

Abstract
Alzheimer’s disease (AD) is a progressive and synaptic failure disease. Despite the many years of research, AD still harbors many secrets. As more of the world’s population grows older, researchers are striving to find greater information on disease progression and pathogenesis. Identifying and treating the markers of this disease, or better yet, preventing it all together, are the hopes of those investing in this field of study. Several years of research revealed that synaptic pathology and mitochondrial oxidative damage are early events in disease progression. Loss of synapses and synaptic damage are the best correlates of cognitive deficits found in AD patients. As the disease progresses, there are significant changes at the synapse. These changes can both shed greater light onto the progression of the disease and serve as markers and therapeutic targets. This article addresses the mechanisms of synaptic action, mitochondrial regulation/dysregulation, resulting synaptic changes caused by amyloid beta and phosphorylated tau in AD progression. This article also highlights recent developments of risk factors, genetics and ApoE4 involvement, factors related to synaptic damage and loss, mislocalization of amyloid beta and phosphorylated tau, mitophagy, microglial activation and synapse-based therapies in AD. Furthermore, impairments in LTD and reactivation of microglia are discussed.
Keywords: Alzheimer’s disease, Synapse, Amyloid Beta, Tau, FYN, Reelin, Microglia, ApoE4, Mitochondria, Amyloid precursor protein, Mitochondrial dynamics, Mitophagy
1. Introduction
First described by a German neuropathologist Alois Alzheimer in 1906, Alzheimer’s Disease (AD) is a neurodegenerative disease that presents with progressive cognitive and behavioral impairments such as the inability to make new memories and the loss of important past memories (Koffie et al., 2011). Both declarative and non-declarative memory is affected in AD. Patients have difficulty reasoning, conceptualizing abstract concepts, and even language impairments (Selkoe, 2002). As a result, patients may also develop depression, difficulty sleeping, and increased anxiety. Through its progression, there is a significant decline in executive functions that eventually renders the patient unable to accomplish daily living activities (Scheff et al., 2014).
Currently, more than 50 million people are affected by AD globally of which more than 5.8 million are from the United States (Alzheimer’s, Association, 2020). These numbers are continuing to rise and by 2050 are expected to quadruple. AD has increased in its mortality rate by 66% over the last decade and is now the 6th leading cause of death in the United States (Koffie et al., 2011). A new case of dementia develops every 3 seconds and more than 85% of AD cases occur in patients over the age of 60 years (Report, 2015). There has been an increase in AD cases in low- and middle-income countries. In 2015, 58% of all people with dementia lived in low- and middle-income countries. However, this percentage is expected to rise to 63% by 2030 and 68% by 2050.
As advancements in healthcare help patients live longer, the prevalence of AD, a disease that primarily affects the elderly, is also increasing (Kashyap et al., 2019). With no current treatments to prevent or reverse AD, the cumulative cost of care for AD patients will place a heavier burden on health care systems worldwide in the years to come (Forner et al., 2017). The total estimated worldwide cost of dementia in 2015 was US$ 818 billion. As cases increase, dementia will become a trillion-dollar disease.
AD occurs in two forms: early-onset familial and late-onset sporadic. Early-onset AD is an extremely uncommon form of the disease seen in one to two percent of all AD cases. Mutations in amyloid beta precursor protein (APP), presenilin 1 (PS1 or PSEN1), and presenilin 2 (PS2 or PSEN2) loci cause early-onset autosomal form of familial AD. Mutations in these genes induce the increased overproduction of amyloid-β (40 or 42), leading to early-onset familial AD. Late-onset sporadic AD is the more common form of the disease and presents after the age of 65 (Report, 2015) and APOE4 genotype. In late-onset sporadic AD, a combination of genetic and environmental factors and lifestyle components play a large role (Amakiri et al., 2019). Other polymorphisms can also contribute to late-onset AD (Amakiri et al., 2019).
Several risk factors are involved in AD, including modifiable and non-modifiable risk factors. Modifiable risk factors for sporadic AD involve type 2 diabetes or obesity and a large number of lifestyle factors such as stress, unhealthy diet, lack of physical exercise, alcohol, smoking, and exposure to environmental toxins (ex. Lead) (George and Reddy, 2019). Furthermore, vascular diseases, depression, stroke, hypertension, and traumatic brain injury are other examples of modifiable risk factors (George and Reddy, 2019). Non-modifiable risk factors include age, sex, or genetic polymorphisms. Apolipoprotein E4 genotype is a major contributor to late-onset sporadic AD.
Several years of intense research on postmortem AD brains and transgenic mouse models of AD have revealed that multiple cellular changes are involved with the disease process, including loss of neurons, synaptic loss/damage, mitochondrial fragmentation, increased free radical production, mitochondrial DNA damage, proliferation of astrocytes and microglia, hormonal imbalance, altered neurotransmitter levels, neurofibrillary tangles, and senile plaques (Han et al., 2020; Reddy et al., 2010; Reddy et al., 2012; Selkoe, 2001; Zhu et al., 2013). These changes primarily were observed in learning and memory regions of the brain, including the entorhinal cortex, and spreads to the hippocampus, temporal cortex, frontoparietal cortex, and finally to subcortical nuclei (Reddy and McWeeney, 2006).
AD brains show an increase in neuronal cell death, neuroinflammation, formation of neurofibrillary tangles (NFT), and formation of amyloid-beta plaques. The genetics, synapse loss, mitochondrial dysfunction, and microRNA dysregulation all contribute to the progression of AD. Figure 1 gives a brief overview of changes in AD brains.
Figure 1.

Synaptic and cellular changes in Alzheimer’s disease. Imaging compares Alzheimer’s disease brain with a healthy brain to show loss of brain mass as a result of disease process.
The inheritance of AD is autosomal dominant. One genetic risk is the Apolipoprotein E4 genotype (ApoE) on chromosome 19 (Selkoe, 2002). ApoE has 3 isoforms and is produced by astrocytes and microglia. It binds to lipoproteins and is taken into nerve cells during the developmental stage of the nervous system or after neuronal damage (Yamazaki et al., 2019). Impaired ApoE affects clearance of amyloid beta (Aβ), a 38-43 amino acid peptide that creates plaques when clumped together in the extracellular space between neurons. The aggregation can lead to an inflammatory response. The triggering receptor expressed on myeloid cells 2 (TREM2) recognizes ApoE and engages in microglial phagocytosis. This was thought to aid in clearance of Aβ but recently has been shown to have an alternate effect in exacerbating tau pathology (Kametani and Hasegawa, 2018). Neurofibrillary tangles, a characteristic of AD brains, are results of hyperphosphorylated tau protein deposited within the neurons.
Reelin is an extracellular signaling protein. It is crucial in the structural layering of the cerebral cortex in mammals by regulating the radial migration of cortical neurons. It is also involved in the maturation and differentiation of dendrites. It is produced by the pyramidal cells of the Piriform and Entorhinal cortex. Reelin is affected by aging and persistent inflammatory responses (Krstic et al., 2013). ApoE receptor 2 and very low-density lipoprotein receptors act as signaling receptors for reelin and regulate neuronal migration during early development. In normal brains, reelin phosphorylates NMDA receptor GluN2 subunits and enhances NMDA and long-term potentiation (LTP) activity. By doing so, reelin plays a role in countering the effects of soluble Aβ. The E4 allelic variant of ApoE isoforms increases the accumulation of intraneural amyloid (Dorostkar et al., 2015). It also leads to increased synaptic loss, modulates inflammatory responses, and increases oligomeric Aβ recruitment near synapses. ApoE4 depletes ApoER2 and NMDA receptors. As a result, reelin activity is reduced, thus disinhibiting the accumulation of Aβ (Sheng et al., 2012). These changes are summarized in Figure 2.
Figure 2.

Activity of Reelin in normal brains inhibits the aggregation of soluble Aβ. However, in Alzheimer’s disease, ApoE4 disrupts the regulatory function of Reelin at NMDA receptors. This leads to disinhibition of soluble Aβ accumulation.
Besides the ApoE gene, Presinilin 1 gene on chromosome 14q, and Presinilin 2 gene on chromosome 1q are also possible aggressors of AD progression. These genes affect the cleavage of Amyloid Precursor Protein (APP), the production of Aβ, and its metabolism (Selkoe, 2002). Presenilins are important for the γ-secretase complex that is involved in the processing of APP. Imbalances in PS1 and PS2 can favor Aβ42 production over that of Aβ40. The favoring of Aβ42 is key in AD progression as will be addressed in this article. Furthermore, chronic impairment of γ-secretase will affect dendritic spine numbers (Dorostkar et al., 2015). Presenilin have a role in pulling calcium out of the endoplasmic reticulum and are important in normal LTP and plasticity (Sheng et al., 2012). Increased PS1 increases LTP in hippocampal synapses and causes calcium dyshomeostasis due to interference with calcium release within the cell (Sheng et al., 2012).
Amongst the indicators of AD, dramatic synaptic loss is the first indicator of AD progression found even in the earliest of stages of AD, Mild Cognitive Impairment (MCI). Researchers noted that the efficiency of transmitting signals and strength of these signals, decreased due to spinal loss. There is also a disintegration of the network as signals no longer carry through due to AD progression. Furthermore, alternate paths for transmission also decrease while the length of compensatory paths taken increases (Kashyap et al., 2019).
The synaptic and cellular changes, such as the distribution of amyloid plaques, contribute to the magnitude of memory deficits. Mice had increased difficulty in the Morris water maze as accumulation of Aβ increased (Manczak et al., 2018). NFT prevalence also correlates strongly with AD progression. Tau mice develop motor and behavioral impairments with the presentation of neurofibrillary pathology (Kandimalla et al., 2018). As NFTs present more, there is increased memory deficits and neuron loss (Pozueta et al., 2013). The plaques and neurofibrillary tangles are found in the hippocampal and basal forebrain-neocortical pathways. These pathways are cholinergic and the development of such plaques and NFTs leads to impairment in acetylcholine production and metabolism.
Mitochondrial dysfunction also occurs early in AD progression (Reddy et al., 2012). Mitochondrial membrane permeability is affected by its interaction with Aβ and results in an increase in free radical generation, an increase in mitochondrial fission, and the beginning of a cascade towards cell death (Moneim, 2015).
The purpose of our article is to highlight the recent developments in synaptic changes disease progression of AD. Our article will focus on the synapse loss and mitochondrial damage as early events in AD progression and will provide an overview of Aβ, phosphorylated tau, and mitochondria pathologies in AD progression. Furthermore, impairments in LTD and reactivation of microglia will be discussed.
2. Alzheimer’s Synapse
Ramon y Cajal was the first to note dendritic spines in the late 19th century. The dendrite is the post synaptic terminal of the synapse and contains NMDA and AMPA receptors. They also have metabotropic receptors and scaffolding proteins. The shape of the spines is controlled by actin and, in some cases such as large CA3 dendrites, other microtubules (Pozueta et al., 2013). Furthermore, the number of dendritic spines is related to actin. F-actin determines the shapes of spines. The loss of F-actin leads to collapse of spines (Kashyap et al., 2019). Dendrites have varying shapes comprised of a spine ‘head’ that is connected to the main shaft or stalk of the dendrite. There are three types of spine heads, including mushroom-shaped, stubby spines, and thin spines (Dorostkar et al., 2015). The necks of these spines are traditionally narrow. Developing neurons have thin necks without bulbous heads, but the shapes of these spines, can change quickly as they age (Pozueta et al., 2013). Increases in spine size allow for more receptors. Shortening of the spine neck, or widening it, reduces electrical resistance. This leads to large excitatory postsynaptic potentials (Dorostkar et al., 2015). Loss of dendritic spines results in neurodegenerative disorders.
Normal aging does result in some cognitive decline. This cognitive decline is due to the reduced maintenance of synaptic contacts. There is also a decrease in spine density that correlates with cognitive decline. The size of the dendritic spine can also correlate with its vulnerability to aging such that small spines are more vulnerable than larger ones. Larger spines are more stable. The synaptic impairment occurs more on the dendrites than the axonal terminals. Mostany and colleagues took images of mice across their lifespan and noted that there were long and thin dendritic spines at juvenile ages, but short and small spines by old age. In mature mice, the large mushrooms occurred more frequently in mature mice. There was a decrease in spine density, spine turnover, as mice aged to adult hood. Between adulthood and old age, there was an increase in spine density and an increase in spine turnover. However, there was a decrease in spine volume. Long term stability of dendritic spines decreases with age. Shrinking of spine volume results in weaker synapses. There is increased homogeneity in the synapses of older populations. This reduces the ability to compensate for brain injury in older ages. Increased spine turnover and smaller spine volume results in cognitive decline. Overall, as one ages, the types of spines may change, but density and spine turnover does not change (Mostany et al., 2013).
2.1. Alteration and Loss of synapses in Alzheimer’s disease
A key change in synapses during AD progression is a loss of synapses. Researchers have noted this decrease in synapses using a variety of visualizing techniques. Some researchers cultured neurons from the cortex of APP/PS1 mice and then stained these samples with phalloidin to visualize spines. This technique showed a marked decrease in spine density (Kashyap et al., 2019). Others, using synaptophysin like immunoreactivity, also noted that there was a 45% decrease in immunoreactivity in AD samples as a result of damaged presynaptic terminals. This technique also revealed dilations in neurites around senile plaques. Using this staining method, the researchers looked at different areas of the brain including the neocortex, the hippocampus, and the entorhinal cortex. They assessed changes in immunoreactivity across layers and noted synaptic loss in layers 2 and 3 of the frontal and parietal cortex as a result of AD progression. Furthermore, they saw greater damage in presynaptic terminals than entire large neurons (Masliah et al., 1991).
Scheff and colleagues counted synapses in the outer molecular layer of the dentate gyrus and noted that there was a statistically significant decrease in synapses between those with cognitive impairments and those without. Furthermore, the dentate gyrus outer layer volume was decreased in early AD patients. They also noted that synapse number and dentate gyrus volume correlated with performance on neuropsychological tests (Scheff et al., 2006).
Dominguez-Alvaro and team used ion beam/scanning electron microscopy to assess the changes in the trans entorhinal cortex (TEC) as a result of AD. The images from 3D modeling and scanning showed that excitatory synapses in AD lacked preference in spine head versus dendritic shaft (50.2% vs 49.1%). However, inhibitory synapses in AD greatly favored the dendritic shaft over the spine heads (90.2% vs 8.8%). Overall, AD samples showed a decreased preference for spine heads. Previous studies, using ion beam/scanning electron microscopy, also noted a decrease in TEC cortical thickness (Dominguez-Alvaro et al., 2019).
Electron microscopy of the hippocampal CA1 stratum lacunosum-moleculare (SLM), showed a reduction in SLM size and a decrease in volume. Researchers also noted that there was a decrease in synaptic density in this area. These two factors were statistically different in 5xADTg mice in comparison to wild-type mice. Furthermore, immunofluorescence array tomography for PSD-95 showed that there was a decrease in SLM volume as 5xADTg mice aged. There was a reduction of small synapses that are very plastic. Even in areas without amyloid plaques, such as the SLM, electron microscopy has shown that there is synapse loss specific to distal dendrites. While synapses are lost, the ones that remain are larger and might have greater strength in connectivity (Neuman et al., 2015).
Researchers, using mathematical models, noted a decrease in coupling strength due to synapse loss. Furthermore, synapse loss results in fewer highest efficiency paths. Researchers have also noted that there is a critical time period, after which the synapse loss rapidly increases (Kashyap et al., 2019).
Overall, the reduced spine density can lead to impaired memory, difficulty in coordinating activities, and lower signal transmission. Increasing evidence also suggests that synapse loss is an early event disease process that occurs due to soluble amyloid beta, phosphorylated tau accumulation, and increased production of mitochondrially generated free radicals at synapses. Therefore, reduced synaptic accumulation of amyloid beta, phosphorylated tau, and mitochondrial free radicals may reduce synaptic loss and enhance cognitive functions in AD patients.
2.2. Loss of synaptic proteins in Alzheimer’s disease
Gonatas and colleagues noted in 1967 that AD brain tissue had abnormal synapses. In early AD models, there is not much synaptic loss. There is more loss of postsynaptic proteins than presynaptic proteins. This was measured by the decrease of a post synaptic actin binding protein drebrin (Gylys et al., 2004). Drebrin is a structural protein in dendritic spines and is important for dendritic morphology. In AD, there is great activation of caspase in neurites and synaptosomes. Caspase activity disorganizes actin filament networks and drebrin (Gylys et al., 2004).
Synaptophysin is a presynaptic vesicle protein. It has been shown to decrease by around 25% in MCI patients. This change can occur well before Aβ plaque formation. Cognitive decline correlates with an impairment of synaptophysin in the hippocampus and other associated cortices (Selkoe, 2002). Loss of synaptophysin is a marker for disease progression in AD patients and AD mice.
Reddy and colleagues extensively studied synaptic proteins in a large number of healthy controls and AD patients (Reddy et al., 2005). To determine whether presynaptic or postsynaptic compartments of neurons are preferentially affected in AD patients, they studied 3 presynaptic vesicle proteins (synaptotagmin, synaptophysin, and Rab 3A), 2 synaptic membrane proteins (Gap 43 and synaptobrevin), and 2 postsynaptic proteins (neurogranin and synaptopodin) in specimens from AD and age-matched healthy control brains (Reddy et al., 2005). Two brain regions, the frontal and parietal cortices were assessed for protein levels by immunoblotting analysis. They found a loss of both presynaptic vesicle proteins and postsynaptic proteins in all brain specimens from AD patients compared to those from age-matched control subjects. Furthermore, they found that the loss of synaptic proteins was more severe in the frontal cortex brain specimens than in the parietal cortex brain specimens from AD subjects compared to those from control subjects, suggesting that the frontal brain may be critical for synaptic function in AD. Using immunohistochemistry techniques, they also determined the distribution pattern of all synaptic proteins in both the frontal and parietal cortices brain specimens from control subjects. Of the 7 synaptic proteins studied, the presynaptic proteins synaptophysin and Rab 3A and the postsynaptic protein synaptopodin were the most down-regulated. They concluded that postsynaptic proteins and presynaptic proteins are important for synaptic function and may be related to cognitive impairments in AD.
De Wilde and colleagues conducted a meta-analysis of synaptic proteins. They identified over 400 publications reporting postmortem synapse and synaptic marker loss from AD patients (de Wilde et al., 2016). Two meta-analyses were performed using a single database of subselected publications. The first meta-analysis confirmed synaptic loss in selected brain regions is an early event in AD pathogenesis. The second meta-analysis of 57 synaptic markers revealed that presynaptic makers were affected more than postsynaptic markers. The present meta-analysis study showed both a consistent synaptic loss across brain regions and an impairment in molecular machinery of endosomal pathways, vesicular assembly mechanisms, glutamate receptors, and axonal transport.
2.3. Synaptic dysfunction in Alzheimer’s disease
As AD progresses, there is an increase in the production of Aβ. This overproduction can reduce the number and plasticity of synapses and resulting synapses may also be abnormal in shape or composition. Aβ affects synaptic vesicle trafficking. SNAP-25, synaptophysin, and synaptotagmin, presynaptic proteins decreased in density in AD patients due the presence of Aβ. Dynamin-1 is altered by Aβ such that it cannot pinch off synaptic vesicles for re-entry into synaptic vesicle pool. Phosphorylated tau also interacts with Drp1 and VDAC1 to impair axonal transport and mitochondrial damage (Rajmohan and Reddy, 2017).
The symptoms of AD grow with the degradation of the glutamatergic and cholinergic synapses. Quantitative morphometric study of temporal and frontal cortical biopsies early into the onset of AD (2-4 years) showed a 25-35% decrease in the synaptic density and a 15-35% decrease in synaptic connections per neuron (Selkoe, 2002). In AD there is particular synapse loss in the neocortex and hippocampus (Jeong, 2017). These synaptic losses increased in areas where there are more amyloid plaques which contain the toxic Aβ (Bakota and Brandt, 2016).
Overall, there is great shrinkage in brain volume due to synaptic damage and cell loss in AD progression. This shrinkage very progressive with disease process and correlate with synaptic and cognitive functions.
3. Amyloid Beta and Alzheimer’s Disease
APP is needed for synaptic activity, formation of synapse and dendritic spines. It has a role in memory and learning (Rajmohan and Reddy, 2017). APP is found in non-neural tissues (Sheng et al., 2012). Aβ is formed from APP found on chromosome 21 (Koffie et al., 2011). Aβ accumulates in the neocortical regions (Dorostkar et al., 2015). The presence of APP gene on chromosome 21 affects patients with Down Syndrome as they have a trisomy 21 that increases the likelihood of Aβ aggregation and early onset AD (Dorostkar et al., 2015).
α-secretase cleaves 90% or even more of the APP. It is the remaining 10% that is cleaved by β and γ secretases. β secretase cleaves APP at the n-terminal to make C99, a membrane bound fragment. The C99 is cleaved by γ secretase to make the c-terminus of the Aβ. However, γ secretase is not accurate at cutting the C99, thus different length fragments of Aβ are formed. PS1 and PS2 mutations lead to increase Aβ42 production (Oakley et al., 2006). Altered products include sAPPα, C83, p3, and APP intracellular domain (AICD). In fact, Aβ is a minor product of the metabolism of APP.
PS1 mutations affects γ secretase complex, thus reducing its activity and unbalancing the Aβ42/Aβ40 ratios by creating fewer Aβ40 (Figure 3). APP c-terminal fragments are also not cleaved by the gamma secretase due to reduced activity. The accumulation of c-terminal fragments causes synaptic failure and memory impairment. This APP c-terminal fragment accumulation and reduced APP metabolism is one of the many possible causes of AD (Kametani and Hasegawa, 2018).
Figure 3.

Under normal conditions, there is very little γ-secretase that cleaves the APP equally into Aβ42 and Aβ40. However, overexpression of Presenilins can cause dysregulation of gamma-secretase resulting in increased production of Aβ42. These Aβ42 fragments can oligomerize and further AD progression.
Aβ42 more commonly aggregates than Aβ40. If there is a cyclized glutamate residue at the N-terminus, there is even greater aggregation of the Aβ. Aβ can be found both extracellularly and intracellularly. Intracellular amyloid accumulation increases with age and is associated with spine loss in APP models (Almeida et al., 2005; Gouras et al., 2005). In other models, it affects synapse structure without affecting spine density. Extracellular amyloid can be taken into neurons by the Receptor for Advanced Glycosylation End products (RAGE). In 5xFAD mouse models, fibrillar amyloid beta can be found in cell bodies and can lead to plaques (Dorostkar et al., 2015).
Magrane and colleagues also investigated the role of intraneuronal Aβ and survival pathways in AD. Using an inducible viral vector system to drive intracellular expression of Aβ42 peptide in primary neuronal cultures, they demonstrated that intraneural Aβ accumulation results in the inhibition of the Akt survival signaling pathway (Magrane et al., 2005). Induction of intraneuronal Aβ42 expression leads to a sequential decrease in levels of phospho-Akt, increase in activation of glycogen synthase kinase-3β, and apoptosis. Downregulation of Akt also paralleled intracellular Aβ accumulation in vivo in the Tg2576 AD mouse model. Overexpression of constitutively active Akt reversed the toxic effects of Aβ through a mechanism involving the induction of heat shock proteins (Hsps). They used a small-interfering RNA approach to explore the possibility of a link between Akt activity and Hsp70 expression and concluded that neuroprotection by Akt could be mediated through downstream induction of Hsp70 expression. They concluded that the early dysfunction associated with intraneuronal Aβ accumulation in AD involve the associated impairments of Akt signaling and suppression of the stress response.
Aβ can also increase its own production through the dephosphorylation (activation) of GSK3β. This interference with normal APP processing leads to an accumulation of Aβ (Pozueta et al., 2013).Overall, these studies indicate that Aβ accumulation induce synaptic damage in AD neurons.
3.1. Amyloid Beta at the Synapse
Aβ can cause dendritic shrinkage and collapse via f-actin remodeling by Rho-GTPases. The Ras superfamily, within the Rho family, modulates cytoskeleton changes by switching between GDP bound and GTP bound forms. RhoA, Rac, and Cdc42 are Rho-GTPases that play a role in dendritic spine formation. RhoA and Rac regulate spine numbers such that activation of RhoA decreases spine density while activation of Rac1 increases spine density. In AD, RhoA may potentially be activated while Rac1 is deactivated. In normal brains, Rac1 binds to serine threonine kinase (PAK) and the resulting autophosphorylation prevents actin-myosin binding thus allowing for actin polymerization. In AD, this process, (PAK), may be defective as a result of Rac1 deactivation (Pozueta et al., 2013).
Extracellular Aβ accumulates around postsynaptic spines and presynaptic spines. This occurs more abundantly at postsynaptic than at presynaptic terminals (Forner et al., 2017). Aβ binds to AMPA receptors and causes the internalization of AMPA receptors thus leading to increased LTD. Binding of Aβ to AMPAR receptors results in ubiquitination and AMPARs are reduced by the ubiquitin ligase Nedd4-1 (Forner et al., 2017). The subsequent impairment of glutamatergic transmission leads to loss of dendritic spines (Sheng et al., 2012).
Aβ can bind to NMDA receptors and cause calcium dyshomeostasis that leads to oxidative stress, production of free radicals, and neuron loss (Pozueta et al., 2013). Aβ can also activate mGluR5 receptors which also leads to increased postsynaptic calcium levels within the cell (Sheng et al., 2012). NMDA receptors and mGluR then increase APP processing which, in a positive feedback loop, leads to increased calcium influx and free radical production (Rajmohan and Reddy, 2017). The many interactions of Aβ are seen in Figure 4.
Figure 4.

1) Aβ enters the mitochondria through Cyclophilin D and binds to alcohol dehydrogenase. 2) Increased Amyloid Beta leads to increased Calcium influx. 3) The Calcium dysregulation is amplified by mitochondrial dysfunction and fission. TCA cycle enzymes, such as α-ketoglutarate dehydrogenase is dysregulated. 4) Further dysfunction leads to increased apoptosis via the caspase cascade and increased ROS production
Researchers have also noted that there is no statistically significant difference in oligomeric Aβ levels between early AD and late AD. However a strong uptick in the amount of oligomeric amyloid beta in the early stages of AD may correlate with dementia associated with AD. Researchers hypothesize that there may be an oligomeric amyloid beta threshold, that when crossed, can lead to increased risk for AD related dementia (Bilousova et al., 2016).
4. Tau and Alzheimer’s Disease
Tau is a hydrophobic protein that focuses on stabilizing neuronal microtubules and regulating axonal transport (Rajmohan and Reddy, 2017). It is a microtubule associated protein that has 16 exons and is encoded on the chromosome 17q21. There are 6 tau isoforms as a result of variable splicing of three exons (Bakota and Brandt, 2016). Tau aggregates in the brainstem and transentorhinal region (Dorostkar et al., 2015). It can be found in dendrites and in postsynaptic terminals of neurons (Sheng et al., 2012).
There are six stages of tau pathology and NFT progression throughout the brain. The first two stages do not cause cognitive impairment. In AD, there is a decrease in the density and shape of dendritic spines, especially in the CA1 region. There is also a loss of neurons, most notably in the CA3 region (Bakota and Brandt, 2016).
Phosphorylation of tau changes over time. Cyclin Dependent Kinase 5 (cdk5) and Glycogen Synthase Kinase 3β (GSK3β) generate diseased tau and regulate Aβ production. Neuregulin-1 (NRG-1) is a member of a family of growth factors that correlates with cdk5 activity. In normal adults, NRG-1 activity and levels are high during the first neonatal week but then is reduced significantly during adult hood. In mice, inhibition of cdk5 activity reduces Aβ but increases tau as GSK3β are active when mice are young. When mice are old, tau levels are not affected (Wen et al., 2008).
Tau, in AD, undergoes translational changes that lead to the characteristic formation of neurofibrillary tangles (NFTs). NFTs are self-assembled paired helical filaments that form inside cell bodies. The tau pathology starts in the transentorhinal region and spreads to limbic and neocortical areas (Kametani and Hasegawa, 2018). As AD progresses, NFTs form in deeper layers of the entorhinal cortex, CA1 of the hippocampus, amygdala and basal magnocellular complex (Krstic et al., 2013). Tau modifications that lead to NFTs may include hyperphosphorylation, truncation or even acetylation. The modifications allow tau to detach from microtubules to affect synaptic function elsewhere within the neuron (Dorostkar et al., 2015).
Hyperphosphorylated tau leads to neurofibrillary tangles and reductions in spine head volumes. Truncation of tau can lead to its toxicity and increased hyperphosphorylation (Dorostkar et al., 2015). Tau is phosphorylated at Serine and Threonine residues. It can also undergo O-glycosylation, ubiquitination, methylation, and acetylation. Truncated tau cannot aggregate into filaments and has irregular cellular localization (Bakota and Brandt, 2016). The different actions of tau are summarized in Figure 5.
Figure 5.

Binding of Aβ to other molecules and structures elicits a variety of detrimental effects on cellular function and disease progression.
4.1. Tau action at the Synapse
When tau is modified, there is a disruption in microtubule-based cellular transport. Synapse loss can occur if mitochondria and receptors are not transported effectively to synapses. This causes diminished mitochondria dependent ATP production and calcium buffering, as well as impaired trafficking of glutamate receptor subunits to the post synaptic membrane. Fewer mitochondria results in an impairment of synaptic vesicle release (Forner et al., 2017).
Acetylated tau proteins reduce Kidney/Brain (KIBRA) protein levels which reduces activity induced postsynaptic actin modification and AMPAR insertion. This impairment affects memory and is associated with AD and Dementia (Forner et al., 2017). Tau is a substrate for Caspase. When it is cleaved by the Caspase, it forms tau 314 and reversibly impairs memory function.
Tau can also be released in the extracellular space where it affects muscarinic acetylcholine receptor (mAChRs). It is also involved in long term plasticity by acting as a substrate for Glycogen Synthase Kinase 3β (GSK-3β) and p38 Mitogen-Activated Protein Kinase (p38MAPK). These two enzymes regulate synaptic function (Forner et al., 2017).
Tau interacts with PSD-95/NMDA receptor complex (NMDAR). A positive response mechanism between tyrosine kinase FYN and NMDAR increases activation of NMDAR during glutamate neurotransmission. This activity has excitotoxic effects on neurons (Forner et al., 2017).
Clinical symptoms of tau aggregation follow that of amyloid plaques. However, it is a strong indicator of neurodegeneration and cognitive decline in AD (Sheng et al., 2012). The prevalence of NFTs correlates with AD cognitive deficits. The accumulation of full-length tau affects cognition as well. Full length tau (hTau) activates Calcineurin, a calcium dependent protein phosphatase, which dephosphorylates or inactivates cAMP response to CREB signaling leading to synaptic impairments (Forner et al., 2017). Calcineurin can induce spine loss (Dorostkar et al., 2015).
4.2. Mislocalization of tau at synapses and synaptic toxicity
Researchers have noted that tau localizes greatly in regions where axons were not myelinated. In layers 4 & 5 of the cerebral cortex, there is strong tau immunoreactivity. Mossy fiber axons of the hippocampus and the inner molecular layer of the dentate gyrus were strongly labeled. Many other areas such as the olfactory bulb, cerebellum, ventral tegmental area, showed strong immunoreactivity for tau. Furthermore, tau can be found in oligodendrocytes but not in microglia and astrocytes (Kubo et al., 2019)
Researchers have also noted that the pre-synaptic release of tau can lead to dysregulation in Alzheimer’s disease. During neuronal death, tau is released. However, researchers have also noted that tau can be released and cross synapses without neuronal death. To further assess the release of tau, researchers employed KCl and glutamate treatment. Such treatment markedly increased tau release without affecting the viability of neurons. This release of tau was calcium dependent. Tetanus toxin experiments also demonstrated that the tau secretion was partially released via pre-synaptic vesicle secretions (Pooler et al., 2013).
Tau may also accumulate in post-synaptic sites. Some studies have shown that phosphorylated tau can be found in the dendritic spines of neurons and affect synaptic transportation and glutamate receptor insertion. These post-synaptic areas may be the target of tau (Yu and Lu, 2012). The increased concentration of tau in the dendrites may disrupt kinesin and dynein. Furthermore, the increased levels of tau in the dendrites affected memory and synaptic plasticity. Mislocalized tau reduced the miniature excitatory postsynaptic currents in rats. When there is a decrease in mEPSCs, there is a silencing of synapses and reduction of AMPARs in the post synaptic site (Hoover et al., 2010).
In APPswe mice that had a mutation at APP670/671 for early-onset familial AD, researchers found that tau was mislocalized to the dendritic spines. Researchers confirmed that soluble Aβ oligomers play a role in the mislocalization of tau at the dendrites. They noted that treatment with Aβ oligomers increased tau expression in the dendrites of wild type mice. By mutating phosphorylation sites on tau, researchers also showed that oligomerized Aβ can only increase mislocalization of tau to the dendrites when tau is phosphorylated (Miller et al., 2014). Tau phosphorylation plays a role in the mislocalization of tau by allowing tau to release off of microtubules and congregate at dendritic shafts and spines (Hoover et al., 2010).
Overall, researchers agree that the presence of P-tau in dendritic spines are early indicators of AD.
4.3. Tau and Amyloid Beta Synergism in Alzheimer’s disease
Accumulation of Aβ in synapses occurs before that of phosphorylated tau. Researchers attribute accumulation of Aβ as a marker for early stage disease and phosphorylated tau as a marker for late stage disease (Bilousova et al., 2016). Researchers have used synaptosomes of the parietal cortex in human subjects and transgenic rat models to understand the sequence of events in AD progression. Synaptosomes are mostly presynaptic structures that contain mitochondria, endosomes, and other structures. Their research noted that phosphorylated tau increased with amyloid beta fraction. As a result, they hypothesize that Aβ may be involved with tau phosphorylation (Bilousova et al., 2016).
Using flow cytometry, researchers have also noted an increased phosphorylated tau at the synaptosomes in AD samples. Furthermore, they noted that, when using the 10G4 antibody to label Aβ, 50% of the synaptosomes in the parietal cortex that contained elevated Aβ, also had elevated phosphorylated Tau. Researchers also noted, in samples from an 80 year old female with AD, that the elevated tau at the terminals was aggregated tau rather than monomer (Henkins et al., 2012).
Oligomeric Aβ stimulates NMDA receptors and results in an upregulation of calcium and redox reactions. This dysfunction leads to downstream effects such as synaptic dysfunction and neuronal loss. Calcineurin, activated by the elevated calcium as a result of Aβ, dephosphorylates target proteins such as actin filaments to cause spine loss at the dendrites. The increased NMDA activity can also lead to increased tau accumulation. Increased phosphorylated tau correlates with, and further amplifies, the dendritic spine loss (Tu et al., 2015).
The timeline of events suggests that Aβ functions upstream of tau. Soluble Aβ activates calcium-dependent phosphatase Calcineurin (PP2B) and through NMDA receptors elevate calcium levels to activate the metabotropic glutamate receptor 5 or the L-type voltage-gated calcium channels (Bakota and Brandt, 2016). However, tau is necessary for Aβ toxicity. In other words, while Aβ initiates synaptic dysfunction brought on by tau pathology, if tau is not present, there will not be resulting memory impairment. Furthermore, there is also protection against synaptic loss, neuron loss, and premature death. There may be a feedback loop between Aβ and tau such that Aβ initiates the accumulation of tau, which in turn affects accumulation of Aβ. FYN mediates the effects of tau by Aβ. FYN regulates NMDA receptor activity. It is brought to the postsynaptic site via interaction with tau. When the localization of FYN to the postsynaptic site by tau was impaired, memory deficits were rescued. Usually there is a distribution of tau amongst the neuron, however, Aβ causes an increased concentration of tau in the post-synaptic sites of the dendrites. This increased concentration attracts FYN, increases NMDA receptor activity, and increases calcium influx that triggers damage due to calcium dyshomeostasis (Bloom, 2014; Yu and Lu, 2012).
Furthermore, Aβ oligomers can lead to tau pathologies such as cytoskeletal impairments, mitochondrial transport defects and neuron death. In addition to FYN, Aβ oligomers activate PKA, and CaMKII to phosphorylate tau and start the cascade of tau pathology. Aβ oligomers are unaffected by the presence of tau and can cause damage such as microtubule disassembly and GSK3β activation that delivers brain-derived neurotrophic factors. Aβ oligomers and tau have prion-like properties that modify each other into toxic forms. Aβ oligomers control post-translational modifications of tau that result in prion-like properties (Bloom, 2014).
Phosphorylation of tau may be mediated by APP (Bakota and Brandt, 2016). Aβ, by altering the levels of a tau ubiquitin ligase (CHIP) that degrades cleaved and hyperphosphorylated tau, increases the concentration of tau molecules. Aβ increases calcium levels which has downstream effects of destabilizing tau from microtubules. The effects continue in impaired transport of mitochondria. Furthermore, the intracellular Aβ can cause AMPK to phosphorylate tau at Serine 262 (Forner et al., 2017). This synergism is summarized in Figure 6.
Figure 6.

Phosphorylation of tau affects many synaptic receptors, proteins, and structures.
5. Defective LTP and LTD in Alzheimer’s disease
Long term potentiation (LTP) defects show up before development of Aβ deposits (especially in the APP mouse line). If there is low APP gene expression but a high Aβ production, the synaptic transmission decay was more evident at early ages of the mice (2-4 months) (Selkoe, 2002). Furthermore, synthetic injection of Aβ into animal models affected working memory and caused memory deficits.
Learning and memory relies on increased synaptic transmission at excitatory synapses. LTP, the underlying synaptic activity that drives memory and learning is calcium and NMDA dependent. Spines must constantly remodel to not only maintain plasticity, but also to encourage memory formation. This pathway is impaired in AD, thus resulting in synaptic dysfunction (Pozueta et al., 2013). Results of LTP are larger dendritic spines and increased synaptic density. Long term depression (LTD) reduces the size of dendritic spines and reduces synaptic density due to reduced synaptic activity (Koffie et al., 2011). LTD and LTP require calcium flow through N-methyl-D-aspartate (NMDA) receptors and metabotropic glutamate receptors (mGluRs). LTP requires a high flow of calcium through the NMDA receptor, while LTD presents itself when there is a low calcium influx through the NMDA receptors. Each of these processes, LTD and LTP, require different subclasses of the NMDA receptors, NR2B and NR2A respectively (Koffie et al., 2011).
Oligomeric Aβ can impair LTP while increasing LTD in a concentration dependent manner. Low amounts of oligomeric Aβ facilitates LTP, but higher concentrations impair it (Pozueta et al., 2013). High levels of Aβ impairs LTP even before plaques form. It does so by effecting calcium channel activity and glutamate receptor dependent signaling pathways (Rajmohan and Reddy, 2017). However, there are a lot of conflicting reports on this particular pathway because in some reports LTP is unaltered (Pozueta et al., 2013).
Aβ unbalances calcium homeostasis, activates Caspases, activates Calcineurin, and modulates excitatory receptors and receptor tyrosine kinases. Through a combination of these molecular mechanisms, Aβ causes synaptic death, LTD and LTP. Calcineurin and Caspase play important intracellular roles in LTD. Aβ binds to 7α-nicotinic acetylcholine receptors, which leads to an internalization of NMDA receptors and LTD as there is reduced calcium influx (Koffie et al., 2011). Aβ also activates mGLuRs which, through downstream effects, internalize AMPA receptors and causes synapse collapse. Calcineurin activation, another effect of Aβ oligomers, also reduces NMDA receptor expression on the surface and leads to greater AMPA receptor internalization. By blocking the activation of calcineurin, these effects can be reversed. Calcineurin activation can activate the NFAT (nuclear factor of activated T-Cells) pathways and the STEP (striatal enriched tyrosine phosphatase) pathways to cause spine loss and initiate LTD through the dephosphorylation of NR2B subunits of NMDA receptors (Koffie et al., 2011). Oligomeric Aβ also bind with tyrosine kinases to modulate NMDA receptor trafficking and reduce LTP. Aβ may also reduce the uptake of glutamate which leads to desensitizes post-synaptic NMDA receptors. Caspase-3 is activated by soluble Aβ, which in turn dephosphorylates AMPA receptors and increases receptor internalization. Unfortunately, chronic inhibition of Caspase-3 resulted in an acceleration of cognitive impairment. Aβ also reduces CREB activation, thus reducing the gene expression of proteins needed for LTP. Difficulty with just removing Aβ altogether is that Aβ, at low levels, does benefit LTP. It is the difficulty of identifying which roles of Aβ are toxic and inhibiting them that has been the continuing challenge in curing AD (Koffie et al., 2011).
In tau mice, only older age brings on difference in LTP. Tau is involved with LTD in the CA1 of the hippocampus by increasing the interaction between GluA2 subunits of AMPARs and PICK1 in order to initiate AMPAR endocytosis, thus hippocampal LTD (Forner et al., 2017). Experiments using tau-null mice show no impairment in LTP when Aβ is applied. Therefore, Aβ may need tau to impair LTP (Pozueta et al., 2013). A second pathway of calcineurin is needed to cause LTD. At the same time, both LTD and AMPA receptor internalization needs Caspase3 activity. AMPA internalization leads to enhanced LTD and reduced LTP (Sheng et al., 2012).
Overall, age-dependent accumulations of Aβ and tau and their interactions at synapse largely impair synaptic activity via altering LTP and LTD levels. Reduced levels of synaptic Aβ and tau are expected to maintain synaptic functions in AD neurons.
6. Mitochondria
A 1987 study focused on analyzing oxygen consumption in the brains of dementia subjects noted that oxygen consumption was elevated in dementia patients over control patients. But, in AD brains there is a decrease in oxygen consumption. Further research showed that synaptic changes, such as altered or fewer mitochondria, can answer this conundrum and may play a significant role in AD progression (Swerdlow, 2018).
6.1. Synaptic Mitochondria and Free Radical production
Mitochondria are primarily synthesized in the cell body of neurons and are then transported to axons or dendrites to supply energy to several synaptic functions, including synaptic transmission, synaptic outgrowth and synaptic vesicle formation (Han et al., 2020; Reddy and Beal, 2008). If mitochondria localized in the cell body are damaged by toxic insults, such as aging, Aβ, or phosphorylated tau, and then transported to synaptic terminals, they may produce low levels of ATP to synapses that can lead to synaptic degeneration.
Synaptic terminals are sites of high energy demand (Reddy and Beal, 2008). Synaptic transmission requires high levels of cellular ATP for neurotransmitter exocytosis and the potentiation of neurotransmitter release. In addition, synaptic terminals require mitochondria for sequestering and releasing Ca2+ for post-tetanic potentiation (Calkins and Reddy, 2011). Therefore, increased transport of healthy mitochondria to synaptic terminals is necessary.
Aβ accumulates at synaptic terminals and impairs synaptic function. Aβ also enters synaptic mitochondria and causes damage. Mitochondrial damage is expected to be greater in synaptic mitochondria than in cell-body mitochondria (Pradeepkiran and Reddy, 2020). The damaged, synaptic mitochondria might not supply high energy for synapses, which might lead to impaired neurotransmission and, ultimately, to cognitive failure.
In the presence of Aβ, there is dysregulation of the mitochondria, particularly at synapses. Several recent studies revealed that Aβ binds to mitochondrial proteins, such as mitochondrial fission protein Drp1 (Manczak et al., 2011), mitochondrial outer-membrane protein VDAC (Manczak and Reddy, 2012), mitochondrial matrix proteins Aβ-binding alcohol dehydrogenase and CypD (Du et al., 2008), these abnormal interactions, induce excessive production of free radicals, enhance fragmentation of mitochondria and affect mitochondrial biogenesis, ultimately leading mitochondrial function. Furthermore, Aβ increases the ability of calcium to enter the cell. This then causes the mitochondria, one of the cell’s calcium regulators, to take in the calcium as well (Swerdlow, 2018). The cascade is depicted in Figure 7.
Figure 7.
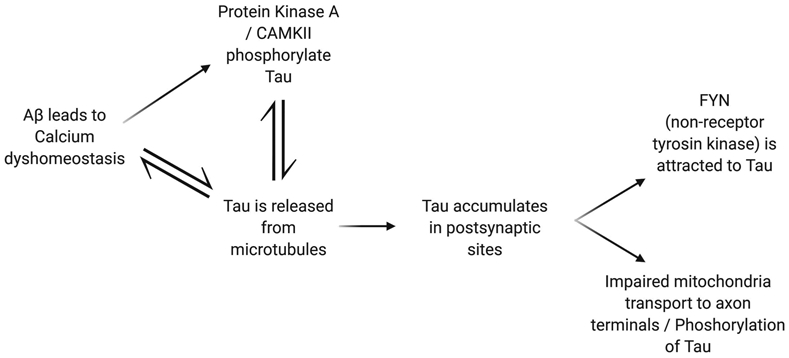
Positive feedback mechanism of tau increases aggregation of Aβ. When the localization of FYN to the postsynaptic site by tau was impaired, memory deficits were rescued.
The mitochondria play a vital role in cell health especially by its tight regulation of calcium. In the presynaptic terminal, calcium concentrations result in vesicular release of neurotransmitter. Mitochondria remove calcium to stop signaling.
Under normal conditions, the calcium influx should equal the calcium efflux. There is low intracellular free calcium and the balance of the cytoplasmic calcium levels is crucial. This homeostasis is maintained by calcium buffers, calcium sensors, and calcium transporters. The mitochondria use an energy dependent influx of calcium. However, excess calcium in the mitochondrial matrix can damage the mitochondria (Nedergaard and Verkhratsky, 2010).
Apoptosis is also mediated by mitochondria. If the cell is in distress or if there is DNA damage, the mitochondria will release cytochrome-c to start apoptosis. In normal cells, mitochondrial distress is prevented from causing cell death. However, in AD, mitochondrial distress may not be stopped from causing cell death (Bonda et al., 2011).
Oxidative phosphorylation, the method by which a proton gradient is created for use in ATP generation, occurs in the mitochondria. The three key calcium activated metabolic enzymes of oxidative phosphorylation include pyruvate, α-ketoglutarate, and isocitrate dehydrogenases (Bonda et al., 2011). Pyruvate dehydrogenase uses calcium dependent dephosphorylation while the other two metabolic enzymes are activated by calcium binding (Bonda et al., 2011).
Mitochondria are dysfunctional when high levels of Aβ interacts with VDAC1 and stops the transport of mitochondrial proteins. This dysfunction creates more free radicals (Rajmohan and Reddy, 2017). The imbalance between the production and clearance of free radicals creates oxidative stress which is marked by the production of 8-hydroxyguanosine. Not only is the stress great on the body due to free radicals, but it damages the mitochondria itself. Mitochondrial DNA is close to the location of reactive oxidative species generation, thus is easily damaged. It is easily damaged because mitochondrial DNA does not have histones nor a repairing mechanism. This form of oxidative stress is characteristic of AD at the synapse (Moneim, 2015).
6.2. Mitochondrial fragmentation in Alzheimer’s disease
In normal cells, mitochondria divide and fuse to reflect metabolic demands. They can organize into tubular networks or divide into rod-like structures to enter narrow areas of the neuron (Wang et al., 2008; Wang et al., 2009; Silva et al., 2013; Kandimalla and Reddy, 2016; Oliver and Reddy, 2019a; Oliver and Reddy 2019b; Reddy and Reddy, 2011, Cai and Thammineni, 2016; Cai and Thammineni, 2017; Cai and Jeang, 2020). Fission and fusion are highly dependent on conditions within the cell such as free radical production, oxidative stress, or even ion homeostasis. Fusion, governed by three GTPases, mitofusin 1, mitofusin 2, and optic atrophy protein 1, allows one mitochondrion to exchange its contents with another. Fission, operated by dynamin-related protein (Drp-1) and Fis1, causes the mitochondria to split. Drp-1 and Fis1 form a ring like structure that constricts the mitochondria until it splits into two (Reddy, 2009; Reddy et al 2011; Bonda et al., 2011).
Using co-immunoprecipitation and colocalization studies, Reddy Lab (Manczak et al, 2011) investigated the molecular links between mitochondrial fission protein, Drp1 and Aβ in in postmortem AD brains, cell and mouse models of APP in disease progression of AD. They found Aβ interacts with Drp1 which causes increased mitochondrial fragmentation and reduced fusion (Manczak et al., 2011), particularly at synapses.
In another study, Reddy Lab (Manczak and Reddy, 2012) also studied phosphorylated tau (p-Tau) interaction with Drp1 in postmortem AD brains and mouse models of tau in disease progression of AD. They found is also reported to interact with Drp1 and increases Drp1 GTPase activity and enhances mitochondrial fragmentation and defective mitochondrial function (Manczak and Reddy 2012; Tu et al., 2015).
Altogether, too much fission leads to increased free radical production (Rajmohan and Reddy, 2017), mitochondrial dysfunction and Defective mitophagy in AD (Reddy and Oliver 2019; Pradeepkiran and Reddy 2020).
6.3. Defective mitophagy in Alzheimer’s disease
Mitophagy is the clearance of damaged mitochondria from the cells. Phosphatase-tensin homologue-induced kinase-1 (PINK-1) and Parkin, are involved in mitophagy. Phosphorylation of Drp-1 at Ser637, mediated by cyclic AMP-dependent kinase (PKA) leads to its inhibition and his leads to defective mitophagy and increased neurodegeneration (Bonda et al., 2011, Kandimalla nd Reddy 2016, Oliver and Reddy 2019a, Oliver and Reddy 2019b).
At synapses from neurons from AD patients, mitochondria have broken cristae and altered size and number when compared to mitochondria in similarly aged control subjects (Reddy and Beal, 2008). Synaptic mitochondrial impairments can be mainly upstream targets of AD. As an upstream target, the mitochondria is affected by Aβ (Bonda et al., 2011). If the mitochondria are too fragmented, they may enter a Caspase cascade. Increased mitochondrial fragmentation reduces mitophagy and leading to mitochondrial dysfunction. Mitochondrial dysfunction releases cytochrome c and activates Caspase 9. Caspase 9 activates Caspase 3 that leads to an apoptotic cascade (Kamat et al., 2016).
For the first time, Ye and colleagues (2015) studied Parkin-mediated mitophagy in mutant hAPP neurons and AD patient brains. They found Parkin-mediated mitophagy is robustly induced in mutant hAPP neurons and AD patient brains. In the absence of Δψm dissipation reagents, hAPP neurons exhibit increased recruitment of cytosolic Parkin to depolarized mitochondria. Under AD-linked pathophysiological conditions, Parkin translocation predominantly occurs in the somatodendritic regions, leading to educed anterograde and increased retrograde mitochondrial axonal transport. Enhanced mitophagy was further confirmed in AD brains, accompanied with depletion of cytosolic Parkin over disease progression. Thus, aberrant accumulation of dysfunctional mitochondria in AD-affected neurons is likely attributable to inadequate mitophagy capacity and unable to clear damaged mitochondria. Altogether, their study provides the first line of evidence that AD-linked chronic mitochondrial stress under in vitro and in vivo pathophysiological conditions effectively triggers Parkin-dependent mitophagy, thus establishing a foundation for further investigations in AD.
Reddy and colleagues (2018) investigated the toxic effects of hippocampal mutant APP (mAPP) and amyloid beta (Aβ) in primary mouse hippocampal neurons (HT22) that express human APP Swedish mutation. Using quantitative reverse transcriptase-polymerase chain reaction, immunoblotting & immunofluorescence and transmission electron microscopy, they assessed mRNA and protein levels of synaptic, autophagy, mitophagy, mitochondrial dynamics, biogenesis, dendritic protein MAP2 and assessed mitochondrial number and length in mAPP-HT22 cells. Mitochondrial function was assessed by measuring the levels of hydrogen peroxide, lipid peroxidation, cytochrome c oxidase activity and mitochondrial adenosine triphosphate. Increased levels of mRNA and protein levels of mitochondrial fission genes (Drp1 and Fis1) and decreased levels fusion (Mfn1, Mfn2 and Opa1) biogenesis (PGC1α, NRF1, NRF2 & TFAM), autophagy (ATG5 & LC3BI, LC3BII), mitophagy (PINK1 & TERT, BCL2 & BNIPBL), synaptic (synaptophysin & PSD95) and dendritic (MAP2) genes were found in mAPP-HT22 cells relative to WT-HT22 cells. Cell survival was significantly reduced mAPP-HT22 cells. GTPase-Drp1 enzymatic activity was increased in mAPP-HT22 cells. Transmission electron microscopy revealed significantly increased mitochondrial numbers and reduced mitochondrial length in mAPP-HT22 cells. These findings suggest that hippocampal accumulation of mAPP and Aβ is responsible for abnormal mitochondrial dynamics and defective biogenesis, reduced MAP2, autophagy, mitophagy and synaptic proteins & reduced dendritic spines and mitochondrial structural and functional changes in mAPP hippocampal cells. These observations strongly suggest that accumulation of mAPP and Aβ causes mitochondrial, synaptic and autophagy/mitophagy abnormalities in hippocampal neurons, leading to neuronal dysfunction.
Reddy Lab (Manczak et al., 2018), also investigated the toxic effects of hippocampal mutant APP and Aβ in 12-month-old APP transgenic mice (Tg2576 strain). Using rotarod and Morris water maze tests, immunoblotting and immunofluorescence, Golgi-cox staining and transmission electron microscopy, they assessed cognitive behavior, protein levels of synaptic, autophagy, mitophagy, mitochondrial dynamics, biogenesis, dendritic protein MAP2 and also quantified dendritic spines and mitochondrial number and length in APP mice that express Swedish mutation. Mitochondrial function was assessed by measuring the levels of hydrogen peroxide, lipid peroxidation, cytochrome c oxidase activity and mitochondrial ATP. Morris water maze and rotarod tests revealed that hippocampal learning and memory and motor learning and coordination were impaired in APP mice relative to wild-type mice. Increased levels of mitochondrial fission proteins and decreased levels of fusion, biogenesis, autophagy, mitophagy, synaptic and dendritic proteins were found in 12-month-old APP mice relative to age-matched non-transgenic WT mice. Golgi-cox staining analysis revealed that dendritic spines are significantly reduced. Transmission electron microscopy revealed significantly increased mitochondrial numbers and reduced mitochondrial length in APP mice. These findings suggest that hippocampal accumulation of mutant APP and Aβ is responsible for abnormal mitochondrial dynamics and defective biogenesis, reduced MAP2, autophagy, mitophagy and synaptic proteins and reduced dendritic spines and hippocampal-based learning and memory impairments in 12-month-old APP mice.
Fang and colleagues (2019) also studied mitophagy in the progression of AD in pluripotent stem cell-derived human AD neurons, in animal AD models and Aβ and tau Caenorhabditis elegans models of AD. They also found mitophagy is impaired in the hippocampus of AD patients, in induced pluripotent stem cell-derived human AD neurons, and in animal AD models. In both Aβ and tau Caenorhabditis elegans models of AD, mitophagy enhancers reverses memory impairment through PINK-1-, Parkinson's disease-related-1; parkin-, or DAF-16/FOXO-controlled germline-tumor affecting-1-dependent pathways. Mitophagy diminishes insoluble Aβ1-42 and Aβ1-40 and prevents cognitive impairment in an APP/PS1 mouse model through microglial phagocytosis of Aβ and suppression of neuroinflammation. Mitophagy enhancement abolishes AD-related tau hyperphosphorylation in human neuronal cells and reverses memory impairment in transgenic tau nematodes and mice. Their findings further support findings of previous studies of Ye et al., 2015, Reddy et al., 2018 and Manczak et al., 2018 that defective mitophagy is a major cellular change in AD progression and pathogenesis.
Overall, these studies proposed that enhancing mitophagy represents a potential therapeutic intervention.
Overall, mitophagy is defective in AD, primarily due to 1) increased production and accumulation of Aβ and phosphorylated tau and 2) abnormal interactions of Aβ and phosphorylated tau with Drp1 and VDAC and reduced PINK1 and Parkin1levels. Therefore, reduced levels of Aβ and phosphorylated tau and Drp1 have been suggested as an effective therapeutic strategy for AD.
7. Microglial activation at the Synapse by Aβ
Microglia are immune cells that react to potential threats and control the neuro-inflammatory response by secreting neurotransmitters, cytokines, and extracellular matrix proteins. Older brains show increased cytokine levels and microglia activation. Microglia can both interact with and exert effects on, neurons. Such effects include synapse formation and pruning. There are two types of activated microglia, M1 (proinflammatory) and M2 (immunosuppressive) (Xie et al., 2017).
There is a non-specific aggregation of microglia in an age-dependent manner. Tumor necrosis factor-α is produced by activated microglia. This leads to AMPA receptor activity, which leads to excitotoxicity and spine loss (Dorostkar et al., 2015). In normal synaptic pruning during development, microglia destroy synapses as a response to CX3CR1. C1q and C3 localize to synapses and trigger microglial elimination of synapses.
In AD progression, ‘dark microglia’, a new myeloid-cell phenotype, destroy dendritic spines, axons, and synapses. These microglia are found abundantly in AD brains but not in normal brains. Soluble Aβ activates microglial elimination of synapses via Cr2 protein localization at synapses. When M2 microglia interact with Aβ, these microglia convert from their immunosuppressive form to M1 (proinflammatory). The conversion rate relates to the relative concentration of soluble Aβ. Aβ activates NLRP3, an inflammasome in microglia. This activation secretes proinflammatory cytokines that further exacerbate the inflammatory response. Inhibition of NLRP3 reduces the deficits caused by Aβ deposition. Activation of NLRP by Aβ is localized around the amyloid plaques (Xie et al., 2017).
It is known that brain microglia are involved in removing dying neurons, pruning nonfunctional synapses, and producing ligands that support neuronal survival (Werneburg et al., 2017). Recently, Badimon and colleagues (2020), investigated the function of brain microglia in healthy and disease states. They demonstrated that microglia are also critical modulators of neuronal activity and associated behavioral responses in mice. Microglia respond to neuronal activation by suppressing neuronal activity, and ablation of microglia amplifies and synchronizes the activity of neurons, leading to seizures. Suppression of neuronal activation by microglia occurs in a highly region-specific fashion and depends on the ability of microglia to sense and catabolize extracellular ATP, which is released upon neuronal activation by neurons and astrocytes. ATP triggers the recruitment of microglial protrusions and is converted by the microglial ATP/ADP hydrolysing ectoenzyme CD39 into AMP; AMP is then converted into adenosine by CD73, which is expressed on microglia as well as other brain cells. Microglial sensing of ATP, the ensuing microglia-dependent production of adenosine, and the adenosine-mediated suppression of neuronal responses via the adenosine receptor A1R are essential for the regulation of neuronal activity and animal behavior. Their findings suggest that microglia-driven negative feedback mechanism operates similarly to inhibitory neurons and is essential for protecting the brain from excessive activation in health and disease.
Overall, microglia play a large role in disease progression, particularly at synapses; however, very little research is done synaptic microglia in relation to mitochondria and energy metabolism. Further research is needed to understand the biology, pathophysiology and therapeutic interventions on synaptic microglia, glia-neuron cross talk in AD.
8. Synapse based therapy in Alzheimer’s disease
Synapse directed therapies for AD may hold a great promise in fighting the disease. However, the contradictory and convoluted disease progression makes creating such solutions very difficult. For example, synaptic activity positively correlates with the creation of Aβ. This shows that the target of Aβ damage is also the regulator of Aβ homeostasis. Researchers explored the effect of chronic inhibition of synaptic activity to see if this would improve or reduce progression of AD. Using mouse models, researchers used whisker ablation to simulate chronic reduction of synaptic activity and noted that there were fewer plaques in the barrel cortex of the mice (Tampellini, 2015). However, the reduced activity resulted in cognitive impairments and worsened memory. While plaque formation may have decreased, Aβ levels within the neuron increased when there was a reduction in activation while synaptophysin levels were lowered. Researchers have tried to flip the experiment by using optogenetics to unilaterally stimulate the performant pathway. While this approach increased plaque burden, there was reduced level of intraneuronal Aβ and had beneficial effects on the neurons. Therefore, the relationship between activity and intraneuronal Aβ levels may be explained by movement of Aβ outside of the cell. As a result, many researchers recommend to maintain intellectually stimulating activities in an effort to slow down the Aβ buildup in the brain (Tampellini, 2015).
To combat tau pathology at the synapse, researchers have suggested using antisense oligonucleotides that reduce tau expression. This would in turn reduce FYN in dendrites. The difficulty with this approach is that the delivery method that will have to pass the blood-brain barrier. Such therapeutic solutions are important to be discovered because Aβ-dependent fibril assembly and neuronal damage rely greatly on tau (Bloom, 2014).
Reelin can suppresses tau phosphorylation. Reduction of reelin leads to increased hyperphosphorylated tau production (Krstic et al., 2013). Reelin inhibits GSK3β by inducing phosphorylation of the adaptor protein disabled-1 and activating the cytosolic kinase pathways that phosphorylate NMDAR subunit NR2 and PI3K by clustering the ApoE receptor 2 and the very low-density lipoprotein receptor (VLDLR). Reelin also affects Aβ production. At low levels of Reelin, Aβ peptide production increases and NFT production increases. Therapies targeted at increasing Reelin or preventing ApoE4 from inhibiting its action may show promise. Reelin also has a role in microtubule assembly by promoting Cdc42-controlled transport of vesicles to increase growth cone motility, filopodia formation, and axonal branching. Increased reelin may increase repair of aging neurons in AD and increase axonal sprouting (Krstic et al., 2013).
The cellular components at the synapse may also show promise at possible therapies. GABAAR agonists may protect neurons form the neurotoxicity of soluble Aβ by activating NMDARs during high frequency synaptic transmission (Rajmohan and Reddy, 2017). Phosphorylation of GSK3β (inactivation) may reduce accumulation of Aβ (Pozueta et al., 2013).
While there may be many synaptic targets for possible new Alzheimer’s therapies, the disease is still an enigma and its own dissonance makes such targeted approaches very difficult. For example, when Aβ was taken from humans and placed in mice, there was no impairment. Reduction of Aβ did not rescue symptoms nor did it affect accumulation of tau in some patients. There are many normal patients with a lot of Aβ and there are AD patients with little Aβ. Despite these findings, the search for a therapy continues (Kametani and Hasegawa, 2018).
9. Conclusions and Future Directions
Tremendous progress has been made in the last several decades in understanding the biology of aging and AD. A large number of studies into Aβ, phosphorylated tau, inflammatory responses, mitochondrial and synaptic activities in disease progression and pathogenesis, have helped researchers get a better understanding of AD. Based on studies of postmortem AD brains, AD mouse models, cell cultures and blood-based markers, therapeutic strategies have been developed and tested in preclinical and clinical trials. The successes of past and current clinical trials are limited. This paper focused on addressing synaptic changes integral to disease progression. A combination of synapse loss and mitochondrial defects, defective mitophagy driven by Aβ, tau and mitochondrial and synaptic pathologies, AD remains a complex disease that requires further investigation. There are some key points to discuss and to explore further in the future:
Can we improve the quality of synaptic mitochondria?
Can we enhance axonal transport of mitochondria to increase synaptic ATP?
Can we reduce abnormal interactions between Aβ, phosphorylated tau with mitochondrial proteins, such as Drp1, VDAC, CypD and ABAD at synapses?
Are synaptic mitochondria more susceptible to mitochondrial fragmentation in AD?
Do synaptic mitochondria cause greater oxidative damage, leading to synaptic damage in AD?
In AD, why are hippocampal and cortical neurons more susceptible to oxidative damage than other neurons such as Purkinje neurons?
What is the impact of microglia and energy metabolism in relation to a synapse in the progression of AD?
Further research is urgently needed to answer above questions. It is also important focus on therapeutic drugs that target mitochondria and synapses. Further research is still needed to understand the biology of mitophagy/autophagy and to enhance autophagy and mitophagy activities in AD-affected brain regions. It is also important to test currently available mitophagy enhancers in both preclinical trials in AD mouse models and clinical trials in MCI and early AD patients.
Highlights.
Alzheimer’s disease (AD) is a progressive and synaptic failure disease.
Several years of research revealed that synaptic pathology and mitochondrial oxidative damage are early events in disease progression.
Loss of synapses and synaptic damage are the best correlates of cognitive deficits found in AD patients.
Reduced amyloid beta and phosphorylated tau, microglial activation and enhanced mitophagy are important for synapse-based therapies in AD.
Acknowledgments
Funding: The research presented in this article was supported by NIH grants AG042178, AG047812, NS105473, AG060767, AG069333 and AG066347 (to PHR).
Abbreviations:
- AD
Alzheimer’s disease
- AICD
Amyloid precursor protein intracellular domain
- APOE4
Apolipoprotein E4 genotype
- APP
Amyloid precursor protein
- Aβ
Amyloid beta
- CDK5
Cyclin Dependent Kinase 5
- Drp1
Dynamin-1-like protein
- Fis1
Fission 1
- GSK3β
Glycogen Synthase Kinase 3β
- HSPs
Heat shock proteins
- LTD
Long-term Depression
- LTP
Long-term Potentiation
- mAPP
Mutant amyloid beta precursor protein
- MCI
Mild Cognitive Impairment
- Mfn1
Mitofusin 1
- Mfn2
Mitofusin 2
- mtDNA
Mitochondrial DNA
- NFAT
Nuclear factor of activated T-Cells
- NFTs
Neurofibrillary tangles
- NMDA receptor
N-methyl-D-aspartate receptor
- NRF1
Nuclear respiratory factor 1
- NRF2
Nuclear respiratory factor 2
- NRG-1
Neuregulin-1
- Opa1
Optic atrophy 1
- PGC-1α
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- PINK1
PTEN-induced kinase 1
- PS1
Presenilin 1
- PS2
Presenilin 2
- ROS
Reactive Oxygen Species
- STEP
Striatal enriched tyrosine phosphatase
- TEC
Trans entorhinal cortex
- TFAM
Transcription factor A, mitochondrial
- TREM2
Triggering receptor expressed on myeloid cells 2
Footnotes
Conflicts of Interest: The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Almeida CG, Tampellini D, Takahashi RH, Greengard P, Lin MT, Snyder EM, Gouras GK, 2005. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol Dis 20, 187–198. [DOI] [PubMed] [Google Scholar]
- Alzheimers Association 2020, Facts and Figures.
- Amakiri N, Kubosumi A, Tran J, Reddy PH, 2019. Amyloid Beta and MicroRNAs in Alzheimer's Disease. Front Neurosci 13, 430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badimon A, Strasburger HJ, Ayata P, Chen X, Nair A, Ikegami A, Hwang P, Chan AT, Graves SM, Uweru JO, Ledderose C, Kutlu MG, Wheeler MA, Kahan A, Ishikawa M, Wang YC, Loh YE, Jiang JX, Surmeier DJ, Robson SC, Junger WG, Sebra R, Calipari ES, Kenny PJ, Eyo UB, Colonna M, Quintana FJ, Wake H, Gradinaru V, Schaefer A, 2020. Negative feedback control of neuronal activity by microglia. Nature. 586, 417–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakota L, Brandt R, 2016. Tau Biology and Tau-Directed Therapies for Alzheimer's Disease. Drugs 76, 301–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilousova T, Miller CA, Poon WW, Vinters HV, Corrada M, Kawas C, Hayden EY, Teplow DB, Glabe C, Albay R 3rd, Cole GM, Teng E, Gylys KH, 2016. Synaptic Amyloid-beta Oligomers Precede p-Tau and Differentiate High Pathology Control Cases. Am J Pathol 186, 185–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom GS, 2014. Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 71, 505–508. [DOI] [PubMed] [Google Scholar]
- Bonda DJ, Smith MA, Perry G, Lee HG, Wang X, Zhu X, 2011. The mitochondrial dynamics of Alzheimer's disease and Parkinson's disease offer important opportunities for therapeutic intervention. Curr Pharm Des 17, 3374–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Jeong YY, 2020. Mitophagy in Alzheimer's Disease and Other Age-Related Neurodegenerative Diseases. Cells. 9(1): 150 Published 2020 January 8. doi: 10.3390/cells9010150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Tammineni P 2017. Mitochondrial Aspects of Synaptic Dysfunction in Alzheimer's=Disease. J Alzheimers Dis. 57(4): 1087–1103. doi: 10.3233/JAD-160726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Tammineni P 2016. Alterations in Mitochondrial Quality Control in Alzheimer's Disease. Front Cell Neurosci. 10:24 Published 2016 February 9. doi: 10.3389/fncel.2016.00024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkins MJ, Reddy PH, 2011. Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer's disease neurons. Biochim Biophys Acta 1812, 507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wilde MC, Overk CR, Sijben JW, Masliah E, 2016. Meta-analysis of synaptic pathology in Alzheimer's disease reveals selective molecular vesicular machinery vulnerability. Alzheimers Dement 12, 633–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Alvaro M, Montero-Crespo M, Blazquez-Llorca L, DeFelipe J, Alonso-Nanclares L, 2019. 3D Electron Microscopy Study of Synaptic Organization of the Normal Human Transentorhinal Cortex and Its Possible Alterations in Alzheimer's Disease. eNeuro 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorostkar MM, Zou C, Blazquez-Llorca L, Herms J, 2015. Analyzing dendritic spine pathology in Alzheimer's disease: problems and opportunities. Acta Neuropathol 130, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD, 2008. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med 14, 1097–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, Lautrup S, Hasan Olive MM, Caponio D, Dan X, Rocktäschel P, Croteau DL, Akbari M, Greig NH, Fladby T, Nilsen H, Cader MZ, Mattson MP, Tavernarakis N, Bohr VA, 2019. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer's disease. Nat Neurosci. 22, 401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forner S, Baglietto-Vargas D, Martini AC, Trujillo-Estrada L, LaFerla FM, 2017. Synaptic Impairment in Alzheimer's Disease: A Dysregulated Symphony. Trends Neurosci 40, 347–357. [DOI] [PubMed] [Google Scholar]
- George EK, Reddy PH, 2019. Can Healthy Diets, Regular Exercise, and Better Lifestyle Delay the Progression of Dementia in Elderly Individuals? J Alzheimers Dis 72, S37–S58. [DOI] [PubMed] [Google Scholar]
- Gouras GK, Almeida CG, Takahashi RH, 2005. Intraneuronal Abeta accumulation and origin of plaques in Alzheimer's disease. Neurobiol Aging 26, 1235–1244. [DOI] [PubMed] [Google Scholar]
- Gylys KH, Fein JA, Yang F, Wiley DJ, Miller CA, Cole GM, 2004. Synaptic changes in Alzheimer's disease: increased amyloid-beta and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am J Pathol 165, 1809–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Jeong YY, Sheshadri P, Su X, Cai Q, 2020. Mitophagy regulates integrity of mitochondria at synapses and is critical for synaptic maintenance. EMBO Rep, e201949801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkins KM, Sokolow S, Miller CA, Vinters HV, Poon WW, Cornwell LB, Saing T, Gylys KH, 2012. Extensive p-tau pathology and SDS-stable p-tau oligomers in Alzheimer's cortical synapses. Brain Pathol 22, 826–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, Ashe KH, Liao D, 2010. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68, 1067–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong S, 2017. Molecular and Cellular Basis of Neurodegeneration in Alzheimer's Disease. Mol Cells 40, 613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamat PK, Kalani A, Rai S, Swarnkar S, Tota S, Nath C, Tyagi N, 2016. Mechanism of Oxidative Stress and Synapse Dysfunction in the Pathogenesis of Alzheimer's Disease: Understanding the Therapeutics Strategies. Mol Neurobiol 53, 648–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kametani F, Hasegawa M, 2018. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer's Disease. Front Neurosci 12, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandimalla R, Manczak M, Yin X, Wang R, Reddy PH, 2018. Hippocampal phosphorylated tau induced cognitive decline, dendritic spine loss and mitochondrial abnormalities in a mouse model of Alzheimer's disease. Hum Mol Genet 27, 30–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandimalla R, Reddy PH, 2016. Multiple faces of dynamin-related protein 1 and its role in Alzheimer's disease pathogenesis. Biochim Biophys Acta 1862, 814–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashyap G, Bapat D, Das D, Gowaikar R, Amritkar RE, Rangarajan G, Ravindranath V, Ambika G, 2019. Synapse loss and progress of Alzheimer's disease -A network model. Sci Rep 9, 6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffie RM, Hyman BT, Spires-Jones TL, 2011. Alzheimer's disease: synapses gone cold. Mol Neurodegener 6, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krstic D, Pfister S, Notter T, Knuesel I, 2013. Decisive role of Reelin signaling during early stages of Alzheimer's disease. Neuroscience 246, 108–116. [DOI] [PubMed] [Google Scholar]
- Kubo A, Misonou H, Matsuyama M, Nomori A, Wada-Kakuda S, Takashima A, Kawata M, Murayama S, Ihara Y, Miyasaka T, 2019. Distribution of endogenous normal tau in the mouse brain. J Comp Neurol 527, 985–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrane J, Rosen KM, Smith RC, Walsh K, Gouras GK, Querfurth HW, 2005. Intraneuronal beta-amyloid expression downregulates the Akt survival pathway and blunts the stress response. J Neurosci 25, 10960–10969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manczak M, Calkins MJ, Reddy PH, 2011. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Hum Mol Genet 20, 2495–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manczak M, Kandimalla R, Yin X, Reddy PH, 2018. Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer's disease. Hum Mol Genet 27, 1332–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manczak M, Reddy PH, 2012. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer's disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum Mol Genet 21, 2538–2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Terry RD, Alford M, DeTeresa R, Hansen LA, 1991. Cortical and subcortical patterns of synaptophysinlike immunoreactivity in Alzheimer's disease. Am J Pathol 138, 235–246. [PMC free article] [PubMed] [Google Scholar]
- Miller EC, Teravskis PJ, Dummer BW, Zhao X, Huganir RL, Liao D, 2014. Tau phosphorylation and tau mislocalization mediate soluble Abeta oligomer-induced AMPA glutamate receptor signaling deficits. Eur J Neurosci 39, 1214–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moneim AE, 2015. Oxidant/Antioxidant imbalance and the risk of Alzheimer's disease. Curr Alzheimer Res 12, 335–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostany R, Anstey JE, Crump KL, Maco B, Knott G, Portera-Cailliau C, 2013. Altered synaptic dynamics during normal brain aging. J Neurosci 33, 4094–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard M, Verkhratsky A, 2010. Calcium dyshomeostasis and pathological calcium signalling in neurological diseases. Cell Calcium 47, 101–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R, 2006. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci 26, 10129–10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver D, Reddy PH. Dynamics of Dynamin-Related Protein 1 in Alzheimer's Disease and Other Neurodegenerative Diseases. Cells. 2019;8(9):961 Published 2019 August 23. doi: 10.3390/cells8090961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver DMA, Reddy PH, 2019. Molecular Basis of Alzheimer's Disease: Focus on Mitochondria. J Alzheimers Dis 72, S95–S116. [DOI] [PubMed] [Google Scholar]
- Pradeepkiran JA, Reddy PH, 2020. Defective mitophagy in Alzheimer's disease. Ageing Res Rev. October 3;64:101191. doi: 10.1016/j.arr.2020.101191. Epub ahead of print. PMID: 33022416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooler AM, Phillips EC, Lau DH, Noble W, Hanger DP, 2013. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep 14, 389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozueta J, Lefort R, Shelanski ML, 2013. Synaptic changes in Alzheimer's disease and its models. Neuroscience 251, 51–65. [DOI] [PubMed] [Google Scholar]
- Rajmohan R, Reddy PH, 2017. Amyloid-Beta and Phosphorylated Tau Accumulations Cause Abnormalities at Synapses of Alzheimer's disease Neurons. J Alzheimers Dis 57, 975–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH, 2009. Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer's disease. Exp Neurol. 218, 286–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH, Beal MF, 2008. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer's disease. Trends Mol Med 14, 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH, Manczak M, Mao P, Calkins MJ, Reddy AP, Shirendeb U, 2010. Amyloid-beta and mitochondria in aging and Alzheimer's disease: implications for synaptic damage and cognitive decline. J Alzheimers Dis 20 Suppl 2, S499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH, Mani G, Park BS, Jacques J, Murdoch G, Whetsell W Jr., Kaye J, Manczak M, 2005. Differential loss of synaptic proteins in Alzheimer's disease: implications for synaptic dysfunction. J Alzheimers Dis 7, 103–117; discussion 173-180. [DOI] [PubMed] [Google Scholar]
- Reddy PH, McWeeney S, 2006. Mapping cellular transcriptosomes in autopsied Alzheimer's disease subjects and relevant animal models. Neurobiol Aging 27, 1060–1077. [DOI] [PubMed] [Google Scholar]
- Reddy PH, Oliver DM, 2019. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer's Disease. Cells. 8(5):488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH, Reddy TP, 2011. Mitochondria as a therapeutic target for aging and neurodegenerative diseases. Curr Alzheimer Res 8, 393–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH, Reddy TP, Manczak M, Calkins MJ, Shirendeb U, Mao P, 2011, Dynamin related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain Res Rev. 67,103–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH, Tripathi R, Troung Q, Tirumala K, Reddy TP, Anekonda V, Shirendeb UP, Calkins MJ, Reddy AP, Mao P, Manczak M, 2012. Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer's disease: implications to mitochondria-targeted antioxidant therapeutics. Biochim Biophys Acta 1822, 639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH, Yin X, Manczak M, Kumar S, Pradeepkiran JA, Vijayan M, Reddy AP, 2018. Mutant APP and amyloid beta-induced defective autophagy, mitophagy, mitochondrial structural and functional changes and synaptic damage in hippocampal neurons from Alzheimer's disease. Hum Mol Genet. 27, 2502–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Neltner JH, Nelson PT, 2014. Is synaptic loss a unique hallmark of Alzheimer's disease? Biochem Pharmacol 88, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, Mufson EJ, 2006. Hippocampal synaptic loss in early Alzheimer's disease and mild cognitive impairment. Neurobiol Aging 27, 1372–1384. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, 2001. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev 81, 741–766. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, 2002. Alzheimer's disease is a synaptic failure. Science 298, 789–791. [DOI] [PubMed] [Google Scholar]
- Sheng M, Sabatini BL, Sudhof TC, 2012. Synapses and Alzheimer's disease. Cold Spring Harb Perspect Biol 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva DF, Selfridge JE, Lu J, et al. 2013. Bioenergetic flux, mitochondrial mass and mitochondrial morphology dynamics in AD and MCI cybrid cell lines. Hum Mol Genet. 22(19):3931–3946. doi: 10.1093/hmg/ddt247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow RH, 2018. Mitochondria and Mitochondrial Cascades in Alzheimer's Disease. J Alzheimers Dis 62, 1403–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tampellini D, 2015. Synaptic activity and Alzheimer's disease: a critical update. Front Neurosci 9, 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu X, Chong WP, Zhai Y, Zhang H, Zhang F, Wang S, Liu W, Wei M, Siu NH, Yang H, Yang W, Cao W, Lau YL, He F, Zhou G, 2015. Functional polymorphisms of the CCL2 and MBL genes cumulatively increase susceptibility to severe acute respiratory syndrome coronavirus infection. J Infect 71, 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Su B, Siedlak SL, et al. 2008. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A. 105(49):19318–19323. doi: 10.1073/pnas.0804871105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Su B, Lee HG, et al. 2009. Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J Neurosci. 2009;29(28):9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, Planel E, Herman M, Figueroa HY, Wang L, Liu L, Lau LF, Yu WH, Duff KE, 2008. Interplay between cyclin-dependent kinase 5 and glycogen synthase kinase 3 beta mediated by neuregulin signaling leads to differential effects on tau phosphorylation and amyloid precursor protein processing. J Neurosci 28, 2624–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werneburg S, Feinberg PA, Johnson KM, Schafer DP 2017. A microglia-cytokine axis to modulate synaptic connectivity and function. Curr Opin Neurobiol. 47:138–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Alzheimer’s Report 2015.
- Xie J, Wang H, Lin T, Bi B, 2017. Microglia-Synapse Pathways: Promising Therapeutic Strategy for Alzheimer's Disease. Biomed Res Int 2017, 2986460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y, Zhao N, Caulfield TR, Liu CC, Bu G, 2019. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol 15, 501–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Sun X, Starovoytov V, Cai Q, 2015. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer's disease patient brains. Hum Mol Genet. 24, 2938–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Lu B, 2012. Synapses and dendritic spines as pathogenic targets in Alzheimer's disease. Neural Plast 2012, 247150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Perry G, Smith MA, Wang X, 2013. Abnormal mitochondrial dynamics in the pathogenesis of Alzheimer's disease. J Alzheimers Dis 33 Suppl 1, S253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]