

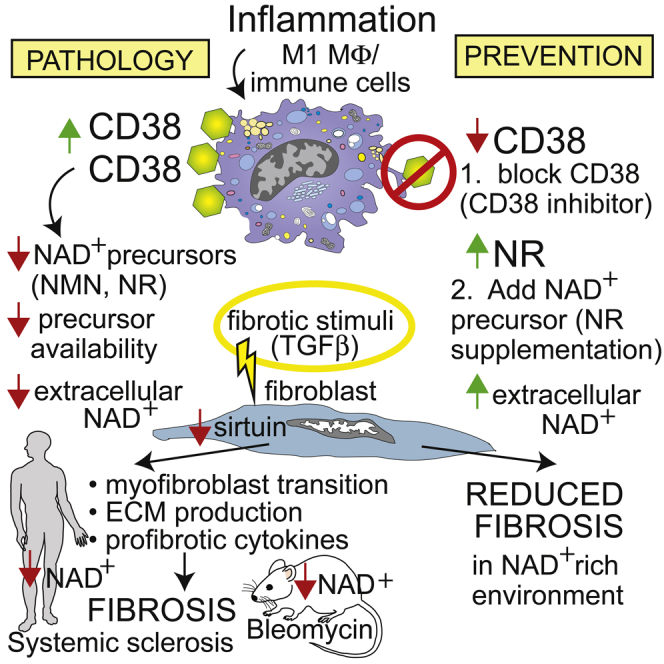
Summary
The processes underlying synchronous multiple organ fibrosis in systemic sclerosis (SSc) remain poorly understood. Age-related pathologies are associated with organismal decline in nicotinamide adenine dinucleotide (NAD+) that is due to dysregulation of NAD+ homeostasis and involves the NADase CD38. We now show that CD38 is upregulated in patients with diffuse cutaneous SSc, and CD38 levels in the skin associate with molecular fibrosis signatures, as well as clinical fibrosis scores, while expression of key NAD+-synthesizing enzymes is unaltered. Boosting NAD+ via genetic or pharmacological CD38 targeting or NAD+ precursor supplementation protected mice from skin, lung, and peritoneal fibrosis. In mechanistic experiments, CD38 was found to reduce NAD+ levels and sirtuin activity to augment cellular fibrotic responses, while inhibiting CD38 had the opposite effect. Thus, we identify CD38 upregulation and resulting disrupted NAD+ homeostasis as a fundamental mechanism driving fibrosis in SSc, suggesting that CD38 might represent a novel therapeutic target.
Subject areas: Human Metabolism, Molecular Biology, Immunology
Graphical Abstract
Highlights
-
•
CD38 shows elevated expression in skin biopsies of patients with systemic sclerosis
-
•
Elevated CD38 is associated with reduced NAD+ and augmented fibrotic responses
-
•
Genetic loss of CD38 is associated with increased NAD+ levels and attenuated fibrosis
-
•
NAD+ boosting via CD38 inhibition or NR supplementation prevents multi-organ fibrosis
Human Metabolism; Molecular Biology; Immunology
Introduction
Systemic sclerosis (SSc), a multisystem disease associated with high mortality, follows unpredictable clinical course and lacks effective therapy (Asano and Varga, 2019; Allanore et al., 2015). Gene expression profiling in SSc has uncovered deregulation of fibrotic and inflammatory/immune pathways, as well as patient-to-patient molecular heterogeneity (Franks et al., 2019). While the pathogenesis remains incompletely understood, a defining SSc hallmark is fibrosis that synchronously affects the skin and lungs and other internal organs (Varga and Abraham, 2007; Distler et al., 2019). Although the extent of fibrosis in SSc and its temporal trajectory are variable between individual patients and across affected organs, intractable fibrosis can lead to lethal organ failure (Bhattacharyya et al., 2011; Ho et al., 2014). Persistent tissue accumulation of myofibroblasts accounts for all forms of fibrosis (Hinz et al., 2012). Activated myofibroblasts originate from tissue-resident progenitors; however, the factors responsible for reprogramming these cells into activated myofibroblasts and of equal importance, preventing myofibroblast reversion toward a metabolically quiescent inactive state, remain largely unknown (Zhao et al., 2020). Thus, the development of effective anti-fibrotic therapies is predicated on comprehensive annotation of disease-specific mechanisms that positively or negatively regulate the differentiation, metabolism, and survival of profibrotic mesenchymal cells (Hinz and Lagares, 2020).
Sirtuins are nicotinamide adenine dinucleotide (NAD+)-dependent deacetylases best known for their fundamental roles in regulation of aging, health span, and longevity (Bonkowski and Sinclair, 2016). Recent studies uncovered an additional important role for sirtuins in regulating fibrotic responses, with suppression of collagen production and myofibroblast differentiation in vivo and in vitro (Wei et al., 2015; Akamata et al., 2016; Sosulski et al., 2017). Moreover, we and others have described impaired sirtuin expression and activity in fibrotic skin and lungs from patients with SSc, implicating sirtuin dysregulation in pathogenesis (Wei et al., 2015; Akamata et al., 2016; Sosulski et al., 2017; Zerr et al., 2016). However, the mechanisms underlying sirtuin dysregulation in patients with SSc are currently unknown. NAD+ is a cofactor for key metabolic processes and a substrate for sirtuins and other enzymes involved in cell signaling and damage repair (Yoshino et al., 2018; Chini et al., 2017). Recent studies have shown that levels of NAD+ as well as its precursors decline during natural aging, as well as in progeroid syndromes (Camacho-Pereira et al., 2016; Frederick et al., 2016; Gomes et al., 2013; Zhu et al., 2015; Tarrago et al., 2018). Notably, sirtuin activity is tightly regulated by NAD+ bioavailability. Indeed, aging- and disease-associated decline in NAD+ is accompanied by sirtuin dysfunction that contributes to pathogenesis (Guarente, 2011). The multifunctional NADase enzyme CD38 is a type II plasma membrane protein expressed on both immune and non-immune cells. CD38 shows ectoenzyme activity with the catalytic site facing the outside of the cell (De Flora et al., 1997; Boslett et al., 2018a; Chatterjee et al., 2018). CD38 is also present on intracellular organelles, including the nucleus and mitochondria (Sun et al., 2002; Aksoy et al., 2006a; Zhao et al., 2012; Peclat et al., 2020). As the main NAD+-hydrolyzing enzyme in mammalian tissues, CD38 plays a key role in age-dependent decline in NAD+ (Camacho-Pereira et al., 2016; Aksoy et al., 2006a, 2006b; Barbosa et al., 2007). Indeed, we showed that tissue levels and activity of CD38 are negatively correlated with NAD+ levels during aging, and CD38 inhibition preserves tissue NAD+ (Camacho-Pereira et al., 2016). The relative roles of CD38 extracellular versus the intracellular enzymatic activity in its biological effects have not been completely defined. However, in the majority of cells, CD38 is expressed mostly as an ecto-enzyme, a phenomenon known as the “topological paradox”. Interestingly, recently we have demonstrated that in addition to controlling NAD+ levels, CD38 also modulates the bioavailability of nicotinamide mononucleotide (NMN) and other extracellular precursors for intracellular NAD+ biosynthesis, its activity governs organismal NAD+ homeostasis (Camacho-Pereira et al., 2016).
We now show that CD38 is substantially elevated in the skin in patients with SSc, and levels are associated with both clinical disease severity and profibrotic signaling activity, while expression of key synthesizing enzymes in the NAD+ salvage pathway are unaltered compared to healthy controls. Genetic and pharmacological targeting of CD38 raised NAD+ levels, leading to attenuation of in vitro and in vivo fibrotic responses in explanted human skin fibroblasts and in preclinical disease models in mice, respectively. Similarly, boosting organismal NAD+ via dietary supplementation with the precursor nicotinamide riboside (NR) prevented fibrosis in the skin, lungs, and peritoneal membrane. Thus, CD38 has a previously unrecognized pathogenic role in multiple organ fibrosis via dysregulation of NAD+ homeostasis and sirtuin dysfunction. Targeting CD38-mediated NAD+ metabolism might therefore represent a novel therapeutic approach to ameliorate chronic fibrosis.
Results
NAD+-consuming enzymes are elevated in SSc
Cellular NAD+ levels are tightly regulated via balanced NAD+ degradation and production via de novo and salvage pathways (Canto et al., 2015; Yoshino et al., 2018; Chini et al., 2017). Dysregulated NAD+ homeostasis, one of the hallmarks of biological aging, is accompanied by age-related metabolic and functional decline and a variety of pathologies in humans and in mice (Johnson and Imai, 2018; Chini et al., 2017). The decline in NAD+ levels that occurs during aging has been shown to be due to changes in NAD+ consumption and production. One of the key enzymes mediating age-related NAD+ catabolism is the NADase CD38 (Camacho-Pereira et al., 2016). To determine if, similar to aging, SSc is also accompanied by altered expression of CD38 and other enzymes involved in maintaining NAD+ homeostasis, we queried a transcriptome data set (GSE76886). Several cellular enzymes associated with either NAD+ consumption or NAD+ production showed differential mRNA expression in SSc skin biopsies, but only differences in expression of CD38, nicotinamide N-methyltransferase (NNMT), and IDO1 reached statistical significance (Figure 1B). In particular, CD38 mRNA was significantly elevated in skin biopsies from patients with SSc and diffuse cutaneous disease (p < 0.0001) but not with limited cutaneous disease (p = 0.479), with levels showing correlation with both clinical disease severity measured by the modified Rodnan skin score (MRSS), as well as with molecular markers of fibrosis (TGF-β and PDGF pathway scores) in the skin (Figures 1C–1E). In marked contrast, expression of CD38 showed negative correlation with SIRT1 pathway scores in the same skin biopsies (Figure 1F), consistent with compromised SIRT1 activity associated with CD38 upregulation. Significantly elevated tissue expression of CD38 in diffuse cutaneous SSc was confirmed by analysis of skin biopsy transcriptome data from two additional independent patient cohorts (Figure S1). The widely expressed cellular enzyme NNMT irreversibly methylates the NAD+ precursor nicotinamide (NAM), thereby reducing its availability for NAD+ salvage via the so-called NAM sink (Eckert et al., 2019; Pissios, 2017). Since upregulation of CD38 in SSc biopsies will generate NAM, which then serves as the substrate for NNMT in the NAD+ clearance pathway (Figure 1A), it was notable that the combined gene score for these two NAD+ consuming enzymes showed even more robust correlation with clinical skin scores and with the fibrotic TGF-β signature (Figure 1G). In contrast to upregulation of NAD+-consuming enzymes observed in the SSc skin biopsies, the principal NAD+ salvage pathway enzymes (nicotinamide phosphoribosyltransferase [NAMPT], NMNAT1, and NMNAT3) (Yoshino et al., 2018; Chini et al., 2017) did not show differential expression, or were suppressed, in SSc (Figure S2A). Although the tissue expression level of NAD+ metabolism genes does not necessarily predict their enzymatic activity, the present results nevertheless suggest that an imbalance in gene expression between NAD+-producing and consuming enzymes might yield altered NAD+ homeostasis in SSc.
Figure 1.

NAD+-consuming enzyme CD38 significantly elevated in SSc skin biopsies
(A) Schematic of cellular NAD+ metabolism, showing key enzymes catalyzing NAD+ consumption and salvage.
(B) Heatmap indicating differential expression of key enzymes catalyzing NAD+ consumption and salvage in healthy control (n = 26) and SSc (n = 68) skin biopsies (GSE76886, red indicates high and green indicates low expression).
(C) CD38 mRNA expression in healthy control and limited and diffuse cutaneous SSc (lcSSc and dcSSc) skin biopsies.
(D–F) Correlation of CD38 mRNA levels with modified Rodnan skin score (MRSS, range 0–51) and with TGF-β, PDGF, and SIRT1 pathway scores in the skin.
(G) Combined CD38 and NNMT gene score correlation with MRSS and with TGF-β signaling. Pearson's correlation.
(H) Immunohistochemistry of skin biopsies using antibodies to CD38; representative images. Arrows indicate CD38+ cells. Scale bar length represents 100 μm.
(I) Quantitation of CD38+ cells in the dermis; horizontal bars, means ± SEM (standard error of the mean) (10 SSc and 4 healthy control biopsies for (H) and (I), clinical data in Table S1 in Transparent Method).
(J) CD38 mRNA levels in explanted SSc (n = 5) and healthy control (n = 4) fibroblasts determined by qPCR.
(K and L) CD38 in human skin fibroblasts. Confluent cultures were incubated for 24 h with TNF-α (10 ng/mL), IL-13 (10 ng/mL), and TGF-β (10 ng/mL). (K) Whole cell lysates were analyzed by immunoblotting. (L) mRNA levels determined by qPCR, mean ± SEM. Experiments were repeated twice.
AMS: alpha-aminomuconate semialdehyde; ACMSD: aminocarboxymuconate semialdehyde decarboxylase; MNAM: N1-methylnicotinamide; NA: nicotinic acid; NAD: nicotinamide adenine dinucleotide; NAM: nicotinamide; NAMPT: nicotinamide phosphoribosyltransferase; NMN: nicotinamide mononucleotide; NMNAT: nicotinamide mononucleotide adenylyltransferase; NNMT: nicotinamide N-methyltransferase; NR: nicotinamide riboside; NRK: nicotinamide riboside kinase; TRP: tryptophan.
We next generated a CD38 co-expression module defined as all genes (n = 194) showing correlation (Spearman r > 0.5) to CD38 in skin biopsies (GSE76886). Hierarchical clustering of skin biopsies using this 194-gene module as a classifier robustly separated the biopsies into two distinct clusters highly enriched for either healthy controls or SSc biopsies (chi-square p = 4.9∗10−6) (Figure S2B). To further evaluate CD38 expression in the skin, we immunostained skin biopsies from patients with SSc and healthy controls (Table S1) using anti-CD38 antibodies. The numbers of CD38-immunopositive interstitial cells in the dermis were significantly elevated in SSc biopsies, with comparable increases noted in both the papillary and reticular dermal layers (Figures 1H and 1I). In explanted SSc skin fibroblasts (n = 5), CD38 mRNA levels were elevated compared to healthy matched control fibroblasts even after their serial ex vivo passages in culture (Figure 1J). Cellular expression of CD38 can be induced by a variety of soluble mediators (Hogan et al., 2019). Incubation of normal skin fibroblasts with TNF-α, IL-13, Toll-like receptor ligands, and TGF-β, each of which are implicated in SSc pathogenesis (Allanore et al., 2015), caused marked stimulation of CD38 expression (Figures 1K and 1L).
We next sought to characterize alterations in NAD+ metabolism pathway enzymes in a bleomycin-induced mouse model of SSc characterized by inflammation and fibrosis in the skin, lung, and other organs (Yue et al., 2018). Interrogating a gene expression data set from bleomycin-treated mice (GSE71999) showed that CD38 mRNA expression was significantly elevated (p < 0.005) in fibrotic skin, and CD38 levels were correlated with NNMT levels (R2 0.433, p = 0.004) (Figure S3A), while mRNA expression of NAD+-synthesizing enzymes was comparable to that in untreated mice of the same age (Sargent et al., 2016). Upregulated expression of CD38 in fibrotic skin was confirmed in an independent mouse experiment (GSE 132869: Figure S3B). Subcutaneous bleomycin injection was accompanied by a substantial increase in CD38+ cells in the lungs, with elevated CD38 expression noted on both peripherally derived CD45-positive leukocytes, as well as tissue-resident CD45-negative stromal cell populations (Figure S3C). In particular, we noted significantly increased numbers of CD38+ inflammatory monocytes, macrophages, plasmacytoid dendritic cells, and neutrophils in the lung from bleomycin-treated mice, whereas no change in B cells was seen (Figure S3D). Consistent with the putative functional linkage between enhanced NAD+ consumption and fibrosis, bleomycin-treated mice demonstrated a significant drop in both circulating and tissue levels of NAD+ (Figure S3E). Thus, fibrosis-associated upregulation of CD38 expression that is uncoupled from concomitant upregulation of NAD+-producing enzymes will trigger pathogenic organismal NAD+ depletion.
Deletion of CD38 ameliorated fibrosis
In view of the consistent coupling of skin fibrosis and increased CD38 expression observed both in patients with SSc as well as in preclinical disease models, together with reduced circulating and tissue NAD+ levels in fibrotic mice, we sought to investigate the potential pathogenic role of CD38 by determining if targeting CD38 will mitigate fibrosis. For this purpose, we first characterized inducible fibrosis in CD38-null mice (Transparent Methods, Experimental Animals) at one year of age, when NAD+ levels in wild-type mice show a significant decline (Figure 2A) (Camacho-Pereira et al., 2016). Absent CD38 expression in these mice was confirmed by immunoblot and flow cytometry in multiple tissues and cell types (Figure 2B). At baseline, the proportions of immune cell populations in the spleen were comparable in wild-type and CD38-null mice (data not shown). Bone-marrow-derived CD38-null macrophages showed unaltered ex vivo M1/M2 polarization; however, compared to wild-type M1 macrophages, CD38-null macrophages showed markedly impaired production of IL-6 and IL-1β (Figure 2C). These findings are consistent with previous reports documenting unaltered maturation and differentiation, but impaired function, of immune cells in CD38-null mice (Cockayne et al., 1998). One-year-old CD38-null mice showed 2- to 3-fold higher NAD+ levels in multiple tissues (skin, lung, liver) compared to age-matched wild-type control mice (Figure 2D). CD38-null mice weighed less than wild-type mice of the same age, as reported previously (Chiang et al., 2015), but no overt behavioral or phenotypic differences were observed.
Figure 2.

Deletion of CD38 ameliorated skin and lung fibrosis
(A) Schematic of experimental design. Twelve-month-old female CD38-null mice or wild-type control mice on standard chow received daily s.c. bleomycin (BLM) injections (10 mg/kg) for 14 d. After 21 d, mice were sacrificed. Five to 7 mice were used for each group, and experiments were repeated 2 times.
(B) CD38 expression undetectable in inguinal lymph nodes (representative immunoblot) and spleen cells (flow cytometry) in CD38-null mice.
(C) Reduced cytokine gene expression (qPCR) in CD38-null bone-marrow-derived M1 macrophages.
(D) Elevated NAD+ levels in skin, lungs, and liver in CD38-null mice.
(E) Attenuated increase in dermal thickness (representative images, trichrome stain; scale bar represents 100 μm) and collagen deposition in CD38-null mice. Results are means ± SEM (standard error of the mean) from an experiment representative of two independent experiments.
(F) Bleomycin treatment-induced weight loss is mitigated in CD38-null mice (n = 5–7 mice/per group).
(G–J) Lung fibrosis induced by bleomycin is attenuated in CD38-null mice. (G) microCT of the lungs (n = 5 mice/group). Representative cross-sectional and 3D images (left) and quantitative lung fibrosis index (right). (H) Lung histology (trichrome stain, scale bar represents 100 μm); representative images (left) and measurement of lung fibrosis (right). (I and J) Collagen accumulation and fibrotic gene expression in the lungs. Error bars in all graphs, means ± SEM.
Chronic subcutaneous bleomycin administration in mice is a widely used approach to model SSc and fibrosis (Marangoni et al., 2016). In this model, bleomycin induces multiple organ fibrosis associated with both innate and adaptive immune response cells, including activation of CD4/8 T cells, B cells, and dendritic cells, thereby recapitulating the principal inflammatory-fibrotic processes that characterize SSc (Allanore et al., 2015). Remarkably, CD38-null mice showed attenuation of the bleomycin-induced fibrotic process, including increase in dermal thickness and collagen accumulation (Figure 2E). Notably, weight loss sustained during the course of bleomycin treatment was significantly attenuated in CD38-null mice (Figure 2F). Furthermore, lungs from CD38-null mice treated with bleomycin also showed that the severity of radiologic and histological fibrosis significantly ameliorated, albeit not fully abrogated (Figures 2G and 2H). Attenuation of fibrosis was accompanied by reduced collagen accumulation and expression of fibrotic genes COL1A1 and COL3A1, as well as the alternately spliced extracellular matrix components fibronectin-EDA and tenascin-C, recognized markers of fibrosis that are abundantly expressed in the fibrotic tissue in SSc (Figures 2I and 2J) (Van Der Straaten et al., 2004; Bhattacharyya et al., 2014, 2016).
NAD+ boosting ameliorated skin and lung fibrosis in mice
In view of the substantial fibrosis protection accompanied by a rise of NAD+ levels that was noted in mice lacking CD38, the principal NAD+-consuming enzyme, we sought to determine if organismal NAD+ boosting by either selectively blocking its CD38-mediated hydrolysis and/or by NAD+ precursor supplementation will mitigate fibrosis. To this end, 18-month-old female mice maintained for 2 weeks on either standard chow diet or chow supplemented with nicotinamide riboside (NR, 3 g/kg chow) were treated with a thiazoloquin(az)olin(on)e compound 78c selective for the CD38 NADase (Tarrago et al., 2018). Treatment with 78c (30 mg/kg weight) administered by daily oral gavage was initiated concomitantly with a two-week course of daily subcutaneous bleomycin injections, and mice remained on NR-supplemented chow diet and continued daily 78c gavage until sacrifice at day 21 (Figure 3A). On standard chow diet, mice treated with bleomycin suffered significant weight loss (maximal 40% at day 11), which however was substantially attenuated in mice on an NR-supplemented chow diet combined with the CD38 inhibitor 78c (Figure 3B). Dietary NR supplementation by itself, or combined with 78c treatment, resulted in a ∼5-fold increase in skin NAD+ levels compared to mice treated with bleomycin only (Figure 3E). Notably, boosting NAD+ in bleomycin-treated mice resulted in significantly improved skin fibrosis, including attenuated increase in dermal thickness, skin collagen content, and fibrotic gene expression, as well as the number of ASMA (alpha smooth muscle actin)-positive myofibroblasts in the lesional dermis (Figures 3C, 3D, 3F, 3G, S4A and S4B). In the skin, the anti-fibrotic efficacy of supplementation with NR was greater compared to the efficacy of CD38 inhibitor treatment. This differential response might reflect limited tissue penetration of the CD38 inhibitor. The expansion of the fibrotic dermis was inversely correlated with tissue levels of NAD+, directly linking NAD+ boosting in the anti-fibrotic effect (Figure 3E). Strikingly, NAD+ boosting by either NR supplementation or via CD38 inhibition also afforded mice robust protection from pulmonary fibrosis, with significantly ameliorated radiological and histological fibrotic changes in the lungs coupled with reduced collagen accumulation and fibrotic gene expression (Figures 3C and 3H–3K). Monocytes and macrophages are thought to play an important role in the pathogenesis of organ fibrosis in SSc and in mouse models of the disease (Bhandari et al., 2020; Marangoni et al., 2016). Significant accumulation of macrophages in both the dermis and the lungs from bleomycin-treated mice was mitigated with NAD+ boosting via treatment of mice with CD38 inhibitor or NR supplementation alone or in combination (Figure S5A and S5B). In particular, the combination treatment markedly attenuated the accumulation of CD11b + macrophages in the skin (Figure S5C) and CD64+CD11b+Sigleclow (myeloid derived) infiltrating macrophages in the lung, while having no significant effect on CD64+CD11b+Siglechigh (tissue resident) macrophages (Figure S5D). In view of the pivotal role that macrophages and other myeloid-derived leukocytes play in SSc pathogenesis (Bhandari et al., 2020), the decrease in macrophage accumulation observed in mice with NAD+ boosting might contribute to the anti-fibrotic effect of this intervention.
Figure 3.

NAD+ boosting ameliorated skin and lung fibrosis in aged mice
(A) Schematic of experimental design. Eighteen-month-old female C57/BL6 mice maintained on standard chow or NAD precursor nicotinamide riboside (NR)-supplemented chow diet. Mice received daily s.c. bleomycin (BLM) injections for 14 d alone or combined with CD38 inhibitor 78c administered by oral gavage. At 21 d, mice were sacrificed and skin and lungs were harvested for analysis. Results from three independent experiments with 6–8 mice per group.
(B–D) NAD+ boosting attenuated bleomycin-induced (B) body weight loss, and (C and D) increase in dermal thickness (representative images, trichrome stain; scale bars represent 100 μm). Results are means ± SEM (standard error of the mean) from an experiment representative of three independent experiments.
(E) NAD+ levels in the skin; results are means ± SEM; negative correlation (Pearson's) of skin NAD+ levels with dermal thickness.
(F–H) (F and G) Skin collagen content (hydroxyproline assays); and gene expression (qPCR); (C) (H) Lung fibrosis quantification (modified Ashcroft score).
(I) Lung collagen content.
(J) Lung fibrosis imaging, representative microCT images (left) and fibrosis quantitation (right).
(K) mRNA levels in the lung determined by qPCR; results are means ± SEM.
Next, we examined how genetic or pharmacological CD38 targeting will modulate the development of the peritoneal membrane fibrosis induced by i.p. chlorhexidine gluconate (CG) injections (Bhattacharyya et al., 2018). Since this inducible model reproduces many fundamental processes underlying the pathogenesis of SSc, including inflammation, myofibroblast accumulation, and extracellular matrix accumulation, it is used widely to investigate pathogenic mechanisms of fibrosis (Costalonga et al., 2020; Kitamura et al., 2015). Mice lacking CD38 suffered less weight loss during the course of CG treatment and showed significantly attenuated peritoneal membrane thickening and collagen deposition (Figures S6A and S6B). Moreover, accumulation of myofibroblasts in the fibrotic peritoneal membrane was also dramatically attenuated in mice lacking CD38 (Figure S6C). Comparable protection from CG-induced peritoneal fibrosis was seen when wild-type mice were treated with the combination of NR plus CD38 inhibitor (Figure S6). Together, these results further highlight the fundamental role of CD38 in governing the intensity of fibrotic processes in multiple organs.
CD38 and NAD+ metabolisms govern cellular fibrotic responses
To investigate how CD38 governs profibrotic cellular responses at a mechanistic level, a series of experiments with explanted human and mouse cells were performed. In order to experimentally recapitulate cellular CD38's outward-facing enzyme function in the in vivo milieu (Tarrago et al., 2018; Hogan et al., 2019), we incubated quiescent skin fibroblasts at confluence with recombinant human CD38. CD38 augmented the profibrotic effects of TGF-β, with increased COL1A1 and ASMA mRNA expression, collagen production, and myofibroblast differentiation, which were accompanied by increased lysine acetylation, an established marker for sirtuin activity (Figures 4A and 4B). In contrast, supplementing fibroblast cultures with NR led to a significant rise in cellular NAD+ levels that was accompanied by attenuation of TGF-β-induced profibrotic responses including stimulation of ASMA and COL1A1 gene expression (Figures 4C–4E). Likewise, incubation of human skin fibroblasts with 78c, which selectively inhibits CD38 NADase activity, suppressed basal collagen gene expression and ASMA levels and prevented their stimulation by TGF-β (Figures 4F and 4H). Moreover, skin fibroblasts explanted from CD38-null mice failed to respond to profibrotic TGF-β stimulation (Figure 4I). In contrast, pharmacological blockade of the cellular NAD+ salvage pathway using the NAMPT inhibitor FK866 resulted in enhanced fibrotic gene expression, coupled to enhanced acetylation of cellular lysines and MnSOD, confirming a reduction in sirtuin activity (Figures 4J and 4K).
Figure 4.

CD38 and NAD+ levels govern fibrotic cellular responses
Confluent foreskin fibroblasts (all except [G] and [I]) or CD38-null mouse fibroblasts (I) were incubated with TGF-β1 (10 ng/mL) in the presence or absence of recombinant human CD38 (rhCD38), 78c CD38 inhibitor (CD38i, 1 μM), FK866 (10 nM), and NR (50 μM or indicated concentrations).
(A) Levels of COL1A1 and ASMA mRNA determined by qPCR.
(B) Collagen production and sirtuin activity determined by immunoblot.
(C–E) (L) NR supplementation reduced collagen and ASMA stimulation, increased cellular NAD+ levels (E), and mitigated stimulation of p300 (L).
(F and I) Collagen and ASMA levels in wild-type and CD38-null mouse fibroblasts detected by immunoblotting and qPCR.
(G) Mouse bone-marrow-derived macrophages were ex vivo differentiated and incubated with LPS to induce endogenous CD38, followed by treatment with CD38i. Levels of lysine acetylation, a validated marker of sirtuin activity, were determined; representative immunoblot.
(H) CD38 inhibitor 78c and NR reduce levels of collagen, ASMA, and Fn-EDA.
(J and K) Incubation with FK866 (NAMPT inhibitor); effects on gene expression and lysine acetylation detected by qPCR and immunoblot.
(M and N) CD38 inhibition and NR mitigated myofibroblast transformation (confocal microscopy), fibrotic gene expression, and Smad2 phosphorylation and nuclear translocation (immunoblot).
(O) NR supplementation reduced Smad-dependent transcriptional activity in transiently transfected fibroblasts.
Experiments were performed in triplicate and repeated three times ([C] [D] [F] [K] [L]) or twice ([A] [B] [M] [N] [O]). Error bars in all graphs, means ± SEM (standard error of the mean).
To examine the impact of CD38 blockade on sirtuin activity, we used bone-marrow-derived macrophages, which show inducible expression of endogenous CD38 upon LPS treatment (Camacho-Pereira et al., 2016). In LPS (lipopolysaccharides)-treated CD38+ macrophages, NAD+ boosting 78c treatment resulted in increased sirtuin, as demonstrated by decreased levels of acetylated lysine (Figure 4G). Sirtuin activation itself had been previously implicated in suppression of fibrotic responses (Wei et al., 2015; Akamata et al., 2016). To further investigate the cellular mechanisms involved in the anti-fibrotic effects of NAD+ boosting, we examined the effect of NR supplementation on mediators of profibrotic cellular fibrotic responses, focusing on the histone acetyltransferase p300 and Smad2. We had shown previously that p300 is a TGF-β-inducible cofactor that is indispensable for Smad-dependent cellular responses and is implicated in SSc pathogenesis (Ghosh et al., 2013; Ghosh and Varga, 2007). Significantly, NR supplementation of fibroblasts prevented the induction of p300 by TGF-β (Figure 4L). Additionally, Smad2 phosphorylation and nuclear translocation, which are induced by TGF-β to mediate its profibrotic effects (Mori et al., 2004), were markedly reduced by NR supplementation, as well as by CD38 inhibition (Figure 4M). Moreover, NR plus CD38 inhibition attenuated, although failed to completely prevent, fibroblast differentiation into myofibroblasts (Figure 4M and 4N). Induction of Smad-dependent transcriptional activity by TGF-β was only partially reduced by NR (Figure 4O). These results together demonstrate that negative and positive modulation of cellular NAD+ homeostasis in fibroblasts via orthogonal loss-of-function and gain-of-function approaches profoundly influence myofibroblast activation, profibrotic cellular signaling, and regulation of fibrotic gene expression.
Discussion
The present results demonstrate that SSc, the prototypic multisystem fibrotic disease, is associated with elevated expression of the NAD+-consuming enzyme CD38 in the absence of parallel increase in NAD+ salvage pathway enzymes, with CD38 levels correlating with both clinical disease severity and with fibrotic signal activity in the skin. Similar alterations in tissue CD38 seen in a mouse model of scleroderma were accompanied by significant reduction in systemic NAD+, reflecting a conserved linkage between disrupted NAD+ homeostasis and fibrosis in both SSc and in mouse models of disease. Blocking CD38 in vivo via either genetic targeting or treatment with a selective NADase inhibitor alone or combined with NAD+ precursor supplementation resulted in boosting NAD+ levels and reduced fibrosis propensity in multiple organs. Fibrotic responses were also attenuated in explanted fibroblasts lacking CD38. Similarly, boosting NAD+ via precursor supplementation of fibroblast cultures attenuated the magnitude of inducible fibrotic cellular responses via increased sirtuin activity and disruption of Smad-dependent canonical TGF-β signaling, while in contrast, ectopic CD38 augmented these fibrotic responses. Together, these observations uncover a previously unsuspected fundamental role for CD38-mediated NAD+ metabolism in SSc fibrosis.
Ample evidence supports the autoimmune nature of SSc, with dysregulated humoral and cellular immunity, both of which prominently contribute to disease pathogenesis (Allanore et al., 2015). Similar to normal wound healing, SSc has an early inflammatory phase where leukocyte-derived mediators trigger fibroblast activation and differentiation into myofibroblasts (Distler et al., 2019; Hinz and Lagares, 2020). In contrast to physiologic self-limited tissue repair, however, unresolving fibrosis in SSc is associated with durable myofibroblast activation and persistence resulting from combination of cell-autonomous metabolic and epigenetic changes coupled with structural and biochemical alterations in the tissue microenvironment. We now demonstrate that the multifunctional NADase CD38 was significantly elevated in skin biopsies from multiple independent SSc patient cohorts. Described originally as a surface marker of activated T cells, CD38 is in fact broadly expressed on lymphoid and myeloid immune cells, as well as stromal cells including endothelial cells and fibroblasts (Reinherz et al., 1979; Malavasi et al., 2008; Tarrago et al., 2018). A principal enzymatic activity of CD38 is NAD+ hydrolysis, which accounts for the key roles of CD38 in several physiologic and disease processes including inflammation, cancer, and metabolic diseases (Barbosa et al., 2007; Malavasi et al., 2008; Chini et al., 2018). Moreover, it is increasingly recognized that decline in NAD+ levels that accompanies chronological aging in both humans and rodents is a key factor underlying the development of frailty and age-related metabolic and health decline (Zhu et al., 2015; Braidy et al., 2011; Massudi et al., 2012; Clement et al., 2019; Tarrago et al., 2018; Camacho-Pereira et al., 2016). Notably, CD38 appears to be the enzyme largely responsible for the organismal NAD+ decline and resultant sirtuin dysfunction that characterize aging (Camacho-Pereira et al., 2016).
Originally identified on immune cells as a cell surface marker, CD38 is a widely expressed multifunctional enzyme that catalyzes the cleavage of a β-glycoside bond between nicotinamide and the ribose moiety of NAD+, with the majority of NADase activity generating nicotinamide (Chini et al., 2018). The cellular localization of CD38 is unusual, with a majority showing a type II membrane orientation with the catalytic site facing the outside of the cell (Da Silva et al., 1998). However, CD38 is also present on intracellular organelles and has endoenzyme activity. The apparent “topological paradox” of CD38 ectoenzyme activity regulating NAD+ homeostasis since NAD+ is predominantly intracellular was recently shown to be attributable, at least, in part, to CD38 regulation of the extracellular levels of NMN as a precursor for intracellular NAD+ synthesis (Chini et al., 2020; Camacho-Pereira et al., 2016; Shrimp et al., 2014; Tarrago et al., 2018). Via these catalytic processes, CD38 regulates organismal NAD+ homeostasis and plays a fundamental role in determining the activity of multiple sirutins along with other NAD+-dependent cellular processes (Aksoy et al., 2006b). In the heart, CD38 expression on both endothelial cells and fibroblasts showed marked induction following ischemia reperfusion injury, and treatment with CD38 inhibitor had a strong cardio-protective effect associated with reversal of NAD+ decline (Reyes et al., 2015; Boslett et al., 2018a, 2018b, 2019). We recently demonstrated upregulated CD38 expression and activity in aging mice and showed that CD38 is largely responsible for age-dependent organismic decline in NAD+ levels (Camacho-Pereira et al., 2016). Both genetic targeting of CD38, as well as treatment with a selective thiazoloquin(az)olin(on)e CD38 inhibitor, reversed age-related NAD+ decline, rescued sirtuin activity, and ameliorated multiple physiological and metabolic parameters of aging, including frailty, declining muscle and cardiac function, and exercise capacity, in mouse models of natural and accelerated aging (Tarrago et al., 2018). We now demonstrate that CD38 expression is elevated in SSc skin biopsies compared to that of healthy controls in a manner analogous to CD38 upregulation seen in aging mice and show that genetic and pharmacological NAD+ boosting has salutary anti-fibrotic effects in mouse models. These observations draw attention to intriguing parallels between the biology of aging and SSc fibrosis attributable to CD38-dependent dysregulation of NAD+ homeostasis.
In human skin, CD38 is detected primarily on T cells, myeloid cells, pericytes, and plasma cells (Tabib et al., 2018). The expression of CD38 is positively regulated in a variety of cell types by inflammatory and profibrotic cytokines, including IL-13, TGF-β, and IL-6, along with the senescence-associated secretory pattern. These extracellular cues, via NF-κΒ- and STAT-mediated intracellular signaling, induce CD38 expression to reduce NAD+ levels (Chini et al., 2018, 2019). Analysis of transcriptome data sets showed increased levels of CD38 in skin biopsies from multiple independent SSc patient cohorts and positive correlation between CD38 expression in the skin and clinical measures and molecular markers of fibrosis. However, whether it is the action IL-6 or other secreted factors that lead to elevated CD38 expression in patients with SSc, and the cell types in the skin that are primarily responsible remain to be established. We had shown previously that the functional and metabolic decline that accompany aging are attenuated in CD38-null mice (Tarrago et al., 2018). We now demonstrate that CD38-null mice were also substantially protected from fibrosis in the skin, lung, and peritoneal lining membrane. Moreover, treatment of aged mice with the selective and potent CD38 NADase inhibitor 78c markedly reduced tissue accumulation of CD38+ inflammatory cells, reversed NAD+ decline, and substantially ameliorated fibrotic and inflammatory changes, while genetic and pharmacological modulation of NAD+ metabolism ex vivo in cultured fibroblasts elicited marked effects on the transcriptional regulation of fibrotic responses. Whether the anti-fibrotic effect of CD38 inhibition we observed in distinct fibrosis models is due primarily to reduced tissue accumulation of macrophages and inflammatory cells, or to a direct anti-fibrotic activity, remains to be conclusively established. Association of disrupted NAD+ homeostasis with altered tissue repair, extracellular matrix remodeling, and fibrosis has been previously recognized, albeit the experimental support coming primarily from animal models (Peclat et al., 2020). For instance, NAD+ boosting via dietary NR supplementation was shown to ameliorate fibrosis of the skeletal muscle and heart in mdx mice (Ryu et al., 2016) and fibrosis of the liver (Zhou et al., 2016; Pham et al., 2019). An alternate NAD+ boosting strategy using NMN supplementation was shown to similarly ameliorate fibrosis in the lung induced by intratracheal bleomycin (Liu et al., 2020). Moreover, we had previously shown that spontaneous fibrosis of the skeletal muscle in aging mice is associated with NAD+ depletion and impaired sirtuin activity and can be mitigated by treatment with the NADase inhibitor 78c (Tarrago et al., 2018). These observations, taken together with the present results in mouse models of fibrosis induced by subcutaneous bleomycin or intraperitoneal chlorhexidine gluconate treatment, further strengthen the link between NAD+ dysregulation and fibrosis and provide compelling support for the potential of NAD+ boosting as a novel therapeutic strategy to prevent, slow the progression, or promoter regression, of fibrosis. These observations notwithstanding, it remains conceivable that CD38 elevation observed in the SSc skin biopsies and in lesional tissue from bleomycin-treated mice in the present report contribute to fibrosis pathogenesis in these conditions through alternate mechanisms that are independent of NAD+ consumption and depletion. These alternate pathogenic mechanisms linking CD38 and fibrosis merit further investigation.
In summary, we demonstrate that in patients with SSc, as well as in preclinical disease models in mice, fibrosis is associated with upregulation of NAD+-consuming enzymes in the absence of concomitant elevated NAD+ production, leading to dysregulated NAD+ homeostasis. Consequent decline in NAD+ levels and functional impairment of sirtuins and other NAD+-dependent metabolic enzymes contributes directly to myofibroblast transition that underlies intractable fibrosis in SSc. Boosting NAD+ levels via genetic or pharmacological CD38 targeting, or via NAD+ precursor supplementation, mitigated multiple organ fibrosis in rodent models and in explanted fibroblasts, at least in part by augmenting sirtuin activity and disrupting Smad-dependent core profibrotic TGF-β signaling. Thus, pharmacological approaches to boost organismal NAD+ via inhibiting CD38 activity, by NAD+ precursor supplementation or by a combination of both, represent potential therapeutic strategies for slowing or reversing fibrosis in SSc.
Limitations of the study
We acknowledge some limitations to our study. First, while we demonstrate elevated expression of the NAD+ consuming enzyme CD38 in the skin biopsies from multiple independent SSc patient cohorts, raising the possibility that CD38 upregulation leads to enhanced NAD+ catabolism and consequent decline in tissue NAD+ bioavailability, we have not provided direct evidence of reduced NAD+ levels in these biopsies. Additional studies will therefore measure levels of NAD+, along with its precursors and metabolites, in both tissue and circulation from patients with SSc and age-matched healthy controls. Second, the present studies do not clearly delineate the principal cell types in the skin responsible for elevated CD38 expression in SSc biopsies. Third, the present studies do not provide unambiguous evidence that the principal biological contributions of CD38 upregulation to SSc pathogenesis are mediated via its outward-facing catalytic activity. The relative role of the ectoenzyme function of CD38 in disease pathogenesis can be addressed in future studies evaluating the impact of agents that selectively target CD38 ectoenzyme activity in disease models.
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, John Varga (vargaj@umich.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate data sets.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (NIH) and National Institute on Aging (NIA) grants 1R56AG054207 (J.V.), AR074523 (J.V. and E.N.C.), AG-26094 (E.N.C), AG58812 (E.N.C), AR070285 (B. K.), AR073371 (S.D.M. and J.V.), P30 AR073371 (J.G.), Scleroderma Foundation Research Grant (D.X.), and the Glenn Foundation for Medical Research via the Paul F. Glenn Laboratories for the Biology of Aging at the Mayo Clinic (E.N.C). We gratefully acknowledge Mary Carns and Kathleen Aren for technical assistance, as well as the Northwestern Mouse Histopathology & Phenotyping Core, Pathology Core, and Nikon Imaging Center and Small Animal Imaging Facility Core for their services. We acknowledge GSK for providing 78c and ChromaDex for providing NR.
Author contributions
J.V., E.N.C., and B.S. generated the hypothesis, designed all experiments, analyzed the results, and prepared the manuscript. B.S., W.X.W., L.K., and S.B. conducted and performed in vivo and the main in vitro experiments. B.K., J.W., and J.G. analyzed gene expression in human and mouse samples. G.C.O., J.M.E.N., C.C.S.C., and E.N.C analyzed NAD metabolism. R.G.M., Q.W., D.G., and B.S. analyzed tissue morphology and expression levels and performed experiments with cultured cells. D.X., S.M., and M.A. performed flow cytometry and data analysis. P.C. determined lung fibrosis score. D.P. performed lung CT imaging and data analysis. All authors contributed to the preparation of the manuscript.
Declaration of interests
J.V. reports being a consultant for TeneoBio and Mitobridge. C.C.S.C. reports holding a patent on the use of CD38 inhibitors for metabolic diseases that is licensed by Elysium Health. E.N.C reports being a consultant for TeneoBio, Calico, Mitobridge, and Cyokinetics. E.N.C is on the advisory board of Eolo pharmaceutical, Argentina. J.G.'s research was support by grants from Lilly, Almirall, AbbVie, Kyowa Kirin, and BMS/Celgene; and he reports serving on the Advisory Board of Lilly, BMS, Novartis, Kyowa Kirin, AnaptysBio, and Almirall.
Published: January 22, 2021
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101902.
Supplemental information
References
- Akamata K., Wei J., Bhattacharyya M., Cheresh P., Bonner M.Y., Arbiser J.L., Raparia K., Gupta M.P., Kamp D.W., Varga J. SIRT3 is attenuated in systemic sclerosis skin and lungs, and its pharmacologic activation mitigates organ fibrosis. Oncotarget. 2016;7:69321–69336. doi: 10.18632/oncotarget.12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksoy P., Escande C., White T.A., Thompson M., Soares S., Benech J.C., Chini E.N. Regulation of SIRT 1 mediated NAD dependent deacetylation: a novel role for the multifunctional enzyme CD38. Biochem. Biophys. Res. Commun. 2006;349:353–359. doi: 10.1016/j.bbrc.2006.08.066. [DOI] [PubMed] [Google Scholar]
- Aksoy P., White T.A., Thompson M., Chini E.N. Regulation of intracellular levels of NAD: a novel role for CD38. Biochem. Biophys. Res. Commun. 2006;345:1386–1392. doi: 10.1016/j.bbrc.2006.05.042. [DOI] [PubMed] [Google Scholar]
- Allanore Y., Simms R., Distler O., Trojanowska M., Pope J., Denton C.P., Varga J. Systemic sclerosis. Nat. Rev. Dis. Primers. 2015;1:15002. doi: 10.1038/nrdp.2015.2. [DOI] [PubMed] [Google Scholar]
- Asano Y., Varga J. Rationally-based therapeutic disease modification in systemic sclerosis: novel strategies. Semin. Cell Dev. Biol. 2019;101:148–160. doi: 10.1016/j.semcdb.2019.12.007. [DOI] [PubMed] [Google Scholar]
- Barbosa M.T., Soares S.M., Novak C.M., Sinclair D., Levine J.A., Aksoy P., Chini E.N. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB J. 2007;21:3629–3639. doi: 10.1096/fj.07-8290com. [DOI] [PubMed] [Google Scholar]
- Bhandari R., Ball M.S., Martyanov V., Popovich D., Schaafsma E., Han S., Eltanbouly M., Orzechowski N.M., Carns M., Arroyo E. Profibrotic activation of human macrophages in systemic sclerosis. Arthritis Rheumatol. 2020;72:1160–1169. doi: 10.1002/art.41243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S., Tamaki Z., Wang W., Hinchcliff M., Hoover P., Getsios S., White E.S., Varga J. FibronectinEDA promotes chronic cutaneous fibrosis through Toll-like receptor signaling. Sci. Transl Med. 2014;6:232ra50. doi: 10.1126/scitranslmed.3008264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S., Wang W., Morales-Nebreda L., Feng G., Wu M., Zhou X., Lafyatis R., Lee J., Hinchcliff M., Feghali-Bostwick C. Tenascin-C drives persistence of organ fibrosis. Nat. Commun. 2016;7:11703. doi: 10.1038/ncomms11703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S., Wang W., Qin W., Cheng K., Coulup S., Chavez S., Jiang S., Raparia K., De Almeida L.M.V., Stehlik C. TLR4-dependent fibroblast activation drives persistent organ fibrosis in skin and lung. JCI Insight. 2018;3:e98850. doi: 10.1172/jci.insight.98850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S., Wei J., Varga J. Understanding fibrosis in systemic sclerosis: shifting paradigms, emerging opportunities. Nat. Rev. Rheumatol. 2011;8:42–54. doi: 10.1038/nrrheum.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonkowski M.S., Sinclair D.A. Slowing ageing by design: the rise of NAD(+) and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 2016;17:679–690. doi: 10.1038/nrm.2016.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boslett J., Helal M., Chini E., Zweier J.L. Genetic deletion of CD38 confers post-ischemic myocardial protection through preserved pyridine nucleotides. J. Mol. Cell Cardiol. 2018;118:81–94. doi: 10.1016/j.yjmcc.2018.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boslett J., Hemann C., Christofi F.L., Zweier J.L. Characterization of CD38 in the major cell types of the heart: endothelial cells highly express CD38 with activation by hypoxia-reoxygenation triggering NAD(P)H depletion. Am. J. Physiol. Cell Physiol. 2018;314:C297–C309. doi: 10.1152/ajpcell.00139.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boslett J., Reddy N., Alzarie Y.A., Zweier J.L. Inhibition of CD38 with the Thiazoloquin(az)olin(on)e 78c Protects the Heart against Postischemic Injury. J. Pharmacol. Exp. Ther. 2019;369:55–64. doi: 10.1124/jpet.118.254557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braidy N., Guillemin G.J., Mansour H., Chan-Ling T., Poljak A., Grant R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS One. 2011;6:e19194. doi: 10.1371/journal.pone.0019194. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Camacho-Pereira J., Tarrago M.G., Chini C.C., Nin V., Escande C., Warner G.M., Puranik A.S., Schoon R.A., Reid J.M., Galina A., Chini E.N. CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab. 2016;23:1127–1139. doi: 10.1016/j.cmet.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C., Menzies K.J., Auwerx J. NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 2015;22:31–53. doi: 10.1016/j.cmet.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S., Daenthanasanmak A., Chakraborty P., Wyatt M.W., Dhar P., Selvam S.P., Fu J.N., Zhang J.Y., Nguyen H., Kang I.H. CD38-NAD(+)axis regulates immunotherapeutic anti-tumor t cell response. Cell Metab. 2018;27:85. doi: 10.1016/j.cmet.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chini C., Hogan K.A., Warner G.M., Tarrago M.G., Peclat T.R., Tchkonia T., Kirkland J.L., Chini E. The NADase CD38 is induced by factors secreted from senescent cells providing a potential link between senescence and age-related cellular NAD(+) decline. Biochem. Biophys. Res. Commun. 2019;513:486–493. doi: 10.1016/j.bbrc.2019.03.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chini C.C.S., Peclat T.R., Warner G.M., Kashyap S., Espindola-Netto J.M., De Oliveira G.C., Gomez L.S., Hogan K.A., Tarragó M.G., Puranik A.S. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD+ and NMN levels. Nat. Metab. 2020;2:1284–1304. doi: 10.1038/s42255-020-00298-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chini C.C.S., Tarrago M.G., Chini E.N. NAD and the aging process: role in life, death and everything in between. Mol. Cell Endocrinol. 2017;455:62–74. doi: 10.1016/j.mce.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang S.H., Harrington W.W., Luo G.Z., Milliken N.O., Ulrich J.C., Chen J., Rajpal D.K., Qian Y., Carpenter T., Murray R. Genetic ablation of CD38 protects against Western diet-induced exercise intolerance and metabolic inflexibility. PLoS One. 2015;10:e0134927. doi: 10.1371/journal.pone.0134927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chini E.N., Chini C.C.S., Espindola Netto J.M., De Oliveira G.C., Van Schooten W. The pharmacology of CD38/NADase: an emerging target in cancer and diseases of aging. Trends Pharmacol. Sci. 2018;39:424–436. doi: 10.1016/j.tips.2018.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement J., Wong M., Poljak A., Sachdev P., Braidy N. The plasma NAD(+) metabolome is dysregulated in "normal" aging. Rejuvenation Res. 2019;22:121–130. doi: 10.1089/rej.2018.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockayne D.A., Muchamuel T., Grimaldi J.C., Muller-Steffner H., Randall T.D., Lund F.E., Murray R., Schuber F., Howard M.C. Mice deficient for the ecto-nicotinamide adenine dinucleotide glycohydrolase CD38 exhibit altered humoral immune responses. Blood. 1998;92:1324–1333. [PubMed] [Google Scholar]
- Costalonga E.C., Fanelli C., Garnica M.R., Noronha I.L. Adipose-derived mesenchymal stem cells modulate fibrosis and inflammation in the peritoneal fibrosis model developed in uremic rats. Stem Cells Int. 2020;2020:3768718. doi: 10.1155/2020/3768718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Silva C.P., Schweitzer K., Heyer P., Malavasi F., Mayr G.W., Guse A.H. Ectocellular CD38-catalyzed synthesis and intracellular Ca2+-signalling activity of cyclic ADP-ribose in T-lymphocytes are not functionally related. FEBS Lett. 1998;439:291–296. doi: 10.1016/s0014-5793(98)01396-9. [DOI] [PubMed] [Google Scholar]
- De Flora A., Guida L., Franco L., Zocchi E. The CD38/cyclic ADP-ribose system: a topological paradox. Int. J. Biochem. Cell Biol. 1997;29:1149–1166. doi: 10.1016/s1357-2725(97)00062-9. [DOI] [PubMed] [Google Scholar]
- Distler J.H.W., Gyorfi A.H., Ramanujam M., Whitfield M.L., Konigshoff M., Lafyatis R. Shared and distinct mechanisms of fibrosis. Nat. Rev. Rheumatol. 2019;15:705–730. doi: 10.1038/s41584-019-0322-7. [DOI] [PubMed] [Google Scholar]
- Eckert M.A., Coscia F., Chryplewicz A., Chang J.W., Hernandez K.M., Pan S., Tienda S.M., Nahotko D.A., Li G., Blazenovic I. Proteomics reveals NNMT as a master metabolic regulator of cancer-associated fibroblasts. Nature. 2019;569:723–728. doi: 10.1038/s41586-019-1173-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks J.M., Martyanov V., Cai G., Wang Y., Li Z., Wood T.A., Whitfield M.L. a machine learning classifier for assigning individual patients with systemic sclerosis to intrinsic molecular subsets. Arthritis Rheumatol. 2019;71:1701–1710. doi: 10.1002/art.40898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick D.W., Loro E., Liu L., Davila A., Jr., Chellappa K., Silverman I.M., Quinn W.J., 3rd, Gosai S.J., Tichy E.D., Davis J.G. Loss of NAD homeostasis leads to progressive and reversible degeneration of skeletal muscle. Cell Metab. 2016;24:269–282. doi: 10.1016/j.cmet.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A.K., Bhattacharyya S., Lafyatis R., Farina G., Yu J.X., Thimmapaya B., Wei J., Varga J. p300 is elevated in systemic sclerosis and its expression is positively regulated by TGF-beta: epigenetic feed-forward amplification of fibrosis. J. Invest. Dermatol. 2013;133:1302–1310. doi: 10.1038/jid.2012.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A.K., Varga J. The transcriptional coactivator and acetyltransferase p300 in fibroblast biology and fibrosis. J. Cell Physiol. 2007;213:663–671. doi: 10.1002/jcp.21162. [DOI] [PubMed] [Google Scholar]
- Gomes A.P., Price N.L., Ling A.J., Moslehi J.J., Montgomery M.K., Rajman L., White J.P., Teodoro J.S., Wrann C.D., Hubbard B.P. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155:1624–1638. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L. Franklin H. Epstein lecture: sirtuins, aging, and medicine. N. Engl. J. Med. 2011;364:2235–2244. doi: 10.1056/NEJMra1100831. [DOI] [PubMed] [Google Scholar]
- Hinz B., Lagares D. Evasion of apoptosis by myofibroblasts: a hallmark of fibrotic diseases. Nat. Rev. Rheumatol. 2020;16:11–31. doi: 10.1038/s41584-019-0324-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz B., Phan S.H., Thannickal V.J., Prunotto M., Desmouliere A., Varga J., De Wever O., Mareel M., Gabbiani G. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am. J. Pathol. 2012;180:1340–1355. doi: 10.1016/j.ajpath.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho Y.Y., Lagares D., Tager A.M., Kapoor M. Fibrosis--a lethal component of systemic sclerosis. Nat. Rev. Rheumatol. 2014;10:390–402. doi: 10.1038/nrrheum.2014.53. [DOI] [PubMed] [Google Scholar]
- Hogan K.A., Chini C.C.S., Chini E.N. The multi-faceted ecto-enzyme CD38: roles in immunomodulation, cancer, aging, and metabolic diseases. Front Immunol. 2019;10:1187. doi: 10.3389/fimmu.2019.01187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson S., Imai S.I. NAD (+) biosynthesis, aging, and disease. F1000Res. 2018;7:132. doi: 10.12688/f1000research.12120.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura M., Nishino T., Obata Y., Oka S., Abe S., Muta K., Ozono Y., Koji T., Kohno S. The kampo medicine Daikenchuto inhibits peritoneal fibrosis in mice. Biol. Pharm. Bull. 2015;38:193–200. doi: 10.1248/bpb.b14-00469. [DOI] [PubMed] [Google Scholar]
- Liu T., Rinke A.E., Wang J., Phan S.H. Cellular NAD+, fibroblast senescence and pulmonary fibrosis. FASEB J. 2020;34:1. [Google Scholar]
- Malavasi F., Deaglio S., Funaro A., Ferrero E., Horenstein A.L., Ortolan E., Vaisitti T., Aydin S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol. Rev. 2008;88:841–886. doi: 10.1152/physrev.00035.2007. [DOI] [PubMed] [Google Scholar]
- Marangoni R.G., Varga J., Tourtellotte W.G. Animal models of scleroderma: recent progress. Curr. Opin. Rheumatol. 2016;28:561–570. doi: 10.1097/BOR.0000000000000331. [DOI] [PubMed] [Google Scholar]
- Massudi H., Grant R., Braidy N., Guest J., Farnsworth B., Guillemin G.J. Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One. 2012;7:e42357. doi: 10.1371/journal.pone.0042357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori Y., Ishida W., Bhattacharyya S., Li Y., Platanias L.C., Varga J. Selective inhibition of activin receptor-like kinase 5 signaling blocks profibrotic transforming growth factor beta responses in skin fibroblasts. Arthritis Rheum. 2004;50:4008–4021. doi: 10.1002/art.20658. [DOI] [PubMed] [Google Scholar]
- Peclat T.R., Shi B., Varga J., Chini E.N. The NADase enzyme CD38: an emerging pharmacological target for systemic sclerosis, systemic lupus erythematosus and rheumatoid arthritis. Curr. Opin. Rheumatol. 2020;32:488–496. doi: 10.1097/BOR.0000000000000737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham T.X., Bae M., Kim M.B., Lee Y., Hu S., Kang H., Park Y.K., Lee J.Y. Nicotinamide riboside, an NAD+ precursor, attenuates the development of liver fibrosis in a diet-induced mouse model of liver fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2019;1865:2451–2463. doi: 10.1016/j.bbadis.2019.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pissios P. Nicotinamide N-methyltransferase: more than a vitamin B3 clearance enzyme. Trends Endocrinol. Metab. 2017;28:340–353. doi: 10.1016/j.tem.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinherz E.L., Kung P.C., Goldstein G., Schlossman S.F. Separation of functional subsets of human t-cells by a monoclonal antibody. Proc. Natl. Acad. Sci. U S A. 1979;76:4061–4065. doi: 10.1073/pnas.76.8.4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes L.A., Boslett J., Varadharaj S., De Pascali F., Hemann C., Druhan L.J., Ambrosio G., El-Mahdy M., Zweier J.L. Depletion of NADP(H) due to CD38 activation triggers endothelial dysfunction in the postischemic heart. Proc. Natl. Acad. Sci. U S A. 2015;112:11648–11653. doi: 10.1073/pnas.1505556112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu D., Zhang H., Ropelle E.R., Sorrentino V., Mazala D.A., Mouchiroud L., Marshall P.L., Campbell M.D., Ali A.S., Knowels G.M. NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation. Sci. Transl Med. 2016;8:361ra139. doi: 10.1126/scitranslmed.aaf5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargent J.L., Li Z., Aliprantis A.O., Greenblatt M., Lemaire R., Wu M.H., Wei J., Taroni J., Harris A., Long K.B. Identification of optimal mouse models of systemic sclerosis by interspecies comparative genomics. Arthritis Rheumatol. 2016;68:2003–2015. doi: 10.1002/art.39658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrimp J.H., Hu J., Dong M., Wang B.S., Macdonald R., Jiang H., Hao Q., Yen A., Lin H. Revealing CD38 cellular localization using a cell permeable, mechanism-based fluorescent small-molecule probe. J. Am. Chem. Soc. 2014;136:5656–5663. doi: 10.1021/ja411046j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosulski M.L., Gongora R., Feghali-Bostwick C., Lasky J.A., Sanchez C.G. Sirtuin 3 deregulation promotes pulmonary fibrosis. J. Gerontol. A. Biol. Sci. Med. Sci. 2017;72:595–602. doi: 10.1093/gerona/glw151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L., Adebanjo O.A., Koval A., Anandatheerthavarada H.K., Iqbal J., Wu X.Y., Moonga B.S., Wu X.B., Biswas G., Bevis P.J. A novel mechanism for coupling cellular intermediary metabolism to cytosolic Ca2+ signaling via CD38/ADP-ribosyl cyclase, a putative intracellular NAD+ sensor. FASEB J. 2002;16:302–314. doi: 10.1096/fj.01-0705com. [DOI] [PubMed] [Google Scholar]
- Tabib T., Morse C., Wang T., Chen W., Lafyatis R. SFRP2/DPP4 and FMO1/LSP1 define major fibroblast populations in human skin. J. Invest Dermatol. 2018;138:802–810. doi: 10.1016/j.jid.2017.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarrago M.G., Chini C.C.S., Kanamori K.S., Warner G.M., Caride A., De Oliveira G.C., Rud M., Samani A., Hein K.Z., Huang R. A potent and specific CD38 inhibitor ameliorates age-related metabolic dysfunction by reversing tissue NAD(+) decline. Cell Metab. 2018;27:1081–1095 e10. doi: 10.1016/j.cmet.2018.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Straaten H.M., Canninga-Van Dijk M.R., Verdonck L.F., Castigliego D., Borst H.P., Aten J., Fijnheer R. Extra-domain-A fibronectin: a new marker of fibrosis in cutaneous graft-versus-host disease. J. Invest Dermatol. 2004;123:1057–1062. doi: 10.1111/j.0022-202X.2004.23474.x. [DOI] [PubMed] [Google Scholar]
- Varga J., Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J. Clin. Invest. 2007;117:557–567. doi: 10.1172/JCI31139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J., Ghosh A.K., Chu H., Fang F., Hinchcliff M.E., Wang J., Marangoni R.G., Varga J. The histone deacetylase sirtuin 1 is reduced in systemic sclerosis and Abrogates fibrotic responses by targeting transforming growth factor beta signaling. Arthritis Rheumatol. 2015;67:1323–1334. doi: 10.1002/art.39061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino J., Baur J.A., Imai S.I. NAD(+) Intermediates: the biology and therapeutic potential of NMN and NR. Cell Metab. 2018;27:513–528. doi: 10.1016/j.cmet.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue X., Yu X., Petersen F., Riemekasten G. Recent advances in mouse models for systemic sclerosis. Autoimmun. Rev. 2018;17:1225–1234. doi: 10.1016/j.autrev.2018.06.013. [DOI] [PubMed] [Google Scholar]
- Zerr P., Palumbo-Zerr K., Huang J., Tomcik M., Sumova B., Distler O., Schett G., Distler J.H. Sirt1 regulates canonical TGF-beta signalling to control fibroblast activation and tissue fibrosis. Ann. Rheum. Dis. 2016;75:226–233. doi: 10.1136/annrheumdis-2014-205740. [DOI] [PubMed] [Google Scholar]
- Zhao X., Kwan J.Y.Y., Yip K., Liu P.P., Liu F.F. Targeting metabolic dysregulation for fibrosis therapy. Nat. Rev. Drug Discov. 2020;19:57–75. doi: 10.1038/s41573-019-0040-5. [DOI] [PubMed] [Google Scholar]
- Zhao Y.J., Lam C.M.C., Lee H.C. The membrane-bound enzyme CD38 exists in two opposing orientations. Sci. Signal. 2012;5:ra67. doi: 10.1126/scisignal.2002700. [DOI] [PubMed] [Google Scholar]
- Zhou C.C., Yang X., Hua X., Liu J., Fan M.B., Li G.Q., Song J., Xu T.Y., Li Z.Y., Guan Y.F. Hepatic NAD(+) deficiency as a therapeutic target for non-alcoholic fatty liver disease in ageing. Br. J. Pharmacol. 2016;173:2352–2368. doi: 10.1111/bph.13513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X.H., Lu M., Lee B.Y., Ugurbil K., Chen W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc. Natl. Acad. Sci. U S A. 2015;112:2876–2881. doi: 10.1073/pnas.1417921112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate data sets.