

SUMMARY
Autophagy is an intracellular degradation system, but its physiological functions in vertebrates are not yet fully understood. Here, we show that autophagy is required for inflation of air-filled organs: zebrafish swim bladder and mouse lung. In wild-type zebrafish swim bladder and mouse lung type II pulmonary epithelial cells, autophagosomes are formed and frequently fuse with lamellar bodies. The lamellar body is a lyso-some-related organelle that stores a phospholipid-containing surfactant complex that lines the air-liquid interface and reduces surface tension. We find that autophagy is critical for maturation of the lamellar body. Accordingly, atg-deficient zebrafish fail to maintain their position in the water, and type-II-pneumocyte-specific Fip200-deficient mice show neonatal lethality with respiratory failure. Autophagy suppression does not affect synthesis of the surfactant phospholipid, suggesting that autophagy supplies lipids and membranes to lamellar bodies. These results demonstrate an evolutionarily conserved role of autophagy in lamellar body maturation.
In Brief
Physiological functions of macroautophagy remain not fully understood in vertebrates. Morishita et al. demonstrate that autophagy is required for inflation of air-filled organs: zebrafish swim bladder and mouse lung. In the swim bladder and type II pulmonary epithelial cells, autophagosomes fuse with lamellar bodies, which drives lamellar body maturation.
Graphical Abstract
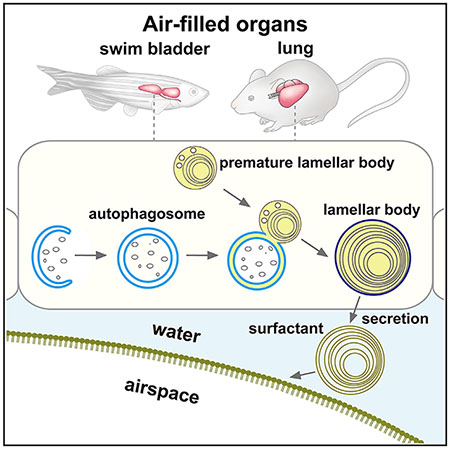
INTRODUCTION
Macroautophagy (hereafter “autophagy”) is an intracellular degradation system that delivers cytoplasmic materials to the lysosome via the autophagosome (Søreng et al., 2018; Tooze and Yoshimori, 2010). Autophagosome formation requires evolutionarily conserved autophagy-related (ATG) proteins that can be classified into six functional units: the UNC-51-like kinase (ULK) complex, ATG9, class III phosphatidylinositol 3-kinase (PI3K) complex I, the WD-repeat protein interacting with phosphoinositide (WIPI)-ATG2 complex, the ATG12 conjugation system, and the ATG8/LC3 conjugation system (Mizushima, 2018; Nakatogawa, 2020). Additional proteins, which are not found in the yeast Saccharomyces cerevisiae, are also required for autophagy, including ATG11, EI24, EPG5, TMEM41B, and vacuole membrane protein 1 (VMP1) (Mizushima, 2018; Moretti et al., 2018; Morita et al., 2018; Shoemaker et al., 2019; Tian et al., 2010). Studies using ATG-deficient cells and organisms have revealed two major functions of autophagy: (1) self-nourishment and metabolic recycling and (2) elimination of unwanted or excess materials (Levine and Kroemer, 2019; Mizushima and Komatsu, 2011; Morishita and Mizushima, 2019). However, the physiological functions of autophagy in vertebrates remain not fully understood.
The lung and swim bladder, an anatomical homolog of the lung in fish, are air-filled organs whose expansion requires a surfactant that lowers the surface tension at the air-liquid interface and prevents collapse of the airspace (Bowman et al., 2019; Chen et al., 2018; Daniels et al., 2004; Olmeda et al., 2017; Pérez-Gil, 2008; Robertson et al., 2014). This surfactant is composed of approximately 90% lipids (mostly phospholipids) and 10% proteins (i.e., surfactant proteins and acid hydrolases) (Agassandian and Mallampalli, 2013). Before secretion, the surfactant is stored in the lamellar body, a lysosome-related organelle, in epithelial cells in the lung (type II pneumocytes) and swim bladder (Bowman et al., 2019; Chen et al., 2018; Robertson et al., 2014). In these cells, surfactant lipids are synthesized in the endoplasmic reticulum (ER) and transported to the lumen of the multivesicular body/late endosome, the origin of the lamellar body, by mechanisms that have not been fully understood (Andreeva et al., 2007; Pérez-Gil, 2008). An ATP-binding cassette transporter A3 (ABCA3), a putative lipid transporter on the limiting membrane of the lamellar body, is critical for lamellar body formation (Ban et al., 2007; Cheong et al., 2007; Fitzgerald et al., 2007; Rindler et al., 2017; Shulenin et al., 2004).
An alternative mechanism for surfactant lipid incorporation into lamellar bodies might occur via autophagy. The possible involvement of autophagy has been suggested by in vitro studies using a transformed mink alveolar epithelial cell line that expresses exogenous β-1,6-N-acetylglucosaminyl-transferase V (GlcNAc-TV) (Hariri et al., 2000; Lajoie et al., 2005). The incorporation of cytoplasmic components and phospholipids into lamellar-body-like structures was inhibited by 3-methyladenine, an inhibitor of class III PI3K (Hariri et al., 2000; Lajoie et al., 2005). However, because class III PI3K is essential not only for autophagy but also for the endocytic pathway involving the formation of the multivesicular body (Funderburk et al., 2010; Futter et al., 2001), it has not yet been directly demonstrated whether autophagy is indeed required for lamellar body formation, particularly in vivo. Therefore, it is imperative to use a direct genetic approach to assess the role of autophagy.
In this study, we performed a phenotypic screening of zebrafish lacking representative major atg genes and found that atg genes are required for inflation of the swim bladder and maturation of lamellar bodies in swim bladder epithelial cells. In these cells, autophagosomes are formed and frequently fuse with lamellar bodies. This role of autophagy is conserved in the lungs of mice. Our results demonstrate an evolutionarily conserved role of autophagy in lamellar body maturation, which is likely the supply of lipids and/or membranes to lamellar bodies.
RESULTS
Loss of atg Genes in Zebrafish Causes Larval Lethality
To reveal the physiological functions of autophagy in vertebrates, we generated a series of zebrafish lacking atg genes using the CRISPR/Cas9 system (Figure S1). We targeted genes corresponding to at least one subunit of each autophagy complex/unit (Figure S1A): fip200/rb1cc1, atg101, and atg13 of the ULK complex; atg9a and atg9b; atg14, a component of PI3K complex I that is essential for autophagy but not the endocytic pathway; atg2a and atg2b of the ATG2–WIPI complex; atg16l1 and atg5 of the ATG conjugation systems; and vmp1. Almost all heterozygous atg-deficient zebrafish were healthy and fertile (Figures 1A and 1B). In contrast, zebrafish with homozygous deficiencies for upstream atg genes that function at the initiation and nucleation steps (fip200−/−, atg13−/−, atg101−/−, atg2a−/−atg2b−/−, atg14atg9a−/−atg9b−/−, and vmp1−/−) generally died by 12 days post-fertilization (dpf) (Figures 1A and 1C). Zebrafish deficient for atg genes encoding the ATG conjugation system components (atg16l1−/− and atg5−/−) also died, but at a slightly later time, around 13 dpf (Figures 1B and 1C). Single deletions of atg2a, atg2b, atg9a, or atg9b did not result in larval lethality (likely due to functional redundancy), as reported in mammalian cell lines (Velikkakath et al., 2012; Yamada et al., 2005). These results suggest that atg genes are essential for survival during larval periods in zebrafish and indicate that zebrafish lacking upstream atg genes died earlier than those lacking genes encoding Atg conjugation system components, as observed in Atg-deficient mice (Kuma et al., 2017).
Figure 1. atg-Gene-Deficient Zebrafish Show Larval Lethality and Defective Inflation of the Swim Bladder.
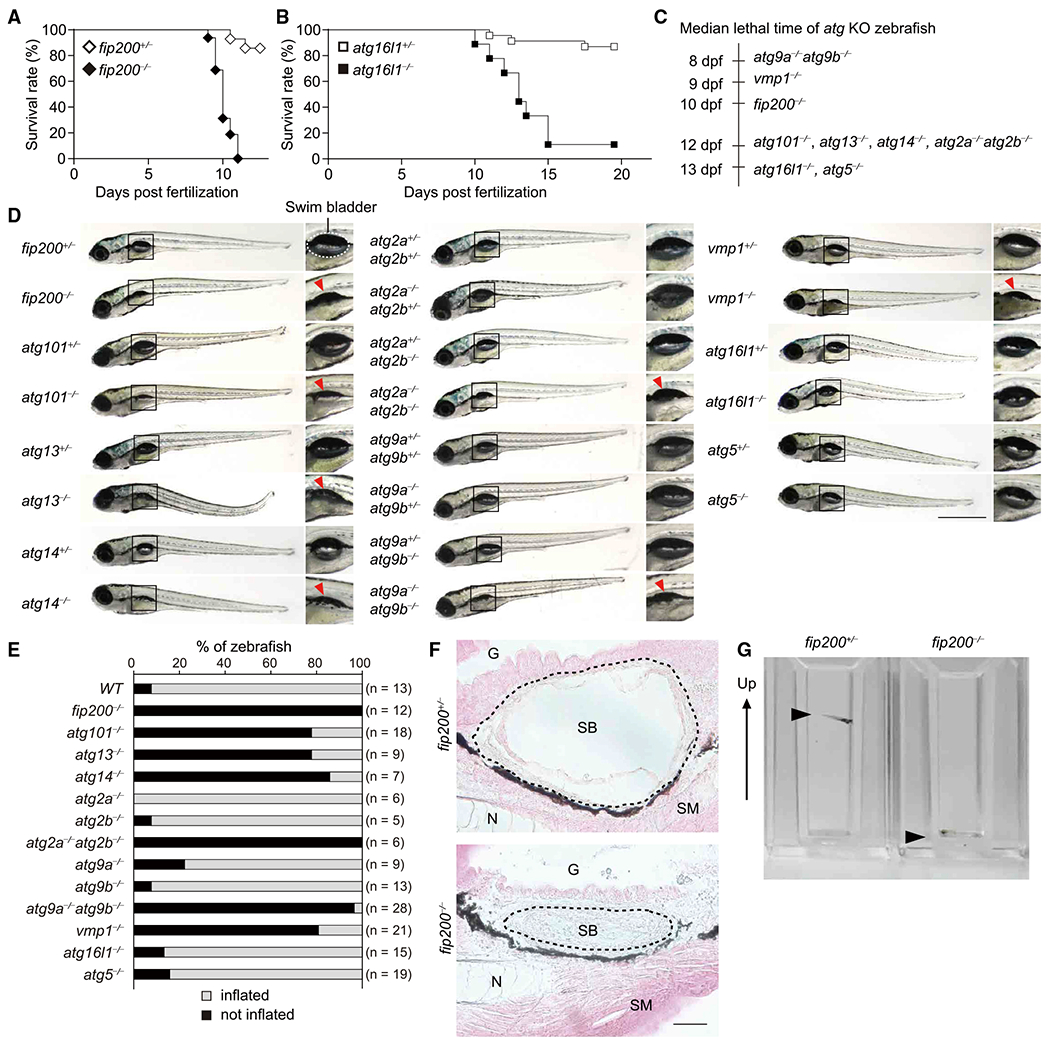
(A and B) Survival rate (%) of fip200+/− (n = 28), fip200−/− (n = 16) (A), atg16l1+/− (n = 23), and atg16l1−/− (n = 9) (B) zebrafish.
(C) Median lethal time of atg9a−/−atg9b−/− (n = 24), vmp1−/− (n = 11), fip200−/− (n = 16), atg101−/− (n = 12), atg13−/− (n = 15), atg14−/− (n = 5), atg2a−/−atg2b−/− (n = 6), atg16l1−/− (n = 9), and atg5−/− (n = 15) zebrafish. dpf, days post-fertilization.
(D) External appearance of atg-deficient zebrafish at 9 dpf(atg9 mutant zebrafish were examined at 6 dpf). Magnified images of the indicated regionsareshown in the right panels. A swim bladder is indicated with a dashed line in fip200+/− zebrafish. Uninflated swim bladders are indicated by red arrowheads. Data are representative of two to four independent experiments. Scale bar, 1 mm.
(E) Rate (%) of inflated (white column) and uninflated (black column) zebrafish swim bladders for each genotype at 9 dpf(atg9 mutant zebrafish were examined at 6 dpf).
(F) Hematoxylin and eosin staining of sagittal sections of 9-dpf fip200+/− and fip200−/− zebrafish. G, gut; N, notochord; SB, swim bladder (indicated by dashed lines); SM, skeletal muscle. Scale bar, 20 μm.
(G) Behavioral analysis of 6-dpf fip200+/− and fip200−/− zebrafish in cuvettes. The arrow indicates the upward direction of the cuvettes. The arrowheads indicate zebrafish. The video from which these stills images were captured is available as Video S1.
Loss of atg Genes in Zebrafish Causes Defective Inflation of the Swim Bladder
Gross examination revealed that atg-deficient zebrafish showed no obvious defects in body growth during development (Figure 1D). Only vmp1−/− zebrafish showed abnormal deposits in the abdomen (Figure 1D), owing to the accumulation of neutral lipids in the intestine and liver (Morishita et al., 2019). Expanded swim bladders, which are inflated between the end of 4 dpf and early 5 dpf just prior to the first feeding (Winata et al., 2009), were observed in wild-type and heterozygous atg mutant zebrafish (Figures 1D and 1E). However, although the swim bladder was developed, defective inflation was observed in zebrafish lacking upstream atg genes (flip200−/−, atg13−/−, atg101−/−, atg2a−/−atg2b−/−, atg14−/−, atg9a−/−atg9b−/−, and vmp1−/−) throughout the larval stages (Figures 1D and 1E). Histological analysis revealed the presence of an airspace in the lumen of the swim bladder in fip200+/− zebrafish (Figure 1F). In fip200−/− zebrafish, however, the airspace surrounded by epithelial cells was collapsed (Figure 1F), suggesting that it was the inflation but not the development of the swim bladder that was impaired. The swim bladder is required for acquisition of buoyancy to maintain a vertical position in the water at a desired depth (Daniels et al., 2004; Winata et al., 2009). Behavioral analysis revealed that fip200+/− zebrafish could float up and maintain their position, but fip200−/− zebrafish could not (Figure 1G; Video S1). Similar behavioral defects were observed in homozygous mutants of the other upstream atg genes that are associated with defective inflation of the swim bladder (data not shown). Zebrafish deficient for genes encoding Atg conjugation system components (atg16l1 and atg5), which are critical but not absolutely essential for autophagosome formation and degradation (Nguyen et al., 2016; Tsuboyama et al., 2016), did not exhibit defects in inflation of the swim bladder (Figures 1D and 1E). These results suggest that complete suppression of autophagic flux owing to depletion of upstream Atg factors but not Atg conjugation systems causes a defect in inflation of the swim bladder.
Defective Maturation of Lamellar Bodies in Swim Bladder Epithelial Cells in fip200-Deficient Zebrafish
We next investigated whether autophagosome formation occurred in swim bladder epithelial cells using microtubule-associated protein light chain 3B (LC3B) as an autophagosome marker (Kabeya et al., 2000). In swim bladder epithelial cells of fip200+/− zebrafish, GFP-LC3B puncta were observed at 4 dpf, when the swim bladder had not yet been inflated (Figure 2A). GFP-LC3B puncta were not detected in the swim bladder of fip200−/− zebrafish (Figure 2A), suggesting that the observed GFP-LC3B puncta in fip200+/− zebrafish are autophagic structures. It is unlikely that these GFP-LC3B puncta are phagosomes formed in the process of LC3-associated phagocytosis (LAP) because LAP does not require the ULK1 complex (FIP200 in mammals [Martinez et al., 2015], Atg13 in zebrafish [Masud et al., 2019], and Atg1/UNC-51 in C. elegans [Fazeli et al., 2016]). These GFP-LC3B puncta were frequently colocalized or attached to large LAMP1-RFP-positive structures (Figure 2B), which likely represent surfactant-containing lamellar bodies (Bowman et al., 2019; Chen et al., 2018; Robertson et al., 2014) because the lamellar bodies are lysosome-related organelles and mostly detected as large LAMP1-positive structures in type II pneumocytes in the lung (Andreeva et al., 2007; Kook et al., 2016; Miklavc et al., 2014; Pérez-Gil, 2008; Wasano and Hirakawa, 1994; Yamano et al., 2001). However, in fip200−/− zebrafish, the size and number of LAMP1-GFP-positive structures in swim bladder epithelial cells decreased relative to wild-type zebrafish (Figure 2C). These results suggest that autophagy is required for maturation of lamellar bodies in swim bladder epithelial cells of zebrafish.
Figure 2. Defective Maturation of Lamellar Bodies in Swim Bladder Epithelial Cells in fip200-Deficient Zebrafish.
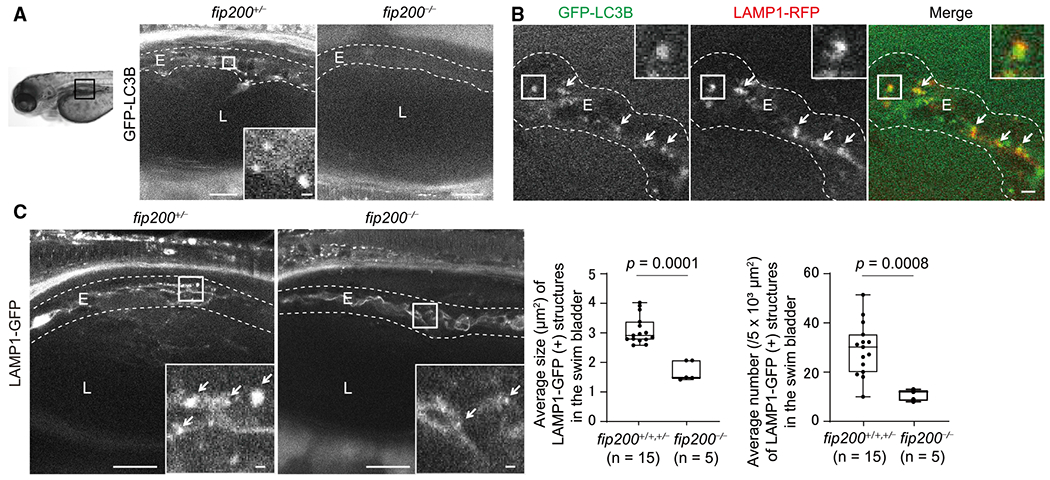
(A) Representative images of GFP-LC3B signals in swim bladders of 4-dpf fip200+/− and fip200−/− zebrafish expressing GFP-LC3B. Scale bar, 20 μm and 1 μm in the inset.
(B) Representative images of GFP-LC3B and LAMP1-RFP (lamellar bodies and endolysosomes) signals in swim bladders of 4-dpf wild-type zebrafish expressing GFP-LC3B and LAMP1-RFP. Arrows indicate LC3B- and LAMP1-positive structures. Scale bar, 2 μm.
(C) Representative images of LAMP1-GFP signals in swim bladders of 4-dpf fip200+/− and fip200−/− zebrafish expressing LAMP1-GFP. Arrows indicate LAMP1-positive structures. Scale bar, 20 μm and 1 μm in the inset. The average size and number of lamellar bodies were quantified and are shown in the graphs. The solid bars and boxes indicate the median and interquartile range (25th to 75th percentile), respectively. The whiskers indicate the largest and smallest values. Differences were statistically analyzed by unpaired two-tailed Mann-Whitney U tests. E, epithelial cells; L, lumen of the swim bladder.
Autophagosomes Fuse with Lamellar Bodies in Type II Pneumocytes in the Mouse Lung
Because the swim bladder is an anatomical homolog of the lung (Bowman et al., 2019; Chen et al., 2018; Daniels et al., 2004; Robertson et al., 2014; Zheng et al., 2011), we next investigated whether autophagy is involved in the maturation of lamellar bodies in type II pneumocytes in the mouse lung. It has previously been reported that autophagosomes are frequently formed under basal conditions in type II pneumocytes in adult mice (Cheong et al., 2014; Mizushima et al., 2004). Additionally, in neonatal mice, GFP-LC3B puncta were formed and frequently fused with or attached to LAMP1 - or ABCA3-positive and ring-shaped lamellar bodies in type II pneumocytes (Figures 3A and 3B). Electron microscopy of type II pneumocytes in mice at 17.5 days post-coitum (dpc) revealed the presence of cytosolic contents such as glycogen particles within some lamellar bodies (Figure 3C). These results indicate that autophagosomes containing cytoplasmic components fuse with lamellar bodies in type II pneumocytes in vivo (Figure 3D), as suggested by results in GlcNAc-TV-expressing culture cells (Hariri et al., 2000; Lajoie et al., 2005).
Figure 3. Autophagosomes Fuse with Lamellar Bodies in Type II Pneumocytes in Mice.
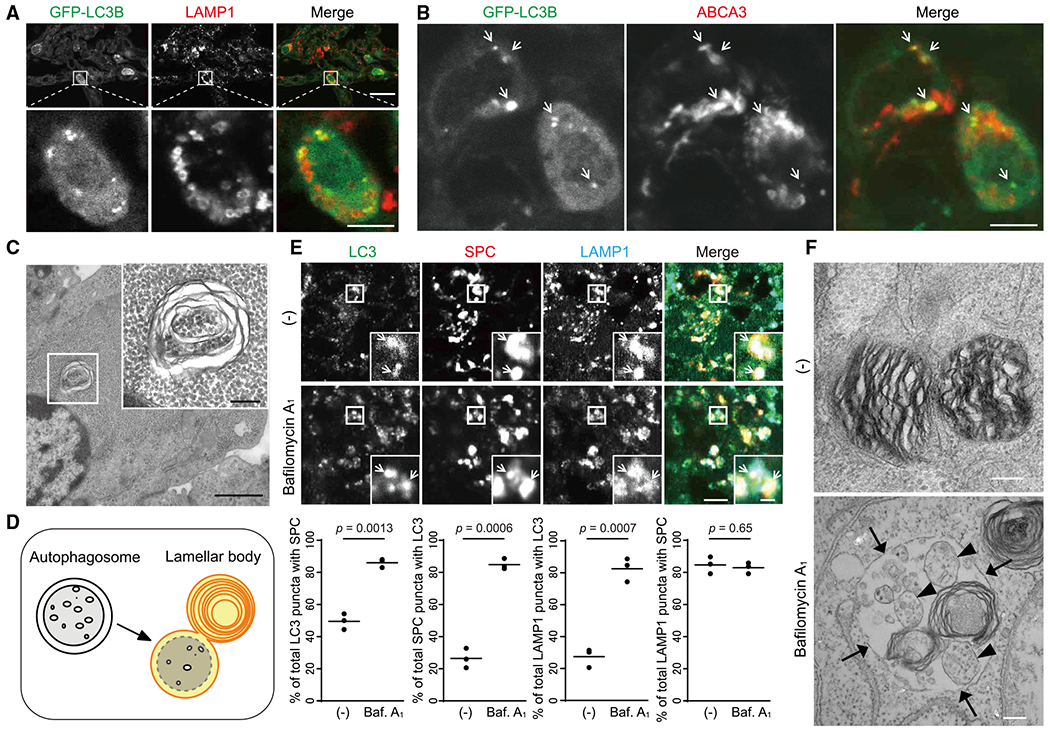
(A and B) Immunohistochemistry of the lung from neonatal GFP-LC3-RFP-LC3ΔG transgenic mouse(1.5 days after birth) using anti-LAMPI (A) or anti-ABCA3 (B) antibodies. Magnified images of type II pneumocytes in the indicated regions are shown in the lower panels. Scale bar, 20 μm and 5 μm in the lower panels.
(C) Transmission electron microscopy of type II pneumocytes in the lung from 17.5-dpc mouse embryos. Magnified images of the indicated regions are shown in the inset. Scale bar, 1 μm and 200 nm in the inset.
(D) A model of fusion between autophagosomes and lamellar bodies.
(E) Immunohistochemistry of lungs from 17.5-dpc mice (n = 3), which were incubated ex vivo with or without 100 nM bafilomycin A1 for 20 h, using anti-LAMP1, anti-LC3, and anti-surfactant protein-C (SPC) antibodies. The percentages of the number of SPC (+) LC3 puncta per total LC3 puncta, LC3 (+) SPC puncta per total SPC puncta, LC3 (+) LAMP1 puncta per total LAMP1 puncta, and SPC (+) LAMP1 puncta per total LAMP1 puncta were analyzed in at least 10 type II pneumocytes (SPC (+)) in each mouse. Dots represent individual mice, and the line indicates the mean. Differences were statistically analyzed using Welch’s t test. Scale bar, 5 μm and 1 μm in the inset.
(F) Transmission electron microscopy of samples prepared as in (E). Arrows indicate the limiting membrane of lamellar bodies. Arrowheads indicate single membrane vesicles containing the cytosol within the lamellar body. Scale bar, 200 nm.
Lamellar bodies possess vacuolar-type ATPase (V-ATPase), which causes them to become acidic, and contain several acid hydrolases (e.g., proteases and glucosidases) (Chander et al., 1986; Hook and Gilmore, 1982). To examine whether materials inside autophagosomes are degraded after fusion with lamellar bodies, we used a lung organ culture system (Sanford et al., 2016). Lung tissues were obtained from 17.5-dpc mice and treated with the V-ATPase inhibitor bafilomycin A1 for 20 h to suppress the function of acid hydrolases. After treatment, LC3-positive structures accumulated and colocalized with LAMP1 and the lamellar body protein surfactant protein-C (SPC) (Figure 3E). Electron microscopy of bafilomycin A1-treated tissues revealed that lamellar bodies incorporated intraluminal vesicles, which are morphologically different from those of multivesicular bodies (Stahlman et al., 2000; Weaver et al., 2002), and likely undigested cytosol surrounded by inner autophagosomal membranes, although some small vesicles might be derived from multivesicular bodies (Figure 3F). These results suggest that autophagosomes frequently fuse with lamellar bodies and that autophagosomal membranes and contents are efficiently degraded in the lamellar bodies of type II pneumocytes.
Defective Maturation of Lamellar Bodies in Type II Pneumocytes in Atg101-Deficient Mice
To examine the requirements of autophagy in the maturation of lamellar bodies in the lung, we generated mice systemically lacking ATG101, a component of the ULK complex (Hosokawa et al., 2009; Mercer et al., 2009) (Figure S2A). Atg101mice exhibited embryonic lethality at around 17.5 dpc (Figure S2B). Atg101−/−embryos were slightly smaller than Atg101+/− embryos (Figure S2C), corresponding to observations in mice lacking ATG13, another component of the ULK complex (Kaizuka and Mizushima, 2015). In type II pneumocytes of 17.5-dpc Atg101−/− mice, lamellar bodies labeled with SPC were smaller than those of Atg101+/− mice (Figure 4A). Electron microscopy revealed the presence of small and lipid-less lamellar bodies in Atg101−/− mice (Figure 4B). Such changes were not observed in Atg5−/− mice (Figure S3), as previously reported (Cheong et al., 2014), probably owing to the residual activity of autophagosome formation in ATG conjugation-deficient cells (Nguyen et al., 2016; Tsuboyama et al., 2016). These results suggest that autophagy is required for maturation of lamellar bodies not only in zebrafish swim bladders, but also in mouse lungs.
Figure 4. Defective Maturation of Lamellar Bodies in Type II Pneumocytes in Atg101- and Fip200-Deficient Mice.
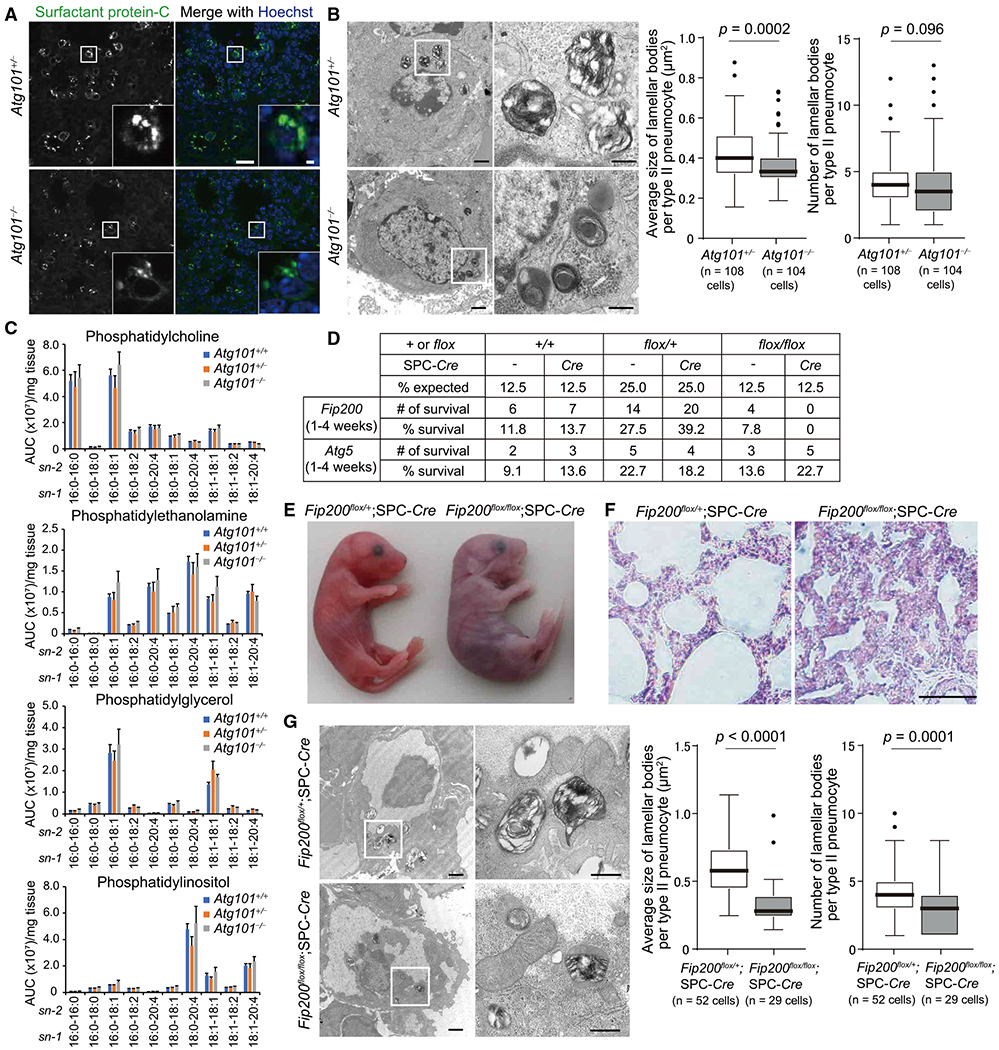
(A) Immunohistochemistry of the lung from 17.5-dpc Atg101+/− and Atg101−/− mice using anti-SPC antibody. Nuclear DNA was stained with Hoechst33342. Scale bar, 20 μm and 2 μm in the inset.
(B) Transmission electron microscopy of type II pneumocytes from 17.5-dpc Atg101+/− and Atg101−/− mice. Magnified images of the indicated regions are shown in the right panels. The average size (cross-sectional area) of each lamellar body was quantified (108 cells and 104 cells for Atg101+/− and Atg101−/− mice, respectively). The solid bars and boxes indicate the median and interquartile range (25th to 75th percentile), respectively. The whiskers indicate the upper and lower quartiles, and outliers are plotted individually. Differences were statistically analyzed by unpaired two-tailed Mann-Whitney U tests. Scale bar, 1 μm and 400 nm in magnified panels.
(C) LC-MS/MS analysis of the number of indicated phospholipids in the lung from 17.0-dpc Atg101+/+ (n = 4), Atg101+/− (n = 5), and Atg101−/− (n = 4) mice. The y axis indicates relative abundance (area under the curve, AUC [arbitrary units]) per mg tissue (means ± SEM).
(D) Survival rate of offspring from intercrosses of Fip200flox/+ and Fip200flox/+;SPC-Cre mice or Atg5flox/+ and Atg5flox/+;SPC-Cre mice.
(E and F) External appearance (E) and hematoxylin and eosin staining of the lung (F) of neonatal Fip200flox/+;SPC-Cre and Fip200flox/flox;SPC-Cre mice at 1 h after birth. The video from which these stills were captured is available as Video S2.
(G) Transmission electron microscopy of type II pneumocytes in the lung from Fip200flox/+;SPC-Cre and Fip200flox/flox;SPC-Cre mice at 1 h after birth was performed and analyzed as in (B) (52 cells and 29 cells for Fip200flox/+;SPC-Cre and Fip200flox/flox;SPC-Cre mice, respectively). Scale bar, 1 μm and 400 nm in magnified panels.
See also Figures S2–S4 and Video S2.
To clarify the role of autophagy in maturation of lamellar bodies, we compared the composition of phospholipids in lung tissues of 17.0-dpc Atg101+/− and Atg101mice (Figure 4C). Among the surfactant phospholipids, phosphatidylcholine is the major phospholipid, comprising 80% of surfactant lipids, and its major surface-active component is present in a disaturated form as dipalmitoylphosphatidylcholine (DPPC; 16:0/16:0) (Agassandian and Mallampalli, 2013). Because we could not collect bronchoalveolar lavage fluid (BALF) from embryonic lungs owing to their small size, we measured the lipid content of whole-lung homogenates. Quantitative liquid chromatography-tandem mass spectrometry (LC-MS/MS) revealed no significant differences in levels of phospholipids, including phosphatidylcholine (i.e., DPPC), phosphatidylethanolamine, phosphatidylglycerol, and phosphatidylinositol, among Atg101+/+, Atg101+/−, and Atg101−/− mice (Figure 4C). These results suggest that delivery of phospholipids from the ER to lamellar bodies, but not synthesis, is defective in the lungs of Atg101−/− mice.
Type-II-Pneumocyte-Specific Fip200-Deficient Mice Show Neonatal Lethality and Respiratory Failure
To circumvent the embryonic lethality of upstream Atg-deficient mice and investigate the role of autophagy in the lung after birth, we generated type-II-pneumocyte-specific F/p200-deficient mice. Mice harboring a Fip200flox allele were crossed with SPC-Cre transgenic mice expressing Cre recombinase under the control of the SPC promoter (Hato et al., 2009). Fip200flox/+;SPC-Cre mice were healthy, fertile, and phenotypically indistinguishable from wild-type littermates. Fip200flox/flox;SPC-Cre mice were also born according to the expected Mendelian ratio but could not survive beyond 24 h after birth (Figure 4D). Normal respiration was initiated at birth in Fip200flox/+;SPC-Cre mice, which became pink and exhibited rapid inflation of their lungs, as observed by the appearance of a white patch on the thorax, signaling lung inflation (Figure 4E). On the other hand, Fip200flox/flox;SPC-Cre mice initiated respiratory effort at birth but showed signs of respiratory distress, such as cyanosis and gasping (Figure 4E; Video S2). Although there was no apparent defect in lung morphogenesis in Fip200flox/flox;SPC-Cre mice (Figure S4A), histological analyses revealed that the airspaces were shrunken, and septal walls were folded (Figure 4F). In contrast to lethal phenotypes observed in Fip200flox/flox;SPC-Cre mice, Atg5flox/flox;SPC-Cre did not show neonatal lethality (Figure 4D). This is consistent with the above observation that maturation of lamellar bodies was normal in Atg5−/− mice (Figure S3). Electron microscopy of the lungs of neonatal Fip200flox/flox;SPC-Cre mice revealed a reduction in the size and number of lamellar bodies (Figure 4G), as observed in 17.5-dpc Atg101−/− mice (Figure 4B). There were no apparent defects in differentiation of type I pneumocytes (the positivity of HOP homeobox [HOPX]) (Figure S4B), supporting the hypothesis that defective maturation of lamellar bodies rather than a developmental delay underlies the respiratory failure in Fip200flox/flox;SPC-Cre mice. Normal maturation of surfactant protein-B (SPB), which is essential for maturation of lamellar bodies and inflation of the lung (Clark et al., 1995), was observed in the lung of P0 Fip200flox/flox;SPC-Cre mice (Figure S4C). Taken together, these results suggest that the defect in lamellar body maturation in Atg101−/− mice was not secondary to the systemic developmental delay and that cell-autonomous autophagy is essential for maturation of lamellar bodies in type II pneumocytes and thus survival after birth.
DISCUSSION
Based on our unbiased phenotypic screening of atg-deficient zebrafish and subsequent investigation of the lungs of atg-deficient mice, we have uncovered an evolutionally conserved function of autophagy in maturation of lamellar bodies. Previous studies using GlcNAc-TV-expressing culture cells and a class III PI3K inhibitor have suggested a potential role of autophagy in lamellar body maturation (Hariri et al., 2000; Lajoie et al., 2005). However, the involvement of autophagy was not studied using specific tools, and the physiological relevance of this function has never been tested. A previous study reported that mice lacking ATG5 or both ULK1 and ULK2 showed mild lung phenotypes (i.e., accumulation of glycogens) but with no observed effect on morphology of lamellar bodies (Cheong et al., 2014). This could be because deletion of ATG5 or both ULK1 and ULK2 may not be sufficient to completely block autophagy (see below). Another recent study using mice deficient for Beclin 1, a component of PI3K complexes I and II, reported that Beclin 1 is required for lung development (Yeganeh et al., 2019). However, this study did not investigate lamellar body maturation in type II pneumocytes. Furthermore, like general PI3K inhibitors, Beclin 1 deletion also suppresses the function of class III PI3K complex II in the endocytic pathway (McKnight et al., 2014), which could also be important for lamellar body maturation. In this study, we demonstrated the fusion of autophagosomes with lamellar bodies in wild-type animals and revealed the critical role of autophagy in maturation of lamellar bodies and inflation of the airspace. Although we do not exclude the possible contribution of a lamellar-body-independent function of autophagy in the lung (e.g., lung development and type II pneumocyte differentiation), the respiratory failure and neonatal death of Fip200flox/flox;SPC-Cre mice are likely caused by defects in lamellar body maturation because similar phenotypes are reported in mice specifically defective in lamellar body maturation (e.g., deficiency of SPB) (Clark et al., 1995; Weng et al., 2019). Further investigations using an inducible knockout system in adult mice will verify these possibilities.
What is the role of autophagy in lamellar body maturation? We observed that autophagosomes frequently fused with lamellar bodies. When autophagy was blunted, only small and lipid-less lamellar bodies were generated. However, the cellular contents of phospholipids were not affected. Based on these findings, we hypothesized that autophagy is important for remobilizing lipids to lamellar bodies rather than promoting biogenesis of surfactant lipids (Ma et al., 2013). Autophagosomes may supply sources of surfactant lipids, which are derived from engulfed endomembranes and the inner autophagosomal membranes. Alternatively, but not mutually exclusively, the fusion of the outer autophagosomal membrane with the limiting membranes of lamellar bodies could expand the size of lamellar bodies, thereby increasing the storage capacity. We do not exclude the possibility that autophagosomes fuse with late endosomes, the origin of lamellar bodies (Perez-Gil and Weaver, 2010; Weaver et al., 2002), to drive lamellar body maturation because lamellar body markers including LAMP1, ABCA3, and SPC also localize to late endosomes (Huotari and Helenius, 2011; Vorbroker et al., 1995; Yamano et al., 2001). This process likely resembles the formation of amphisomes (Gordon and Seglen, 1988; Strømhaug and Seglen, 1993). There are several factors that are critical for lamellar body maturation, such as SPB. Although we observed the presence of mature SPB in the lung of neonatal Fp200-deficient mice, biosynthesis, maturation, and functions of these factors might also be altered in the absence of autophagy. As autophagy has several functions in the lung (e.g., intracellular quality control and glycogen metabolism) (Cheong et al., 2014; Inoue et al., 2011; Yoshii et al., 2016), the role of autophagy in lamellar body maturation will need further investigation in the future.
The molecular mechanism by which autophagosomes fuse with lamellar bodies remains unclear. The machinery used for autophagosome-endolysosome fusion is likely shared because lamellar bodies originate from multivesicular bodies/late endosomes. Indeed, a recent study suggests that zebrafish lacking Snap29, a soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) protein essential for fusion between autophagosomes and endolysosomes (Itakura et al., 2012; Matsui et al., 2018), show a defect in inflation of the swim bladder (Mastrodonato et al., 2019). However, as SNAP29 also plays an important role in the endocytic and exocytotic pathways (Morelli et al., 2014), autophagic defects may not solely contribute to the phenotype. Accordingly, it is important to reveal whether there is a specific fusion machinery responsible for fusion between autophagosomes and surfactant-containing lamellar bodies.
In this study, we also revealed that upstream ATG proteins, but not ATG conjugation systems, are required for maturation of lamellar bodies. In addition, zebrafish lacking upstream ATGs die slightly earlier than those lacking Atg conjugation system components. This is similar to what has been observed in mice; mice lacking upstream Atg genes (Fip200−/−, Atg13−/−, Atg9a−/−, Beclin 1−/−, and Vps34/Pik3c3−/−) die in utero, while those lacking ATG conjugation systems (atg3−/−, atg5−/−, atg7−/−, atg12−/−, and atg16l1−/−) die within 1 day after birth (Kuma et al., 2017; Yoshii et al., 2016). These phenotypic differences could be explained by the formation of autophagosomes even in the absence of the ATG conjugation systems, although at a reduced rate (Nguyen et al., 2016; Tsuboyama et al., 2016). Although ULK1 and ULK2 are upstream genes, lamellar body maturation is unaffected in mice lacking both ULK1 and ULK2, at least at morphological levels (Cheong et al., 2014). Because ULK1 and ULK2 may not be absolutely required for autophagy (Alers et al., 2011), a remaining low level of autophagic activity in these mice could produce a milder phenotype, as observed in mice lacking ATG conjugation components. An alternative interpretation of the observed differences among ATG-depleted animals could be that the swim bladder and lung phenotypes are caused by defects of autophagy-independent functions of upstream ATG proteins (Chen et al., 2016; Kaizuka and Mizushima, 2015; Velikkakath et al., 2012). However, the defective inflation of the swim bladder was observed in zebrafish lacking all classes of upstream ATG proteins essential for autophagy, suggesting that the phenotype is most likely caused by autophagy deficiency rather than by a defect in autophagy-independent functions. Further comprehensive investigations using animals lacking all classes of ATG proteins will be important to exclude the possible involvement of autophagy-independent functions in lamellar body maturation in the swim bladder and lung. Previous reports showed that zebrafish embryos with morpholino-mediated knockdown of atg5 demonstrated severe defects in morphogenesis (e.g., twisted body shapes and pericardial edema) (Hu et al., 2011; Lee et al., 2014). However, we did not observe such aberrant morphogenesis in atg knockout zebrafish embryos. These phenotypic differences may be due to differences in genetic backgrounds and experimental conditions or off-target effects by morpholinos (Kok et al., 2015).
Multilamellar bodies are formed not only in the lung and swim bladder, but also in keratinocytes and other cell types under physiological and pathological conditions, including cancer, lysosomal storage diseases, and infectious diseases (Handerson and Pawelek, 2003; Lajoie et al., 2005; Omi et al., 2020; Schmitz and Muller, 1991). Thus, autophagy could also be involved in the maturation of multilamellar bodies in these cell types. Accordingly, a key objective of future research is the identification of molecular mechanisms underlying the activation of autophagy during lamellar body maturation, which may modulate the functions of multilamellar bodies in several physiological and disease conditions.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Noboru Mizushima (nmizu@m.u-tokyo.ac.jp).
Materials availability
New reagents generated in this study are available via the lead contact.
Data and Code Availability
This study did not generate/analyze datasets/code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Zebrafish
Wild-type zebrafish (RIKEN Wako) were obtained from the Zebrafish National Bioresource Project of Japan and maintained in 14-h light/10-h dark conditions at 28.5°C according to established protocols (Kimmel et al., 1995). The CRISPR/Cas9 system (Jao et al., 2013), including pT7-gRNA, which was graciously provided by Wenbiao Chen (plasmid #46759, Addgene), and recombinant Cas9 protein (1074181, IDT), was used to generate atg-deficient zebrafish. Target regions with low off-target effects were decided based on CRISPRscan analysis (Moreno-Mateos et al., 2015) (target sequences are shown in Figure S1); gRNA fragments targeting each gene were synthesized using a MEGAshortscript T7 transcription kit (AM1354, Thermo Fisher Scientific) and purified using a mirVana miRNA Isolation Kit (AM1560, Thermo Fisher Scientific). Wild-type embryos at the one-cell stage were microinjected with approximately 100 pg of sgRNAand 300 pg of Cas9 proteins using FemtoJet (Eppendorf) equipped with a Femtotip II injection capillary (Ep-pendorf). Heteroduplex mobility assays (Ota et al., 2014) were performed to genotype atg-deficient zebrafish using genomic DNA, primers flanking the target site (forward and reverse primer sequences are shown in Figure S1), PrimeSTAR Max DNA polymerase (R045A, TaKaRa Bio Inc.), and 10% polyacrylamide gels. Homozygous afg-deficient zebrafish were generated by intercrossing heterozygous afg-deficient zebrafish. Methods to generate vmpl-deficient zebrafish have been reported previously (Morishita et al., 2019). Defects in autophagy in fip200/rb1cc1-deficient and vmp1-deficient zebrafish have been confirmed previously (Kaizuka et al., 2016; Morishita et al., 2019).
Mice
The experimental procedures used to produce Afg5flox/+ (Hara et al., 2006), Fip200flox/+ (Gan et al., 2006), SPC-Cre (Hato et al., 2009), and GFP-LC3-RFP-LC3ΔG transgenic (Kaizuka et al., 2016) mice have been described previously. The CRISPR/Cas9 system was used to generate an Afg101+/− mouse line. In brief, pcDNA3-hCas9 plasmid (Kaizuka and Mizushima, 2015) was linearized by Notl, purified by phenol-chloroform extraction, transcribed to mRNA using the mMessage mMachine T7 Ultra kit (AM1345, Thermo Fisher Scientific), and purified using an RNeasy Mini Kit (74104, QIAGEN). Similarly, pT7-Atg101 gRNA plasmid was linearized by BamHI, purified by phenol-chloroform extraction, transcribed to RNA by using a MEGAshortscript T7 transcription kit (AM1354, Thermo Fisher Scientific), and purified using a mirVana miRNA Isolation Kit (AM1560, Thermo Fisher Scientific). Cas9 mRNA (100 ng/μl) and Atg101 gRNA (50 ng/μl) were injected into the pronuclei of fertilized eggs collected from C57BL/6J mice (CLEA Japan, Inc.) to obtain heterozygous mutant mice. To genotype Atg101−/− mice, heteroduplex mobility assays were performed using primers flanking the target site (forward primer, 5′- GAGCAGGAAGTGGACAGGAC-3′ reverse primer, 5′- GGCCACAAACCTACCTTGAA-3′). Mice and lungs were externally imaged using a stereoscopic microscope (SZX10, Olympus). Experiments were conducted in male and female mice at the age of 13.5 dpc to six months.
All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Tokyo (Medical-P17-084).
METHOD DETAILS
Antibodies and reagents
For immunostaining and immunoblotting, mouse monoclonal anti-LC3B (CTB-LC3-2-IC, Cosmo Bio), anti-ABCA3 (ab24751, Abcam), anti-HOPX (sc-398703, Santa Cruz Biotechnology), anti-HSP90 (610419, BD Transduction Laboratories), rat monoclonal anti-LAMP1 (ab25245, Abcam), and rabbit polyclonal anti-SPC (ABC99, EMD Millipore), anti-SPB (07-619, EMD Millipore), anti-p62 (PM045, MBL) antibodies were used as primary antibodies. Alexa Fluor 488-conjugated anti-mouse IgG (A11001, Thermo Fisher Scientific), Alexa Fluor 568-conjugated anti-rabbit IgG (A11011, Thermo Fisher Scientific), Alexa Fluor 568-conjugated anti-ratbbit IgG (A11077, Thermo Fisher Scientific), Alexa Fluor 633-conjugated anti-rat IgG (A11094, Thermo Fisher Scientific), Alexa Fluor 660-conjugated anti-mouse IgG (A-21055, Thermo Fisher Scientific), Alexa Fluor 660-conjugated anti-rabbit IgG (A21074, Thermo Fisher Scientific), HRP-conjugated goat polyclonal anti-rabbit IgG (111-035-144, Jackson ImmunoResearch Laboratories), and HRP-conjugated goat polyclonal anti-mouse IgG (115-035-003, Jackson ImmunoResearch Laboratories) antibodies were used as secondary antibodies. Hoechst33342 (H342, Dojindo Molecular Technologies) was used to stain DNA. Bafilomycin A1 (B1793) was purchased from Sigma-Aldrich.
Plasmids
The pT7-gRNA (plasmid #46759, Addgene) (Jao et al., 2013), pcDNA3-hCas9 (Kaizuka and Mizushima, 2015), and pMXs-IP-GFP-rat LC3B plasmids (Hara et al., 2008) have previously been described. Rat LAMP1 cDNA (Tsuboyama et al., 2016) was inserted into pCS2 vector together with EGFP or mRFP.
Gross and survival analysis of zebrafish
Zebrafish larvae were externally imaged with a stereoscopic microscope (SZX10). The lack of swim bladder inflation was recognized as a visible absence of air space in the swim bladder. A video of swimming fish in a cuvette was recorded using a Nikon D90 camera. To perform a survival assay of zebrafish larvae, progeny from intercrosses of atg-deficient zebrafish in the same nursery environment without food were observed twice a day. Dead larvae were collected and frozen. At 14 dpf, the remaining larvae were sacrificed, and all larvae, including dead zebrafish, were genotyped. The survival of atg16l1-deficient zebrafish was also analyzed underfeeding conditions. Results of survival rates of fip200-deficient and atg16l1-deficient zebrafish are shown as Kaplan-Meier survival curves.
Live imaging of zebrafish embryos
Zebrafish eggs at the one-cell stage were microinjected with approximately 50 pg of either GFP-rat LC3B and rat LAMP1-RFP mRNA or rat LAMP1-GFP mRNA, which were synthesized from plasmids using the mMESSAGE mMACHINE SP6 Transcription Kit (AM1340, Thermo Fisher Scientific) and purified using RNeasy Mini Kit (74104, QIAGEN). From 1 dpf, zebrafish embryos were maintained in water with 1-phenyl-2-thiourea (P7629, Sigma-Aldrich), which inhibits melanogenesis. Zebrafish were anesthetized with 0.03% tricaine (A5040, Sigma-Aldrich), placed in water on a glass-bottomed dish, and viewed using a confocal microscope (FV1000 IX81, Olympus) with an objective lens (UPLSAPO30XS, Olympus).
Histology and immunohistochemistry
The zebrafish larvae or mouse lung were fixed in 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) overnight, infiltrated with 15% and 30% sucrose in PBS, embedded in Tissue-TekOCT Compound (Sakura Japan Co.), frozen, and sectioned (into 7-pm thick slices) using a cryostat (CM3050 S, Leica Microsystems). Cryosections were mounted on slides. For hematoxylin and eosin staining, cryosections were stained with hematoxylin and eosin and photographed using a microscope (BX51, Olympus) equipped with a digital camera (DP70, Olympus). For immunohistochemistry, cryosections were washed with PBS, treated with PBS containing 0.05% Triton X-100 for 15 min, blocked with 3% bovine serum albumin in PBS for 30 min, and incubated with primary antibodies for 1 h. Then, sections were washed with PBS, incubated with secondary antibodies for 1h. For nuclear DNA staining, samples were treated with Hoechst33342 in PBS for 10 min and washed three times with PBS. The coverslips were mounted with SlowFade antifade reagents (S36936, Thermo Fisher Scientific), observed using a confocal laser microscope (FV1000 IX81), and images were captured with FluoView software (Olympus) and edited with Photoshop CS6 software (Adobe systems). The number of punctate structures was quantified using FIJI software (threshold, binarization, and particle-analysis functions).
To prepare samples for immunohistochemistry of ex vivo cultured mouse lung, 17.5-dpc mouse lungs were dissected, cut into approximately 20 mm3 pieces, washed with PBS, and cultured for 20 h in Dulbecco’s modified Eagle’s medium (DMEM; D6546, Sigma-Aldrich) supplemented with 10% fetal bovine serum (172012, Sigma-Aldrich), 2 mM l-glutamine (25030-081, GIBCO), 50 μg/ml penicillin, and streptomycin in a 5% CO2 incubator.
Electron microscopy
The zebrafish larvae or mouse lungs were fixed with 2% PFA and 2% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) overnight. Tissues were then post-fixed with 1.0% osmium tetroxide in 0.1 M phosphate buffer for 2 h, dehydrated in a graded series of ethanol, and embedded in epoxy resin. Ultra-thin sections were made using an ultramicrotome (Reichert). Sections were stained with uranyl acetate and lead citrate and observed under an H7100 electron microscope (Hitachi).
Immunoblotting
Mouse lungs were lysed with lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, and complete EDTA-free protease inhibitor cocktail [19543200, Roche]). The supernatants were collected by centrifugation at 15,000 × g for 10 min, and the protein concentrations were adjusted using the bicinchoninic acid method (23228, Thermo Fisher Scientific). The lysates were solubilized with immunoblot sample buffer (46.7 mM Tris-HCl [pH 6.8], 1.67% sodium dodecyl sulfate [SDS], 5% glycerol, 1.55% dithiothreitol, and 0.003% bromophenol blue), subjected to SDS-polyacrylamide gel electrophoresis, transferred to an Immobilon-P polyvinylidene difluoride membrane (IPVH00010, Millipore), and blotted with antibodies. Super-Signal West Pico Chemiluminescent substrate (1856136, Thermo Fisher Scientific) was used for detection of each protein signal. Signals were captured using FUSION SOLO7S (Vilber-Lourmat). The images were processed using Photoshop CS6.
Phospholipid quantification of the lung
Quantification of phospholipids in the lung taken from 17.0-dpc mouse embryos was performed as previously described (Yamamoto et al., 2016). In brief, the lung was dissected and homogenized on ice with 10 volumes of PBS. Lipids were extracted from the homogenates by the Bligh and Dyer method (Yamamoto et al., 2016). A hybrid triple quadrupole-linear ion-trap hybrid mass spectrometer (4000Q-TRAP, Sciex) equipped with a liquid chromatograph (Nexera X2 system, Shimadzu) was used for LC-MS/MS analysis; 500 pmol of d5-labeled eicosapentaenoic acid and PE 14:0-14:0 were added to each sample as an internal standard. Samples were injected by an autosampler and separated using a step gradient with mobile phase A (1:1:1 acetonitrile:methanol:water [v/v/v] solution containing 1 mM ammonium formate and 5 μM phosphoric acid) and mobile phase B (2-propanol containing 1 mM ammonium formate and 5 μM phosphoric acid) at a flow rate of 0.2 mL/min at 50°C. Identification was conducted using multiple reaction monitoring (MRM) transition and retention times. MRM data were analyzed by MultiQuant and Analyst software (Sciex). Quantification was performed based on the peak area of the extracted ion chromatograms for each lipid.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
Mann–Whitney U-tests or Welch’s t tests were performed to compare means between two groups. Statistical analysis was performed using GraphPad Prism 8 software (GraphPad software).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-ABCA3 | Abcam | Cat#ab24751; RRID: AB_448287 |
| Rat monoclonal anti-LAMP1 | Abcam | Cat#ab25245; RRID: AB_449893 |
| Mouse monoclonal anti-LC3B | Cosmo Biometrics | Cat#CTB-LC3-2-IC: AB_10707197 |
| Mouse monoclonal anti-HSP90 | 3D Transduction Laboratories | Cat#610419; RRID: AB_397799 |
| Rabbit polyclonal anti-SPC | EMD Millipore | Cat#ABC99; RRID: AB_2755001 |
| Rabbit polyclonal anti-SPB | EMD Millipore | Cat#07-614; RRID: AB_310753 |
| Rabbit polyclonal anti-p62 | MBL | Cat#PM045; RRID: AB_1279301 |
| Mouse monoclonal anti-HOPX | Santa Cruz Biotechnology | Cat#sc-398703; RRID: AB_2687966 |
| Alexa Fluor 488-conjugated anti-mouse IgG | Thermo Fisher Scientific | Cat#A-11001; RRID: AB_2534069 |
| Alexa Fluor 568-conjugated anti-rabbit IgG | Thermo Fisher Scientific | Cat#A-11011; RRID: AB_143157 |
| Alexa Fluor 568-conjugated anti-rat IgG | Thermo Fisher Scientific | Cat#A-11077; RRID: AB_141874 |
| Alexa Fluor 660-conjugated anti-mouse IgG | Thermo Fisher Scientific | Cat#A-21055; RRID: AB_2535722 |
| Alexa Fluor 633-conjugated anti-rat IgG | Thermo Fisher Scientific | Cat#A-21094; RRID: AB_141553 |
| Alexa Fluor 660-conjugated anti-rabbit IgG | Thermo Fisher Scientific | Cat#A-21074; RRID: AB_2535735 |
| HRP-conjugated goat polyclonal anti-rabbit IgG | Jackson ImmunoResearch Laboratories | Cat#111-035-144; RRID: AB_2307391 |
| HRP-conjugated goat polyclonal anti-mouse IgG | Jackson ImmunoResearch Laboratories | Cat#115-035-003; RRID: AB_10015289 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Alt-R S.p. Cas9 Nuclease 3NLS | IDT | Cat#1074181 |
| PrimeSTAR Max DNA polymerase | Takara Bio Inc. | Cat#R045A |
| Hoechst33342 | Dojindo Molecular Technologies | Cat#H342 |
| SlowFade antifade reagents | Thermo Fisher Scientific | Cat#S36936 |
| 1-phenyl 2-thiourea | Sigma-Aldrich | Cat#P7629 |
| tricaine | Sigma-Aldrich | Cat#A5040 |
| Critical Commercial Assays | ||
| mMESSAGE mMACHINE SP6 Transcription Kit | Thermo Fisher Scientific | Cat#AM1340 |
| MEGAshortscript T7 Transcription Kit | Thermo Fisher Scientific | Cat#AM1354 |
| mirVana miRNA Isolation Kit | Thermo Fisher Scientific | Cat#AM1560 |
| RNeasy Mini Kit | QIAGEN | Cat#74104 |
| Experimental Models: Organisms/Strains | ||
| Zebrafish: RIKEN Wako wild-type strain | Zebrafish National Bioresource | ZFIN ID: ZDB-GENO-070802-4 |
| Zebrafish: atg101+/− | This study | N/A |
| Zebrafish: atg13+/− | This study | N/A |
| Zebrafish: atg14+/− | This study | N/A |
| Zebrafish: atg2a+/− | This study | N/A |
| Zebrafish: atg2b+/− | This study | N/A |
| Zebrafish: atg5+/− | This study | N/A |
| Zebrafish: atg9a+/− | This study | N/A |
| Zebrafish: atg9b+/− | This study | N/A |
| Zebrafish: atg16l1+/− | This study | N/A |
| Zebrafish: fip200/rb1cc1+/− | (Kaizuka et al., 2016) | ZFIN ID: ZDB-ALT-171018-3 |
| Zebrafish: vmp1+/− | (Morishita et al., 2019) | ZFIN ID: ZDB-ALT-200609-11 |
| Mouse: C57BL/6J | Sankyo Labo Service | N/A |
| Mouse: Afg101+/− | This study | N/A |
| Mouse: Afg5+/− | (Kuma et al., 2004) | BRC No.: RBRC02231 |
| Mouse: Fip200flox/+ | (Gan et al., 2006) | N/A |
| Mouse: Afg5flox/+ | (Hara et al., 2006) | BRC No.: RBRC02975 |
| Mouse: SPC-Cre | (Hato et al., 2009) | CARD ID: 2191 |
| Mouse: GFP-LC3-RFP-LC3ΔG | (Kaizuka et al., 2016) | BRC No: RBRC09930 |
| Recombinant DNA | ||
| pMXs-IP-GFP-LC3B | (Hara et al., 2008) | Addgene #38195 |
| pCS2-LAMP1-GFP | This study | N/A |
| pCS2-LAMP1-RFP | This study | N/A |
| Software and Algorithms | ||
| CRISPRscan | (Moreno-Mateos et al., 2015) | https://www.crisprscan.org/ |
| ImageJ | (Schindelin et al., 2012) | RRID: SCR_002285 |
| Photoshop CS6 | Adobe Systems | N/A |
| GraphPad Prism 8 software | GraphPad software | N/A |
Highlights.
Autophagy is required for larval survival and swim bladder inflation in zebrafish
Autophagy is required for maturation of surfactant-containing lamellar bodies
Autophagosomes fuse with lamellar bodies in the swim bladder and lung
FIP200 in the lung is essential for neonatal survival and respiration in mice
ACKNOWLEDGMENTS
We thank Yuichi Oike for providing SPC-Cre mice (obtained through Yu Mi-kami), Motoki Goto for help with the generation of Atg101-deficient, mice, Yuriko Sakamaki and Chieko Saito for help with electron microscopy, Nozomi Sato and Tomoya Eguchi for care of the zebrafish and mice, Yoko Ishida for help with the production of schematic diagrams, and Keiko Igarashi for help with the histological assays. This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas (grant 25111005 to N.M.) and a Grant-in-Aid for Young Scientists (grant 18K14694 to H.M.) from the Japan Society for the Promotion of Science; Exploratory Research for Advanced Technology (ERATO; grant JPMJER1702 to N.M.) from the Japan Science and Technology Agency (JST); AMED-FORCE and CREST (grants JP19gm4010005 and JP18gm0710006 to M.M.) from the Japan Agency for Medical Research and Development; and National Institutes of Health grants (grant R01NS094144to J.-L.G.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108477.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Agassandian M, and Mallampalli RK (2013). Surfactant phospholipid metabolism. Biochim. Biophys. Acta 1831, 612–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alers S, Löffler AS, Paasch F, Dieterle AM, Keppeler H, Lauber K, Campbell DG, Fehrenbacher B, Schaller M, Wesselborg S, and Stork B (2011). Atg13 and FIP200 act independently of Ulk1 and Ulk2 in autophagy induction. Autophagy 7, 1423–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreeva AV, Kutuzov MA, and Voyno-Yasenetskaya TA (2007). Regulation of surfactant secretion in alveolar type II cells. Am. J. Physiol. Lung Cell. Mol. Physiol 293, L259–L271. [DOI] [PubMed] [Google Scholar]
- Ban N, Matsumura Y, Sakai H, Takanezawa Y, Sasaki M, Arai H, and Inagaki N (2007). ABCA3 as a lipid transporter in pulmonary surfactant biogenesis. J. Biol. Chem 282, 9628–9634. [DOI] [PubMed] [Google Scholar]
- Bowman SL, Bi-Karchin J, Le L, and Marks MS (2019). The road to lyso-some-related organelles: Insights from Hermansky-Pudlak syndrome and other rare diseases. Traffic 20, 404–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chander A, Johnson RG, Reicherter J, and Fisher AB (1986). Lung lamellar bodies maintain an acidic internal pH. J. Biol. Chem 261, 6126–6131. [PubMed] [Google Scholar]
- Chen S, Wang C, Yeo S, Liang CC, Okamoto T, Sun S, Wen J, and Guan JL (2016). Distinct roles of autophagy-dependent and -independent functions of FIP200 revealed by generation and analysis of a mutant knock-in mouse model. Genes Dev. 30, 856–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Song G, Yang H, Mao L, Cui Z, and Huang K (2018). Development of the Swimbladder Surfactant System and Biogenesis of Lysosome-Related Organelles Is Regulated by BLOS1 in Zebrafish. Genetics 208, 1131–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong N, Zhang H, Madesh M, Zhao M, Yu K, Dodia C, Fisher AB, Savani RC, and Shuman H (2007). ABCA3 is critical for lamellar body biogenesis in vivo. J. Biol. Chem 282, 23811–23817. [DOI] [PubMed] [Google Scholar]
- Cheong H, Wu J, Gonzales LK, Guttentag SH, Thompson CB, and Lindsten T (2014). Analysis of a lung defect in autophagy-deficient mouse strains. Autophagy 10, 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark JC, Wert SE, Bachurski CJ, Stahlman MT, Stripp BR, Weaver TE, and Whitsett JA (1995). Targeted disruption of the surfactant protein B gene disrupts surfactant homeostasis, causing respiratory failure in newborn mice. Proc. Natl. Acad. Sci. USA 92, 7794–7798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels CB, Orgeig S, Sullivan LC, Ling N, Bennett MB, Schürch S, Val AL, and Brauner CJ (2004). The origin and evolution of the surfactant system in fish: insights into the evolution of lungs and swim bladders. Physiol. Biochem. Zool 77, 732–749. [DOI] [PubMed] [Google Scholar]
- Fazeli G, Trinkwalder M, Irmisch L, and Wehman AM (2016). C. elegans midbodies are released, phagocytosed and undergo LC3-dependent degradation independent of macroautophagy. J. Cell Sci 129, 3721–3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald ML, Xavier R, Haley KJ, Welti R, Goss JL, Brown CE, Zhuang DZ, Bell SA, Lu N, McKee M, et al. (2007). ABCA3 inactivation in mice causes respiratory failure, loss of pulmonary surfactant, and depletion of lung phosphatidylglycerol. J. Lipid Res 48, 621–632. [DOI] [PubMed] [Google Scholar]
- Funderburk SF, Wang QJ, and Yue Z (2010). The Beclin 1-VPS34 complex–at the crossroads of autophagy and beyond. Trends Cell Biol. 20, 355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futter CE, Collinson LM, Backer JM, and Hopkins CR (2001). Human VPS34 is required for internal vesicle formation within multivesicular endosomes. J. Cell Biol 155, 1251–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan B, Peng X, Nagy T, Alcaraz A, Gu H, and Guan JL (2006). Role of FIP200 in cardiac and liver development and its regulation of TNFalpha and TSC-mTOR signaling pathways. J. Cell Biol 175, 121–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon PB, and Seglen PO (1988). Prelysosomal convergence of autophagic and endocytic pathways. Biochem. Biophys. Res. Commun 151, 40–47. [DOI] [PubMed] [Google Scholar]
- Handerson T, and Pawelek JM (2003). Beta1,6-branched oligosaccharides and coarse vesicles: a common, pervasive phenotype in melanoma and other human cancers. Cancer Res. 68, 5363–5369. [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Mi-gishima R, Yokoyama M, Mishima K, Saito I, Okano H, and Mizushima N (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889. [DOI] [PubMed] [Google Scholar]
- Hara T, Takamura A, Kishi C, lemura S, Natsume T, Guan JL, and Mizushima N (2008). FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 181, 497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariri M, Millane G, Guimond MP, Guay G, Dennis JW, and Nabi IR (2000). Biogenesis of multilamellar bodies via autophagy. Mol. Biol. Cell 11, 255–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hato T, Kimura Y, Morisada T, Koh GY, Miyata K, Tabata M, Kadomatsu T, Endo M, Urano T, Arai F, et al. (2009). Angiopoietins contribute to lung development by regulating pulmonary vascular network formation. Biochem. Biophys. Res. Commun 381, 218–223. [DOI] [PubMed] [Google Scholar]
- Hook GE, and Gilmore LB (1982). Hydrolases of pulmonary lysosomes and lamellar bodies. J. Biol. Chem 257, 9211–9220. [PubMed] [Google Scholar]
- Hosokawa N, Sasaki T, lemura S, Natsume T, Hara T, and Mizushima N (2009). Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 5, 973–979. [DOI] [PubMed] [Google Scholar]
- Hu Z, Zhang J, and Zhang Q (2011). Expression pattern and functions of autophagy-related gene atg5 in zebrafish organogenesis. Autophagy 7, 1514–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huotari J, and Helenius A (2011). Endosome maturation. EMBO J. 30, 3481–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue D, Kubo H, Taguchi K, Suzuki T, Komatsu M, Motohashi H, and Yamamoto M (2011). Inducible disruption of autophagy in the lung causes airway hyper-responsiveness. Biochem. Biophys. Res. Commun 405, 13–18. [DOI] [PubMed] [Google Scholar]
- Itakura E, Kishi-itakura C, and Mizushima N (2012). The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151, 1256–1269. [DOI] [PubMed] [Google Scholar]
- Jao LE, Wente SR, and Chen W (2013). Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc. Natl. Acad. Sci. USA 110, 13904–13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, and Yoshimori T (2000). LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaizuka T, and Mizushima N (2015). Atg13 Is Essential for Autophagy and Cardiac Development in Mice. Mol. Cell. Biol 36, 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaizuka T, Morishita H, Hama Y, Tsukamoto S, Matsui T, Toyota Y, Kodama A, Ishihara T, Mizushima T, and Mizushima N (2016). An autophagic flux probe that releases an internal control. Mol. Cell 64, 835–849. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, and Schilling TF (1995). Stages of embryonic development of the zebrafish. Dev. Dyn 203, 253–310. [DOI] [PubMed] [Google Scholar]
- Kok FO, Shin M, Ni CW, Gupta A, Grosse AS, van Impel A, Kirchmaier BC, Peterson-Maduro J, Kourkoulis G, Male I, et al. (2015). Reverse genetic screening reveals poor correlation between morpholino-induced and mutant phenotypes in zebrafish. Dev. Cell 32, 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kook S, Wang P, Young LR, Schwake M, Saftig P, Weng X, Meng Y, Neculai D, Marks MS, Gonzales L, et al. (2016). Impaired Lysosomal Integral Membrane Protein 2-dependent Peroxiredoxin 6 Delivery to Lamellar Bodies Accounts for Altered Alveolar Phospholipid Content in Adaptor Protein-3-deficient pearl Mice. J. Biol. Chem 291, 8414–8427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, and Mizushima N (2004). The role of autophagyduring the early neonatal starvation period. Nature 432, 1032–1036. [DOI] [PubMed] [Google Scholar]
- Kuma A, Komatsu M, and Mizushima N (2017). Autophagy-monitoring and autophagy-deficient mice. Autophagy 13, 1619–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lajoie P, Guay G, Dennis JW, and Nabi IR (2005). The lipid composition of autophagic vacuoles regulates expression of multilamellar bodies. J. Cell Sci 118, 1991–2003. [DOI] [PubMed] [Google Scholar]
- Lee E, Koo Y, Ng A, Wei Y, Luby-Phelps K, Juraszek A, Xavier RJ, Cleaver O, Levine B, and Amatruda JF (2014). Autophagy is essential for cardiac morphogenesis during vertebrate development. Autophagy 10, 572–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, and Kroemer G (2019). Biological FunctionsofAutophagyGenes: A Disease Perspective. Cell 176, 11–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D, Molusky MM, Song J, Hu CR, Fang F, Rui C, Mathew AV, Pennathur S, Liu F, Cheng JX, et al. (2013). Autophagy deficiency by hepatic FIP200 deletion uncouples steatosis from liver injury in NAFLD. Mol. Endocrinol 27, 1643–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez J, Malireddi RK, Lu Q, Cunha LD, Pelletier S, Gingras S, Orchard R, Guan JL, Tan H, Peng J, et al. (2015). Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat. Cell Biol 17, 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Mastrodonato V, Beznoussenko G, Mironov A, Ferrari L, Deflorian G, and Vaccari T (2019).Agenetic model of CEDNIK syndrome in zebrafish highlights the role of the SNARE protein Snap29 in neuromotor and epidermal development. Sci. Rep 9, 1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masud S, Prajsnar TK, Torraca V, Lamers GEM, Benning M, Van Der Vaart M, and Meijer AH (2019). Macrophages target Salmonella by Lc3-associated phagocytosis in a systemic infection model. Autophagy 15, 796–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Jiang P, Nakano S, Sakamaki Y, Yamamoto H, and Mizushima N (2018). Autophagosomal YKT6 is required for fusion with lysosomes independently of syntaxin 17. J. Cell Biol 217, 2633–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight NC, Zhong Y, Wold MS, Gong S, Phillips GR, Dou Z, Zhao Y, Heintz N, Zong WX, and Yue Z (2014). Beclin 1 is required for neuron viability and regulates endosome pathways via the UVRAG-VPS34 complex. PLoS Genet. 10, e1004626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer CA, Kaliappan A, and Dennis PB (2009). A novel, human Atg13 binding protein, Atg101, interactswith ULK1 and is essential for macroautophagy. Autophagy 5, 649–662. [DOI] [PubMed] [Google Scholar]
- Miklavc P, Ehinger K, Thompson KE, Hobi N, Shimshek DR, and Frick M (2014). Surfactant secretion in LRRK2 knock-out rats: changes in lamellar body morphology and rate of exocytosis. PLoS ONE 9, e84926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N (2018). A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol 20, 521–527. [DOI] [PubMed] [Google Scholar]
- Mizushima N, and Komatsu M (2011). Autophagy: renovation of cells and tissues. Cell 147, 728–741. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, and Ohsumi Y (2004). In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 15, 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli E, Ginefra P, Mastrodonato V, Beznoussenko GV, Rusten TE, Bilder D, Stenmark H, Mironov AA, and Vaccari T (2014). Multiple functions of the SNARE protein Snap29 in autophagy, endocytic, and exocytic trafficking during epithelial formation in Drosophila. Autophagy 10, 2251–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Mateos MA, Vejnar CE, Beaudoin JD, Fernandez JP, Mis EK, Khokha MK, and Giraldez AJ (2015). CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Methods 12, 982–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti F, Bergman P, Dodgson S, Marcellin D, Claerr I, Goodwin JM, DeJesus R, Kang Z, Antczak C, Begue D, et al. (2018). TMEM41B is a novel regulator of autophagy and lipid mobilization. EMBO Rep. 19, e45889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita H, and Mizushima N (2019). Diverse Cellular Roles of Autophagy. Annu. Rev. Cell Dev. Biol 35, 453–475. [DOI] [PubMed] [Google Scholar]
- Morishita H, Zhao YG, Tamura N, Nishimura T, Kanda Y, Sakamaki Y, Okazaki M, Li D, and Mizushima N (2019). Acritical role of VMP1 in lipoprotein secretion. eLife 8, e48834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita K, Hama Y, Izume T, Tamura N, Ueno T, Yamashita Y, Sakamaki Y, Mimura K, Morishita H, Shihoya W, et al. (2018). Genome-wide CRISPR screen identifies TMEM41B as a gene required for autophagosome formation. J. Cell Biol 217, 3817–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatogawa H (2020). Mechanisms governing autophagosome biogenesis. Nat. Rev. Mol. Cell Biol 21, 439–458. [DOI] [PubMed] [Google Scholar]
- Nguyen TN, Padman BS, Usher J, Oorschot V, Ramm G, and Lazarou M (2016). Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J. Cell Biol 215, 857–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmeda B, Martínez-Calle M, and Pérez-Gil J (2017). Pulmonary surfactant metabolism in the alveolar airspace: Biogenesis, extracellular conversions, recycling. Ann. Anat 209, 78–92. [DOI] [PubMed] [Google Scholar]
- Omi J, Watanabe-Takahashi M, Igai K, Shimizu E, Tseng CY, Miyasaka T, Waku T, Hama S, Nakanishi R, Goto Y, et al. (2020). The inducible amphisome isolates viral hemagglutinin and defends against influenza A virus infection. Nat. Commun 11, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota S, Hisano Y, Ikawa Y, and Kawahara A (2014). Multiple genome modifications by the CRISPR/Cas9 system in zebrafish. Genes Cells 19, 555–564. [DOI] [PubMed] [Google Scholar]
- Pérez-Gil J (2008). Structure of pulmonary surfactant membranes and films: the role of proteins and lipid-protein interactions. Biochim. Biophys. Acta 1778, 1676–1695. [DOI] [PubMed] [Google Scholar]
- Perez-Gil J, and Weaver TE (2010). Pulmonary surfactant pathophysiology: current models and open questions. Physiology (Bethesda) 25, 132–141. [DOI] [PubMed] [Google Scholar]
- Rindler TN, Stockman CA, Filuta AL, Brown KM, Snowball JM, Zhou W, Veldhuizen R, Zink EM, Dautel SE, Clair G, et al. (2017). Alveolar injury and regeneration following deletion of ABCA3. JCI Insight 2, e97381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson GN, Croll RP, and Smith FM (2014). The structure of the caudal wall of the zebrafish (Danio rerio) swim bladder: evidence of localized lamellar body secretion and a proximate neural plexus. J. Morphol 275, 933–948. [DOI] [PubMed] [Google Scholar]
- Sanford EL, Choy KW, Donahoe PK, Tracy AA, Hila R, Loscertales M, and Longoni M (2016). MiR-449a Affects Epithelial Proliferation during the Pseudoglandular and Canalicular Phases of Avian and Mammal Lung Development. PLoS ONE 11, e0149425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz G, and Müller G (1991). Structure and function of lamellar bodies, lipid-protein complexes involved in storage and secretion of cellular lipids. J. Lipid Res 32, 1539–1570. [PubMed] [Google Scholar]
- Shoemaker CJ, Huang TQ, Weir NR, Polyakov NJ, Schultz SW, and Denic V (2019). CRISPR screening using an expanded toolkit of autophagy reporters identifies TMEM41B as a novel autophagy factor. PLoS Biol. 17, e2007044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulenin S, Nogee LM, Annilo T, Wert SE, Whitsett JA, and Dean M (2004). ABCA3 gene mutations in newborns with fatal surfactant deficiency. N. Engl. J. Med 350, 1296–1303. [DOI] [PubMed] [Google Scholar]
- Søreng K, Neufeld TP, and Simonsen A (2018). Membrane Trafficking in Autophagy. Int. Rev. Cell Mol. Biol 336, 1–92. [DOI] [PubMed] [Google Scholar]
- Stahlman MT, Gray MP, Falconieri MW, Whitsett JA, and Weaver TE (2000). Lamellar body formation in normal and surfactant protein B-deficient fetal mice. Lab. Invest 80, 395–403. [DOI] [PubMed] [Google Scholar]
- Strømhaug PE, and Seglen PO (1993). Evidence for acidity of prelysosomal autophagic/endocytic vacuoles (amphisomes). Biochem. J 291, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Li Z, Hu W, Ren H, Tian E, Zhao Y, Lu Q, Huang X, Yang P, Li X, et al. (2010). C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell 141, 1042–1055. [DOI] [PubMed] [Google Scholar]
- Tooze SA, and Yoshimori T (2010). The origin of the autophagosomal membrane. Nat. Cell Biol 12, 831–835. [DOI] [PubMed] [Google Scholar]
- Tsuboyama K, Koyama-Honda I, Sakamaki Y, Koike M, Morishita H, and Mizushima N (2016). The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science 354, 1036–1041. [DOI] [PubMed] [Google Scholar]
- Velikkakath AK, Nishimura T, Oita E, Ishihara N, and Mizushima N (2012). Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol. Biol. Cell 23, 896–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorbroker DK, Voorhout WF, Weaver TE, and Whitsett JA (1995). Post-translational processing of surfactant protein C in rat type II cells. Am. J. Physiol 269, L727–L733. [DOI] [PubMed] [Google Scholar]
- Wasano K, and Hirakawa Y (1994). Lamellar bodies of rat alveolar type 2 cells have late endosomal marker proteins on their limiting membranes. Histochemistry 102, 329–335. [DOI] [PubMed] [Google Scholar]
- Weaver TE, Na CL, and Stahlman M (2002). Biogenesis of lamellar bodies, lysosome-related organelles involved in storage and secretion of pulmonary surfactant. Semin. Cell Dev. Biol 13, 263–270. [DOI] [PubMed] [Google Scholar]
- Weng JS, Nakamura T, Moriizumi H, Takano H, Yao R, and Takekawa M (2019). MCRIP1 promotes the expression of lung-surfactant proteins in mice by disrupting CtBP-mediated epigenetic gene silencing. Commun. Biol 2, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winata CL, Korzh S, Kondrychyn I, Zheng W, Korzh V, and Gong Z (2009). Development of zebrafish swimbladder: The requirement of Hedgehog signaling in specification and organization of the three tissue layers. Dev. Biol 331, 222–236. [DOI] [PubMed] [Google Scholar]
- Yamada T, Carson AR, Caniggia I, Umebayashi K, Yoshimori T, Nakabayashi K, and Scherer SW (2005). Endothelial nitric-oxide synthase antisense (NOS3AS) gene encodes an autophagy-related protein (APG9-like2) highly expressed in trophoblast. J. Biol. Chem 280, 18283–18290. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Miki Y, Sato H, Nishito Y, Gelb MH, Taketomi Y, and Murakami M (2016). Expression and Function of Group IIE Phospholipase A2 in Mouse Skin. J. Biol. Chem 291, 15602–15613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano G, Funahashi H, Kawanami O, Zhao LX, Ban N, Uchida Y, Morohoshi T, Ogawa J, Shioda S, and Inagaki N (2001). ABCA3 is a lamellar body membrane protein in human lung alveolar type II cells. FEBS Lett. 508, 221–225. [DOI] [PubMed] [Google Scholar]
- Yeganeh B, Lee J, Ermini L, Lok I, Ackerley C, and Post M (2019). Autophagy is required for lung development and morphogenesis. J. Clin. Invest 129, 2904–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii SR, Kuma A, Akashi T, Hara T, Yamamoto A, Kurikawa Y, Itakura E, Tsukamoto S, Shitara H, Eishi Y, and Mizushima N (2016). Systemic Analysis of Atg5-Null Mice Rescued from Neonatal Lethality by Transgenic ATG5 Expression in Neurons. Dev. Cell 39, 116–130. [DOI] [PubMed] [Google Scholar]
- Zheng W, Wang Z, Collins JE, Andrews RM, Stemple D, and Gong Z (2011). Comparative transcriptome analyses indicate molecular homology of zebrafish swimbladder and mammalian lung. PLoS ONE 6, e24019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets/code.