and Ser319 59. Homeodomain-interacting protein kinase 3 (HIPK3) mediates Runx2 phosphorylation on Ser301, Ser319, and threonine 326 (Thr326), which is critical for Runx2 transactivation and the expression of Runx2-targeted osteogenic marker, osteocalcin, in C3H10T1/2 multipotent osteoprogenitors60. In addition, protein kinase B (AKT) induces Runx2 phosphorylation on Ser203, Thr205, and Thr207 within the RHD, which increases Runx2 binding to DNA and promotes Runx2 transcriptional activity61. Furthermore, protein kinase Cδ (PKCδ) has been shown to phosphorylate Runx2 on Ser247, which plays an important role in mediating fibroblast growth factor-increased Runx2 transcriptional activity and Runx2 expression62.
On the other hand, phosphorylation on Runx2 at Ser104 inhibits Runx2 transcriptional activity, by abolishing the heterodimerization of Runx2 with CBFβ and reducing Runx2 stability63, 64. In addition, the phosphorylation of Ser451 resides within the C-terminal transcription inhibition domain of Runx2 is associated with reduced Runx2 activity, as mutation of Ser451 did not affect Runx2 protein levels but markedly enhances Runx2 transcriptional activity63. Similarly, glycogen synthase kinase 3 beta (GSK3β)-regulated Runx2 phosphorylation on Ser369, Ser373, and Ser377 inhibits Runx2 activity and bone formation65. Furthermore, phosphorylation of Ser540 in the C-terminus of Runx2 is required for the Sox9-mediated degradation of Runx266. Taken together, Runx2 phosphorylation in the different Runx2 functional domains, including RHD, PST and the transcriptional inhibitory domain, differentially affect Runx2 stability, and Runx2 binding to its targeting DNA and other proteins that are critical for Runx2 transcriptional activity. Therefore, it is important to understand how the different kinases are coordinated to regulate Runx2 in a spatiotemporal manner to execute its function as the master osteogenic regulator.
- Runx2 Acetylation
Protein PTM by acetylation is a reversible modification catalyzed by lysine acetyltransferases, which transfers acetyl group from acetyl coenzyme A (Ac-CoA) onto lysine residues of histones, transcription factors and other proteins67. Acetylation of histone proteins, controlled by the histone acetyltransferases (HATs) that add acetyl group onto protein and histone deacetylases (HDACs) that remove the acetyl group, and reduces DNA affinity of histones within the chromatin structure, leading to increased DNA binding to transcription factors. In addition, HATs catalyzes lysine acetylation of non-histone proteins, such as transcription factors, which regulates the stability and DNA binding of the transcription factors. Acetylation of Runx2 by the p300, a transcriptional co-activator with HAT activity, has been shown to increase Runx2 stability as well as enhance Runx2 osteogenic transcriptional activity and osteoblast differentiation. In bone morphogenetic protein 2(BMP-2)-induced osteoblast differentiation, BMP-2-activated Smads promote binding of Runx2 to the p300, resulting Runx2 acetylation by p300 that protects Runx2 from degradation by proteasome68. In addition, BMP-2 activated ERK/MAPK signaling pathway increases the expression of p300, which in turn induces Runx2 acetylation that enhances Runx2 stability and transactivity53. Similarly, FGF2-induced ERK/MAPK kinase activation promotes Runx2 phosphorylation at S301 residue that facilitates Runx2 acetylation and increases Runx2 stability and activity69. In addition, parathyroid hormone (PTH)-induced Runx2 phosphorylation increases Runx2-mediated recruitment of p300 to the promoter of MMP-13, which facilitates acetylation of histone 4 (H4) and H3 by p300 that further promotes MMP-13 gene expression70.
On the other hand, HDACs can bind directly to Runx2 and act as corepressors of Runx2 for its transcriptional activities. Several HDACs, such as HDAC 1, 3 and 6, catalyze deacetylation of Runx2 that mediates Runx2 ubiquitination and degradation, leading to reduced Runx2 transcriptional activity and inhibited osteoblast differentiation 71–73. Similarly, deacetylation of Runx2 by HDAC 4 and 5 facilitates ubiquitination-mediated Runx2 degradation68. Consistently, inhibitors for HDACs are shown to increase Runx2 acetylation and promote osteoblast differentiation in vitro as well as bone formation in vivo68, 74. Therefore, acetylation represents one of the key PTMs on Runx2 that regulates Runx2 stability and transcriptional activity, which is essential for the differentiation of osteoblasts and skeleton development.
- Runx2 ubiquitination
Ubiquitination-mediated protein degradation plays an important role in maintaining Runx2 protein stability in osteoblasts75. Regulated by a cascade of E1 ubiquitin-activating enzymes, E2 ubiquitin-conjugating enzymes, and E3 ubiquitin ligases that bind to specific target proteins, protein polyubiquitination mediates the degradation by the proteasome76. Among several identified Runx2-associated E3 ubiquitin ligases (Figure 2), Smad ubiquitin regulatory factor 1 (Smurf1) plays a major role in promoting Runx2 polyubiquitination and subsequent proteasomal degradation in osteoblasts77, 78. Overexpression of Smurf1 in osteoblasts in mice inhibits osteoblast differentiation and postnatal bone formation77. In contrast, Smurf1 deficiency in mice renders increased osteoblast activities in response to BMP-2 signaling and age-dependent increase in bone mass78. These studies have determined the molecular regulation and function of Runx2 PTM by ubiquitination in osteoblasts, and uncovered Smurf1 as the first E3 ubiquitin ligase in regulating Runx2 degradation in osteoblasts and bone formation. Additional E3 ubiquitin ligases that are associated with poly-ubiquitination-mediated Runx2 degradation and inhibited osteoblast differentiation include the homologous to E6AP C-terminus (HECT) domain E3 ligase Smurf2 and WWP1, the U-box E3 ligase C-terminus of Hsc70-interacting protein (CHIP), and the RING E3 ligase SCFSkp268, 79,80, 81. In contrast, a recent report has shown that WWP2 E3 ligase-mediated Runx2 mono-ubiquitination is linked to increased Runx2 transcription activity during osteoblast differentiation 82.
Runx2 phosphorylation on Ser472 by cyclin D1-CDK4 also mediates Runx2 ubiquitination and degradation, suggesting a link between Runx2 phosphorylation and its ubiquitination83. In addition, Runx2 acetylation by p300 inhibits Smurf1-mediated ubiquitination and degradation, possibly by competing for the same lysine residues on Runx2, supporting a link between Runx2 acetylation and ubiquitination68. Furthermore, Runx2 phosphorylation on Ser294 promotes Runx2 acetylation, leading to inhibition of ubiquitination-mediated Runx2 degradation, increased Runx2 stability and Runx2 transcriptional activity 69. Therefore, complex and dynamic PTMs crosstalk in the regulation of Runx2. The spatiotemporal subcellular localization, proximity and activity of the respective catalytic PTM enzymes may collectively determine the effects of the Runx2 PTMs on Runx2 stability and its transcriptional activity during osteoblast differentiation and bone formation. Uncovering the mechanistic details should guide the design of precise modulation of Runx2, thus providing new targets for precision medicine.
- Runx2 O-GlcNAcylation
Protein PTM by O-linked β-N-acetylglucosamine (O-GlcNAc), O-GlcNAcylation, has emerged as an important PTM mechanism, which regulates multiple cellular signals and functions in different tissues to maintain cellular homeostasis in response to changes in nutrients and environmental cues84, 85. Different from the classic N-glycosylation and O-glycosylation, O-GlcNAcylation is a dynamic modification on serine or threonine residues of proteins by a single sugar moiety GlcNAc, which is tightly controlled by two specific enzymes, the sugar adding O-GlcNAc transferase (OGT) and the sugar-removing O-GlcNAcase (OGA). Overall protein O-GlcNAcylation levels are elevated during in vitro osteoblast differentiation of MC3T3-E1 cells; and increased O-GlcNAcylation by OGA inhibition enhances Runx2 transcriptional activity and expression of the Runx2 target gene, osteocalcin86. Similarly, OGA inhibition also enhances BMP-2-induced osteoblast differentiation and Runx2 transcriptional activity in MC3T3-E1 cells87. In vivo administration of OGA inhibitor increases expansion in the growth plate height and in the hypertrophic zone height, supporting a role played by increased O-GlcNAcylation in promoting chondrogenic differentiation88. Of note, O-GlcNAcylation positively regulates osteoblast differentiation but not osteoclast differentiation in vitro as demonstrated by using inhibitors for OGA and OGT respectively89. Immunoprecipitation of Runx2 with a pan-O-GlcNAc antibody has determined a direct O-GlcNAc modification on Runx2, which may contribute to increased Runx2 transcriptional activity in MC3T3-E1 cells with elevated O-GlcNAcylation87. Three potential O-GlcNAc modification sites, including Ser32, Ser33, and Ser371 on human RUNX2 protein expressed in the HEK293 cells, have been identified using tandem mass spectrometry87. The impact of O-GlcNAcylation on these potential residues on Runx2 structure and function remains unknown, which merits further investigation and validation in a relevant biological systems. As mentioned above, multiple Runx2 PTMs by phosphorylation, acetylation and ubiquitination may interplay to collectively affect Runx2 stability and Runx2 transcriptional activity. Accordingly, the putative O-GlcNAcylation at Ser371 residue, located in the C-terminal PST region that mediates multiple Runx2 PTMs and Runx2 interactions with several important co-transcriptional factors, may also contribute to the regulation of Runx2 by other PTMs and thus becomes a central regulatory hub. In particular, as O-GlcNAcylation and phosphorylation may occur on the same or adjacent serine or threonine residues on many proteins90, it is likely that Runx2 O-GlcNAcylation at Ser371 may affect Runx2 phosphorylation on several sites identified within the Runx2 PST domain. Furthermore, increased O-GlcNAcylation has been shown to activate many important signaling pathways for Runx2 PTMs, such as ERK/MAPK, p38 MAPK and AKT, highlighting a potentially complex and dynamic PTM regulatory network. Additional studies are required to elucidate the precise O-GlcNAcylation of Runx2 and their impact on other PTMs, and the cumulative effects on Runx2 transcriptional activity, which will uncover novel regulatory mechanisms of Runx2 and Runx2-dependent biological function.
Overall, multifaceted modifications of Runx2 by PTMs crosstalk to fine-tune Runx2 dynamic interactions with other proteins spatiotemporally, which may be executed by forming distinct functional complexes that regulate Runx2 stability, nuclear localization, binding to target genes and transcriptional co-factors that are critical for the Runx2-directed osteogenic transcriptional activity.
Functional regulation of Runx2 in arteriosclerosis
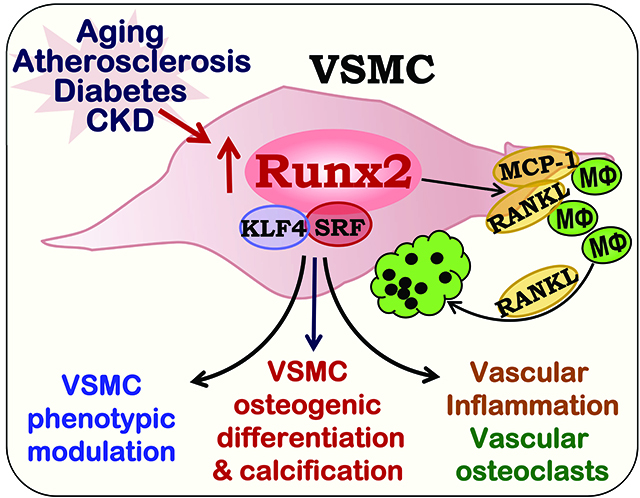
An important role of Runx2 in the cardiovascular system has begun to be uncovered, as emerging studies have revealed a positive correlation of Runx2 upregulation with vascular calcification, stiffness and atherosclerosis in humans and mouse models14–16. In vitro cell culture studies have further determined an essential role of the Runx2 upregulation in calcification of multiple vascular cells, including calcifying vascular cells, VSMC, endothelial cells and vascular progenitor cells12, 91–93. In VSMC, increased expression Runx2 alone is sufficient to promote VSMC osteogenic differentiation calcification12. Although the precise Runx2 domains that are essential for its pro-calcification function in VSMC, the Runx2 interacting proteins and the mode of action remain elusive, lessons learned from bone biology have been instrumental to guide our understanding of the signaling pathways that contribute to the Runx2 upregulation in the vasculature. In particular, the major signaling pathways that converge to the Runx2 upregulation to stimulate osteoblast differentiation, such as the BMP-2, ERK/MAPK and PI3K/AKT signaling pathways, induce expression, PTMs and transcriptional activity of Runx2 in VSMC that promote the pathogenesis of vascular calcification and arteriosclerosis. The regulation of Runx2 in VSMC and their contribution to Runx2-dependent VSMC osteogenic differentiation and calcification, vascular inflammation and formation of bone resorbing osteoclast-like cells, as well as VSMC phenotypic switch are discussed below (Figure 3).
Figure 3. Upregulation of Runx2 in vascular cells promotes vascular calcification, inflammation, smooth muscle cell phenotypic switch that accelerates arteriosclerosis.

In response to local and circulating stimuli, activation of pro-osteogenic signals induce upregulation of the Runt-related transcription factor 2 (Runx2) and its post-translational modifications (PTMs), which increase Runx2 transcriptional activity. Upregulation of Runx2 in vascular smooth muscle cells (VSMC) induces osteogenic differentiation and VSMC calcification; and increases the expression of inflammatory cytokines that promotes infiltration of macrophages and the differentiation of vascular osteoclasts. In addition, upregulation of Runx2 in VSMC represses the expression of SMC specific marker genes, enhancing SMC phenotypic switch. Ultimately, Runx2-induced molecular and cellular changes in VSMC promotes the development of vascular calcification, atherosclerosis and arterial stiffness.
• Runx2 upregulation and PTMs promote vascular calcification
Induction of Runx2 by multiple local and circulating factors, including oxidized lipids, high glucose, and imbalanced phosphate/calcium contents from metabolic disorders promote VSMC calcification, which is associated with the progression of atherosclerosis, diabetes mellitus and CKD6, 12, 92, 94, 95. A common feature underlying the Runx2 upregulation by these stimuli is the increase in oxidative stress, leading to activation of multiple pro-osteogenic signals, including AKT, MAPK and BMP2 signaling pathways. Activation of AKT signaling pathway plays a major role in upregulating Runx2 and promoting VSMC calcification in vitro and vascular calcification in atherosclerosis, diabetes and CKD12, 16, 17, 96, 97. Inhibition of AKT signaling by a pharmacological inhibitor blocks oxidative stress and high glucose-induced Runx2 upregulation and calcification in VSMC12. Similarly, inhibiting AKT activation by blocking O-GlcNAcylation of AKT attenuates the Runx2 upregulation, Runx2 osteogenic transcriptional activity and VSMC calcification 17. Furthermore, Runx2 knockdown attenuates constitutively activated AKT-promoted VSMC calcification, supporting a definitive role of AKT-induced Runx2 upregulation in driving osteogenic differentiation and calcification in VSMC. Increased forkhead box protein O (FoxO) 1/3 phosphorylation and nuclear exclusion by AKT activation mediate AKT-induced Runx2 upregulation via inhabiting Runx2 ubiquitination and subsequent degradation12, 97. Although the regulatory mechanisms and the specific E3 ligase(s) that are responsible for poly-ubiquitination-mediated Runx2 degradation in VSMC await further investigations, these studies have established an important role of AKT/FoxO1/3 signaling pathways in regulating Runx2 PTM by ubiquitination, which contributes to the osteogenic transcription function of Runx2 in VSMC. Additionally, Runx2 modulation by the small ubiquitin-like modifiers (SUMOylation), a process similar to ubiquitination, is associated with AMP kinase alpha1-regulated VSMC calcification98.
Induction of Runx2 transcriptional activity by the activation of p38 MAPK in VSMC has been shown to promote VSMC calcification in vitro and vascular calcification in atherosclerosis-prone ApoE−/− mice in vivo99. Increased binding of Runx2 to phosphorylated p38 MAPK is associated with increased Runx2 transcriptional activity, suggesting p38 MAPK may directly phosphorylate Runx2 and thus enhance Runx2 transcriptional activity99. Importantly, knockdown of p38 MAPK inhibits Runx2 transcriptional activity and VSMC calcification99. As a variety of environmental and intrinsic signals, including oxidative stress, extracellular matrix, mechanical loading, and BMP-2 signals activate the p38 MAPK signaling pathway in VSMC, the p38 MAPK-induced upregulation and transcriptional activity of Runx2 may represent an important regulatory mechanism for Runx2 osteogenic function in VSMC and Runx2-driven VSMC calcification.
The potent pro-osteogenic effects by the BMP-2 signalling axis have also been well established in VSMC. Activation of BMP2 signalling by lipid, glucose metabolism and inflammatory cytokines upregulates Runx2 and promotes osteogenic differentiation of VSMC, as well as medial and intimal calcification in animal models of diabetes and atherosclerosis100–103. Studies from the Towler’s group have demonstrated the important regulation of the BMP-Msx2-Wnt signaling axis in promoting vascular calcification104. However, the mechanisms underlying BMP-2-induced Runx2 upregulation in VSMC is not fully understood. In osteoblasts, direct interaction of Runx2 with the BMP-2-activated Smads, Smad 1, 5 and co-Smad, Smad4 via the Runx2 NMTS domain directs the stable nuclear matrix localization of the Runx2 complexes that is essential for Runx2 osteogenic transactional activities51, 52. The Smad1 and Smad5 also mediate Runx2 acetylation which protects against ubiquitination-dependent Runx2 degradation and thus increases Runx2 transcriptional activity53. Additionally, BMP-2 induces activation of the ERK/MAPK signaling pathways that are important for Runx2 phosphorylation. The MAPK signaling pathways-mediated Runx2 phosphorylation enhances binding of Runx2 to other co-factors, including the coregulatory Smad proteins in osteoblast differentiation105, 106; while pharmacological inhibitors of the MEK/ERK abolish Runx2-Smad1 binding53, indicating the importance of Runx2 phosphorylation for its binding to the BMP-2 regulated Smads. Given the similarity between Runx2-regulated osteogenic differentiation process in bone and VSMC, it is possible that the effects of BMP-2-induced Runx2 upregulation may be mediated by enhanced Runx2 phosphorylation that controls Runx2 stability and its interactions with the BMP-2 specific Smad 1 and 5. Identifying such regulation is necessary to define the BMP-2 regulated Runx2 upregulation and Runx2 osteogenic transcriptional activity in VSMC that determines vascular calcification.
The essential functional domains that are responsible for Runx2-dependent VSMC calcification have not been fully elucidated. By generating SMC-specific Runx2 exon 8 deletion mice, we have determined the definitive role of Runx2 exon 8 in regulating VSMC osteogenic differentiation and vascular calcification in the atherosclerosis-prone ApoE−/− mice 16. These results are consistent with the critical function of Runx2 exon 8 in regulating osteoblast differentiation and bone formation51. Therefore, future investigations to elucidate the mechanisms underlying Runx2 exon 8-mediated osteogenic transcriptional activity of Runx2 in VSMC will provide precise molecular insights into Runx2-dependent vascular calcifications.
• Runx2 upregulation induces vascular inflammatory and mineral resorbing signals
Besides its osteogenic transcriptional activity to induce the expression of bone and matrix proteins that directly promotes VSMC osteogenic differentiation and calcifications, the Runx2 transcription factor has been shown to regulate inflammatory molecules and osteoclast regulators in VSMC, suggesting multilateral regulations of vascular Runx2 on arteriosclerosis. This is particularly enlightening since macrophage infiltration and inflammation play a central role in the pathogenesis of atherosclerosis107, 108.
In response to oxidative stress and inflammatory cytokines by infiltrated inflammatory cells, VSMC produce increased amount of pro-inflammatory molecules, such as monocyte chemoattractant protein-1 (MCP-1), its primary receptor C-C chemokine receptor type 2 (CCR2), and osteopontin. Concurrent with VSMC osteogenic differentiation and calcification, Runx2 induces the expression of osteopontin, an important inflammatory regulator in the vasculature109, 110. We have shown that SMC-specific Runx2 deletion inhibits oxidative stress-induced expression of MCP-1 and CCR2 in VSMC16, supporting the regulation of MCP-1/CCR2 expression by the Runx2 transcription factor. The MCP-1/CCR2 signal axis is critical for infiltration of monocytes/macrophages in arterial walls in mice111, 112. Consistently, macrophage infiltration is blocked by the SMC-specific Runx2 ablation in the atherosclerosis-prone ApoE−/− mice16. We have further uncovered the regulation of Runx2 on the expression of receptor activator of nuclear factor kappa-B ligand (RANKL) in VSMC via its binding to the promoter of RANKL gene. Using a co-culture system, we demonstrated a direct effect of VSMC-expressed RANKL in inducing macrophage infiltration. Consistently, increased macrophage infiltration is shown in atherosclerotic lesions in the ApoE−/− mice with deficiency of osteoprotegrin (OPG), a RANKL decoy receptor, supporting a role of RANKL in accelerating macrophage infiltration in vivo113. As infiltrated monocytes/macrophages produce inflammatory cytokines and oxidative stress that can further stimulate the Runx2 expression by VSMC, the inhibition of both macrophage infiltration and inflammatory molecules in the SMC-specific Runx2 deficient mice highlights an important role of VSMC-derived Runx2 in regulating the interplay between VSMC and macrophages that exuberate vascular inflammation.
The pro-inflammatory feature of the increased RANKL in VSMC contributes to enhanced vascular calcification in atherosclerosis, although RANKL deficiency does not block oxidative stress-induced calcification of VSMC114. In the RANKL transgenic mice, increased RANKL expression induces soft tissue calcification, while inhibition of RANKL by a monoclonal antibody against RANKL inhibits vascular calcification115. Additionally, the RANKL/RANK system is crucial for the formation of bone-resorbing osteoclasts from monocytic precursors116. We have demonstrated that increased RANKL in the calcified atherosclerotic lesions is associated with increased osteoclast-like cells in the atherosclerosis-prone ApoE−/− mice16, 114. Consistently, the OPG deficient mice exhibit accelerated vascular calcification and increased osteoclast-like cells in the calcified lesions117, 118. Inhibition of Runx2-dependent RANKL expression in VSMC attenuates vascular osteoclast formation in the SMC-specific Runx2 deficient mice, as well as differentiation of osteoclast-like cells from macrophages in a VSMC and macrophage co-culture system16, 114. Accordingly, RANKL may not only serve as a chemoattractant that induces macrophage infiltration but also act as a signal to facilitate macrophage differentiation into osteoclast-like cells in the vicinity of calcified VSMC. The presence of osteoclast-like cells in close apposition to the calcified lesions in human and mouse atherosclerotic arteries16, 114, 117, 119, 120 supports the coupling between Runx2-regulated VSMC calcification and RANKL-induced formation of vascular osteoclasts in the calcified atherosclerotic lesions, resembling the interplay of osteoblasts and osteoclasts in regulating bone remodeling.
Therefore, upregulation of Runx2 in VSMC greatly accelerates the development and progression of arteriosclerosis via multilateral mechanisms, including directly promoting osteogenic differentiation and calcification of VSMC, as well as inducing formation of mineral-resorbing vascular osteoclast-like cells via macrophage infiltration and vascular inflammation, which further accelerate vascular calcification and arterial stiffness.
• Runx2 upregulation enhances SMC phenotypic switch
The pathophysiological function of Runx2 in VSMC remains elusive. Runx2 regulation on cell lineage commitment has been linked to the binding of Runx2 to the promoters of cell fate-regulated target genes that exhibit distinct Runx2-dependent modification in histone acetylation and methylation121. In addition, Runx2 also control cell proliferation and lineage commitment by repressing RNA Pol I-mediated ribosomal RNA (rRNA) synthesis via forming complexes with the RNA Pol I transcription factors and thus affecting local chromatin histone modifications122. The observations of both enhanced rRNA transcription and protein synthesis by the Runx2 deficiency and repressed rRNA synthesis by the Runx2 upregulation suggest that Runx2 may control lineage-specific ribosomal biogenesis122. In normal and healthy arteries and isolated VSMC, the expression of Runx2 is very low12, 16, 123. Furthermore, Runx2 deficiency does not affect either the basal expression of SMC marker genes in VSMC, or vascular structure and function in mice12, 16, 18. However, the upregulated Runx2 is evident in atherosclerosis and vascular calcification, which is inversely correlated with the expression of SMC markers12, 14–16, 18, 124, suggesting that the Runx2 upregulation plays a key regulatory role in vascular pathology.
In cultured VSMC, upregulated Runx2 directly induces VSMC calcification, concomitantly with decreased expression of SMC markers, indicating that the Runx2 upregulation drives VSMC phenotypic switch and trans-differentiation of VSMC into “bone-like” cells. In support of this notion, overexpression of Runx2 has been shown to repress myocardin-induced SMC differentiation125; and expression of myocardin-induced SMC genes is enhanced in the Runx2 null cells125. Consistently, we have demonstrated that Runx2 deficiency in VSMC blocks oxidative stress-induced repression of SMC markers, indicating the role of the Runx2 upregulation in mediating oxidative stress-induced VSMC de-differentiation16.
The inhibitory effects of the Runx2 upregulation on myocardin-induced expression of SMC marker genes in C3H10T1/2 cells is mediated via direct binding of Runx2 to the serum response factor (SRF). The Runx2/SRF binidng disrupts the formation of the myocardin/SRF complex, which is the master transcription complex that determines the expression of SMC marker genes125. In VSMC, oxidative stress induces downregulation of myocardin and upregulation of Runx212, leading to reduced myocardin/SRF binding that decrease myocardin-dependent expression of SMC marker genes, but enhanced Runx2/SRF binding increases expression of Runx2-dependent osteogenic genes126. Therefore, SRF serves as an important co-transcription factor for Runx2 in regulating SMC phenotypic switch and osteogenic transdifferntation, via tilting the balance of SRF/myocardin binding toward SRF/Runx2 binding.
Runx2 also interacts with krüppel-like factor 4 (KLF4), and such interaction increases osteogenic transcription activity of Runx2 and promotes VSMC calcification127. KLF4 is a member of the zinc-finger class of transcriptional regulators, it represses SMC phenotype by inhibiting the binding of myocardin and SRF to the promoters of SMC marker genes128–130. Increased KLF4 has been shown to promote high phosphate-induced VSMC osteogenic differentiation and calcification129. Accordingly, upregulation of both KLF4 and Runx2 in SMC contributes to SMC phenotypic switch via repressing the activity of the master SMC transcription factors myocardin/SRF; and their interaction further facilitates the osteogenic transdifferentation of VSMC.
It is important to note that Runx287 and the master SMC regulators, myocardin and SRF131, can be directly modified by O-GlcNAcylation. In VSMC and arteries from diabetic mice, increased O-GlcNAcylation upregulates Runx2 and downregulates SMC markers17. Studies in human SMC show that the expressions of SRF/myocardin and KLF4 are inversely regulated by O-GlcNAcylation inhibition132. Accordingly, increased O-GlcNAcylation may promote SMC phenotypic switch by modulating these key transcription regulators of SMC marker genes. As O-GlcNAcylation appears to be the “integrated sensor” for oxidative and metabolic stress in the vasculature6, it is necessary to define the precise O-GlcNAc modificaiton domains on Runx2, SRF/myocardin and KLF4 that are essential for their precise regulations of SMC phenotype. In addition, KLF4 can modulate histone acetylation via the recruitment of p300133; and Runx2 acetylation by p300 enhances Runx2 stability and transactivity53. Therefore, further studies of the spaciotemporal regulation of the Runx2 and KLF4 by PTMs, including O-GlcNAcylation, and their interplay in modulating chromation structure as well as the recruitment of SMC-specific transcription complex on the promoters of the SMC marker genes will provide new molecualr insights into the function of Runx2 in regulating SMC phenotype switch and the development of vascular disease.
Conclusions and Prospective
The Runx2 upregulation in the cardiovascular system has emerged as an important mediator for adverse cellular events that drive vascular pathology. Lessons learned from the bone biology have facilitated our understanding of the Runx2 gene, Runx2 protein structure and the multilateral genetic, epigenetic and PTM regulatory mechanisms that control Runx2 expression and transcriptional activity. Studies in the cardiovascular system have now established the indispensable function of upregulated vascular Runx2 in promoting vascular calcification, a Runx2-dependent cell-driven process resembling osteogenic differentiation of the bone-forming osteoblasts. As elucidated in the osteoblast-mediated osteogenic differentiations, the Runx2–P1 promoter that drives the osteogenic specific differentiation displays conserved three-dimensional chromatin structure with the syntenic Supt3h promoter. Therefore, comprehensive analysis of Supt3h expression and its interaction with Runx2-P1 on the chromatin structure map during VSMC osteogenic differentiation and calcification should reveal fundamental regulation of Runx2-P1 expression in VSMC.
Activation of osteogenic signaling pathways in the vascular system, including ERK/MAPK, AKT and BMP-2, induces Runx2 upregulation and PTMs, such as phosphorylation, acetylation, ubiquitination, O-GlcNAcylation. The Runx2 PTMs crosstalk spatiotemporally to control Runx2 stability, nuclear localization as well as the Runx2 binding to its co-factors and target DNA, which is critical for Runx2 osteogenic transcriptional activity and vascular calcification. It has also been recognized that opposite regulations of Runx2 in bone and vascular cells by local and circulating factors and the microenvironments contribute to increased vascular calcification and decreased bone mineral density, the bone-vascular mineral paradox often seen in patients with CKD and atherosclerosis (reviewed by Chen et al)7. For instance, Runx2 expression and transcriptional activity is inversely regulated by oxidative stress in bone cells and vascular cells, leading to opposite mineralization outcomes12, 134, 135. Therefore, it is important to dissect the distinct signaling pathways that uniquely upregulate Runx2, and maybe more importantly, the Runx2 PTMs and its transcription co-factors in the vascular cells that determines vascular calcification, which may provide molecular insight into new targets for therapeutic strategies to prevent the Runx2 upregulation in vascular cells.
Uncovering the role of Runx2 in regulating expression of inflammatory molecules and infiltration of macrophages in the vasculature has shed light on Runx2-mediated vascular inflammation that accelerates the pathogenesis of vascular disease. The SMC-specific Runx2 deletion in the ApoE−/− mice attenuates infiltration of CD68-positive macrophages into the atherosclerotic lesions, which has not only supported the significant contribution of VSMC to vascular inflammation, but also highlighted the function of VSMC-expressed Runx2 in regulating the expression of inflammatory cytokines that mediate the crosstalk between VSMC and macrophages. Of particular interest, Runx2-regulated expression of RANKL by VSMC functions as a chemoattractant that induces macrophage/monocyte migration towards VSMC; and further induces the differentiation of the macrophage/monocyte into bone-resorbing osteoclast-like cells. It is unknown whether the vascular osteoclasts are derived form a specific subset of macrophages, and how these vascular osteoclasts may contribute to the pathogenesis of vascular calcification and atherosclerosis. The advance in the single-cell technologies, such as single-cell RNA sequencing, offers a powerful tool and a new opportunity to dissect Runx2-mediated crosstalk between VSMC, its surrounding specific macrophage subsets and other immune cells or unknown cell types in arteriosclerosis.
There is an increasing appreciation of a new role of Runx2 in regulating SMC phenotypic switch, as compelling evidence has revealed the repressing of SMC contractile markers by the Runx2 upregulation, although Runx2 deficiency in SMC does not affect basal expression of SMC marker genes and SMC phenotype. t is conceivable that low expression level of Runx2 in normal VSMC restricts its function, while upregulation of Runx2 by the pro-osteogenic signals in the vasculature enables its transcriptional reprogramming. Runx2 has been shown to regulate cell proliferation and lineage commitment by binding to the promoters of specific cell fate-regulated target genes as well as controlling lineage-specific ribosomal biogenesis. The identification of the binding of Runx2 with key SMC phenotype regulators, such as SRF and KLF4, and its repression of the expression of SMC marker genes via disrupting the function of the master SMC transcription complex SRF/myocardin has provided molecular insights into the regulation of Runx2 on SMC phenotype switch. It remains to be determined how the Runx2 upregulation in VSMC and Runx2 PTMs, particularly O-GlcNAcylation of Runx2, may affect chromatin structure, histone acetylation/methylation, and the recruitment of SMC-specific transcription factors that determine SMC-specific gene expression. Studies to uncover Runx2-dependent genome-wide chromatin accessibility and formation of the transcriptional complex on SMC-specific marker genes will provide important new insights into the molecular regulation of Runx2 on SMC phenotypic switch.
Lastly, it is also important to recognize the heterogeneities of VSMC, as several subpopulations of VSMC have been observed in the atherosclerotic plaques. Whether Runx2 is upregulated in all VSMC or exclusively in selective VSMC subpopulations is unknown. How the specific VSMC subpopulations drive phenotypic switch, vascular calcification, and production of inflammatory cytokines remain to be elucidated. VSMC phenotypic transformation is linked to a wide variety of vascular pathologies, including vascular calcification, stiffness and atherosclerosis, leading to increased cardiovascular morbidity and mortality. Further investigations are warranted to address these unanswered questions, which should provide new breakthroughs in the understanding of Runx2-dependent transcriptional reprogramming of vascular cells and their contributions to the development of arteriosclerosis.
HIGHLIGHTS.
This review covers the Runx2 regulatory mechanisms and its function in modulating VSMC osteogenic differentiation, vascular inflammation and VSMC phenotypic switch that contribute to the pathogenesis of arteriosclerosis, and highlights:
The key transcriptional regulation of Runx2, the critical Runx2 functional domains and their essential role in osteogenic differentiation and bone formation.
The dynamic and complex cross-talks between a wide variety of posttranslational modifications (PTMs), including phosphorylation, acetylation, ubiquitination and O-GlcNAcylation, in regulating Runx2 stability, cellular localization and osteogenic transcriptional activity.
The activation of ERK/MAPK/p38, BMP-2 and AKT signaling cascades in the upregulation of Runx2-mediated osteogenic differentiation and calcification of VSMC.
VSMC-derived Runx2 in the regulation of inflammatory cytokines, infiltration of macrophages and formation of vascular osteoclast-like cells.
The emerging role of upregulation of Runx2 in modulating VSMC phenotypic switch.
ACKNOWLEDGEMENTS
Due to the scope and space limitation, we apologize for not being able to include all the important work in the field.
SOURCES OF FUNDING
The original research of the authors has been supported by grants from the National Institutes of Health HL092215, HL136165, HL146103 and DK100847 as well as United States Department of Veterans Affairs BX003617 and BX004426 to YC.
ABBREVIATIONS:
- Ac-CoA
acetyl coenzyme A
- AKT
protein kinase B
- ALP
alkaline phosphatase
- ApoE
apolipoprotein E
- BMP-2
bone morphogenetic protein-2
- CCD
cause cleidocranial dysplasia
- CKD
chronic kidney disease
- CBFβ
core binding factor β
- Col 1
type 1 collagen
- ERK
extracellular signal-regulated kinase
- FoxO
forkhead box protein O
- GSK3β
glycogen synthase kinase 3 beta
- HATs
histone acetyltransferases
- HDACs
histone deacetylases
- HIPK3
Homeodomain-interacting protein kinase 3
- KLF4
krüppel-like factor 4
- MAPK
mitogen-activated protein kinases
- MMP 13
matrix metalloproteinases-13
- NLS
nuclear localization signal
- NMTS
nuclear matrix targeting signal
- OGA
O-GlcNAcase
- OGT
O-GlcNAc transferase
- O-GlcNAcylation
O-linked β-N-acetylglucosamine modification
- OPN
osteopontin
- OPG
osteoprotegrin
- PKCδ
protein kinase Cδ
- PST
proline-serine-threonine
- PTMs
posttranslational modifications
- PTH
parathyroid hormone
- RANKL
receptor activator of nuclear factor kappa-B ligand
- RHD
runt-homology domain
- rRNA
ribosomal RNA
- RANKL
receptor activator of nuclear factor kappa-B ligand
- Runx2
Runt-related transcription factor 2
- SMA
smooth muscle α-actin
- SMMHC
smooth muscle cell myosin heavy chain
- Smurf1
Smad ubiquitin regulatory factor 1
- Sox9
sex-determining region Y-box 9
- SRF
serum response factor
- Supt3h
suppressor of Ty3 homolog
- STAT1
signal transducer and activator of transcription 1
- TLE2
transducin-like Enhancer of split 2
- VSMC
vascular smooth muscle cells
- YAP
Yes-associated protein
Footnotes
DISCLOSURES
The authors have no potential conflicts of interests to disclose.
REFERENCES
- 1.Lakatta EG. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises. 2003;107:490–497 [DOI] [PubMed] [Google Scholar]
- 2. Chirinos JA, Segers P, Hughes T, Townsend R. Large-artery stiffness in health and disease: Jacc state-of-the-art review. J Am Coll Cardiol. 2019;74:1237–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. London GM, Guerin AP, Marchais SJ, Metivier F, Pannier B, Adda H. Arterial media calcification in end-stage renal disease: Impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant. 2003;18:1731–1740 [DOI] [PubMed] [Google Scholar]
- 4. Shanahan CM, Cary NR, Salisbury JR, Proudfoot D, Weissberg PL, Edmonds ME. Medial localization of mineralization-regulating proteins in association with monckeberg’s sclerosis: Evidence for smooth muscle cell-mediated vascular calcification. Circulation. 1999;100:2168–2176 [DOI] [PubMed] [Google Scholar]
- 5. Raffield LM, Cox AJ, Criqui MH, Hsu FC, Terry JG, Xu J, Freedman BI, Carr JJ, Bowden DW. Associations of coronary artery calcified plaque density with mortality in type 2 diabetes: The diabetes heart study. Cardiovasc Diabetol. 2018;17:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen Y, Zhao X, Wu H. Metabolic stress and cardiovascular disease in diabetes mellitus: The role of protein o-glcnac modification. Arterioscler Thromb Vasc Biol. 2019;39:1911–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen Y, Zhao X, Wu H. Arterial stiffness: A focus on vascular calcification and its link to bone mineralization. Arterioscler Thromb Vasc Biol. 2020;40:1078–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lacolley P, Regnault V, Avolio AP. Smooth muscle cell and arterial aging: Basic and clinical aspects. Cardiovascular research. 2018;114:513–528 [DOI] [PubMed] [Google Scholar]
- 9. Lanzer P, Boehm M, Sorribas V, Thiriet M, Janzen J, Zeller T, St Hilaire C, Shanahan C. Medial vascular calcification revisited: Review and perspectives. European heart journal. 2014;35:1515–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Steitz SA, Speer MY, Curinga G, Yang HY, Haynes P, Aebersold R, Schinke T, Karsenty G, Giachelli CM. Smooth muscle cell phenotypic transition associated with calcification: Upregulation of cbfa1 and downregulation of smooth muscle lineage markers. Circ Res. 2001;89:1147–1154 [DOI] [PubMed] [Google Scholar]
- 11. Tintut Y, Parhami F, Bostrom K, Jackson SM, Demer LL. Camp stimulates osteoblast-like differentiation of calcifying vascular cells. Potential signaling pathway for vascular calcification. J Biol Chem. 1998;273:7547–7553 [DOI] [PubMed] [Google Scholar]
- 12. Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM, McDonald JM, Chen Y. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor runx2 by akt signaling. J Biol Chem. 2008;283:15319–15327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shao JS, Cai J, Towler DA. Molecular mechanisms of vascular calcification: Lessons learned from the aorta. Arterioscler Thromb Vasc Biol. 2006;26:1423–1430 [DOI] [PubMed] [Google Scholar]
- 14. Engelse MA, Neele JM, Bronckers AL, Pannekoek H, de Vries CJ. Vascular calcification: Expression patterns of the osteoblast-specific gene core binding factor alpha-1 and the protective factor matrix gla protein in human atherogenesis. Cardiovasc. Res. 2001;52:281–289 [DOI] [PubMed] [Google Scholar]
- 15. Tyson KL, Reynolds JL, McNair R, Zhang Q, Weissberg PL, Shanahan CM. Osteo/chondrocytic transcription factors and their target genes exhibit distinct patterns of expression in human arterial calcification. Arterioscler. Thromb. Vasc. Biol. 2003;23:489–494 [DOI] [PubMed] [Google Scholar]
- 16. Sun Y, Byon CH, Yuan K, Chen J, Mao X, Heath JM, Javed A, Zhang K, Anderson PG, Chen Y. Smooth muscle cell-specific runx2 deficiency inhibits vascular calcification. Circ Res. 2012;111:543–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Heath JM, Sun Y, Yuan K, Bradley WE, Litovsky S, Dell’Italia LJ, Chatham JC, Wu H, Chen Y. Activation of akt by o-linked n-acetylglucosamine induces vascular calcification in diabetes mellitus. Circ Res. 2014;114:1094–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lin ME, Chen T, Leaf EM, Speer MY, Giachelli CM. Runx2 expression in smooth muscle cells is required for arterial medial calcification in mice. Am J Pathol. 2015;185:1958–1969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen BR, Selby PB, Owen MJ. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–771 [DOI] [PubMed] [Google Scholar]
- 20. Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T. Targeted disruption of cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–764 [DOI] [PubMed] [Google Scholar]
- 21. Ducy P, Starbuck M, Priemel M, Shen J, Pinero G, Geoffroy V, Amling M, Karsenty G. A cbfa1-dependent genetic pathway controls bone formation beyond embryonic development. Genes Dev. 1999;13:1025–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee B, Thirunavukkarasu K, Zhou L, Pastore L, Baldini A, Hecht J, Geoffroy V, Ducy P, Karsenty G. Missense mutations abolishing DNA binding of the osteoblast-specific transcription factor osf2/cbfa1 in cleidocranial dysplasia. Nature genetics. 1997;16:307–310 [DOI] [PubMed] [Google Scholar]
- 23. Mundlos S, Otto F, Mundlos C, Mulliken JB, Aylsworth AS, Albright S, Lindhout D, Cole WG, Henn W, Knoll JH, Owen MJ, Mertelsmann R, Zabel BU, Olsen BR. Mutations involving the transcription factor cbfa1 cause cleidocranial dysplasia. Cell. 1997;89:773–779 [DOI] [PubMed] [Google Scholar]
- 24. Ducy P, Karsenty G. Two distinct osteoblast-specific cis-acting elements control expression of a mouse osteocalcin gene. Mol. Cell Biol. 1995;15:1858–1869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/cbfa1: A transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–754 [DOI] [PubMed] [Google Scholar]
- 26. Jiménez MJ, Balbín M, López JM, Alvarez J, Komori T, López-Otín C. Collagenase 3 is a target of cbfa1, a transcription factor of the runt gene family involved in bone formation. Molecular and cellular biology. 1999;19:4431–4442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Levanon D, Negreanu V, Bernstein Y, Bar-Am I, Avivi L, Groner Y. Aml1, aml2, and aml3, the human members of the runt domain gene-family: Cdna structure, expression, and chromosomal localization. Genomics. 1994;23:425–432 [DOI] [PubMed] [Google Scholar]
- 28. Bae SC, Ogawa E, Maruyama M, Oka H, Satake M, Shigesada K, Jenkins NA, Gilbert DJ, Copeland NG, Ito Y. Pebp2 alpha b/mouse aml1 consists of multiple isoforms that possess differential transactivation potentials. Molecular and cellular biology. 1994;14:3242–3252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu JC, Lengner CJ, Gaur T, Lou Y, Hussain S, Jones MD, Borodic B, Colby JL, Steinman HA, van Wijnen AJ, Stein JL, Jones SN, Stein GS, Lian JB. Runx2 protein expression utilizes the runx2 p1 promoter to establish osteoprogenitor cell number for normal bone formation. J Biol Chem. 2011;286:30057–30070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Harada H, Tagashira S, Fujiwara M, Ogawa S, Katsumata T, Yamaguchi A, Komori T, Nakatsuka M. Cbfa1 isoforms exert functional differences in osteoblast differentiation. J Biol Chem. 1999;274:6972–6978 [DOI] [PubMed] [Google Scholar]
- 31. Robertson AJ, Larroux C, Degnan BM, Coffman JA. The evolution of runx genes ii. The c-terminal groucho recruitment motif is present in both eumetazoans and homoscleromorphs but absent in a haplosclerid demosponge. BMC research notes. 2009;2:59–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barutcu AR, Tai PWL, Wu H, Gordon JAR, Whitfield TW, Dobson JR, Imbalzano AN, Lian JB, van Wijnen AJ, Stein JL, Stein GS. The bone-specific runx2-p1 promoter displays conserved three-dimensional chromatin structure with the syntenic supt3h promoter. Nucleic acids research. 2014;42:10360–10372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Drissi H, Luc Q, Shakoori R, Chuva De Sousa Lopes S, Choi JY, Terry A, Hu M, Jones S, Neil JC, Lian JB, Stein JL, Van Wijnen AJ, Stein GS. Transcriptional autoregulation of the bone related cbfa1/runx2 gene. Journal of cellular physiology. 2000;184:341–350 [DOI] [PubMed] [Google Scholar]
- 34. Zambotti A, Makhluf H, Shen J, Ducy P. Characterization of an osteoblast-specific enhancer element in the cbfa1 gene. J Biol Chem. 2002;277:41497–41506 [DOI] [PubMed] [Google Scholar]
- 35. Lee MH, Kim YJ, Yoon WJ, Kim JI, Kim BG, Hwang YS, Wozney JM, Chi XZ, Bae SC, Choi KY, Cho JY, Choi JY, Ryoo HM. Dlx5 specifically regulates runx2 type ii expression by binding to homeodomain-response elements in the runx2 distal promoter. J. Biol. Chem. 2005;280:35579–35587 [DOI] [PubMed] [Google Scholar]
- 36. Kammerer M, Gutzwiller S, Stauffer D, Delhon I, Seltenmeyer Y, Fournier B. Estrogen receptor α (erα) and estrogen related receptor α (errα) are both transcriptional regulators of the runx2-i isoform. Mol Cell Endocrinol. 2013;369:150–160 [DOI] [PubMed] [Google Scholar]
- 37. Yang DC, Tsai CC, Liao YF, Fu HC, Tsay HJ, Huang TF, Chen YH, Hung SC. Twist controls skeletal development and dorsoventral patterning by regulating runx2 in zebrafish. PloS one. 2011;6:e27324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huang G, Shigesada K, Ito K, Wee HJ, Yokomizo T, Ito Y. Dimerization with pebp2beta protects runx1/aml1 from ubiquitin-proteasome-mediated degradation. The EMBO journal. 2001;20:723–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ogawa E, Maruyama M, Kagoshima H, Inuzuka M, Lu J, Satake M, Shigesada K, Ito Y. Pebp2/pea2 represents a family of transcription factors homologous to the products of the drosophila runt gene and the human aml1 gene. Proc Natl Acad Sci U S A. 1993;90:6859–6863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Golling G, Li L, Pepling M, Stebbins M, Gergen JP. Drosophila homologs of the proto-oncogene product pebp2/cbf beta regulate the DNA-binding properties of runt. Molecular and cellular biology. 1996;16:932–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Thirunavukkarasu K, Mahajan M, McLarren KW, Stifani S, Karsenty G. Two domains unique to osteoblast-specific transcription factor osf2/cbfa1 contribute to its transactivation function and its inability to heterodimerize with cbfbeta. Molecular and cellular biology. 1998;18:4197–4208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bialek P, Kern B, Yang X, Schrock M, Sosic D, Hong N, Wu H, Yu K, Ornitz DM, Olson EN, Justice MJ, Karsenty G. A twist code determines the onset of osteoblast differentiation. Dev. Cell. 2004;6:423–435 [DOI] [PubMed] [Google Scholar]
- 43. Zhou G, Zheng Q, Engin F, Munivez E, Chen Y, Sebald E, Krakow D, Lee B. Dominance of sox9 function over runx2 during skeletogenesis. Proc Natl Acad Sci U S A. 2006;103:19004–19009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yagi R, Chen LF, Shigesada K, Murakami Y, Ito Y. A ww domain-containing yes-associated protein (yap) is a novel transcriptional co-activator. The EMBO journal. 1999;18:2551–2562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang YW, Yasui N, Ito K, Huang G, Fujii M, Hanai J, Nogami H, Ochi T, Miyazono K, Ito Y. A runx2/pebp2alpha a/cbfa1 mutation displaying impaired transactivation and smad interaction in cleidocranial dysplasia. Proc. Natl. Acad. Sci. U. S. A. 2000;97:10549–10554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee KS, Kim HJ, Li QL, Chi XZ, Ueta C, Komori T, Wozney JM, Kim EG, Choi JY, Ryoo HM, Bae SC. Runx2 is a common target of transforming growth factor beta1 and bone morphogenetic protein 2, and cooperation between runx2 and smad5 induces osteoblast-specific gene expression in the pluripotent mesenchymal precursor cell line c2c12. Molecular and cellular biology. 2000;20:8783–8792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Aronson BD, Fisher AL, Blechman K, Caudy M, Gergen JP. Groucho-dependent and -independent repression activities of runt domain proteins. Molecular and cellular biology. 1997;17:5581–5587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Quack I, Vonderstrass B, Stock M, Aylsworth AS, Becker A, Brueton L, Lee PJ, Majewski F, Mulliken JB, Suri M, Zenker M, Mundlos S, Otto F. Mutation analysis of core binding factor a1 in patients with cleidocranial dysplasia. American journal of human genetics. 1999;65:1268–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim S, Koga T, Isobe M, Kern BE, Yokochi T, Chin YE, Karsenty G, Taniguchi T, Takayanagi H. Stat1 functions as a cytoplasmic attenuator of runx2 in the transcriptional program of osteoblast differentiation. Genes Dev. 2003;17:1979–1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zaidi SK, Javed A, Choi JY, van Wijnen AJ, Stein JL, Lian JB, Stein GS. A specific targeting signal directs runx2/cbfa1 to subnuclear domains and contributes to transactivation of the osteocalcin gene. J. Cell Sci. 2001;114:3093–3102 [DOI] [PubMed] [Google Scholar]
- 51. Choi JY, Pratap J, Javed A, Zaidi SK, Xing L, Balint E, Dalamangas S, Boyce B, van Wijnen AJ, Lian JB, Stein JL, Jones SN, Stein GS. Subnuclear targeting of runx/cbfa/aml factors is essential for tissue-specific differentiation during embryonic development. Proc Natl Acad Sci U S A. 2001;98:8650–8655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lian JB, Stein GS, Javed A, van Wijnen AJ, Stein JL, Montecino M, Hassan MQ, Gaur T, Lengner CJ, Young DW. Networks and hubs for the transcriptional control of osteoblastogenesis. Rev Endocr Metab Disord. 2006;7:1–16 [DOI] [PubMed] [Google Scholar]
- 53. Jun JH, Yoon WJ, Seo SB, Woo KM, Kim GS, Ryoo HM, Baek JH. Bmp2-activated erk/map kinase stabilizes runx2 by increasing p300 levels and histone acetyltransferase activity. J Biol Chem. 2010;285:36410–36419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xiao G, Jiang D, Thomas P, Benson MD, Guan K, Karsenty G, Franceschi RT. Mapk pathways activate and phosphorylate the osteoblast-specific transcription factor, cbfa1. J Biol Chem. 2000;275:4453–4459 [DOI] [PubMed] [Google Scholar]
- 55. Greenblatt MB, Shim JH, Zou W, Sitara D, Schweitzer M, Hu D, Lotinun S, Sano Y, Baron R, Park JM, Arthur S, Xie M, Schneider MD, Zhai B, Gygi S, Davis R, Glimcher LH. The p38 mapk pathway is essential for skeletogenesis and bone homeostasis in mice. J Clin Invest. 2010;120:2457–2473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ge C, Xiao G, Jiang D, Franceschi RT. Critical role of the extracellular signal-regulated kinase-mapk pathway in osteoblast differentiation and skeletal development. J Cell Biol. 2007;176:709–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shui C, Spelsberg TC, Riggs BL, Khosla S. Changes in runx2/cbfa1 expression and activity during osteoblastic differentiation of human bone marrow stromal cells. J Bone Miner Res. 2003;18:213–221 [DOI] [PubMed] [Google Scholar]
- 58. Ge C, Xiao G, Jiang D, Yang Q, Hatch NE, Roca H, Franceschi RT. Identification and functional characterization of erk/mapk phosphorylation sites in the runx2 transcription factor. J Biol Chem. 2009;284:32533–32543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zou W, Greenblatt MB, Shim JH, Kant S, Zhai B, Lotinun S, Brady N, Hu DZ, Gygi SP, Baron R, Davis RJ, Jones D, Glimcher LH. Mlk3 regulates bone development downstream of the faciogenital dysplasia protein fgd1 in mice. J Clin Invest. 2011;121:4383–4392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sierra OL, Towler DA. Runx2 trans-activation mediated by the msx2-interacting nuclear target requires homeodomain interacting protein kinase-3. Mol Endocrinol. 2010;24:1478–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pande S, Browne G, Padmanabhan S, Zaidi SK, Lian JB, van Wijnen AJ, Stein JL, Stein GS. Oncogenic cooperation between pi3k/akt signaling and transcription factor runx2 promotes the invasive properties of metastatic breast cancer cells. Journal of cellular physiology. 2013;228:1784–1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kim HJ, Kim JH, Bae SC, Choi JY, Kim HJ, Ryoo HM. The protein kinase c pathway plays a central role in the fibroblast growth factor-stimulated expression and transactivation activity of runx2. J Biol Chem. 2003;278:319–326 [DOI] [PubMed] [Google Scholar]
- 63. Wee HJ, Huang G, Shigesada K, Ito Y. Serine phosphorylation of runx2 with novel potential functions as negative regulatory mechanisms. EMBO Rep. 2002;3:967–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Huang YF, Lin JJ, Lin CH, Su Y, Hung SC. C-jun n-terminal kinase 1 negatively regulates osteoblastic differentiation induced by bmp2 via phosphorylation of runx2 at ser104. J Bone Miner Res. 2012;27:1093–1105 [DOI] [PubMed] [Google Scholar]
- 65. Kugimiya F, Kawaguchi H, Ohba S, Kawamura N, Hirata M, Chikuda H, Azuma Y, Woodgett JR, Nakamura K, Chung UI. Gsk-3beta controls osteogenesis through regulating runx2 activity. PloS one. 2007;2:e837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cheng A, Genever PG. Sox9 determines runx2 transactivity by directing intracellular degradation. Journal of Bone and Mineral Research. 2010;25:2680–2689 [DOI] [PubMed] [Google Scholar]
- 67. Yang XJ. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic acids research. 2004;32:959–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jeon EJ, Lee KY, Choi NS, Lee MH, Kim HN, Jin YH, Ryoo HM, Choi JY, Yoshida M, Nishino N, Oh BC, Lee KS, Lee YH, Bae SC. Bone morphogenetic protein-2 stimulates runx2 acetylation. J Biol Chem. 2006;281:16502–16511 [DOI] [PubMed] [Google Scholar]
- 69. Park OJ, Kim HJ, Woo KM, Baek JH, Ryoo HM. Fgf2-activated erk mitogen-activated protein kinase enhances runx2 acetylation and stabilization. J Biol Chem. 2010;285:3568–3574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Boumah CE, Lee M, Selvamurugan N, Shimizu E, Partridge NC. Runx2 recruits p300 to mediate parathyroid hormone’s effects on histone acetylation and transcriptional activation of the matrix metalloproteinase-13 gene. Mol Endocrinol. 2009;23:1255–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhang Z, Deepak V, Meng L, Wang L, Li Y, Jiang Q, Zeng X, Liu W. Analysis of hdac1-mediated regulation of runx2-induced osteopontin gene expression in c3h10t1/2 cells. Biotechnol Lett. 2012;34:197–203 [DOI] [PubMed] [Google Scholar]
- 72. Schroeder TM, Kahler RA, Li X, Westendorf JJ. Histone deacetylase 3 interacts with runx2 to repress the osteocalcin promoter and regulate osteoblast differentiation. J Biol Chem. 2004;279:41998–42007 [DOI] [PubMed] [Google Scholar]
- 73. Westendorf JJ, Zaidi SK, Cascino JE, Kahler R, van Wijnen AJ, Lian JB, Yoshida M, Stein GS, Li X. Runx2 (cbfa1, aml-3) interacts with histone deacetylase 6 and represses the p21(cip1/waf1) promoter. Molecular and cellular biology. 2002;22:7982–7992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bae HS, Yoon WJ, Cho YD, Islam R, Shin HR, Kim BS, Lim JM, Seo MS, Cho SA, Choi KY, Baek SH, Kim HG, Woo KM, Baek JH, Lee YS, Ryoo HM. An hdac inhibitor, entinostat/ms-275, partially prevents delayed cranial suture closure in heterozygous runx2 null mice. J Bone Miner Res. 2017;32:951–961 [DOI] [PubMed] [Google Scholar]
- 75. Tintut Y, Parhami F, Le V, Karsenty G, Demer LL. Inhibition of osteoblast-specific transcription factor cbfa1 by the camp pathway in osteoblastic cells. Ubiquitin/proteasome-dependent regulation. J Biol Chem. 1999;274:28875–28879 [DOI] [PubMed] [Google Scholar]
- 76. Scheffner M, Nuber U, Huibregtse JM. Protein ubiquitination involving an e1-e2-e3 enzyme ubiquitin thioester cascade. Nature. 1995;373:81–83 [DOI] [PubMed] [Google Scholar]
- 77. Zhao M, Qiao M, Harris SE, Oyajobi BO, Mundy GR, Chen D. Smurf1 inhibits osteoblast differentiation and bone formation in vitro and in vivo. J Biol Chem. 2004;279:12854–12859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yamashita M, Ying SX, Zhang GM, Li C, Cheng SY, Deng CX, Zhang YE. Ubiquitin ligase smurf1 controls osteoblast activity and bone homeostasis by targeting mekk2 for degradation. Cell. 2005;121:101–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Choi YH, Kim YJ, Jeong HM, Jin YH, Yeo CY, Lee KY. Akt enhances runx2 protein stability by regulating smurf2 function during osteoblast differentiation. The FEBS journal. 2014;281:3656–3666 [DOI] [PubMed] [Google Scholar]
- 80. Li X, Huang M, Zheng H, Wang Y, Ren F, Shang Y, Zhai Y, Irwin DM, Shi Y, Chen D, Chang Z. Chip promotes runx2 degradation and negatively regulates osteoblast differentiation. J Cell Biol. 2008;181:959–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Thacker G, Kumar Y, Khan MP, Shukla N, Kapoor I, Kanaujiya JK, Lochab S, Ahmed S, Sanyal S, Chattopadhyay N, Trivedi AK. Skp2 inhibits osteogenesis by promoting ubiquitin-proteasome degradation of runx2. Biochim Biophys Acta. 2016;1863:510–519 [DOI] [PubMed] [Google Scholar]
- 82. Zhu W, He X, Hua Y, Li Q, Wang J, Gan X. The e3 ubiquitin ligase wwp2 facilitates runx2 protein transactivation in a mono-ubiquitination manner during osteogenic differentiation. J Biol Chem. 2017;292:11178–11188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Shen R, Wang X, Drissi H, Liu F, O’Keefe RJ, Chen D. Cyclin d1-cdk4 induce runx2 ubiquitination and degradation. J Biol Chem. 2006;281:16347–16353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wells L, Vosseller K, Hart GW. Glycosylation of nucleocytoplasmic proteins: Signal transduction and o-glcnac. Science. 2001;291:2376–2378 [DOI] [PubMed] [Google Scholar]
- 85. Ong Q, Han W, Yang X. O-glcnac as an integrator of signaling pathways. Frontiers in endocrinology. 2018;9:599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kim SH, Kim YH, Song M, An SH, Byun HY, Heo K, Lim S, Oh YS, Ryu SH, Suh PG. O-glcnac modification modulates the expression of osteocalcin via ose2 and runx2. Biochem Biophys Res Commun. 2007;362:325–329 [DOI] [PubMed] [Google Scholar]
- 87. Nagel AK, Ball LE. O-glcnac modification of the runt-related transcription factor 2 (runx2) links osteogenesis and nutrient metabolism in bone marrow mesenchymal stem cells. Molecular & cellular proteomics : MCP. 2014;13:3381–3395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Andrés-Bergós J, Tardio L, Larranaga-Vera A, Gómez R, Herrero-Beaumont G, Largo R. The increase in o-linked n-acetylglucosamine protein modification stimulates chondrogenic differentiation both in vitro and in vivo. J Biol Chem. 2012;287:33615–33628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Koyama T, Kamemura K. Global increase in o-linked n-acetylglucosamine modification promotes osteoblast differentiation. Experimental cell research. 2015;338:194–202 [DOI] [PubMed] [Google Scholar]
- 90. Zeidan Q, Hart GW. The intersections between o-glcnacylation and phosphorylation: Implications for multiple signaling pathways. J Cell Sci. 2010;123:13–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Parhami F, Morrow AD, Balucan J, Leitinger N, Watson AD, Tintut Y, Berliner JA, Demer LL. Lipid oxidation products have opposite effects on calcifying vascular cell and bone cell differentiation. A possible explanation for the paradox of arterial calcification in osteoporotic patients. Arterioscler Thromb Vasc Biol. 1997;17:680–687 [DOI] [PubMed] [Google Scholar]
- 92. Jono S, McKee MD, Murry CE, Shioi A, Nishizawa Y, Mori K, Morii H, Giachelli CM. Phosphate regulation of vascular smooth muscle cell calcification. Circ Res. 2000;87:E10–17 [DOI] [PubMed] [Google Scholar]
- 93. Bostrom K, Demer LL. Regulatory mechanisms in vascular calcification. Crit Rev. Eukaryot. Gene Expr. 2000;10:151–158 [PubMed] [Google Scholar]
- 94. Kapustin AN, Davies JD, Reynolds JL, McNair R, Jones GT, Sidibe A, Schurgers LJ, Skepper JN, Proudfoot D, Mayr M, Shanahan CM. Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circ Res. 2011;109:e1–12 [DOI] [PubMed] [Google Scholar]
- 95. Giachelli CM. Vascular calcification mechanisms. J Am Soc Nephrol. 2004;15:2959–2964 [DOI] [PubMed] [Google Scholar]
- 96. Byon CH, Chen Y. Molecular mechanisms of vascular calcification in chronic kidney disease: The link between bone and the vasculature. Curr Osteoporos Rep. 2015;13:206–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Deng L, Huang L, Sun Y, Heath JM, Wu H, Chen Y. Inhibition of foxo1/3 promotes vascular calcification. Arterioscler Thromb Vasc Biol. 2015;35:175–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Cai Z, Ding Y, Zhang M, Lu Q, Wu S, Zhu H, Song P, Zou MH. Ablation of adenosine monophosphate-activated protein kinase alpha1 in vascular smooth muscle cells promotes diet-induced atherosclerotic calcification in vivo. Circ Res. 2016;119:422–433 [DOI] [PubMed] [Google Scholar]
- 99. Yang Y, Sun Y, Chen J, Bradley WE, Dell’Italia LJ, Wu H, Chen Y. Akt-independent activation of p38 map kinase promotes vascular calcification. Redox Biol. 2018;16:97–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Hruska KA, Mathew S, Saab G. Bone morphogenetic proteins in vascular calcification. Circ. Res. 2005;97:105–114 [DOI] [PubMed] [Google Scholar]
- 101. Bostrom KI, Jumabay M, Matveyenko A, Nicholas SB, Yao Y. Activation of vascular bone morphogenetic protein signaling in diabetes mellitus. Circ Res. 2011;108:446–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Shao JS, Aly ZA, Lai CF, Cheng SL, Cai J, Huang E, Behrmann A, Towler DA. Vascular bmp msx2 wnt signaling and oxidative stress in arterial calcification. Ann. N. Y. Acad. Sci. 2007;1117:40–50 [DOI] [PubMed] [Google Scholar]
- 103. Stabley JN, Towler DA. Arterial calcification in diabetes mellitus: Preclinical models and translational implications. Arteriosclerosis, thrombosis, and vascular biology. 2017;37:205–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Shao JS, Cheng SL, Pingsterhaus JM, Charlton-Kachigian N, Loewy AP, Towler DA. Msx2 promotes cardiovascular calcification by activating paracrine wnt signals. J. Clin. Invest. 2005;115:1210–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Afzal F, Pratap J, Ito K, Ito Y, Stein JL, van Wijnen AJ, Stein GS, Lian JB, Javed A. Smad function and intranuclear targeting share a runx2 motif required for osteogenic lineage induction and bmp2 responsive transcription. J. Cell Physiol. 2005;204:63–72 [DOI] [PubMed] [Google Scholar]
- 106. Franceschi RT, Xiao G, Jiang D, Gopalakrishnan R, Yang S, Reith E. Multiple signaling pathways converge on the cbfa1/runx2 transcription factor to regulate osteoblast differentiation. Connect. Tissue Res. 2003;44 Suppl 1:109–16.: 109–116 [PMC free article] [PubMed] [Google Scholar]
- 107. Swirski FK, Weissleder R, Pittet MJ. Heterogeneous in vivo behavior of monocyte subsets in atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29:1424–1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Potteaux S, Gautier EL, Hutchison SB, van Rooijen N, Rader DJ, Thomas MJ, Sorci-Thomas MG, Randolph GJ. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of apoe−/− mice during disease regression. J Clin Invest. 2011;121:2025–2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Yoshida T, Yamashita M, Hayashi M. Kruppel-like factor 4 contributes to high phosphate-induced phenotypic switching of vascular smooth muscle cells into osteogenic cells. J Biol Chem. 2012;287:25706–25714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Lund SA, Giachelli CM, Scatena M. The role of osteopontin in inflammatory processes. J Cell Commun Signal. 2009;3:311–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Gu L, Okada Y, Clinton SK, Gerard C, Sukhova GK, Libby P, Rollins BJ. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol. Cell. 1998;2:275–281 [DOI] [PubMed] [Google Scholar]
- 112. Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in ccr2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–897 [DOI] [PubMed] [Google Scholar]
- 113. Bennett BJ, Scatena M, Kirk EA, Rattazzi M, Varon RM, Averill M, Schwartz SM, Giachelli CM, Rosenfeld ME. Osteoprotegerin inactivation accelerates advanced atherosclerotic lesion progression and calcification in older apoe−/− mice. Arterioscler. Thromb. Vasc. Biol. 2006;26:2117–2124 [DOI] [PubMed] [Google Scholar]
- 114. Byon CH, Sun Y, Chen J, Yuan K, Mao X, Heath JM, Anderson PG, Tintut Y, Demer LL, Wang D, Chen Y. Runx2-upregulated receptor activator of nuclear factor kappab ligand in calcifying smooth muscle cells promotes migration and osteoclastic differentiation of macrophages. Arterioscler Thromb Vasc Biol. 2011;31:1387–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Helas S, Goettsch C, Schoppet M, Zeitz U, Hempel U, Morawietz H, Kostenuik PJ, Erben RG, Hofbauer LC. Inhibition of receptor activator of nf-kappab ligand by denosumab attenuates vascular calcium deposition in mice. Am. J. Pathol. 2009;175:473–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Takeshita S, Kaji K, Kudo A. Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts. J Bone Miner Res. 2000;15:1477–1488 [DOI] [PubMed] [Google Scholar]
- 117. Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, Boyle WJ, Simonet WS. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12:1260–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Collin-Osdoby P Regulation of vascular calcification by osteoclast regulatory factors rankl and osteoprotegerin. Circ. Res. 2004;95:1046–1057 [DOI] [PubMed] [Google Scholar]
- 119. Jeziorska M, McCollum C, Wooley DE. Observations on bone formation and remodelling in advanced atherosclerotic lesions of human carotid arteries. Virchows Archiv : an international journal of pathology. 1998;433:559–565 [DOI] [PubMed] [Google Scholar]
- 120. Doherty TM, Uzui H, Fitzpatrick LA, Tripathi PV, Dunstan CR, Asotra K, Rajavashisth TB. Rationale for the role of osteoclast-like cells in arterial calcification. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2002;16:577–582 [DOI] [PubMed] [Google Scholar]
- 121. Young DW, Hassan MQ, Yang XQ, Galindo M, Javed A, Zaidi SK, Furcinitti P, Lapointe D, Montecino M, Lian JB, Stein JL, van Wijnen AJ, Stein GS. Mitotic retention of gene expression patterns by the cell fate-determining transcription factor runx2. Proc Natl Acad Sci U S A. 2007;104:3189–3194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Young DW, Hassan MQ, Pratap J, Galindo M, Zaidi SK, Lee SH, Yang X, Xie R, Javed A, Underwood JM, Furcinitti P, Imbalzano AN, Penman S, Nickerson JA, Montecino MA, Lian JB, Stein JL, van Wijnen AJ, Stein GS. Mitotic occupancy and lineage-specific transcriptional control of rrna genes by runx2. Nature. 2007;445:442–446 [DOI] [PubMed] [Google Scholar]
- 123. Chen NX, Duan D, O’Neill KD, Wolisi GO, Koczman JJ, Laclair R, Moe SM. The mechanisms of uremic serum-induced expression of bone matrix proteins in bovine vascular smooth muscle cells. Kidney Int. 2006;70:1046–1053 [DOI] [PubMed] [Google Scholar]
- 124. Shanahan CM, Proudfoot D, Tyson KL, Cary NR, Edmonds M, Weissberg PL. Expression of mineralisation-regulating proteins in association with human vascular calcification. Z. Kardiol. 2000;89 Suppl 2:63–68 [DOI] [PubMed] [Google Scholar]
- 125. Tanaka T, Sato H, Doi H, Yoshida CA, Shimizu T, Matsui H, Yamazaki M, Akiyama H, Kawai-Kowase K, Iso T, Komori T, Arai M, Kurabayashi M. Runx2 represses myocardin-mediated differentiation and facilitates osteogenic conversion of vascular smooth muscle cells. Mol. Cell Biol. 2008;28:1147–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Chen J, Yuan K, Mao X, Miano JM, Wu H, Chen Y. Serum response factor regulates bone formation via igf-1 and runx2 signals. J Bone Miner Res. 2012;27:1659–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Zhu L, Zhang N, Yan R, Yang W, Cong G, Yan N, Ma W, Hou J, Yang L, Jia S. Hyperhomocysteinemia induces vascular calcification by activating the transcription factor runx2 via kruppel-like factor 4 up-regulation in mice. J Biol Chem. 2019;294:19465–19474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Salmon M, Gomez D, Greene E, Shankman L, Owens GK. Cooperative binding of klf4, pelk-1, and hdac2 to a g/c repressor element in the sm22α promoter mediates transcriptional silencing during smc phenotypic switching in vivo. Circ Res. 2012;111:685–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Yoshida T, Hayashi M. Role of krüppel-like factor 4 and its binding proteins in vascular disease. Journal of atherosclerosis and thrombosis. 2014;21:402–413 [DOI] [PubMed] [Google Scholar]
- 130. Ghaleb AM, Yang VW. Krüppel-like factor 4 (klf4): What we currently know. Gene. 2017;611:27–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Ni Z, Deng J, Potter CMF, Nowak WN, Gu W, Zhang Z, Chen T, Chen Q, Hu Y, Zhou B, Xu Q, Zhang L. Recipient c-kit lineage cells repopulate smooth muscle cells of transplant arteriosclerosis in mouse models. Circ Res. 2019;125:223–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Paredes F, Williams HC, Quintana RA, San Martin A. Mitochondrial protein poldip2 (polymerase delta interacting protein 2) controls vascular smooth muscle differentiated phenotype by o-linked glcnac (n-acetylglucosamine) transferase-dependent inhibition of a ubiquitin proteasome system. Circ Res. 2020;126:41–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Evans PM, Zhang W, Chen X, Yang J, Bhakat KK, Liu C. Kruppel-like factor 4 is acetylated by p300 and regulates gene transcription via modulation of histone acetylation. J Biol Chem. 2007;282:33994–34002 [DOI] [PubMed] [Google Scholar]
- 134. Byon CH, Heath JM, Chen Y. Redox signaling in cardiovascular pathophysiology: A focus on hydrogen peroxide and vascular smooth muscle cells. Redox Biol. 2016;9:244–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Mody N, Tintut Y, Radcliff K, Demer LL. Vascular calcification and its relation to bone calcification: Possible underlying mechanisms. J. Nucl. Cardiol. 2003;10:177–183 [DOI] [PubMed] [Google Scholar]