

Abstract
Background and Purpose:
Ischemic injury triggers multiple pathological responses in the brain tissue, including spreading depolarizations across the cerebral cortex (CSD). Microglia have been recently shown to play a significant role in the propagation of CSD. However, the intracellular responses of myeloid cells during ischemic stroke have not been investigated.
Methods:
We have studied intracellular calcium activity in cortical microglia in the stroke model of the middle cerebral artery occlusion, using the murine reporter line PC∷G5-tdT. High-speed two-photon microscopy through cranial windows was employed to record signals from genetically encoded indicators of calcium. Inflammatory stimuli and pharmacological inhibition were used to modulate microglial calcium responses in the somatosensory cortex.
Results:
In vivo imaging revealed periodical calcium activity in microglia during the hyperacute phase of ischemic stroke. This activity was more frequent during the first 6 hours after occlusion, but the amplitudes of calcium transients became larger at later time points. Consistent with CSD nature of these events, we reproducibly triggered comparable calcium transients with microinjections of potassium chloride (KCl) into adjacent cortical areas. Further, lipopolysaccharide (LPS)-induced peripheral inflammation, mimicking sterile inflammation during ischemic stroke, produced significantly greater microglial calcium transients during CSD. Finally, in vivo pharmacological analysis with calcium release-activated channel (CRAC) inhibitor CM-EX-137 demonstrated that CSD-associated microglial calcium transients after KCl microinjections are mediated at least in part by the CRAC mechanism.
Conclusions:
Our findings demonstrate that microglia participate in ischemic brain injury via previously undetected mechanisms, which may provide new avenues for therapeutic interventions.
Keywords: ischemic stroke, microglia, calcium imaging, two-photon, PC∷G5-tdT, CRAC
Graphical Abstract
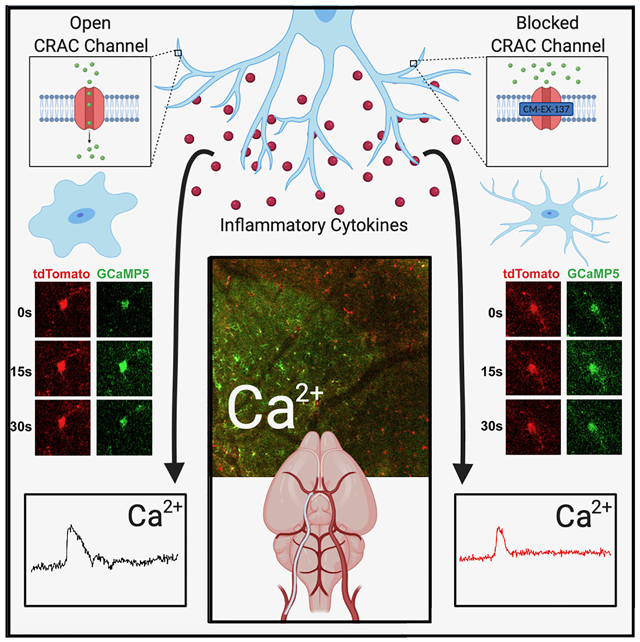
Introduction
Ischemic stroke causes ionic imbalance and electrical instability in the brain due to the hypoxic environment. Without sufficient oxygen, neurons are unable to maintain homeostasis and ultimately apoptose.1 These neurons and other dying cells release inflammatory cytokines into the extracellular environment, triggering a vicious cycle that further damages surrounding tissues and leads to the clinical symptoms associated with ischemic stroke.2
Evolution of ischemic injury is frequently associated with a wave-like propagation of abnormal neural activity, termed cortical spreading depolarizations (CSD).3 CSD initiate from a pathologic source and spread across the cortex, disrupting the metabolic equilibrium and reducing cerebral blood flow. After the initial wave passes, synaptic activity is silenced for 5-15 minutes before the tissue stabilizes and returns to its baseline function.3 In vaso-occlusive stroke, a depolarization propagates around the edge of the initial ischemic core, where cells are most susceptible to disruption.3 The tissue affected by the CSD experiences significant vasoconstriction, which inhibits sufficient membrane repolarization.3 This leads to an increased area of infarcted core as well as a larger circumference of tissue susceptible to further CSD.
As damaged cerebral neurons undergo reactive depolarizations of their membranes, a significant influx of calcium occurs, activating the intrinsic apoptosis pathway.4-6 Intracellular calcium overload is closely tied to the propagation of CSD, as has been shown in rodent models.4, 7 Cell-surface receptors on microglia receive signals of tissue damage and trigger an intracellular release of calcium from the endoplasmic reticulum (ER). The subsequent fall in calcium concentration within the ER leads to the opening of store-operated Calcium Release-Activated Calcium (CRAC) channels, which are the principal route of calcium entry from the extracellular space into immune cells.8, 9 Elevation of cytosolic calcium through CRAC channels mediates a host of cellular responses including secretion of proinflammatory cytokines.10, 11
Very little is presently known about the role of microglial calcium activity in ischemic stroke. Here, we used a genetically encoded Ca2 indicator in a murine model,12, 13 in the attempt to clarify the exact role of microglial calcium signaling in stroke.
Methods
The data supporting the conclusions of this experiment are available from the corresponding author upon reasonable request. A detailed Methods section can be found in the online-only Data Supplement.
Mice
Generation of the Cre-dependent GCaMP5- and tdTomato-expressing (PC∷G5-tdT) mouse line has been described previously.12 The Aif1(Iba1)-IRES-Cre (Iba1-Cre) strain has been also described and characterized earlier.13 The Cx3cr1-CreER animals14 were purchased from the Jackson laboratories. Focal cerebral ischemia was induced in 8-to-12-week-old mice by permanent MCA occlusion with silicon-coated filaments.15 Further details on breeding, surgical procedures and histological staining with 2,3,5-triphenyltetrazolium chloride (TTC) are provided in the online-only Data Supplement. All experiments were reviewed and approved by the University of Utah and University of Virginia IACUC Committees.
Cranial window surgery and two-photon imaging
In deeply anesthetized animals, skin and periosteum were removed from the skull and a 3-4 mm diameter circle was drilled in the right parietal bone. The skull flap was gently removed with fine forceps and a circular glass coverslip was placed over the exposed dura and sealed around the edges with superglue. A metal pin headpost was attached to the parietal bone and the exposed bone was covered with dental cement.15 Next, some animals were subjected to MCAo or sham procedures approximately 2 h prior to imaging. Lightly anesthetized animals were then mounted on the microscope stage and cortical microglia were imaged with Prairie Ultima IV at 0.2 Hz and 1,000 × 1,000 pixel resolution, or Olympus FVMPE-RS at 2.5 Hz and 500 × 500 pixel resolution. In both settings, the pulsed infrared lasers were tuned to 920 nm and emitted light was captured by green and red channel detectors.
Induction of CSD and Drug Administration
Chemical induction of CSD was performed through burr holes drilled over the visual cortex, posterior to the cranial window. After mounting the mouse on the imaging platform, a fine-tip glass micropipette filled with 1M KCl was loaded in the programmable Nanoject injector and lowered into the burr hole using a precise micromanipulator. One hundred nL of 1M KCl was expelled into the cortex 1 min after the initiation of high-speed imaging and the resulting CSD were recorded with 1.05 NA, 25X lens and 2x zoom. Some animals received lipopolysaccharides (LPS) i.p. at 1 mg/kg 12 h before imaging, or saline. Another group received CRAC channel inhibitor CM-EX-137-SDD at 25 mg/kg or vehicle via p.o. gavage 4 hours before imaging. The details on drug preparation are provided in the online-only Data Supplement.
Image processing and Analysis
The two-photon imaging T-series were analyzed with Imaris 9.5. The mean green (GCaMP5) fluorescence intensities, indicating intracellular calcium ([Ca2+]i) changes over time, were calculated in microglial somata detected with red (tdTomato) fluorescence. The traces were analyzed with custom scripts written in Python and plotted with OriginPro 7.5. These methods are explained further in the online-only Data Supplement.
Statistical analysis
Unless otherwise stated, all results are reported as mean ± SD and statistical tests were considered significant when p < 0.05. Statistical calculations (Student’s t-test, ANOVA) were performed as appropriate with GraphPad Prism.
Results
A robust in vivo reporter system to monitor microglial calcium activity
To investigate intracellular calcium levels in cortical microglia, we used crosses of Iba1(Aif1)-IRES-Cre (hereafter referred to as Iba1-Cre) mice with Polr2a-inserted GCaMP5G reporter, PC∷G5-tdT 12, 13. We hypothesized that acute ischemic stroke dysregulates intracellular microglial calcium activity, with calcium transients corresponding with and contributing to CSD. To address this question, we used an in vivo imaging approach through sealed craniotomy over the somatosensory cortex of the right hemisphere. In this setting, the Iba1-Cre; PC∷G5-tdT reporter system conveniently labels the Aif1/Iba1 cell lineage, involving meningeal macrophages and parenchymal microglia, with the structural reporter tdTomato and calcium indicator GCaMP5G (Figure 1A-C).12 Sparse neuronal labeling observed in deep cortical layers does not appreciably interfere with microglial cell specificity in the cortical plate.13
Figure 1. Waves of calcium activity in the ischemic brain.

A, Low magnification recording (0.2 Hz) acquired in the cortical parenchyma. Left panels show cells labeled with tdTomato in the Iba1-Cre; PC∷G5-tdT mouse. The panels on the right show the progression of calcium waves triggered by ischemic injury. The wavefront is contoured with a dotted white line. B, Higher power recording in the meningeal plane, consisting of meningeal macrophages. Macrophages did not display marked calcium activity in response to the underlying mild calcium wave in the cortex. C, In contrast, microglia in the cortical plate showed strong calcium transients during the wave progression, especially in the later stages of ischemic injury. D, Diagram showing the distribution of calcium waves detected with two photon imaging in 3 stroke mice (MCAo 1-3) and 3 sham controls. The occurrence of waves is indicated with vertical ticks, the duration of imaging sessions is denoted with open boxes. A representative section of an infarcted brain and a control brain stained with TTC at the end of the imaging session are to the right of each group.
Ischemic stroke induces waves of calcium activity in microglia
After the cranial window implantation, we performed middle cerebral artery occlusion (MCAo) using intraluminal filaments inserted in the right internal carotid artery. Histological staining performed at the end of each experiment determined that infarcts developed in the right hemisphere, striatum, thalamus and other brain regions of the MCAo-treated mice, but not in sham controls (Figure 1D and Figure I in the online-only Data Supplement). The lightly anesthetized mice were then mounted on the stage of a multiphoton microscope and intravital recordings were performed in a series of imaging sessions corresponding to the early hyperacute (0 - 6 h) and late hyperacute (6 – 24 h) ischemic stroke.16 Initial recordings revealed periodical calcium events that propagated through the field of view in a directional manner. These waves of calcium activity were clearly observed in the parenchymal microglia (Figure 1A and C) but did not elicit calcium activity in meningeal macrophages in a directionally progressive fashion (Figure 1B). In order to quantify the frequency of microglial calcium waves during the acute stroke, we imaged three mice subjected to MCAo over 20 hours after occlusion. Through this period, the recording sessions were evenly distributed in three imaging blocks, totaling 5 hours of imaging per animal. In the stroked animals, 18 wave-like calcium activities were recorded during the combined 15 hours of imaging, while no such events were observed during an equivalent recording period in the sham controls (Figure 1D). Thus, the average interval between calcium waves in the stroked animals was 50 minutes. In these experiments, microglial calcium events had a slightly decreasing frequency (8 events during the first 6 hours, 6 events during 6 - 12 hours after occlusion, and 4 events during 12-20 hours after stroke. In contrast, calcium amplitudes in later on tended to be more pronounced (Figure 1C and Videos I-III in the online-only Data Supplement). However, insufficient numbers at each time period precluded rigorous statistical analysis.
High speed imaging reveals temporal dynamics of microglial calcium transients
In order to achieve greater temporal resolution of the calcium transients during the wave activity, we used multiphoton imaging system with a high-speed resonant scanner.15 Similar to the previous experiment, the Iba1-Cre; PC∷G5-tdT reporter animals were permanently occluded with intraluminal filaments to induce ischemic injury, and imaged via sealed craniotomy over the ipsilateral somatosensory cortex. High speed recordings of spreading calcium activity in the late hyperacute phase were acquired (Figure 2). In the settings of these imaging experiments, the shifts of baseline green fluorescence along the propagating wavefront were clearly detectable (Figure 2A, C).17 It has been previously shown that these shifts are intrinsic optical signals (IOS) associated with hemodynamic responses to CSD.18 The correlation between electrophysiological and IOS signals during CSD has been addressed,19 and further refinements suggested that the delay between the peak of depolarization and the associated cerebral blood flow response is at the order of several tens of seconds.20 We noticed that the peak of microglial calcium loading ([Ca2+]i transients) always displayed a further delay relative to the IOS wavefront. We measured the kinetics of this lag in two independent animals undergoing late hyperacute CSD and found it to range between 1.95 s and 3.91 s (Figure 2B and D). It was followed by robust calcium transients averaging 15 s duration. In agreement with previous experiments, these calcium amplitudes were more pronounced than calcium activity recorded during the early hyperacute phase.
Figure 2. High-speed recordings (2.5 Hz) of microglial calcium responses to the wavefront of depolarization during late hyperacute ischemic stroke.

A, Time-stamped images (in milliseconds) of a microglial cell cluster undergoing a wave of calcium activity, documenting key phases of the pathology. tdTomato (red) and GCaMP5G (green) signals are shown here in overlay. The dashed white line in the first frame indicates the wavefront of elevated intrinsic fluorescence and the white arrow shows the direction of progression. Three cells selected for analysis of calcium activity are numbered 1-3 and circled. A nearby reference area free of microglial processes was used to calculate baseline fluorescence intensity (b). Scale bar, 50 μm. B, ΔF/F0 traces calculated over the areas indicated above. The baseline trace is shown in overlay with each of the selected cells. The time difference between the peak of the baseline fluorescence and microglial somas was found to be 3.91 s in this event. C and D, Similar analysis performed in a different animal undergoing late hyperacute ischemic stroke (approx. 12 hours after permanent occlusion of MCA). Here, the delay between the rise of intrinsic fluorescence baseline and intracellular calcium peak in microglia was between 1.95 s and 3.51 s.
Microglial calcium waves can be induced with KCl microinjections
Because the patterns of calcium waves were consistent with CSD pathology, we tested if microglial calcium can be induced with potassium chloride (KCl) application. Microinjections of KCl in the cerebral cortex have been shown in numerous studies to reliably induce CSD.4, 7, 20, 21 To study CSD-associated microglial calcium in a greater detail, we stimulated cortex with microinjections of KCl through burr holes in the visual cortex, and recorded the responses in the somatosensory cortex area (Figure 3A). These stimuli elicited a similar calcium waves in the imaging area with approximately 60 s delay after the application. These waves, detected as IOS wavefronts, were propagating at the velocity of 6.7 ± 1.99 mm/min. In comparison, stroke induced CSD wavefronts were propagating at similar but somewhat slower speeds (4.9 ± 1.71 mm/min) (Figure 1B and Video III and IV in the online-only Data Supplement). Unlike stroke-associated CSD waves, KCl-induced wavefronts traveled along the same trajectory from the site of microinjection. As additional evidence that the calcium waves are linked to CSD pathology, we measured voltage with extracellular micropipette inserted close to the field of two-photon imaging and found that microglial calcium waves correlated with direct-current potential deflection (data not shown).
Figure 3. Chemical induction of CSD with KCl allows accurate analysis of microglial calcium activity in the normal and inflamed state.

A, Relative positions of the cranial window and burr hole over the somatosensory cortex and visual cortex, respectively, as employed in these experiments. Also shown is the headpost attached to the occipital bone. B, Microinjections of KCl induced reproducible CSD waves at mean velocities of 6 mm/min. As shown in the bar chart on the right, the wavefront velocity was unchanged after LPS treatment, but differed somewhat from the stroke-induced CSD which tended to propagate at a slower speed. C, Two-photon images of microglial cells undergoing CSD-associated calcium transient after saline (left) or LPS (right) injections. Tmax indicates the time frame of the greatest signal amplitude in the GCaMP5 channel, the frames to the left show time frames captured 10 s earlier. The microglial soma size increased following LPS injection by 27 % (graph on the right, p = 0.102, NS). Scale bar, 50 μm. D, Changes in CSD-related microglial calcium activity following LPS analyzed with ΔF/F0, Peak Duration and AOC metrics. * indicates p < 0.05, ** p < 0.01; unpaired, two-tailed t-test; n=15 cells from 3 mice per group. Bars represent means ± SD.
LPS stimulus amplifies microglial calcium transients during CSD
Next, we asked if peripheral inflammation can mimic the effects of stroke-associated sterile inflammation on progressive increase of microglial calcium activity. The mice were injected with LPS 12 hours before the imaging experiment. CSD were induced with KCl as before, and the elicited waves were recorded and analyzed. The wavefront velocity in the LPS-treated animals did not significantly differ from saline controls, or stroke mice (6.9 ± 1.10 mm/min; p = 0.06, one-way ANOVA; n=6 to 9 cells from 3 mice per group) (Figure 3B, Video III-V in the online-only Data Supplement). Microglial cell soma size increased by 27% after LPS treatment, although not significantly (269.6 ± 30.45 μm3 to 343.7 ± 31.46 μm3; p = 0.1) (Figure 3C). Calcium transients in 15 LPS-affected microglia responding to CSD in three different animals were analyzed and compared to controls. We found that peripheral inflammation significantly increased microglial calcium activity by three different metrics. The maximum peak intensity (F/F0) increased in activated microglia by 47% (2.06 to 3.03, p = 0.025), the peak duration increased by 16% (14.9 ± 0.92 s to 17.3 ± 0.68 s; p = 0.04) and the area under the curve (AOC) measure increased by 59% (28.8 ± 4.14 AU to 45.8 ± 4.06 AU; p = 0.007). (Figure 3D). These results demonstrate that an inflammatory state drives enhanced microglial calcium activity in vivo.
CSD-induced microglial calcium transients are sensitive to CRAC channel inhibition
Finally, we tested if the calcium influx in CSD-affected microglia is mediated by the store-operated calcium channels (SOCE), also known as calcium-release activated calcium (CRAC) channels. These channels open after depletion of calcium from intracellular stores, permitting calcium entry and sustained [Ca2+]i signaling in numerous cell types especially in the immune system.8 We selected the highly selective inhibitor developed by CalciMedica, CM-EX-137.9 This compound has been previously shown to reduce microglial activation and microglial-induced cell death in traumatic brain injury,22 but in vivo imaging experiments on microglial calcium activity have not been conducted. To increase specificity in this study, we performed these measurements in mice of Cx3cr1-CreER; PC∷G5-tdT genotype, as the tamoxifen-inducible Cx3cr1-CreER driver is highly specific for microglia.14 The healthy, naive animals received CM-EX-137 per oral gavage 4 hours before the induction of CSD and imaging. Three CSD were induced in the cortex of each animal, using the same KCl microinjection paradigm as before. The 3 most highly responding cells were analyzed during each CSD, every time in a different location in the cranial window. The depth of imaging ranged from 100 μm to 200 μm below the meningeal surface. The results are summarized in Figure 4, and Figure II and Video VI-VII in the online-only Data Supplement. We found a 20% decrease in microglial [Ca2+]i peak intensity (F/F0) in the CM-EX-137 treated animals compared to controls (0.61 ± 0.05 to 0.49 ± 0.03, p = 0.04). The duration of calcium transients also decreased by 10% (16.4 ± 0.84 s to 14.9 ± 0.63 s), although not significantly. Furthermore, the AOC values were decreased in CM-EX-137 treated mice by 26.7% (10.64 ± 1.01AU to 7.80 ± 0.57 AU) at a significance level of p = 0.014 (unpaired two-tailed t-test; n=26 to 30 cells from 3 mice; two males and one female in each group). Thus, these results demonstrate that the CRAC mechanism is at least in part responsible for calcium activity in microglia during CSD.
Figure 4. Partial inhibition of CSD-induced calcium transients in microglia with selective CRAC channel blocker CM-EX-137.
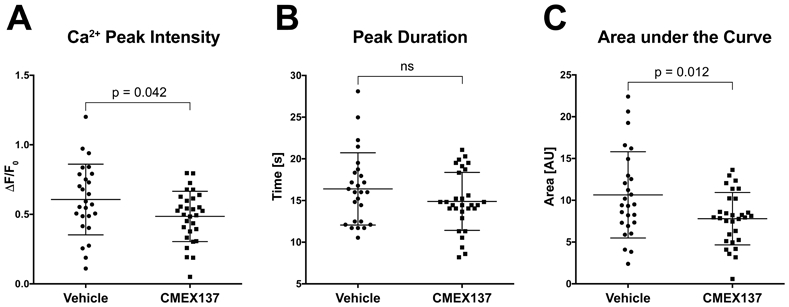
A, Mice received 25 mg/kg CM-EX-137, or vehicle, in 200 μL p.o. The mean peak intensity of calcium transients during KCl-evoked CSD was reduced by 20% in the CM-EX-137 treated animals. B, The average duration of calcium transients tended to be shorter in the animals receiving drug (14.89 s vs.16.39 s in controls), but the difference was not statistically significant. C, The mean area under the curve calculated from the GCaMP5 fluorescence peaks was found to be significantly reduced by 26% (p = 0.012, unpaired two-tailed t-test. n=26 cells from 3 mice for vehicle controls; n=30 cells from 3 animals for drug treated mice, in all panels). Horizontal lines in scatter plots indicate means ± SD.
Discussion
In this study, we provide evidence that ischemic injury, produced by MCAo in the mouse brain, triggers CSD in a recurring and progressive fashion. Moreover, we show for the first time that these CSD bring about frequent calcium transients in microglia (Figure 1). These novel findings and the underlying experimental framework open up new possibilities to isolate and evaluate this aspect of stroke pathology, as a first step towards new medical interventions.
CSD are well-documented clinical complications of stroke, subarachnoid hemorrhage, traumatic brain injury and migraine.3, 23-25 First described in human patients almost twenty years ago,26 CSD are becoming better characterized and understood in intensive care units.27-31 Although anecdotal evidence exists for several mechanisms of CSD suppression in the clinical setting,32, 33 a more in-depth investigation of the cellular and molecular aspects underlying this pathology will have to turn to animal models.
Mouse lines expressing genetically encoded indicators of calcium have enabled major advances in understanding brain cell activity in normal function and disease. The characterization of microglial intracellular activity has trailed behind other cells types. Two major reasons have been the lack of suitable tools and the generally lower frequency of microglial signals.13 Several groups have successfully recorded microglial calcium activity in vivo with other indicators of calcium,34, 35 but the Polr2a-based PC∷G5-tdT reporter possesses several distinct advantages including the robustness of GCaMP5 expression and inclusion of structural reporter tdTomato in the Cre-positive cells.12 With two different induction strategies, using the constitutive Iba1-Cre and tamoxifen-inducible Cx3cr1-CreER, we have characterized microglial responses to CSD. Reproducibly, these cells display a distinct delay of [Ca2+]i rises relative to the passing wavefront detected as IOS shift (Figure 2). The shifts in IOS are due to hemodynamic changes associated with depression following CSD.20 Further delay of microglial calcium activity after this wavefront, 2 to 4 seconds in our measurements, reveals that microglial calcium transients undoubtedly lag behind neuronal [Ca2+]i influx. Neuronal calcium was shown to precede the wave of glutamate release following CSD, but lagged behind interstitial K+ elevation.4 Conversely, astrocytic calcium activity was also delayed relative to neuronal responses, trailing the glutamate release.4 It is thus possible that microglia are more similar to astrocytes with regard to timing of CSD-associated [Ca2+]i dynamics. The primary stimulus triggering microglial [Ca2+]i activity remains to be determined, but ATP released from neurons and/or astrocytes is a likely candidate. With respect to [Ca2+]i duration, microglial cell soma transients had relatively homogeneous distribution, peaking around 15 s (Figure 3D and Figure 4A through C). In contrast, CSD-induced neuronal [Ca2+]i transients last more than one minute.4
We also demonstrate that peripheral inflammation increased microglial calcium activity by more than 50% during CSD (Figure 3D). This is in line with our previous work showing increased frequency of spontaneous [Ca2+]i transients in inflamed microglia.13 The progression from the early to the late phase of hyperacute ischemic stroke appears to provoke a similar amplification of microglial calcium activity (compare Videos I and II in the online-only Data Supplement). The precise mechanism of this exacerbation is presently not understood, but it might be due to upregulation of CRAC channel subunits such as Stim1 and Orai1.22 Further molecular profiling will be required to establish the exact mechanisms underlying enhanced calcium influx in activated microglia.
The patterns of microglial [Ca2+]i transients during stroke-induced CSD, especially in the late hyperacute phase, represent the most widespread intracellular activity observed in microglia thus far. In this setting, even if the transients are initiated in the distal processes, they spread across the entire microglial cell, almost invariably including the soma (Videos II, III and V in the online-only Data Supplement). We have therefore focused our [Ca2+]i analyses on GCaMP signals in microglial cell bodies, which we have determined most accurately represented microglial responses in this pathology. Others have recently noted that microglial somatic calcium activity is rare, even under seizure conditions.35 This underscores the importance of blocking the CSD-associated [Ca2+]i activity that has been shown to activate calcineurin, leading to nuclear translocation of the NFAT transcription factor, which in turn orchestrates multiple inflammatory responses and phagocytic transformation.22
The role of microglia in stroke and CSD has been investigated by several other authors. Szalay and colleagues have studied the effect of microglia ablation following MCAo.36 They found that a near-complete ablation of microglia with PLX3397 resulted in a 60% increase in infarct size following MCAo.36 Cserep et al. recently proposed that microglia protect ischemic neurons by extending processes to their somata in a P2Y12 receptor–dependent mechanism.37 The same group also showed that selective elimination of microglia decreased the incidence and evolution of CSD in the mouse brain.36, 38 Although very interesting, these studies provide limited insight into targetable microglial pathways which might be beneficial in stroke management.
We posit that microglial calcium activity in hyperacute stroke may underlie many detrimental effects of activated microglia. For example, it has been shown that CSD stimulates microglial secretion of Il1 39 and TNF 40 TNF lowers the threshold for CSD induction, promoting perpetuation of CSD induction through positive feedback.40, 41 Elevated microglial calcium in vitro has been implicated by many studies in microglial activation, leading to secretion of inflammatory molecules inducing neuronal death or injury.10, 42 Here we show in vivo that CSD-associated [Ca2+]i activity in microglia is, at least in part, due to calcium influx through the CRAC channels. A significant reduction of 26% in CSD-elicited calcium activity was achieved 4 hours after oral gavage with a CRAC channel inhibitor, which is compatible with hyperacute stroke interventions (Figure 4). The data supports the concept that treatment with a brain penetrant CRAC channel inhibitor can provide protection from microglial calcium overload in an animal model of stroke. This effect could then translate into a clinical benefit in humans who suffer an acute ischemic episode like stroke, a key therapeutic goal for CalciMedica, who developed a number of therapeutic candidates that could be adopted for stroke indication. However, additional preclinical studies are needed to support the clinical development of CRAC channel inhibitors for a variety of acute ischemic and inflammatory conditions.
We cannot exclude the possibility that the inhibitory effect of CM-EX-137 on CSD-associated microglial calcium rises are mediated to some degree by other cell types. For example, Stegner and colleagues have recently shown that cortical neurons express CRAC/SOCE channels, particularly the Stim2 and Orai2 subunits, that contribute to calcium overload and neuronal cell death in acute ischemic stroke.43 It is thus conceivable that neuronal CRAC channel inhibition may indirectly mitigate microglial calcium responses during CSD. Further, the incomplete inhibition of microglial [Ca2+]i transients with CM-EX-137 may reflect the presence of other calcium influx mechanisms, such as the calcium permeable purinergic P2X7 channel which is highly expressed in microglia, and further upregulated in cerebral ischemia.44 To unequivocally address the role of CRAC-mediated calcium overload in ischemic stroke pathology, it is essential to perform genetic ablations of the key CRAC subunits in specific cell types, which we will pursue in future work.
Together, the present work lays the foundation for precise dissection of the sources and consequences of microglial calcium overload in stroke, in the context of the neurovascular unit and other cell types associated with this pathology.
Summary/ Conclusions
Distinct and modifiable microglial calcium activity is a hitherto unrecognized component of CSD pathology. We have demonstrated that recurrent calcium waves are common in peri-infarct CSD. Using the KCl-evoked CSD paradigm, we have characterized calcium dynamics in the naïve and activated microglia. Pharmacological blockade of KCl-evoked CSD has indicated that CRAC channel-dependent mechanisms contribute to microglial calcium overload. More research is warranted to determine whether CRAC channel inhibition can improve outcomes in preclinical models of ischemic stroke and ultimately in human stroke patients.
Supplementary Material
Acknowledgements
The Brain Immunology and Glia (BiG) Center at the University of Virginia School of Medicine has provided access to high-level imaging equipment, enabling this study. We are grateful to the Bitplane support team for advice on image analysis. CM-EX-137 was a gift from Kenneth Stauderman. We thank David Krizaj for stimulating discussions and advice on extracellular electrophysiology.
Sources of Funding
The generation of the PC∷G5-tdT reporter system and development of the experimental framework was by supported by NIH grant R21 OD016562 to P.T. He also received research grants from the Focused Ultrasound Foundation.
Abbreviations
- CRAC
calcium-release activated calcium channels
- IOS
intrinsic optical signals
- MCAo
middle cerebral artery occlusion
- PC∷G5-tdT
Polr2a-based GCaMP5 and tdTomato Cre-dependent reporter
- SOCE
store-operated calcium channels
Footnotes
Disclosures
K.A. Stauderman is a Chief Scientific Officer of CalciMedica, Inc., and a stockholder in the company. CalciMedica filed a patent for the use of CRAC channel inhibitors including CM-EX-137 for the treatment of stroke and traumatic brain injury. For the remaining authors, no competing financial interests exist.
Contributor Information
Lei Liu, Department of Neurosurgery, University of Virginia..
Kathryn N. Kearns, Department of Neurosurgery, University of Virginia..
Ilyas Eli, Department of Neurosurgery, University of Utah.
Khadijeh A. Sharifi, Department of Neurosurgery and Neuroscience, University of Virginia..
Sauson Soldozy, Department of Neurosurgery, University of Virginia..
Elizabeth W. Carlson, Department of Neurosurgery, University of Utah.
Kyle W. Scott, Department of Neurosurgery, University of Virginia..
M. Filip Sluzewski, Department of Electrical and Computer Engineering, University of Virginia..
Scott T. Acton, Department of Electrical and Computer Engineering, University of Virginia..
Kenneth A. Stauderman, CalciMedica.
M. Yashar S. Kalani, Departments of Neurosurgery, and Neuroscience, University of Virginia..
Min Park, Department of Neurosurgery, University of Virginia..
Petr Tvrdik, Departments of Neurosurgery and Neuroscience, University of Virginia..
References
- 1.Karsy M, Brock A, Guan J, Taussky P, Kalani MY, Park MS. Neuroprotective strategies and the underlying molecular basis of cerebrovascular stroke. Neurosurg Focus. 2017;42:E3. [DOI] [PubMed] [Google Scholar]
- 2.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999;22:391–397 [DOI] [PubMed] [Google Scholar]
- 3.Lauritzen M, Dreier JP, Fabricius M, Hartings JA, Graf R, Strong AJ. Clinical relevance of cortical spreading depression in neurological disorders: Migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J Cereb Blood Flow Metab. 2011;31:17–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Enger R, Tang W, Vindedal GF, Jensen V, Johannes Helm P, Sprengel R, Looger LL, Nagelhus EA. Dynamics of ionic shifts in cortical spreading depression. Cereb Cortex. 2015;25:4469–4476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kramer DR, Fujii T, Ohiorhenuan I, Liu CY. Cortical spreading depolarization: Pathophysiology, implications, and future directions. J Clin Neurosci. 2016;24:22–27 [DOI] [PubMed] [Google Scholar]
- 6.Wainsztein N, Rodriguez Lucci F. Cortical spreading depression and ischemia in neurocritical patients. Neurosurg Clin N Am. 2018;29:223–229 [DOI] [PubMed] [Google Scholar]
- 7.Chuquet J, Hollender L, Nimchinsky EA. High-resolution in vivo imaging of the neurovascular unit during spreading depression. J Neurosci. 2007;27:4036–4044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cahalan MD, Zhang SL, Yeromin AV, Ohlsen K, Roos J, Stauderman KA. Molecular basis of the crac channel. Cell Calcium. 2007;42:133–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stauderman KA. Crac channels as targets for drug discovery and development. Cell Calcium. 2018;74:147–159 [DOI] [PubMed] [Google Scholar]
- 10.Färber K, Kettenmann H. Functional role of calcium signals for microglial function. Glia. 2006;54:656–665 [DOI] [PubMed] [Google Scholar]
- 11.Hanisch UK, Kettenmann H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394 [DOI] [PubMed] [Google Scholar]
- 12.Gee JM, Smith NA, Fernandez FR, Economo MN, Brunert D, Rothermel M, Morris SC, Talbot A, Palumbos S, Ichida JM, et al. Imaging activity in neurons and glia with a polr2a-based and cre-dependent gcamp5g-ires-tdtomato reporter mouse. Neuron. 2014;83:1058–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pozner A, Xu B, Palumbos S, Gee JM, Tvrdik P, Capecchi MR. Intracellular calcium dynamics in cortical microglia responding to focal laser injury in the pc∷G5-tdt reporter mouse. Front Mol Neurosci. 2015;8:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38:79–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tvrdik P, Kearns KN, Sharifi KA, Sluzewski MF, Acton ST, Kalani MYS. Calcium imaging of microglial network activity in stroke. Methods Mol Biol. 2019;2034:267–279 [DOI] [PubMed] [Google Scholar]
- 16.Allen LM, Hasso AN, Handwerker J, Farid H. Sequence-specific mr imaging findings that are useful in dating ischemic stroke. Radiographics. 2012;32:1285–1297; discussion 1297–1289 [DOI] [PubMed] [Google Scholar]
- 17.Sluzewski MF, Tvrdik P, Acton ST. Segmentation of cortical spreading depression wavefronts through local similarity metric. 2019 Ieee International Conference on Image Processing (Icip). 2019;eess.IV:1485–1489 [Google Scholar]
- 18.Ba AM, Guiou M, Pouratian N, Muthialu A, Rex DE, Cannestra AF, Chen JW, Toga AW. Multiwavelength optical intrinsic signal imaging of cortical spreading depression. J Neurophysiol. 2002;88:2726–2735 [DOI] [PubMed] [Google Scholar]
- 19.Peixoto NL, Fernandes de Lima VM, Hanke W. Correlation of the electrical and intrinsic optical signals in the chicken spreading depression phenomenon. Neurosci Lett. 2001;299:89–92 [DOI] [PubMed] [Google Scholar]
- 20.Obrenovitch TP, Chen S, Farkas E. Simultaneous, live imaging of cortical spreading depression and associated cerebral blood flow changes, by combining voltage-sensitive dye and laser speckle contrast methods. Neuroimage. 2009;45:68–74 [DOI] [PubMed] [Google Scholar]
- 21.Wendt S, Wogram E, Korvers L, Kettenmann H. Experimental cortical spreading depression induces nmda receptor dependent potassium currents in microglia. J Neurosci. 2016;36:6165–6174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mizuma A, Kim JY, Kacimi R, Stauderman K, Dunn M, Hebbar S, Yenari MA. Microglial calcium release-activated calcium channel inhibition improves outcome from experimental traumatic brain injury and microglia-induced neuronal death. J Neurotrauma. 2019;36:996–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dreier JP. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med. 2011;17:439–447 [DOI] [PubMed] [Google Scholar]
- 24.Dreier JP, Reiffurth C. The stroke-migraine depolarization continuum. Neuron. 2015;86:902–922 [DOI] [PubMed] [Google Scholar]
- 25.Hartings JA. Spreading depolarization monitoring in neurocritical care of acute brain injury. Curr Opin Crit Care. 2017;23:94–102 [DOI] [PubMed] [Google Scholar]
- 26.Strong AJ, Fabricius M, Boutelle MG, Hibbins SJ, Hopwood SE, Jones R, Parkin MC, Lauritzen M. Spreading and synchronous depressions of cortical activity in acutely injured human brain. Stroke. 2002;33:2738–2743 [DOI] [PubMed] [Google Scholar]
- 27.Dohmen C, Sakowitz OW, Fabricius M, Bosche B, Reithmeier T, Ernestus RI, Brinker G, Dreier JP, Woitzik J, Strong AJ, et al. Spreading depolarizations occur in human ischemic stroke with high incidence. Ann Neurol. 2008;63:720–728 [DOI] [PubMed] [Google Scholar]
- 28.Dreier JP, Fabricius M, Ayata C, Sakowitz OW, Shuttleworth CW, Dohmen C, Graf R, Vajkoczy P, Helbok R, Suzuki M, et al. Recording, analysis, and interpretation of spreading depolarizations in neurointensive care: Review and recommendations of the cosbid research group. J Cereb Blood Flow Metab. 2017;37:1595–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dreier JP, Lemale CL, Kola V, Friedman A, Schoknecht K. Spreading depolarization is not an epiphenomenon but the principal mechanism of the cytotoxic edema in various gray matter structures of the brain during stroke. Neuropharmacology. 2018;134:189–207 [DOI] [PubMed] [Google Scholar]
- 30.Eriksen N, Rostrup E, Fabricius M, Scheel M, Major S, Winkler MKL, Bohner G, Santos E, Sakowitz OW, Kola V, et al. Early focal brain injury after subarachnoid hemorrhage correlates with spreading depolarizations. Neurology. 2019;92:e326–e341 [DOI] [PubMed] [Google Scholar]
- 31.Hartings JA, Wilson JA, Hinzman JM, Pollandt S, Dreier JP, DiNapoli V, Ficker DM, Shutter LA, Andaluz N. Spreading depression in continuous electroencephalography of brain trauma. Ann Neurol. 2014;76:681–694 [DOI] [PubMed] [Google Scholar]
- 32.Hoffmann U, Dilekoz E, Kudo C, Ayata C. Gabapentin suppresses cortical spreading depression susceptibility. J Cereb Blood Flow Metab. 2010;30:1588–1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sakowitz OW, Kiening KL, Krajewski KL, Sarrafzadeh AS, Fabricius M, Strong AJ, Unterberg AW, Dreier JP. Preliminary evidence that ketamine inhibits spreading depolarizations in acute human brain injury. Stroke. 2009;40:e519–522 [DOI] [PubMed] [Google Scholar]
- 34.Brawek B, Liang Y, Savitska D, Li K, Fomin-Thunemann N, Kovalchuk Y, Zirdum E, Jakobsson J, Garaschuk O. A new approach for ratiometric in vivo calcium imaging of microglia. Sci Rep. 2017;7:6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Umpierre AD, Bystrom LL, Ying Y, Liu YU, Worrell G, Wu LJ. Microglial calcium signaling is attuned to neuronal activity in awake mice. Elife. 2020;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szalay G, Martinecz B, Lenart N, Kornyei Z, Orsolits B, Judak L, Csaszar E, Fekete R, West BL, Katona G, et al. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat Commun. 2016;7:11499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cserep C, Posfai B, Lenart N, Fekete R, Laszlo ZI, Lele Z, Orsolits B, Molnar G, Heindl S, Schwarcz AD, et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science. 2020;367:528–537 [DOI] [PubMed] [Google Scholar]
- 38.Varga DP, Menyhart A, Posfai B, Csaszar E, Lenart N, Cserep C, Orsolits B, Martinecz B, Szlepak T, Bari F, et al. Microglia alter the threshold of spreading depolarization and related potassium uptake in the mouse brain. J Cereb Blood Flow Metab. 2020:271678X19900097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jander S, Schroeter M, Peters O, Witte OW, Stoll G. Cortical spreading depression induces proinflammatory cytokine gene expression in the rat brain. J Cereb Blood Flow Metab. 2001;21:218–225 [DOI] [PubMed] [Google Scholar]
- 40.Grinberg YY, Dibbern ME, Levasseur VA, Kraig RP. Insulin-like growth factor-1 abrogates microglial oxidative stress and tnf-alpha responses to spreading depression. J Neurochem. 2013;126:662–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grinberg YY, van Drongelen W, Kraig RP. Insulin-like growth factor-1 lowers spreading depression susceptibility and reduces oxidative stress. J Neurochem. 2012;122:221–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoffmann A, Kann O, Ohlemeyer C, Hanisch UK, Kettenmann H. Elevation of basal intracellular calcium as a central element in the activation of brain macrophages (microglia): Suppression of receptor-evoked calcium signaling and control of release function. J Neurosci. 2003;23:4410–4419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stegner D, Hofmann S, Schuhmann MK, Kraft P, Herrmann AM, Popp S, Hohn M, Popp M, Klaus V, Post A, et al. Loss of orai2-mediated capacitative ca(2+) entry is neuroprotective in acute ischemic stroke. Stroke. 2019;50:3238–3245 [DOI] [PubMed] [Google Scholar]
- 44.Ye X, Shen T, Hu J, Zhang L, Zhang Y, Bao L, Cui C, Jin G, Zan K, Zhang Z, et al. Purinergic 2×7 receptor/nlrp3 pathway triggers neuronal apoptosis after ischemic stroke in the mouse. Exp Neurol. 2017;292:46–55 [DOI] [PubMed] [Google Scholar]
- 45.Morris GP, Wright AL, Tan RP, Gladbach A, Ittner LM, Vissel B. A comparative study of variables influencing ischemic injury in the longa and koizumi methods of intraluminal filament middle cerebral artery occlusion in mice. PLoS One. 2016;11:e0148503. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.