

Abstract
Objective:
Coronary artery disease (CAD) is associated with a compensatory switch in mechanism of flow-mediated dilation (FMD) from nitric oxide (NO) to H2O2. The underlying mechanism responsible for the pathological shift is not well understood, and recent reports directly implicate telomerase and indirectly support a role for autophagy. We hypothesize that autophagy is critical for shear stress-induced release of NO and is a crucial component of for the pathway by which telomerase regulates FMD.
Approach and Results:
Human left ventricular (LV), atrial, and adipose resistance arterioles were collected for videomicroscopy and immunoblotting. FMD and autophagic flux were measured in arterioles treated with autophagy modulators alone, and in tandem with telomerase-activity modulators. LC3B II/I was higher in LV tissue from CAD patients compared to non-CAD (2.8±0.2 vs 1.0±0.2-fold change; p<0.05), while p62 was similar between groups. Shear stress increased Lysotracker fluorescence in non-CAD arterioles, with no effect in CAD arterioles. Inhibition of autophagy in non-CAD arterioles induced a switch from NO to H2O2, while activation of autophagy restored NO-mediated vasodilation in CAD arterioles. In the presence of an autophagy activator, telomerase inhibitor prevented the expected switch (Control: 82±4%; L-NAME: 36±5%; PEG-Cat: 80±3). Telomerase activation was unable to restore NO-mediated FMD in the presence of autophagy inhibition in CAD arterioles (Control: 72±7%; L-NAME: 79±7%; PEG-Cat: 38±9%).
Conclusions:
We provide novel evidence that autophagy is responsible for the pathological switch in dilator mechanism in CAD arterioles, demonstrating that autophagy acts downstream of telomerase as a common denominator in determining the mechanism of FMD.
Keywords: autophagy, CAD, FMD, lysosome, nitric oxide, p62, shear stress, telomerase, Endothelium/Vascular Type/Nitric Oxide, Translational Studies, Coronary Artery Disease, Physiology
Graphical Abstract
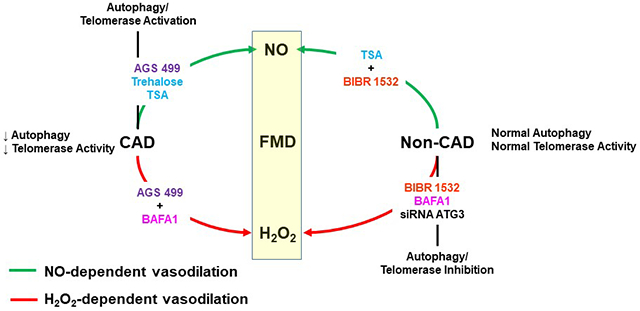
Introduction
The microcirculation is increasingly recognized as playing a crucial role in a variety of cardiovascular diseases 1. Resistance arterioles (~50-200 um) are responsible for maintaining blood pressure and flow to vital organs, thereby preserving a delicate physiological homeostasis 1, 2. The health of the vasculature is dependent upon the health of the endothelium, which is continuously exposed to mechanical (e.g. shear stress) and chemical stimuli (e.g. oxidized low-density lipoproteins). In response to shear stress, the endothelium releases substances, most notably nitric oxide (NO), that stimulate relaxation of the adjacent vascular smooth muscle (vasodilation). This vasodilation to shear stress, termed flow-mediated dilation (FMD), is a powerful barometer of overall vascular health 3.
In healthy participants, microvascular FMD is primarily due to endothelial release of NO generated by endothelial NO synthase (eNOS), in addition to endothelial hyperpolarizing factors. However, in subjects with coronary artery disease (CAD) the maintained FMD no longer involves NO 4. Rather, an age-independent compensatory switch in the mechanism of dilation occurs in the setting of CAD, such that mitochondrial-derived hydrogen peroxide (H2O2) becomes the primary endothelium-derived relaxing factor in response to flow 5. Although both NO and H2O2 are vasodilators, NO promotes vascular quiescence, while H2O2 promotes inflammation, proliferation, and atherosclerosis 1, 6. Thus, understanding the underlying mechanism for this switch from NO to H2O2-mediated dilation is essential for minimizing tissue stress and injury from vascular paracrine redox toxicity, and might help explain the large influence of microvascular vasodilator impairment on cardiovascular events.
Macro-autophagy (autophagy) is a systematic and controlled process by which cells degrade their cellular components to maintain physiological homeostasis 7. Autophagy recycles oxidative byproducts in response to stress and heightened metabolism. By regulating the production of ROS, NO-dependent vascular homeostasis in response to shear stress is maintained 8, 9. However, the role of autophagy in pathological switch from NO to H2O2-mediated FMD has not been explored. Within the vasculature, autophagy is important for maintaining redox balance, and has been demonstrated to be critical in maintenance of NO bioavailability 8, 9. In this context, both human and animal data demonstrate that advancing age reduces endothelium-dependent vasodilation that is paralleled by reductions in autophagy 10,11. The effects of autophagy in promoting endothelial NO formation and in reducing ROS production makes it an ideal candidate as a regulator of the switch in mediator of FMD. However, the role of autophagy in determining the mechanism of microvascular dilation has not been examined.
We have previously demonstrated that extranuclear telomerase plays an important role in the disease-associated shift in the mediator of FMD, even before the detectable onset of telomere shortening. This non-canonical effect of telomerase may function by activating autophagy and reversing the vascular dysfunction associated with CAD 12–14. The primary role of telomerase is to maintain telomere length 15, but more recent evidence shows several different functions of TERT, the catalytic subunit of telomerase 12–14, 16. Evidence for cross-talk between telomerase and autophagy stems from murine embryonic fibroblasts, where overexpression of TERT increases markers of autophagy, evidenced by the increased conversion of LC3B I to LC3B II, indicative of increased autophagosome formation 17. Reciprocally, inhibition of TERT activity reduces the conversion of LC3 I to LC3 II 17.
Collectively, autophagy plays a critical role in maintaining vascular homeostasis, positioning this process as a potential central regulator of the mechanism of microvascular FMD. In this study, we seek to establish whether autophagy is necessary and sufficient to regulate the mechanism of FMD in the human microvascular circulation and define whether autophagy and telomerase activity work in tandem to maintain physiological vascular environment.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Tissue collection –
Fresh, surgically discarded human atrial and adipose tissues were collected in cold 4°C HEPES (NaCl 275 mmol/L, KCL 7.99 mmol/L, MgSO4 4.9 mmol/L, CaCl2·2H2O 3.2 mmol/L, KH2PO4 2.35 mmol/L, EDTA 0.07 mmol/L, glucose 12 mmol/L, HEPES acid 20 mmol/L). Whole human heart tissue was collected from the Wisconsin Donor Network in HEPES buffer, sectioned into labeled Eppendorf tubes (Left ventricle, right atrium, etc.), and flash frozen in liquid nitrogen then stored at −80°C. De-identified patient information was saved into a secure database. Tissue from patients with no more than one known cardiovascular risk factor and no clinical diagnosis of CAD were used for non-CAD groups. Patient characteristics for heart and vessel samples are listed in Tables 1 and 2, respectively). All protocols were approved by the Medical College of Wisconsin/Froedtert Memorial Lutheran Hospital Institutional Review Board.
Table 1:
Patient Characteristics for Heart Tissue
| Variable | Non-CAD | CAD |
|---|---|---|
| Sex, M/F | 0/5 | 3/2 |
| Age, yr | 47±5 | 56±2 |
| BMI | 35.2±0.7 | 35.1±3.9 |
| Disease/Risk Factors | ||
| Coronary Artery Disease | 0 | 5 |
| Hypertension | 0 | 4 |
| Hypercholesterolemia | 0 | 1 |
| Diabetes Mellitus | 0 | 2 |
| Heart Failure | 0 | 0 |
| Tobacco Use | 3 | 2 |
| Previous MI | 0 | 3 |
BMI, body mass index; CAD, coronary artery disease; MI, myocardial infarction
Table 2:
Patient Characteristics for Vessels
| Variable | Non-CAD | CAD |
|---|---|---|
| Sex, M/F | 9/25 | 19/5 |
| Age, yr | 47±2 | 65±1* |
| BMI | 29.5±1.0 | 29.9±1.9 |
| Disease/Risk Factors | ||
| Coronary Artery Disease | 0 | 24 |
| Hypertension | 4 | 16 |
| Hypercholesterolemia | 1 | 13 |
| Diabetes Mellitus | 2 | 10 |
| Heart Failure | 0 | 3 |
| Tobacco Use | 0 | 3 |
| Previous MI | 0 | 7 |
BMI, body mass index; CAD, coronary artery disease; MI, myocardial infarction
P < 0.05 vs. non-CAD
Videomicroscopy –
Human adipose and atrial resistance arterioles (average internal diameter: 123±8 μm; min: 105 μm; max: 140 μm) were isolated from the freshly collected tissue (used within 24–48 hours of receipt). Isolated arterioles were cannulated at both ends with glass micropipettes (of similar internal minimal diameter) in an organ chamber filled with Krebs buffer, pressurized to 60 mmHg and pre-constricted with endothelin-1 (ET-1; 0.1–1 nmol/L) to achieve a stable 25%-50% reduction in passive diameter18. Flow through the cannulated arterioles was achieved by changing the height of each reservoir in equal amounts in opposite directions, thus minimizing any changes in intraluminal pressure. In response to flow, internal diameters were measured using on-screen calibrated calipers after 5 minutes at a given pressure gradient. Two flow-response curves were generated; first with vehicle, then a second with inhibitor(s), including L-NAME (100 μM) or PEG-cat (500U/mL). Time controls were generated with no intervention between the two flow-response curves. At the end of each experiment, papaverine (100 μM) was added to assess vasodilator capacity/smooth muscle function18, 19. Data are reported as percent change (%) from pre-constricted diameter to the stimulated diameter (FMD or pharmacological).
Modulation of Autophagy
Isolated adipose and atrial arterioles were incubated for 15-20 hours in endothelial cell growth media containing 5% serum (Lonza), containing either vehicle (DMSO 2% by volume), Bafilomycin A1 (BAFA1; Sigma-Aldrich, 100 nmol/L), trichostatin A (TSA; Cayman Chemical, 100 nmol/L), ATG3 siRNA (Ambion, AM16708; 10 nmol/L), trehalose (Research Products International, 10 mmol/L), BIBR 1532 (Tocris Bioscience; 10 umol/L), or AGS 499 (Ester Priel, PhD, Israel; 20 nmol/L). TSA and trehalose were independently used to induce autophagy in arterioles, while BAFA1 and ATG3 siRNA were used to inhibit or prevent completion of autophagy in arterioles. Previous work from our lab has demonstrated that modulation of TERT with the BIBR 1532 and AGS 499 induce a switch in mechanism of dilation in human atrial and adipose arterioles from NO to H2O2, and H2O2 to NO, respectively12. Following incubation, arterioles were cannulated and underwent stimulation of FMD as described above.
Protein Quantification -
Western blot analysis was performed on lysates from heart tissue in MOPS buffer (4.2 g/L MOPS, 0.8 g/L EGTA, 1.6 g/L EDTA, 1.3 g/L NaF, 8.6 g/L B-glycerophosphate, 2.2 g/L Na-pyrophosphate, 10 mL/L Na-orthovanadate, 5mL/L IGEPAL CA-30 Sigma I3021) with 1x phosphatase/protease inhibitor cocktail. Protein concentration determined by BCA assay and activated with 4x BME. 50 ug of protein were loaded into BioRad precast criterion gel 4–20% tris-HCl (p62, GAPDH) and BioRad precast criterion 4–20% TGX gel (LC3B). After separation by electrophoresis (90 volts for 30 min, then 160 volts for 70 min), proteins were transferred to nitrocellulose membranes using 7:2:1 v/v/v Water/Methanol/tris-glycine (100 volts for 70 min) using a cold tank transfer. Membranes were washed with TBS-T (TBS + 0.1% Tween) and then blocked 5% non-fat milk (GAPDH and p62) or 5% BSA (LC3B) for 1 hour at room temperature. After blocking, membranes were quickly rinsed 3 times using TBS-T. Primary antibodies were diluted in 3% IgG-free BSA (1:1000, LC3B; Cell Signaling Technology Cat# 2775; p62/SQSTM1, 1:1000 Abnova Corporation Cat# H00008878-M01; GAPDH, 1:20000; Abcam Cat# ab8245) at 4°C overnight. The following day, membranes were rinsed 4–5 times in TBS-T for 5–8 min, and then secondary antibodies were added (5% non-fat milk, 1:2500 LC3B; Cell Signaling Technologies anti-rabbit IgG-HRP (Cat. #7074P2), 1:80,000 GAPDH, 1:5000 p62; Millipore anti-mouse IgG-HRP (AP308P)). Clarity (BioRad) substrate was then applied to membranes (GAPDH and p62). The Clarity substrate was spiked with SuperSignal West Femto (ThermoFisher, 2 % of the volume of the Clarity substrate). Bands were quantified using ImageJ.
Measurement of Autophagic Flux within Resistance Arterioles
Resistance arterioles were cannulated in HEPES buffer (pH 7.4 at 37°C) and gradually pressurized to 60 mmHg over 60 minutes. Following pressurization, baseline images were captured using fluorescence microscopy. Shear was induced by gravity, moving buffer filled reservoirs in equal and opposite vertical directions to create a gradient of 100 cmH2O. LysoTracker Red DND-99 (Invitrogen L7528; 50 nM) was added prior to pressurization to buffer filled reservoirs so the probe was perfused intraluminally. Lysotracker was used to quantify shear-induced autophagic flux in the presence and absence of lysosome inhibition (BAFA1; 100 nmol/L). Images were captured every 15 min for an hour using a fluorescent microscope (model Olympus IX73 with X-cite FIRE LED lamp). Fold change in fluorescence intensity from static values at each time point of max flow was compared between vessels.
Shear-Induced Mitochondria Hydrogen Peroxide Production
Resistance arterioles were cannulated in HEPES buffer as described above. Following pressurization, baseline images were captured using fluorescence microscopy. Shear was induced by moving buffer filled reservoirs in equal and opposite vertical increments to create a gradient of 100 cmH2O. Mito Peroxy Yellow 1 (MitoPY1, Tocris Bioscience; 10 μM, 1 h) was added prior to pressurization to buffer filled reservoirs so the probe was perfused intraluminally. MitoPY1 was used to quantify mitochondrial H2O2 production in the presence and absence of autophagy inhibitors and activators, as well as PEG-cat. Images were captured every minute for 10 minutes using fluorescence microscopy (model Olympus IX73 with X-cite FIRE LED lamp). The % increase in fluorescence intensity from static values to 5 min of max flow (% increase to flow) was compared between intact control and experimental vessels.
Endothelial Cell Collection and Quantitative Immunofluorescence Microscopy
In order to examine whether autophagy markers differ between tissue, venous endothelial cells were collected and examined for classic markers of autophagy. A total of 13 non-CAD subjects (< 1 risk factor) and 10 subjects with a clinical diagnosis of CAD underwent an endothelial cell biopsy from a forearm vein. All participants gave written informed consent and the study protocol for endothelial cell collection was approved by the Boston Medical Center Institutional Review Board and conform to the scientific ethical principles outlined in the Declaration of Helsinki. Venous endothelial cells were collected as previously described 20, 21. Briefly, a 0.018-inch J-wire (Arrow International, Reading, PA) was inserted through a 20 or 22-gauge intravenous catheter in a forearm vein and used to gently rub the endothelial surface. Endothelial cells were then recovered from the wire in a red blood cell lysis/dissociation buffer, centrifuged, and applied to poly-L-lysine coated slides (Sigma, St. Louis, MO). The cells were fixed onto slides using 4% paraformaldehyde, dried, and frozen at −80°C prior to staining. Protein levels of LC3B and p62 were quantified using immunofluorescence microscopy. Cells were permeabilized with 0.1% triton-X in 50 mM glycine for 10 minutes, washed three times with 50 mM glycine in 1X PBS, and blocked for 10 minutes with 0.5% bovine serum albumin in 50 mM glycine/1X PBS. Slides were stained with primary antibody against either LC3B (1:200 dilution) or p62 (1:200 dilution) for three hours at 37°C. Following three washes with 50 mM glycine/1X PBS, the cells were incubated with secondary antibodies for 45 minutes at 37°C. The fluorescence intensity was quantified in 20 endothelial cells per participant and averaged. The fluorescence intensity was normalized to the intensity in human aortic endothelial cells, which were stained simultaneously, to control for batch-to-batch variability. Final intensity was calculated by dividing the average fluorescence intensity for the participant sample by the average fluorescence intensity of the human aortic endothelial cell sample and multiplying by 100. The intensity is expressed as arbitrary units (AU). For LC3B, the number of puncta per cell were counted for 20 cells per participant and averaged. All quantification was performed blinded to participant identity and CAD status. Confocal images were obtained using a Leica SP5 microscope (Solms, Germany).
Statistical Analysis
All data are expressed as mean ± SEM. FMD is expressed as a percentage of maximal dilation. 2-way repeated measures ANOVA was used with pressure gradient and intervention as factors to assess vascular function. When a significant difference was observed between control and inhibitor curves, responses at individual concentrations were compared using a Tukey post hoc analysis. Differences in protein levels were assessed with a Student’s t-test. Analyses was performed using Graphpad. Statistical significance was defined as p < 0.05.
Results
Autophagy in Human Left Ventricle Tissue from CAD and non-CAD subjects
We evaluated markers of autophagy in subjects with and without CAD via Western blots for LC3B and p62, commonly used markers for autophagy (participant characteristics in Table 1). The ratio of LC3 II/I, an indicator of autophagosome formation, was elevated in heart tissue (left ventricle) from patients with CAD vs. non-CAD (Supplemental Figs. 1 & 2; P < 0.05). Levels of p62, an indicator of autolysosome degradation were not different between CAD and non-CAD (Supplemental Fig. 1B; P = 0.38). To confirm endothelial specificity of our observation and to evaluate autophagic flux, immunofluorescence (IF) was used to stain for LC3B and p62 levels in isolated primary endothelial cells from subjects with and without CAD (other CV risk factors present). No difference between non-CAD and CAD cells was observed in expression of LC3B (Supplemental Fig. 3A), number of LC3-bound puncta (non-CAD: 18±1, CAD: 16±1; p = 0.25), or expression of p62 (Supplemental Fig. 3B).
Exposure to Shear Stress Initiates Autophagy in Human Adipose Resistance Arterioles
Shear-induced autophagic flux was measured in cannulated, pressurized adipose arterioles using fluorescence microscopy. In response to 60 min of shear stress (100 cmH2O gradient), intraluminal Lysotracker Red DND-99 fluorescence increased in adipose arterioles from non-CAD subjects, while there was no change in CAD adipose arterioles (Fig 1A & B). This indicates an elevation in autophagic lysosomal acidification indicative of heightened autophagic flux. Importantly, this increase in shear-induced autophagic flux was blocked by incubating non-CAD adipose arterioles overnight with BAFA1 (Fig. 1C). Activation of autophagy with TSA restored the ability of CAD adipose arterioles to initiate autophagy in response to flow stimulus (Fig. 1D).
Figure 1: Shear stress induces autophagy within non-CAD adipose resistance arterioles, while presence of CAD blunts shear-induced autophagy.
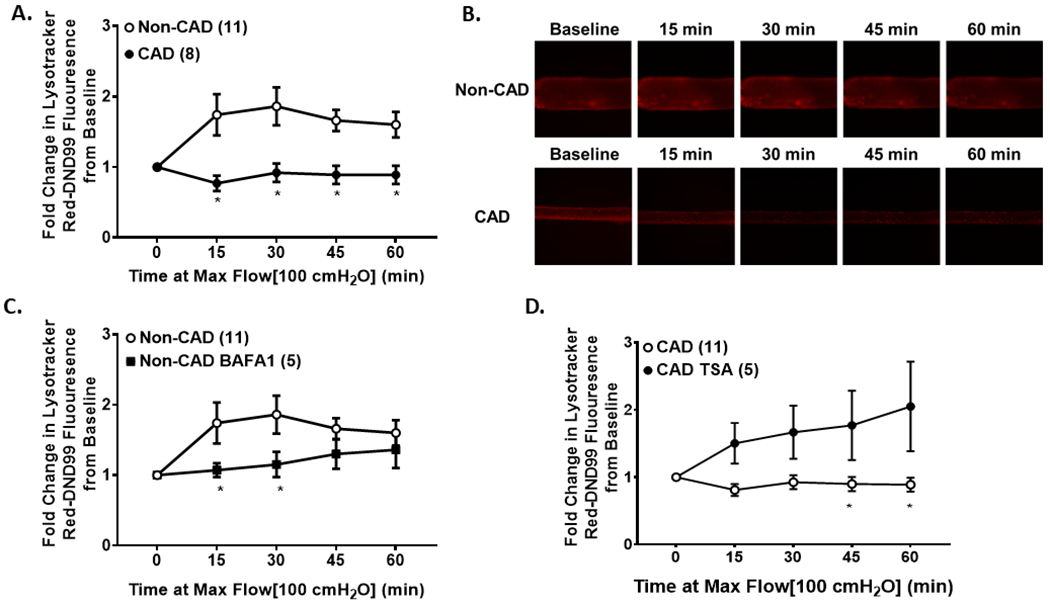
(A) Lysotracker Red DND-99 fluorescence in response to 60 minutes of shear stress (100 cmH2O); (B) Representative images of shear-induced changes in Lysotracker Red DND-99 fluorescence in response to shear; (C) Overnight incubation of non-CAD adipose arterioles with BAFA1 blunts shear-induced autophagy; (D) Overnight incubation of CAD adipose arterioles with TSA enhances shear-induced autophagy. * p < 0.05 vs. non-CAD.
Autophagy Modulates Mitochondrial H2O2 Production in Human Adipose Resistance Arterioles in Response to Shear Stress
Exposure to shear stress did not increase mitochondria H2O2 production in non-CAD arterioles, however inhibition of autophagy (BAFA1 100 nmol/L O/N) increased mitochondria H2O2 production, which was prevented in the presence of PEG-cat (Fig. 2A&B). Conversely, exposure to shear stress increased mitochondria H2O2 production in CAD adipose arterioles, and this was abolished in the presence of autophagy activation (TSA; 100 nmol/L O/N) in addition to PEG-cat (Fig. 2C&D).
Figure 2: Autophagy modulates mitochondria H2O2 production in response to shear stress.
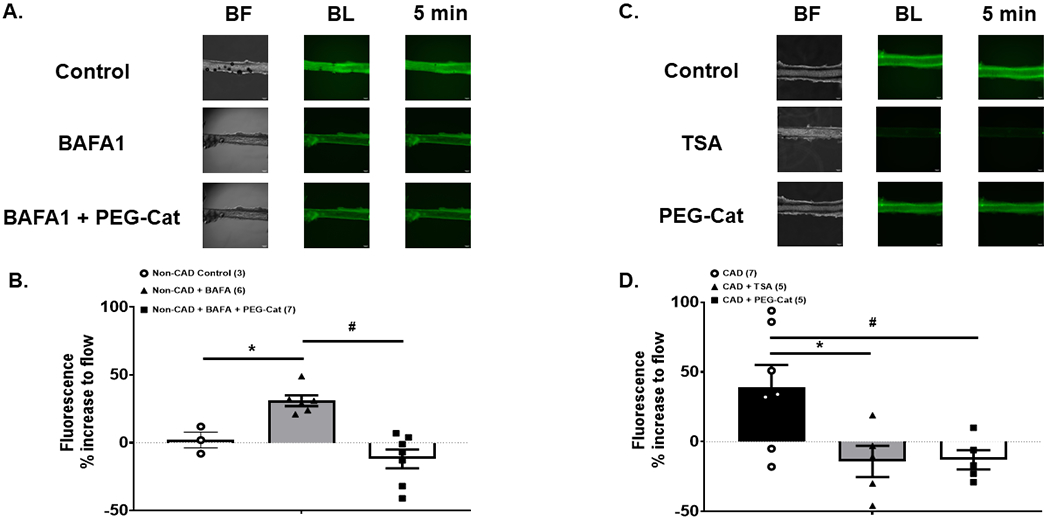
Overnight inhibition of autophagy via the lysosomal inhibitor BAFA1 increases mitochondria H2O2 production in response to shear stress that is negated in the presence of PEG-Catalase in non-CAD adipose resistance arterioles (A & B). Mitochondria H2O2 production in response to shear stress is elevated in CAD adipose resistance arterioles and negated with overnight activation of autophagy (TSA), or by treating with the H2O2 scavenger PEG-Catalase (C & D). * p < 0.05 vs. control; # p < 0.05 vs. peg-cat condition.
Autophagy Initiates a Switch in the Mechanism of FMD in Human Atrial and Adipose Resistance Arterioles
Adipose arterioles from CAD patients demonstrate a blunted autophagic response to shear stress. We hypothesized that this impairment in autophagy contributes to the CAD-associated switch in FMD from NO to H2O2.To test the role of autophagy on the mechanism of FMD, autophagy was inhibited in non-CAD (BAFA1) or activated (TSA and trehalose) in CAD atrial and adipose arterioles either acutely (30 min) or overnight (15-20 hours), and the mechanism of FMD was interrogated. Both acute and overnight inhibition of autophagy switched the mechanism of dilation from NO to H2O2 as evidenced by a reduction in vasodilation to shear stress in the presence of PEG-catalase, but normal dilation in the presence of the eNOS inhibitor, L-NAME (Acute: Supplemental Fig. 4A; Overnight: Fig 3A&B). The magnitude of dilation was independent of the mediator of dilation. siRNA mediated knockdown of ATG3 resulted in a similar switch from NO to H2O2-mediated FMD, while scramble siRNA-maintained NO-dependent dilation (Fig. 3C & 3D). Acute activation of autophagy with TSA (100 nM) in arterioles from non-CAD subjects had no effect on the mechanism of dilation, however overnight incubation with TSA elicited an incomplete switch in the mechanism of dilation from NO to H2O2 (Supplemental Fig. 5A&B). Maximal smooth muscle dilation was not affected by inhibition of autophagy (Fig. 3E).
Figure 3: Inhibition of autophagy in non-CAD arterioles switches the mechanism of FMD.
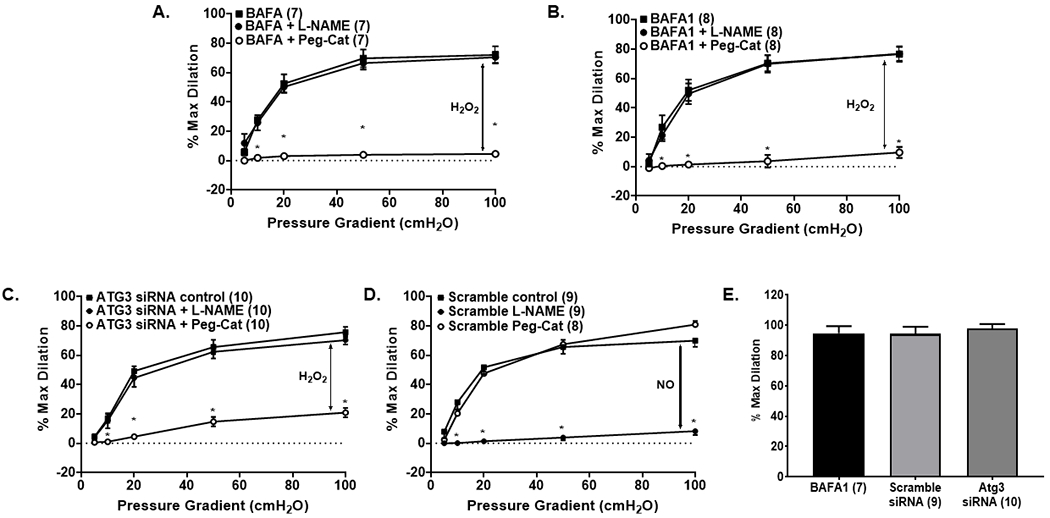
(A, B) Overnight inhibition of autophagy via the lysosomal inhibitor BAFA1 switches the mechanism of FMD in atrial and adipose arterioles from NO to H2O2; (C) Overnight incubation with Atg3 siRNA switches the mechanism of dilation in adipose arterioles from NO to H2O2; (D) scramble ATG3 siRNA does not affect the mechanism of dilation in non-CAD adipose arterioles. (E) Smooth muscle responses to papaverine are not affected by autophagy inhibition. * P < 0.05 vs. control; BAFA1, bafilomycin A1; L-NAME, NG-Nitro-l-arginine methyl ester; Peg-Cat; polyethylene glycol-catalase.
Acute activation of autophagy in CAD arterioles with TSA maintained H2O2 as the primary mechanism of dilation (Supplemental Fig. 6A&B); however overnight incubation of CAD arterioles with TSA (Fig. 4A) or trehalose (Fig. 4B) switched the mechanism of dilation from H2O2 to NO, as evidenced by inhibition of dilation in the presence of L-NAME but FMD was not changed when the arterioles were incubated with PEG-cat. Smooth muscle responses to papaverine were not different after autophagy activation (Fig. 4C).
Figure 4: Activation of autophagy in CAD arterioles switches the mechanism of FMD.
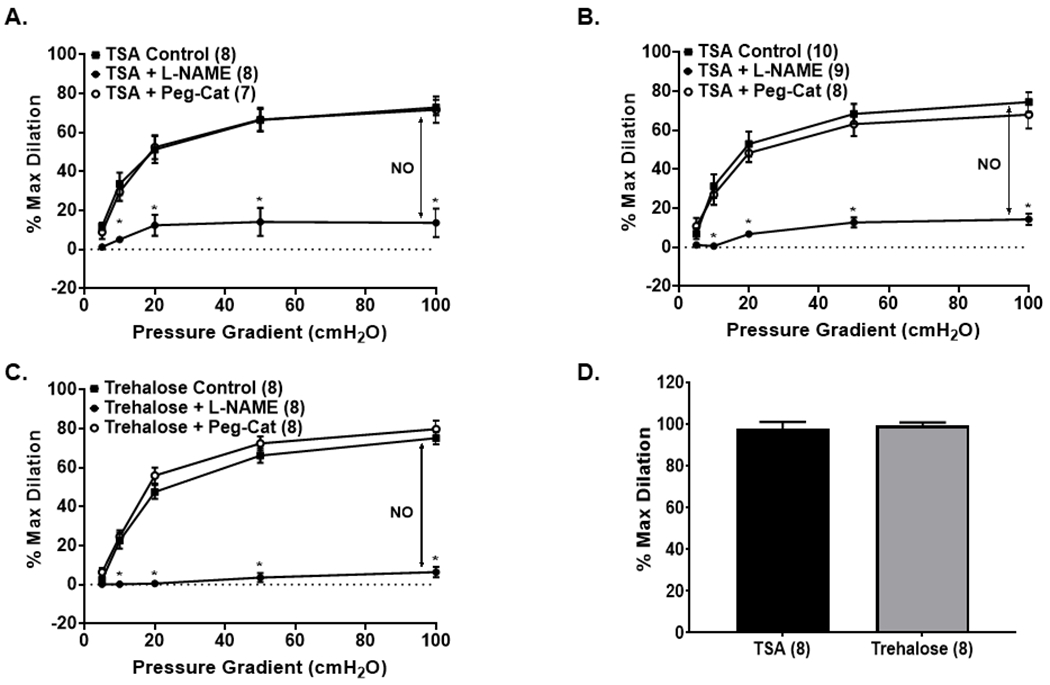
(A, B) Overnight activation of autophagy with TSA in atrial (A) or adipose (B) arterioles switches the mechanism of dilation from H2O2 to NO; (C) In CAD atrial arterioles, activation of autophagy with trehalose, induces a switch from H2O2 to NO. (D) Smooth muscle responses to papaverine are not affected by autophagy activation. * P < 0.05 vs. control; TSA, trichostatin A; L-NAME, NG-Nitro-l-arginine methyl ester; Peg-Cat; polyethylene glycol-catalase.
Autophagy Mediates the Effects of Telomerase on FMD in Human Arterioles.
We examined the cross-talk between TERT and autophagy in the context of vascular function. TERT inhibits mitochondrial-derived ROS and induces a switch in the mechanism of dilation from H2O2 to NO in CAD arterioles 12. Overnight incubation of non-CAD adipose arterioles with the TERT inhibitor BIBR 1532 reduced Lysotracker Red DND-99 fluorescence intensity in response to shear-stress relative to vehicle (P < 0.05), but co-incubation of BIBR 1532 and TSA increased fluorescence intensity during shear (P < 0.05; Fig. 5A). Furthermore, overnight incubation of CAD arterioles with the TERT expression activator AGS 499 increased fluorescence intensity (index of autophagic flux) in response to shear-stress (P < 0.05), and this was blocked with co-incubation of BAFA1 (P < 0.05; Fig. 5B). Activation of autophagy using either TSA or trehalose maintained NO as the primary mechanism of vasodilation in non-CAD arterioles that had been treated overnight with BIBR 1532, an inhibitor of TERT activity (P < 0.05; Fig. 6A&B). Conversely, increasing TERT expression (AGS 499) but inhibiting autophagy (BAFA1) maintained H2O2 as the mechanism of vasodilation in CAD arterioles (P < 0.05; Fig. 6C). The smooth muscle response to papaverine was unaffected by treatment with the combination of telomerase inhibition and autophagy activation, or autophagy inhibition and telomerase activation in non-CAD and CAD arterioles respectively. (Fig. 6D).
Figure 5: Autophagy signaling is required for shear-induced telomerase-induced responses.
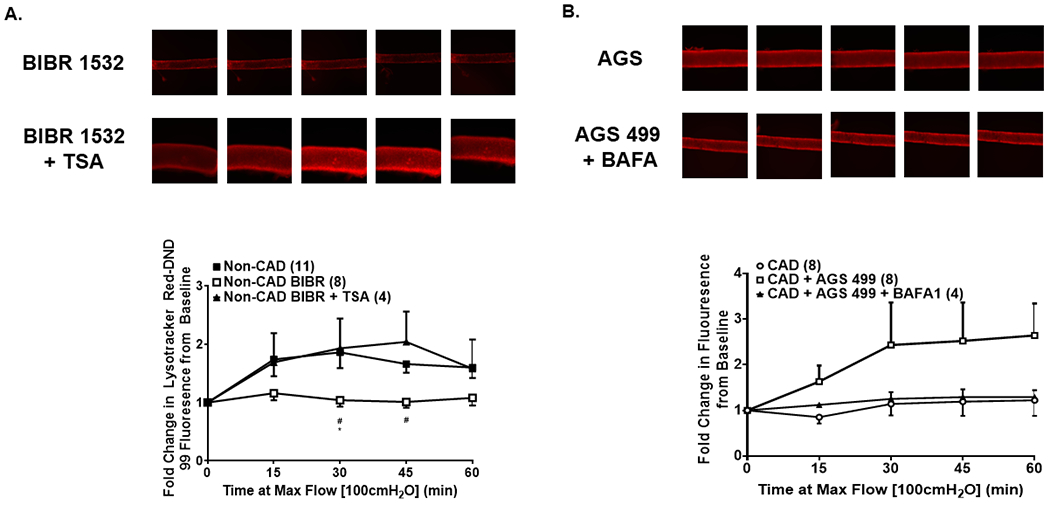
(A) Inhibition of TERT activity in non-CAD adipose arterioles with BIBR 1532 blunts shear-induced autophagy, but co-incubation with TSA rescues this response. (B) Activation of TERT expression in CAD adipose arterioles with AGS 499 enhances shear-induced autophagy, however, co-incubation with the lysosomal inhibitor BAFA1 abolishes this response. * P < 0.05 vs. control; TSA, trichostatin A; BAFA1, bafilomycin A1.
Figure 6: Modulation of autophagy independent of changes in TERT activity determines the mechanism of FMD in non-CAD and CAD arterioles.
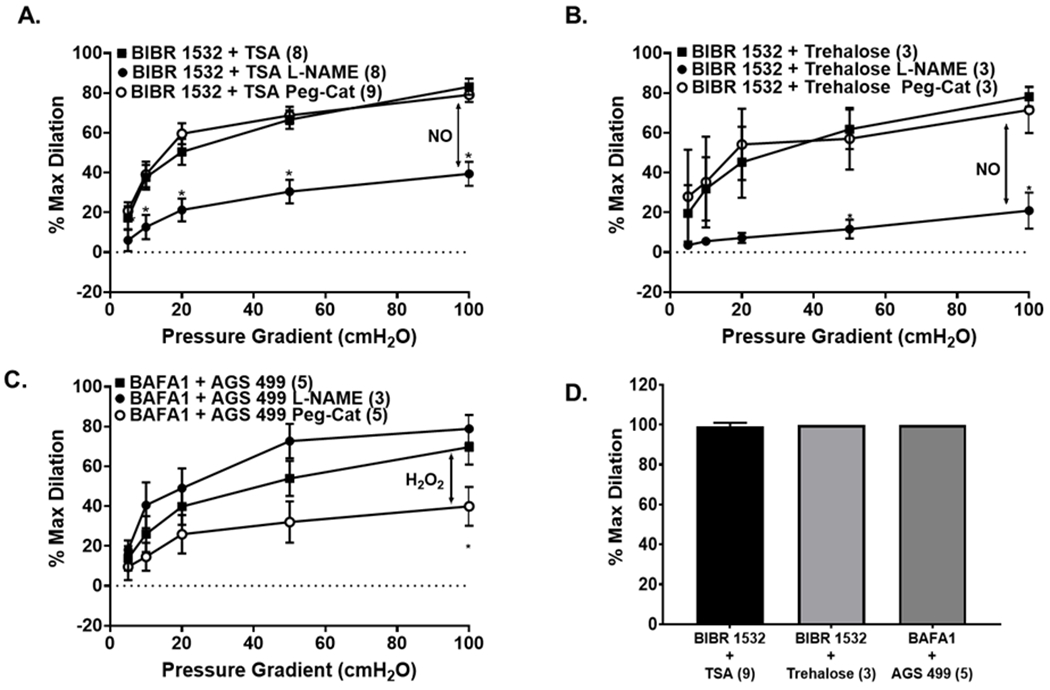
(A, B) Activation of autophagy with TSA (A) or trehalose (B), despite inhibition of TERT activity with BIBR 1532, maintains NO as the primary mechanism of dilation in non-CAD adipose arterioles; (C) Inhibition of autophagy with BAFA1 despite activation of TERT activity with AGS 499 maintains H2O2 as the primary mechanism of dilation in CAD arterioles. (D) Smooth muscle responses to papaverine are not different with incubation with modulators of autophagy and telomerase. * P < 0.05 vs. control; TSA, trichostatin A; BAFA1, bafilomycin A1; L-NAME, NG-Nitro-l-arginine methyl ester; Peg-Cat; polyethylene glycol-catalase.
Discussion
There are 3 major new findings from this work. First, we found that shear stress increased autophagy in isolated human adipose resistance arterioles, while lysosomal inhibition or Atg3 siRNA block shear-induced autophagy and switches the mechanism of dilation from NO to H2O2. Conversely, in CAD arterioles, shear stress does not increase autophagy, and agonist-activation of autophagy reverts the mechanism of dilation from H2O2 to NO. Modulation of autophagy in both non-CAD and CAD arterioles maintains native mechanism of dilation, independent of alterations to TERT activity. In cardiac tissue from patients with CAD, autophagosome formation (LC3 II/I) was elevated relative to tissue from non-CAD patients. However, the classic marker of autolysosome clearance, p62, was not different in tissue from patients with or without CAD. Collectively, these results highlight the critical role of autophagy in maintaining NO-mediated FMD in human resistance arterioles and provide a fundamental mechanistic link between autophagy and telomerase-mediated changes in human microvascular reactivity.
Role of Autophagy in the Heart
Autophagy plays an important role in the health of cardiac tissue 22. Previously, autophagy has been studied in the context of cardiomyocytes and the development of heart failure. Within the context of the current study, we found that LC3 II/I, a marker of autophagosome formation, was increased in LV tissue from patients with CAD relative to non-CAD LV tissue, suggesting a greater formation of autophagosomes. Importantly, p62 was not different between non-CAD and CAD samples (Supplemental Fig. 1B), suggesting that, while autophagosome formation is elevated in CAD, the rate of autophagosome clearance is similar between CAD and non-CAD. Collectively, these observations suggest a greater burden of damaged material designated for autophagy resulting in a greater formation of autophagosomes within CAD subjects. One key concept that has emerged is that autophagy has a dichotomous nature, such that autophagy may be adaptive or maladaptive. Both upregulation (excessive) and downregulation (insufficient) of autophagy can be pathological in either the inability to respond to metabolic stressors (insufficient) or hyperactivation in response to metabolic stress. It should be emphasized that protein quantification of autophagy only offers a “snap-shot” of the dynamic nature of autophagy. Interestingly, within primary endothelial cells sampled from participants with and without CAD, we found no differences in LC3B or p62. This could be due to a few factors including that these primary endothelial cells were sampled from the antecubital vein, which is not exposed to high levels of shear stress relative to arteries, nor were LC3-II differentiated from LC3-I. In contrast, Park, et. al.23 recently demonstrated that LC3B increases, while p62 decreases primary endothelial cells sampled from the radial artery exposed to shear stress via handgrip exercise. Taken together, it may be that a certain level of “stress” may be required to observe differences in autophagy within primary endothelial cells in humans.
Shear-Induced Autophagic Flux
A second novel finding of the present study is that shear stress increased autophagic flux in non-CAD resistance arterioles with no change in CAD arterioles. We observed that isolated arterioles from non-CAD subjects exposed to shear stress for 60 minutes exhibited marked increases in Lysotracker Red DND-99 fluorescence, indicative of an increase in size or number of acidic lysosomes (Fig 1A&C), and thus, proper initiation and progression of autophagy. Importantly, this increase in fluorescence was blocked when arterioles were incubated with BAFA1, which inhibits lysosome acidification and autophagosome-lysosome fusion. In CAD arterioles, exposure to shear stress resulted in an attenuated Lysotracker Red DND-99 fluorescence response (relative to non-CAD), which was ameliorated after overnight incubation with the autophagy activator TSA (Fig. 1D). These findings extend what has been previously demonstrated within cell culture models 8, 9, 24–27 and provide novel evidence that shear stress increases autophagy within isolated adipose arterioles.
There are several lines of evidence supporting a role for shear-stress in modulating autophagic responses within the vasculature to help maintain endothelial integrity. Bharath, et. al. 8 demonstrated in bovine aortic endothelial cells that 3-hours of exposure to physiological shear stress (~20 dyn·cm2) increased LC3-II/I ratio, a classic marker of autophagosome formation, that was paralleled by an increase in NO and ROS. Importantly, inhibition of autophagy drastically decreased shear-induced NO production while exaggerating ROS production. Within the current study, we demonstrate that inhibition of autophagy in non-CAD arterioles increases mitochondria H2O2 production in response to shear that is abolished in the presence of PEG-cat. Furthermore, in CAD arterioles, H2O2 production is increased in response to shear stress, an effect that is abolished with activation of autophagy. Together, these data indicate that autophagy modulates redox status within the microvasculature. It should be noted that not all shear stress promotes autophagy. Indeed, laminar (5-20 dyn/cm2), but not oscillatory (± 5 dyn/cm2 @ 1 Hz), turbulent, or low shear (< 5 dyn/cm2) promotes autophagy as evidenced by increases in mRNA and protein expression of beclin-1 and LC3B, and decreases in p62 that are paralleled by increases in eNOS expression 8, 24–27. Relevant to the microcirculation, virtually all flow is laminar since Reynolds number is rarely achieved in pathophysiological conditions. Interestingly, the protective effects of autophagy may result from its critical role in maintaining endothelial cell structural stability. It has been demonstrated that intact autophagic flux is necessary for proper endothelial cell alignment in response to shear stress 27 and in maintaining eNOS expression 24, 28.
Autophagy as the Mediator of FMD, and Cross-Talk between Autophagy and Telomerase
Previous work from our group showed that microvascular FMD is predominately due to NO, while this mechanism switches to H2O2 with the presence of CAD 4. Here we demonstrate that autophagy plays a critical role in determining the mechanism for FMD. In non-CAD arterioles, overnight inhibition of autophagy via two independent methods (BAFA1 and Atg3 siRNA) switched the mediator of FMD from NO to H2O2 (Fig. 3, Supplemental Fig. 4), that is, treated arterioles did not dilate in the presence of PEG-cat, but dilated normally in the presence of L-NAME. Conversely, overnight activation of autophagy with two independent methods in CAD arterioles switched the mechanism of dilation from H2O2 to NO (Fig. 4A&B, Supplemental Fig 6). Thus, autophagy is a critical element in regulating the mediation of FMD in the microcirculation.
We have previously demonstrated that several factors contribute to the mediator of FMD, including telomerase, lysophosphatidic acid, and ceramide 5, 12, 18, 19, 29, 30, however it is not known whether these seemingly independent pathways might be linked in regulating vascular endothelial function. We hypothesized that autophagy acts downstream of other factors involved in the switch in mechanism of dilation. Consistent with this notion, autophagy raises NO and reduces ROS in endothelial cells 8. To test this hypothesis, we examined whether autophagy regulates the vasomotor response to telomerase. Resistance arterioles from non-CAD subjects were co-incubated overnight with the telomerase inhibitor BIBR 1532 as well as TSA, an autophagy activator. If autophagy acts downstream of telomerase we would predict that TSA would restore the mediator of FMD from H2O2 to NO. If telomerase acts downstream of autophagy, TSA would not change the mediator in presence of BIBR 1532. If both pathways are independent, then TSA should restore NO as the mediator of FMD in presence of BIBR 1532 and in separate experiments we would anticipate that AGS 499 would restore NO as the mediator of FMD in presence of the autophagy inhibitor BAFA1. Our observations provide novel evidence for the cross-talk between telomerase and autophagy, and indicate that autophagy may act as a final common signaling pathway to determine the mechanism of FMD in the microcirculation (Fig. 6) acting to moderate the detrimental effects of DNA damage, as well as mitochondrial membrane depolarization, which would specify a role for mitophagy within vascular health and function.
Previous evidence for the cross-talk between autophagy and telomerase comes from cell culture. Ali, et. al. 17 demonstrated in murine embryonic fibroblasts that transgenic repression of telomerase reverse transcriptase (TERT) reduced autophagic activity at baseline and with amino acid starvation (autophagy stimulator). Transgenic overexpression of TERT induces autophagy in an mTORC1-dependent manner. In a similar fashion, Cheng, et. al. 31 demonstrated the functional effects of this cross-talk by showing that repression of telomerase impairs autophagic responses to ischemia-reperfusion within the murine kidney as evidenced by a reduced expression of LC3II and accumulation of p62, effects that were ameliorated by treatment with the mTOR inhibitor, rapamycin. The current study extends these findings to the human microvasculature. Healthy tissue, in response to shear stress-induced autophagic activity, maintains NO bioavailability and mitigates ROS. Within CAD arterioles, shear stress fails to induce autophagy (via Lysotracker Red DND-99), potentially reducing NO bioavailability and tipping the redox balance to a more oxidative stress state (Fig. 1&2) resulting in H2O2 as the predominant mechanism of dilation in response to shear stress.
Experimental Considerations
Our study has several limitations. First, we are limited in our ability to separate CAD from non-CAD subjects as we are dependent upon de-identified, surgically discarded and donor tissue that is reviewed by trained pathology staff for diagnosis of CAD in addition to individual risk factors. However, it is possible that some non-CAD subjects had sub-clinical CAD. Second, we examined “basal” autophagy within the left ventricle and primary isolated endothelial cells sampled from different patient populations. Current guidelines for examination of autophagy suggest this is an imperfect measurement, and lysosomal inhibition within samples would provide a better indicator of autophagic flux 32. Finally, previous reports have demonstrated that autophagy declines with advancing age 10, 11, 33; however, due to our sample size, we do not have sufficient power to evaluate the relations of age or sex with the autophagy measures. Previous evidence from our lab has demonstrated that the switch in dilator mechanism is independent of age and is seen specifically in the presence of CAD 4.
Perspectives
We identify a critical and fundamental role for autophagy in the disease-associated switch in endothelial mediator of FMD in human microcirculation. Utilizing both pharmacological and genetic interventions, we demonstrate that in response to shear-stress, resistance arteriolar autophagic flux increases however, this response is blunted in subjects with CAD and contributes to the compensatory switch to H2O2 as the mechanism of dilation. Additionally, autophagy acts downstream of an established modulator of FMD mechanism, telomerase, and may serve as a fundamental pathway to ultimately determine the central mechanism of flow-induced dilation in the human microcirculation. Collectively, this data indicates that autophagy plays a central role in maintaining the functionality of the microvasculature in health and disease.
Supplementary Material
Highlights.
Shear stress fails to induce autophagy in resistance arterioles from subjects with CAD.
Failure to initiate autophagy in response to shear stress increases mitochondrial derived H2O2 in CAD that is rescued by activation of autophagy. Inhibition of autophagy in non-CAD increases mitochondrial H2O2.
Inhibition of autophagy switches the mechanism of microvascular FMD from NO to H2O2 in non-CAD, while activation of autophagy in CAD switches the mechanism of dilation from H2O2 to NO
Telomerase appears to signal through autophagy to determine the mechanism of microvascular dilation in health and disease
Acknowledgements:
A) Acknowledgements: We would like to thank the following locations for their assistance in providing tissue for this study: Wisconsin Donor Network and Versiti, Froedtert Memorial Lutheran Hospital, and Wheaton Franciscan Healthcare’s Elmbrook Memorial Hospital.
B) Sources of Funding: This work was supported by the National Institutes of Health Grants T32GM089586 and AHA 20POST35050017 (W.E.H); K01-HL143142 (JLF); R01HL133029 (A.M. Beyer); R01-HL-135901-01 and Northwestern Mutual professorship (to D.D. Gutterman).
Abbreviations
- BAFA1
Bafilomycin A1
- BIBR 1532
Telomerase Inhibitor
- CAD
Coronary Artery Disease
- eNOS
Endothelial Nitric Oxide Synthase
- FMD
Flow-Mediated Dilation
- H2O2
Hydrogen Peroxide
- LC3B
Microtubule-associated proteins 1A/1B light chain 3B
- NO
Nitric Oxide
- PEG-Cat
polyethylene glycol catalase
- TSA
Trichostatin A
Footnotes
Conflicts of Interest/Disclosures: The authors have no conflicts of interest or disclosures
References
- 1.Gutterman DD, Chabowski DS, Kadlec AO, et al. The human microcirculation: Regulation of flow and beyond. Circulation research. 2016;118:157–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poole DC, Behnke BJ, Padilla DJ. Dynamics of muscle microcirculatory oxygen exchange. Medicine and science in sports and exercise. 2005;37:1559–1566 [DOI] [PubMed] [Google Scholar]
- 3.Cooper LL, Palmisano JN, Benjamin EJ, et al. Microvascular function contributes to the relation between aortic stiffness and cardiovascular events: The framingham heart study. Circulation. Cardiovascular imaging. 2016;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beyer AM, Zinkevich N, Miller B, et al. Transition in the mechanism of flow-mediated dilation with aging and development of coronary artery disease. Basic research in cardiology. 2017;112:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Phillips SA, Hatoum OA, Gutterman DD. The mechanism of flow-induced dilation in human adipose arterioles involves hydrogen peroxide during cad. American journal of physiology. Heart and circulatory physiology. 2007;292:H93–100 [DOI] [PubMed] [Google Scholar]
- 6.Cai H Hydrogen peroxide regulation of endothelial function: Origins, mechanisms, and consequences. Cardiovascular research. 2005;68:26–36 [DOI] [PubMed] [Google Scholar]
- 7.Nussenzweig SC, Verma S, Finkel T. The role of autophagy in vascular biology. Circulation research. 2015;116:480–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bharath LP, Mueller R, Li Y, et al. Impairment of autophagy in endothelial cells prevents shear-stress-induced increases in nitric oxide bioavailability. Canadian journal of physiology and pharmacology. 2014;92:605–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bharath LP, Cho JM, Park SK, et al. Endothelial cell autophagy maintains shear stress-induced nitric oxide generation via glycolysis-dependent purinergic signaling to endothelial nitric oxide synthase. Arteriosclerosis, thrombosis, and vascular biology. 2017;37:1646–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.LaRocca TJ, Henson GD, Thorburn A, et al. Translational evidence that impaired autophagy contributes to arterial ageing. The Journal of physiology. 2012;590:3305–3316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaplon RE, Hill SD, Bispham NZ, et al. Oral trehalose supplementation improves resistance artery endothelial function in healthy middle-aged and older adults. Aging. 2016;8:1167–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beyer AM, Freed JK, Durand MJ, et al. Critical role for telomerase in the mechanism of flow-mediated dilation in the human microcirculation. Circulation research. 2016;118:856–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ait-Aissa K, Heisner JS, Norwood Toro LE, et al. Telomerase deficiency predisposes to heart failure and ischemia-reperfusion injury. Frontiers in cardiovascular medicine. 2019;6:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ait-Aissa K, Kadlec AO, Hockenberry J, et al. Telomerase reverse transcriptase protects against angiotensin ii-induced microvascular endothelial dysfunction. American journal of physiology. Heart and circulatory physiology. 2018;314:H1053–h1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Sullivan RJ, Karlseder J. Telomeres: Protecting chromosomes against genome instability. Nature reviews. Molecular cell biology. 2010;11:171–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qing H, Aono J, Findeisen HM, et al. Differential regulation of telomerase reverse transcriptase promoter activation and protein degradation by histone deacetylase inhibition. Journal of cellular physiology. 2016;231:1276–1282 [DOI] [PubMed] [Google Scholar]
- 17.Ali M, Devkota S, Roh JI, et al. Telomerase reverse transcriptase induces basal and amino acid starvation-induced autophagy through mtorc1. Biochemical and biophysical research communications. 2016;478:1198–1204 [DOI] [PubMed] [Google Scholar]
- 18.Chabowski DS, Kadlec AO, Ait-Aissa K, et al. Lysophosphatidic acid acts on lpa1 receptor to increase h2 o2 during flow-induced dilation in human adipose arterioles. British journal of pharmacology. 2018;175:4266–4280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kadlec AO, Chabowski DS, Ait-Aissa K, et al. Pgc-1alpha (peroxisome proliferator-activated receptor gamma coactivator 1-alpha) overexpression in coronary artery disease recruits no and hydrogen peroxide during flow-mediated dilation and protects against increased intraluminal pressure. Hypertension. 2017;70:166–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fetterman JL, Holbrook M, Flint N, et al. Restoration of autophagy in endothelial cells from patients with diabetes mellitus improves nitric oxide signaling. Atherosclerosis. 2016;247:207–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tabit CE, Shenouda SM, Holbrook M, et al. Protein kinase c-beta contributes to impaired endothelial insulin signaling in humans with diabetes mellitus. Circulation. 2013;127:86–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sciarretta S, Maejima Y, Zablocki D, et al. The role of autophagy in the heart. Annual review of physiology. 2018;80:1–26 [DOI] [PubMed] [Google Scholar]
- 23.Park SK, La Salle DT, Cerbie J, et al. Elevated arterial shear rate increases indices of endothelial cell autophagy and nitric oxide synthase activation in humans. American journal of physiology. Heart and circulatory physiology. 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo F, Li X, Peng J, et al. Autophagy regulates vascular endothelial cell enos and et-1 expression induced by laminar shear stress in an ex vivo perfused system. Annals of biomedical engineering. 2014;42:1978–1988 [DOI] [PubMed] [Google Scholar]
- 25.Yao P, Zhao H, Mo W, et al. Laminar shear stress promotes vascular endothelial cell autophagy through upregulation with rab4. DNA and cell biology. 2016;35:118–123 [DOI] [PubMed] [Google Scholar]
- 26.Liu J, Bi X, Chen T, et al. Shear stress regulates endothelial cell autophagy via redox regulation and sirt1 expression. Cell death & disease. 2015;6:e1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vion AC, Kheloufi M, Hammoutene A, et al. Autophagy is required for endothelial cell alignment and atheroprotection under physiological blood flow. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:E8675–e8684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang JX, Qu XL, Chu P, et al. Low shear stress induces vascular enos uncoupling via autophagy-mediated enos phosphorylation. Biochimica et biophysica acta. Molecular cell research. 2018;1865:709–720 [DOI] [PubMed] [Google Scholar]
- 29.Freed JK, Beyer AM, LoGiudice JA, et al. Ceramide changes the mediator of flow-induced vasodilation from nitric oxide to hydrogen peroxide in the human microcirculation. Circulation research. 2014;115:525–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beyer AM, Durand MJ, Hockenberry J, et al. An acute rise in intraluminal pressure shifts the mediator of flow-mediated dilation from nitric oxide to hydrogen peroxide in human arterioles. American journal of physiology. Heart and circulatory physiology. 2014;307:H1587–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng H, Fan X, Lawson WE, et al. Telomerase deficiency delays renal recovery in mice after ischemia-reperfusion injury by impairing autophagy. Kidney international. 2015;88:85–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.LaRocca TJ, Gioscia-Ryan RA, Hearon CM Jr., et al. The autophagy enhancer spermidine reverses arterial aging. Mechanisms of ageing and development. 2013;134:314–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.