

Summary
Inborn errors of human IFN-γ immunity underlie mycobacterial disease. We report a patient with mycobacterial disease due to inherited deficiency of the transcription factor T-bet. The patient has extremely low counts of circulating Mycobacterium-reactive NK, invariant NKT (iNKT), mucosal-associated invariant T (MAIT), and Vδ2+ γδ T lymphocytes, and of Mycobacterium-non reactive classic TH1 lymphocytes, with the residual populations of these cells also producing abnormally small amounts of IFN-γ. Other lymphocyte subsets develop normally, but produce low levels of IFN-γ, with the exception of CD8+ αβ T and non-classic CD4+ αβ TH1* lymphocytes, which produce IFN-γ normally in response to mycobacterial antigens. Human T-bet deficiency thus underlies mycobacterial disease by preventing the development of innate (NK) and innate-like adaptive lymphocytes (iNKT, MAIT, and Vδ2+ γδ T cells) and IFN-γ production by them, with mycobacterium-specific, IFN-γ-producing, purely adaptive CD8+ αβ T and CD4+ αβ TH1* cells unable to compensate for this deficit.
Graphical Abstract

Introduction
In the course of primary infection, life-threatening disease in otherwise healthy children, adolescents, and even adults, can result from monogenic inborn errors of immunity (IEI), which display genetic heterogeneity and physiological homogeneity (Casanova, 2015a, 2015b). Mendelian susceptibility to mycobacterial disease (MSMD) is characterized by a selective, inherited predisposition to clinical disease caused by weakly virulent mycobacteria, such as Mycobacterium bovis Bacille Calmette-Guérin (BCG) vaccines and environmental mycobacteria (EM) (Bustamante, 2020; Kerner et al., 2020). Patients are also vulnerable to bona fide tuberculosis. Patients with typical, “isolated” MSMD are rarely prone to other infectious agents, with the exception of Salmonella and occasionally other intra-macrophagic bacteria, fungi, and parasites. Patients with atypical, “syndromic” MSMD often display other clinical phenotypes, infectious or otherwise. MSMD, both “isolated” and “syndromic”, displays a high level of genetic heterogeneity, with causal mutations in 16 genes, and additional allelic heterogeneity, resulting in 31 different disorders. However, there is also physiological homogeneity, as all known genetic causes of MSMD affect interferon gamma (IFN-γ)-dependent immunity (Bustamante, 2020; Kerner et al., 2020; Rosain et al., 2019). Mutations of IFNG, IL12B, IL12RB1, IL12RB2, IL23R, TYK2, ISG15, RORC, IKBKG (NEMO), IRF8, and SPPL2A impede IFN-γ production by innate and adaptive immune cells, whereas mutations of IFNGR1, IFNGR2, STAT1, JAK1, and CYBB impair cellular responses to IFN-γ (Fig. S1A). The clinical penetrance and severity of MSMD depend strongly on genetic etiology and increase with decreasing levels of IFN-γ activity (Dupuis et al., 2000). Seven etiologies also result in an impairment of immunological circuits other than the IFN-γ circuit, accounting for “syndromic” MSMD in the corresponding patients (Bustamante, 2020). Collectively, these studies revealed the crucial role of human IFN-γ in antimycobacterial immunity and its redundancy for immunity against many other pathogens.
The cellular basis of MSMD in patients with impaired responses to IFN-γ involves mononuclear phagocytes. The ability of these cells to contain the ingested mycobacteria depends on their activation by IFN-γ (Nathan et al., 1983). The cellular basis of MSMD in patients with impaired IFN-γ production is poorly understood, as most types of lymphocytes can produce IFN-γ (Schoenborn and Wilson, 2007). Some genetic etiologies of MSMD affect some lymphocyte subsets more than others. ISG15 deficiency preferentially impairs the production of IFN-γ by NK cells (Bogunovic et al., 2012; Zhang et al., 2015). IL-12Rβ2 deficiency preferentially impairs the production of IFN-γ by NK, B, γδ T, classic αβ T cells, type 1 innate lymphoid cells (ILC1s), and ILC2s, whereas IL-23R deficiency preferentially impairs the production of this cytokine by invariant NKT (iNKT) and mucosal-associated invariant T (MAIT) cells, and both IL-12Rβ1 and TYK2 deficiencies impair both the IL-12- and IL-23-dependent subsets (Boisson-Dupuis et al., 2018; Martínez-Barricarte et al., 2018). SPPL2a and IRF8 deficiencies selectively impair the production of IFN-γ by CD4+ CCR6+ TH1 (TH1*) cells (Hambleton et al., 2011; Kong et al., 2018), a TH1 cell subset enriched in Mycobacterium-specific effector cells, whereas CCR6− TH1 cells do not respond to mycobacteria (Acosta-Rodriguez et al., 2007). RORγ/RORγT deficiency impairs the development of iNKT and MAIT cells, and also compromises the production of IFN-γ by γδ T and αβ TH1* cells (Okada et al., 2015). Interestingly, although the lack of both αβ T and γδ T cells in patients with severe combined immunodeficiency (SCID) underlies BCG disease (Casanova et al., 1995), most, if not all other deficits of antigen-specific αβ T-cell responses, whether affecting only CD4+ or CD8+ T cells, such as MHC class II and MHC class I deficiencies, respectively, typically do not (Aluri et al., 2018; Ardeniz et al., 2015; Dimitrova et al., 2014; Elhasid and Etzioni, 1996; Hanalioglu et al., 2017; Hanna and Etzioni, 2014; De La Salle et al., 1994; Lum et al., 2019, 2020; Morgan et al., 2011; Saleem et al., 2000; Zimmer et al., 2005). Moreover, selective deficiencies of NK or iNKT cells do not confer a predisposition to mycobacterial disease (Casey et al., 2012; Cottineau et al., 2017; Hughes et al., 2012; Latour, 2007; Locci et al., 2009; Tangye et al., 2017). The nature of the IFN-γ-producing innate, innate-like adaptive, and purely adaptive lymphocyte subsets indispensable for antimycobacterial immunity, either alone or in combination, therefore remains largely unknown. No genetic cause has yet been identified for half the MSMD patients. We therefore sought to discover a new genetic etiology of MSMD that would expand the molecular circuit controlling human IFN-γ immunity while better delineating the cellular network involved.
Results
Identification of an MSMD patient homozygous for an indel variant of TBX21
We studied a three-year-old boy (“P”, II.2) born to first-cousin Moroccan parents (Fig. 1A). He suffered from disseminated BCG disease (BCG-osis) following vaccination at the age of three months. He also had persistent reactive airway disease (RAD), but was otherwise healthy (see STAR Methods - Case Report). He did not suffer from any other severe clinical infectious diseases despite documented (VirScan) infection with various viruses, including Epstein-Barr virus (EBV), human cytomegalovirus (CMV), roseola virus, adenoviruses A, B, C and D, influenza virus A, rhinovirus A, and bacteria, such as Streptococcus pneumoniae and Staphylococcus aureus (Fig. S1B). We hypothesized that P had an autosomal recessive (AR) defect. We performed whole-exome sequencing (WES) on P, his unaffected brother, and both parents. Genome-wide linkage (GWL) analysis revealed 32 linked regions (LOD score >1.3 and size >500 kb) under a model of complete penetrance (Supplemental Data S1). In these linked regions, there were 15 rare homozygous non-synonymous or essential splicing variants of 15 different genes (minor allele frequency, MAF < 0.003 in gnomAD v2.1 and 1000 Genomes Project, including for each major ancestry) with a combined annotation-dependent depletion (CADD) score above their mutation significance cutoffs (MSC) (Auton et al., 2015; Itan et al., 2016; Karczewski et al., 2020; Kircher et al., 2014; Zhang et al., 2018) (Table S1). After the exclusion of genes with other predicted loss-of-function (LOF) variants with a frequency greater than 0.5% in gnomAD and of genes not expressed in leukocytes, only four candidate genes (TBX21, PSMD9, ARHGAP27 and ERN1) remained. The c.466_471delGAGATGinsAGTTTA insertion and/or deletion (indel) variant of TBX21 (T-box protein 21, or T-box, expressed in T cells, T-bet) was the candidate variant predicted to be the most damaging (Kircher et al., 2014). Moreover, based on connectivity to IFNG, the central gene of the entire network of all known MSMD-causing genes (Itan et al., 2013, 2014), TBX21 was the most plausible candidate gene (Table S1). T-bet is a transcription factor that governs the development or function of several IFN-γ-producing lymphocytes in mice, including TH1 cells (Lazarevic et al., 2013; Szabo et al., 2000, 2002). These findings suggested that homozygosity for the rare indel variant c.466_471delGAGATGinsAGTTTA of TBX21 causes MSMD. We investigated this variant according to the guidelines for genetic studies of single patients (Casanova et al., 2014). Sanger sequencing confirmed that P was homozygous for the indel variant of exon 1 of TBX21, whereas his unaffected brother was homozygous wild-type (WT/WT) and both parents were heterozygous (WT/M) (Fig. S1C). A closely juxtaposed 12-nucleotide (nt) region identical to this variant sequence was detected 8-nt upstream from the variant, and may have served as a template for its generation (Fig. S1D). The variant did not alter the exon 1-exon 2 junction of the TBX21 mRNA in EBV-transformed B (EBV-B) cells or peripheral blood mononuclear cells (PBMCs) (Supplemental databases S1). The variant present in P thus resulted in the replacement of E156 and M157, two amino acids that are highly conserved across different species and among other paralogs of T-box transcription factors, with S156 and L157 (Fig. 1B and Fig. S1E).
Figure 1. Discovery of an MSMD patient with a homozygous TBX21 variant and the corresponding molecular characterization.
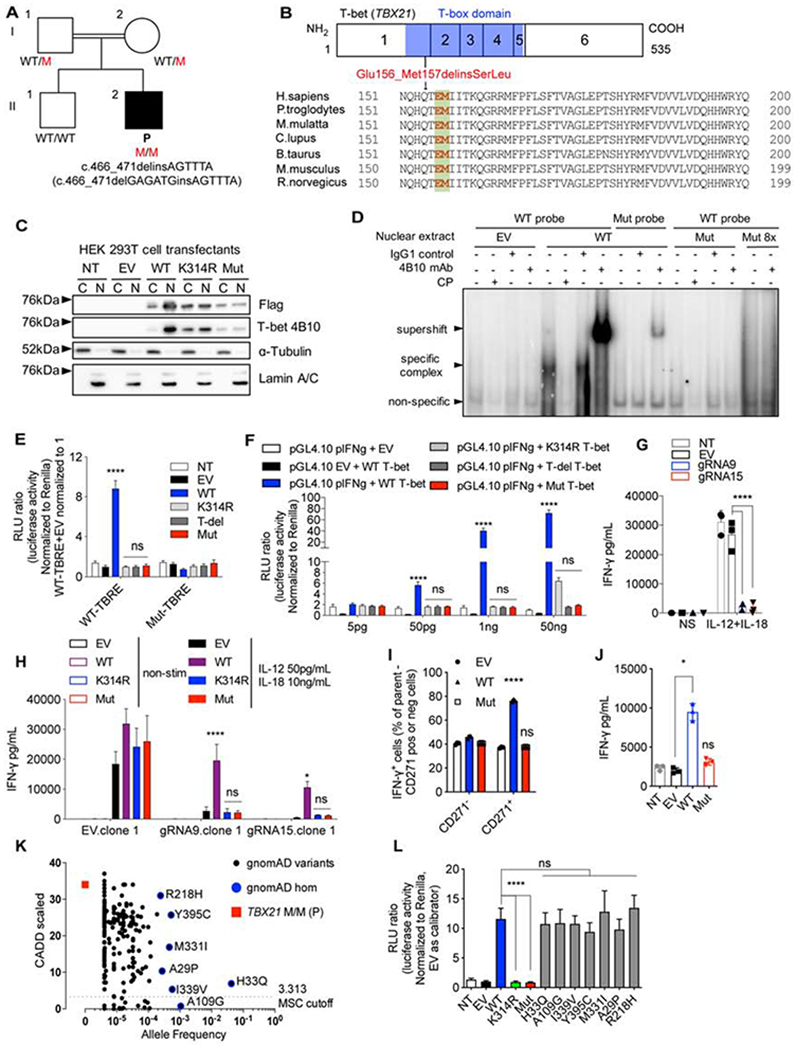
(A) Pedigree of a consanguineous family with the mutant TBX21 allele. (B) Schematic representation of the mutation. (C) Nuclear and cytoplasmic fractions from HEK 293T cells transfected with indicated TBX21 cDNA-containing vectors subjected to immunoblotting against indicated proteins. (D) EMSA on nuclear extracts of HEK293T cells transfected with empty vector (EV), WT or Mut TBX21 alleles. (E) TBRE-reporter luciferase assay testing WT or Mut T-bet in HEK 293T cells. (F) Human IFNG reporter luciferase assay testing HEK 293T cells transfected with WT or Mut TBX21 cDNA-containing vectors, at the indicated concentrations. (G) IFN-γ production in response to stimulation with IL-12 and IL-18 in T-bet knock-out (KO) NK-92 cells. (H) IFN-γ production in response to stimulation with IL-12 and IL-18 in T-bet KO NK-92 cells complemented with EV, WT, K314R or Mut TBX21-containing plasmids. (I) Intracellular IFN-γ production in response to phorbol 12-myristate 13-acetate (PMA) and ionomycin (P/I) in expanding TH0 cells transduced with EV, WT or Mut TBX21 cDNA. (J) IFN-γ production from cells transduced as in (I) in response to stimulation with anti-CD3/CD28 Ab. (K) Graph of CADD score against minor allele frequency (MAF), for TBX21 variants reported in the gnomAD. Mutation significance cutoff (MSC) was shown. (L) TBRE-reporter luciferase assay testing indicated variants of TBX21 as in (E). See also Figure S1, Table S1, and Supplemental Data S1.
In Fig. 1E - J and L, bars represent the mean and standard deviation. Dots represent individual samples or technical replicates. Two-way ANOVA was used for analysis in (E – I). One-way ANOVA was used for analysis in (J and L). In (E – J and L), * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, ns = not significant.
Overexpressed mutant T-bet is LOF
We investigated the expression of the mutant allele (Mut), by overexpressing an empty vector (EV), or vectors containing the WT or Mut allele, or negative controls with the T-box domain deleted (T-del) or K314R, the human ortholog of a known LOF mouse mutant, in human embryonic kidney (HEK) 293T cells (Jang et al., 2013). The production and nuclear translocation of Mut T-bet were impaired (Fig. 1C and Fig. S1F). An assessment of its binding to consensus T-box regulatory elements (TBRE) in DNA revealed that nuclear proteins from WT-transfected cells bound the WT-TBRE, but not the Mut-TBRE. Furthermore, this specific complex was super-shifted by an anti-T-bet antibody (Ab) and inhibited by a competitor probe (CP). However, Mut T-bet did not bind WT-TBRE DNA (Fig. 1D). We also assessed the ability of WT and Mut T-bet to induce a luciferase transgene under the control of the TBRE or human IFNG proximal promoter (Janesick et al., 2012; Soutto et al., 2002). WT T-bet induced high levels of luciferase activity with WT-TBRE but not with Mut-TBRE. Mut T-bet and the negative controls (T-del and K314R) did not induce luciferase activity (Fig. 1E). Mut T-bet was also LOF for transactivation of the IFNG promoter, whereas the negative control, K314R, was markedly hypomorphic (Fig. 1F). We investigated the amino-acid substitution responsible for the abolition of transcriptional activity. We tested the effects of the WT and Mut forms of T-bet and of T-bet forms with single-residue substitutions (E156S and M157L), or alanine substitutions (E156A and M157A) on T-bet protein production and transcriptional activity. The loss of E156 (E156S and E156A) abolished transcriptional activity, but the production of the T-bet protein was unaffected. By contrast, a loss of methionine residues (M157L and M157A) preserved transcriptional activity but decreased the levels of T-bet protein (Fig. S1G and H). T-bet is required for optimal IFN-γ production in NK, ILCs, γδ, and CD4+ T cells in mice and is sufficient for IFN-γ production in NK and CD4+ T cells in humans (Chen et al., 2007; Lazarevic et al., 2013; Powell et al., 2012; Szabo et al., 2002; Yu et al., 2006). We investigated the impact of the T-bet mutation on the induction of IFNG expression, by generating CRISPR/Cas9 gene-edited human NK-92 cell lines lacking TBX21 (Supplemental Data S1). Upon stimulation with IL-12 + IL-18, these TBX21 knockout (KO) NK-92 cells displayed a strong impairment of IFN-γ production (Fig. 1G and Fig. S1I). We re-introduced the WT or Mut TBX21-containing plasmid into TBX21 KO NK-92 cells. We found that the WT TBX21 rescued IFN-γ production, whereas the Mut TBX21 did not (Fig. 1H and Fig. S1J). Finally, the overexpression of WT, but not of Mut T-bet increased IFN-γ levels to values above those for endogenous production in expanding naïve CD4+ T cells from healthy donors (Fig. 1I and J, Fig. S1K). Thus, overexpression of Mut T-bet abolished DNA binding, and the mutant protein had no transactivation activity and could not induce IFN-γ production in an NK cell line or CD4+ T cells. It can, therefore, reasonably be considered a LOF allele.
TBX21 variants in the general population are functional
The TBX21 indel variant in P was not found in the gnomAD v2.1.1, v3, Bravo, or Middle Eastern cohort databases, or our in-house database of more than 8,000 exomes, including > 1,000 for individuals of North African origin (Karczewski et al., 2020; Scott et al., 2016; Taliun et al., 2019). The CADD score of 34 obtained for this allele is well above the MSC of 3.313 (Fig. 1K) (Itan et al., 2016; Kircher et al., 2014; Zhang et al., 2018). The TBX21 gene has a low tolerance of deleterious variations, with a low gene damage index (GDI) score of 3.493 (Itan et al., 2015) and a low residual variation intolerance score (RVIS: −0.74) (Petrovski et al., 2015). Moreover, only two predicted LOF variants (variants: 17:47745045 A/AGCTG, and 17:47744913 C/G) were found in the heterozygous state in gnomAD. As their MAFs were <5x10-6, homozygosity rates for any of these three variants are well below the prevalence of MSMD (about 1/50,000). In the general population covered by the gnomAD database, seven variants have been identified in the homozygous state: A109G has a low CADD score, below the MSC, whereas H33Q, I339V, Y395C, M331I, R218H, and A29P have CADD scores above the MSC (Fig. 1K and Supplemental Data S1). The mouse ortholog of H33Q (H32Q) has been shown to be functionally neutral (Tantisira et al., 2004). None of these seven alleles affected transactivation of the WT-TBRE promoter (Fig. 1L). Thus, all the TBX21 variants present in the homozygous state in gnomAD are functionally neutral. In our in-house cohort of >8,000 exomes from patients with various infectious phenotypes, P is the only patient carrying a rare biallelic variant at the TBX21 locus. We investigated whether any of the other 36 non-synonymous variants of TBX21 in our in-house database could underlie infections in the heterozygous state, by testing each of them experimentally (Supplemental Data S1). None had any functional impact on transcriptional activity (Fig. S1L and M). Therefore, the data for P and his family, our in-house cohort, and the general population suggest that inherited T-bet deficiency, whether complete or partial, is exceedingly rare in the general population (< 5.8 x 10-8). These findings also suggest that homozygosity for the Mut LOF variant of TBX21 is responsible for MSMD in P.
Homozygosity for the TBX21 mutation underlies complete T-bet deficiency
We investigated the production and function of endogenous Mut T-bet in Herpes saimiri virus-transformed T cells (HVS-T) and primary CD4+ T cells from P. Levels of TBX21 mRNA were normal, but endogenous T-bet protein levels were low in P’s cells (Fig. 2A and B, Fig. S2A and B). Together with the observation of low levels of Mut T-bet protein on overexpression (Fig. S1F), these findings suggest that the TBX21 mutation decreases T-bet protein levels by a post-transcriptional mechanism. T-bet transactivates IFNG and TNF by directly binding to their regulatory promoter or enhancer (Garrett et al., 2007; Kanhere et al., 2012; Soutto et al., 2002; Szabo et al., 2000; Tong et al., 2005). Levels of spontaneous IFN-γ and TNF-α production by P’s HVS-T cells were much lower than those for HVS-T cells from all the healthy donors and heterozygous relatives tested (Fig. 2C - F). This cytokine production defect in TBX21 mutant HVS-T cells was rescued by WT T-bet complementation (Fig. 2G and Fig. S2C). We investigated whether homozygous variants of PSMD9, ARHGAP27, or ERN1 contributed to the low levels of IFN-γ production, by transducing HVS-T cells from P with the WT cDNAs of PSMD9, ARHGAP27, or ERN1. Overexpression of the WT PSMD9, ARHGAP27, or ERN1 did not rescue the IFN-γ deficit (Fig. S2D - G). Moreover, the overexpression of these genes in HVS-T cells from P that had already been transduced with WT TBX21 did not further enhance IFN-γ production. These data suggest that PSMD9, ARHGAP27, and ERN1 variants make no significant contribution to the patient’s immunological and clinical phenotype (Fig. S2H and I).
Figure 2. The T-bet variant in patient-derived cells is loss-of-function.

(A) Whole-cell lysates from expanding CD4+ T cells from healthy donors (CTL), a heterozygous parent (WT/M) and the patient (M/M) subjected to immunoblotting. (B) Immunoblotting against indicated proteins of whole-cell lysates of HVS-T cells derived from CTL, M/M and WT/M. (C and D) Spontaneous production of IFN-γ (C) and TNF-α (D) from cultures of HVS-T cells from CTL, M/M and WT/M. (E and F) The IFNG (E) and TNF (F) expression of HVS-T cells, as in (C), was assessed by RT-qPCR. (G) HVS-T cells from M/M were rescued with EV or WT TBX21. The percentage of intracellular IFN-γ-producing cells with or without P/I stimulation is shown. (H) CD4+ T cells from CTL, M/M and WT/M were expanded under TH0 or TH1 conditions. M/M cells were transduced as described in (G). Cells were subjected to ICS for IFN-γ and TNF-α in response to P/I. (I) Percentage of IFN-γ- and TNF-α-producing cells in (H). (J) Transduced TH0 cells, as in (H), were isolated and restimulated, and were subjected to RNA-seq, along with non-transduced cells. Pathway enrichment among differentially regulated genes is shown. (K) The numbers of T-bet-dependent differentially expressed immune genes in RNA-seq are shown. (L) Heat-map of selected T-bet-dependent downstream genes, as in (K). See also Figure S2, Table S2, and Supplemental Data S1.
In Fig. 2C – G and I, bars represent the mean and the standard error of the mean. Dots represent individual samples. One-way ANOVA was used for analysis in (C and D). Mann-Whitney tests were used for analysis in (E and F), comparing WT/M or M/M to CTL. In (C - F), * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, and ns = not significant.
T-bet deficiency alters the expression of downstream target genes
The functional impact of the T-bet mutation was also investigated in primary CD4+ T cells. Upon stimulation with P/I, IFN-γ production was almost entirely abolished in P’s expanded TH0 cells and TNF-α production was impaired; the production of both these molecules was rescued by WT T-bet (Fig. 2H and I, Fig. S2J and K). In TH1 conditions, exogenous IL-12 bypassed T-bet and induced moderate IFN-γ production by P’s cells. However, the levels of IFN-γ were still ~60-70% lower than those in healthy donors (Fig. 2H and I). We investigated other T-bet-dependent transcriptional targets, by performing RNA-seq to compare TH0 cells from 4 healthy donors, P, and P’s cells complemented with WT T-bet after incubation with anti-CD3/28 Ab beads. We found that 415 genes were downregulated and 489 genes were upregulated in P’s cells relative to healthy donors (Supplemental Data S1, Table S2). The complementation of P’s cells with WT T-bet reversed the differential expression of 99 of the downregulated and 161 of the upregulated genes (Supplemental Data S1). Thus, only a subset of differentially regulated genes were rescued, possibly due to differences in genetic variants other than TBX21 between controls and P. Alternatively, T-bet may control the differential expression of some genes early in activation, and the genetic rescue may have occurred too late to reverse this effect. These targets were enriched in cytokine signaling pathway genes (Fig. 2J). We therefore decided to focus on genes involved in immunological signaling. Only 32 such genes were upregulated or downregulated in P’s cells, but these differences in expression relative to controls were reversed by WT T-bet (Fig. 2K). Known T-bet-dependent targets, such as IFNG, CCL3 and CXCR3, were downregulated in CD4+ T cells from the T-bet-deficient patient, whereas IFNGR2 expression was upregulated (Fig. 2L) (Iwata et al., 2017; Jenner et al., 2009). A set of new T-bet target genes was also identified, including CCL1, CCL13, CCL4, CSF2, CXCR5, GZMM, CD40, CD86, IL7R, IL10, LTB, ITGA5, and ITGB5 (Fig. 2L). Collectively, our data indicate that the patient had AR complete T-bet deficiency, affecting the expression of a set of T-bet-dependent target genes, including IFNG.
T-bet induces permissive chromatin accessibility and CpG methylation in IFNG
We analyzed the molecular mechanisms by which T-bet controls transcription. Epigenetically, T-bet is known to induce a permissive environment for transcription at the IFNG promoter through histone modifications and the suppression of CpG methylation (Lewis et al., 2007; Miller and Weinmann, 2010; Tong et al., 2005). However, it remains unknown whether T-bet directly regulates chromatin accessibility in mice or humans. The regulation of CpG methylation at the genome-wide scale by T-bet has never been studied. We performed omni-ATAC-seq analysis and EPIC DNA CpG methylation array analysis with TH0 cells derived from P and controls. In P’s cells, a gain of chromatin accessibility was observed at 1,661 loci and a loss of chromatin accessibility was observed at 3,675 loci (Fig. 3A and B, Supplemental Data S1, Table S3). We found that 662 and 1,642 of these loci, respectively, were subject to strict regulation by T-bet, as their gains and losses of chromatin accessibility were reversed by WT T-bet (Fig. 3B, Table S3). The chromatin regions opened up by T-bet were heavily occupied by bound T-bet, whereas those closed by T-bet did not typically require T-bet binding (Fig. 3C) (Kanhere et al., 2012). An enrichment in T-box binding elements was observed in loci for which chromatin accessibility was increased by T-bet, whereas an enrichment in Forkhead box elements was observed at loci for which chromatin accessibility was decreased by T-bet (Fig. 3D and E). The chromatin accessibility of 181 immunological genes (247 loci), including IFNG, CYBB, and TYK2, three known MSMD-causing genes, was increased by T-bet, whereas that of 73 immunological genes (82 loci) was decreased by T-bet (Fig. 3F, Supplemental Data S1, and Table S3). Three known T-bet-dependent targets, IFNG, TNF and CXCR3, were among the top hits for the differentially regulated loci. The transcription start sites (TSS) of IFNG and TNF, the proximal promoter of IFNG, and the enhancers of CXCR3 were inaccessible in T-bet-deficient cells, and this inaccessibility was rescued by WT T-bet (Fig. 3G - I). The EPIC DNA CpG methylation array analysis identified 644,236 CpG sites that were differentially regulated (Table S3). Three CpG loci within IFNG were hypermethylated in conditions of T-bet deficiency, whereas their methylation was reduced to levels similar to those in controls on complementation with WT T-bet (Fig. 3J and K). NR5A2, TIMD4, ATXN2, ZAK, SLAMF8, TBKBP1, CD247, HDAC4 and several other genes were also regulated in a similar manner (Fig. 3J and Table S3). Interestingly, the methylation of six CpG loci within ENTPD1 not previously linked to T-bet also increased in a T-bet-dependent manner (Fig. 3J and L). By contrast, IL10 was a top target for which CpG methylation was drastically reduced in T-bet deficiency but rescued by WT T-bet (Fig. 3M). Taken together, these results demonstrate that T-bet orchestrates the expression of target genes by modulating both their chromatin accessibility and CpG methylation. Genome-wide omni-ATAC-seq and CpG methylation array analyses identified new epigenetic targets of T-bet (Table S3). They also showed that chromatin accessibility at IFNG was increased by T-bet at both the TSS and promoter sites, whereas the CpG methylation of IFNG was decreased by T-bet at three different positions.
Figure 3. An altered spectrum of epigenetic regulation in CD4+ T cells in T-bet deficiency.

(A) Expanded CD4+ TH0 cells from CTL, IL-12Rβ1-deficient MSMD patients (IL12RB1 M/M), WT/M, M/M, and M/M cells complemented with empty vector (M/M +EV) or with WT T-bet (M/M +WT) were subjected to omni-ATAC-seq. PCA analysis of the peaks called in the various samples, plotted in ggplot2. (B) Number of called peaks differentially regulated as indicated. (C) Heatmap of all loci opening or closing in a T-bet-dependent manner as in (B). (D and E) The most significantly enriched DNA binding motifs at loci displaying T-bet-dependent increases (D) and loci displaying T-bet-dependent decreases (E), as in (B and C). (F) Chromatin accessibility heatmap for all the loci displaying the most significant differential regulation of immune genes (CTL – M/M, adjusted P-value < 1 x 10−5), abolished by WT T-bet (M/M+EV - M/M+WT, adjusted P-value < 1 x 10−5). (G - I) Chromatin accessibility of transcription start site (TSS), proximal promoter and distal enhancer regions of IFNG, TNF and CXCR3. (J) The differential methylation (beta-value) of CpG sites between M/M and CTL or between M/M+EV and M/M+WT is shown. (K - M) CpG methylation status of CpG sites in IFNG (K), ENTPD1 (L) and IL10 (M). See also Table S3 and Supplemental Data S1.
T-bet deficiency impairs NK cell maturation
We then investigated the role of T-bet in the development of leukocyte lineages. Complete blood counts for fresh samples from P showed that the numbers of lymphocytes, neutrophils, and monocytes were normal. We studied the PBMC subsets of P after recovery from mycobacterial disease, by mass cytometry (cytometry by time-of-flight, CyTOF). A simultaneous analysis of the expression of 38 different cell surface markers was performed to compare the leukocyte subsets of P and his parents, healthy donors, and patients with IL-12Rβ1 deficiency, the most common genetic etiology of MSMD, as controls (Fig. 4A and Fig. S3A) (Van Gassen et al., 2015; Korsunsky et al., 2019). The frequencies of the plasmacytoid DC (pDC) and conventional DC 1 and 2 (cDC1 and cDC2) subsets were not affected by human T-bet deficiency (Fig. S3B). All major myeloid lineages were normal in P. We therefore focused on the development of lymphoid lineages. Total NK cells (defined as Lin−CD7+CD16+ or CD94+) were present in normal numbers in P (Fig. S3C and D). However, CD16+ and CD56bright NK cells levels were much lower in P than in controls (Fig. 4A and B). Moreover, P had an abnormally high frequency of CD56−CD127− NK cells (Fig. S3D); this NK cell subset has low levels of cytotoxicity and is rare in healthy and normal individuals (Björkström et al., 2010). The frequencies of ILC precursors (ILCPs) and ILC2s in P were similar to those in healthy donors and IL-12Rβ1-deficient patients (Fig. S3E and F). In stringent analyses, ILC1 and ILC3 are too rare for quantification in human peripheral blood (Lim et al., 2017). Thus, human T-bet is required for the correct development or maturation of NK cells, but not monocytes, DCs, ILC2 or ILCP.
Figure 4. Impaired in vivo development of NK, invariant NKT, MAIT, Vδ2+ γδ T, and TH1 cells in T-bet deficiency.

(A) UMAP representation shows immunophenotyping of 40,000 CD45+CD66b−DNA+ live PBMCs from age-matched controls (Age CTL) and M/M, by CyTOF. (B) Frequencies of CD56bright NK cells and CD56dimCD16+ NK cells by CyTOF. (C - E) Percentages of iNKT (C), MAIT (D), and Vδ2+ γδ T cells (E) among live single cells from indicated individuals, including P’s healthy brother (WT/WT), by flow cytometry. (F) viSNE map of 10,000 CD45RA− memory CD4+ T cells from indicated individuals, with expression of CXCR3 and CCR5 shown. (G and H) Frequencies of CCR6− TH1 (CCR6−CXCR3+CCR4−) (G) and CCR6+ TH1* (CCR6+CXCR3+CCR4−) cells (H) among live CD45+CD66b− cells are shown. (I) A bubble graph is presented to show genes for which the proportion of cells displaying expression is altered in the M/M cells in comparison with PBMCs from the TBX21 WT/M father, as a control, by scRNA-seq. The size of the bubble indicates the proportion of cells expressing the gene and the color scale indicates the log2-transformed ratios. The genes highlighted in red were identified as T-bet-regulated by ATAC-seq and/or RNA-seq. LD (low level of detection). ND (not detected). See also Figure S3 - S4 and Supplemental Data S1.
In Fig. 4B – E, G and H, bars represent the mean and the standard error of the mean. Dots represent individual samples or technical replicates. Mann-Whitney nonparametric tests were used for analysis in (C – E). In (C - E), * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, ns = not significant.
Low levels of the iNKT, MAIT, and Vδ2+ γδ T-cell lineages in T-bet deficiency
iNKT, MAIT, and γδ T cells are “innate-like” adaptive T-cell lineages with less T-cell receptor (TCR) diversity than conventional, “purely” adaptive αβ T cells (Chien et al., 2014; Crosby and Kronenberg, 2018; Godfrey et al., 2019). iNKT cells constitute a group of T cells with invariant TCRs combining properties from both T cells and NK cells (Crosby and Kronenberg, 2018). The iNKT cell levels of P were low (present at a level ~27-fold lower than that in controls) (Fig. 4C, Fig. S3G and H). MAIT cells express invariant Vα7.2-Jα33 TCRα restricted by a monomorphic class I-related MHC molecule, along with ligands derived from vitamin B synthesis (Kjer-Nielsen et al., 2012; Xiao and Cai, 2017). P also had a lower frequency of MAIT cells than healthy donors (~15-fold) (Fig. 4D and Fig. S3I). Total γδ T-cell frequency was normal in P. However, the frequency of the Vδ2+ subset, a group of γδ T cells that recognize phosphoantigen (pAgs) (Gu et al., 2018; Harly et al., 2012; Vavassori et al., 2013), was low (~6-fold lower than control levels) in P, whereas the levels of the Vδ1+ subset of γδ T cells seemed to be slightly high (Fig. 4E, Fig. S3J and K). Mild abnormalities of B-cell development and antibody production unrelated to the patient’s mycobacterial disease were observed and will be reported in a separate study (Yang R, in preparation). CD4+ and CD8+ αβ T cells are the two most prevalent blood lineages of adaptive lymphocytes expressing a highly diverse αβ TCR repertoire. Antigen-driven CD8+ T-cell effector responses and the optimal induction of memory CD8+ T cells in mice are controlled by T-bet (Bettelli et al., 2004; Juedes et al., 2004; Sullivan et al., 2003). In the T-bet-deficient patient, total CD8+ T cells and the composition of naïve, central memory, effector memory and TEMRA cells were normal (Fig. S4A). We further investigated CD8+ T cells in an unbiased manner, by automatic viSNE clustering with a panel of surface markers, including chemokine receptors (Amir el et al., 2013). We found no apparent difference between the memory CD8+ T cells of P and controls. However, a small subset of naïve CD8+ T cells (CD45RA+CD38intCXCR3intCCR6−CCR5−CD27highCD127high) was absent from P (Fig. S4B). Thus, the development of iNKT, MAIT cells, Vδ2+ γδ T cells, and a small subset of naïve CD8+ T cells is impaired in T-bet deficiency.
Selective depletion of CCR6− TH1 cells from CD4+ T cells in T-bet deficiency
Both P and his heterozygous parents had normal distributions of naïve and memory CD4+ T cells (Fig. S4C). We further analyzed individual CD4+ T cell subsets by viSNE clustering on antigen-experienced memory cells. Several memory CD4+ T cell subsets typically present in healthy donors were missing in P. In particular, most of the CXCR3+ cells, corresponding to TH1 cells in humans, and CCR5+ cells (Groom and Luster, 2011; Loetscher et al., 1998; Sallusto et al., 1998), were missing in P (Fig. 4F, Fig. S4D and E). The frequency of classic CXCR3+CCR6− TH1 cells was lower than that in all healthy adult and age-matched donors examined, whereas the frequency of CXCR3+CCR6+ non-classic TH1* cells, which are known to be mostly Mycobacterium-specific, was comparable to that of age-matched healthy donors (Fig. 4G and H). The frequencies of human TH2, TH17, and follicular helper (TFH) cells in peripheral blood were normal (Fig. S4F - H). However, levels of CXCR3+ TFH cells, a group of TH1-biased TFH cells that produce IFN-γ together with IL-21 in germinal centers (Ma et al., 2015; Velu et al., 2016; Zhang et al., 2019a), were low in P (Fig. S4H). CXCR3+ regulatory T cells (Tregs), a group of TH1-skewed Tregs (Koch et al., 2009; Levine et al., 2017; Tan et al., 2016) were also present at abnormally low levels, but the proportion of total Tregs was normal (Fig. S4I and J). Thus, human T-bet deficiency selectively impairs development of the classic CXCR3+CCR6− TH1, CXCR3+ TFH, and CXCR3+ Treg CD4+ T-cell subsets, but has no effect on the TFH, TH2, TH17, CCR6+ TH1*, and total Treg subsets, as shown by CyTOF and flow cytometry.
Single-cell transcriptomic profile in vivo is altered by T-bet deficiency
We investigated the development and phenotype of leukocyte subsets in the patient further, by performing single-cell RNA-seq (scRNA-seq) with PBMCs from P and his father. The clustering of the various immune subsets yielded eight distinct major subsets: NK cells, pDCs, monocytes, B cells, CD8+ cytotoxic T lymphocytes (CD8+ CTLs), CD8+ naïve, CD4+ naïve and CD4+ effector/memory T (TEM) cells (Becht et al., 2019). Consistent with the CyTOF results, normal frequencies of pDC, CD8+ CTLs, CD4+ naïve, and TEM cells, were obtained, together with a low frequency of NK cells, on scRNA-seq (Supplemental Data S1). We investigated the transcriptomic changes at single-cell level associated with T-bet deficiency, by filtering to select all genes with expression detected in > 5% of cells in at least one cluster, with at least a four-fold change in expression. We identified 34 genes as differentially regulated in T-bet-deficient cells relative to a heterozygous control. As for the RNA-seq data, some targets with expression known to be dependent on T-bet, including CXCR3 in TEM, CD8+ CTLs, and NK cells, and CCL4 and CCL3 in all cell types, were downregulated in T-bet-deficient cells (Fig. 4I). The expression of XCL1, STAT4, SOX4, LMNA and ANXA1 was also impaired in at least one subset of T-bet-deficient cells (Fig. 4I). In humans and mice, XCL1, STAT4, SOX4, LMNA and ANXA1 are known to be involved in TH1 immunity (Dorner et al., 2002, 2003, 2004; Gavins and Hickey, 2012; Kroczek and Henn, 2012; Nishikomori et al., 2002; Thieu et al., 2008; Toribio-Fernández et al., 2018; Yoshitomi et al., 2018). NKG7 is involved in the initiation of human TH1 commitment and its genetic locus is tightly occupied by T-bet (Jenner et al., 2009; Kanhere et al., 2012; Lund et al., 2005). NKG7 expression in naive CD4+, CD8+ and B cells was dependent on functional T-bet, whereas PRDM1 was downregulated in CD4+ T and CD8+ CTLs cells from P (Fig. 4I). The IFNG gene was weakly expressed across lymphocyte populations, as shown by scRNAseq, and its expression did not seem to be dependent on T-bet in basal conditions (data not shown). In addition, the expression of several genes not previously linked to T-bet was also altered in at least one cell subset in P (Fig. 4I). Thus, in addition to CXCR3, NKG7, CCL3 and CCL4, which were weakly expressed in at least one leukocyte subset from this patient with human T-bet deficiency, consistent with the findings of RNA-seq and omni-ATAC-seq, the expression of a set of previously unknown target genes in immune subsets is also controlled by T-bet (Fig. 4I).
Impaired IFN-γ production by NK, MAIT, Vδ2+ γδ T, and CD4+ T cells
Human IFN-γ is essential for antimycobacterial immunity, as all 31 known genetic etiologies of MSMD affect IFN-γ-dependent immunity. The in vivo development of NK, iNKT, MAIT, Vδ2+ γδ T, and classic TH1 cells was found to be impaired in P, but it remained possible that the IFN-γ production capacity of the remaining lymphocytes could compensate, thereby contributing to antimycobacterial immunity. We assessed the response of P’s PBMCs to ex vivo stimulation with P/I. They secreted significantly less IFN-γ secretion than control cells (Fig. 5A). The frequencies of IFN-γ- or TNF-α-producing total lymphocytes were also low in P (Fig. 5B - D). We assessed the IFN-γ and TNF-α production of lymphocyte subsets by spectral flow cytometry (Fig. S5A). Upon P/I stimulation, the frequencies of IFN-γ- or TNF-α-producing CD56+ NK cells were low in P (Fig. 5E, Fig. S5B and C). We then assessed of the response of P’s NK cells to ex vivo stimulation with IL-12, IL-15, and IL-18. Total NK cells from P displayed impaired degranulation, with low levels of CD107a expression, and almost no IFN-γ production (Fig. 5F, Fig. S5D and E). T-bet deficiency not only reduced the overall frequencies of iNKT, MAIT, and Vδ2+ γδ T cells, but also compromised the production of IFN-γ and TNF-α ex vivo by the residual activated iNKT, MAIT, and Vδ2+ γδ T cells detected in P (Fig. 5G - K, Fig. S5F). Despite an increase in the frequency of Vδ1+ γδ T cells in vivo, the levels of IFN-γ- or TNF-α-producing-Vδ1+ γδ T cells were also low ex vivo (Fig. 5G, Fig. 5L and M). By contrast, we observed no difference in IFN-γ and TNF-α production by Vδ1−Vδ2− γδ T cells between P and controls (Fig. 5G, Fig. S5G and H). IFN-γ-producing cells from the various lymphocyte subsets examined in healthy donors expressed high levels of T-bet, but all the residual IFN-γ-producing cells from the various subsets in P tested negative for T-bet (Fig. 5G). We also analyzed the response of αβ T cells to P/I. CD8+ T cells secrete substantial amounts of IFN-γ upon microbial challenge (Schoenborn and Wilson, 2007). The frequencies of CD8+ T cells producing IFN-γ or TNF-α in response to P/I did not differ between P and age-matched healthy donors (Fig. S5I and J). By contrast, the frequencies of IFN-γ- or TNF-α-producing CD4+ T cells were slightly low in P (Fig. 5N and O). Thus, among the residual circulating lymphocytes of P, NK, iNKT, MAIT, Vδ2+, Vδ1+ γδ T, and CD4+ T cells had impaired IFN-γ production ex vivo in response to stimulation with IL-12, IL-15, IL-18 or P/I, whereas Vδ1−Vδ2− γδ and CD8+ αβ T cells did not.
Figure 5. Impaired IFN-γ ex vivo production from the remaining lymphocytes of the T-bet-deficient patient.

(A) IFN-γ production in culture supernatants of PBMCs from indicated individuals in response to stimulation with P/I is shown. (B) Dot-plots showing the expression of T-bet and IFN-γ in total live lymphocytes as in (A). (C and D) Frequency of IFN-γ (C) or TNF-α-producing (D) cells in response to P/I as in (B). (E) Frequency of IFN-γ-producing cells among NK cells in response to P/I. (F) PBMCs were stimulated with IL-12, IL-15 and IL-18. The expression of CD107a and IFN-γ among NK cells were assessed by flow cytometry. (G) Dot-plots showing the expression of T-bet and IFN-γ by indicated subsets in response to P/I. (H - O) Frequency of IFN-γ- (H, J, L, N) or TNF-α (I, K, M, O)-producing cells among MAIT cells, Vδ2+ γδ T cells, V1+ γδ T cells and CD4+ T cells in response to P/I. (P – U) FACS sorted naïve and memory CD4+ T cells were activated/expanded. The production of IFN-γ was assessed intracellularly (P) and in culture supernatants (Q). The production of TNF-α was assessed intracellularly (R) and in culture supernatants (S). The production of IL-22 (T) and IL-17A (U) was assessed in culture supernatants. (V - Y) Naïve CD4+ T cells were subjected to activation under TH0 or TH1 conditions. The production of IFN-γ (V and W) and TNF-α (X and Y) was assessed. (Z) Memory CD4+ T cells were activated under TH0 or TH1 conditions. The production of IFN-γ and TNF-α was assessed. See also Figure S5.
In Fig. 5A, C – F, H - Z, bars represent the mean and the standard error of the mean. Dots represent individual samples or technical replicates. Mann-Whitney nonparametric tests were used for analysis in (A, C – E, H - O). In (A, C – E, H - O), * p < 0.05, ** p < 0.01, *** p <0.001, **** p < 0.0001, ns = not significant.
Selective impairment of IFN-γ production by TH cells in T-bet deficiency
T-bet was first discovered and has been most extensively studied in CD4+ T cells in the context of mouse TH1 cells (Szabo et al., 2000, 2002). This discovery, together with that of GATA3 (Zheng and Flavell, 1997), revealed the molecular determinism of TH1/TH2 CD4+ T-cell differentiation and paved the way for an understanding of TH17, iTreg, TH22, TFH, and TH9 cell lineage determination (Finotto et al., 2002; Zhu et al., 2010). We therefore investigated the impact of T-bet deficiency on primary CD4+ T cells. IFN-γ production by memory CD4+ T cells was impaired by T-bet deficiency (Fig. 5P and Q). Another TH1 cytokine, TNF-α, was also produced in smaller amounts by memory CD4+ T cells from P than by those of most of the healthy donors (Fig. 5R and S). By contrast, the memory CD4+ T cells of P produced larger amounts of the TH17 effector cytokines IL-17A and IL-22 than did those of healthy donors (Fig. 5T and U, Fig. S5K). Unlike previous studies (Gökmen et al., 2013; Zhang et al., 2019b), we found that T-bet deficiency had no effect on IL-9 production ex vivo (Fig. S5L and M). Surprisingly, ex vivo TH2 cytokines from memory CD4+ T cells were also not affected by human T-bet deficiency (Fig. S5N - Q). P’s memory CD4+ cells produced less IL-21 ex vivo than the memory CD4+ cells of most of the healthy donors (Fig. S5R). We then investigated the role of T-bet in human TH cell differentiation in vitro. Naïve CD4+ T cells from P or healthy donors were allowed to differentiate in TH0, TH1, TH2, TH9, or TH17 conditions. The induction of IFN-γ production in naïve CD4+ T cells was abolished by T-bet deficiency under TH1 conditions (Fig. 5V and W). Similarly, T-bet-deficient naïve CD4+ T cells produced less TNF-α than the corresponding cells from most controls (Fig. 5X and Y). Furthermore, the induction of IL-9 under various conditions in vitro was weaker in naïve CD4+ T cells from P than in the corresponding cells from most controls (Fig. S5S and T). In vitro-induced TH2 cells from P produced more IL-10, but not IL-13, than control cells (Fig. S5U and V). Even memory CD4+ T cells from P displayed impaired IFN-γ and TNF-α production under TH1 polarizing conditions (Fig. 5Z). Thus, AR T-bet deficiency leads not only to defective IFN-γ and TNF-α production ex vivo and in vitro, but also to a moderate upregulation of the production of IL-17A and IL-22, two cytokines characteristic of TH17 cells (Lazarevic et al., 2011).
Poor cellular response to BCG infection in vitro in T-bet deficiency
We investigated the molecular and cellular basis of BCG disease in P, by identifying the leukocyte subsets producing the largest amounts of IFN-γ in a T-bet-dependent manner during acute BCG infection in vitro. The infection of PBMCs with BCG induced IFN-γ production, which was further increased by stimulation with exogenous IL-12 (Fig. 6A). PBMCs from P had low levels of IFN-γ and TNF-α production, but the level of IL-5 production in response to BCG infection was higher than that in most of the controls (Fig. 6A, Fig. S6A and B). Almost all the IFN-γ-producing cells in controls had high levels of T-bet expression (Fig. S6C). Thus, T-bet+ IFN-γ+ double-positive (DP) cells were the major Mycobacterium-responsive cells, with a function potentially dependent on T-bet. However, T-bet+ IFN-γ DP cell levels were low during acute infection in P (Fig. 6B, Fig. S6C). T-bet+ IFN-γ DP cells induced by exogenous IL-12 alone comprised predominantly NK cells, whereas those induced by IL-23 alone also contained a large proportion of MAIT cells, in addition to NK cells (Fig. S6D). Among the T-bet+ IFN-γ+ cells of healthy donors, NK, MAIT, Vδ2+ γδ T, CD4+ and CD8+ αβ T cells were the dominant responders during BCG infection, whereas iNKT, Vδ1+ γδ T, Vδ1−Vδ2− γδ T and B cells were minority responders (Fig. 6C and D). However, these subsets of T-bet+ IFN-γ+ cells were almost entirely depleted from P’s PBMCs following BCG infection (Fig. 6D). We then investigated each leukocyte subset separately (Supplemental Data S1). Fewer than 2% of Vδ1+ γδ T cells, Vδ1−Vδ2− γδ T cells, B cells, CD4+ or CD8+ αβ T cells became T-bet+ IFN-γ+ during BCG infection. However, among innate or innate-like lymphocytes, up to 10% of NK cells, 40% of iNKT, and 50% of Vδ2+ γδ T cells from healthy donors, but not from P, became T-bet+ IFN-γ+ in response to BCG infection, and the frequency of these cells was further increased by exogenous IL-12 (Fig. 6E - G, Fig. S6E and F). Up to 50% of MAIT cells became T-bet+ IFN-γ+ during BCG infection, but no difference was observed between age-matched healthy donors and P (Fig. 6E and Fig. S6E). Upon acute BCG infection in vitro, despite a less robust activation of CD4+ T cells than of innate or innate-like lymphocytes, the frequency of T-bet+ IFN-γ+ cells was low among CD4+ but not among CD8+ αβ T cells of P (Fig. 6H, Fig. S6G and H). Thus, the IFN-γ production controlled by T-bet during acute BCG infection in vitro takes place mostly in NK, iNKT, Vδ2+ γδ T and CD4+ αβ T cells, but not in Vδ1+ γδ T cells, Vδ1−Vδ2− γδ T cells, or CD8+ αβ T cells. The development of MAIT cells is also dependent on T-bet, whereas the IFN-γ production of these cells during BCG infection is not. These experimental findings in vitro do not exclude a contribution of other subsets in vivo. NK, iNKT, MAIT, and Vδ2+ γδ T cells responded robustly to acute BCG infection in vitro, but these subsets were absent or functionally deficient in the patient with inherited T-bet deficiency.
Figure 6. T-bet deficiency leads to defective NK, iNKT and Vδ2+ γδT cells, resulting in susceptibility to mycobacteria.

(A) IFN-γ secretion by PBMCs from indicated individuals stimulated with and without live M. bovis BCG in the presence and absence of IL-12 and IL-23. (B) Percentages of T-bet+ IFN-γ+ double-positive (DP) cells in the indicated samples, cultured as in (A). (C) Percentages of indicated immune subsets among the DP cells of both CTL and Age CTL individuals. (D) viSNE plots showing different clusters of immune cells among DP cells. (E) Percentages of DP cells within each immune subset, as indicated. (F and G) Dot-plots showing the expression of T-bet and IFN-γ in NK (F) and Vδ2+ γδ T cells (G), cultured as in (A). (H) Percentages of DP cells within each indicated adaptive immune subset. (I and J) PBMCs from indicated individuals were stimulated with lysates of M. bovis BCG (BCG-lysates) for 14 days. Dot plots of Vγ9δ2 γδT cells (I) and the total number of Vγ9δ2 γδT cells (J) are shown. (K) The level of IFN-γ in culture supernatants from (J) is shown. (L - N) The intracellular IFN-γ production of antigen-specific CD4+ or CD8+ T cells expanded from PPD or BCG-lysates was measured. The percentages of IFN-γ-producing cells among PPD-responsive CD4+ T (L), BCG-lysate-responsive CD4+ (M) and CD8+ T (N) cells are shown. (O) Memory CCR6+CD4+ T-cell clones responding to the peptide pool from M. bovis BCG were identified. IFN-γ production in culture supernatants from these BCG-specific clones was shown. See also Figure S6, Table S4, and Supplemental Data S1.
In Fig. 6A, B, E, H, K, L, M and N, bars represent the mean and the standard error of the mean. Dots represent individual samples and technical replicates. In (O), bars represent the mean and the standard deviation. Dots represent individual T-cell clones. In (C), the numbers indicate the mean and the standard error of the mean. Mann-Whitney nonparametric tests were used for analysis in (A, B, E and H). In (A, B, E and H), * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, and ns = not significant.
Defective prolonged anti-BCG immunity mediated by Vδ2+ γδ T cells
The stimulation of PBMCs in vitro with live BCG involves both antigens specifically recognized by Mycobacterium-specific cognate αβ and γδ T cells and many other stimuli. BCG infection in vitro mimics acute infection in vivo, but may not be robust enough for investigations of the antigen-specific adaptive immune response, particularly as concerns prolonged adaptive immunity. We therefore studied Vδ2+ γδ T cells and CD4+ αβ T cells, two adaptive immune lymphocyte subsets that produced substantial amounts of IFN-γ during BCG infection in vitro and are known to function in an antigen-specific manner. In PBMCs from healthy donors, Vδ2+ γδ T cells responded most robustly during BCG infection (Fig. 6E). Vδ2+ γδ T cells, a major subset of γδ T cells recognizing pAg from microbial sources (Gu et al., 2018; Harly et al., 2012; Vavassori et al., 2013), are also known to play an essential role in the recall response to mycobacterial re-infection in humans and non-human primates (Chen, 2005; Shen et al., 2002). Vδ2+ γδ T cells proliferate vigorously in response to mycobacterial infection in vivo and can expand robustly in response to pAg-rich lysates of mycobacterial species in vitro (Hoft et al., 1998; Modlin et al., 1989; Panchamoorthy et al., 1991; Parker et al., 1990; Tsukaguchi et al., 1995). We investigated whether the function of Vδ2+ γδ T cells was affected in the patient with T-bet deficiency, as these cells represented a small, but important proportion (~ 0.2%) of P’s peripheral lymphocytes. The populations of Vδ2+ T cells from all controls and relatives of P expanded vigorously following prolonged stimulation with BCG lysates. By contrast, no such expansion was observed for T-bet-deficient Vδ2+ T cells (Fig. 6I and J, Fig. S6I). After two weeks of expansion, the levels of IFN-γ production by T-bet-deficient cells were lower than those for healthy donors’ cells (Fig. 6K).
Redundant role of T-bet in IFN-γ production by BCG-specific cognate TH1* cells
IFN-γ production by CD4+ αβ T cells upon BCG infection ex vivo was subtly affected by T-bet deficiency, but it remains unclear whether there is a difference in the mycobacterial antigen-specific CD4+ αβ T-cell response. It also remains unclear whether the memory adaptive immunity to mycobacteria elicited by antigen-specific CD4+ T cells is dependent on T-bet. We addressed both these unresolved issues by analyzing Mycobacterium-responsive CD4+ T cells expanded through prolonged stimulation with BCG-lysates or tuberculin purified protein derivative (PPD) (Supplemental Data S1). Like those of BCG-vaccinated healthy donors, P’s CD4+ T cells proliferated equally well in response to both BCG-lysates and PPD (Supplemental Data S1). No difference in IFN-γ production from PPD-responsive CD4+ T cells was observed between healthy donors and P (Fig. 6L and Fig. S6J). Unlike PPD, which activated only CD4+ T cells, BCG-lysates activated both CD4+ T cells and CD8+ T cells, albeit to a lesser extent (Supplemental Data S1). Like PPD stimulation, IFN-γ production from both BCG-lysate-responsive CD4+ and CD8+ T cells was similar between controls and P (Fig. 6M and N, Fig. S6K and L). We confirmed these findings by screening antigen-reactive T-cell libraries established from the CD4+CCR6− (containing classic TH1 cells) and CD4+CCR6+ (containing TH1* Mycobacterium-responsive cells) memory subsets (Geiger et al., 2009). Consistent with our in vivo findings, the T cells in the CD4+CCR6− and CD4+CCR6+ libraries had low levels of CXCR3 or IFN-γ (Supplemental Data S1). P’s CD4+CCR6+ T-cell library responded robustly to BCG, tetanus toxoid and C. albicans, and his CD4+CCR6− T-cell library responded normally to influenza virus, CMV, and EBV. Despite intact proliferation, IFN-γ production from T-bet-deficient T cells responding to influenza virus, EBV, tetanus toxoid, and C. albicans was weak. However, P’s CCR6+ T cells, consisting almost entirely of Mycobacterium-specific memory TH1* cells (Acosta-Rodriguez et al., 2007; Becattini et al., 2015), proliferated robustly in response to BCG peptides. Moreover, their IFN-γ production was normal, and their levels of IL-10 production were slightly higher (Fig. 6O and Supplemental Data S1). The normal levels of IFN-γ production could not be attributed to cells with a revertant genotype, as reported in other T-cell primary immunodeficiency diseases (PIDs) (Davis et al., 2008; Revy et al., 2019), because the IFN-γ+ BCG-specific T-cell clones still carried the TBX21 indel variant. Thus, the prolonged immunity to BCG infection mediated by Vδ2+ γδ T and memory αβ T cells was divergently controlled by T-bet, as T-bet was required for the generation of long-term immunity due to Vδ2+ γδ T cells, but redundant for IFN-γ production by BCG-specific cognate CD4+ TH1* cells or Mycobacterium-responsive CD8+ αβ T cells (Table S4).
Discussion
We report here the identification and study of the first patient discovered to have inherited, complete T-bet deficiency resulting clinically in MSMD. Key observations made in T-bet-deficient mice were validated in this human patient with T-bet deficiency: 1) the development of TH1 cells and their production of effector cytokines, including IFN-γ in particular, requires T-bet (Szabo et al., 2000, 2002); 2) the development of NK and iNKT cells is dependent on T-bet (Townsend et al., 2004); and 3) the regulation of T-bet-dependent targets, including CXCR3, TNF and IFNG, involves both direct transactivation and epigenetic modulation (Miller and Weinmann, 2010). Accordingly, T-bet-deficient mice are highly vulnerable to mycobacteria, including M. tuberculosis and M. avium (Matsuyama et al., 2014; Sullivan et al., 2005), like mice deficient for other genes that govern IFN-γ immunity (Casanova, 1999). By contrast, despite the requirement of T-bet for immunity to a broad spectrum of pathogens following experimental inoculation in mice (Bakshi et al., 2006; Harms Pritchard et al., 2015; Hultgren et al., 2004; Kao et al., 2011; Lebrun et al., 2015; Oakley et al., 2013; Ravindran et al., 2005; Rosas et al., 2006; Svensson et al., 2005; Szabo et al., 2002), the only apparent infectious phenotype of the T-bet-deficient patient is MSMD. Our study provides compelling evidence that inherited T-bet deficiency is a genetic etiology of MSMD due to the disruption of IFN-γ immunity (Casanova et al., 2014). This experiment of nature suggests that T-bet is required for protective immunity to intramacrophagic mycobacteria but largely redundant for immunity to most intracellular pathogens, including viruses in particular. This is at odds with findings in mice, but consistent with other genetic etiologies of MSMD, all of which are inborn errors of IFN-γ immunity (Bustamante, 2020; Kerner et al., 2020). Our observation further suggests that the functions of T-bet unrelated to IFN-γ are redundant in humans. The identification of additional T-bet-deficient patients is required to draw firm conclusions. However, it is striking that humans genetically deprived of key immunological molecules other than T-bet or IFN-γ often show a much greater redundancy than the corresponding mutant mice (Casanova and Abel, 2004, 2018).
Our study also suggests unexpected immunological abnormalities not documented in T-bet-deficient mice that also contribute to the development of MSMD: 1) T-bet was apparently required for the optimal development of two innate-like adaptive lineages of immune cells, MAIT and Vδ2+ γδ T cells; 2) T-bet was also apparently required for the optimal production of IFN-γ by the few NK, iNKT, MAIT, and Vδ2+ γδ T lymphocytes that were able to develop (Bettelli et al., 2004; Daussy et al., 2014; Garrido-Mesa et al., 2019; Intlekofer et al., 2007; Lugo-Villarino et al., 2003; Pearce et al., 2003; Rankin et al., 2013; Sullivan et al., 2003; Tang et al., 2010). Unexpectedly, IFN-γ production by cognate purely adaptive Mycobacterium-specific TH1* CD4+ T cells, and by PPD-, or BCG-lysate-responsive CD4+ or CD8+ αβ T cells, was unaffected by T-bet deficiency. The compensatory mechanisms underlying intact IFN-γ production by these antigen-specific CD4+ T cells remain to be discovered. Taken together, impaired IFN-γ production by NK and iNKT cells, as in mice, and by MAIT and Vδ2+ γδ T cells, as shown here, accounts for MSMD in this patient with T-bet deficiency, despite normal TH1* development and function. By contrast, inborn errors of immunity that disrupt IFN-γ production by the selective depletion of NK, iNKT, CD4+, or CD8+ αβ T cells do not underlie mycobacterial disease, because of the compensation provided by other subsets. Conversely, the loss of all T-cell subsets in SCID patients does result in predisposition to mycobacterial disease. Interestingly, a different combination of deficits accounts for MSMD in patients with RORγT deficiency, who lack iNKT and MAIT cells and whose γδ T and TH1* cells do not produce IFN-γ, and whose NK cells are unable to compensate (Okada et al., 2015). T-bet and RORγT deficiencies are characterized by iNKT, MAIT, and γδ T-cell deficiencies, whereas an NK deficit is observed only in T-bet deficiency and a deficit of TH1* cells is observed only in RORγT deficiency. We found that human T-bet was essential for both innate (NK cells) and innate-like (iNKT, MAIT, and Vδ2+ γδ T cells) adaptive immunity to mycobacteria, but, surprisingly, redundant for classical, purely adaptive immunity (CD4+ TH1* and CD8+ αβ T cells) to mycobacteria.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jean-Laurent Casanova (casanova@mail.rockefeller.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement with Inserm or the Rockefeller University.
Data and Code Availability
The whole-exome sequencing, RNA-seq and omni-ATAC-seq datasets generated during this study are available at the Sequence Read Archive (SRA) (PRJNA641463). The scRNA-seq datasets generated during this study are available at the EGA European Genome-Phenome Archive (EGAS00001004504). This study did not generate any unique code. Any other piece of data will be available upon reasonable request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human subjects
The patient and the relatives studied here were living in and followed up in Morocco. The study was approved by and performed in accordance with the requirements of the institutional ethics committee of Necker Hospital for Sick Children, Paris, France, and the Rockefeller University, New York, USA. Informed consent was obtained for the patient, his relatives, and healthy control volunteers enrolled in the study. This study was also approved by the Sydney Local Health District RPAH Zone Human Research Ethics Committee and Research Governance Office, Royal Prince Alfred Hospital, Camperdown, New South Wales, Australia (protocol X16-0210/LNR/16/RPAH/257). Written informed consent was obtained from participants or their guardians. Informed consent from participants in Switzerland was approved by the local ethical committee (CE3428 authorized by Comitato Etico Cantonale, www.ti.ch/CE). Experiments using samples from human subjects were conducted in the United States, France, Australia and Switzerland, in accordance with local regulations and with the approval of the IRBs of corresponding institutions. Plasma samples from unrelated healthy subjects used as controls for antibody profiling by phage immunoprecipitation-sequencing (PhIP-Seq) were collected at Sidra Medicine in accordance with a study protocol approved by the Clinical Research Ethics Board of Sidra Medicine.
Case report of the patient (P) - P was born in 2015 to Moroccan first-cousin parents. He was vaccinated with BCG at the age of three months. After vaccination, he developed fever and left axillary lymphadenopathy accompanied by a cutaneous eruption. He received amoxicillin for 10 days, resulting in clinical improvement. At the age of six months, he was hospitalized for persistent fever, weight loss, cutaneous erythema, ear drainage and persistent oral thrush. Axillary adenopathy was present, with fistulation associated with hepatosplenomegaly and multiple abdominal adenopathies. The patient was treated with four antimycobacterial drugs; he displayed a good clinical response to 18 months of treatment and has been off antibiotics for 15 months. He was also found to have mild cytomegalovirus (CMV) viremia at the time (CMV PCR 3.74 log/mL, threshold < 2.4 log/mL), and was treated with ganciclovir. Serological PCR tests for CMV were negative at the age of two years, but weakly positive at the age of three years (IgM anti-CMV weakly positive, CMV PCR 3.62 log/mL, threshold < 2.4 log/mL). However, the patient showed no characteristic features of CMV infection and did not present clinically confirmed CMV disease. At the age of 11 months, he was hospitalized for four days due to airway hyperresponsiveness requiring treatment with inhaled steroids and albuterol. The patient had a high blood eosinophil count, documented since the age of since months (1,460/mm3). By the age of 17 months, he had persistent upper respiratory tract inflammation, with manifestations of persistent rhinorrhea, difficult respiration, wheezing and coughing that improved on treatment with oral steroid and salbutamol. The patient has since been treated with inhaled steroids for persistent upper respiratory tract inflammation. The clinical manifestations affecting the upper respiratory tract persisted at the age of three years. No trigger was identified for these symptoms and signs, which were not triggered by exercise, activity, feeding or parasitic infections. A serum IgE allergen screen was negative for more than 250 allergens tested. The patient requires inhaled steroid therapy (fluticasone spray) every morning and evening. He also frequently visits the emergency room, and is hospitalized about once per month for exacerbations requiring oral steroid (beta-methasone) and inhaled salbutamol. The patient has been vaccinated for Tetanus, Diphtheria, and Haemophilus B, for all of which positive serum IgG result have been obtained. He also tested positive for anti-HSV-1 IgG, anti-EBV IgG, anti-CMV IgG, anti-mumps IgG and anti-parainfluenza IgG in clinical serology examinations. No anatomical abnormalities of the airways were noted. The patient has a brother who is healthy, with no history of severe infectious disease or airway hyperresponsiveness. The genetic cause of the patient’s airway hyperresponsiveness will be studied separately.
Cell lines
HEK293T and Phoenix A retroviral packaging cells were cultured in IMDM (Gibco) supplemented with 10% fetal bovine serum (FBS). B cells from the patient or controls were immortalized in-house with Epstein Barr virus (EBV-B cells) and cultured in RPMI 1640 (Gibco) supplemented with 10% FBS. NK-92 cells were cultured in RPMI 1640 (Gibco) supplemented with 10% FBS, 1 mM sodium pyruvate, 200 U/mL recombinant proleukin IL-2 (PROMETHEUS). Herpesvirus saimiri-transformed T (HVS-T) cells were generated with either H. saimiri strain C488 for transformation or the TERT transformation system (Wang et al., 2016). HVS-T cells were cultured in Panserin/RPMI 1640 (ratio 1:1) supplemented with 20% FBS, L-glutamine, gentamycin and 20 U/mL human rIL-2 (Roche). Isolated CD4+ T cells were cultured in X-vivo 15/gentamycin/L-glutamine (Lonza) supplemented with human AB Serum (GemCell). Peripheral blood mononuclear cells (PBMCs) were cultured in RPMI 1640 supplemented with 10% FBS for stimulation experiments. All cell lines used tested negative for mycoplasma. HEK293T, Phoenix A and NK-92 cells were purchased from the ATCC.
METHOD DETAILS
Phage immunoprecipitation-sequencing (PhIP-Seq)
Patient serum and control samples were analyzed in duplicate by phage immunoprecipitation-sequencing (Hernandez et al., 2018; Xu et al., 2015). We used 10% liquid IVIg from pooled human plasma (Privigen® CSL Behring AG), human IgG-depleted serum (Supplier No HPLASERGFA5ML, Molecular Innovations, Inc.), and plasma samples from two unrelated healthy adult subjects and one unrelated healthy three-year-old boy for comparison. PhIP-Seq was carried out as previously described (Xu et al., 2015), but with the following modifications. We determined levels of total IgG in the serum or plasma samples with the Human IgG total ELISA Ready-SET-Go kit (Thermo Fisher Scientific) and incubated diluted samples containing approximately 4 μg of total IgG at 4°C overnight with 2 × 1010 plaque-forming units (PFUs) of a modified version of the original VirScan phage library (Xu et al., 2015). Specifically, the T7 phage library used here for peptide display contained the same viral peptides as the original VirScan phage library plus additional peptides derived from protein sequences of various microbial B-cell antigens available from the IEDB (www.iedb.org). For the computational analysis and background correction, we also sequenced the phage library before (input library sample) and after immunoprecipitation with beads alone (mock IP). We performed a number (n = 46) pf technical repeats. Single-end sequencing was performed with the NextSeq500 system (Illumina), to generate approximately two million reads per sample and ~20 million reads for the input library samples. As previously described (Xu et al., 2015), reads were mapped onto the original library sequences with Bowtie 2 and read counts were adjusted according to library size. A zero-inflated generalized Poisson model was used to estimate the p-values, to reflect enrichment for each of the peptides. We considered peptides to be significantly enriched only if the −log10 p-value was at least 2.3 in all replicates. Species-specific score values were computed for each serum or plasma sample, by counting the significantly enriched peptides for a given species with less than a continuous seven-residue subsequence, the estimated size of a linear epitope, in common. We corrected for unspecific binding of peptides to the capture matrix, by also calculating species-specific background score values by counting the peptides displaying enrichment to the 90th percentile of the mock IP samples. These peptides were used for background subtraction.
Whole-exome sequencing (WES), genotyping and genome-wide linkage (GWL) analysis
All four members of the family tested were subjected to genomic DNA extraction followed by WES. Exome capture was performed with the SureSelect Human All Exon V4+UTR and SureSelect Human All Exon V6 (Agilent Technologies). Paired-end sequencing was performed on a HiSeq 2000 (Illumina), generating 100-base reads. We used the Genome Analysis Software Kit (GATK) (version 3.4–46) best-practice pipeline to analyze our WES data (Li et al., 2009; McKenna et al., 2010). Reads were aligned with the human reference genome hg19, with BWA (Li and Durbin, 2009). PCR duplicates were removed with Picard tools (picard.sourceforge.net/). The GATK base quality score recalibrator was applied to correct sequencing artifacts. The GATK HaplotypeCaller was used to identify variant calls (Depristo et al., 2011; McKenna et al., 2010) Variants were annotated with SnpEff (snpeff. sourceforge.net/) (Cingolani et al., 2012).
All four members of the family were genotyped with Genome-Wide Human SNP Array 6.0 data. Genotype calling was achieved with the Affymetrix Power Tools Software Package (https://www.affymetrix.com/support/developer/powertools/changelog/index.html). Parametric multipoint linkage analysis was performed with MERLIN 1.1.2 software (Abecasis et al., 2002), assuming autosomal recessive inheritance with complete penetrance and a damaging allele frequency of 1 x 10−4. Allele frequencies were estimated for 562,685 SNP markers, with family founders and HapMap CEU trios. Markers were clustered using an r2 threshold (--rsq parameter) of 0.4. The genetic variant of interest was confirmed by PCR amplification of the region (forward primer: 5’- gtgaggactacgcgctacc-3’, and reverse primer: 5’-cagaagcattgtcgagccag-3’) followed by Sanger sequencing.
cDNA sequencing
We used cDNA sequencing to characterize the consequences of the patients’ variant. In brief, total RNA was extracted from EBV-B cells or PBMCs from the patient and healthy donors (Qiagen). We synthesized cDNA from mRNA, using Superscript III (Thermo Fisher Scientific). A region spanning the mutation site was amplified by PCR with the forward (5’- tagaagccaggcgtcagagc-3’) and reverse R4 (5’- ctcggcattctggtaggcag-3’), R5 (5’- gcaatgaactgggtttcttgg-3’), or R6 (5’- gactcaaagttctcccggaatcc-3’) primers. PCR amplicons were sequenced with the BigDye Terminator Cycle Sequencing Kit (Thermo Fisher Scientific). Alternatively, cDNA generated from patient or control PBMCs was amplified by PCR with the forward primer 5’- tctacactctttccctacacgacgctcttccgatctgtgaggactacgcgctacc-3’ and reverse primer 5’- gtgactggagttcagacgtgtgctcttccgatctgcaatgaactgggtttcttgg-3’, to amplify a region spanning the mutation in a first round of PCR. A second round of PCR was then performed on the PCR products from the first PCR as a template. We used the forward primer 5’- aatgatacggcgaccaccgagatctacactctttccctacacgac -3’ and the reverse primers 5’- caagcagaagacggcatacgagatcgtattcggtgactggagttcagacgtgtg -3’, 5’-caagcagaagacggcatacgagatctcctagagtgactggagttcagacgtgtg -3’, and 5’- caagcagaagacggcatacgagattagttgcggtgactggagttcagacgtgtg -3’, to barcode two healthy control cDNAs and the patient’s cDNA. Adaptor and barcoded PCR amplicons were mixed and purified, and used for next-generation sequencing by Nano Format 350 paired-end MiSeq sequencing (illumina). We aligned MiSeq data to the reference genome with RNA-STAR aligner (Afgan et al., 2016; Dobin et al., 2013). The aligned sequencing data were then viewed with Integrative Genome Browser (Robinson et al., 2011).
Reverse transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted from cells with the RNeasy Kit (Qiagen) or the Quick-RNA kit (Zymo Research). Reverse transcription was performed with the Superscript III enzyme (Thermo Fisher Scientific). Messenger RNAs were quantified with the cDNA as a template and the TBX21 (Probe 1: Hs00894393 and Probe 2: Hs00894391), IFNG (Hs99999041), TNF (Hs01113624), IL5 (Hs01548712) (Thermo Fisher), GUSB (Thermo Fisher) probes.
T-bet/TBX21 overexpression
A wild-type (WT) TBX21 plasmid with a pCMV6 backbone was purchased from Origene (RC207902). The genetic variants of TBX21 studied here, and the variants described in gnomAD (http://gnomad.broadinstitute.org/) or in our in-house database were introduced into pCMV6-WT TBX21 by site-directed mutagenesis PCR with the CloneAmp HiFi PCR Premix (Takara Bio). TBX21 WT plasmids or plasmids containing variants were used to transfect HEK293T cells in the presence of Lipofectamine 2000 reagent (Thermo Fisher Scientific) to achieve overexpression. The WT or mutant (Mut) TBX21 was overexpressed in in vitro-expanded T cells, HVS-T cells, or NK-92 cells, as previously described (Martínez-Barricarte et al., 2016). In brief, the TBX21 plasmid was subcloned into pLZRSire-ΔNGFR (Addgene) (Van De Vosse et al., 2009). We then used pLZRS containing the WT or Mut TBX21 to transfect Phoenix A cells, and retroviruses were produced and concentrated with Retro-X (Takara Bio). Concentrated retrovirus preparations were used for retroviral transduction.
Immunoblotting
Proteins were solubilized in RIPA buffer containing 10 mM Tris-Cl (pH 8.0), 1 mM EDTA, 0.5 mM EGTA, 1% Triton X100, 0.1% sodium deoxycholate, 0.1% SDS and 150 mM NaCl supplemented with protease-inhibitor cocktail (Complete mini, Roche). Alternatively, nuclear proteins from HEK293T transfectants were purified with the NE-PER Nuclear Extraction kit in accordance with the manufacturer’s procedures (Thermo Fisher Scientific). Proteins were quantified with the Bradford assay. Equal amounts of lysate (5 μg to 50 μg of) were mixed with SDS sample buffer and boiled for 5min at 95°C before being subjected to electrophoresis in 12% acrylamide SDS-PAGE gels. The following antibodies were used for immunoblotting: mouse anti-human T-bet 4B10 monoclonal Ab (BioxCell), rabbit anti-human T-bet polyclonal Ab (Proteintech), anti-Flag-M2 HRP-conjugated Ab (Sigma-Aldrich), anti-α-tubulin HRP-conjugated Ab (Proteintech), anti-lamin A/C Ab (Santa Cruz), anti-GAPDH HRP-conjugated Ab (Santa Cruz), anti-STAT1 monoclonal Ab (BD Biosciences), sheep ECL anti-mouse IgG HRP-conjugated Ab (GE Healthcare), and ECL anti-rabbit IgG HRP-conjugated Ab (GE Healthcare).
Electrophoretic mobility shift assays (EMSA)
HEK293T cells were transfected with empty plasmid (EV), WT or mutant TBX21 pCMV6 plasmids. After 2 days, nuclear extracts were obtained from the cells with the NE-PER Nuclear Extraction Kit (Thermo Fisher Scientific). EMSA was performed as previously described (Liu et al., 2011). In brief, 32P was used to label consensus T-box responsive element (TBRE) duplexes of DNA oligos (WT oligo: 5’-gataatttcacacctaggtgtgaaatt-3’, Mut Oligo as control: 5’-gataatttcacgtctaggtacgaaatt-3’ and 5’-gataatttcgtacctagacgtgaaatt-3’). Unlabeled WT TBRE duplex was used as non-radioactive competitive probe. We mixed 10 μg of nuclear extract from each condition or 80 μg (8x) of nuclear extract from Mut TBX21 transfectants with or without unlabeled probe, 2 μg mouse IgG1 control (BioxCell) or 2 μg mouse anti-T-bet 4B10 monoclonal Ab (BioxCell) and incubated for 30 min. 32P-labeled WT or Mut TBRE duplex was then added and the mixture was incubated for 30 min on ice in the presence of poly-dIdC (Sigma-Aldrich). Samples were subjected to polyacrylamide gel electrophoresis. The gel was dried and placed against X-ray film for 2 days, after which the radioactive signal was read on a Typhoon platform.
Luciferase reporter assays
HEK293T cells were transfected with the indicated expression plasmids, firefly luciferase plasmids under the control of WT or Mut TBRE (Janesick et al., 2012), as previously described, or with human IFNG promoters in a pGL4.10 backbone (−565 - +85 of IFNG), and with a constitutively expressed Renilla luciferase plasmid for normalization (pRL-SV40). Cells were transfected in the presence of Lipofectamine 2000 (Thermo Fisher Scientific) for 3 days. Luciferase levels were measured with Dual-Glo reagent, according to the manufacturer’s protocol (Promega). Firefly luciferase values were normalized against Renilla luciferase values, and fold-induction is reported relative to controls transfect with empty plasmid.
Generation and analysis of T-bet-deficient NK-92 cells
T-bet-deficient NK-92 cells were generated according to a protocol described elsewhere (Sanjana et al., 2014). We used oligonucleotides for gRNA9 and gRNA15 (gRNA9_Forward: 5’-caccgtccaacaatgtgacccaggt-3’, gRNA_Reverse: 5’-aaacacctgggtcacattgttggac-3’; gRNA15_Forward: 5’-caccgccgcactcaccgtccctgct-3’, gRNA15_reverse: 5’-aaacagcagggacggtgagtgcggc-3’). DNA duplex was annealed from the oligonucleotides above, and was inserted into pLenti-CRISPR-V2 plasmids (Addgene) (Sanjana et al., 2014). We then used pLenti-CRISPR plasmids, VSV-G envelope, and psPAX2 plasmids to transfect HEK293T cells in the presence of Lipofectamine 2000. Supernatants containing lentiviruses were harvested and concentrated with Lenti-X concentrator (Takara Bio). We resuspended 106 NK-92 cells with 20x concentrated lentiviruses for gRNA9, gRNA15 or empty plasmid, in the presence of proleukin IL-2 (200 U/mL) (PROMETHEUS). After incubation for 2 days, we added 3.75 μg/mL puromycin to the culture for the selection of stably transduced NK-92 cells. After a further two weeks, cells were harvested for the Surveyor Nuclease Assay (Integrated DNA Tech) and a three-primer PCR-based screening system (Harayama and Riezman, 2017) to confirm successful DNA editing. Cells were plated at a density of 0.7 cells in 200μL per well in a U-bottomed 96-well plate. Two weeks later, single-cell clones were screened with a three-primer PCR-based system, with Phire Direct PCR Master Mix (Thermo Fisher Scientific). The primers used for this screening assay were gRNA9_Forward: 5’- agcccctacccctaattcct-3’, gRNA9_Reverse: 5’-ggcatctattccctgggacc-4’, gRNA9_Reverse_in: 5’-cttttgaagagcaggtcctacct-3’ and gRNA15_Forward: 5’-GCCTGAATATGACCCCCGTC-3’, gRNA15_Reverse: 5’-caccactaccaccactaaagc-3’; gRNA15_Forward_in: 5’-acagagatgatcatcaccaagca-3’. Single-cell clones with biallelic disruptions were selected for validation by RT-qPCR and western blotting. EV-transduced single-cell clones were randomly picked as controls. Cells were then transduced with retroviruses generated from pLZRS-ires-ΔD271 plasmids containing EV, WT, Mut or K314R TBX21, as described above (Martínez-Barricarte et al., 2016). Stably transduced cells were isolated with anti-CD271 antibody-coated beads (Miltenyi Biotec). Cells were stimulated with 50 pg/mL IL-12 (R&D) and 10 ng/mL IL-18 (InvivoGen), or were left unstimulated, and all cells were incubated for one day. The IFN-γ content of the supernatants was determined by ELISA. Intracellular IFN-γ production was measured with flow cytometry with intracellular staining (ICS). In brief, cells were fixed and permeabilized (BioLegend), and were subjected to ICS staining with FcBlock (Miltenyi Biotec), anti-T-bet BV421 (BioLegend) and anti-IFN-γ PE (BioLegend) before their use for ICS flow cytometry analysis.
Cytokine determinations in TBX21-transduced primary CD4+ T cells
PMBCs from three healthy donors were stained with FcBlock (Miltenyi Biotec), anti-CD4-APC-Cy7, anti-CD45RA-Alexa488, anti-CCR7-Alexa647 and Live-Dead exclusion Aqua. Naïve CD4+ T cells were isolated by FACS. Cells were stimulated with anti-CD3/CD28 Ab Dynabeads (Thermo Fisher Scientific) at a 1:1 cell:bead ratio under TH0 or TH2 (recombinant human IL-4 12.5 ng/mL, R&D) polarizing conditions, in the presence of 100 U/mL proleukin IL-2 (PROMETHEUS). Fresh medium (X-vivo 15, 5% human AB serum) and cytokines were added every 3 days. After 7 days, cells were restimulated with Dynabeads at a 1:2 ratio (bead:cell). One day after restimulation, cells were transduced with retroviruses generated from pLZRS-ires-ΔCD271 plasmids containing WT or Mut TBX21, as previously described (Martínez-Barricarte et al., 2016). Transduced cells were isolated with anti-CD271 antibody-coated beads (Miltenyi Biotec) 14 days after stimulation. Intracellular cytokine production was determined by ICS. In brief, cells were restimulated with 25 ng/mL phorbol 12-myristate 13-acetate (PMA) and 0.5 μM ionomycin for 6 h in the presence of GolgiPlug (BD Bioscences). Cells were stained with Zombie-NIR live-dead exclusion dye (BioLegend), FcBlock (Miltenyi Biotec), and FITC-conjugated anti-CD271 antibody (BioLegend). Cells were then fixed, permeabilized and subjected to ICS staining with anti-TNF-α-BV510 (BioLegend), anti-IFN-γ-PERCP/Cy5.5 (BioLegend), anti-IL-4-PE, anti-IL-13-APC antibodies, followed by flow cytometry analysis with an Aurora cytometer (Cytek).
HVS-T cell analysis
We plated 200,000 HVS-T cells from healthy donors or P in a 96-well U-bottomed plate at a density of 1x106 cells/mL. The plate was incubated for 2 days, and the culture supernatants and cells were harvested for ELISA or RT-qPCR for IFNG and TNF. HVS-T cells from P were transduced with retroviruses generated from pLZRS-ires-ΔCD271 plasmids containing WT TBX21 or EV, as previously described (Martínez-Barricarte et al., 2016). After incubation for 5 days, the HVS-T cells were stimulated with 25 ng/mL PMA and 500 nM ionomycin or were left untreated; they were then incubated in the presence of GolgiPlug (BD Biosciences) for 6-8 h. Cells were stained with Zombie-NIR live-dead dye(BioLegend), FcBlock (Miltenyi Biotec), anti-CD4-BV750 (BD Biosciences), anti-CD3-BV650 (BD Biosciences), anti-CD8a-Pacific Blue (BioLegend), and anti-CD271-FITC (BioLegend) antibodies, fixed and permeabilized for ICS staining with anti-T-bet-PE (BioLegend), anti-IFN-γ-BV711 (BioLegend), anti-TNF-α-BV510 (BioLegend), anti-IL-13-APC (BioLegend), anti-IL-10-PE-Dazzle594 (BioLegend) antibodies. ICS analysis was performed on an Aurora cytometer (Cytek).
HVS-T cells were also analyzed for IFN-γ production in response to PSMD9, ARHGAP27 and ERN1 cDNA overexpression. In brief, pTRIP-mCherry-2A-PSMD9 and ARHGAP27 were generated by subcloning from MGC clones BC004184 and BC067345 (transOMIC). A C-terminal V5 tag was added upstream from the stop codons. The pLenti-Cter-GFP-2A-puro empty vector plasmid was obtained from OriGene. The pCMV6-ERN1-Cter-Myc-Flag plasmid was obtained from OriGene. pLenti-ERN1-Cter-GFP (fusion)-2A-puro was generated by the double digestion, with EcoRI and XhoI, of pCMV6-ERN1 and pLenti-Cter-GFP-2A-puro, followed by the isolation of the ERN1 fragment by gel extraction, and T4 ligation. Lentiviruses containing cDNAs encoding EV, PSMD9, ARHGAP27, ERN1-GFP were generated in HEK293T cells by transfection with pTRIP or pLenti-GFP-2A-puro, VSV.G and Pax2 packaging plasmids. Transfected HEK293T cells were analyzed for the expression of PSMD9 and ARHGAP27 by assessing the expression of the C-terminal V5 tag and ERN1 by western blotting with anti-GFP Ab or anti-ERN1 Ab (Proteintech). Lentiviruses containing the cDNAs of EV, PSMD9, ARHGAP27 and ERN1-GFP were used to transduce HVS-T cells derived from the cells of P (M/M), or M/M HVS-T cells retrovirally transduced with WT T-bet (M/M+WT). After 2 – 4 days of transduction, cells were stimulated with 100 ng/mL PMA and 2 μM ionomycin for 8 h and harvested for staining for flow cytometry. ERN1-transduced HVS-T cells and their control HVS-T cells were stained with Zombie NIR Fixable Viability Dye (BioLegend) and surface staining was then performed with FcBlock, anti-CD271-PE/Dazzle594 (BioLegend), anti-CD4-BUV563 (BD Biosciences) and anti-CD8-BUV737 (BD Biosciences) antibodies. Cells were fixed and permeabilized with the FOXP3/Transcription Factor Buffer kit (Thermo Fisher Scientific) and stained intracellularly with anti-T-bet-PE/Cy7 (BioLegend), anti-IFN-γ-BV711 (BioLegend) and anti-TNF-α-BV510 (BioLegend) antibodies. For PSMD9- and ARHGAP27-transduced HVS-T cells and their control HVS-T cells, cells were stained with Zombie NIR Fixable Viability Dye (BioLegend) and then surface stained with FcBlock and anti-CD271-Alexa647 antibody (BD Pharmingen). Cells were fixed with 2.8% PFA and permeabilized with the FOXP3/Transcription Factor Buffer kit (Thermo Fisher Scientific). They were then subjected to intracellular staining with anti-T-bet-PE (BioLegend) and anti-IFN-γ-BV711 (BioLegend) antibodies. ICS analysis was performed on an Aurora cytometer (Cytek).
Functional analysis of expanded T-bet-deficient CD4+ T cells in vitro
CD4+ T cells were isolated from PBMCs from five healthy controls (CTL), IL-12Rβ1-deficient (IL12RB1 M/M) patients, the T-bet WT/M heterozygous father (TBX21 WT/M), and the T-bet M/M patient (TBX21 M/M), with anti-CD4 antibody-coated beads (Miltenyi Biotec). They were then expanded with anti-CD3/CD28 antibody-coated Dynabeads (Thermo Fisher Scientific) at a 1:1 (cell:bead) ratio under TH0 or TH1 (2.5 ng/mL IL-12 R&D and anti-human IL-4 neutralizing Ab InVivoMab, BioXcell) conditions, in the presence of 100 U/mL proleukin IL-2 (PROMETHEUS) and anti-human IL-10 neutralizing Ab (Thermo Fisher Scientific). Medium (X-vivo15 5% human AB serum) and cytokine cocktails were refreshed every 3 – 4 days. After 7 days, the culture consisted of >95% CD4+ T cells. Cells were restimulated with anti-CD3/CD28 Ab-coated Dynabeads every 7-8 days. P’s CD4+ T cells were transduced with retroviruses obtained from pLZRS-ires-ΔCD271 plasmids with or without WT TBX21 or EV, as previously described (Martínez-Barricarte et al., 2016). Six days after transduction, cells were restimulated with 25 ng/mL PMA and 500 nM ionomycin in the presence of GolgiPlug (BD Biosciences) for 6 h. Cells were stained with Zombie NIR Live-Dead exclusion reagents (BioLegend), anti-CD271-FITC antibody and were then fixed and permeabilized. They were stained with anti-TNF-α-BV510, anti-IL-13-APC, anti-IL-4-PE, anti-IL-17A-BV785, anti-IL-10-PE-Dazzle594, anti-IL-5-BV421 and anti-IFN-γ-PerCp-Cy5.5 antibodies and subjected to flow cytometry in an Aurora cytometer (Cytek). Three weeks after their initial expansion, transduced CD271+ CD4+ T cells from P were isolated with anti-CD271 antibody-coated beads (Miltenyi Biotec). The cells isolated, and expanded CD4+ TH0 cells from healthy donors or T-bet wt/m individuals were restimulated with anti-CD3/CD28 antibody-coated beads in the absence of cytokine for 16 h or 48 h. Cytokine levels were determined in 13-Legendplex assays (BioLegend) on culture supernatants from cultures restimulated for 48 h . Total RNA was extracted from cells restimulated for 16 h for RNA-seq analysis (Qiagen). In brief, 100 ng of total RNA was used to generate RNA-Seq libraries with the Illumina TruSeq stranded mRNA LT kit (Cat# RS-122-2101). Libraries prepared with unique barcodes were pooled at equal molar ratios. The pool was denatured and sequenced on an Illumina NextSeq 500 sequencer with high-output V2 reagents and NextSeq Control Software v1.4 to generate 75 bp single reads, according to the manufacturer’s protocol (Illumina, Cat# 15048776).
RNA-seq FASTQ files were obtained, and inspected by fastqc for quality control of the raw data. The sequencing reads of each FASTQ file were then mapped to the GENCODE human reference genome GRCh38.p13 (Frankish et al., 2019) with STAR aligner v2.7 (Dobin et al., 2013), and the quality of each mapped alignment in the BAM file was evaluated by RSeQC (Wang et al., 2012). Reads were quantified to generate gene-level read counts from the read alignment, with featureCounts v1.6.0 (Liao et al., 2014), based on GENCODE GRCh38.p13 gene annotation. The gene-level read counts were then normalized and log2-transformed by DESeq2 (Love et al., 2014), to obtain the gene expression value for all genes and all samples. An analysis of the differential expression of all genes and immune genes was performed to compare gene expression in the four TBET samples (TBX21 WT/M, TBX21 M/M, M/M+EV, and M/M+WT) with that in five control samples, with each sample having two technical replicates. The 1,984 immune-related genes were identified from the immune system process gene set in the Molecular Signature Database (MSigDB) (Liberzon et al., 2015). By applying the trimmed mean of M values (TMM), normalization and gene-wise generalized linear model regression were performed in edgeR (Robinson et al., 2010). The genes displaying significant differential expression were selected on the basis of a p-value ≤ 0.05 and a FDR ≤ 0.05. The differential gene expression data were plotted as a heatmap by ComplexHeatmap (Gu et al., 2016), and genes and samples were clustered on the basis of complete linkage and Euclidean distance of gene expression values.
Omni-ATAC-seq on CD4+ T cells and analysis
CD4+ T cells derived from healthy controls, IL12RB1 M/M patients, a TBX21 WT/M parent and the TBX21 M/M P were expanded under TH0 conditions, as described above. Nine days after the retroviral transduction of CD4+ T cells from P, as described above, CD271+ cells were isolated with anti-CD271 antibody-coated beads (Miltenyi Biotec). Omni-ATAC-seq library preparation was performed as previously described (Corces et al., 2017). In brief, 50,000 cells from three healthy donors (CTL), two IL12RB1 M/M patients, technical duplicates for a TBX21 WT/M individual, the TBX21 M/M P and P cells complemented with EV (M/M+EV) or WT (M/M+WT) TBX21 were harvested, washed with 50 μl cold PBS, and centrifuged to obtain a pellet. Pellets were lysed with 50 μl cold lysis buffer consisting of 48.5 μl resuspension buffer (10 mM Tris-HCl pH 7.5, 10 mM NaCl, 3 mM MgCl2 in water), 0.5 μl 10% NP-40, 0.5 μl 10% Tween-20, and 0.5 μl 1% digitonin. Lysates were incubated on ice and then washed with 0.1% Tween-20 resuspension buffer. Lysates were centrifuged and the nuclei in the pellet were subjected to Tn5 transposition with 50 μL of a mixture of 25 μL 2 x TD buffer, 16.5 μL PBS, 0.5 μL 10% Tween-20, 0.5 μL 1% digitonin, 2.5 μL Tn5 transposase, 5 μL nuclease-free water at 37 °C in a thermomixer operating at 1,000 rpm for 30 min. DNA fragments were extracted with the MinElute PCR purification kit and eluted in 30 μl EB buffer (Qiagen). We used 10 μL of eluted DNA for the amplification of DNA fragments with the NEBNext PCR master mix (New England BioLab), over six cycles, with the Ad1_forward and 15 indexed Ad2_reverse primers, as previously described (Buenrostro et al., 2013, 2015). The partially amplified library was subjected to quantitative PCR analysis with the Ad1 and Ad2 primers, Sybr Green reagents and NEBNext PCR master mix for 25 cycles. Additional PCR cycles were performed, based on 1/3 the maximal fluorescence for each sample, as previously described (Buenrostro et al., 2013, 2015). A left-sided isolation of PCR products was performed with AMPure beads (Beckman Coulter). DNA was quantified with a Qubit fluorimeter. Equal amounts of each sample were pooled and subjected to paired-end sequencing on a NextSeq high-output sequencer generating 75 bp reads.
The ATAC-seq reads were aligned with the hg38 genome from the BSgenome.Hsapiens.UCSC.hg38 Bioconductor package (version 1.4.1), with Rsubread's alignment method in paired-end mode, with fragments of 1 to 5,000 base pairs in length considered correctly paired (Liao et al., 2014). Normalized, fragment signal bigWigs were created with the rtracklayer package. Peak calls were made with MACS2 software in BAMPE mode (Feng et al., 2012; Zhang et al., 2008) and peak summits were used in MEME-ChIP software the identification of known and novel motifs (Ma et al., 2014). Differential ATAC-seq signals were identified with the DESeq2 package (Love et al., 2014). Fragment length distribution plots and nucleosome-free/mono-nucleosome meta-TSS plots were produced with the profileplyr package (Carroll and Barrows, 2019). Heatmaps of ChIP-seq and ATAC-seq signals were created from the normalized bigWigs signal, with DeepTools2 software (Ramírez et al., 2016).
T-bet ChIP-seq data (SRR332104) and its input control (SRR332102) were retrieved as unaligned sequences from the European Nucleotide Archive (Kanhere et al., 2012). Sequences were aligned with the hg38 genome from the Bsgenome.Hsapiens.UCSC.hg38 Bioconductor package (version 1.4.1), with Rsubread's alignment method, and predicted fragment lengths calculated with the ChIPQC package (Carroll et al., 2014; Liao et al., 2014; de Santiago and Carroll, 2018). Normalized, fragment-extended bigWigs signals were created with the rtracklayer package.
EPIC DNA methylation arrays and quantification of DNA methylation
CD4+ T cells derived from healthy controls, a TBX21 WT/M individual and the TBX21 M/M P were expanded under TH0 conditions, as described above. Seventeen days after the retroviral transduction of CD4+ T cells from P as described above, CD271+ cells were isolated with anti-CD271 antibody-coated beads (Miltenyi Biotec). DNA from non-transduced or CD271+ isolated CD4+ T cells was extracted with a DNA mini kit (Qiagen). DNA was subjected to bisulfite conversion with the EZ DNA Methylation Kit (ZymoResearch). We quantified the DNA in all samples by performing EPIC arrays (Illumina), following the manufacturer’s instructions. Technical replicates were included for several samples during EPIC processin, to ensure that the measurements were reliable. All microarrays were scanned with the Illumina HiScan system. Data files were processed and converted to beta-values, corresponding to the percentage CpG methylation for a given CpG site. We wrote scripts to filter all loci, retaining those with higher levels of methylation in M/M and M/M+EV than the maximum methylation level in controls, and lower levels of methylation in M/M+WT than in controls (group 1), and those with lower levels of methylation in M/M and M/M+EV than the minimum level of methylation in controls, and higher levels of methylation in M/M+WT than the minimum level in controls (group 2). We then measured and plotted the difference in CpG methylation between the patient and controls, by subtracting the mean methylation level for the five controls from that for M/M and comparing the value obtained with the level of restored CpG methylation by subtracting the level of methylation in M/M+WT from the methylation in M/M+EV. On the plot, red dots represent the loci with higher levels of methylation in the patient than in controls, and green dots represent loci with lower levels of methylation in the patient than in controls. Dot size is proportional to the fold difference in methylation in patients relative to the mean value for controls. The genes of our interest are highlighted on the scatter plot.
Immunophenotyping of leukocytes
In the CyTOF experiment, PBMCs from 26 healthy adults of diverse ethnicities, nine child or adolescent controls (age range: 1 to 17 years, with diverse ancestries, including one age-matched child from Morocco), six IL-12Rβ1-deficient patients (recruited from previous studies), and the T-bet-deficient patient (M/M). We recruited 26 healthy adult controls locally in New York City. We analyzed the PBMCs of four individuals on two separate dates, as duplicates. We obtained 30 vials of cells from these adult controls, 10 of which were frozen after being allowed to rest at room temperature for 24 – 48 h to mimic travel conditions. The vials of cells from the six IL-12Rβ1-deficient patients and the age-matched controls were typically frozen after centrifugation on Ficoll following the overnight shipment of the blood sample. For this experiment, the PBMCs of P (M/M) were prepared from blood that had traveled overnight from Morocco to Paris. Metal-conjugated mAbs were obtained from Fluidigm according to the manufacturer’s instructions. The following antibodies were included in the staining panel: anti-CD45-89Y, anti-CD57-113In, anti-CD11c-115In, anti-CD33-141Pr, anti-CD19-142Nd, anti-CD45RA-143Nd, anti-CD141-144Nd, anti-CD4-145Nd, anti-CD8-146Nd, anti-CD20-147Sm, anti-CD16-148Nd, anti-CD127-149Sm, anti-CD1c-150Nd, anti-CD123-151Eu, anti-CD66b-152Sm, anti-PD-1-153Eu, anti-CD86-154Sm, anti-CD27-155Gd, anti-CCR5-156Gd, anti-CD117-158Gd, anti-CD24-159Tb, anti-CD14-160Gd, anti-CD56-161Dy, anti-gdTCR-162Dy, anti-DRTH2-163Dy, anti-CLEC12A-164Dy, anti-CCR6-165Ho, anti-CD25-166Er, anti-CCR7-167Er, anti-CD3-168Er, anti-CX3CR1-169Tm, anti-CD38-170Er, anti-CD161-171Yb, anti-CD209-172Yb, anti-CXCR3-173Yb, anti-HLA-DR-174Yb, anti-CCR4-176Yb, anti-CD11b-209Bi.
For the identification of immunophenotypes unique to the TBX21 patient, we compared data from six batches of CyTOF experiments including age-matched pediatric controls. In addition to manually inspecting data from each batch separately, we also compared immunophenotypes across batches in an objective and unbiased manner, with a computerized system (Ogishi et al. Submitted). This method involved a combination of high-dimensional batch-correction, unsupervised clustering, and batch-specific cell-type classification through machine learning. We first used a computational approach to normalize batch effects and identify phenotypes unique to the TBX21 patient through an unbiased clustering approach (Manuscript in preparation). Each of the FCS files was processed to exclude dead cells and doublets automatically. Cells from healthy local controls were used for batch correction with Harmony (Korsunsky et al., 2019). Data were down-sampled to 200,000 cells per batch before batch correction. Unsupervised clustering was then performed with batch-corrected marker expression values and the FlowSOM and ConsensusClusterPlus packages in Bioconductor (Van Gassen et al., 2015; Wilkerson and Hayes, 2010). We retained 40 clusters after metaclustering. Each cluster was manually inspected and annotated on the basis of its marker expression profile. A batch-specific cell-type classifier was then trained on the uncorrected expression values and cell-type annotations. We sampled a maximum of 100 cells per cell type at random, to improve computation efficiency and to alleviate class imbalance issues, as some of the cell types are much rarer than others. A highly randomized tree algorithm was used, with tuning of the hyperparameters mtry and numRandomCuts for each batch (Geurts et al., 2006). Classifiers were trained through five repeats of 10-fold cross-validation with internal upsampling. Internal accuracy during cross-validation always exceeded ~95%. Finally, the trained classifier was applied to the rest of the cells, including the patient’s cells, to predict their cell types. With this approach, any cell could be classified probabilistically as belonging to one of the predefined cell types, making it possible to avoid a strong skewing of patient data, which would affect the batch-correction and clustering steps. For the purposes of visualization, batch correction was performed with Harmony on cells from all controls and patients. Data were down-sampled to 40,000 cells per batch before batch correction. Finally, the trained classifier was applied to all cells, including the patient's cells, to predict their cell types and, thus, to identify immunophenotypes for each individual. After harmonization, dimension reduction was performed by Uniform Manifold Approximation and Projection (UMAP) (Becht et al., 2019). The figures are color-coded on the basis of the cell type identified by the unsupervised clustering approach described above. All analyses were performed with R ver.3.6.3.
We also deconvoluted 10,000 memory CD4+ T cells (DNA+Live_CD45+CD66b−CD123−CD3+CD20−CD56−γδTCR−CD4+CD8−CD45RA−) into viSNE plots based on 20 surface markers (113ln_CD57, 115ln_CD11c, 149sm_CD127, 150Nd_CD1c, 154Sm_CD86, 155Gd_CD27, 156Gd_CCR5, 158Gd_CD117, 159Tb_CD24, 163Dy_CRTH2, 165Ho_CCR6, 166Er_CD25, 167Er_CCR7, 169Tm_CX3CR1, 170Er_CD38, 171Yb_CD161, 173Yb_CXCR3, 174Yb_HLA-DR, 176Yb_CCR4, 209Bi_CD11b) (Amir el et al., 2013). Analyses were performed by manually gating populations for NK cells, naïve and memory subsets of CD4+ and CD8+ T cells, innate lymphoid cells (ILCs), Tregs and dendritic cell subsets. For T helper cell analysis, we included an additional four healthy donors from a separate experiment (same staining panel) in the analysis. Manual gating was used for TH analysis. We also deconvoluted 10,000 CD8+ T cells into viSNE plots based on 113ln_CD57, 115ln_CD11c, 143Nd-CD45RA, 144Nd-CD141, 149sm_CD127, 150Nd_CD1c, 154Sm_CD86, 155Gd_CD27, 156Gd_CCR5, 158Gd_CD117, 159Tb_CD24, 163Dy_CRTH2, 165Ho_CCR6, 166Er_CD25, 167Er_CCR7, 169Tm_CX3CR1, 170Er_CD38, 171Yb_CD161, 173Yb_CXCR3, 174Yb_HLA-DR, 176Yb_CCR4, 209Bi_CD11b (Amir el et al., 2013).
Immunophenotyping for MAIT, invariant NKT cells, Vδ2 and Vδ1 cells was performed in three different batches. In the first batch, PBMCs from 18 healthy controls (CTL), three IL-12Rβ1-deficient individuals (IL12RB1 M/M), two T-bet-heterozygous parents (TBX21 WT/M), P (TBX21 M/M) and P’s healthy brother (TBX21 WT/WT) were included. They were stained with the following panel: FcBlock (Miltenyi Biotec), Zombie-NIR live-dead exclusion dye (BioLegend), anti-CD3-Alexa532 (Thermo Fisher Scientific), anti-γδTCR-FITC (Thermo Fisher Scientific), anti-Vδ2-APC-Fire750 (BioLegend), anti-CD56-BV605 (BioLegend), anti-CD4-BV750 (BD Biosciences), anti-CD8a-BV510 (BioLegend), anti-Vα7.2-BV711 (BioLegend), anti-Vα24-Jα18-PE-Cy7 (BioLegend), anti-Vδ1-Vioblue (Miltenyi Biotec), anti-CD161-PE (BioLegend) and anti-Vβ11-APC (Miltenyi Biotec) antibodies. Cells were analyzed with an Aurora cytometer (Cytek). In the 2nd batch, 31 adult control samples, seven samples from age-matched controls (age CTL), including a Moroccan child, and samples from P (M/M) and his mother sample were included. In the 3rd batch, 16 adult control samples, five samples from age-matched controls (age CTL) and a sample from P were included. In the 3rd batch, 16 adult control samples, five samples from age-matched controls (age CTL) and P’s sample were included. For the three batches considered together, we obtained 65 data points for healthy adults, with PBMCs from 20 healthy adults undergoing two analyses in different batches, PBMCs from three samples undergoing analyses three times, in different batches and PBMCs from 16 individuals analyzed only once. All 12 age-matched controls were different individuals. Three vials of PBMCs from P, isolated from fresh blood and frozen in the same batch, were analyzed in three separate batches. P’s parents and P’s brother’s PBMCs were isolated from fresh blood and frozen. The age matched controls were of various ethnic backgrounds, and all were aged 1 – 17 years at the time of blood sampling. The PBMCs of most of these age-matched controls were prepared from blood that had been transported overnight. PBMCs from three IL-12Rβ1-deficient individuals were prepared from blood transported overnight. In the second and third batches, immunophenotyping for MAIT, invariant NKT cells, Vδ2 and Vδ1 cells was performed with a flow cytometry staining panel containing FcBlock (Miltenyi Biotec), Zombie-NIR live-dead exclusion dye (BioLegend), anti-γδTCR-alexa 647 (BioLegend), anti-CD3-V450 (BD Biosciences), anti-CD56-BV605 (BioLegend), anti-CD4-BUV563 (BD Biosciences), anti-Vδ1TCR-FITC (Miltenyi), anti-CD8-BUV737 (BD Biosciences), anti-Vδ2TCR-APC/Fire750 (BioLegend), anti-CD20-BV785 (BioLegend), anti-Vα7.2-BV711 (BioLegend), anti-CD161-PE (BioLegend), MR1-5-OP-RU-tetramer (NIH tetramer core facility), anti-Vβ11-APC (Miltenyi), anti-iNKT-BV480 (BD Biosciences) antibodies. Cells were analyzed with an Aurora cytometer (Cytek). Three batches of data were compiled for the final analysis. The percentages of the various immune subsets were determined by manual gating.
Immunophenotyping for innate lymphoid cells and NK cells was confirmed separately, by conventional flow cytometry. In brief, biotinylated anti-human anti-CD1a (HI149), anti-CD3 (OKT3), anti-CD5 (UCHT2), anti-CD14 (61D3), anti-CD19 (HIB19), anti-CD34 (4H11), anti-CD123 (6H6), anti-CD203c (FR3-16A11), anti-CD303 (AC144), anti-TCRαβ (IP26), anti-TCRγδ (B1) and anti-FcεRIα (AER-37(CRA-1)) antibodies were used in combination with streptavidin BUV661, and conjugated anti-CD62L-FITC (Dreg-56), anti-CD94-PerCP-Vio700 (REA113), anti-TCRVα24Jα18-PE (6B11), anti-CD26-PE-CF594 (M-A261), anti-CD127-PE-Cy7 (eBioRDR5), anti-CRTH2-Alexa Fluor 647 (BM16), anti-CD161-Alexa Fluor 700 (HP-3G10), anti-EOMES-APC-eFluor 780 (WD1928), anti-NKp46-BV421 (9E2/NKp46), anti-CD45RA-BV570 (HI100), anti-CD117-BV605 (104D2), anti-RORγt-BV650 (Q21-559), anti-CD7-BV711(M-T701), anti-CD16-BUV496 (3G8), anti-CD25-BUV563 (2A3), anti-CD56-BUV737 (NCAM16.2) and anti-CD45-BUV805 (HI30); anti-T-bet-BV786 (O4-46), anti-GATA3-BUV395 (L50-823) antibodies. Antibodies were purchased from BioLegend, Thermo Fisher Scientific, BD Biosciences, or Miltenyi Biotec. Fc receptors were blocked with IgG from human serum (Sigma-Aldrich). Surface membrane staining was performed in Brilliant Stain Buffer (BD Biosciences). Transcription factors were stained with the Foxp3 staining kit (Thermo Fisher Scientific), according to the manufacturer’s instructions. The fixable viability dye eFluor 506 (Thermo Fisher Scientific) was used to exclude dead cells. Samples were acquired on a BD Symphony A5 cytometer (BD Biosciences) with FACSDiva 8 software and were analyzed with FlowJo 10 (BD Biosciences).
Immunophenotyping was confirmed separately for the T- and B-cell subsets by conventional flow cytometry. In brief, PBMCs from healthy controls and P were labeled with mAbs against CD3, CD4, CD8, CD45RA, CCR7, CD127, CD25, CXCR5, CXCR3, CCR6, PD-1, and CD56. The CD4+ and CD8+ T-cell subsets were separated into naïve (TNaive: CCR7+CD45RA+), central memory (TCM: CCR7+CD45RA−), effector memory (TEM: CCR7−CD45RA−), and terminally differentiated effector memory cells (TEMRA: CCR7−CD45RA+) after initial gating on CD8−CD4+ or CD8+CD4− cells. Proportions of regulatory T cells (CD4+CD127loCD25hi), total memory (CD4+CD45RA−), and circulating Tfh (cTfh; CD4+CD45RA−CXCR5+) cells, and subsets of total memory and cTfh cells defined on the basis of CXCR3 and CCR6 expression (TH1: CCR6−CXCR3+, TH17: CCR6+CXCR3−, TH1*/TH1-17: CCR6+CXCR3+, TH2: CCR6−CXCR3−) were also determined.
Single-cell (sc) RNA-seq and analysis
We performed scRNA-seq on PBMCs obtained from the TBX21 WT/M heterozygous father and the TBX21 M/M patient. PBMCs were quickly thawed at 37°C and gently resuspended by serial additions of DMEM + 10% heat-inactivated (HI) FBS, according to the recommendations from the 10x Genomics “Sample preparation Fresh Frozen Human PBMC” protocol Rev C. Cells were centrifuged at 400 x g for 5 min, washed with DMEM + 10% HI-FBS, and cells were then counted and viability was assessed with the LIVE/DEA Viability kit (Thermo Fisher Scientific) according to the manufacturer’s guidelines. Following centrifugation at 400 x g for 5 min, cells were resuspended at a concentration of 1000 cells/μl in PBS + 0.04% BSA and loaded onto a 10 x Genomics Chromium chip for single-cell capture. Reverse transcription and library preparation were performed with Chromium Single Cell 3′ Reagent Kits (v2) in accordance with the manufacturer’s guidelines, and library quality was assessed with a Bioanalyzer DNA chip. Each library was sequenced on two lanes of an Illumina HiSeq 4000 sequencer in a 28 bp/98 bp paired-end configuration.
The sequence-read quality of individual sequencing lanes was assessed with BVAtools (https://bitbucket.org/mugqic/bvatools). Cell Ranger v3.0.1 was used to aggregate the sequences from two lanes/samples, to map reads to the hg38 human reference genome assembly, for filtering, and counting barcodes and UMIs; a list of UMI counts was thus obtained for each gene and each cell. Cells outside the [5%,95%] interval for these metrics or with >10% mitochondrial genes were excluded. The DoubletFinder package was used to identify and filter out cell doublets (McGinnis et al., 2019). After the removal of dead cells and doublets, 10,409 cells for the T-bet wt/m heterozygous father and 11,741 cells for the T-bet m/m P were analyzed together with the Seurat v3 R package (Stuart et al., 2019), and cell clustering was performed by the Uniform Manifold Approximation and Projection dimension reduction method (Becht et al., 2019), using the most variable genes, but excluding mitochondrial and ribosomal protein genes. This analysis identified eight distinct cell clusters; the marker genes for each cluster were identified with Seurat and used to determine cell types. Differential expression was analyzed by comparing UMI gene counts between the patient and the control for each cluster, with the MAST approach (Finak et al., 2015). Genes with a p-value < 10−25 for differential expression were analyzed in more detail. For each selected gene, cells in which the expression of the gene was detectable were counted for the index case and the control in each cluster, to reduce the dropout bias inherent to droplet scRNA approaches. The number of cells expressing the gene was normalized according to the number of cells in each cluster and the fold-difference in expression was calculated by comparing the index case and the control. For the figures, cluster annotations and the levels of expression of genes of interest were projected onto the UMAP clustering, but with the cells of the patient and the control shown separately.
Stimulation of NK cells ex vivo
PBMCs from five healthy donors including a travel control, the T-bet WT/M mother, the T-bet WT/WT brother and the T-bet M/M P were left untreated or stimulated with IL-12 (Miltenyi Biotec), IL-15 (Miltenyi Biotec) and IL-18 (MBL/CliniSciences) for 18 h. Cells were treated with GolgiPlug and GolgiStop (BD Biosciences), and stained with anti-CD107a-PE antibody (eBioH4A4, Thermo Fisher Scientific) for 4 h. They were then stained with eFluor506 live-dead reagents (Thermo Fisher Scientific), followed by antibodies binding to surface antigens, including anti-CD1a-FITC (H1149), anti-CD3-FITC (OKT3), anti-CD5-FITC (UCHT2), anti-CD14-FITC (TUK4), anti-CD19-FITC (HIB19), anti-CD34-FITC (581), anti-CD123-FITC (6H6), anti-CD203c (NP4D6), anti-CD303-FITC (201A), anti-FcεRIα-FITC (AER-37(CRA-1)), anti-TCRαβ-FITC (IP26), anti-TCRγδ-FITC (B1), anti-CD94-PerCP-Vio700 (REA113), anti-CD127-PE-Cy7 (eBioRDR5), anti-NKp46-APC (9E2/NKp46), anti-CD7-BV711 (M-T701), anti-CD56-BV786 (NCAM16.2), anti-CD16-BUV737 (3G8), and anti-CD45-BUV805 (HI30) antibodies. All the markers targeted by FITC-conjugated Abs were considered to be Lineage-specific markers (Lin). Cells were fixed and permeabilized (Fix & Perm; Thermo Fisher Scientific) and subjected to intracellular staining with anti-granzyme B-PE-CF594 (GB11), anti-EOMES-APC-eFluor 780 (WD1928), anti-perforin-BV421 (δG9), anti-TNF-α-BV605 (Mab11) and anti-IFN-γ-BUV395 (B27) antibodies. Antibodies were purchased from BioLegend, Thermo Fisher Scientific, BD Biosciences, or Miltenyi Biotec. We analyzed ICS and surface staining with a BD LSRFortessa flow cytometer (BD Biosciences).
CD4+ T-cell isolation and functional characterization ex vivo and TH differentiation in vitro
The healthy controls (CTL) for this experiment were adults recruited locally in Australia, and PBMCs for P (M/M) were prepared from two separate overnight shipments of blood from Casablanca to Paris and frozen. The two dots in these figure panels represent data from two independent experiments on two batches. PBMCs from healthy donors and P were incubated with mAbs against CD4, CD45RA, CCR7, CXCR5, CD127 and CD25. Naïve and memory CD4+ T cells were isolated by first excluding Tregs (CD25hiCD127lo) and then sorting CD4+CD45RA+CCR7+CXCR5− and CD4+CD45RA−CCR7± cells, respectively. Isolated naïve and memory CD4+ T cells were then cultured in 96-well round-bottomed well plates (30-40 x103 cells/well) with T-cell activation and expansion (TAE) beads (anti-CD2/CD3/CD28; Miltenyi Biotech) alone (TH0) or under TH1 (50 ng/mL IL-12), TH2 (100 U/mL, IL-4), TH9 (2.5 ng/mL TGF-β, 100 U/mL IL-4), or TH17 (2.5 ng/mL TGF-β, 50 ng/mL IL-1-β, 50 ng/mL IL-6, 50 ng/mL IL-21, 50 ng/mL IL-23, 50 ng/mL PGE2) polarizing conditions. After 5 days, supernatants were harvested and the production of IL-4, IL-5, IL-9, IL-10, IL-13, IL-17A, IL-17F, IFN-γ and TNF-β was assessed with cytometric bead arrays (Becton Dickinson); IL-22 secretion was measured by ELISA (eBioscience). For cytokine expression, activated CD4+ T cells were restimulated with PMA (100 ng/mL)/ionomycin (750 ng/mL) for 6 h, with brefeldin A (10 μg/mL) added after two hours. Cells were then fixed and the intracellular expression of IL-4, IL-9, IL-13, IL-10, IL-17A, IL-17F, IL-22, IL-21 and IFN-γ was determined.
Stimulation of PBMCs with PMA/ionomycin for intracellular staining and flow cytometry analysis
This experiment was performed twice, on two different dates. The two batches of data were compiled for the final analysis. Fourteen healthy adult donors were recruited at the Rockefeller University (New York, USA). Two healthy adult controls, four age-matched controls, P and his relatives were recruited at Necker Hospital (Paris, France). PBMCs from two of 16 adult controls were frozen after being allowed to rest at room temperature for 24 – 48 h to mimic transport conditions. The PBMCs from the other 14 adult controls were prepared directly from freshly collected blood and frozen. Two vials of PBMCs from P were used in this experiment: one prepared directly from fresh blood before freezing, and the other prepared and frozen after overnight shipment of the blood samples. The PBMCs of four age-matched controls were prepared from blood that had been shipped overnight. The PBMCs of P’s relatives were prepared from blood that had been shipped overnight. PBMCs from six healthy adult controls were stimulated twice, in two separate batches, as technical replicates. The second vial of P’s PBMCs was stimulated in two sets of wells, as technical duplicates. The PBMCs of three age-matched controls were stimulated in two sets of wells, as technical duplicates. The PBMCs of P’s WT/M mother were stimulated in two sets of wells, as technical duplicates.
In both batches of experiments, we plated 300,000 cells per well in 96-well U-bottomed plates, at a density of 1.5 x 106 cells/ml. Cells were cultured for 40 h in RPMI. They were then stimulated with 50 ng/mL PMA and 1 μM ionomycin. After 1 h, Golgiplug was added to the culture, in accordance with the manufacturer’s protocol. After another 7 h, cells were harvested for staining and flow cytometry. In brief, cells were first stained with Zombie NIR Viability kit (BioLegend) for 15 min. They were then stained with FcBlock (Miltenyi), anti-γδTCR-alexa 647 (BioLegend), anti-CD3-V450 (BD Biosciences), anti-CD56-BV605 (BioLegend), anti-CD4-BUV563 (BD Biosciences), anti-Vδ1TCR-FITC (Miltenyi), anti-CD8-BUV737 (BD Biosciences), anti-Vδ2TCR-APC/Fire750 (BioLegend), anti-CD20-BV785 (BioLegend), anti-Vα7.2-Alexa 700 (BioLegend), MR1-5-OP-RU-tetramer (NIH tetramer core facility), anti-Vβ11-APC (Miltenyi), and anti-iNKT-BV480 (BD Biosciences) antibodies for 30 min. The stained cells were fixed with the FOXP3/Transcription Factor kit (Thermo Fisher Scientific) and stained intracellularly with anti-T-bet-PE/Cy7 (BioLegend), anti-IFN-γ-BV711 (BioLegend), anti-TNF-α-BV510 (BioLegend), anti-IL-17A-PERCP/Cy5.5, anti-RORγT-PE (BD Biosciences), anti-CD3-V450 (BD Biosciences), anti-CD4-BUV563 (BD Biosciences) and anti-CD8-BUV737 (BD Biosciences) antibodies in Perm/Wash buffer. Cells were then analyzed in a CyTek Aurora spectral flow cytometer.
Stimulation of PBMCs with PMA/ionomycin for cytokine secretion
This experiment was done in three batches, on three separate dates. The data from the three batches were compiled for the final analysis. Thirty healthy adult controls were recruited at the Rockefeller University (New York, USA) and one healthy adult control was recruited at Necker Hospital (Paris, France). PBMCs from eight of the 31 adult controls were frozen after being allowed to rest at room temperature for 24 – 48 h to mimic travel conditions. The PBMCs of all the other 23 adult controls were isolated from fresh blood and frozen. PBMCs from 18 adult controls were analyzed once. Those from 13 adult controls were analyzed twice, in two separate batches. Three vials of PBMCs from P were used in this experiment: two vials of PBMCs isolated from fresh blood and then frozen, and a vial of PBMCs isolated from blood that had been transported overnight, which were then frozen. These three vials of PBMCs from P were used separately, in three batches. One vial of PBMCs prepared from fresh blood was plated in two sets of wells as technical duplicates. Thus, four data points were measured for P. The PBMCs of P’s relatives were prepared from blood that had been shipped overnight. PBMCs from P’s healthy brother, mother and father were each analyzed once. Five age-matched controls were recruited from around the world, including one matched for both age and ethnicity from Morocco. PBMCs were isolated from the blood of these patients after overnight shipment and frozen. PBMCs from three age-matched controls were plated in two sets of wells, as technical duplicates. The PBMCs of the age- and ethnicity-matched control were plated in technical triplicates. In three batches of experiments, we plated 300,000 cells per well in 96-well U-bottomed plates, at a density of 1.5 x 106 cells/mL. Cells were cultured for 40 h in RPMI. They were then stimulated by incubation with 50 ng/mL PMA and 1 μM ionomycin for 8 h. Supernatants were harvested for human TH cytokine analysis with the 13-plex LegendPlex kit (BioLegend).
Stimulation of PBMCs with live BCG infection for cytokine production
This experiment was performed in three batches, on three dates. The data from the three batches were compiled for the final analysis. Thirty healthy adult controls were recruited at the Rockefeller University (New York, USA) and one healthy adult control was recruited at Necker Hospital (Paris, France). PBMCs from eight of the 31 adult controls were frozen after being allowed to rest at room temperature for 24 – 48 h to mimic travel conditions. The PBMCs of all the other 23 adult controls were isolated from fresh blood and frozen. PBMCs from 19 adult controls were analyzed once, and those from 10 adult controls were assayed twice, in two separate batches. PBMCs from one control were plated in three sets, as technical triplicates, in one batch. PBMCs from one control were assayed five times in three separate batches, as technical replicates. In total, 47 data points were obtained for adult controls. Five age-matched controls were recruited globally, including one matched for both age and ethnicity from Morocco. Their PBMCs were obtained from blood that had been shipped overnight and were frozen. PBMCs from three age-matched controls were plated in two sets of wells, as technical duplicates. PBMCs from the other age-matched controls were plated in single sets. Eight data points in total were obtained for age-matched controls. Three vials of PBMCs from P were used in this experiment: two vials of PBMCs isolated from fresh blood and then frozen, and a vial of PBMCs isolated from blood that had been transported overnight, which were then frozen. These three vials of PBMCs from P were used separately, in three batches. In total three data points were generated for P. PBMCs from P’s relatives were isolated from blood that had been shipped overnight. PBMCs from P’s healthy brother, mother and father were each analyzed once each.
In all three batches of experiments, we plated 300,000 cells per well in 96-well U-bottomed plates, at a density of 1.5 x 106 cells/ml. For each sample, cells were plated in the presence and absence of live M. bovis-BCG at a MOI=1 (gift from Carl Nathan), and in the presence and absence of recombinant IL-12 (5 ng/mL, R&D) or recombinant IL-23 (10 ng/mL, R&D) (Vogt and Nathan, 2011). Golgiplug (BD Biosciences) was added to each well after 40 h of stimulation. Eight hours later, supernatants were harvested for cytokine determinations in a 13-plex Legendplex assay (BioLegend), and cells were collected by centrifugation for FACS staining. In brief, cells were first stained with the Zombie NIR Viability kit (BioLegend) for 15 minutes. They were then stained with FcBlock (Miltenyi Biotec), anti-γδTCR-alexa 647 (BioLegend), anti-CD3-V450 (BD Biosciences), anti-CD56-BV605 (BioLegend), anti-CD4-BUV563 (BD Biosciences), anti-Vδ1TCR-FITC (Miltenyi Biotec), anti-CD8-BUV737 (BD Biosciences), anti-Vδ2TCR-APC/Fire750 (BioLegend), anti-CD20-BV785 (BioLegend), anti-Vα7.2-Alexa 700 (BioLegend), MR1-5-OP-RU-tetramer (NIH tetramer core facility), anti-Vβ11-APC (Miltenyi Biotec), and anti-iNKT-BV480 (BD Biosciences) antibodies for 30 minutes. The stained cells were fixed with the FOXP3/Transcription Factor Buffer set (Thermo Fisher Scientific) and intracellularly stained with anti-T-bet-PE/Cy7 (BioLegend), anti-IFN-γ-BV711 (BioLegend), anti-TNF-α-BV510 (BioLegend), anti-IL-17A-PERCP/Cy5.5 and anti-RORγT-PE (BD Biosciences) antibodies in Perm/Wash buffer. Cells were acquired on a CyTek Aurora spectral flow cytometer and the data were analyzed with Cytobank. T-bet+ IFN-γ+ double-positive cells were gated manually from the live single-lymphocyte population. The viSNE algorithm was then used to cluster these T-bet+ IFN-γ+ double-positive cells with surface lineage-determining markers (Amir el et al., 2013). Automated viSNE clusters were plotted with tSNE1 and tSNE2. Clusters were then overlaid on manually gated immune cell subsets for illustration.
Vδ2+ γδ T cell stimulation
Lysates of M. bovis-BCG were prepared and provided by Dr. Carl F. Nathan. PBMCs from adult controls, IL-12Rβ1 m/m patients, the T-bet M/M P and his relatives were resuspended at a density of 1x106 cells/mL. We plated 200 μL cells per well for each condition, in a 96-well U-bottomed plate. We added BCG lysate to each well at a final concentration of 5 μg/mL. Recombinant IL-2 (Roche) was added to a final concentration of 10 U/mL. Fresh medium and IL-2 were added to the culture every three to four days. Eleven days after stimulation, the cells were fully confluent in the 96-well plate. Cells were then transferred to a 48-well plate and each well was topped up to500 μL per well with medium. After 14 days of stimulation, the cells were stained with FcBlock (Miltenyi Biotec), Zombie-NIR live/dead (BioLegend), anti-CD3-Alexa532 (Thermo Fisher Scientific), anti-γδTCR-FITC (Thermo Fisher Scientific), anti-Vδ2 TCR-APCFire750 (BioLegend), anti-CD56-BV605 (BioLegend), anti-CD4-BV750 (BD Biosciences), anti-CD8a-BV510 (BioLegend), anti-β11TCR-APC (Miltenyi Biotec), anti-CD161-PE (BioLegend), anti- Vα7.2 TCR-BV711 (BioLegend), anti-Vα24-Jα18 (iNKT)-PE/Cy7 (BioLegend), anti-Vδ1-Vioblue (Miltenyi Biotec), anti-CD16-BV650 (BioLegend), anti-CD20-BV785 (BioLegend), anti-αβTCR-Alexa700 (BioLegend) antibodies and analyzed by flow cytometry on an Aurora cytometer (Cytek). Supernatants were harvested for human TH cytokine analysis with the13-plex LegendPlex kit (BioLegend).
Stimulation of αβ T cells with PPD and BCG-lysate
PBMCs from four BCG-vaccinated healthy controls and P were stained with the CellTrace Far-Red Cell Proliferation Kit (Thermo Fisher Scientific) at a dilution of 1:25,000. Cells were washed and resuspended at a density of 2 x 106 cells/mL in RPMI 10% human AB serum (Gemini). Cells were plated in 96-well U-bottomed plates and stimulated with BCG-lysate at a final concentration of 5 μg/mL or with tuberculin purified protein derivative (PPD) at a final concentration of 5 μg/mL (STATEN SERUM INSTITUT). Cells were cultured for 6 days and then harvested and stained with the Zombie NIR Viability kit (BioLegend) for 15 minutes. Cells were then surface-stained with FcBlock, anti-CD3-V450 (BD Biosciences), anti-CD4-BUV563 (BD Biosciences), anti-CD8-BUV737 (BD Biosciences), anti-Vδ2TCR-APC/Fire750 (BioLegend), anti-CD56-BV605 (BioLegend) and anti-CD20-BV785 (BioLegend) antibodies for 30 minutes. Cells were fixed with the FOXP3/Transcription Factor Buffer kit (Thermo Fisher Scientific) and stained by overnight incubation with anti-T-bet-PE/Cy7 (BioLegend), anti-IFN-γ-BV711 (BioLegend), anti-TNF-α-BV510 (BioLegend) and anti-human/mouse-Ki-67 (BioLegend) antibodies in Perm/Wash buffer. Data were acquired in a CyTek Aurora spectral flow cytometer.
Isolation and screening of CD4+ T cell lines
The T-cell clone experiment was performed as previously described (Geiger et al., 2009). In brief, CCR6+ and CCR6− CD4+ memory T cells were sorted from CD14− PBMCs with a FACS Aria (BD Biosciences), excluding CD45RA+CCR7+ naïve cells, and CD25+, CD19+, and CD8+ cells. Sorted T cells (500 cells/well) were polyclonally stimulated with 1 μg/mL PHA (Remel) in the presence of irradiated (45 Gy) allogeneic feeder cells (104 per well) and IL-2 (500 IU/mL) in RPMI complete medium. After expansion for at least 20 days, the T-cell lines were analyzed by surface or intracellular staining with the following monoclonal antibodies: anti-CCR6-PE (BD Biosciences; clone: 11A9); anti-CXCR3-APC (BD Biosciences; clone: 1C6); anti-IFN-γ-FITC or anti-IFN-γ-PE (BD Biosciences, clone: B27); anti-IL-17A-eFluor660 FITC (eBiosciences, clone: eBio64DEC17); anti-IL-4-PE (BD Biosciences, clone: MP4-25D2); anti-IL-22-PerCP-eFluor-710 (Thermo Fisher Scientific, clone: 22URTI). For identification of the antigen-responding T-cell clones, the T-cell lines were also screened by culturing thoroughly washed cells (2.5×105 cells/well) with autologous irradiated B cells (2.5×104 cells/well), with or without a three-hour pulse of the following antigens: M. tuberculosis peptide pool (0.5 μg/mL/peptide, comprising 207 15-mer peptides), BCG peptide pool (0.5 μg/mL/peptide, comprising 211 15-mer peptides), HCMV peptide pool (0.5 μg/mL/peptide, comprising 76 15-20-mer peptides), EBV peptide pool (0.5 μg/mL/peptide, comprising 46 15-20-mer peptides), Influenza virus HA peptide pool (2 μg/mL/peptide, comprising 351 15-mer peptides), Candida albicans peptide pool (0.5 μg mL/peptide, comprising 252 15-mer peptides) or tetanus toxoid peptide pool (1 μg/mL/peptide, comprising 125 15-20-mer peptides). These peptide pools were kindly provided by Cecilia Lindestam Arlehamn and Alessandro Sette at the La Jolla Institute for Immunology (Lindestam Arlehamn et al., 2016). Proliferation was assessed on day 4, after incubation for 16 h with 1 μCi/mL [3H]-thymidine (GE Healthcare). The concentrations of cytokines in the culture supernatants produced by the antigen-responding T-cell clones were measured after 48 h of stimulation with cytometric bead arrays (eBioscience).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using PRISM software (Graphpad). All values are expressed as Mean ± SD or SEM as indicated in the figure legends. Student’s t-test, one-way ANOVA, two-way ANOVA, or non-parametric tests were performed tailored to the hypotheses tested, and were explained in figure legends. Statistical significance was accepted at α < 0.05.
Supplementary Material
(A) Schematic view of the 16 MSMD-causing genes within myeloid and lymphoid cells in the context of host antimycobacterial immunity. (B) We tested for serum IgG against various viral peptides or peptides derived from bacteria or other pathogens by phage immunoprecipitation-sequencing. Serum samples from an unrelated healthy three-year-old boy (CTL1), two healthy adults (CTL2 and CTL3), and a TBX21 M/M P, and IgG-depleted serum and intravenous immunoglobulin (IVIg) were assayed. The heatmap (left) shows the counts of peptides significantly enriched for a given species with less than a continuous seven-residue subsequence, the estimated size of a linear epitope, in common. The species for which the patient had a score greater than our set threshold (>3) are shown. Count values above the set threshold are shown on a color scale from green (≥4) to red (≥40) and dark-red (≥80). (C) Genomic DNA from P (M/M) and his parents (WT/M) was subjected to Sanger sequencing to confirm the TBX21 variant. (D) Comparison between the WT and Mut sequences of the region in which the variant is located within exon 1 of TBX21. An alignment of the WT and Mut sequences revealed a juxtaposed region with a sequence identical to that of the variant sequence. (E) Alignment of peptide fragments from human T-box domain-containing transcription factors. (F) pCMV6 plasmids containing EV, WT T-bet (WT), the K314R variant of T-bet (K314R), T-bet with a deletion of the T-box domain (T-del), and mutant T-bet (Mut) were used to transfect HEK293T cells. T-bet expression was characterized by immunoblotting with anti-T-bet 4B10 monoclonal Ab and anti-Flag Ab. Anti-GAPDH antibody was used as an endogenous control. (G) Mutations of individual residues in the T-bet cDNA (E156S, M157L, E156A, and E157A) were generated in pCMV6 plasmids by site-directed mutagenesis. The plasmids were used to transfect HEK293T cells for overexpression. T-bet levels were measured by immunoblotting with anti-T-bet 4B10 Ab and anti-Flag Ab. Anti-GAPDH Ab was used as endogenous control. (H) The plasmids described in (G) were used to transfect HEK293T cells, together with the IFNG reporter plasmids and Renilla luciferase pRL-SV40 plasmids. After 3 days, the luciferase signal was assessed in a Dual-Glo assay. (I) Single-cell CRISPR/Cas9-edited NK-92 clones were stimulated with 50 pg/mL IL-12 and 10 ng/mL IL-18 or were left untreated (NS). Intracellular IFN-γ production was determined by ICS flow cytometry. (J) T-bet-deficient or pLenti-Crispr-V2-EV-transduced NK-92 single-cell clones were transduced with retroviruses generated from pLZRS-ireΔNGFR plasmids containing no insert (EV), or the WT, K314R or Mut TBX21 cDNA with a C-terminal Flag-tag. They were stimulated with 50 pg/mL IL-12 and 10 ng/mL IL-18 or were left untreated. Intracellular IFN-γ production was determined by ICS flow cytometry. (K) Naïve CD4+ T cells were expanded under TH0 conditions, and were then transduced with retroviruses generated from pLZRS-ireΔNGFR plasmids containing no insert (EV), or the WT, or Mut TBX21 cDNA with a C-terminal Flag-tag. Transduced cells were positive for the bicistronic marker CD271. Cells were surface-stained for CD271 and intracellularly stained for IFN-γ and TNF-α and subjected to flow cytometry analysis. (L) All variants (including the only homozygous variant – Mut, and 36 heterozygous variants, as indicated) from the HGID in-house database are represented on the schematic illustration. (M) All in-house TBX21 variants, as in (L), were tested for transcriptional activity in the human IFNG luciferase reporter assay. pCMV6 plasmids containing variants were used to transfect cells, together with the human IFNG reporter plasmids or the control EV pGL4.10-EV luciferase reporter plasmid, and the pRL-SV40 Renilla plasmid for luciferase activity measurement.
In (H, I and M), bars represent the mean and the standard deviation. Dots represent individual samples or technical replicates. One-way ANOVA was used for analysis in (H). Two-way ANOVA was used for analysis in (I). A non-parametric analysis for comparison with pGL4.10-pIFNG + pCMV6-WT TBX21 transfection condition was performed in (M). In (H, I and M), * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
(A) CD4+ T cells from healthy donors (CTL), the heterozygous father (WT/M) or P (M/M) were expanded with anti-CD3/CD28 antibody-coated beads. They were subjected to RT-qPCR for TBX21 with two different probes. Data are displayed as 2−ΔΔCt after normalization relative to GUS (endogenous control) expression, using the control mean as a calibrator. (B) TBX21 expression was assessed in immortalized T cells (HVS-T) from healthy donors (CTL), the heterozygous father (WT/M) and the patient (M/M), by RT-qPCR, as in (A). (C) P-derived HVS-T cells (M/M) were retrovirally transduced with EV or WT TBX21 containing a bicistronic CD271 surface marker. Intracellular T-bet and IFN-γ production by these cells and by CTL and heterozygous TBX21 WT/M HVS-T cells was assessed by flow cytometry in the presence and absence of P/I stimulation. (D) Expression of PSMD9 and ARHGAP27 (SH3 domain-containing 20 protein) in HEK293T cells transfected with pTRIP-mCherry-2A-PSMD9 or ARHGAP27-V5 plasmids along with VSVG and Pax2 plasmids, as assessed by immunoblotting with an anti-V5 tag Ab. (E) Expression of ERN1 in HEK293T cells transfected with pLenti-EV or WT-ERN1-Cter-GFP plasmids along with VSVG and Pax2 plasmids, as assessed by immunoblotting with anti-GFP or anti-ERN1Ab. (F) Flow cytometry analysis of the intracellular production of IFN-γ in control (CTL) HVS-T cells, M/M HVS-T cells from P, and in M/M HVS-T cells transduced with pTRIP-mCherry-2A-plasmids containing WT PSMD9 or WT ARHGAP27, or empty vector.(G) Flow cytometry analysis of IFN-γ intracellular production in control (CTL) cells, P’s HVS-T cells (M/M), and P’s HVS-T cells transduced with pLenti-Cter-GFP-2A-puro containing the WT ERN1 or empty vector. (H and I) Flow cytometry analysis of intracellular IFN-γ production in control (CTL) cells, M/M HVS-T cells from P, or M/M HVS-T cells retrovirally transduced with EV or WT T-bet (MM+EV or MM+WT) or MM+WT HVS-T cells lentivirally transduced with the cDNA of EV or WT PSMD9, ARHGAP27 (H) and ERN1 (I). (J) Isolated CD4+ T cells from a healthy donor, a TBX21 WT/M parent and the TBX21 homozygous M/M P were expanded with anti-CD3/CD28 antibody-coated beads under TH0 or TH1 conditions. After 7d, EV or WT T-bet plasmids were used to transduce P’s cells in the presence of a bicistronic CD271 surface reporter. Cells were subjected to ICS for IFN-γ and TNF-α in response to P/I restimulation. The gating strategy for the experiment is shown. (K) Cells transduced as in (J) were isolated by MACS with anti-CD271 antibody-coated beads. Together with non-transduced cells, they were re-stimulated with anti-CD3/CD28 antibody-coated beads, and their culture supernatants were subjected to ELISA for IFN-γ and TNF-α.
In (A and B), bars represent the mean and the standard deviation; in (K), bars represent the mean and the standard error of the mean. Dots represent individual samples. One-way ANOVA was used for analysis in (B), comparing WT/M or M/M to CTL. ns = not significant.
(A) PBMCs were immunophenotyped with CyTOF. The gating strategy for different immune subsets in a healthy control is indicated. (B) Percentages of pDCs, cDC1 cells and cDC2 cells among adult healthy donors (CTL), age-matched controls (Age-matched CTL), IL-12Rβ1-deficient MSMD patients (IL12RB1 M/M) and P (TBX21 M/M), as indicated in (A). (C) PBMCs from P and a healthy control were subjected to the FACS immunophenotyping of innate lymphoid cells (ILCs) and NK cells. CD117+ ILCs, consisting principally of ILC precursors (ILCP) and type 2 ILC (ILC2) cells, were gated manually on conventional flow cytometry. The gating strategy is shown. (D) Total NK cells (CD16+ or CD94+Lin−CD7+ cells) were plotted with CD56 against CD127 for manual gating on different NK cell subsets. (E) Percentage of ILC precursor and ILC2 cells in adult healthy donors (CTL), age-matched controls (Age CTL), IL-12Rβ1-deficient MSMD patients (IL12RB1 M/M) and P (TBX21 M/M), summarized from CyTOF data, as in (A). (F) Immunophenotyping of ILC2 and ILCP cells, analyzed by fluorescence flow cytometry. Plot of ILC2 and ILCP, and the percentage of ILC2 and ILCP (within live CD45+ cells) in a healthy donor (CTL), the T-bet-deficient patient (M/M) and his age/ethnicity-matched healthy brother (WT/WT bro). (G) PBMCs from a CTL, IL-12Rβ1-deficient MSMD patients (IL12RB1 M/M), the TBX21 WT/M parents (WT/M mo or fa), the T-bet-deficient P (M/M) and his age/ethnicity-matched healthy brother (WT/WT bro) were stained with surface markers for lymphocytes involved innate immunity (MAIT and iNKT cells) or both adaptive and innate immunity (γδ T cells, in particular Vδ1 and Vδ2 γδ T cells). The gating strategy for flow cytometry is shown. (H - J) iNKT (H), MAIT (I) and Vδ2+ γδ T (J) cells are shown for P and his relatives. (K) Percentages of Vδ1+ γδ T cells in healthy adult donors (CTL), age-matched controls (Age CTL), IL-12Rβ1-deficient MSMD patients (IL12RB1 M/M) and P (TBX21 M/M), his age/ethnicity-matched healthy brother (TBX21 WT/WT) and his healthy parents (TBX21 WT/M).
In (B and E), bars represent the mean and the standard deviation. In (K), bars represent the mean and the standard error of the mean. Dots represent individual samples. Mann-Whitney nonparametric tests were used for analysis in (K). In (K), ** p < 0.01.
(A) Naïve (CD45RA+CCR7+), central memory (CM, CD45RA− CCR7+), effector memory (EM, CD45RA−CCR7−) and TEMRA (CD45RA+CCR7−) cells were manually gated from CD8+ T cells on CyTOF data. The percentages of these cells are shown. (B) 10,000 total CD8+ T cells were clustered by viSNE, based on a selection of surface markers. Manual gating of naïve and memory subsets overlaid on clusters of CD8+ T cells. CXCR3 and CD38 expression are indicated on this plot. (C) Naïve (CD45RA+CCR7+), central memory (CM, CD45RA−CCR7+), effector memory (EM, CD45RA−CCR7−) and TEMRA (CD45RA+CCR7−) cells were manually gated from CD4+ T cells on the basis of CyTOF data. The percentages of these cells are shown. (D) The gating strategy for the CyTOF analysis of memory CD4+ T cells and TH1, TH2, Th17 and TH1* cells is shown. (E) Memory CD4+ T cells (CD45RA−) from indicated individuals were gated out manually, and further subjected to viSNE deconvolution with T-cell chemokine markers for 10,000 randomly selected events. The expression of CXCR3, CCR5, CCR6 and CCR4 is shown. (F and G) Percentages of TH2 (CCR6−CXCR3−CCR4+) (F) and TH17 cells (CCR6+CXCR3−CCR4+) (G) among memory CD4+ T cells. (H) Frequencies of the circulating follicular T helper (cTFH) cells and their subsets (CXCR3+, double-negative as DN, CCR6+, or CXCR3+CCR6+ TFH) from healthy donors (CTL) and the T-bet-deficient patient (M/M) gated from conventional flow cytometry data. (I) Percentages of Treg cells gated from CyTOF data. (J) Percentages of CXCR3+ and CCR5+CXCR3− Tregs gated from CyTOF data. In (A, C, F, G, H, I and J), bars represent the mean and the standard error of the mean. Dots represent individual samples.
(A) Gating strategy for the surface and intracellular staining of PBMCs ex vivo stimulated with P/I, for an age-matched control sample. (B) Intracellular staining of IFN-γ and T-bet in NK cells, as shown in (A), from an age-matched control (Age CTL), P (M/M) and P’s healthy brother (WT/WT) stimulated with P/I. (C) Frequencies of TNF-α-producing cells among CD56+ NK cells, as gated in (A), in the indicated individuals. (D) The gating strategy for gating total NK cells from a travel control (CTL), P (M/M), P’s brother (WT/WT) and P’s heterozygous mother (WT/M), in the presence and absence of stimulation with IL-12, IL-15 and IL-18, is shown. (E) Expression of CD107a and IFN-γ among total NK cells, as in (D). (F) Frequencies of IFN-γ-producing cells among iNKT cells, as gated in (A) for the indicated individuals. (G and H) Vδ1−Vδ2− γδ T cells were gated from PBMCs prepared as shown in (A). Intracellular staining for IFN-γ (G) and TNF-α (H) is shown. (I and J) CD8+ T cells were gated from PBMCs prepared as shown in (A). Intracellular staining for IFN-γ (I) and TNF-α (J) is shown. (K - R) Naïve and memory CD4+ T cells from controls (CTL) and P (M/M) were sorted by FACS and activated/expanded. Intracellular staining of IL-22 (K), IL-9 (L) and IL-21 (R). Production of IL-9 (M), IL-4 (N), IL-5 (O), IL-10 (P) and IL-13 (Q), as measured in culture supernatants. (S and T) Naïve CD4+ T cells were isolated by FACS and subjected to activation under TH0, TH9, TH17 or TH9 conditions combined with TH17 conditions. The production of IL-9 was assessed intracellularly (S) and in culture supernatants (T). (U) Naïve CD4+ T cells were isolated by FACS and subjected to activation under TH0, TH1 or TH2 conditions. IL-10 production, determined with culture supernatants, is shown. (V) Naïve CD4+ T cells were isolated by FACS and subjected to activation under TH0, TH2 or TH9 conditions. IL-13 production, as measured on culture supernatants.
In (C, F - V), bars represent the mean and the standard error of the mean. Dots represent individual samples. Mann-Whitney non-parametric tests were used for analysis in (C, F - J). In (C, F - J), * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, ns = not significant.
(A and B) PBMCs from adult controls (CTL), age-matched controls (Age CTL), P (M/M), P’s healthy brother (WT/WT), and P’s healthy parents (WT/M) were left unstimulated or stimulated with live M. bovis BCG in the presence and absence of IL-12 and IL-23. The production of TNF-α (A) and IL-5 (B) in culture supernatants was assessed with Legendplex cytometric bead arrays. (C) PBMCs from P (M/M), P’s healthy brother (WT/WT) and one of P’s healthy parents (WT/M), stimulated as in (A), were analyzed for the intracellular expression of T-bet and IFN-γ by flow cytometry. Percentages of T-bet+ IFN-γ+ cells within live single lymphocytes are highlighted. (D) Pie-charts of the percentages of the indicated immune subsets in PBMCs from healthy controls stmulated with IL-12 or IL-23 alone. (E and F) PBMCs from adult controls (CTL), age-matched controls (Age CTL), P (M/M), P’s healthy brother (WT/WT), and P’s healthy parents (WT/M) were left unstimulated or were stimulated with live M. bovis BCG in the presence and absence of IL-12 and IL-23. Frequencies of T-bet+ IFN-γ+ cells gated on each individual immune subset, including NK, iNKT, MAIT, Vδ2+ γδ T (Vd2), Vδ1+ γδ T (Vd1) and Vδ1−Vδ2− γδ T (Vd1-Vd1-gdT) cells from different groups of individuals, stimulated with BCG (E) and BCG + IL-23 (F), are shown. (G) Frequencies of T-bet+ IFN-γ+ cells gated on adaptive immune subsets, including B, CD4+ T and CD8+ T cells from different groups of individuals, stimulated with BCG + IL-23. (H) Dot-plots showing the expression of intracellular T-bet and IFN-γ gated on CD4+ T cells with and without BCG stimulation in the presence and absence of IL-12 and IL-23. (I) Frequency of Vδ2+ γδ T cells in PBMCs from different groups of individuals expanded with BCG-lysates for 14 d. (J) Dot-plots showing intracellular T-bet and IFN-γ levels in non-responsive and PPD-responsive CD4+ T cells following PPD stimulation. (K and L) Dot-plots showing intracellular T-bet and IFN-γ levels in non-responsive and BCG-lysates-responsive CD4+ T (K) and CD8+ T cells (L).
In (A, B, E, F, G and I), bars represent the mean and the standard error of the mean. In (D), numbers represent the mean and the standard deviation. Dots represent individual samples. Mann-Whitney nonparametric tests were used for analysis in (A, B, E, F and G), * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, ns = not significant.
Cells were restimulated with anti-CD3/CD28 Ab-coated beads for 16 h. Lists of DEG_Status, DEG_FoldChange and DEG_ZScore identifying differentially regulated target genes genome-wide. Slim_DEG identifies differentially regulated target immune genes.
Related data were divided into five sub-sheets. Sheets titled ATAC_Closed by T-bet, ATAC_Opened by T-bet, and ATAC_61 genes above cutoff represent related data from Omni-ATAC-seq dataset generated from CD4+ T cells. Sheets titled Epic_array_MM<CTL and Epic_array_MM>CTL represent related data from the Epic-methylation CpG array.
Highlights (CELL-D-20-00152R1).
Human inherited T-bet deficiency underlies severe mycobacterial disease.
The development of a variety of IFN-γ-producing lymphocyte subsets is impaired.
The production of IFN-γ by innate and innate-like lymphocytes is impaired.
Mycobacterium-specific αβ CD4 TH1* and CD8 cells produce IFN-γ normally.
Human T-bet is involved in the regulation of distinct immune subsets compared to its mouse counterpart, with its deficiency associated with severe mycobacterial disease
Acknowledgments
We thank patients and families; members of the laboratory; Cecilia Lindestam Arlehamn and Alessandro Sette; NIH Tetramer Core Facility (NTCF); James McCluskey, Jamie Rossjohn, and David Fairlie; the Genomics and the Flow Cytometry Resource Center of Rockefeller University; National Center for Advancing Translational Sciences of the National Institutes of Health (UL1TR001866 to Rockefeller University); Steven Elledge; National Institute of Allergy and Infectious Diseases (R37AI095983 to J.L.C and U19AI118626 to F.S.); Sackler Center for Biomedicine and Nutrition at the Center for Clinical and Translational Science (CCTS), Shapiro-Silverberg Fund for the Advancement of Translational Research at CCTS, Rockefeller University (to R.Y.), Research Grant Program of the Immune Deficiency Foundation (to R.Y.), Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence (ANR-10-LABX-62-IBEID), French National Research Agency under the “Investments for the future” program (ANR-10-IAHU-01), ANR-GENMSMD (ANR-16-CE17-0005-01 to J.B.), ANR-LTh-MSMD-CMCD (ANR-18-CE93-0008-01 to A.P), Fonds de Recherche en Santé Respiratoire (SRC2017 to J.B.), French Foundation for Medical Research (FRM) (EQU201903007798), the SCOR Corporate Foundation for Science, ECOS Nord (C19S01-63407 to J.B.), Swiss National Science Foundation (310030L_182728 to F.S.), French Foundation for Medical Research (FRM) (EQU201903007798), Helmut Horten Foundation; Canadian Center for Computational Genomics (C3G), Genomics Technology Platform (GTP) and Genome Canada. J. R. was supported by INSERM Poste d’accueil; T.K. by Qatar National Research Fund (PPM1-1220-150017). S.G.T. by Program grants (1113904), Principal Research Fellowship (1042925), and Leadership 3 Investigator grant (1176665) from the National Health and Medical Research Council of Australia, C.S.M. by an Early/Mid-Career Development Research Fellowship, Ministry of Health, NSW Government; R.Y. by Stony Wold-Herbert Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
Dr. Glimcher serves on the Board of Directors of GlaxoSmithKline Pharmaceutical Company and the Analog Device Corporation and formerly served on the Boards of Bristol Myers Squibb Pharmaceutical Company and the Waters Corporation. She is also on the Scientific Advisory Boards of Abpro, Kaleido, and Repare biotechnology companies. Dr. Casanova serves on the Scientific Advisory Boards of ADMA Biologics Inc., Celgene and Kymera Therapeutics, Inc.. He also consults for Elixiron Immunotherapeutics. Other authors declare no competing interests.
References
- Abecasis GR, Cherny SS, Cookson WO, and Cardon LR (2002). Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet 30, 97–101. [DOI] [PubMed] [Google Scholar]
- Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, Sallusto F, and Napolitani G (2007). Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat. Immunol 8, 639–646. [DOI] [PubMed] [Google Scholar]
- Afgan E, Baker D, van den Beek M, Blankenberg D, Bouvier D, Čech M, Chilton J, Clements D, Coraor N, Eberhard C, et al. (2016). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 44, W3–W10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aluri J, Gupta M, Dalvi A, Mhatre S, Kulkarni M, Hule G, Desai M, Shah N, Taur P, Vedam R, et al. (2018). Clinical, immunological, and molecular findings in five patients with Major histocompatibility complex class II deficiency from India. Front. Immunol 9, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir el AD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP, and Pe’er D (2013). viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol 31, 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardeniz Ö, Unger S, Onay H, Ammann S, Keck C, Cianga C, Gerçeker B, Martin B, Fuchs I, Salzer U, et al. (2015). β2-Microglobulin deficiency causes a complex immunodeficiency of the innate and adaptive immune system. J. Allergy Clin. Immunol 136, 392–401. [DOI] [PubMed] [Google Scholar]
- Auton A, Abecasis GR, Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, Donnelly P, Eichler EE, et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakshi CS, Malik M, Carrico PM, and Sellati TJ (2006). T-bet deficiency facilitates airway colonization by Mycoplasma pulmonis in a murine model of asthma. J Immunol 177, 1786–1795. [DOI] [PubMed] [Google Scholar]
- Becattini S, Latorre D, Mele F, Foglierini M, De Gregorio C, Cassotta A, Fernandez B, Kelderman S, Schumacher TN, Corti D, et al. (2015). T cell immunity. Functional heterogeneity of human memory CD4(+) T cell clones primed by pathogens or vaccines. Science (80-. ). 347, 400–406. [DOI] [PubMed] [Google Scholar]
- Becht E, McInnes L, Healy J, Dutertre C-A, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2019). Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol 37, 38–44. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, and Kuchroo VK (2004). Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med 200, 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björkström NK, Ljunggren H-G, and Sandberg JK (2010). CD56 negative NK cells: origin, function, and role in chronic viral disease. Trends Immunol. 31, 401–406. [DOI] [PubMed] [Google Scholar]
- Bogunovic D, Byun M, Durfee LA, Abhyankar A, Sanal O, Mansouri D, Salem S, Radovanovic I, Grant AV, Adimi P, et al. (2012). Mycobacterial disease and impaired IFN-gamma immunity in humans with inherited ISG15 deficiency. Science (80-. ). 337, 1684–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisson-Dupuis S, Ramirez-Alejo N, Li Z, Patin E, Rao G, Kerner G, Lim CK, Krementsov DN, Hernandez N, Ma CS, et al. (2018). Tuberculosis and impaired IL-23-dependent IFN-γ immunity in humans homozygous for a common TYK2 missense variant. Sci. Immunol 3, eaau8714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109, 21 29 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante J (2020). Mendelian susceptibility to mycobacterial disease: recent discoveries. Hum. Genet 139, 993–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll T, and Barrows D (2019). profileplyr: Visualization and annotation of read signal over genomic ranges with profileplyr. [Google Scholar]
- Carroll TS, Liang Z, Salama R, Stark R, and de Santiago I (2014). Impact of artifact removal on ChIP quality metrics in ChIP-seq and ChIP-exo data. Front. Genet 5, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova JL (1999). IL-12 and IFN-γ in host defense against mycobacteria and salmonella in mice and men. Curr. Opin. Immunol 11, 346–351. [DOI] [PubMed] [Google Scholar]
- Casanova JL (2015a). Human genetic basis of interindividual variability in the course of infection. Proc. Natl. Acad. Sci. U. S. A 112, E7118–E7127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova JL (2015b). Severe infectious diseases of childhood as monogenic inborn errors of immunity. Proc. Natl. Acad. Sci. U. S. A 112, E7128–E7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova JL, and Abel L (2004). The human model: A genetic dissection of immunity to infection in natural conditions. Nat. Rev. Immunol 4, 55–66. [DOI] [PubMed] [Google Scholar]
- Casanova JL, and Abel L (2018). Human genetics of infectious diseases: Unique insights into immunological redundancy. Semin. Immunol 36, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova J-L, Conley ME, Seligman SJ, Abel L, and Notarangelo LD (2014). Guidelines for genetic studies in single patients: lessons from primary immunodeficiencies. J. Exp. Med 211, 2137–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova JL, Jouanguy E, Lamhamedi S, Blanche S, and Fischer A (1995). Immunological conditions of children with BCG disseminated infection. Lancet (London, England) 346, 581. [DOI] [PubMed] [Google Scholar]
- Casey JP, Nobbs M, McGettigan P, Lynch S, and Ennis S (2012). Recessive mutations in MCM4/PRKDC cause a novel syndrome involving a primary immunodeficiency and a disorder of DNA repair. J. Med. Genet 49, 242–245. [DOI] [PubMed] [Google Scholar]
- Chen ZW (2005). Immune regulation of gammadelta T cell responses in mycobacterial infections. Clin. Immunol 116, 202–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, He W, Kim ST, Tao J, Gao Y, Chi H, Intlekofer AM, Harvey B, Reiner SL, Yin Z, et al. (2007). Epigenetic and Transcriptional Programs Lead to Default IFN-γ Production by γδ T Cells. J. Immunol 178, 2730–2736. [DOI] [PubMed] [Google Scholar]
- Chien Y, Meyer C, and Bonneville M (2014). γδ T Cells: First Line of Defense and Beyond. Annu. Rev. Immunol 32, 121–155. [DOI] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, and Ruden DM (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 6, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottineau J, Kottemann MC, Lach FP, Kang Y-H, Vély F, Deenick EK, Lazarov T, Gineau L, Wang Y, Farina A, et al. (2017). Inherited GINS1 deficiency underlies growth retardation along with neutropenia and NK cell deficiency. J. Clin. Invest 127, 1991–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby CM, and Kronenberg M (2018). Tissue-specific functions of invariant natural killer T cells. Nat. Rev. Immunol 18, 559–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daussy C, Faure F, Mayol K, Viel S, Gasteiger G, Charrier E, Bienvenu J, Henry T, Debien E, Hasan UA, et al. (2014). T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J. Exp. Med 211, 563–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BR, Dicola MJ, Prokopishyn NL, Rosenberg JB, Moratto D, Muul LM, Candotti F, and Michael Blaese R (2008). Unprecedented diversity of genotypic revertants in lymphocytes of a patient with Wiskott-Aldrich syndrome. Blood 111, 5064–5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, Del Angel G, Rivas MA, Hanna M, et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet 43, 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova D, Ong PY, O’Gorman MRG, and Church JA (2014). Major Histocompatibility Complex Class II Deficiency Complicated by Mycobacterium avium Complex in a Boy of Mixed Ethnicity. J. Clin. Immunol 34, 677–680. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorner BG, Scheffold A, Rolph MS, Huser MB, Kaufmann SHE, Radbruch A, Flesch IEA, and Kroczek RA (2002). MIP-1 , MIP-1 , RANTES, and ATAC/lymphotactin function together with IFN- as type 1 cytokines. Proc. Natl. Acad. Sci 99, 6181–6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorner BG, Steinbach S, Hüser MB, Kroczek RA, and Scheffold A (2003). Single-cell analysis of the murine chemokines MIP-1alpha, MIP-1beta, RANTES and ATAC/lymphotactin by flow cytometry. J. Immunol. Methods 274, 83–91. [DOI] [PubMed] [Google Scholar]
- Dorner BG, Smith HRC, French AR, Kim S, Poursine-Laurent J, Beckman DL, Pingel JT, Kroczek RA, and Yokoyama WM (2004). Coordinate Expression of Cytokines and Chemokines by NK Cells during Murine Cytomegalovirus Infection. J. Immunol 172, 3119–3131. [DOI] [PubMed] [Google Scholar]
- Dupuis S, Döffinger R, Picard C, Fieschi C, Altare F, Jouanguy E, Abel L, and Casanova JL (2000). Human interferon-gamma-mediated immunity is a genetically controlled continuous trait that determines the outcome of mycobacterial invasion. Immunol. Rev 178, 129–137. [DOI] [PubMed] [Google Scholar]
- Elhasid R, and Etzioni A (1996). Major histocompatibility complex class II deficiency: a clinical review. Blood Rev. 10, 242–248. [DOI] [PubMed] [Google Scholar]
- Feng J, Liu T, Qin B, Zhang Y, and Liu XS (2012). Identifying ChIP-seq enrichment using MACS. Nat. Protoc 7, 1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, et al. (2015). MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finotto S, Neurath MF, Glickman JN, Qin S, Lehr HA, Green FH, Ackerman K, Haley K, Galle PR, Szabo SJ, et al. (2002). Development of spontaneous airway changes consistent with human asthma in mice lacking T-bet. Science (80-. ). 295, 336–338. [DOI] [PubMed] [Google Scholar]
- Frankish A, Diekhans M, Ferreira A-M, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, et al. (2019). GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett WS, Lord GM, Punit S, Lugo-Villarino G, Mazmanian SK, Ito S, Glickman JN, and Glimcher LH (2007). Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell 131, 33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido-Mesa N, Schroeder J-H, Stolarczyk E, Gallagher AL, Lo JW, Bailey C, Campbell L, Sexl V, MacDonald TT, Howard JK, et al. (2019). T-bet controls intestinal mucosa immune responses via repression of type 2 innate lymphoid cell function. Mucosal Immunol. 12, 51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, and Saeys Y (2015). FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytom. Part A 87, 636–645. [DOI] [PubMed] [Google Scholar]
- Gavins FNE, and Hickey MJ (2012). Annexin A1 and the regulation of innate and adaptive immunity. Front. Immunol 3, 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger R, Duhen T, Lanzavecchia A, and Sallusto F (2009). Human naive and memory CD4+ T cell repertoires specific for naturally processed antigens analyzed using libraries of amplified T cells. J. Exp. Med 206, 1525–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geurts P, Ernst D, and Wehenkel L (2006). Extremely randomized trees. Mach. Learn 63, 3–42. [Google Scholar]
- Godfrey DI, Koay H-F, McCluskey J, and Gherardin NA (2019). The biology and functional importance of MAIT cells. Nat. Immunol 20, 1110–1128. [DOI] [PubMed] [Google Scholar]
- Gökmen MR, Dong R, Kanhere A, Powell N, Perucha E, Jackson I, Howard JK, Hernandez-Fuentes M, Jenner RG, and Lord GM (2013). Genome-Wide Regulatory Analysis Reveals That T-bet Controls Th17 Lineage Differentiation through Direct Suppression of IRF4. J. Immunol 191, 5925–5932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groom JR, and Luster AD (2011). CXCR3 in T cell function. Exp. Cell Res 317, 620–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu S, Borowska MT, Boughter CT, and Adams EJ (2018). Butyrophilin3A proteins and Vγ9Vδ2 T cell activation. Semin. Cell Dev. Biol 84, 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. [DOI] [PubMed] [Google Scholar]
- Hambleton S, Salem S, Bustamante J, Bigley V, Boisson-Dupuis S, Azevedo J, Fortin A, Haniffa M, Ceron-Gutierrez L, Bacon CM, et al. (2011). IRF8 mutations and human dendritic-cell immunodeficiency. N. Engl. J. Med 365, 127–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanalioglu D, Ayvaz DC, Ozgur TT, van der Burg M, Sanal O, and Tezcan I (2017). A novel mutation in TAP1 gene leading to MHC class I deficiency: Report of two cases and review of the literature. Clin. Immunol 178, 74–78. [DOI] [PubMed] [Google Scholar]
- Hanna S, and Etzioni A (2014). MHC class i and II deficiencies. J. Allergy Clin. Immunol 134, 269–275. [DOI] [PubMed] [Google Scholar]
- Harayama T, and Riezman H (2017). Detection of genome-edited mutant clones by a simple competition-based PCR method. PLoS One 12, e0179165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harly C, Guillaume Y, Nedellec S, Peigne CM, Monkkonen H, Monkkonen J, Li J, Kuball J, Adams EJ, Netzer S, et al. (2012). Key implication of CD277/butyrophilin-3 (BTN3A) in cellular stress sensing by a major human gammadelta T-cell subset. Blood 120, 2269–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms Pritchard G, Hall AO, Christian DA, Wagage S, Fang Q, Muallem G, John B, Glatman Zaretsky A, Dunn WG, Perrigoue J, et al. (2015). Diverse roles for T-bet in the effector responses required for resistance to infection. J Immunol 194, 1131–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez N, Melki I, Jing H, Habib T, Huang SSY, Danielson J, Kula T, Drutman S, Belkaya S, Rattina V, et al. (2018). Life-threatening influenza pneumonitis in a child with inherited IRF9 deficiency. J. Exp. Med 215, 2567–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoft DF, Brown RM, and Roodman ST (1998). Bacille Calmette-Guerin vaccination enhances human gamma delta T cell responsiveness to mycobacteria suggestive of a memory-like phenotype. J Immunol 161, 1045–1054. [PubMed] [Google Scholar]
- Hughes CR, Guasti L, Meimaridou E, Chuang CH, Schimenti JC, King PJ, Costigan C, Clark AJL, and Metherell LA (2012). MCM4 mutation causes adrenal failure, short stature, and natural killer cell deficiency in humans. J. Clin. Invest 122, 814–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultgren OH, Verdrengh M, and Tarkowski A (2004). T-box transcription-factor-deficient mice display increased joint pathology and failure of infection control during staphylococcal arthritis. Microbes Infect 6, 529–535. [DOI] [PubMed] [Google Scholar]
- Intlekofer AM, Takemoto N, Kao C, Banerjee A, Schambach F, Northrop JK, Shen H, Wherry EJ, and Reiner SL (2007). Requirement for T-bet in the aberrant differentiation of unhelped memory CD8+ T cells. J Exp Med 204, 2015–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itan Y, Zhang S-Y, Vogt G, Abhyankar A, Herman M, Nitschke P, Fried D, Quintana-Murci L, Abel L, and Casanova J-L (2013). The human gene connectome as a map of short cuts for morbid allele discovery. Proc. Natl. Acad. Sci 110, 5558–5563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itan Y, Mazel M, Mazel B, Abhyankar A, Nitschke P, Quintana-Murci L, Boisson-Dupuis S, Boisson B, Abel L, Zhang S-Y, et al. (2014). HGCS: an online tool for prioritizing disease-causing gene variants by biological distance. BMC Genomics 15, 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itan Y, Shang L, Boisson B, Patin E, Bolze A, Moncada-Velez M, Scott E, Ciancanelli MJ, Lafaille FG, Markle JG, et al. (2015). The human gene damage index as a gene-level approach to prioritizing exome variants. Proc Natl Acad Sci U S A 112, 13615–13620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itan Y, Shang L, Boisson B, Ciancanelli MJ, Markle JG, Martinez-Barricarte R, Scott E, Shah I, Stenson PD, Gleeson J, et al. (2016). The mutation significance cutoff: gene-level thresholds for variant predictions. Nat Methods 13, 109–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata S, Mikami Y, Sun H-W, Brooks SR, Jankovic D, Hirahara K, Onodera A, Shih H-Y, Kawabe T, Jiang K, et al. (2017). The Transcription Factor T-bet Limits Amplification of Type I IFN Transcriptome and Circuitry in T Helper 1 Cells. Immunity 46, 983–991.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janesick A, Shiotsugu J, Taketani M, and Blumberg B (2012). RIPPLY3 is a retinoic acid-inducible repressor required for setting the borders of the pre-placodal ectoderm. Development 139, 1213–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang EJ, Park HR, Hong JH, and Hwang ES (2013). Lysine 313 of T-box is crucial for modulation of protein stability, DNA binding, and threonine phosphorylation of T-bet. J Immunol 190, 5764–5770. [DOI] [PubMed] [Google Scholar]
- Jenner RG, Townsend MJ, Jackson I, Sun K, Bouwman RD, Young RA, Glimcher LH, and Lord GM (2009). The transcription factors T-bet and GATA-3 control alternative pathways of T-cell differentiation through a shared set of target genes. Proc. Natl. Acad. Sci. U.S. A 106, 17876–17881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juedes AE, Rodrigo E, Togher L, Glimcher LH, and von Herrath MG (2004). T-bet controls autoaggressive CD8 lymphocyte responses in type 1 diabetes. J Exp Med 199, 1153–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanhere A, Hertweck A, Bhatia U, Gökmen MR, Perucha E, Jackson I, Lord GM, and Jenner RG (2012). T-bet and GATA3 orchestrate Th1 and Th2 differentiation through lineage-specific targeting of distal regulatory elements. Nat. Commun 3, 1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao C, Oestreich KJ, Paley MA, Crawford A, Angelosanto JM, Ali MA, Intlekofer AM, Boss JM, Reiner SL, Weinmann AS, et al. (2011). Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat Immunol 12, 663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerner G, Rosain J, Guérin A, Al-Khabaz A, Oleaga-Quintas C, Rapaport F, Massaad MJ, Ding JY, Khan T, Ali F. Al, et al. (2020). Inherited human IFN-γ deficiency underlies mycobacterial disease. J. Clin. Invest 130, 3158–3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, and Shendure J (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46, 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjer-Nielsen L, Patel O, Corbett AJ, Le Nours J, Meehan B, Liu L, Bhati M, Chen Z, Kostenko L, Reantragoon R, et al. (2012). MR1 presents microbial vitamin B metabolites to MAIT cells. Nature 491, 717–723. [DOI] [PubMed] [Google Scholar]
- Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, and Campbell DJ (2009). The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat. Immunol 10, 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong XF, Martinez-Barricarte R, Kennedy J, Mele F, Lazarov T, Deenick EK, Ma CS, Breton G, Lucero KB, Langlais D, et al. (2018). Disruption of an antimycobacterial circuit between dendritic and helper T cells in human SPPL2a deficiency. Nat Immunol 19, 973–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh P. ru, and Raychaudhuri S (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroczek RA, and Henn V (2012). The Role of XCR1 and its Ligand XCL1 in Antigen Cross-Presentation by Murine and Human Dendritic Cells. Front. Immunol 3, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Salle H, Hanau D, Fricker D, Urlacher A, Kelly A, Salamero J, Powis SH, Donato L, Bausinger H, Laforet M, et al. (1994). Homozygous human TAP peptide transporter mutation in HLA class I deficiency. Science (80-. ). 265, 237–241. [DOI] [PubMed] [Google Scholar]
- Latour S (2007). Natural killer T cells and X-linked lymphoproliferative syndrome. Curr. Opin. Allergy Clin. Immunol 7, 510–514. [DOI] [PubMed] [Google Scholar]
- Lazarevic V, Chen X, Shim JH, Hwang ES, Jang E, Bolm AN, Oukka M, Kuchroo VK, and Glimcher LH (2011). T-bet represses TH 17 differentiation by preventing Runx1-mediated activation of the gene encoding RORγt. Nat. Immunol 12, 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarevic V, Glimcher LH, and Lord GM (2013). T-bet: a bridge between innate and adaptive immunity. Nat Rev Immunol 13, 777–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrun A, Portocarrero C, Kean RB, Barkhouse DA, Faber M, and Hooper DC (2015). T-bet Is Required for the Rapid Clearance of Attenuated Rabies Virus from Central Nervous System Tissue. J Immunol 195, 4358–4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AG, Mendoza A, Hemmers S, Moltedo B, Niec RE, Schizas M, Hoyos BE, Putintseva EV, Chaudhry A, Dikiy S, et al. (2017). Stability and function of regulatory T cells expressing the transcription factor T-bet. Nature 546, 421–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis MD, Miller SA, Miazgowicz MM, Beima KM, and Weinmann AS (2007). T-bet’s ability to regulate individual target genes requires the conserved T-box domain to recruit histone methyltransferase activity and a separate family member-specific transactivation domain. Mol. Cell. Biol 27, 8510–8521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim AI, Li Y, Lopez-Lastra S, Stadhouders R, Paul F, Casrouge A, Serafini N, Puel A, Bustamante J, Surace L, et al. (2017). Systemic Human ILC Precursors Provide a Substrate for Tissue ILC Differentiation. Cell 168, 1086–1100.e10. [DOI] [PubMed] [Google Scholar]
- Lindestam Arlehamn CS, McKinney DM, Carpenter C, Paul S, Rozot V, Makgotlho E, Gregg Y, van Rooyen M, Ernst JD, Hatherill M, et al. (2016). A Quantitative Analysis of Complexity of Human Pathogen-Specific CD4 T Cell Responses in Healthy M. tuberculosis Infected South Africans. PLOS Pathog. 12, e1005760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Okada S, Kong X-F, Kreins AY, Cypowyj S, Abhyankar A, Toubiana J, Itan Y, Audry M, Nitschke P, et al. (2011). Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J. Exp. Med 208, 1635–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locci M, Ghici ED, Marangoni F, Bosticardo M, Catucci M, Aiuti A, Cancrini C, Marodi L, Espanol T, Bredius RGM, et al. (2009). The Wiskott-Aldrich syndrome protein is required for iNKT cell maturation and function. J. Exp. Med 206, 735–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loetscher P, Uguccioni M, Bordoli L, Baggiolini M, Moser B, Chizzolini C, and Dayer J-M (1998). CCR5 is characteristic of Th1 lymphocytes. Nature 391, 344–345. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugo-Villarino G, Maldonado-Lopez R, Possemato R, Penaranda C, and Glimcher LH (2003). T-bet is required for optimal production of IFN-gamma and antigen-specific T cell activation by dendritic cells. Proc Natl Acad Sci U S A 100, 7749–7754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum SH, Neven B, Slatter MA, and Gennery AR (2019). Hematopoietic Cell Transplantation for MHC Class II Deficiency. Front. Pediatr 7, 516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum SH, Anderson C, McNaughton P, Engelhardt KR, MacKenzie B, Watson H, Al-Mousa H, Al-Herz W, Al-Saud B, Mohammed R, et al. (2020). Improved transplant survival and long-term disease outcome in children with MHC class II deficiency. Blood 135, 954–973. [DOI] [PubMed] [Google Scholar]
- Lund R, Ahlfors H, Kainonen E, Lahesmaa A-M, Dixon C, and Lahesmaa R (2005). Identification of genes involved in the initiation of human Th1 or Th2 cell commitment. Eur. J. Immunol 35, 3307–3319. [DOI] [PubMed] [Google Scholar]
- Ma CS, Wong N, Rao G, Avery DT, Torpy J, Hambridge T, Bustamante J, Okada S, Stoddard JL, Deenick EK, et al. (2015). Monogenic mutations differentially affect the quantity and quality of T follicular helper cells in patients with human primary immunodeficiencies. J. Allergy Clin. Immunol 136, 993–1006.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Noble WS, and Bailey TL (2014). Motif-based analysis of large nucleotide data sets using MEME-ChIP. Nat. Protoc 9, 1428–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Barricarte R, de Jong SJ, Markle J, de Paus R, Boisson-Dupuis S, Bustamante J, van de Vosse E, Fleckenstein B, and Casanova J-L (2016). Transduction of Herpesvirus saimiri -Transformed T Cells with Exogenous Genes of Interest In Current Protocols in Immunology, (Hoboken, NJ, USA: John Wiley & Sons, Inc.), pp. 7.21C.1–7.21C.12. [DOI] [PubMed] [Google Scholar]
- Martínez-Barricarte R, Markle JG, Ma CS, Deenick EK, Ramírez-Alejo N, Mele F, Latorre D, Mahdaviani SA, Aytekin C, Mansouri D, et al. (2018). Human IFN- immunity to mycobacteria is governed by both IL-12 and IL-23. Sci. Immunol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama M, Ishii Y, Yageta Y, Ohtsuka S, Ano S, Matsuno Y, Morishima Y, Yoh K, Takahashi S, Ogawa K, et al. (2014). Role of Th1/Th17 balance regulated by T-bet in a mouse model of Mycobacterium avium complex disease. J Immunol 192, 1707–1717. [DOI] [PubMed] [Google Scholar]
- McGinnis CS, Murrow LM, and Gartner ZJ (2019). DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 8, 329–337.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. (2010). The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SA, and Weinmann AS (2010). Molecular mechanisms by which T-bet regulates T-helper cell commitment. Immunol Rev 238, 233–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modlin RL, Pirmez C, Hofman FM, Torigian V, Uyemura K, Rea TH, Bloom BR, and Brenner MB (1989). Lymphocytes bearing antigen-specific γδ T-cell receptors accumulate in human infectious disease lesions. Nature 339, 544–548. [DOI] [PubMed] [Google Scholar]
- Morgan NV, Goddard S, Cardno TS, McDonald D, Rahman F, Barge D, Ciupek A, Straatman-Iwanowska A, Pasha S, Guckian M, et al. (2011). Mutation in the TCRα subunit constant gene (TRAC) leads to a human immunodeficiency disorder characterized by a lack of TCRαβ+ T cells. J. Clin. Invest 121, 695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan CF, Murray HW, Wiebe ME, and Rubin BY (1983). Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J Exp Med 158, 670–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikomori R, Usui T, Wu C-Y, Morinobu A, O’Shea JJ, and Strober W (2002). Activated STAT4 has an essential role in Th1 differentiation and proliferation that is independent of its role in the maintenance of IL-12R beta 2 chain expression and signaling. J. Immunol 169, 4388–4398. [DOI] [PubMed] [Google Scholar]
- Oakley MS, Sahu BR, Lotspeich-Cole L, Solanki NR, Majam V, Pham PT, Banerjee R, Kozakai Y, Derrick SC, Kumar S, et al. (2013). The transcription factor T-bet regulates parasitemia and promotes pathogenesis during Plasmodium berghei ANKA murine malaria. J Immunol 191, 4699–4708. [DOI] [PubMed] [Google Scholar]
- Okada S, Markle JG, Deenick EK, Mele F, Averbuch D, Lagos M, Alzahrani M, Al-Muhsen S, Halwani R, Ma CS, et al. (2015). Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science (80-. ). 349, 606–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchamoorthy G, McLean J, Modlin RL, Morita CT, Ishikawa S, Brenner MB, and Band H (1991). A predominance of the T cell receptor V gamma 2/V delta 2 subset in human mycobacteria-responsive T cells suggests germline gene encoded recognition. J. Immunol 147, 3360–3369. [PubMed] [Google Scholar]
- Parker CM, Groh V, Band H, Porcelli SA, Morita C, Fabbi M, Glass D, Strominger JL, and Brenner MB (1990). Evidence for extrathymic changes in the T cell receptor gamma/delta repertoire. J. Exp. Med 171, 1597–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce EL, Mullen AC, Martins GA, Krawczyk CM, Hutchins AS, Zediak VP, Banica M, DiCioccio CB, Gross DA, Mao C-A, et al. (2003). Control of Effector CD8+ T Cell Function by the Transcription Factor Eomesodermin. Science (80-. ). 302, 1041–1043. [DOI] [PubMed] [Google Scholar]
- Petrovski S, Gussow AB, Wang Q, Halvorsen M, Han Y, Weir WH, Allen AS, and Goldstein DB (2015). The Intolerance of Regulatory Sequence to Genetic Variation Predicts Gene Dosage Sensitivity. PLoS Genet 11, e1005492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell N, Walker AW, Stolarczyk E, Canavan JB, Gokmen MR, Marks E, Jackson I, Hashim A, Curtis MA, Jenner RG, et al. (2012). The Transcription Factor T-bet Regulates Intestinal Inflammation Mediated by Interleukin-7 Receptor+ Innate Lymphoid Cells. Immunity 37, 674–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin LC, Groom JR, Chopin M, Herold MJ, Walker JA, Mielke LA, McKenzie ANJ, Carotta S, Nutt SL, and Belz GT (2013). The transcription factor T-bet is essential for the development of NKp46+ innate lymphocytes via the Notch pathway. Nat. Immunol 14, 389–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindran R, Foley J, Stoklasek T, Glimcher LH, and McSorley SJ (2005). Expression of T-bet by CD4 T cells is essential for resistance to Salmonella infection. J Immunol 175, 4603–4610. [DOI] [PubMed] [Google Scholar]
- Revy P, Kannengiesser C, and Fischer A (2019). Somatic genetic rescue in Mendelian haematopoietic diseases. Nat. Rev. Genet 20, 582–598. [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosain J, Kong XF, Martinez-Barricarte R, Oleaga-Quintas C, Ramirez-Alejo N, Markle J, Okada S, Boisson-Dupuis S, Casanova JL, and Bustamante J (2019). Mendelian susceptibility to mycobacterial disease: 2014–2018 update. Immunol. Cell Biol 97, 360–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas LE, Snider HM, Barbi J, Satoskar AA, Lugo-Villarino G, Keiser T, Papenfuss T, Durbin JE, Radzioch D, Glimcher LH, et al. (2006). Cutting edge: STAT1 and T-bet play distinct roles in determining outcome of visceral leishmaniasis caused by Leishmania donovani. J Immunol 177, 22–25. [DOI] [PubMed] [Google Scholar]
- Saleem MA, Arkwright PD, Davies EG, Cant AJ, and Veys PA (2000). Clinical course of patients with major histocompatibility complex class II deficiency. Arch. Dis. Child 83, 356–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F, Lenig D, Mackay CR, and Lanzavecchia A (1998). Flexible Programs of Chemokine Receptor Expression on Human Polarized T Helper 1 and 2 Lymphocytes. J. Exp. Med 187, 875–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Santiago I, and Carroll T (2018). Analysis of ChIP-seq Data in R/Bioconductor. pp. 195–226. [DOI] [PubMed] [Google Scholar]
- Schoenborn JR, and Wilson CB (2007). Regulation of Interferon-γ During Innate and Adaptive Immune Responses. Adv. Immunol 96, 41–101. [DOI] [PubMed] [Google Scholar]
- Scott EM, Halees A, Itan Y, Spencer EG, He Y, Azab MA, Gabriel SB, Belkadi A, Boisson B, Abel L, et al. (2016). Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat. Genet 48, 1071–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Zhou D, Qiu L, Lai X, Simon M, Shen L, Kou Z, Wang Q, Jiang L, Estep J, et al. (2002). Adaptive immune response of Vgamma2Vdelta2+ T cells during mycobacterial infections. Science (80-. ). 295, 2255–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soutto M, Zhang F, Enerson B, Tong Y, Boothby M, and Aune TM (2002). A minimal IFN-gamma promoter confers Th1 selective expression. J Immunol 169, 4205–4212. [DOI] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan BM, Juedes A, Szabo SJ, Von Herrath M, and Glimcher LH (2003). Antigen-driven effector CD8 T cell function regulated by T-bet. Proc. Natl. Acad. Sci. U. S. A 100, 15818–15823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan BM, Jobe O, Lazarevic V, Vasquez K, Bronson R, Glimcher LH, and Kramnik I (2005). Increased susceptibility of mice lacking T-bet to infection with Mycobacterium tuberculosis correlates with increased IL-10 and decreased IFN-gamma production. J Immunol 175, 4593–4602. [DOI] [PubMed] [Google Scholar]
- Svensson A, Nordstrom I, Sun JB, and Eriksson K (2005). Protective immunity to genital herpes simplex [correction of simpex] virus type 2 infection is mediated by T-bet. J Immunol 174, 6266–6273. [DOI] [PubMed] [Google Scholar]
- Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, and Glimcher LH (2000). A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100, 655–669. [DOI] [PubMed] [Google Scholar]
- Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, and Glimcher LH (2002). Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science (80-. ). 295, 338–342. [DOI] [PubMed] [Google Scholar]
- Taliun D, Harris DN, Kessler MD, Carlson J, Szpiech ZA, Torres R, Taliun SAG, Corvelo A, Gogarten SM, Kang HM, et al. (2019). Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. BioRxiv 563866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan TG, Mathis D, and Benoist C (2016). Singular role for T-BET+CXCR3+ regulatory T cells in protection from autoimmune diabetes. Proc. Natl. Acad. Sci. U. S. A 113, 14103–14108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Desierto MJ, Chen J, and Young NS (2010). The role of the Th1 transcription factor T-bet in a mouse model of immune-mediated bone-marrow failure. Blood 115, 541–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tangye SG, Palendira U, and Edwards ESJ (2017). Human immunity against EBV— lessons from the clinic. J. Exp. Med 214, 269–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tantisira KG, Hwang ES, Raby BA, Silverman ES, Lake SL, Richter BG, Peng SL, Drazen JM, Glimcher LH, and Weiss ST (2004). TBX21: a functional variant predicts improvement in asthma with the use of inhaled corticosteroids. Proc Natl Acad Sci U S A 101, 18099–18104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thieu VT, Yu Q, Chang H-C, Yeh N, Nguyen ET, Sehra S, and Kaplan MH (2008). Signal transducer and activator of transcription 4 is required for the transcription factor T-bet to promote T helper 1 cell-fate determination. Immunity 29, 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Aune T, and Boothby M (2005). T-bet antagonizes mSin3a recruitment and transactivates a fully methylated IFN-gamma promoter via a conserved T-box half-site. Proc Natl Acad Sci U S A 102, 2034–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toribio-Fernández R, Zorita V, Rocha-Perugini V, Iborra S, Martínez del Hoyo G, Chevre R, Dorado B, Sancho D, Sanchez-Madrid F, Andrés V, et al. (2018). Lamin A/C augments Th1 differentiation and response against vaccinia virus and Leishmania major. Cell Death Dis. 9, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend MJ, Weinmann AS, Matsuda JL, Salomon R, Farnham PJ, Biron CA, Gapin L, and Glimcher LH (2004). T-bet regulates the terminal maturation and homeostasis of NK and Valpha14i NKT cells. Immunity 20, 477–494. [DOI] [PubMed] [Google Scholar]
- Tsukaguchi K, Balaji KN, and Boom WH (1995). CD4+ alpha beta T cell and gamma delta T cell responses to Mycobacterium tuberculosis. Similarities and differences in Ag recognition, cytotoxic effector function, and cytokine production. J. Immunol 154, 1786–1796. [PubMed] [Google Scholar]
- Vavassori S, Kumar A, Wan GS, Ramanjaneyulu GS, Cavallari M, El Daker S, Beddoe T, Theodossis A, Williams NK, Gostick E, et al. (2013). Butyrophilin 3A1 binds phosphorylated antigens and stimulates human gammadelta T cells. Nat Immunol 14, 908–916. [DOI] [PubMed] [Google Scholar]
- Velu V, Mylvaganam GH, Gangadhara S, Hong JJ, Iyer SS, Gumber S, Ibegbu CC, Villinger F, and Amara RR (2016). Induction of Th1-Biased T Follicular Helper (Tfh) Cells in Lymphoid Tissues during Chronic Simian Immunodeficiency Virus Infection Defines Functionally Distinct Germinal Center Tfh Cells. J. Immunol 197, 1832–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt G, and Nathan C (2011). In vitro differentiation of human macrophages with enhanced antimycobacterial activity. J. Clin. Invest 121, 3889–3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van De Vosse E, Verhard EM, De Paus RA, Platenburg GJ, Van Deutekom JCT, Aartsma-Rus A, and Van Dissel JT (2009). Antisense-mediated exon skipping to correct IL-12Rβ1 deficiency in T cells. Blood 113, 4548–4555. [DOI] [PubMed] [Google Scholar]
- Wang L, Wang S, and Li W (2012). RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185. [DOI] [PubMed] [Google Scholar]
- Wang Y, Ma CS, Ling Y, Bousfiha A, Camcioglu Y, Jacquot S, Payne K, Crestani E, Roncagalli R, Belkadi A, et al. (2016). Dual T cell- and B cell-intrinsic deficiency in humans with biallelic RLTPR mutations. J. Exp. Med 213, 2413–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson MD, and Hayes DN (2010). ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 26, 1572–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, and Cai J (2017). Mucosal-associated invariant T cells: New insights into antigen recognition and activation. Front. Immunol 8, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu GJ, Kula T, Xu Q, Li MZ, Vernon SD, Ndung’u T, Ruxrungtham K, Sanchez J, Brander C, Chung RT, et al. (2015). Comprehensive serological profiling of human populations using a synthetic human virome. Science (80-. ). 348, aaa0698–aaa0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshitomi H, Kobayashi S, Miyagawa-Hayashino A, Okahata A, Doi K, Nishitani K, Murata K, Ito H, Tsuruyama T, Haga H, et al. (2018). Human Sox4 facilitates the development of CXCL13-producing helper T cells in inflammatory environments. Nat. Commun 9, 3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Wei M, Becknell B, Trotta R, Liu S, Boyd Z, Jaung MS, Blaser BW, Sun J, Benson DM Jr., et al. (2006). Pro- and antiinflammatory cytokine signaling: reciprocal antagonism regulates interferon-gamma production by human natural killer cells. Immunity 24, 575–590. [DOI] [PubMed] [Google Scholar]
- Zhang J, Liu W, Wen B, Xie T, Tang P, Hu Y, Huang L, Jin K, Zhang P, Liu Z, et al. (2019a). Circulating CXCR3+ Tfh cells positively correlate with neutralizing antibody responses in HCV-infected patients. Sci. Rep. 9, 10090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Bigio B, Rapaport F, Zhang S-Y, Casanova J-L, Abel L, Boisson B, and Itan Y (2018). PopViz: a Webserver for visualizing minor allele frequencies and damage prediction scores of human genetic variations. Bioinformatics 34, 4307–4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, Volpi S, Li Z, Sanal O, Mansouri D, et al. (2015). Human intracellular ISG15 prevents interferon-alpha/beta over-amplification and auto-inflammation. Nature 517, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Siegel AM, Sun G, Dimaggio T, Freeman AF, and Milner JD (2019b). Human TH9 differentiation is dependent on signal transducer and activator of transcription (STAT) 3 to restrain STAT1-mediated inhibition. J. Allergy Clin. Immunol 143, 1108–1118.e4. [DOI] [PubMed] [Google Scholar]
- Zheng W, and Flavell RA (1997). The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 89, 587–596. [DOI] [PubMed] [Google Scholar]
- Zhu J, Yamane H, and Paul WE (2010). Differentiation of Effector CD4 T Cell Populations. Annu. Rev. Immunol 28, 445–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer J, Andrés E, Donato L, Hanau D, Hentges F, and de la Salle H (2005). Clinical and immunological aspects of HLA class I deficiency. QJM - Mon. J. Assoc. Physicians 98, 719–727. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Schematic view of the 16 MSMD-causing genes within myeloid and lymphoid cells in the context of host antimycobacterial immunity. (B) We tested for serum IgG against various viral peptides or peptides derived from bacteria or other pathogens by phage immunoprecipitation-sequencing. Serum samples from an unrelated healthy three-year-old boy (CTL1), two healthy adults (CTL2 and CTL3), and a TBX21 M/M P, and IgG-depleted serum and intravenous immunoglobulin (IVIg) were assayed. The heatmap (left) shows the counts of peptides significantly enriched for a given species with less than a continuous seven-residue subsequence, the estimated size of a linear epitope, in common. The species for which the patient had a score greater than our set threshold (>3) are shown. Count values above the set threshold are shown on a color scale from green (≥4) to red (≥40) and dark-red (≥80). (C) Genomic DNA from P (M/M) and his parents (WT/M) was subjected to Sanger sequencing to confirm the TBX21 variant. (D) Comparison between the WT and Mut sequences of the region in which the variant is located within exon 1 of TBX21. An alignment of the WT and Mut sequences revealed a juxtaposed region with a sequence identical to that of the variant sequence. (E) Alignment of peptide fragments from human T-box domain-containing transcription factors. (F) pCMV6 plasmids containing EV, WT T-bet (WT), the K314R variant of T-bet (K314R), T-bet with a deletion of the T-box domain (T-del), and mutant T-bet (Mut) were used to transfect HEK293T cells. T-bet expression was characterized by immunoblotting with anti-T-bet 4B10 monoclonal Ab and anti-Flag Ab. Anti-GAPDH antibody was used as an endogenous control. (G) Mutations of individual residues in the T-bet cDNA (E156S, M157L, E156A, and E157A) were generated in pCMV6 plasmids by site-directed mutagenesis. The plasmids were used to transfect HEK293T cells for overexpression. T-bet levels were measured by immunoblotting with anti-T-bet 4B10 Ab and anti-Flag Ab. Anti-GAPDH Ab was used as endogenous control. (H) The plasmids described in (G) were used to transfect HEK293T cells, together with the IFNG reporter plasmids and Renilla luciferase pRL-SV40 plasmids. After 3 days, the luciferase signal was assessed in a Dual-Glo assay. (I) Single-cell CRISPR/Cas9-edited NK-92 clones were stimulated with 50 pg/mL IL-12 and 10 ng/mL IL-18 or were left untreated (NS). Intracellular IFN-γ production was determined by ICS flow cytometry. (J) T-bet-deficient or pLenti-Crispr-V2-EV-transduced NK-92 single-cell clones were transduced with retroviruses generated from pLZRS-ireΔNGFR plasmids containing no insert (EV), or the WT, K314R or Mut TBX21 cDNA with a C-terminal Flag-tag. They were stimulated with 50 pg/mL IL-12 and 10 ng/mL IL-18 or were left untreated. Intracellular IFN-γ production was determined by ICS flow cytometry. (K) Naïve CD4+ T cells were expanded under TH0 conditions, and were then transduced with retroviruses generated from pLZRS-ireΔNGFR plasmids containing no insert (EV), or the WT, or Mut TBX21 cDNA with a C-terminal Flag-tag. Transduced cells were positive for the bicistronic marker CD271. Cells were surface-stained for CD271 and intracellularly stained for IFN-γ and TNF-α and subjected to flow cytometry analysis. (L) All variants (including the only homozygous variant – Mut, and 36 heterozygous variants, as indicated) from the HGID in-house database are represented on the schematic illustration. (M) All in-house TBX21 variants, as in (L), were tested for transcriptional activity in the human IFNG luciferase reporter assay. pCMV6 plasmids containing variants were used to transfect cells, together with the human IFNG reporter plasmids or the control EV pGL4.10-EV luciferase reporter plasmid, and the pRL-SV40 Renilla plasmid for luciferase activity measurement.
In (H, I and M), bars represent the mean and the standard deviation. Dots represent individual samples or technical replicates. One-way ANOVA was used for analysis in (H). Two-way ANOVA was used for analysis in (I). A non-parametric analysis for comparison with pGL4.10-pIFNG + pCMV6-WT TBX21 transfection condition was performed in (M). In (H, I and M), * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
(A) CD4+ T cells from healthy donors (CTL), the heterozygous father (WT/M) or P (M/M) were expanded with anti-CD3/CD28 antibody-coated beads. They were subjected to RT-qPCR for TBX21 with two different probes. Data are displayed as 2−ΔΔCt after normalization relative to GUS (endogenous control) expression, using the control mean as a calibrator. (B) TBX21 expression was assessed in immortalized T cells (HVS-T) from healthy donors (CTL), the heterozygous father (WT/M) and the patient (M/M), by RT-qPCR, as in (A). (C) P-derived HVS-T cells (M/M) were retrovirally transduced with EV or WT TBX21 containing a bicistronic CD271 surface marker. Intracellular T-bet and IFN-γ production by these cells and by CTL and heterozygous TBX21 WT/M HVS-T cells was assessed by flow cytometry in the presence and absence of P/I stimulation. (D) Expression of PSMD9 and ARHGAP27 (SH3 domain-containing 20 protein) in HEK293T cells transfected with pTRIP-mCherry-2A-PSMD9 or ARHGAP27-V5 plasmids along with VSVG and Pax2 plasmids, as assessed by immunoblotting with an anti-V5 tag Ab. (E) Expression of ERN1 in HEK293T cells transfected with pLenti-EV or WT-ERN1-Cter-GFP plasmids along with VSVG and Pax2 plasmids, as assessed by immunoblotting with anti-GFP or anti-ERN1Ab. (F) Flow cytometry analysis of the intracellular production of IFN-γ in control (CTL) HVS-T cells, M/M HVS-T cells from P, and in M/M HVS-T cells transduced with pTRIP-mCherry-2A-plasmids containing WT PSMD9 or WT ARHGAP27, or empty vector.(G) Flow cytometry analysis of IFN-γ intracellular production in control (CTL) cells, P’s HVS-T cells (M/M), and P’s HVS-T cells transduced with pLenti-Cter-GFP-2A-puro containing the WT ERN1 or empty vector. (H and I) Flow cytometry analysis of intracellular IFN-γ production in control (CTL) cells, M/M HVS-T cells from P, or M/M HVS-T cells retrovirally transduced with EV or WT T-bet (MM+EV or MM+WT) or MM+WT HVS-T cells lentivirally transduced with the cDNA of EV or WT PSMD9, ARHGAP27 (H) and ERN1 (I). (J) Isolated CD4+ T cells from a healthy donor, a TBX21 WT/M parent and the TBX21 homozygous M/M P were expanded with anti-CD3/CD28 antibody-coated beads under TH0 or TH1 conditions. After 7d, EV or WT T-bet plasmids were used to transduce P’s cells in the presence of a bicistronic CD271 surface reporter. Cells were subjected to ICS for IFN-γ and TNF-α in response to P/I restimulation. The gating strategy for the experiment is shown. (K) Cells transduced as in (J) were isolated by MACS with anti-CD271 antibody-coated beads. Together with non-transduced cells, they were re-stimulated with anti-CD3/CD28 antibody-coated beads, and their culture supernatants were subjected to ELISA for IFN-γ and TNF-α.
In (A and B), bars represent the mean and the standard deviation; in (K), bars represent the mean and the standard error of the mean. Dots represent individual samples. One-way ANOVA was used for analysis in (B), comparing WT/M or M/M to CTL. ns = not significant.
(A) PBMCs were immunophenotyped with CyTOF. The gating strategy for different immune subsets in a healthy control is indicated. (B) Percentages of pDCs, cDC1 cells and cDC2 cells among adult healthy donors (CTL), age-matched controls (Age-matched CTL), IL-12Rβ1-deficient MSMD patients (IL12RB1 M/M) and P (TBX21 M/M), as indicated in (A). (C) PBMCs from P and a healthy control were subjected to the FACS immunophenotyping of innate lymphoid cells (ILCs) and NK cells. CD117+ ILCs, consisting principally of ILC precursors (ILCP) and type 2 ILC (ILC2) cells, were gated manually on conventional flow cytometry. The gating strategy is shown. (D) Total NK cells (CD16+ or CD94+Lin−CD7+ cells) were plotted with CD56 against CD127 for manual gating on different NK cell subsets. (E) Percentage of ILC precursor and ILC2 cells in adult healthy donors (CTL), age-matched controls (Age CTL), IL-12Rβ1-deficient MSMD patients (IL12RB1 M/M) and P (TBX21 M/M), summarized from CyTOF data, as in (A). (F) Immunophenotyping of ILC2 and ILCP cells, analyzed by fluorescence flow cytometry. Plot of ILC2 and ILCP, and the percentage of ILC2 and ILCP (within live CD45+ cells) in a healthy donor (CTL), the T-bet-deficient patient (M/M) and his age/ethnicity-matched healthy brother (WT/WT bro). (G) PBMCs from a CTL, IL-12Rβ1-deficient MSMD patients (IL12RB1 M/M), the TBX21 WT/M parents (WT/M mo or fa), the T-bet-deficient P (M/M) and his age/ethnicity-matched healthy brother (WT/WT bro) were stained with surface markers for lymphocytes involved innate immunity (MAIT and iNKT cells) or both adaptive and innate immunity (γδ T cells, in particular Vδ1 and Vδ2 γδ T cells). The gating strategy for flow cytometry is shown. (H - J) iNKT (H), MAIT (I) and Vδ2+ γδ T (J) cells are shown for P and his relatives. (K) Percentages of Vδ1+ γδ T cells in healthy adult donors (CTL), age-matched controls (Age CTL), IL-12Rβ1-deficient MSMD patients (IL12RB1 M/M) and P (TBX21 M/M), his age/ethnicity-matched healthy brother (TBX21 WT/WT) and his healthy parents (TBX21 WT/M).
In (B and E), bars represent the mean and the standard deviation. In (K), bars represent the mean and the standard error of the mean. Dots represent individual samples. Mann-Whitney nonparametric tests were used for analysis in (K). In (K), ** p < 0.01.
(A) Naïve (CD45RA+CCR7+), central memory (CM, CD45RA− CCR7+), effector memory (EM, CD45RA−CCR7−) and TEMRA (CD45RA+CCR7−) cells were manually gated from CD8+ T cells on CyTOF data. The percentages of these cells are shown. (B) 10,000 total CD8+ T cells were clustered by viSNE, based on a selection of surface markers. Manual gating of naïve and memory subsets overlaid on clusters of CD8+ T cells. CXCR3 and CD38 expression are indicated on this plot. (C) Naïve (CD45RA+CCR7+), central memory (CM, CD45RA−CCR7+), effector memory (EM, CD45RA−CCR7−) and TEMRA (CD45RA+CCR7−) cells were manually gated from CD4+ T cells on the basis of CyTOF data. The percentages of these cells are shown. (D) The gating strategy for the CyTOF analysis of memory CD4+ T cells and TH1, TH2, Th17 and TH1* cells is shown. (E) Memory CD4+ T cells (CD45RA−) from indicated individuals were gated out manually, and further subjected to viSNE deconvolution with T-cell chemokine markers for 10,000 randomly selected events. The expression of CXCR3, CCR5, CCR6 and CCR4 is shown. (F and G) Percentages of TH2 (CCR6−CXCR3−CCR4+) (F) and TH17 cells (CCR6+CXCR3−CCR4+) (G) among memory CD4+ T cells. (H) Frequencies of the circulating follicular T helper (cTFH) cells and their subsets (CXCR3+, double-negative as DN, CCR6+, or CXCR3+CCR6+ TFH) from healthy donors (CTL) and the T-bet-deficient patient (M/M) gated from conventional flow cytometry data. (I) Percentages of Treg cells gated from CyTOF data. (J) Percentages of CXCR3+ and CCR5+CXCR3− Tregs gated from CyTOF data. In (A, C, F, G, H, I and J), bars represent the mean and the standard error of the mean. Dots represent individual samples.
(A) Gating strategy for the surface and intracellular staining of PBMCs ex vivo stimulated with P/I, for an age-matched control sample. (B) Intracellular staining of IFN-γ and T-bet in NK cells, as shown in (A), from an age-matched control (Age CTL), P (M/M) and P’s healthy brother (WT/WT) stimulated with P/I. (C) Frequencies of TNF-α-producing cells among CD56+ NK cells, as gated in (A), in the indicated individuals. (D) The gating strategy for gating total NK cells from a travel control (CTL), P (M/M), P’s brother (WT/WT) and P’s heterozygous mother (WT/M), in the presence and absence of stimulation with IL-12, IL-15 and IL-18, is shown. (E) Expression of CD107a and IFN-γ among total NK cells, as in (D). (F) Frequencies of IFN-γ-producing cells among iNKT cells, as gated in (A) for the indicated individuals. (G and H) Vδ1−Vδ2− γδ T cells were gated from PBMCs prepared as shown in (A). Intracellular staining for IFN-γ (G) and TNF-α (H) is shown. (I and J) CD8+ T cells were gated from PBMCs prepared as shown in (A). Intracellular staining for IFN-γ (I) and TNF-α (J) is shown. (K - R) Naïve and memory CD4+ T cells from controls (CTL) and P (M/M) were sorted by FACS and activated/expanded. Intracellular staining of IL-22 (K), IL-9 (L) and IL-21 (R). Production of IL-9 (M), IL-4 (N), IL-5 (O), IL-10 (P) and IL-13 (Q), as measured in culture supernatants. (S and T) Naïve CD4+ T cells were isolated by FACS and subjected to activation under TH0, TH9, TH17 or TH9 conditions combined with TH17 conditions. The production of IL-9 was assessed intracellularly (S) and in culture supernatants (T). (U) Naïve CD4+ T cells were isolated by FACS and subjected to activation under TH0, TH1 or TH2 conditions. IL-10 production, determined with culture supernatants, is shown. (V) Naïve CD4+ T cells were isolated by FACS and subjected to activation under TH0, TH2 or TH9 conditions. IL-13 production, as measured on culture supernatants.
In (C, F - V), bars represent the mean and the standard error of the mean. Dots represent individual samples. Mann-Whitney non-parametric tests were used for analysis in (C, F - J). In (C, F - J), * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, ns = not significant.
(A and B) PBMCs from adult controls (CTL), age-matched controls (Age CTL), P (M/M), P’s healthy brother (WT/WT), and P’s healthy parents (WT/M) were left unstimulated or stimulated with live M. bovis BCG in the presence and absence of IL-12 and IL-23. The production of TNF-α (A) and IL-5 (B) in culture supernatants was assessed with Legendplex cytometric bead arrays. (C) PBMCs from P (M/M), P’s healthy brother (WT/WT) and one of P’s healthy parents (WT/M), stimulated as in (A), were analyzed for the intracellular expression of T-bet and IFN-γ by flow cytometry. Percentages of T-bet+ IFN-γ+ cells within live single lymphocytes are highlighted. (D) Pie-charts of the percentages of the indicated immune subsets in PBMCs from healthy controls stmulated with IL-12 or IL-23 alone. (E and F) PBMCs from adult controls (CTL), age-matched controls (Age CTL), P (M/M), P’s healthy brother (WT/WT), and P’s healthy parents (WT/M) were left unstimulated or were stimulated with live M. bovis BCG in the presence and absence of IL-12 and IL-23. Frequencies of T-bet+ IFN-γ+ cells gated on each individual immune subset, including NK, iNKT, MAIT, Vδ2+ γδ T (Vd2), Vδ1+ γδ T (Vd1) and Vδ1−Vδ2− γδ T (Vd1-Vd1-gdT) cells from different groups of individuals, stimulated with BCG (E) and BCG + IL-23 (F), are shown. (G) Frequencies of T-bet+ IFN-γ+ cells gated on adaptive immune subsets, including B, CD4+ T and CD8+ T cells from different groups of individuals, stimulated with BCG + IL-23. (H) Dot-plots showing the expression of intracellular T-bet and IFN-γ gated on CD4+ T cells with and without BCG stimulation in the presence and absence of IL-12 and IL-23. (I) Frequency of Vδ2+ γδ T cells in PBMCs from different groups of individuals expanded with BCG-lysates for 14 d. (J) Dot-plots showing intracellular T-bet and IFN-γ levels in non-responsive and PPD-responsive CD4+ T cells following PPD stimulation. (K and L) Dot-plots showing intracellular T-bet and IFN-γ levels in non-responsive and BCG-lysates-responsive CD4+ T (K) and CD8+ T cells (L).
In (A, B, E, F, G and I), bars represent the mean and the standard error of the mean. In (D), numbers represent the mean and the standard deviation. Dots represent individual samples. Mann-Whitney nonparametric tests were used for analysis in (A, B, E, F and G), * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, ns = not significant.
Cells were restimulated with anti-CD3/CD28 Ab-coated beads for 16 h. Lists of DEG_Status, DEG_FoldChange and DEG_ZScore identifying differentially regulated target genes genome-wide. Slim_DEG identifies differentially regulated target immune genes.
Related data were divided into five sub-sheets. Sheets titled ATAC_Closed by T-bet, ATAC_Opened by T-bet, and ATAC_61 genes above cutoff represent related data from Omni-ATAC-seq dataset generated from CD4+ T cells. Sheets titled Epic_array_MM<CTL and Epic_array_MM>CTL represent related data from the Epic-methylation CpG array.
Data Availability Statement
The whole-exome sequencing, RNA-seq and omni-ATAC-seq datasets generated during this study are available at the Sequence Read Archive (SRA) (PRJNA641463). The scRNA-seq datasets generated during this study are available at the EGA European Genome-Phenome Archive (EGAS00001004504). This study did not generate any unique code. Any other piece of data will be available upon reasonable request.