

Abstract
It is now appreciated that in addition to their role in humoral immunity B cells also exert regulatory mechanisms that lead to attenuation of inflammatory responses. The concept of B cell regulation became well-recognized when mice deficient in B cells due to genetic disruption were shown to be refractory to recovery from the signs of experimental autoimmune encephalomyelitis (EAE), the mouse model of multiple sclerosis. This seminal study spurred the search for B cell regulatory phenotypes and mechanisms of action. Our approach was to utilize differential B cell depletion with anti-CD20 to retain B cells whose presence were required to achieve EAE recovery. Utilizing flow cytometry, adoptive cell therapy and genetic approaches, we discovered a new B cell subset that upon adoptive transfer into B cell-deficient mice, was sufficient to promote EAE recovery. This B cell subset is IgM+, but due to low/negative IgD cell surface expression it was named B cell IgD low (BDL). Mechanistically, we found that in the absence of BDL, the absolute cell number of CD4+Foxp3+ T regulatory cells (Treg), essential for immune tolerance, were significantly reduced. Furthermore, we found that BDL expression of glucocorticoid-induced tumor necrosis factor ligand (GITRL) was essential for induction of Treg proliferation and maintenance of their homeostasis. Thus, we have identified a new B cell subset that is critical for immunological tolerance through interactions with Treg.
Keywords: B cell IgD low, Glucocorticoid-induced tumor necrosis factor ligand, CD4+Foxp3+ T regulatory cell, Experimental autoimmune encephalomyelitis, Immune tolerance
Introduction
The historical view of B cells is that they present antigens to T cells that in turn provide crucial signaling to induce their differentiation into plasma cells that secrete antibodies. While antibodies are the backbone of humoral immunity leading to pathogen clearance, it is now appreciated that B cells are also important in regulating the extent of the immune response. The first concrete evidence that B cells have the potential to control the extent of the adaptive immune response was in a seminal 1996 study utilizing the mouse model of multiple sclerosis (MS), experimental autoimmune encephalomyelitis (EAE). We found that mice deficient in B cells were unable to recover from the signs of EAE, although disease onset and progression were not altered [1]. This was an unexpected finding at the time that ruled out a role for both B cell antigen presentation and antibody production in driving the onset of EAE. The mechanisms responsible for recovery were yet to be discovered.
Over the last 24 years, extensive research efforts have gone into investigating B cell regulatory mechanisms. Given the complexity of the immune system, in retrospect it is not surprising that multiple regulatory mechanisms have been described including IL-10 and IL-35 production, CD73, FasL, PD-L1 among others [2]. While much of the field concentrated on finding a novel B cell subset that regulated via IL-10 production, our studies pointed us in the direction of IL-10-independent mechanisms [3]. The key to our studies was the discovery of the elusive CD4+Foxp3+ T regulatory cell (Treg), which is critical for the maintenance of immune tolerance and the suppression of autoimmunity [4, 5]. Here, we provide the history from our first realization that B cell regulation is linked to Treg, to discovering the mechanism and then finally to the characterization of a new B cell subset that interacts with Treg to promote their homeostasis thereby maintaining immune tolerance [3, 6, 7].
Discovery of B cell Regulation in Autoimmunity
To study a role for B cells in EAE, early studies depleted B cells in rats from birth with anti-IgM antibodies, in which EAE was induced by myelin basic protein (MBP) or spinal cord homogenate [8]. These rats were resistant to EAE induction, which upon further examination was due to the generation of anti-MBP antibodies [9]. A pathogenic role for antibodies in EAE induction was independently confirmed [10, 11]. The conclusion from these collective studies was that B cells were functioning as antigen presenting cells facilitating the priming of myelin-specific encephalitogenic T cells leading the subsequent production of myelin-specific pathogenic antibodies [12].
To further investigate the necessity for B cell antigen presentation in EAE induction, we utilized mice deficient in B cells due to genetic targeting of the membrane exon of the IgM heavy chain (μMT) [13]. Since expression of the IgM heavy chain in complex with the surrogate light chain on the pre-B cell surface is required for B cell development in the bone marrow, mature B cells are not generated [13, 14]. B10.PL (H-2u) WT and B10.PLμMT mice immunized with the MBP immunodominant peptide Ac1–11 exhibited similar EAE incidence and day of onset and progression to peak of disease [1]. These data indicated that neither B cell antigen presentation nor antibody production was required for the priming of CD4+ T cells and subsequent EAE onset. B10.PL mice undergo spontaneous recovery from EAE, but to our surprise, B10.PLμMT mice did not undergo recovery and exhibited an acute, but chronic disease course (Fig. 1A) [1]. This seminal study, was the first to demonstrate a regulatory role for B cells in autoimmunity without the need for antibody production. Our findings were later confirmed in C57BL/6μMT mice immunized with the myelin oligodendrocyte glycoprotein immunodominant peptide 35–55, which implicated a role for IL-10 in B cell regulation [15]. These two studies combined were responsible for sparking an interest in B cell regulation and the realization that B cells play complex roles in adaptive immunity beyond antibody production.
Figure 1. Graphic representation of EAE disease courses in WT and μMT mice with various B cell and anti-CD20 treatments.
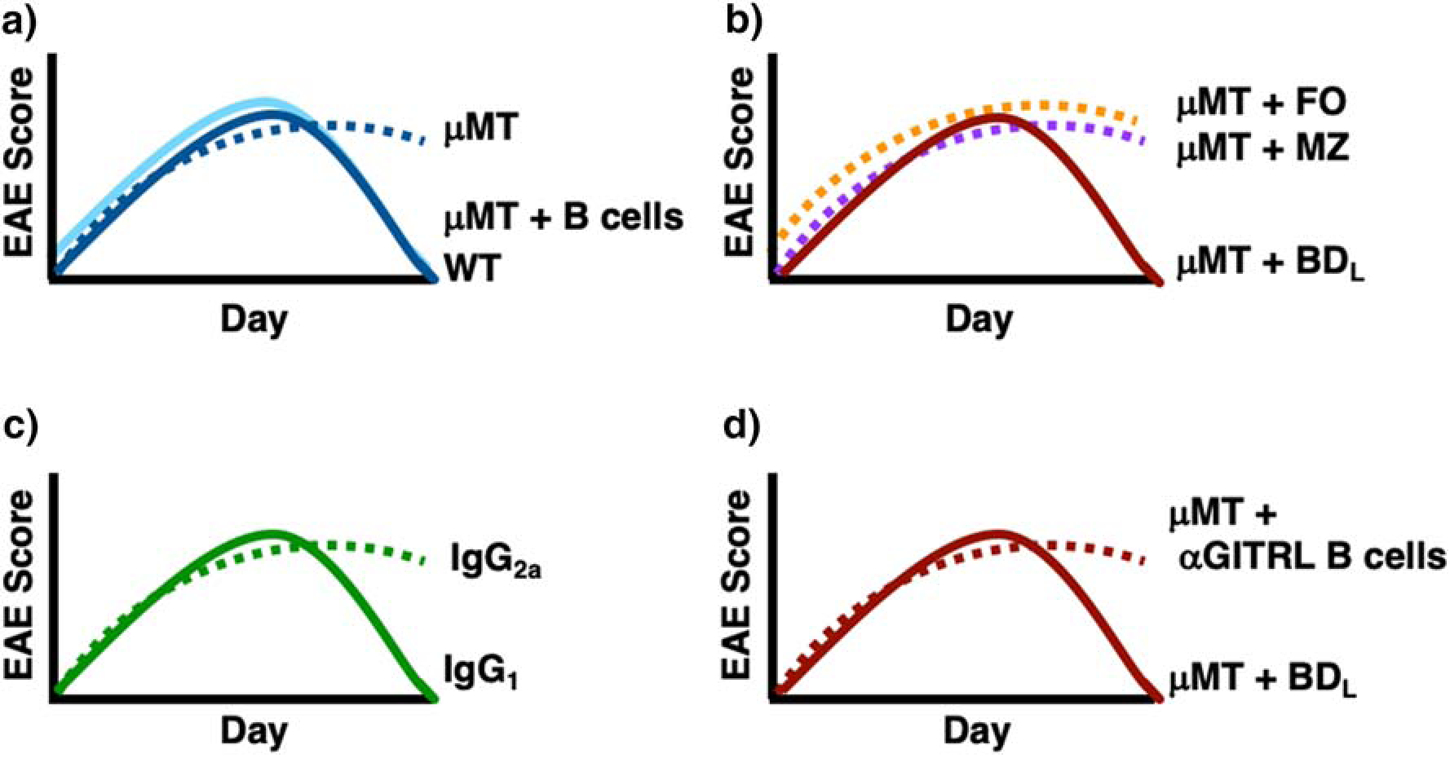
Graphic EAE disease courses are shown for WT mice (A, dark blue line) that undergo recovery as compared to μMT mice (A, dark blue dotted line) that do not. μMT mice transferred either total splenic B cells (A, solid light blue line) or BDL (B,D, solid red line) recover from EAE, while those transferred FO (B, dotted orange line), MZ (B, dotted purple line) or anti-GITRL blocked B cells (D, dotted red line) do not. B cell depletion with anti-CD20 IgG2a (C, solid green line) preserves BDL and Treg numbers leading to B cell recovery, while total B cell depletion with anti-CD20 IgG1 (C, dotted green line) leads to chronic EAE.
Investigation of B Cell Regulatory Mechanisms
In exploring the mechanism whereby B cells promote EAE recovery, we first focused on proteins required for cognate interactions. We examined the B7 family proteins B7.1 (CD80) and B7.2 (CD86) utilizing mixed bone marrow (BM) chimeras generated by transplanting donor BM deficient in both B7.1 and B7.2 (B7−/−) into sublethally irradiated B10.PLμMT recipient mice (B7−/−→μMT). The control WT→WT and B7−/−→WT chimeras recovered from EAE, whereas the μMT→μMT and B7−/−→μMT chimeras did not [6]. These data indicated that mice deficient in B7 in the immune system were unable to recover from the signs of EAE. Because these studies were conducted prior to commercially available antibodies to Foxp3, we utilized quantitative real-time PCR to assess the presence of Treg in the spinal cord and lymph nodes at the peak of EAE. Consistent with the lack of EAE recovery, Treg transcripts were reduced in the spinal cord in the μMT→μMT and B7−/−→μMT chimeras as compared to the controls [6]. Interestingly, Treg transcripts were not reduced in the lymph nodes [6]. These data were the first that suggested B cells crossed talk with Treg to facilitate recovery from EAE.
In our early studies, we also investigated whether any IgM+ B cell could be regulatory and drive recovery from EAE. First, we utilized hen egg lysozyme (HEL) BCR transgenic mice bred to B10.PLμMT mice (HELμMT) so that only a single BCR was expressed thereby limiting diversity, and determined whether they would recover from EAE [16]. We found that expression of a single BCR was not sufficient to drive recovery from EAE [17]. We then increased BCR diversity by breeding a IgM heavy chain BCR transgenic mouse to B10.PLμMT mice (Vh186.2μMT [18]. In these mice, the heavy chain is fixed but can pair with any light chain, thereby generating a BCR repertoire with limited diversity [18]. Interestingly, the limited diversity mice exhibited intermediate recovery between the B10.PLμMT and B10.PL WT control mice [17]. These BCR diversity studies provided the first evidence that the regulatory B cell we sought was a unique B cell subset [17].
Because IL-10 production by B cells was shown to be an important effector regulatory pathway [15], we investigated whether a similar mechanism existed in our B10.PL EAE model and found that it was IL-10-independent [3, 19]. We contributed this largely to using a passive induction EAE model that does not require immunization with complete Freund’s adjuvant composed of heat-killed Mycobacterium tuberculosis that contains toll-like receptor (TLR) ligands for TLR2, TLR4 and TLR9 [20]. Stimulation of both TLR4 and TLR9 on B cells has been shown to be potent inducers of their IL-10 production [21]. Thus, by inducing EAE in the absence of CFA, we avoided potential bystander activation of B cells and subsequent IL-10 production [3, 19].
Strategy Utilized to Identify BDL as a New B Cell Subset with Regulatory Activity
As a cell lineage B cells are quite complex being composed of numerous developmental stages and four known mature B cell subsets: B1a, B1b, MZ and FO B cells (Fig. 2). B1a B cells express CD5 and CD11b as a distinguishing markers, develop from the fetal liver are enriched within the peritoneal and pleural cavities in mice and dominate T-independent immune responses [22]. Little is known regarding B1b B cells, but they are BM-derived and provide protective antibodies following infection [23]. MZ and FO B cells are referred to as B2 B cells. MZ B cells provide a first-line defense against blood-borne pathogens and largely contribute to T-independent immune responses [24]. FO B cells circulate throughout the body and participate in germinal center responses generating high affinity protective antibodies responses and long-term memory in the form of long-lived plasma cells and memory B cells [25].
Figure 2. B cell development schematic.
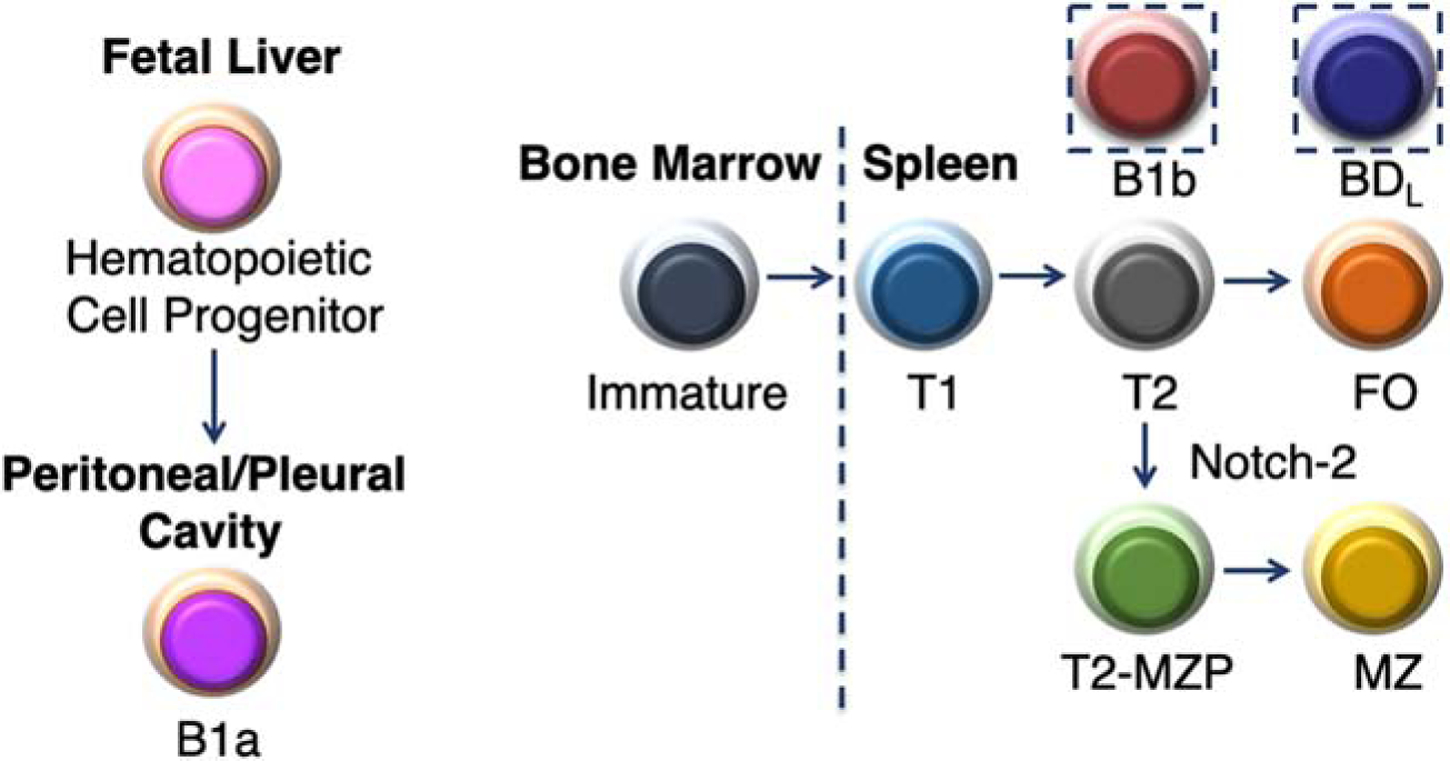
B cell development begins in the fetal liver which gives rise to the B1a subset. All other known B cell subsets differentiate in the bone marrow (BM) where immature B cells migrate to the spleen to complete their development first transitioning from transitional (1) and then T2, which upon receiving a Notch-2 signal differentiates into a T2-margianl zone precursor (MZP) and then into mature MZ B cells. The T2 subset also differentiates into mature FO B cells. The exact developmental pathway for B1b and BDL is not known and are thus placed in dotted boxes.
One of the first questions we addressed was the identity of the B cell subset exhibiting immune regulation in our studies. B cell development begins in the yolk sac/fetal liver from which the B1a population of innate-like B cells emerge [26–28]. B1a cells in mice are largely found in the peritoneal and pleural cavities of mice (Fig. 2), but they also circulate and can be found in the spleen and lymph nodes and persist for the life of the animal [29]. Human B1a cells have also been described with similar functional characteristics as mice [30, 31]. B1a B cells are major producers of natural IgM in the serum and are responsible for early antibody responses following infection because they can become activated and differentiate into short-term antibody secreting plasmablasts in the absence of T cell help in what are termed T cell-independent antibody responses [32]. Because B1a B cells do not originate from the BM they are not reconstituted following irradiation and subsequent BM transplantation (T). In our early studies, we found that reconstitution of B cells in B10.PLμMT mice by BMT resulted in recovery from EAE [6]. Although at the time it was not known that B1a cells emerged from the fetal liver, in retrospect, it indicated that the regulatory B cell we sought was not a B1a B cell. This was later confirmed in studies in which we transferred highly purified total splenic B cells into μMT mice that lacked the B1a subset (Fig. 1A) [3, 7].
In addition, to B1a B cells, the term B1 is also used to identify a second B cell subset termed B1b. Phenotypically, both B1a and B1b are B220+CD23−CD11b+ [33]. What distinguishes the two subsets is expression of CD5 by B1a, but not, B1b B cells. B1b B cells are considered a mature B cell subset that is BM-derived (Fig. 1) [34]. Although not well studied both functionally and developmentally (Fig. 2), B1b B cells were shown to exhibit an innate ability to recognize protective antigens in bacteria [23]. In our B cell adoptive transfer studies, our purification strategy included gating on CD23, thereby eliminating both CD23− B1a and B1b B cells from our studies, thus they were excluded from further evaluation [7].
We then turned our attention to mature B2 cells composed of follicular (FO) and marginal zone (MZ) B cells that develop in the spleen from immature B cells that migrate from the BM (Fig. 2). Immature B cells differentiate into transitional (T) 1 and then T2, which diverges to give rise to FO and MZ B cells (Fig. 2). We first eliminated MZ B cells (Fig. 2), which were aptly named because they are located within the MZ of the spleen. The MZ separates the white pulp from the red pulp and the termination of arterial vessels in the MZ allows blood to pass through an open system of reticular cells exposing resident macrophages and MZ B cells to blood-borne pathogens prior to collection by the red pulp venules [24, 35]. MZ B cells are enriched for specificity to microbial polysaccharides and express high levels of TLR and the complement receptor CD21/CD35 [24]. These characteristics along with existing in a quasi-activated state, allow them to rapidly respond to antigens with repeating subunits and differentiate into antibody secreting cells [24, 36]. Commitment to the MZ lineage requires Notch-2 signaling (Fig. 2) [37, 38]. MZ B cells are CD21hiCD23−, while FO B cells are CD21intCD23+ [24]. This differential cell surface expression was utilized for fluorescent activated cell sorting (FACS) to obtain highly pure populations of B220+ (Fig. 3A) MZ (B220+IgMhiCD21hiCD23−CD93−) (Fig. 3B, D) and FO (B220+IgM+CD21intCD23+CD93−) (Fig. 3B, E) B cells [7, 39, 40]. The T1 and T2 subsets are eliminated from the gating of FO and MZ B cells by their CD93 expression (Fig. 3C). Upon adoptive transfer into B10.PLμMT mice, only the FO subset was able to drive recovery from EAE (Fig. 1B) [7]. To further confirm that MZ B cells were not the regulatory subset of interest, we repeated the B cell transfer studies using Notch-2-deficient donors lacking MZ B cells, which supported recovery from EAE [7].
Figure 3. Representative B cell gating strategy.
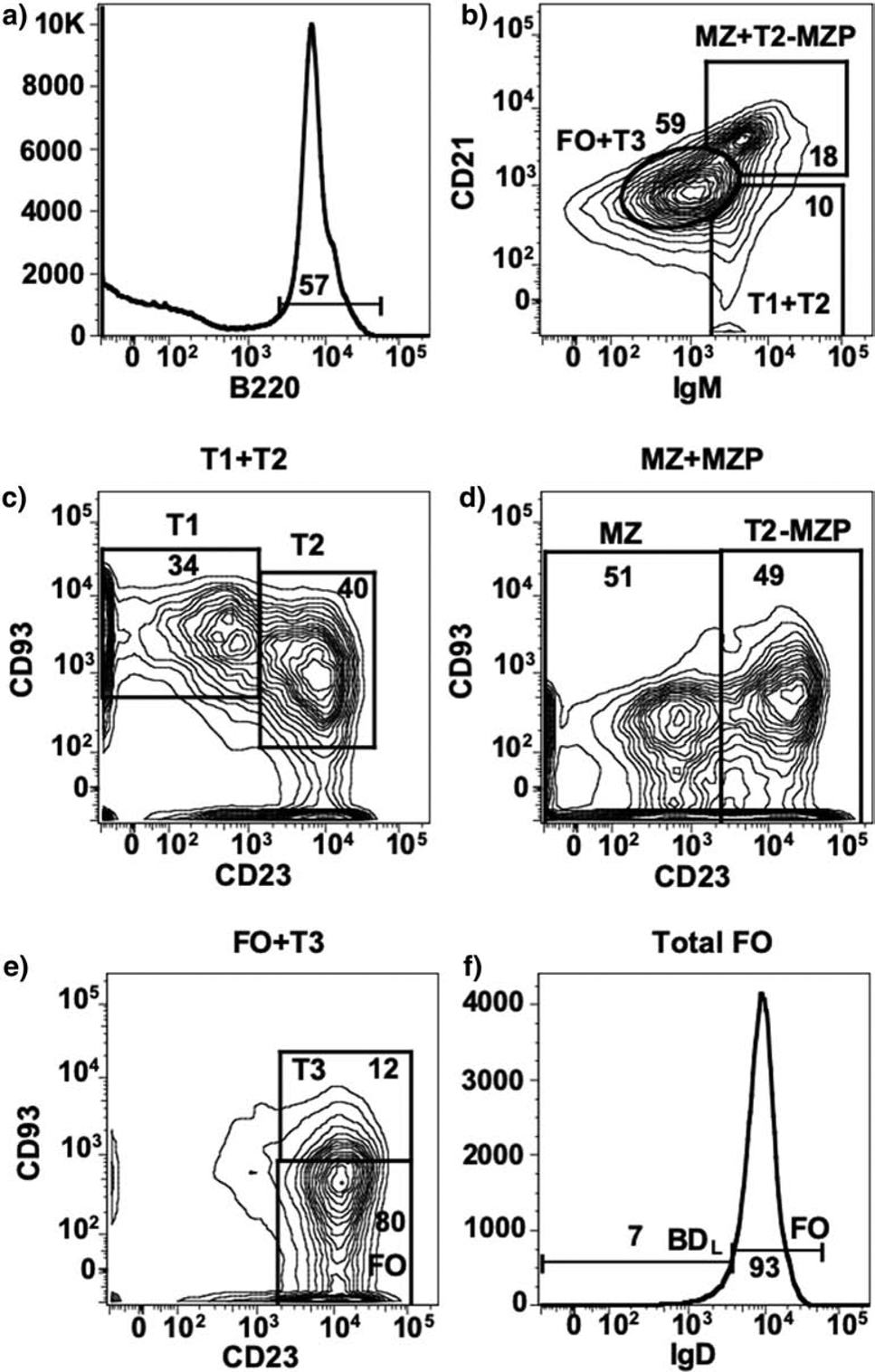
Splenic B220+ B cells (A) are differentially gated on IgM and CD21 to separate splenic B cells subsets into FO + T3, MZ + T2-MZP and T1 + T2 (B). C) T1 and T2 subsets are CD93+ and are separated by CD23 expression. D) MZ and T2-MZP subsets are separated by CD23 expression. E) FO B cells are separated from T3 by lack of CD93 expression. F) FO B cells are separated into IgDlow/− BDL and IgDhi FO B cells. The number on the contour plots indicates the percentage of cells in each gate.
The above series of studies highly suggested that the regulatory B cell subset we sought resided within the gating strategy utilized to identify the FO B cell subset (Fig. 1B, 3). FO B cells reside within the B cell follicles of secondary lymphoid organs and produce the majority of high affinity isotype class switched antibodies produced in a T cell-dependent manner within the germinal center [41]. The identification of FO B cells as regulatory was surprising because they had not been previously implicated in immune regulation. Because the majority of B cells in the spleen fall within the FO B cell phenotype and subsets had not been previously identified in this B cell lineage, we required a strategy to enrich for B cells with the capacity of driving EAE recovery.
B Cell Depletion Strategy to Identify BDL
The strategy that we developed to identify BDL was to utilize anti-CD20 differential B cell depletion efficiency. We obtained two anti-mouse CD20 mAb from Biogen (Cambridge, MA) with identical antigen binding variable regions, but different Fc regions. The anti-CD20 IgG2a isotype depleted the majority of B cells in the spleen and phenocopied μMT mice in that recovery from EAE did not occur leading to chronic disease (Fig. 1C) [3]. In contrast, the IgG1 isotype did not deplete MZ B cells and depleted ~90% of FO B cells in the spleen [7]. Of importance to our studies, the anti-CD20 IgG1 B cell depleted mice recovered from EAE (Fig. 1C) [7]. These data indicated that the regulatory B cell subset we sought was retained in the partially depleted mice. Because adoptive transfer studies indicated that MZ B cells were not the regulatory B cell we sought (Fig. 1B), we concentrated on further investigating the phenotype of the remaining FO B cells. What we observed was an enrichment of B cells expressing low/negative levels of IgD from ~7% in isotype control-treated mice (Fig. 3F) to ~34% in anti-CD20 IgG1-treated mice [7]. Adoptive transfer experiments demonstrated that the IgDlow/neg (BDL) subset drove recovery from EAE, while the FO (IgDhi) did not (Fig. 1B) [7]. Due to IgD being a definitive cell surface marker, the B subset exhibiting regulatory activity was named B cell IgD low (BDL) [7].
To determine whether BDL were a unique B cell subset, we performed RNA sequencing analysis comparing FO IgDhi, BDL and MZ B cells. Using principle component analysis (PCA) plotted in three dimensions, we confirmed that MZ B cells were distinct from both FO IgDhi and BDL B cells in all three dimensions [7]. When we compared FO IgDhi and BDL, we found that they clustered similarly in two dimensions, but clustered separately in the third [7]. When we compared transcriptional profiles, BDL as compared to FO IgDhi, exhibited significantly higher expression of 343 transcripts and lower expression of 121 transcripts [7]. Genes associated with cell cycle/proliferation, developmental processes and cell surface receptor signaling were upregulated in BDL [7]. These cumulative data demonstrated that BDL are a unique B cell subset that can be identified and purified by their B220+IgM+IgDlow/−CD21intCD23+CD93int cell surface phenotype (Fig. 3) [7]. However, how they developmentally fit into the B cell lineage still needs further exploration (Fig. 2).
Mice Deficient in B Cells have Reduced Numbers of Treg
In the course of our studies to identify BDL, we investigated the mechanism whereby B cells contribute to recovery from EAE. Early in these studies, the discovery of Treg as being essential to preventing the onset of autoimmunity and our observation that Foxp3 transcripts in the spinal cord were lower in μMT →μMT and μMT→WT chimeras [6], prompted us to examine whether they played a role in driving recovery in our studies. When we investigated the link between B cells and Treg, we found that as compared to WT mice, μMT mice had an ~3-fold reduction in Treg numbers in the spleen (Fig. 4) [3]. Interestingly, mice with total B cell depletion with anti-CD20 IgG2a, which do not recover from EAE, had a significant reduction in the absolute number of Treg in the spleen (Fig. 4) [3]. In contrast, mice partially depleted of B cells (anti-CD20 IgG1) that retain BDL, did not exhibit a decrease in Treg numbers (Fig. 4) [7]. Thus, our studies indicated a direct correlation between the number of Treg and BDL and the ability to recover from EAE (Fig. 5). To explore that link further, we adoptively transferred total B cells with or without dye labelled Treg into μMT mice and found that Treg numbers significantly increased due to proliferation (Fig. 4) [3]. In a follow up study, we showed that BDL, but not FO or MZ B cells, drove the expansion of Treg following adoptive transfer into μMT mice (Fig. 4) [7]. These cumulative studies indicated that direct cell-cell interactions between BDL and Treg are required to maintain Treg homeostasis. Of importance to the translatability of our findings, is that on both the B10.PL and C57BL/6 backgrounds, μMT mice had a significant reduction in Treg that were significantly increased by the adoptive transfer of syngenic BDL [7].
Figure 4. Graphic representation of Treg numbers in WT and μMT mice with various B cell and anti-CD20 treatments.
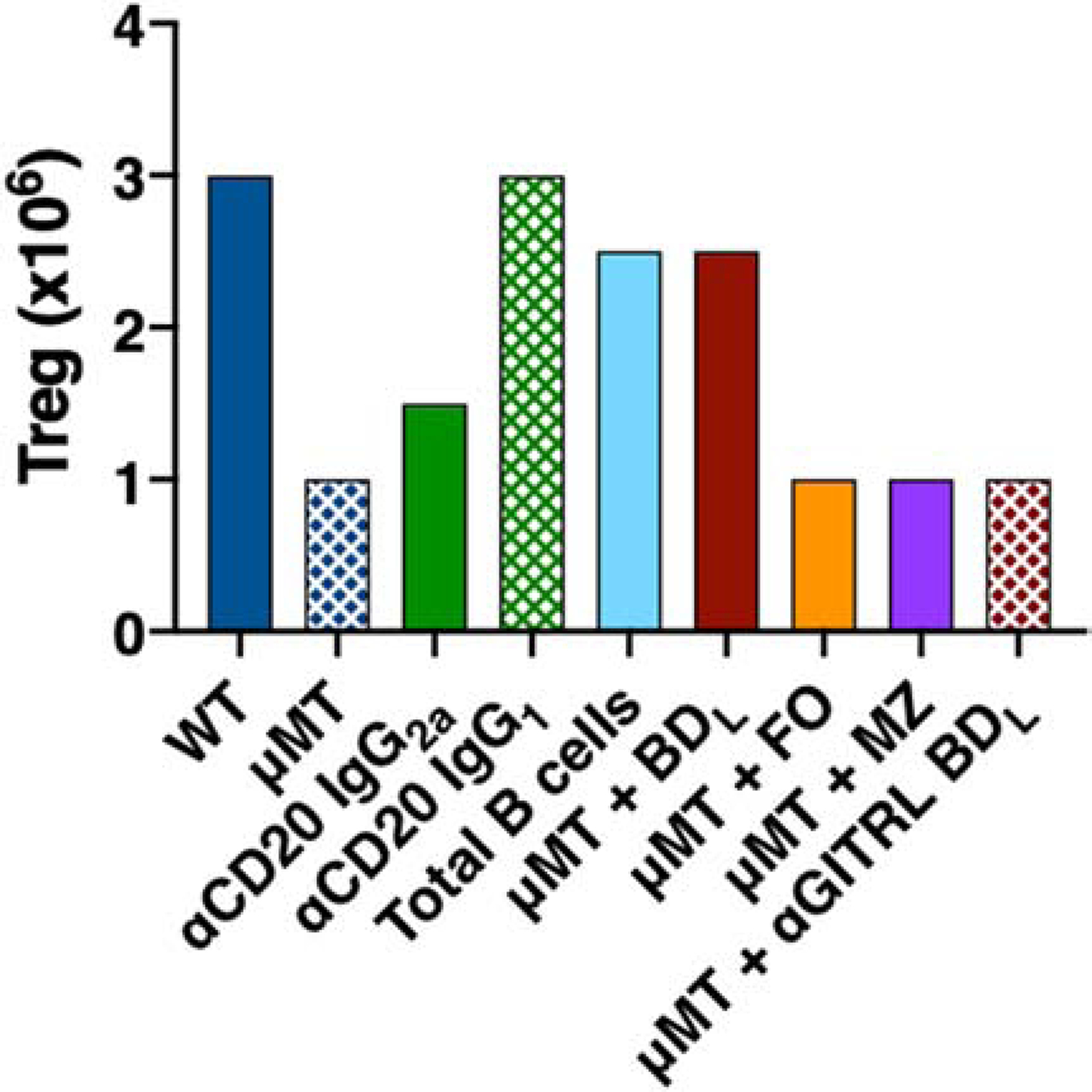
Based on published studies, the relative number of Treg in the spleen of WT mice treated with PBS or isotype control antibodies or administered anti-CD20 IgG2a or IgG1; and μMT mice treated with PBS or adoptively transferred FACS purified total B cells, BDL, FO, MZ or anti-GITRL-blocked BDL B cells are shown.
Figure 5. BDL promote immune tolerance by inducing Treg proliferation via GITRL.
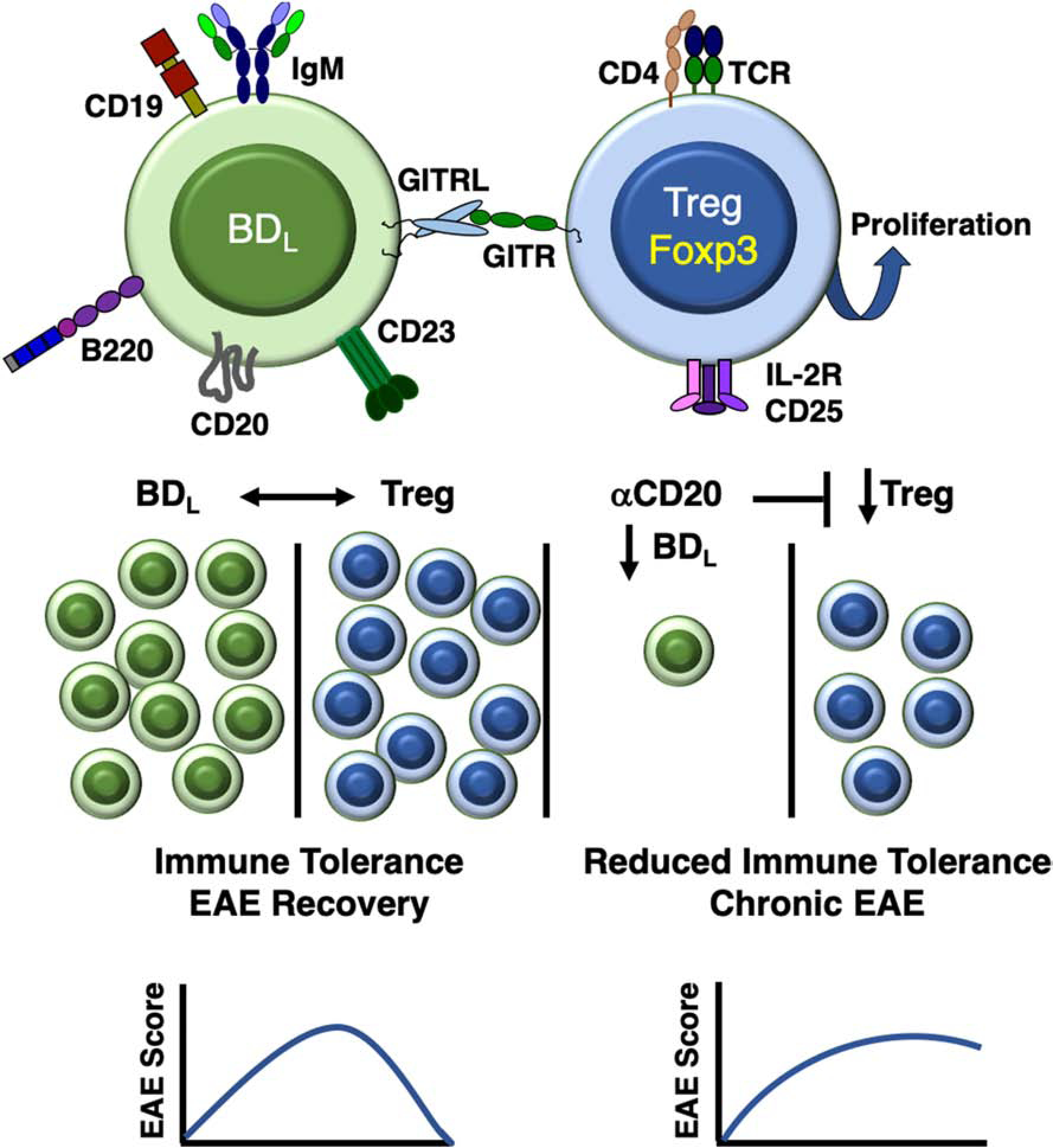
BDL are a unique splenic B cell subset that expresses IgM, but not IgD, and interact with Treg via GITRL leading to their proliferation through engagement of GITR. A balance between BDL and Treg is required to maintain Treg numbers at a sufficient level to maintain a high level of immune tolerance leading to recovery from EAE. If BDL are depleted by anti-CD20, Treg numbers are reduced leading to reduced immune tolerance and chronic EAE.
Investigations of B Cell Mechanisms Driving Treg Expansion
Throughout the course of our studies, we investigated various mechanism whereby BDL interact with and promote Treg expansion. The following studies were conducted prior to the identification of the BDL phenotype with total splenic B cells, but nevertheless allowed us to eliminate several potential mechanisms. To determine whether B7 expression was required, we repeated the B cell adoptive transfer experiment using B7−/− B cells, which were able to drive Treg expansion, but not to the same level as WT B cells [3]. Using B7.1−/− and B7.2−/− single knockout B cells, revealed that it was B7.2 that contributed to the reduced Treg expansion [3]. The requirement for antigen presentation was examined using C2ta-deficient mice that lack MHC class II expression due its role as a transactivator required for MHC class II transcription [42]. Similar to CD86−/− mice, μMT mice adoptively transferred C2ta−/− B cells exhibited Treg expansion that was reduced as compared to mice that received WT B cells [3]. While these studies indicate that cognate interactions between B cells and Treg could occur and contribute to Treg expansion, we have not repeated these studies with BDL. Thus, we cannot exclude the possibility that other B cell subsets are interacting with Treg via cognate interactions resulting in either Treg expansion or survival. In addition, both CD86 and MHC class II could engage Treg without the need for antigen presentation. A subset of Treg express cytotoxic T lymphocyte-associated antigen (CTLA)-4 that could interact with CD86 expressed by BDL [43, 44]. In support of this possibility is the finding that BDL express ~2-fold higher levels of CD86 as compared to FO B cells [7]. In addition, Treg subsets have been reported to express lymphocyte activation gene (LAG)-3, which binds to MHC class II and was shown to be important for Treg control of T cell homeostasis [45, 46].
BDL Promote Treg Homeostasis via Glucocorticoid-induced TNF Ligand (GITRL)
The finding that B cells/BDL induce Treg proliferation was the key observation that led to the discovery that BDL maintain Treg homeostasis via their expression of GITRL. GITR, the receptor for GITRL, is highly expressed by Treg and was shown to induce their proliferation [47–49]. Immune cells shown to express GITRL include dendritic cells, macrophages as well as B cells [3, 7, 50, 51]. An additional clue that B cells could potentially drive Treg proliferation via GITR:GITRL is the finding that transgenic mice overexpressing GITRL specifically in B cells had a significant increase in Treg and attenuated EAE severity [51]. Antibody blocking of GITRL prior to adoptive transfer of total B cells or BDL into μMT mice significantly attenuated their ability to induce Treg expansion (Fig. 4) [3, 7]. This finding was confirmed utilizing GITRL-deficient BDL [7]. In addition, GITRL blocking of B cells also abrogated their ability to promote recovery from EAE (Fig. 1D) [3]. These data provide strong evidence that BDL promote immune tolerance by maintenance of Treg homeostasis via their expression of GITRL (Fig. 5).
BDL Future Perspectives
While GITRL is an important effector molecule for BDL many questions still remain regarding their development, localization and function. Of particular interest is whether the IgM+IgDlow/− phenotype is critical for their function. Although the function of IgD is still not fully clear, one elucidated role is to attenuate the response to self in anergic IgM+ B cells while promoting their accumulation. The lack of IgD expression by IgM+ B cells led to a reduction in expression of self-reactive BCR [52, 53]. Interestingly, IgD was also shown to facilitate prompt responsiveness to foreign antigens by promoting recognition of multimeric antigens [52–54]. Thus, one could speculate that while IgM+IgDlow/− BDL would be not be self-reactive they also would exhibit poor responses to foreign antigens. This phenotype is uniquely suited for the role of BDL in Treg homeostasis whereby their lack of response to self- and foreign-antigens would prevent their activation and subsequent Treg activation. This would allow BDL to induce the proliferation of all Treg without introducing a bias towards reactivity to self or non-self. However, additional studies are required to fully understand whether BDL participate in immune responses leading to germinal center formation and antibody production or whether they are anergic and/or self-reactive.
BDL Activity Exists in Humans
Treg in humans are essential for the dampening of autoimmune responses as profoundly shown in babies with mutations in the Foxp3 gene that quickly succumb to the disease immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX). IPEX is driven by activated CD4+ T cells leading to multi-organ damage as the result of autoimmunity and other life threatening inflammatory disorders [55, 56]. To determine whether BDL exist in humans, we developed an in vitro assay to measure Treg proliferation in the presence and absence of B cells [7]. As in mice, we found that splenic B cells (CD19+CD20+CD24low/int) with low/neg IgD expression induced Treg proliferation [7]. This finding was confirmed using peripheral blood B cells, in which we included an IgDhi subset, and again found that only the IgDlow/neg B cells induced Treg proliferation [7]. While these findings indicate that BDL exist in humans, additional studies are required to further refine the BDL phenotype since there are a number of human B cell subsets that exist beyond BDL that have a IgDlow/neg phenotype including isotype-class switched memory B cells that are present in both the spleen and peripheral blood [57].
Besides BDL, others have described human B cell subsets exhibiting regulatory activity [58]. The best characterized express both IgM and IgD and are further defined by a CD24hiCD38hi immature phenotype [59]. In addition to expressing IgD they differ from BDL by utilizing an IL-10-dependent mechanism to suppress T helper differentiation and induce T cell conversion to Treg [59, 60]. In contrast, BDL induce Treg proliferation in an IL-10-independent manner [3, 7]. Other human B cell regulatory mechanisms reported include granzyme B, IgG4, adenosine, TGF-β and indoleamine 2,3-dioxygenase [58]. Additional cell surface markers are required to generate a more definitive BDL phenotype to allow for their specific identification and isolation to further our understanding of this novel B cell subset in humans.
Summary
In our studies, we uncovered the novel BDL B cell subset that interacts with Treg via GITR:GITRL thereby inducing Treg proliferation such that their cell numbers exist at a sufficient level to drive recovery from EAE (Fig. 5). It is now clear that Treg cell number is a critical factor in their ability to suppress inflammatory immune responses [61]. In our studies, this was demonstrated by the onset of spontaneous onset of EAE in MBP-TCR transgenic mice and enterocolitis in IL-10−/− mice following total B cell depletion [3]. Although numerous challenges and limitations exist that need to be overcome to implement widespread Treg adoptive cell therapy, increasing Treg numbers is considered a viable therapeutic strategy for the treatment of MS and other autoimmune diseases [62, 63]. As an alternative to expensive Treg adoptive cell therapy, we propose that BDL could be harnessed to promote Treg expansion naturally in in vivo for the treatment of autoimmunity.
Research Highlights.
B cells may negatively regulate the extent of the inflammatory response.
B cell IgD low (BDL) are a new mouse splenic B cell subset that plays a critical role in immune tolerance.
BDL interact with CD4+Foxp3+ T regulatory cells (Treg) via glucocorticoid-induced tumor necrosis factor ligand (GITRL) and induce their proliferation.
BDL maintenance of Treg homeostasis keeps immune tolerance high and inflammation from becoming chronic.
Acknowledgments
We thank this work was supported in part by National Institutes of Health grants R56AI122655, R56AI12938 and AI069358; National Multiple Sclerosis Society research grant RG 1501-03034 and the Versiti Blood Research Institute Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Wolf SD, Dittel BN, Hardardottir F, Janeway CA Jr. Experimental autoimmune encephalomyelitis induction in genetically B cell-deficient mice. J Exp Med. 1996;184:2271–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ray A, Dittel BN. Mechanisms of Regulatory B cell Function in Autoimmune and Inflammatory Diseases beyond IL-10. J Clin Med. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ray A, Basu S, Williams CB, Salzman NH, Dittel BN. A novel IL-10-independent regulatory role for B cells in suppressing autoimmunity by maintenance of regulatory T cells via GITR ligand. J Immunol. 2012;188:3188–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–61. [DOI] [PubMed] [Google Scholar]
- [5].Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–6. [DOI] [PubMed] [Google Scholar]
- [6].Mann MK, Maresz K, Shriver LP, Tan Y, Dittel BN. B cell regulation of CD4+CD25+ T regulatory cells and IL-10 via B7 is essential for recovery from experimental autoimmune encephalomyelitis. J Immunol. 2007;178:3447–56. [DOI] [PubMed] [Google Scholar]
- [7].Ray A, Khalil MI, Pulakanti KL, Burns RT, Gurski CJ, Basu S, et al. Mature IgD(low/−) B cells maintain tolerance by promoting regulatory T cell homeostasis. Nature communications. 2019;10:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Willenborg DO, Prowse SJ. Immunoglobulin-deficient rats fail to develop experimental allergic encephalomyelitis. J Neuroimmunol. 1983;5:99–109. [DOI] [PubMed] [Google Scholar]
- [9].Willenborg DO, Sjollema P, Danta G. Immunoglobulin deficient rats as donors and recipients of effector cells of allergic encephalomyelitis. J Neuroimmunol. 1986;11:93–103. [DOI] [PubMed] [Google Scholar]
- [10].Myers KJ, Sprent J, Dougherty JP, Ron Y. Synergy between encephalitogenic T cells and myelin basic protein-specific antibodies in the induction of experimental autoimmune encephalomyelitis. J Neuroimmunol. 1992;41:1–8. [DOI] [PubMed] [Google Scholar]
- [11].Schluesener HJ, Sobel RA, Linington C, Weiner HL. A monoclonal antibody against a myelin oligodendrocyte glycoprotein induces relapses and demyelination in central nervous system autoimmune disease. J Immunol. 1987;139:4016–21. [PubMed] [Google Scholar]
- [12].Ray A, Mann MK, Basu S, Dittel BN. A case for regulatory B cells in controlling the severity of autoimmune-mediated inflammation in experimental autoimmune encephalomyelitis and multiple sclerosis. J Neuroimmunol. 2010;230:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kitamura D, Roes J, Kuhn R, Rajewsky K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature. 1991;350:423–6. [DOI] [PubMed] [Google Scholar]
- [14].Tsubata T, Tsubata R, Reth M. Crosslinking of the cell surface immunoglobulin (mu-surrogate light chains complex) on pre-B cells induces activation of V gene rearrangements at the immunoglobulin kappa locus. Int Immunol. 1992;4:637–41. [DOI] [PubMed] [Google Scholar]
- [15].Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol. 2002;3:944–50. [DOI] [PubMed] [Google Scholar]
- [16].Mason DY, Jones M, Goodnow CC. Development and follicular localization of tolerant B lymphocytes in lysozyme/anti-lysozyme IgM/IgD transgenic mice. Int Immunol. 1992;4:163–75. [DOI] [PubMed] [Google Scholar]
- [17].Mann MK, Ray A, Basu S, Karp CL, Dittel BN. Pathogenic and regulatory roles for B cells in experimental autoimmune encephalomyelitis. Autoimmunity. 2012;45:388–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hannum LG, Haberman AM, Anderson SM, Shlomchik MJ. Germinal center initiation, variable gene region hypermutation, and mutant B cell selection without detectable immune complexes on follicular dendritic cells. J Exp Med. 2000;192:931–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ray A, Wang L, Dittel BN. IL-10-independent regulatory B-cell subsets and mechanisms of action. Int Immunol. 2015;27:531–6. [DOI] [PubMed] [Google Scholar]
- [20].Dittel BN, Merchant RM, Janeway CA, Jr. Evidence for Fas-dependent and Fas-independent mechanisms in the pathogenesis of experimental autoimmune encephalomyelitis. J Immunol. 1999;162:6392–400. [PubMed] [Google Scholar]
- [21].Barr TA, Brown S, Ryan G, Zhao J, Gray D. TLR-mediated stimulation of APC: Distinct cytokine responses of B cells and dendritic cells. Eur J Immunol. 2007;37:3040–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yoshimoto M The ontogeny of murine B-1a cells. Int J Hematol. 2020;111:622–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cunningham AF, Flores-Langarica A, Bobat S, Dominguez Medina CC, Cook CN, Ross EA, et al. B1b cells recognize protective antigens after natural infection and vaccination. Front Immuno. 2014;5:535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pillai S, Cariappa A, Moran ST. Marginal zone B cells. Annu Rev Immunol. 2005;23:161–96. [DOI] [PubMed] [Google Scholar]
- [25].Shlomchik MJ, Weisel F. Germinal center selection and the development of memory B and plasma cells. Immunol Rev. 2012;247:52–63. [DOI] [PubMed] [Google Scholar]
- [26].Owen JJ, Cooper MD, Raff MC. In vitro generation of B lymphocytes in mouse foetal liver, a mammalian ‘bursa equivalent’. Nature. 1974;249:361–3. [DOI] [PubMed] [Google Scholar]
- [27].Cumano A, Furlonger C, Paige CJ. Differentiation and characterization of B-cell precursors detected in the yolk sac and embryo body of embryos beginning at the 10- to 12-somite stage. Proc Natl Acad Sci USA. 1993;90:6429–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kobayashi M, Shelley WC, Seo W, Vemula S, Lin Y, Liu Y, et al. Functional B-1 progenitor cells are present in the hematopoietic stem cell-deficient embryo and depend on Cbfβ for their development. Proc Natl Acad Sci USA. 2014;111:12151–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Montecino-Rodriguez E, Dorshkind K. B-1 B cell development in the fetus and adult. Immunity. 2012;36:13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Griffin DO, Holodick NE, Rothstein TL. Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+ CD27+ CD43+ CD70. J Exp Med. 2011;208:67–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Griffin DO, Rothstein TL. Human b1 cell frequency: isolation and analysis of human b1 cells. Front Immunol. 2012;3:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Baumgarth N B-1 Cell Heterogeneity and the Regulation of Natural and Antigen-Induced IgM Production. Front Immunol. 2016;7:324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ghosn EE, Yang Y, Tung J, Herzenberg LA, Herzenberg LA. CD11b expression distinguishes sequential stages of peritoneal B-1 development. Proc Natl Acad Sci USA. 2008;105:5195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ghosn EE, Yamamoto R, Hamanaka S, Yang Y, Herzenberg LA, Nakauchi H, et al. Distinct B-cell lineage commitment distinguishes adult bone marrow hematopoietic stem cells. Proc Natl Acad Sci USA. 2012;109:5394–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kraal G, Mebius R. New insights into the cell biology of the marginal zone of the spleen. Int Rev Cytol. 2006;250:175–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Oliver AM, Martin F, Gartland GL, Carter RH, Kearney JF. Marginal zone B cells exhibit unique activation, proliferative and immunoglobulin secretory responses. Eur J Immunol. 1997;27:2366–74. [DOI] [PubMed] [Google Scholar]
- [37].Hozumi K, Negishi N, Suzuki D, Abe N, Sotomaru Y, Tamaoki N, et al. Delta-like 1 is necessary for the generation of marginal zone B cells but not T cells in vivo. Nat Immunol. 2004;5:638–44. [DOI] [PubMed] [Google Scholar]
- [38].Saito T, Chiba S, Ichikawa M, Kunisato A, Asai T, Shimizu K, et al. Notch2 is preferentially expressed in mature B cells and indispensable for marginal zone B lineage development. Immunity. 2003;18:675–85. [DOI] [PubMed] [Google Scholar]
- [39].Basu S, Ray A, Dittel BN. Cannabinoid receptor 2 is critical for the homing and retention of marginal zone B lineage cells and for efficient T-independent immune responses. J Immunol. 2011;187:5720–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Basu S, Campbell HM, Dittel BN, Ray A. Purification of specific cell population by fluorescence activated cell sorting (FACS). J Vis Exp. 2010;(41):1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zhang Y, Garcia-Ibanez L, Toellner KM. Regulation of germinal center B-cell differentiation. Immunol Rev. 2016;270:8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chang CH, Fontes JD, Peterlin M, Flavell RA. Class II transactivator (CIITA) is sufficient for the inducible expression of major histocompatibility complex class II genes. J Exp Med. 1994;180:1367–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Read S, Malmstrom V, Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J Exp Med. 2000;192:295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192:303–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in regulatory T cells. Immunity. 2004;21:503–13. [DOI] [PubMed] [Google Scholar]
- [46].Workman CJ, Vignali DA. Negative regulation of T cell homeostasis by lymphocyte activation gene-3 (CD223). J Immunol. 2005;174:688–95. [DOI] [PubMed] [Google Scholar]
- [47].Liao G, Nayak S, Regueiro JR, Berger SB, Detre C, Romero X, et al. GITR engagement preferentially enhances proliferation of functionally competent CD4+CD25+FoxP3+ regulatory T cells. Int Immunol. 2010;22:259–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ephrem A, Epstein AL, Stephens GL, Thornton AM, Glass D, Shevach EM. Modulation of Treg cells/T effector function by GITR signaling is context-dependent. Eur J Immunol. 2013;43:2421–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ronchetti S, Ricci E, Petrillo MG, Cari L, Migliorati G, Nocentini G, et al. Glucocorticoid-induced tumour necrosis factor receptor-related protein: a key marker of functional regulatory T cells. J Immunol Res. 2015;2015:171520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Nocentini G, Riccardi C. GITR: a modulator of immune response and inflammation. Adv Exp Med Biol. 2009;647:156–73. [DOI] [PubMed] [Google Scholar]
- [51].van Olffen RW, Koning N, van Gisbergen KP, Wensveen FM, Hoek RM, Boon L, et al. GITR triggering induces expansion of both effector and regulatory CD4+ T cells in vivo. J Immunol. 2009;182:7490–500. [DOI] [PubMed] [Google Scholar]
- [52].Sabouri Z, Perotti S, Spierings E, Humburg P, Yabas M, Bergmann H, et al. IgD attenuates the IgM-induced anergy response in transitional and mature B cells. Nat Commun. 2016;7:13381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Gutzeit C, Chen K, Cerutti A. The enigmatic function of IgD: some answers at last. Eur J Immunol. 2018;48:1101–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ubelhart R, Hug E, Bach MP, Wossning T, Duhren-von Minden M, Horn AH, et al. Responsiveness of B cells is regulated by the hinge region of IgD. Nat Immunol. 2015;16:534–43. [DOI] [PubMed] [Google Scholar]
- [55].Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20–1. [DOI] [PubMed] [Google Scholar]
- [56].Wildin RS, Freitas A. IPEX and FOXP3: clinical and research perspectives. J Autoimmun. 2005;25 Suppl:56–62. [DOI] [PubMed] [Google Scholar]
- [57].Pauli NT, Henry Dunand CJ, Wilson PC. Exploiting human memory B cell heterogeneity for improved vaccine efficacy. Frontiers in immunology. 2011;2:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Mauri C, Menon M. Human regulatory B cells in health and disease: therapeutic potential. J Clin Invest. 2017;127:772–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Blair PA, Norena LY, Flores-Borja F, Rawlings DJ, Isenberg DA, Ehrenstein MR, et al. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic Lupus Erythematosus patients. Immunity. 2010;32:129–40. [DOI] [PubMed] [Google Scholar]
- [60].Flores-Borja F, Bosma A, Ng D, Reddy V, Ehrenstein MR, Isenberg DA, et al. CD19+CD24hiCD38hi B cells maintain regulatory T cells while limiting TH1 and TH17 differentiation. Sci Transl Med. 2013;5:173ra23. [DOI] [PubMed] [Google Scholar]
- [61].Spence A, Klementowicz JE, Bluestone JA, Tang Q. Targeting Treg signaling for the treatment of autoimmune diseases. Curr Opin Immunol. 2015;37:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Duffy SS, Keating BA, Moalem-Taylor G. Adoptive Transfer of Regulatory T Cells as a Promising Immunotherapy for the Treatment of Multiple Sclerosis. Front Neurosci. 2019;13:1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Bluestone JA, Tang Q, Sedwick CE. T regulatory cells in autoimmune diabetes: past challenges, future prospects. J Clin Immunol. 2008;28:677–84. [DOI] [PubMed] [Google Scholar]