

Abstract
Background:
The therapeutic effects of antipsychotic drugs (APDs) are mainly attributed to their postsynaptic inhibitory functions on the dopamine D2 receptor, which, however, cannot explain the delayed onset of full therapeutic efficacy. It was previously shown that APDs accumulate in presynaptic vesicles during chronic treatment and are released like neurotransmitters in an activity-dependent manner triggering an auto-inhibitory feedback mechanism. Although closely mirroring therapeutic action onset, the functional consequence of the APD accumulation process remained unclear.
Aims:
Here we tested whether the accumulation of the APD haloperidol (HAL) is required for full therapeutic action in psychotic-like rats.
Methods:
We designed a HAL analog compound (HAL-F), which lacks the accumulation property of HAL, but retains its postsynaptic inhibitory action on dopamine D2 receptors.
Results/outcomes:
By perfusing LysoTracker fluorophore-stained cultured hippocampal neurons, we confirmed the accumulation of HAL and the non-accumulation of HAL-F. In an amphetamine hypersensitization psychosis-like model in rats, we found that subchronic intracerebroventricularly delivered HAL (0.1 mg/kg/day), but not HAL-F (0.3–1.5 mg/kg/day), attenuates psychotic-like behavior in rats.
Conclusions/interpretation:
These findings suggest the presynaptic accumulation of HAL may serve as an essential prerequisite for its full antipsychotic action and may explain the time course of APD action. Targeting accumulation properties of APDs may, thus, become a new strategy to improve APD action.
Keywords: Haloperidol, presynaptic accumulation, schizophrenia, behavior
Introduction
Schizophrenia is a life-long neuropsychiatric disorder, causing highly reduced quality of life and/or suicide for the affected individuals. Aberrations in various brain regions and neurotransmitter systems are believed to be involved in the pathogenesis of schizophrenia, yet dopaminergic hyperactivity within the striatum is the strongest dysfunction detected in schizophrenic patients, associated with the psychotic symptoms of the disease (Kapur et al., 2005; van Rossum, 1966). In order to ameliorate these symptoms, antipsychotic drugs (APDs), which antagonize postsynaptic dopamine D2 receptors, are commonly used. Haloperidol (HAL), a typical APD, has been shown to be beneficial for the treatment of psychosis in patients (Ayd, 1978; Miyamoto et al., 2005), as well as for the reversal of psychosis-like disturbances in animal models (Samaha et al., 2008; Uzuneser et al., 2018). However, these effects emerge only after repeated treatment for several weeks, an effect that cannot be fully explained with pharmacological or psychological models of action (Carey, 1987). These restorative effects are attributed to the postsynaptic receptor antagonism property of HAL, yet a presynaptic action might also be involved. The weak-base property of APDs, including HAL, enables them to be accumulated in synaptic vesicles in all brain regions, resulting in prolonged clinical effects weeks after cessation of intake. Such accumulations, which are mediated by a pharmacokinetic process called acidic trapping (de Duve et al., 1974; Trapp et al., 2008), have been detected in post mortem tissue samples (Kornhuber et al., 1999), tissue slices from rats (Gemperle et al., 2003), and cultured neurons (Tischbirek et al., 2012). The accumulated drugs are released, both in cultured neurons and freely moving rats, in a use-dependent manner via exocytosis, resulting in elevated drug concentrations in the synaptic cleft (Tischbirek et al., 2012). Subsequently, they bind not only to the postsynaptic D2 receptors, but also to presynaptic voltage-gated sodium channels, inhibiting further neurotransmission by an accumulation-dependent auto-inhibitory mechanism (Tischbirek et al., 2012). Furthermore, cyamemazine, an atypical APD, accumulates in presynaptic striatal neurons and is co-released with dopamine by action potential or amphetamine administration in tissue slices (Tucker et al., 2015). Thereby, the release of vesicular APDs may enhance dopaminergic synaptic transmission (Walters and Levitan, 2019). This co-release ensures postsynaptic D2 receptor antagonism and presynaptic auto-inhibition during salient events when phasic dopaminergic neurotransmission occurs.
The presynaptic APD accumulation, despite having been repeatedly reported, has not been thoroughly investigated in terms of its functional effects. The possible benefits of this process to the antipsychotic efficacy of the drugs remain unanswered. In order to address this issue, we investigated the potential contribution of the vesicular accumulation property of HAL to its therapeutic efficacy in an animal model showing psychosis-like symptoms. For this purpose, we designed a HAL analog compound (HAL-F) that lacks the weak-base property of HAL, thus is unable to accumulate in synaptic vesicles, yet has comparable affinity to HAL for the postsynaptic D2 receptors (Figure 1(a)). We firstly examined whether HAL-F indeed does not accumulate in neuronal cultures using LysoTracker Green (LTG) staining. After confirming non-accumulation, we tested the possible antipsychotic efficacy of HAL-F in vivo, using rats. Using an escalating amphetamine (AMPH) sensitization regimen, we induced psychosis-like symptoms in the animals (Peleg-Raibstein et al., 2006; Uzuneser et al., 2018). Using mini pumps and brain infusion kits to enable continuous delivery within the lateral ventricle, we locally administered therapeutic doses of HAL and HAL-F, the latter with two different doses, and compared their antipsychotic efficacy.
Figure 1.
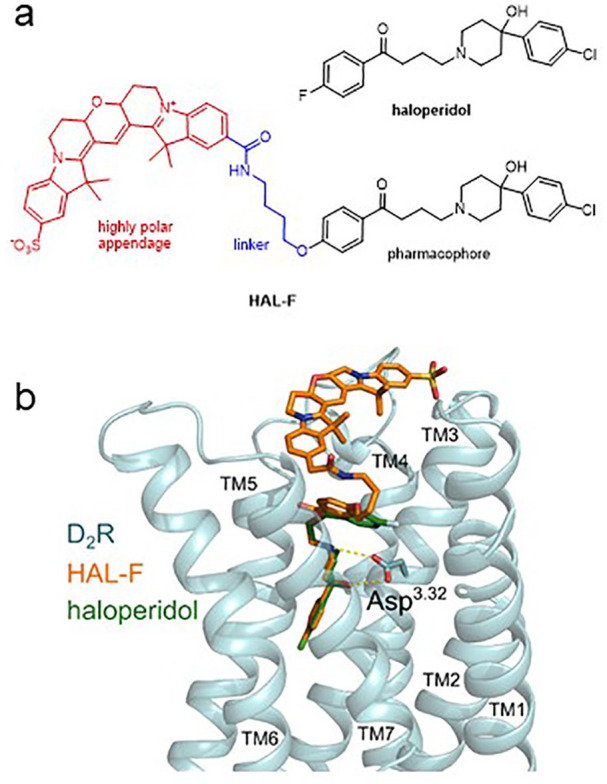
Development and validation of the non-accumulating haloperidol (HAL) analog HAL-F with preserved dopamine D2 receptor affinity: (a) chemical structure of HAL-F; (b) ligand receptor interactions of HAL (green) and HAL-F (orange) with the dopamine D2 receptor (blue). HAL-F binds to dopamine receptors in the same manner as HAL, despite carrying the large polar appendage attached to the HAL scaffold.
Experimental procedures
Docking studies with HAL-F
Docking studies were performed using the recently published crystal structure of the D2 receptor (Protein Data Bank entry 6CM4; Wang et al., 2018). The Dock Prep module of UCSF Chimera (Pettersen et al., 2004) was utilized to prepare the protein structure for docking. Coordinates of the co-crystallized ligand, lipids and water molecules were removed, missing side chains completed, and hydrogens added. The 3D coordinates of the docked compounds were created and optimized using Avogadro (Version 1.2.0) (Hanwell et al., 2012), an open-source molecular builder and visualization tool. The subsequent docking was performed using AutoDock Vina (Trott and Olson, 2010) applying a search space of 28 Å × 28 Å × 38 Å to ensure a complete coverage of the binding pocket and the extracellular side of the receptor. The ligands were subjected to the docking procedure using an exhaustiveness value of 16 and a randomly selected starting position. Twenty conformations of each ligand were obtained and inspected manually. We selected one final conformation for each ligand, based on the scoring function of AutoDock Vina and the canonical salt bridge to Asp3.32. Visualization was performed using the PyMOL Molecular Graphics System, Version 2.1.1 (Schrödinger, LLC).
Receptor binding profile of the non-accumulating HAL analog HAL-F
Radioligand binding studies with the dopamine receptor subtypes D1–D4 were performed as described previously (Hübner et al., 2000, 2016). In brief, competition binding experiments were done using membranes of Chinese hamster ovary (CHO) cells stably expressing the human D2L and D2S (Hayes et al., 1992), D3 (Sokoloff et al., 1990), or D4.4 receptor (Asghari et al., 1995). Radioligand displacement assays were run in binding buffer (50 mM Tris, 5 mM MgCl2, 1 mM EDTA, 100 µg/mL bacitracin, and 5 µg/mL soybean trypsin inhibitor at pH 7.4) with [3H]spiperone (specific activity = 68 Ci/mmol, PerkinElmer, Rodgau, Germany) at final concentrations of 0.15–0.30 nM and membrane concentrations of 1–6 µg protein per assay tube. The assays were carried out at specific binding affinities characterized by a Bmax value of 1100 fmol/µg and a KD value of 0.10 nM for D2L, a Bmax value of 8500 fmol/µg and a KD value of 0.060 nM for D2S, a Bmax value of 8500 fmol/µg, and a KD value of 0.15 nM for D3, and a Bmax value of 1300 fmol/µg and a KD value of 0.25 nM for D4.4. Competition binding tests with the dopamine D1 subtype were done analogously with membranes from HEK293T cells transiently transfected with the cDNA for D1 (cDNA Resource Center, Bloomsburg, PA, USA) by using the Mirus TransIT-293 transfection reagent (PeqLab, Erlangen, Germany) (Möller et al., 2017). Binding experiments were performed with the radioligand [3H]SCH23390 (80 Ci/mmol, Biotrend, Cologne, Germany) at a final concentration of 0.40 nM, KD of 0.30 nM, Bmax of 2700 fmol/mg, and 3 µg protein per well. Unspecific binding was determined in the presence of 10 µM HAL, and protein concentration was established by the method of Lowry et al. (1951) using bovine serum albumin as standard.
The resulting competition curves of the receptor binding experiments were analyzed by nonlinear regression using the algorithms in Prism 6.0 (GraphPad Software, San Diego, CA, USA). The data were initially fit using a sigmoid model to provide an IC50 value, representing the concentration corresponding to 50% of maximal inhibition. IC50 values were transformed to absolute inhibition constant Ki values according to the equation of Cheng and Prusoff (1973).
Vesicular accumulation: imaging solutions, dyes and pharmacological reagents
For imaging experiments, staining solution was prepared by dissolving LTG (Invitrogen Molecular Probes, Eugene, OR, USA) to a concentration of 50 nM in imaging solution buffer (ISB). ISB was prepared by dissolving 84.16 mg/mL sodium chloride, 1.88 mg/mL potassium chloride, 18.02 mg/mL glucose (Sigma-Aldrich, St. Louis, MO, USA), 23.84 mg/m 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 2.776 mg/mL calcium chloride, and 5.08 mg/mL magnesium chloride in water, and was adjusted to a pH of 7.5.
HAL (purchased from Sigma-Aldrich, Taufkirchen, Germany) and its analog compound (HAL-F) were both dissolved in dimethyl sulfoxide (DMSO) and diluted to a concentration of 5 µM in staining solution. Unless HAL and HAL-F are not present in staining solution, a corresponding volume of DMSO was added. All chemicals and substances were from Carl Roth (Karlsruhe, Germany) unless otherwise stated.
Vesicular accumulation: cell culture
Dissociated hippocampal neuronal culture was established from P0 to P3 old unsexed Wistar rats (Charles Rivers, Wilmington, MA). Dissection was conducted according to the guidelines of the State of Bavaria and with approval by the ethics committee of the Friedrich-Alexander University of Erlangen-Nuremberg, Germany. Hanks balanced salt solution (HBSS, Biochrom GmbH, Berlin, Germany) was used for collecting tissues and washing them. Dissection solution for tissue digestion consisted of 20 µg DNAse (Sigma-Aldrich, St. Louis, MO, USA) and 5 mg/mL trypsin (Sigma-Aldrich, St. Louis, MO, USA) dissolved in HBSS. To stop enzymatic reactions, digestion solution consisted of HBSS with 20% fetal calf serum (Biochrom GmbH, Berlin, Germany), supplemented with 8 mg/mL sodium chloride and 0.037 mg/mL potassium chloride. MatrigelTM, used to coat coverslips (Karl Hecht, Sondheim vor der Rhön, Germany) for enhanced cell attachment, was purchased from Omnilab (Munich, Germany). The starting medium, used for the first day of culturing, consisted of minimum essential medium (MEM, Gibco, Life Technologies, Carlsbad) supplemented with 5 mg/mL glucose (Sigma-Aldrich, St. Louis, MO, USA), 0.2 mg/mL sodium bicarbonate (Merck, Darmstadt, Germany), 0.01 mg/mL transferrin (Merck, Darmstadt, Germany), 10% fetal bovine serum (Biochrom GmbH, Berlin, Germany), 1% l-glutamine solution (2M, Biochrom GmbH, Berlin, Germany), and 0.2% insulin solution (Sigma-Aldrich, St. Louis, MO, USA). Growing medium consisted of MEM supplemented with 5 mg/mL glucose, 0.2 mg/mL sodium bicarbonate, 0.01 mg/mL transferrin, 5% fetal bovine serum, 0.25% l-glutamine solution (2M, Biochrom GmbH, Berlin, Germany), 2% B27 (Gibco, Life Technologies, Carlsbad, CA, USA), 6 µM AraC (Sigma-Aldrich, St. Louis, MO, USA), and 0.1% penicillin-streptomycin (100X, Merck, Darmstadt, Germany).
Newborn rats were decapitated and the brains were removed. The cerebellum was cut off and the hemispheres were separated. The hippocampi were isolated and collected in a culture dish containing ice-cold HBSS. After breaking and washing the tissue, enzymatic dissociation was initialized by incubating in dissection solution for 15 min at 37°C. Cells were then washed with digestion solution. The cells were transferred to dissection solution and gently separated by trituration. Cells were then washed with digestion solution and centrifuged. Subsequently, cells were resuspended in starting medium. A 50 µL cell suspension was plated on Matrigel-coated coverslips. After 1 h incubation at 37°C with 5% CO2, 950 µL starting medium was added. The next day, 500 µL of the medium in each well was removed followed by the addition of 1 mL growing medium. Neurons were incubated at 37°C with 5% CO2 until experiments were performed between DIV25 and DIV30. All chemicals and substances not stated were from Carl Roth (Karlsruhe, Germany).
Live cell imaging
To record the fluorescence intensity (FI), a Nikon TI-Eclipse inverted fluorescence microscope equipped with a 10×, 0.45 NA objective (Nikon Instruments Europe, Düsseldorf, Germany) and a water-cooled EM-CCD camera (iXon Ultra 897, Andor, Belfast, Northern Ireland) were used. Experiments were conducted in the dark and at room temperature. Fluorescence was excited by a Nikon Intensilight C-HGFI using dichroic long-pass mirrors with a cutoff wavelength of 495 nm. Light was previously centered by passing an excitation filter ranging from 442 to 502 nm. Emitted light passed emission band-pass filters ranging from 485 to 555 nm (Semrock, Rochester, NY, USA). Exposure time of 150 ms and frame rate of 0.1 Hz were adjusted by Andor Solis software. Data were exported as tagged image file format (tiff) containing 512 × 512 pixels of 16-bit monochromatic intensity values.
Coverslips with cultured neurons were posed into a measure chamber suitable for bathing and were constantly perfused during recording. A piezo-controlled stepper device (SF-77B, Warner Instruments) enabled a fast exchange of solutions. The perfusion rate was kept constant at 0.2 mL/min using the Fast-Step Valve Control Perfusion System (VC-77SP8, Warner Instruments). For each experiment, the sample was perfused with staining solution containing a corresponding volume of DMSO to record baseline. Subsequently, perfusion was switched to bathe the cells respectively with test compound or DMSO vehicle for 15 min.
Image processing
Feature point detection was automatically conducted by a non-maximum suppression algorithm (Sbalzerini and Koumoutsakos, 2005) implemented as a MATLAB routine that selected circular regions of interest (ROIs) around detected centroids representing the location of acidic compartments. For further analysis of recorded image stacks, custom-written software was coded in MATLAB. Analysis comprised read outs of fluorescence traces from individual ROIs represented as the mean value over the intensity value of each pixel within the ROI for each frame in the image stack. The background value for each frame was determined by applying a Gaussian filter followed by thresholding of the gray level histogram. Bright pixels that exceeded the threshold level were excluded. Finally, the averaged remaining pixels gave the final background value of the corresponding frame that was subtracted from the original. The reference value was calculated from the FI baseline right before perfusion switch (average over values derived from frame 25–29) and set to 100%, from which the relative FI was calculated. A one-sided, unpaired t-test within a moving window along fluorescence traces starting from frame 60 was applied to determine the location when a plateau was reached and gave the end of fluorescence decrease. The final result was computed by taking the average over the next five frames beyond the last location of the t-test window.
Animals
Male Sprague-Dawley rats (Charles River, Germany; 300–350 g) were used as subjects. They were housed in colonies (four animals per cage) in a temperature- (22±2°C) and humidity- (55±10%) controlled room under a light-dark cycle (light on from 07:00 to 19:00). The behavioral tests were conducted during the light phase of the cycle. Food and water were provided ad libitum. All experiments were carried out in agreement with the Animal Protection Law of the Federal Republic of Germany and the European Communities Council Directive of 24 November 1986 (86/609/EEC), and were approved by the local authority Regierung von Mittelfranken.
Drugs and treatment procedure
D-amphetamine sulfate (AMPH; 1 mL/kg; Fagron) or 0.9% saline (SAL; 1 mL/kg) was administered intraperitoneally (i.p.). AMPH was dissolved in SAL. The sensitization procedure was adapted from Peleg-Raibstein et al. (2006) and had been established in our laboratory (Möller et al., 2017; Uzuneser et al., 2018). Animals were AMPH-sensitized for six consecutive days, receiving three AMPH (or SAL) injections per day with escalating doses of AMPH; from 1 mg/kg to 8 mg/kg with an increment of 1 mg/kg after each injection. The dose was sustained at 8 mg/kg for the last 10 injections. HAL and HAL-F were dissolved in a vehicle (VEH) solution containing distilled water with 0.3% ascorbic acid, 10% cyclodextrin, and 20% dimethyl sulfoxide. Drugs were administered continuously and locally via Alzet osmotic mini pumps (model 2001; 7-day delivery; DURECT Corporation) connected to brain infusion kits (Brain Infusion Kit 2; DURECT Corporation). HAL is a small, weakly charged lipophilic molecule, thus it can freely cross the blood–brain barrier (Banks, 2016). The significantly larger volume of HAL-F possibly disables it from crossing the barrier. Therefore, both drugs were locally delivered into the brain. In some earlier studies, HAL was administered locally into the lateral ventricle and was shown to abolish grooming behavior (0.1 mg/kg; Versteeg et al., 1993), and not induce tardive dyskinesia (0.0375 mg/kg; Andreassen and Jorgensen, 1994). However, in these studies, HAL was delivered not continuously by mini pumps, but rather by daily injections. The delivery method of a drug determines its postsynaptic receptor occupancy (Kapur et al., 2003). Therefore, we performed a pilot study to determine the concentration of HAL that would be therapeutically effective when administered continuously into the lateral ventricle. According to this pilot study, 0.1 mg/kg/day HAL, without causing extrapyramidal side effects, was significantly more effective on the alleviation of psychotic-like symptoms than 0.01 or 0.03 mg/kg/day (data not shown). Because the molecular weight of HAL-F was approximately three times the molecular weight of HAL (Figure 1(a)), the concentration of 0.3 mg/kg/day HAL-F (HAL-F-0.3) was prepared to deliver an equal amount of substance. As the affinity of HAL-F for D2 receptors is less compared with HAL, we also tested a 5-fold increased dose of HAL-F (1.5 mg/kg/day; HAL-F-1.5). Mini pumps and catheter tubes were filled with these solutions, and the pumps were connected to the brain infusion cannulas via 3-cm long catheter tubes. Using a spacer, the cannula depth was altered to 4.5 mm. Before implantation, filled brain infusion assemblies were incubated in sterile SAL overnight for priming (Sanchez-Mendoza et al., 2016).
Surgical procedure
Surgeries were performed 1–2 days after the offset of AMPH sensitization. Animals were anesthetized with a solution containing distilled water with 30% ketamine (Ketaset; 0.1 g/mL) and 15% medetomidine (Dorbene; 1 mg/mL), administered i.p. (1 mL/kg). Animals were fixed to the stereotaxic frame and a sagittal cut was made behind the eyes towards the neck by a scalpel, exposing the skull. Using the reference points on the skull, the coordinates of the lateral ventricle were assessed (0.8 mm posterior, 1.4 mm lateral, 4.5 mm ventral from the bregma; Paxinos and Watson, 2014; Figure 1(b)). Using a microdrill, a hole was made on the right hemisphere. A pocket was made on the neck area by a hemostat, through which the pump was inserted. Cyanoacrylate adhesive was applied to the base of the cannula. The cannula was inserted through the skull using a microdrive, and the scalp wound was closed using wound clips. Animals were single housed after the surgeries. Behavioral tests started after a recovery of five days, with daily inspection of animal recovery.
Behavioral procedures
Sixty animals were randomly divided into five groups, with one group receiving SAL and the other four receiving AMPH administration. SAL-administered animals were implanted with VEH-containing pumps, whereas the AMPH-administered animals were implanted with VEH-, HAL-, HAL-F-0.3- or HAL-F-1.5-containing pumps. AMPH-induced locomotion and pre-pulse inhibition (PPI) tests were conducted 6 and 7 days after the pump implantation, respectively.
The AMPH-induced locomotion test was conducted in a cubic gray open field arena (50 × 50 × 50 cm). A 20-min baseline period was followed by a 40-min AMPH-induced period, prior to which animals received an i.p. injection of AMPH (1.5 mg/kg). Locomotor activity was analyzed using Biobserve Viewer III (Biobserve GmbH, Germany) software. Rearing was analyzed manually, using the videotapes (Amato et al., 2020; Möller et al., 2017; Uzuneser et al., 2018, 2019)
PPI of the acoustic startle response test was conducted 24 h later after the AMPH-induced locomotion test, using a paradigm adapted from Peleg-Raibstein et al. (2006). Animals were placed into the soundproof boxes within restraining metal cages (27 × 9 × 10 cm). Two loudspeakers, which were mounted inside the soundproof boxes, presented the acoustic stimuli. Two pre-pulse alone trials (80 and 86 dB), three pulse alone trials (100, 110, and 120 dB), and six pre-pulse + pulse trials were repeated 10 times in a pseudo-randomized fashion, and the startle responses were measured using the TSE startle response system (TSE Systems, Bad Homburg, Germany). The percentage PPI was calculated with the formula %PPI=100 – (100 × (startle amplitude of pre-pulse + pulse trials / startle amplitude of pulse alone trials)) (Amato et al., 2020; Fendt and Fanselow, 1999; Uzuneser et al., 2018, 2019).
Post mortem brain imaging
Coverslips with sections of the brain cut on a microtome (20 µm) were stored at −20°C until imaging to analyze accumulation of fluorescent HAL-F in brain tissue. All pictures were recorded at room temperature and in the dark using a Nikon TI-Eclipse inverted fluorescence microscope equipped with a 10×, 0.45 NA objective (Nikon Instruments Europe, Düsseldorf, Germany) and a water-cooled EM-CCD camera (iXon Ultra 897, Andor, Belfast, Northern Ireland). Emitted light from a LedHUB lamp (Omicron-Laserage Laserprodukte GmbH, Rodgau, Germany) passed an excitation filter ranging from 521 to 565 nm (Semrock, Rochester, NY, USA) and was directed to the probe with a dichroic long-pass mirror with a cutoff wavelength of 562 nm (HCBS562). Emitted fluorescence light from the probe passed emission band-pass filter ranging from 554 to 633 nm. Exposure time was constantly set to 130 ms. Pictures were taken as 16-bit monochromatic intensity values and stored as tiff. VisiView software (Visitron System GmbH) controlled the microscope and camera.
For image processing, the stored tiff files were loaded to MATLAB and analyzed by a custom-written program. The images were backgrounded by averaging the intensity from the 2% darkest pixels. Therefore, a Gaussian filter was applied to reduce noise and the lighter pixels were cut off by thresholding the gray level histogram from the smoothed image. In the end the program returned the mean intensity value of emitting tissue excluding the dark regions (“tissue holes”) by segmentation. Therefore, the intensity histogram of the gray level image was used to create an intensity profile. The two intensity populations were divided by a threshold defined by the minimum in the intensity profile between the two populations. The threshold was detected by estimating the first derivative using a gradient filter over the intensity profile and subsequent detection of zero crossings. After cutting off the dark pixels, the mean intensity was calculated by averaging the intensity of the remaining pixels. We grouped the results by animals treated with the same concentration (n=3 per group). An unpaired Student’s t-test was performed to check for significant differences.
Data analysis
The data are shown as mean ± SEM. For the life cell imaging, the data were statistically analyzed using Microsoft Excel or built-in routines in MATLAB. Error bars indicate SEM. Statistical significance was tested by comparing the treatment groups (HAL and HAL-F) with the control group (DMSO) using two-sided, unpaired t-tests. For the behavioral analysis, the locomotion and rearing were analyzed by a two-way analysis of variance (ANOVA) with treatment combination and time as factors. Pre-planned analyses were calculated to compare group differences using Bonferroni-corrected least significant difference (LSD) tests. Differences were calculated as both activity at single time points of 5-min blocks, and as overall activity, using area under the curve (AUC) analysis. For PPI analysis, a three-way ANOVA was conducted, using factors treatment, pre-pulse intensity, and pulse intensity. Differences between groups were calculated using pre-planned Bonferroni-corrected LSD tests. Differences at single pre-pulse–pulse pairings were also analyzed separately using one-way ANOVA. When sphericity was violated, the Greenhouse–Geisser correction method was used.
Results
Development of the non-accumulating HAL analog HAL-F
To develop a high affinity HAL derivative showing no vesicular accumulation, we planned to attach a polar and reasonably large appendage, which significantly decreases the logP value and the overall size of the ligand. These changes in the physicochemical properties should significantly hamper the vesicular accumulation. As an appendage of choice, we picked the fluorescence dye Cy3B, which enabled us to validate brain parenchyma distribution after treatment by fluorescence-based methods (Figure 1(a)).
As the retention of high dopamine D2 receptor affinity was required for our studies, the point of attachment of the appendage to the pharmacophore core was crucial to avoid negative interactions with the receptor binding pocket. Based on extensive structure–affinity relation studies of dopamine D2 receptor ligands we identified the most promising position of the fluoro substituent for the attachment of a handle for Cy3B. We planned to introduce a butylene linker which would be connected to the arene moiety by an ether unit (Figure 2). This linker would enable the appendage to reach outside the binding pocket circumventing sterical clashes or negative electrostatic interactions. Additionally, we planned to synthesize in the first step a “dummy” ligand which features the linker unit but not the Cy3B moiety. Comparing the binding properties of this dummy with HAL and HAL-F would allow insights into the contributions of the linker and the polar appendage to the receptor affinity.
Figure 2.

Synthesis of the haloperidol analog HAL-F.
Before entering the chemical synthesis, we confirmed our empirical data-based design by structure-based molecular modeling. Thus, we performed docking studies with the D2 receptor crystal structure, HAL and HAL-F. Applying the Dock Prep module chimera (Pettersen et al., 2004; Shapovalov and Dunbrack, 2011) and AutoDock Vina (Trott and Olson, 2010), we could create detailed insights into the binding poses of these ligands. Our analysis showed an overlay of HAL and HAL-F in the D2 receptor binding pocket (Figure 1(b)). The pharmacophoric elements displayed a highly similar binding pose, whereas the linker and the Cy3B unit pointed outwards of the D2 receptor. These results confirmed that we had chosen the right point of attachment and linker length to retain a high binding affinity.
Chemical synthesis of HAL-F
The synthesis of HAL-F is depicted in Figure 2. Starting from commercially available 4-(4-chlorobutanoyl)phenol (1), the 4-phenylpiperidin-4-ol moiety was attached by nucleophilic substitution leading to the HAL derivative (2). The linker was introduced by the reaction of (2) with 1,-4-dibromobutane yielding the bromoalkyl derivative (3), which was converted to the respective azide (4). Finally, catalytic hydrogenation followed by acylation with commercially available Cy3B-OSu gave HAL-F. The chemical and photosensitive HAL-F was purified by preparative high-performance liquid chromatography and characterized by liquid chromatography–mass spectrometry (for details see supplemental material).
HAL-F and HAL D2 receptor affinity
Receptor binding affinities of the HAL-F and the reference ligand HAL were determined by radioligand competition binding experiments with the dopamine receptor subtypes D1, D2L, D2S, D3, and D4.4. Affinities to the receptors of the D2 family (D2–D4) were investigated with membranes from CHO cells stably expressing D2L, D2S, D3, or D4.4 and the radioligand [3H]spiperone. D1 affinity was measured with membranes from transiently transfected HEK293T cells and the radioligand [3H]SCH23390. HAL-F which carrys an additional Cy3 fluorophore attached to the linker showed Ki values in the low nanomolar range. HAL-F binding affinities were 13 nM for D2S and 30 nM for D2L, whereas HAL affinities were determined with 0.83 nM for D2S and to 0.64 nM for D2L. Ki values for D4.4 (2800 nM) and D1 (3600 nM) were clearly reduced relative to HAL resulting in an increased selectivity of HAL-F for the D2 and D3 receptor subtypes (Table 1).
Table 1.
Receptor binding profile of haloperidol (HAL) and the non-accumulating analog HAL-F for human dopamine receptor subtypes.
| Ki value (nM), mean ± SEMa | |||||
|---|---|---|---|---|---|
| [3H]SCH23390 |
[3H]spiperone |
||||
| Compound | D1 | D2L | D2S | D3 | D4.4 |
| HAL | 5.9 ± 1.2 | 0.64 ± 0.42 | 0.83 ± 0.27 | 3.1 ± 0.89 | 6.1 ± 1.4 |
| HAL-F | 3600 ± 1000 | 30 ± 5.4 | 13 ± 4.1 | 68 ± 13 | 2800 ± 430 |
Ki values derived from 4 to 9 individual experiments, each done in triplicate.
HAL-F does not accumulate in the acidic vesicles of cultured neurons
To examine the contribution of HAL accumulation property on the therapeutic efficacy of APDs, HAL-F was designed as a HAL analog compound that largely retains its postsynaptic inhibitory action on the dopamine D2 receptors, but without the property to accumulate in synaptic vesicles. To confirm that HAL-F lacks the property to accumulate in subcellular departments, LTG, a fluorescent model substance that has similar physiochemical properties to HAL and HAL-F, was applied (Kornhuber et al., 2010; Rayport and Sulzer, 1995). LTG has the ability to enrich in acidic compartments. If the test compound is also capable of accumulating, it will displace LTG leading to a decrease in the fluorescence signal that is derived from the LTG-stained compartments (Figure 3(a)).
Figure 3.

Haloperidol (HAL) analog (HAL-F) lacks the property to accumulate in acidic compartments: (a) experimental procedure in hippocampal neurons; (b) representative images of living hippocampal neurons stained with LysoTracker Green (LTG) before (left panel) and after (right panel) HAL; (c) fluorescence intensity (FI). Left: relative FI in relation to before perfusion switch (set as 100%; mean±SEM); Right: light curves represent mean curves over all regions. Bold curves represent mean curves over all measurements per treatment (n=15 per group; ***p< 0.001, two-tailed, unpaired t-test versus dimethyl sulfoxide (DMSO) control).
Previously, we have already shown that HAL accumulates in acidic compartments as well as in synaptic vesicles by staining hippocampal neuronal culture with LysoTracker Red and αSyt1-cypHer5 antibodies specific for the acidic lumen of synaptic vesicles (Tischbirek et al., 2012). To test if HAL-F accumulates in any acidic compartments, we stained hippocampal neurons cultured on a coverslip between DIV25 and DIV30 with 50 nM LTG. Then fluorescence was recorded under permanent perfusion with the LTG-containing solution. After a 5-min baseline recording, the sample was perfused with either 5 µM HAL, 5 µM HAL-F or a corresponding volume of DMSO as a control (Figure 3(b)).
During perfusion with HAL, relative FI decreased significantly (p<0.001; Figure 3(c)) confirming LTG displacement from acidic compartments by HAL. In contrast, perfusion with HAL-F did not affect relative FI proving that HAL-F lacks the property to accumulate in any acidic compartments in the used concentration (Figure 3(c)).
HAL, but not HAL-F, reverses AMPH-induced hyperactivity
During the 20-min baseline, animals habituated to the test environment (Figure 4(a)). Their locomotion and rearing activities declined over time (locomotion: F(1.865,102.576)=91.098, p<0.001; rearing: F(2.307,126.901)=76.053, p<0.001). A treatment × time interaction was not observed for either behavior (locomotion: F(7.46,102.576)=1.44, p>0.05; rearing: F(9.229,126.901)=1.385, p>0.05). A significant effect of treatment was observed, but only for rearing (locomotion: F(4,55)=1.454, p>0.05; rearing: F(4,55)=2.633, p=0.044). However, pre-planned comparisons did not reveal significant effects of either treatment on overall baseline locomotor or rearing activity (Figure 4(b) to (e)).
Figure 4.

Haloperidol (HAL), but not its non-accumulating analog HAL-F has antipsychotic effects in rodent models of psychosis: (a) treatment plan for induction of amphetamine (AMPH) hypersensitization–induced psychotic-like state and its treatment with osmotic mini pump intracerebroventricular application. Schizophrenia-like behavior was tested by AMPH-induced (intraperitoneally) hyperlocomotion (AIH) and pre-pulse inhibition (PPI); (b) time course of horizontal locomotion; (c) area under the curve (AUC) for locomotor activity during 30 min after the treatment; (d) time course of rearing; (e) AUC for rearing activity during 30 min after the treatment (mean±SEM; n=7–15 per group; *p<0.05, #p<0.01, $p<0.001 versus AMPH-VEH; §p<0.05 versus saline-VEH; VEH – vehicle).
An AMPH challenge induced a rapid increase in locomotion and rearing in all treatment groups, but the rate of elevation was not uniform between groups (locomotion: F(4,55)=6.065, p<0.001; rearing: F(4,55)=3.808, p=0.008; Figure 4(b) to (e)). A significant treatment × time interaction was observed for both locomotion (F(13.961,191.967)=2.287, p=0.007) and rearing (F(14.373,197.629)=3.362, p<0.001). Compared to the AMPH-naïve animals, AMPH-sensitized animals showed amplified locomotion and rearing at single time points and at 30 min AUC level; locomotion: 10 min: p=0.005, 15 min: p=0.008, AUC: p=0.024; rearing: 5 min: p=0.024, 10 min: p=0.001, 15 min: p=0.001, AUC: p=0.032). Therapeutic HAL treatment reversed the locomotor activity to the level of AMPH-naïve animals, and diminished the rearing activity even more robustly compared with VEH-treated AMPH-sensitized animals (locomotion: 5 min: p=0.012, 10 min: p=0.015, 20 min: p=0.045, AUC: p=0.015; rearing: 5 min: p<0.001, 10 min: p<0.001, 15 min: p=0.001, 20 min: p=0.012, AUC: p=0.001).
Importantly, we have not observed such an attenuating effect by HAL-F treatment. Either dose of HAL-F (0.3 or 1.5) failed to restore the hyperlocomotor activity of AMPH-sensitized animals, which was supported by lack of significant effects at single time points and at 30 min AUC level (p>0.05). However, we observed a significant reversal effect of low dose HAL-F-0.3 treatment on the rearing behavior (5 min: p=0.016, 10 min: p=0.013; Figure 4(b) to (e)).
Altogether, these findings suggest that continuous local HAL administration into the ventricle produces similar therapeutic effects to continuous peripheral HAL administration in terms of attenuating hyperlocomotor response to AMPH after short-term treatment. Such therapeutic effects were absent after continuous local HAL-F treatment for either dose tested, suggesting a lack of antipsychotic activity. Notably, our findings, the locomotor-enhancing effect of AMPH in AMPH-sensitized animals and its reversal by therapeutic short-term HAL treatment, are identical to our previous findings, where 0.5 mg/kg/day HAL was delivered subcutaneously (Uzuneser et al., 2018). A 0.5 mg/kg/day subcutaneous HAL delivery has been shown to occupy 73% striatal dopamine D2 receptors, which is within the therapeutic range and below the extrapyramidal side effect induction threshold (Kapur et al., 2003). Thus, this replication confirms that 0.1 mg/kg/day intracerebroventricular (i.c.v.) HAL delivery occupies therapeutically effective levels of striatal dopamine D2 receptors.
HAL, but not HAL-F, improves impaired PPI
Figure 5 shows the mean PPI of animals with all possible pre-pulse × pulse pairings. The amount of inhibition increased with increased pre-pulse stimulus intensity, which was evident by a main effect of the pre-pulse (F(1,55)=248.987, p<0.001). There was a significant effect of treatment (F(4,55)=2.734, p=0.038). Pre-planned comparisons failed to show a disruption in PPI by AMPH sensitization, shown by a lack of significant effect on the inhibition at single pairings and on overall inhibition (p>0.05). However, compared with AMPH-VEH group, continuous i.c.v. HAL treatment clearly improved PPI, indicated by a significantly increased inhibition at 86–110 pairing (p=0.024) and a tendency for increased inhibition at 80–120 pairing (p=0.086; Figure 5(a) and (b)). Overall inhibition analyses also supported this improvement (p=0.009). Although a visual inspection suggests an improved inhibition by HAL-F treatment, especially by the higher dose of HAL-F (1.5) treatment, pre-planned comparisons did not reveal a significant effect of HAL-F on PPI (p>0.05). Altogether, these findings suggest that continuous local HAL administration into the ventricle at a therapeutic dose improves sensorimotor gating. Such an improvement was less pronounced after continuous local HAL-F administration, suggesting reduced therapeutic effect of HAL-F compared with HAL.
Figure 5.

Haloperidol (HAL), but not its non-accumulating analog HAL-F, has antipsychotic effects in rodent models of psychosis: (a) amphetamine (AMPH)-hypersensitized animals show reduced pre-pulse inhibition (PPI) of an acoustic startle reflex. HAL enhances PPI in these animals, suggesting antipsychotic efficacy. Less PPI enhancement was observed after subchronic treatment with HAL-F as shown by percentage inhibition for each pre-pulse–pulse pair; (b) overall percentage PPI (background noise = 68 dB; p100–120: pulse intensities 100–120 dB; mean±SEM; n=7–15 per group; *p<0.05, #p<0.01 versus AMPH/VEH; VEH – vehicle); (c) HAL-F reached the brain parenchyma in a dose dependent way (**p<0.01); (d) Fluorescent labelled HAL-F (red) accumulating in the dorsal striatum after chronic treatment.
Dose-dependent brain penetrance of HAL-F in vivo
Although HAL-F does not accumulate in vesicles in vitro, for its D2 receptor action in vivo, it was necessary to reach the brain parenchyma. In order to test this, we used the fluorescent properties of HAL-F and imaged tissue distribution (Figure 5(c) and (d)). Results show that HAL-F reached the brain parenchyma at the dorsal striatum in a dose-dependent way with a higher concentration achieved by 1.5 compared with 0.3 mg/kg/day dose (t=−4.026, p=0.002).
Discussion
In this study, we asked whether the observed presynaptic accumulation and activity-dependent synaptic release of APDs after chronic treatment is required for their usually delayed therapeutic efficacy. We designed the HAL analog compound, HAL-F, which did not accumulate in the presynaptic vesicles of cultured neurons, but largely retained postsynaptic D2 receptor binding. We then compared its antipsychotic efficacy with therapeutic HAL treatment using rats in an induced psychotic-like state. We found that continuous HAL-F treatment is significantly less effective than HAL in the remission of psychotic-like symptoms in two behavioral tests in rats. These findings suggest that the presynaptic accumulation of HAL might play a significant role in its antipsychotic efficacy, and may explain the time course of its optimal action.
To the best of our knowledge, this is the first study that shows the contribution of presynaptic activities of APDs on their functional effects. We believe that the less pronounced therapeutic efficacy of HAL-F, compared with HAL, on psychosis-like behavior is caused by its inability to accumulate in presynaptic vesicles. We firstly confirmed the lack of presynaptic accumulation of HAL-F using LTG, a fluorescent surrogate for APDs (Freundt et al., 2007). Similar to APDs, LTG accumulated in the presynaptic vesicle in hippocampal cultured neurons, and was displaced by HAL, but not by HAL-F. Mechanistically, accumulated APDs have been shown to be released by action potential via exocytosis, which would inhibit calcium influx and further neurotransmitter release by inhibiting the upstream voltage-gated sodium channels (Ogata and Narahashi, 1989; Tischbirek et al., 2012). Furthermore, accumulated APDs have been shown to be co-released with dopamine at striatal terminals following activity (Tucker et al., 2015). Such bursts of dopamine neuron activity modulate salience and goal-directed behavior (Wightman and Heien, 2006). During acute psychosis, hyperdopaminergia within the striatum causes excessive salience being attributed to everyday stimuli, such as an eye contact or an unexpected sound. Such trivial events gain great meaning for the affected individuals, who attempt to explain these occurrences by mistaken beliefs (Kapur, 2003). Therefore, the co-release of dopamine and accumulated APDs ensures the inhibition of further presynaptic dopamine exocytosis and postsynaptic D2 receptor activity during salient events to prevent psychosis, as well as reduces off-target effects of APDs. Our behavioral findings are in line with these notions.
A possible limitation of this study is that HAL-F is a newly designed test compound with yet unknown affinity to non-dopaminergic receptors. Although it binds with high affinity to D2 receptor and prefers them largely over D1 receptors, the D2 receptor binding was attenuated compared with HAL. Although we tried to balance the difference in molecular weight and binding affinities for the treatment study, we cannot rule out that higher doses of HAL-F may yield antipsychotic efficacy in the behavioral tests. Furthermore, unlike HAL, HAL-F has a 2-fold higher affinity for D2S receptor compared with D2L receptor (Table 1). As D2S receptor is predominantly presynaptic (Beaulieu and Gainetdinov, 2011), its antagonism by HAL-F could cause a disinhibition of presynaptic dopaminergic neurons, causing an elevated dopamine release, thus, reducing the therapeutic efficacy of the analog compound. Alternatively, HAL-F showing negligible affinity for D4 receptor could also influence its therapeutic efficacy as D4 receptors are elevated in schizophrenia (Seeman et al., 1993) and many prescribed APDs, including HAL, antagonize it with varying affinities. These limitations should be taken into consideration in future studies.
AMPH sensitization–induced locomotor hyperactivity has been repeatedly utilized to model the psychotic symptoms of schizophrenia. Our sensitization paradigm caused locomotor hyperactivity, which was limited to the initial 20–30 min after the administration of the AMPH challenge, as we previously reported (Uzuneser et al., 2018). We believe that this limitation is associated with temporally distinct signaling cascades that are activated by the dopamine D2 receptors. The D2 receptor–mediated canonical G-protein–dependent cascade has been shown to induce short-lasting effects (15–25 min), whereas the D2 receptor–mediated non-canonical signaling cascade, which leads to the inhibition of protein kinase B (Akt) and resulting disinhibition of glycogen synthase kinase 3 (GSK-3), induces progressive and longer-lasting effects (up to 2 h) on dopamine-related behavior (Beaulieu and Gainetdinov, 2011; Beaulieu et al., 2005, 2007). Both of these cascades are regulated by APDs (Sutton et al., 2007; Sutton and Rushlow, 2011). Because our AMPH-induced locomotion test duration was 40 min, we cannot make conclusions on the effects of HAL or HAL-F on the longer-lasting D2 receptor–mediated non-canonical cascade. Still, our findings show that the short-lasting AMPH-induced locomotor and rearing activities were attenuated by ventricular HAL delivery. Our previous findings demonstrated a similar attenuation in psychotic-like animals after 7-day peripheral HAL (Uzuneser et al., 2018), aripiprazole (Möller et al., 2017), and olanzapine (Groos et al., 2018) administration in rodent models. At either dose tested, HAL-F delivery did not attenuate the AMPH-induced motor behavior, implying deficient antipsychotic efficacy on the G-protein–dependent cascade. Such a deficiency hints to the involvement of APD accumulation and an activity-dependent APD co-release with dopamine on the attenuation of short-lasting canonical dopamine D2 receptor–mediated signaling mechanisms.
Attenuation of the PPI of the acoustic startle response is widely considered as an endophenotype of schizophrenia with a high translational appeal (Powell et al., 2012). We observed a partial recovery of PPI by HAL-F treatment, which appeared to be dose dependent. Still, HAL treatment more robustly improved PPI, suggesting an inferior antipsychotic effect of HAL-F on the aversively motivated sensorimotor gating mechanism. Our previous post mortem neurochemical findings hinted dorsal hippocampal dopaminergic neurotransmission as a modulator of PPI (Uzuneser et al., 2018). Additionally, gating of auditory-evoked responses is mediated by dorsal hippocampus, and modulated by AMPH, quinpirole, and HAL in rats (Bickford-Wimer et al., 1990; Ellenbroek et al., 2002). Thus, the improvement of PPI by HAL or HAL-F could be mediated by the normalization of dorsal hippocampal dopaminergic activity. The dorsal hippocampus receives dopaminergic afferents mainly from locus coeruleus, which is activated in response to environmental novelty that habituates over time (Takeuchi et al., 2016; Vankov et al., 1995). Therefore, it is possible that the lack of phasic dorsal hippocampal dopamine activity during the PPI test rendered the activity-dependent co-release of accumulated APDs unnecessary. This putative scenario could explain the partial and dose-dependent recovery of PPI by the non-accumulating HAL-F.
Clinical findings suggest that chronically HAL-administered patients, even after a prolonged discontinuation period, still show clinical effects (Kornhuber et al., 1999). Furthermore, HAL can still be detected in their plasma after a month of drug-free time (de Leon et al., 2004). Accumulation of HAL and its action potential–dependent release can explain this phenomenon. Furthermore, the augmentation of APD responses by combined use of electroconvulsive therapy improves the symptoms of treatment-resistant schizophrenia patients (Falkai et al., 2005; Petrides et al., 2014), further emphasizing the significance of action potential–dependent release of accumulated APDs.
APDs have long been considered to lead to substantial clinical improvements after several weeks of intake, an observation named as “delayed onset of antipsychotic efficacy.” This delayed efficacy was explained by the “depolarization block” hypothesis, which suggests that the firing of dopaminergic neurons in the brain is inactivated after repeated APD administration, which is crucial for mediating the antipsychotic response (Grace, 1992; Grace et al., 1997). APD efficiency has been shown to correlate with the induction of chronic depolarization block in dopaminergic neurons in animal models (Grace and Bunney, 1986; Valenti et al., 2011). Depolarization block in substantia nigra neurons has been shown to be modulated by the inhibition of somatic voltage-gated sodium channels (Tucker et al., 2012), which are also inhibited by APDs after their accumulation and activity-dependent release (Tischbirek et al., 2012). Although the idea of delayed onset of APDs was later challenged by studies claiming that antipsychotic response starts within the first few days after administration (Agid et al., 2003, 2006; Leucht et al., 2005), it is still widely accepted that the initial benefit increases over weeks of treatment (Pickar et al., 1984). Such gradual development of antipsychotic efficacy can be caused by the necessity of tissue accumulation of APDs for maximal therapeutic action, possibly contributing to depolarization block. Our findings are doubtlessly in line with these observations. Alternatively, altered gene expression or remodeled circuitries in critical brain regions by APD treatment might also explain slow development of full clinical action by APDs. It is possible that reduced therapeutic efficacy of HAL-F is caused by its failure to alter gene expression or to induce synaptic adaptations as HAL does. It should also be noted that although APDs gradually improve their efficacy, APD-induced treatment failure occurs after prolonged treatment (Amato et al., 2020; Samaha et al., 2007; Uzuneser et al., 2018). The mechanism of failure is still debatable. It is intriguing to speculate the possible involvement of disrupted APD accumulation after prolonged treatment as a novel mechanism of failure.
In conclusion, our findings indicate that presynaptic accumulation of the APD HAL may be a critical determinant of its antipsychotic efficacy. This finding is in line with the depolarization block hypothesis, suggesting that APDs function not only by blocking the postsynaptic dopamine D2 receptors, but also by inhibiting the depolarization of presynaptic dopamine neurons. Furthermore, accumulation and action potential–dependent release of APDs can explain clinical phenomena such as prolonged clinical effects of APDs after the offset of their intake, improved therapeutic effects of APDs against treatment-resistant schizophrenia when combined with electroconvulsive therapy, and slow development of full clinical action of APDs. Modulating accumulation properties of APDs with preserved dopamine D2 receptor action, may, thus, be a new strategy to improve antipsychotic efficacy.
Supplemental Material
Supplemental material, sj-pdf-1-jop-10.1177_0269881120965908 for Presynaptic vesicular accumulation is required for antipsychotic efficacy in psychotic-like rats by Taygun C Uzuneser, Eva-Maria Weiss, Jana Dahlmanns, Liubov S Kalinichenko, Davide Amato, Johannes Kornhuber, Christian Alzheimer, Jan Hellmann, Jonas Kaindl, Harald Hübner, Stefan Löber, Peter Gmeiner, Teja W Grömer and Christian P Müller in Journal of Psychopharmacology
Acknowledgments
We thank Benedikt Quinger for his excellent technical support. The present work was conducted in partial fulfilment of the requirements to obtain the degree “Dr. hum. biol.” for TCU.
Footnotes
Author contributions: TWG and CPM conceived the study. TWG, CPM, SL, and PG designed the HAL-F compound. JH, SL, and HH synthesized the compound and evaluated binding affinities. J Kaindl performed the molecular modeling studies. EMW, JD, J Kornhuber, CA, and TWG evaluated presynaptic accumulation. TCU, DA, LSK, and CPM tested behavioral effects. TCU, SL, HH, and CPM interpreted the experiments and wrote the manuscript.
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the German Research Foundation (Deutsche Forschungsgemeinschaft) grants MU 2789/7-1, MU2789/7-2, GR 4549/1-1, and AL 294/10-1.
ORCID iDs: Christian Alzheimer  https://orcid.org/0000-0003-3910-5072
https://orcid.org/0000-0003-3910-5072
Jan Hellmann
https://orcid.org/0000-0002-6710-431X
Christian P Müller
https://orcid.org/0000-0002-5325-9900
Supplemental material: Supplemental material for this article is available online.
References
- Agid O, Seeman P, Kapur S. (2006) The “delayed onset” of antipsychotic action—An idea whose time has come and gone. J Psychiatry Neurosci 31: 93–100. [PMC free article] [PubMed] [Google Scholar]
- Agid O, Kapur S, Arenovich T, et al. (2003) Delayed-onset hypothesis of antipsychotic action: A hypothesis tested and rejected. Arch Gen Psychiatry 60: 1228–1235. [DOI] [PubMed] [Google Scholar]
- Amato D, Canneva F, Cumming P, et al. (2020) A dopaminergic mechanism of antipsychotic drug efficacy, failure, and failure reversal: The role of dopamine transporter. Mol Psychiatry 25: 2101–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreassen OA, Jorgensen HA. (1994) Tardive dyskinesia: Behavioural effects of repeated intracerebroventricular haloperidol injections in rats do not confirm the kindling hypothesis. Pharmacol Biochem Behav 49: 309–312. [DOI] [PubMed] [Google Scholar]
- Asghari V, Sanyal S, Buchwaldt S, et al. (1995) Modulation of intracellular cyclic AMP levels by different human dopamine D4 receptor variants. J Neurochem 65: 1157–1165. [DOI] [PubMed] [Google Scholar]
- Ayd FJ. (1978) Haloperidol: Twenty years’ clinical experience. J Clin Psychiatry 39: 807–814. [PubMed] [Google Scholar]
- Banks WA. (2016) From blood-brain barrier to blood-brain interface: New opportunities for CNS drug delivery. Nat Rev Drug Discov 15: 275–292. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Gainetdinov RR. (2011) The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev 63: 182–217. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Gainetdinov RR, Caron MG. (2007) The Akt-GSK-3 signaling cascade in the actions of dopamine. Trends Pharmacol Sci 28: 166–172. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Sotnikova TD, Marion S, et al. (2005) An Akt/β-arrestin2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122: 261–273. [DOI] [PubMed] [Google Scholar]
- Bickford-Wimer PC, Nagamoto H, Johnson R, et al. (1990) Auditory sensory gating in hippocampal neurons: A model system in the rat. Biol Psychiatry 27: 183–192. [DOI] [PubMed] [Google Scholar]
- Carey RJ. (1987) Conditioning and the delayed onset of a haloperidol-induced behavioral effect. Biol Psychiatry 22: 269–277. [DOI] [PubMed] [Google Scholar]
- Cheng YC, Prusoff WH. (1973) Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 22: 3099–3108. [DOI] [PubMed] [Google Scholar]
- de Duve C, de Barsy T, Poole B, et al. (1974) Commentary: Lysosomotropic agents. Biochem Pharmacol 23: 2495–2531. [DOI] [PubMed] [Google Scholar]
- de Leon J, Diaz FJ, Wedlund P, et al. (2004) Haloperidol half-life after chronic dosing. J Clin Psychopharmacol 24: 656–660. [DOI] [PubMed] [Google Scholar]
- Ellenbroek BA, Lubbers LJ, Cools AR. (2002) The role of hippocampal dopamine receptors in prepulse inhibition. Eur J Neurosci 15: 1237–1243. [DOI] [PubMed] [Google Scholar]
- Falkai P, Wobrock T, Lieberman J, et al. (2005) World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 1: Acute treatment of schizophrenia. World J Biol Psychiatry 6: 132–191. [DOI] [PubMed] [Google Scholar]
- Fendt M, Fanselow MS. (1999) The neuroanatomical and neurochemical basis of conditioned fear. Neurosci Biobehav Rev 23: 743–760. [DOI] [PubMed] [Google Scholar]
- Freundt EC, Czapiga M, Lenardo MJ. (2007) Photoconversion of Lysotracker Red to a green fluorescent molecule. Cell Res 17: 956–958. [DOI] [PubMed] [Google Scholar]
- Gemperle AY, Enz A, Pozza MF, et al. (2003) Effects of clozapine, haloperidol and iloperidone on neurotransmission and synaptic plasticity in prefrontal cortex and their accumulation in brain tissue: An in vitro study. Neuroscience 117: 681–695. [DOI] [PubMed] [Google Scholar]
- Grace AA. (1992) The depolarization block hypothesis of neuroleptic action: Implications for the etiology and treatment of schizophrenia. J Neural Transm Suppl 36: 91–131. [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. (1986) Induction of depolarization block in midbrain dopamine neurons by repeated administration of haloperidol: Analysis using in vivo intracellular recording. J Pharmacol Exp Ther 238: 1092–1100. [PubMed] [Google Scholar]
- Grace AA, Bunney BS, Moore H, et al. (1997) Dopamine-cell depolarization block as a model for the therapeutic actions of antipsychotic drugs. Trends Neurosci 20: 31–37. [DOI] [PubMed] [Google Scholar]
- Groos D, Zheng F, Kornhuber J, et al. (2018) Chronic antipsychotic treatment targets GIRK current suppression, loss of long-term synaptic depression and behavioral sensitization in a mouse model of amphetamine psychosis. J Psychopharmacol 33: 74–85. [DOI] [PubMed] [Google Scholar]
- Hanwell MD, Curtis DE, Lonie DC, et al. (2012) Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J Cheminform 4: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes G, Biden TJ, Selbie LA, et al. (1992) Structural subtypes of the dopamine D2 receptor are functionally distinct: Expression of the cloned D2A and D2B subtypes in a heterologous cell line. Mol Endocrinol 6: 920–926. [DOI] [PubMed] [Google Scholar]
- Hübner H, Haubmann C, Utz W, et al. (2000) Conjugated enynes as nonaromatic catechol bioisosteres: Synthesis, binding experiments, and computational studies of novel dopamine receptor agonists recognizing preferentially the D3 subtype. J Med Chem 43: 756–762. [DOI] [PubMed] [Google Scholar]
- Hübner H, Schellhorn T, Gienger M, et al. (2016) Structure-guided development of heterodimer-selective GPCR ligands. Nat Commun 7: 12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur S. (2003) Psychosis as a state of aberrant salience: A framework linking biology, phenomenology, and pharmacology in schizophrenia. Am J Psychiatry 160: 13–23. [DOI] [PubMed] [Google Scholar]
- Kapur S, Mizrahi R, Li M. (2005) From dopamine to salience to psychosis—Linking biology, pharmacology and phenomenology of psychosis. Schizophr Res 79: 59–68. [DOI] [PubMed] [Google Scholar]
- Kapur S, Vanderspek SC, Brownlee BA, et al. (2003) Antipsychotic dosing in preclinical models is often unrepresentative of the clinical condition: A suggested solution based on in vivo occupancy. J Pharmacol Exp Ther 305: 625–631. [DOI] [PubMed] [Google Scholar]
- Kornhuber J, Henkel AW, Groemer TW, et al. (2010) Lipophilic cationic drugs increase the permeability of lysosomal membranes in a cell culture system. J Cell Physiol 224: 152–164. [DOI] [PubMed] [Google Scholar]
- Kornhuber J, Schultz A, Wiltfang J, et al. (1999) Persistence of haloperidol in human brain tissue. Am J Psychiatry 156: 885–890. [DOI] [PubMed] [Google Scholar]
- Leucht S, Busch R, Hamann J, et al. (2005) Early onset hypothesis of antipsychotic drug action: A hypothesis tested, confirmed and extended. Biol Psychiatry 57: 1543–1549. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, et al. (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193: 265–275. [PubMed] [Google Scholar]
- Miyamoto S, Duncan GE, Marx CE, et al. (2005) Treatments for schizophrenia: A critical review of pharmacology and mechanisms of action of antipsychotic drugs. Mol Psychiatry 10: 79–104. [DOI] [PubMed] [Google Scholar]
- Möller D, Banerjee A, Uzuneser TC, et al. (2017) Discovery of G protein-biased dopaminergics with a pyrazolo[1,5-a]pyridine substructure. J Med Chem 60: 2908–2929. [DOI] [PubMed] [Google Scholar]
- Ogata N, Narahashi T. (1989) Block of sodium channels by psychotropic drugs in single guinea-pig cardiac myocytes. Br J Pharmacol 97: 905–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. (2014) The Rat Brain in Stereotaxic Coordinates. San Diego: Academic Press. [Google Scholar]
- Peleg-Raibstein D, Sydekum E, Feldon J. (2006) Differential effects on prepulse inhibition of withdrawal from two different repeated administration schedules of amphetamine. Int J Neuropsychopharmacol 9: 737–749. [DOI] [PubMed] [Google Scholar]
- Petrides G, Malur C, Braga RJ. (2014) Electroconvulsive therapy augmentation in clozapine-resistant schizophrenia: A prospective, randomized study. Am J Psychiatry 172: 52–58. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, et al. (2004) UCSF Chimera—A visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612. [DOI] [PubMed] [Google Scholar]
- Pickar D, Labarca R, Linnoila M, et al. (1984) Neuroleptic-induced decrease in plasma homovanillic acid and antipsychotic activity in schizophrenic patients. Science 225: 954–957. [DOI] [PubMed] [Google Scholar]
- Powell SB, Weber M, Geyer MA. (2012) Genetic models of sensorimotor gating: Relevance to neuropsychiatric disorders. Curr Top Behav Neurosci 12: 251–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayport S, Sulzer D. (1995) Visualization of antipsychotic drug binding to living mesolimbic neurons reveals D2 receptor, acidotropic, and lipophilic components. J Neurochem 65: 691–703. [DOI] [PubMed] [Google Scholar]
- Samaha AN, Reckless GE, Seeman P, et al. (2008) Less is more: Antipsychotic efficacy is greater with transient rather than continuous drug delivery. Biol Psychiatry 64: 145–152. [DOI] [PubMed] [Google Scholar]
- Samaha AN, Seeman P, Stewart J, et al. (2007) “Breakthrough” dopamine supersensitivity during ongoing antipsychotic treatment leads to treatment failure over time. J Neurosci 27: 2979–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Mendoza EH, Carballo J, Longart M, et al. (2016) Implantation of miniosmotic pumps and delivery of tract tracers to study brain reorganization in pathophysiological conditions. J Vis Exp 18: e52932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sbalzerini IF, Koumoutsakos P. (2005) Feature point tracking and trajectory analysis for video imaging in cell biology. J Struct Biol 151: 182–195. [DOI] [PubMed] [Google Scholar]
- Seeman P, Guan HC, van Tol HHM. (1993) Dopamine D4 receptors elevated in schizophrenia. Nature 365: 441–445. [DOI] [PubMed] [Google Scholar]
- Shapovalov MV, Dunbrack RL., Jr (2011) A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 19: 844–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokoloff P, Giros B, Martres MP, et al. (1990) Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 347: 146–151. [DOI] [PubMed] [Google Scholar]
- Sutton LP, Rushlow WJ. (2011) The dopamine D2 receptor regulates Akt and GSK-3 via Dvl-3. Int J Neuropsychopharmacol 22: 1–15. [DOI] [PubMed] [Google Scholar]
- Sutton LP, Honardoust D, Mouyal J, et al. (2007) Activation of the canonical Wnt pathway by the antipsychotics haloperidol and clozapine involves dishevelled-3. J Neurochem 102: 153–169. [DOI] [PubMed] [Google Scholar]
- Takeuchi T, Duszkiewicz AJ, Sonneborn A, et al. (2016) Locus coeruleus and dopaminergic consolidation of everyday memory. Nature 537: 357–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischbirek CH, Wenzel EM, Zheng F, et al. (2012) Use-dependent inhibition of synaptic transmission by the secretion of intravesicularly accumulated antipsychotic drugs. Neuron 74: 830–844. [DOI] [PubMed] [Google Scholar]
- Trapp S, Rosania GR, Horobin RW, et al. (2008) Quantitative modeling of selective lysosomal targeting for drug design. Eur Biophys J 37: 1317–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O, Olson AJ. (2010) AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31: 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker KR, Block ER, Levitan ES. (2015) Action potentials and amphetamine release antipsychotic drug from dopamine neuron synaptic VMAT vesicles. Proc Natl Acad Sci USA 112: 4485–4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker KR, Huertas MA, Horn JP, et al. (2012) Pacemaker rate and depolarization block in nigral dopamine neurons: A somatic sodium channel balancing act. J Neurosci 32: 14519–14531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzuneser TC, Schindehütte M, Dere E, et al. (2018) Schizophrenia dimension-specific antipsychotic drug action and failure in amphetamine-sensitized psychotic-like rats. Eur Neuropsychopharmacol 28: 1382–1393. [DOI] [PubMed] [Google Scholar]
- Uzuneser TC, Speidel J, Kogias G, et al. (2019) Disrupted-in-schizophrenia 1 (DISC1) overexpression and juvenile immune activation cause sex-specific schizophrenia-related psychopathology in rats. Front Psychiatry 10: 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenti O, Cifelli P, Gill KM, et al. (2011) Antipsychotic drugs rapidly induce dopamine neuron depolarization block in a developmental rat model of schizophrenia. J Neurosci 31: 12330–12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vankov A, Herve-Minvielle A, Sara SJ. (1995) Response to novelty and its rapid habituation in locus coeruleus neurons of the freely exploring rat. Eur J Neurosci 7: 1180–1187. [DOI] [PubMed] [Google Scholar]
- van Rossum JM. (1966) The significance of dopamine-receptor blockade for the action of neuroleptic drugs. Arch Int Pharmacodyn Ther 160: 492–494. [PubMed] [Google Scholar]
- Versteeg DH, Florijn WJ, Holtmaat AJ, et al. (1993) Synchronism of pressor response and grooming behavior in freely moving, conscious rats following intracerebroventricular administration of ACTH/MSH-like peptides. Brain Res 631: 265–269. [DOI] [PubMed] [Google Scholar]
- Walters SH, Levitan ES. (2019) Vesicular antipsychotic drug release evokes an extra phase of dopamine transmission. Schizophr Bull. Epub ahead of print 29 July 2019. DOI: 10.1093/schbul/sbz085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Che T, Levit A, et al. (2018) Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone. Nature 555: 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wightman RM, Heien ML. (2006) Phasic dopamine signaling during behavior, reward, and disease states. CNS Neurol Disord Drug Targets 5: 99–108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-pdf-1-jop-10.1177_0269881120965908 for Presynaptic vesicular accumulation is required for antipsychotic efficacy in psychotic-like rats by Taygun C Uzuneser, Eva-Maria Weiss, Jana Dahlmanns, Liubov S Kalinichenko, Davide Amato, Johannes Kornhuber, Christian Alzheimer, Jan Hellmann, Jonas Kaindl, Harald Hübner, Stefan Löber, Peter Gmeiner, Teja W Grömer and Christian P Müller in Journal of Psychopharmacology