

Abstract
Metastatic cancer involving spread to the peritoneal cavity is referred to as peritoneal carcinomatosis and has a very poor prognosis. Our previous study demonstrated a toll-like receptor (TLR) and C-type lectin receptor (CLR) agonist pairing of monophosphoryl lipid A (MPL) and trehalose-6,6’-dicorynomycolate (TDCM) effectively inhibits tumor growth and ascites development following TA3-Ha and EL4 challenge through a mechanism dependent upon B-1a cell-produced natural IgM and complement. In the current study, we investigated additional players in the MPL/TDCM-elicited response. MPL/TDCM treatment rapidly increased type I IFN levels in the peritoneal cavity along with myeloid cell numbers, including macrophages and Ly6Chi monocytes. Type I IFN receptor (IFNAR1−/−) mice produced tumor-reactive IgM following MPL/TDCM treatment, but failed to recruit Ly6C+ monocytes and were not afforded protection during tumor challenges. Clodronate liposome depletion of phagocytic cells, as well as targeted depletion of Ly6C+ cells, also ablated MPL/TDCM-induced protection. Cytotoxic mediators known to be produced by these cells were required for effects. TNFα was required for effective TA3-Ha killing and nitric oxide was required for EL4 killing. Collectively, these data reveal a model whereby MPL/TDCM-elicited anti-tumor effects strongly depend on innate cell responses, with B-1a cell-produced tumor-reactive IgM and complement pairing with myeloid cell-produced cytotoxic mediators to effectively eradicate tumors in the peritoneal cavity.
Introduction
Metastatic spread of cancers is associated with significant morbidity and mortality. In particular, metastasis of tumor cells into the peritoneal cavity is often associated with very poor prognosis, especially when associated with development of malignant ascites(1). This condition, referred to as peritoneal carcinomatosis, is most often caused by cancers of the abdominal region, including ovarian, endometrial, colorectal, gastric, and pancreatic cancers(2). However, extra-abdominal tumors causing peritoneal carcinomatosis also occur and include breast cancer, lung cancers and lymphomas(2). Currently, treatment of advanced stages of ascites-forming peritoneal carcinomatosis involves surgical debulking of solid tumors and serial paracentesis of ascitic fluid as a means of palliative care(3). However, recent procedures including pressurized intraperitoneal aerosol chemotherapy (PIPEC) and hyperthermic intraperitoneal chemotherapy (HIPEC) have had recent success in extending median patient survival up to 1.3 and 3.9 years(4,5), respectively, but at the expense of morbidity and quality of life. Therefore, due to the abysmal prognosis associated with this disease and the lack of curative treatments available, it is essential that more effective treatments be explored.
Relative to other tissues enriched with immune cells, the peritoneal cavity can be considered an immunosuppressive environment. Although exchange of plasma components supplies the peritoneal fluid with many of the proteins found in the circulation (6), additional soluble factors produced in the cavity, including IL-10 constitutively produced by resident B-1 cells as well as prostaglandins, indoleamine 2,3-dioxygenase, and nitric oxide produced by macrophages, tip the balance towards immunosuppression in the peritoneal cavity (7,8). In patients suffering from peritoneal carcinomatosis, ascites contains high levels of IL-10 and TGF-beta, along with regulatory T cells and immunosuppressive macrophages (9), suggesting the suppression within the peritoneal cavity is maintained, if not augmented, under conditions of peritoneal carcinomatosis. Overcoming immune suppression in the peritoneal cavity may provide opportunities for effective treatment of peritoneal carcinomatosis. One strategy that may overcome immune suppression involves activating the immune system through use of pathogen associated molecular patterns (PAMPs). We previously demonstrated a treatment consisting of monophosphoryl lipid A (MPL), a Toll-like receptor 4 (TLR4) agonist, and trehalose-6,6’-dicorynomycolate (TDCM), a ligand for macrophage inducible C-type lectin (Mincle/MCL), provides significant protection in mouse models of breast cancer-induced peritoneal carcinomatosis and peritoneal lymphomatosis(10). In response to this combinational treatment (referred to as MPL/TDCM), B-1a cells are activated to secrete high levels of tumor-reactive IgM that leads to complement-dependent elimination of tumor cells. Thus, despite their role as an immune suppressive population through IL-10 production, MPL/TDCM induces B-1a cells to adapt a tumor-protective, as opposed to tumor-promoting, role.
In the current study, we examined further changes MPL/TDCM causes within the peritoneal environment, as complement-dependent tumor cell lysis did not appear to be the sole mechanism of tumor cell killing. Our previous in vivo studies exhibited the possibility of complement dependent cytotoxicity (CDC)-independent mechanisms of killing within the peritoneal cavity(10). Therefore, we examined alterations in leukocytes and soluble factors within the peritoneal cavity following MPL/TDCM treatment. We identified treatment induces significant increases in peritoneal levels of type I IFN, which in turn, results in Ly6C+ monocyte recruitment and increases in small peritoneal macrophages. In the absence of Ly6C+ cells, phagocytic cells, or the type I IFN receptor, MPL/TDCM treatment was rendered ineffective during tumor challenge. Thus, both innate B-1a cells and Ly6C+ cells are critical for MPL/TDCM-induced protection against peritoneal carcinomatosis. These findings may help us better understand how to optimally engage the appropriate leukocytes to elicit potent anti-tumor activity in the inherently immunosuppressive environment of the peritoneal cavity.
Materials and Methods
Mice
Wild-type (WT), μMT (Ighmtm1Cgn), and CD19Cre(B6.129P2(C)-Cd19tm1(cre)Cgn/J) mice were on a C57BL/6 background and from Jackson Laboratories. IFNAR−/− and IFNARfl/fl (Ifnar1tm1Uka) were kindly provided by Dr. Erik Barton (originally from Drs. Herbert Virgin and Ulrich Kalinke, respectively). Animal experiments were approved by Wake Forest’s Animal Care and Use Committee. Mice were housed in a specific pathogen-free animal facility and were fed standard chow and housed in autoclaved cages. Experiments were conducted using 8- to 12- week old age-matched mice.
Tumor challenges and treatments
TA3-Ha cells were obtained from Dr. Richard Lo-Man (Pasteur Institute, Paris, France) in 2010, as previously described(10,11). EL4 cells were as previously described(10). Ascites stocks were tested negative for rodent pathogens, including Mycoplasma (IMPACT IV testing, IDEXX-RADIL; EL4 in 2016 and TA3-Ha in 2012). An aliquot of each tested ascites stock was passaged twice in C57BL/6 mice to make stock aliquots (stored under liquid nitrogen) to be used in experiments. Cells were expanded in culture for 3 days prior to injection. Mice were given 2–2.5 × 104 TA3-Ha cells in 200 μl PBS i.p. Mice developing ascites with signs of distress were humanely euthanized. The main signs of distress accompanying abdominal swelling (ascites development) were hunching with piloerection and/or reduced movement about the cage (lethargy).
Mice were injected intraperitoneally (i.p.) with Sigma adjuvant system containing 10 μg monophosphoryl lipid A (MPL) and 10 μg synthetic trehalose dicorynomycolate (TDCM) mixed in 0.4% squalene (Sigma) in a 200 μl volume as previously described (10). In the EL4 model, treatment was given 24 hr post challenge, and in the TA3-Ha model, treatment was given on the same day as tumor challenge (within 2 hours of challenge). In some experiments, mice were given 3 mg/kg of N-acetylcysteine (NAC; Sigma) or 400 mg/kg aminoguanidine in 200 ul PBS i.p. as indicated. Anti-Ly6C (Monts 1; BioXcell) or control mAb (LTF-2; BioXcell) were administered i.p. on days −1, 1, and 3 post-tumor challenge (200 μg/dose). Anti-TNFα antibody (XT3.11; BioXcell) was administered i.p. on days 2 (200 μg), 4 (200 μg) and 6 (100 μg) post-tumor challenge. For phagocytic cell depletion, clodronate or control liposomes (FormuMax Ltd.) were given i.p. (100–200 μ1) to mice one day prior to tumor challenge. Adoptive transfers of B-1a cells into μMT mice were performed as previously described(10).
In vitro cytotoxicity assay
Sera was harvested from MPL/TDCM-treated mice on day 14 and incubated with TA3-Ha or EL4 cells (1–2 ×106 cells/ml RPMI + 2% fetal calf serum (FCS)) at a 1:10 dilution with or without 2–10 % standard rabbit complement (Cedarlane, CL3111) for 18–24 hours in a 37°C CO2 incubator. Recombinant murine TNF-alpha (Biolegend) was added to cultures at 10 ng/ml. Viability was measured using live/dead blue aqua (Invitrogen) and annexin V (BD Horizon) staining according to manufacturer’s instructions. Samples were analyzed using a BD LSR Fortessa X20 (Becton Dickenson) and Flowjo analysis software.
Flow cytometry
Peritoneal cells were harvested using 10 ml of DPBS to lavage the peritoneal cavity. Cells were resuspended in staining buffer (PBS containing 2% newborn calf serum) and pre-incubated with 0.5 μg/ml FcBlock (eBioscience) and stained with biotin-conjugated Helix Pomatia agglutinin (HPA: Sigma) and mAbs and streptavidin-conjugated to fluorochromes (Biolegend, eBioscience, and BD Biosciences): CD5 (53–7.3), Ly6G (Gr-1), CD86 (GL-1), CD19 (1D3), CD11b (M1/70), F4/80 (BM8), CD11c (N418), Ly6C (HK1.4), MHC II IA-IE (M5/114.15.2), and CD138 (281–2). Small and large peritoneal macrophages with distinguished as previously described by Cain et al(12). Viability was measured using live/dead blue aqua (Invitrogen) and annexin V (BD Horizon) staining according to manufacturer’s instructions. TA3-Ha cells were identified as HPA+CD11bnegCD138+CD19neg(10,11). Serum Ig, IgM, and complement C3b binding (1:10 dilution) to tumor cells were assessed as previously described(10,11). Cells were washed once with Dulbecco’s PBS (DPBS) containing 2% FCS and stained with goat anti-mouse C3-fluorescein (Cappel; 1/200) and goat anti-mouse Ig(H+L)-Alexa 647 (Invitrogen) for 30 minutes at room temperature. Alternatively, goat anti-mouse IgM F(ab’)2-FITC was incubated with tumor cells that had either been isolated from the peritoneal cavity of challenged mice or cultured cells that had been preincubated with sera (1:10) isolated from MPL/TDCM-treated mice. Samples were washed once with DPBS + 2% FCS and fixed in 1.5% buffered formaldehyde. Intracellular caspase-3 (C92–605) staining was conducted using an intracellular staining protocol in conjunction with eBioscience Fix/Perm kit. Cells were analyzed using a BD LSR Fortessa X20 (Becton Dickinson), with the exception of Annexin V stained cells which were run in the absence of fixation.
Type I IFN Bioassay
Six hours post-MPL/TDCM treatment, the peritoneal cavity was lavaged with 2 ml of DPBS. Each sample was spun down at 330 rcf for 5 minutes and the supernatant was harvested. Supernatant or universal interferon (PBL #11200–2; Pestka Biomedical Laboratories) was incubated with 2 × 105 cells/ml EL4 cells at multiple dilutions for 24 hours at 37°C in a 24-well plate. GFP-expressing vesicular stomatitis virus (kindly provided by Dr. Douglas Lyles) was added to cells at a multiplicity of infection of 3.0(13). Cells were incubated at 37°C for 5 hours. Cells were washed twice with DPBS + 2 % FCS and fixed with 1.5% buffered formaldehyde. Samples were run on BD LSR Fortessa X20.
Statistical analysis
Data are shown as means ± SEM with differences assessed using unpaired Student’s t test unless otherwise indicated. Differences between multiple groups were determined using one-way ANOVA. Differences in Kaplan-Meier survival curves were assessed using the Log Rank test.
Results
TA3-Ha cells are moderately sensitive to complement dependent cytotoxicity (CDC)
Our previous study demonstrated the efficacy of MPL/TDCM treatment against peritoneal carcinomatosis depended on the production of tumor-specific IgM by B-1a cells and classical complement activation(10). Consistent with this, tumor cells recovered from treated mice have high levels of surface-bound IgM and C3 fragments(10). However, some treatment efficacy was also noted in mice lacking C5(10), suggesting additional mechanisms may be involved in tumor cell killing. Incubation of TA3-Ha cells with sera harvested from MPL/TDCM-treated mice along with complement for 24 hours significantly decreased viability (60% viable cells recovered) relative to that observed with sera (80%) or complement (90%) alone (Fig. 1A). Interestingly, viable (annexin VnegLive/deadneg) TA3-Ha cells recovered at 24 hours post culture had substantial surface staining of Ig and C3 deposition (Fig. 1B–C), suggesting there may be some resistance to complement-dependent lysis. Because CDC induces death via both programmed cell death and necrosis(14), we measured caspase-3 staining in tumor cells harvested from mice 5 days post challenge. As shown in Fig. 1D, there was a 4-fold increase in the frequency of caspase-3+ TA3-Ha cells in treated mice. Nonetheless, the majority of recovered cells from treated mice were viable (Live/Deadnegcaspase3neg) yet had robust complement and antibody deposition (Fig. 1E–F), as we previously reported(10). Therefore, our data suggests TA3-Ha cells are partially sensitive to CDC. However, incomplete lysis in vitro and in vivo along with MPL/TDCM efficacy in C5-deficient mice (10) suggests additional mechanisms may contribute to tumor cell killing in the context of MPL/TDCM treatment.
Figure 1. TA3-Ha cells are moderately susceptible to CDC.
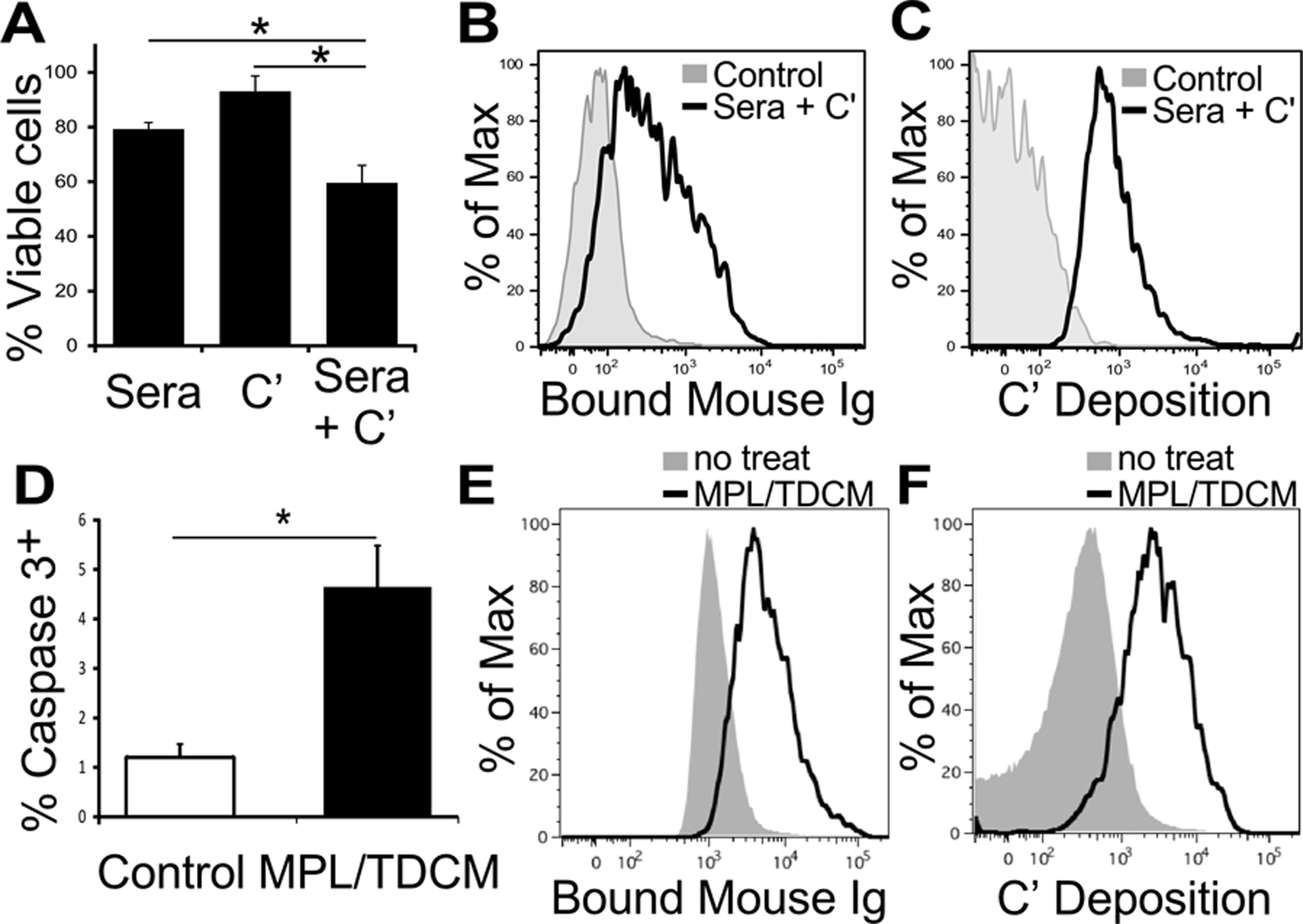
(A-C) TA3-Ha cells were incubated with sera harvested from mice injected with MPL/TDCM (day 14), 10% rabbit complement, or both for 24 hours. (A) The percentage of viable cells (FSChiSSChi live/deadneg annexin Vneg cells) recovered for each culture condition relative to untreated TA3-Ha cells as determined by flow cytometry. Results are pooled data from 3 independent experiments. Significant differences were determined for this pooled data using one-way ANOVA. (B-C) Ig (B) and C3 deposition (C) on treated and untreated (control) TA3-Ha cells at 24 post-incubation. Representative staining results from 3 independent experiments are shown. (D) Frequencies of caspase-3+ TA3-Ha cells among cells recovered from peritoneal cavities of mice 5 days post TA3-Ha challenge (2.5 × 104 i.p.) ± MPL/TDCM treatment on d0. (E-F) Ig (E) and C3 (F) deposition on FSChiSSChi live/deadnegcaspase3neg TA3-Ha cells recovered from mice on d5. Asterisks indicates significance (p<0.05, n=4 mice/group).
Myeloid-derived leukocytes increase in the peritoneal cavity in response to MPL/TDCM
We previously reported that MPL and TDCM rapidly induced B cell expansion, activation and differentiation into antibody secreting cells, with moderate effects on other lymphocyte populations (10). However, MPL and TDCM are potent stimuli that activate multiple immune cell types(15,16). T cell frequencies were not significantly altered by treatment as we had previously reported (10) but NK cell frequencies were increased (Supplemental Fig. 1A, 2A). Notably, β2m−/− mice (lacking MHC class I, CD1, and FcRn), CD1d−/−, as well as WT mice treated with an NK cell depleting mAb were significantly protected by MPL/TDCM treatment (Supplemental Fig. 1B–C and 2B). Protection in β2m−/− mice was not to the same degree as WT mice, suggesting a partial or modifying role for MHC class I or another β2m-bearing receptor with respect to TA3-Ha susceptibility and/or responsiveness to MPL/TDCM-elicited protection. Nonetheless, β2m expression was not an absolute requirement for MPL/TDCM-elicited protection against TA3-Ha challenge.
Given the potential for additional effector leukocytes to contribute to rapid tumor cell clearance following treatment, we examined changes in non-lymphoid cells. MPL has been shown to induce recruitment of myeloid-derived leukocytes to the peritoneum(17). Consistent with this, at 24 hours post-treatment, significant increases in peritoneal Ly6Chi monocyte (5-fold), macrophage, and CD11c+CD11b+ and CD11c+CD11b− dendritic cell numbers were observed (Fig. 2A and Supplemental Fig. 3). Of note, intraperitoneal injection of LPS, which also provides protection against peritoneal carcinomatosis(10), causes significant influx of inflammatory monocytes into the peritoneal cavity, which then differentiate into small peritoneal macrophages (SPMs)(18,19). Consistent with this, MPL/TDCM treatment caused a significant increase in SPM and decrease in large peritoneal macrophage (LPM) numbers at 24 hours (Fig. 2A). These changes in numbers corresponded to significant and preferential increases in monocyte and SPM, and decreases in LPM, frequencies among leukocytes (Supplemental Fig. 4A, C). At 3 days post-treatment, monocyte and SPM numbers and their frequencies among leukocytes were further increased (Fig. 2B and Supplemental Fig. 3B) and these cells had significantly higher expression of CD86, as well as increased representation of MHC II+ cells (Fig. 2C), indicating a more classically activated phenotype(19). Increased numbers of DCs expressing MHC class II+ were also present with treatment, although CD86 expression was not further increased (Fig. 2C and Supplemental Fig. 3B–C). Increases in neutrophils were also observed post treatment (Supplemental Fig. 5). B cells were not required for MPL/TDCM-induced increases in monocytes, macrophage or dendritic cell populations as recruitment was normal in B cell deficient mumt mice (Supplemental Fig. 6). Thus, MPL/TDCM treatment significantly increases the number of myeloid-derived leukocytes in the peritoneal cavity.
Figure 2. Myeloid-derived leukocyte populations are increased in the peritoneal cavity post MPL/TDCM treatment.
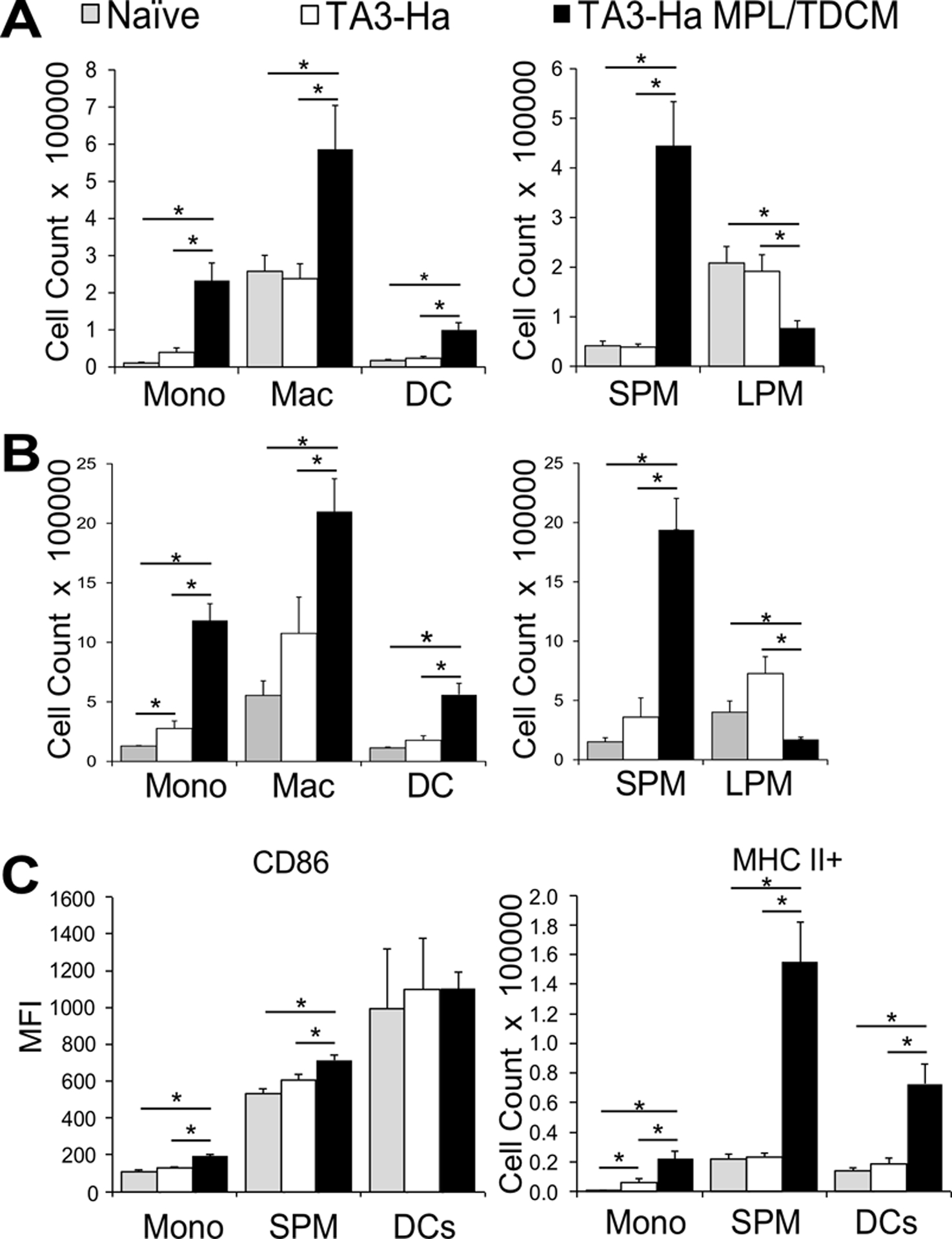
(A-B) Inflammatory monocytes (CD11b+ Ly6G− CD11c− B220− Ly6C+ SSClow), macrophages (CD11b+ Ly6G− CD11c− B220− Ly6Clow SSClow) and dendritic cells (cDC2; CD11b+ Ly6G− CD11c+ B220−) in peritoneal cavities on day 1 (A) and day 3 (B) post TA3-Ha cell challenge ± MPL/TDCM treatment on d0. Small peritoneal macrophages (SPM; CD11bmidF4/80mid) and large peritoneal macrophages (LPM; CD11bhiF4/80hi) were further quantified. (C) CD86 expression level (MFI) and MHC II+ monocyte and SPM cell numbers on day 1 post-tumor challenge. Values represent means ± SEM. Asterisks (*) indicate significant differences between indicated groups (p<0.05; n=3–5 mice/group.) (Mono: monocyte, Mac: macrophage, DC: dendritic cell, SPM: small peritoneal macrophage, LPM: large peritoneal macrophage). Results and statistical analysis for one of two representative experiments are shown.
IFN-I is rapidly produced following MPL/TDCM treatment and is required anti-tumor effects
Type I interferon (IFN-I) has the potential to be activated through TRIF signaling from the TLR4 pathway as well as through CLR signaling and is known to promote rapid leukocyte recruitment(20). ELISAs were not sensitive enough to detect type I IFN in diluted peritoneal lavage samples. We therefore assessed whether type I IFN was produced in response to MPL/TDCM treatment using a virus infectivity bioassay as a readout of IFN I activity(21) (Supplemental Fig. 7A). Significant increases in IFN-I were evident in the peritoneal cavity 6 hours post-MPL/TDCM treatment (Fig. 3A). The release of IFN-I was only detectable at 6 hours post-treatment, with time points extending to 48 hours (Supplemental Fig. 7B). Importantly, MPL/TDCM treatment did not provide protection to IFNAR1−/− mice challenged with TA3-Ha cells (Fig. 3B) or EL4 cells (Supplemental Fig. 8A) demonstrating a critical role for type I IFN signaling in MPL/TDCM-elicited protection.
Figure 3. MPL/TDCM anti-tumor responses are type I interferon-dependent.
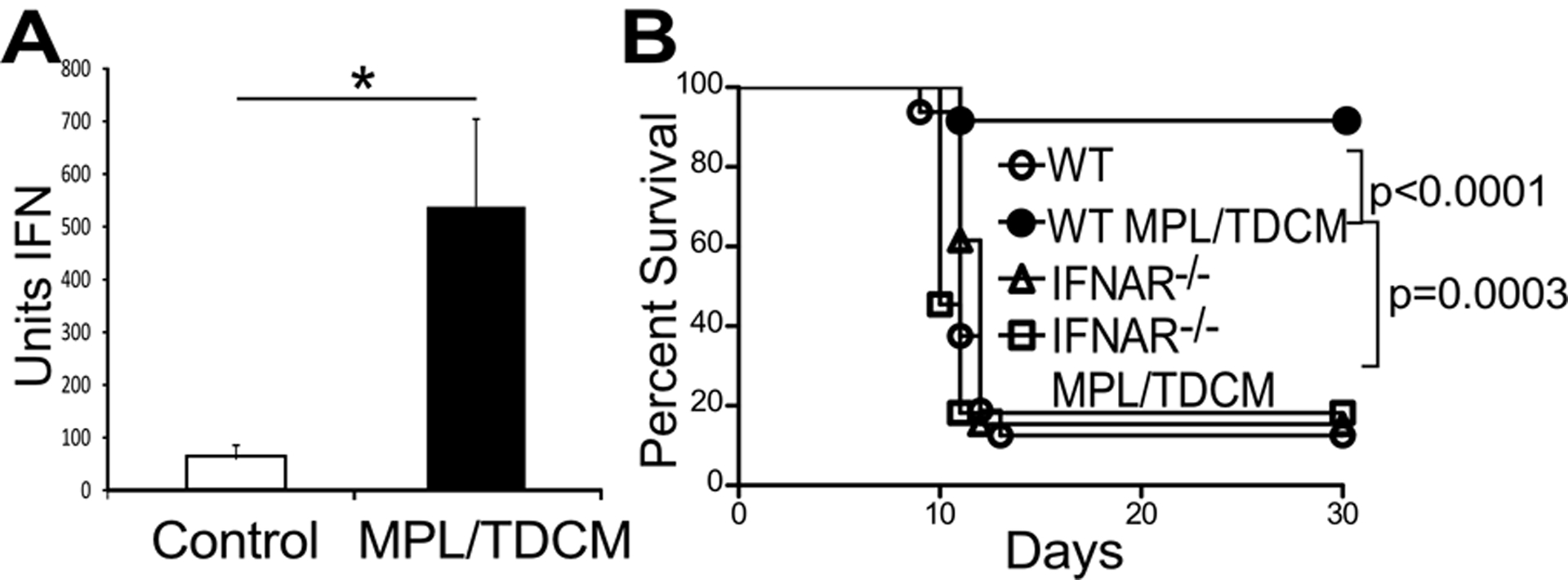
(A-B) Mice were challenged with 2.5 × 104 TA3-Ha cells ± MPL/TDCM on d0. (A) Peritoneal lavage fluid was harvested from WT mice at 6 hours post-treatment and type I interferon was measured using a bioassay. Values represent means ± SEM. Asterisks (*) indicate significant differences (p<0.05, n=3 mice per group). Results and statistical analysis for one of two representative experiments are shown. (B) WT and IFNAR−/− mice were monitored for morbidity requiring euthanasia (n=11–16 mice per group). Statistical analysis (Log rank) for survival results of 3 pooled experiments are shown with calculated p values.
B cell anti-tumor effector functions are independent of type I interferon signaling
We examined the effect of IFNAR deficiency on MT-elicited activation of B cells. We first examined production of tumor-specific IgM, given its key role in protection. MPL/TDCM treatment elicited comparable IgM deposition on recovered peritoneal TA3-Ha cells from WT and IFNAR−/− mice (Fig. 4A) and serum IgM levels binding to cultured TA3-Ha cells was also similar from MT-treated mice (Fig. 4B), indicative that IFN-I signaling does not contribute to MPL/TDCM-induced tumor-reactive IgM production. To determine if other critical B cell functions may be perturbed in IFNAR−/− mice, we adoptively transferred WT or IFNAR−/− B-1a cells into μMT recipient mice. However, WT and IFNAR−/− B-1a cells both conferred significant protection against tumor challenge (Fig. 4C–D). Furthermore, mice that conditionally lacked IFN-I receptor on B cells using the Cre/flox system had comparable survival to control mice (Fig 4E). Hence, the requirement for type I IFN in MPL/TDCM-elicited protection against peritoneal carcinomatosis is independent of B cell functions.
Figure 4. IFN-I functions independently of B cells and their effector functions.
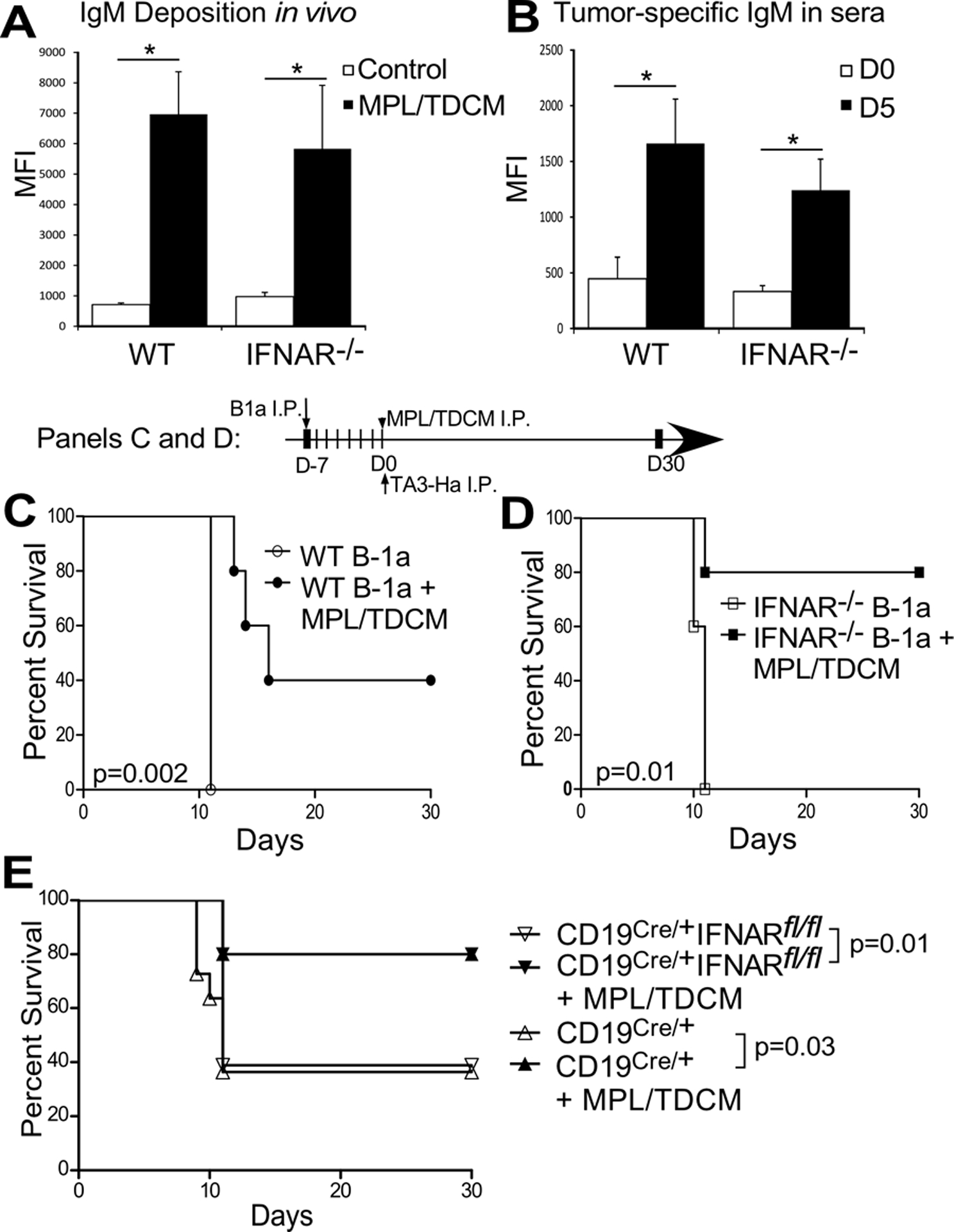
(A) Ig deposition on TA3-Ha cells recovered from WT and IFNAR−/− mice 3 days post TA3-Ha challenge (n=5/group). (B) TA3-Ha-specific IgM in sera harvested from WT and IFNAR−/− mice on d0 and d5 post MPL/TDCM treatment (n=5/group). In A-B, values represent means ± SEM. Asterisks (*) indicate significant differences in MFI as analyzed using paired t-tests (p<0.05). Results and statistical analysis for one of two representative experiments are shown. (C-D) μMT mice were reconstituted with either 1×106 WT (C) or IFNAR−/− (D) B-1a cells on d-7 and challenged with TA3-Ha cells on d0 (n=5/group). P values indicate Log rank analysis results from 1 experiment. (E) Survival of CD19Cre/+ (WT) and CD19Cre/+ IFNARfl/fl mice conditionally lacking IFNAR on B cells following TA3-Ha challenge ± MPL/TDCM treatment on d0 (n=10–20 mice/group; pooled results from 3 independent experiments). P values indicate Log-rank results for analysis of pooled experimental survival results.
IFN-I contributes to MPL/TDCM-induced protection against peritoneal carcinomatosis via Ly6C+ monocyte recruitment
Type I IFN causes the recruitment of myeloid derived cells, including Ly6Chi inflammatory monocytes, into the peritoneal cavity in response to inflammation(22). Consistent with this, monocyte, macrophage, or dendritic cell numbers were not optimally increased in IFNAR1−/− mice at 3 days post MPL/TDCM treatment (Fig. 5A and Supplemental Fig. 3D), although neutrophils were significantly increased (Supplemental Fig. 5). When the macrophage populations were separated out further, LPMs were similarly decreased in WT and IFNAR−/− mice, whereas SPMs numbers were significantly lower in IFNAR−/− mice compared to WT mice (Fig. 5B), which is consistent with significantly lower monocyte numbers in treated IFNAR−/− mice (Fig. 5A).
Figure 5. Ly6Chi monocyte recruitment into the peritoneal cavity depends on type I IFN and is essential in the MPL/TDCM-elicited anti-tumor response.
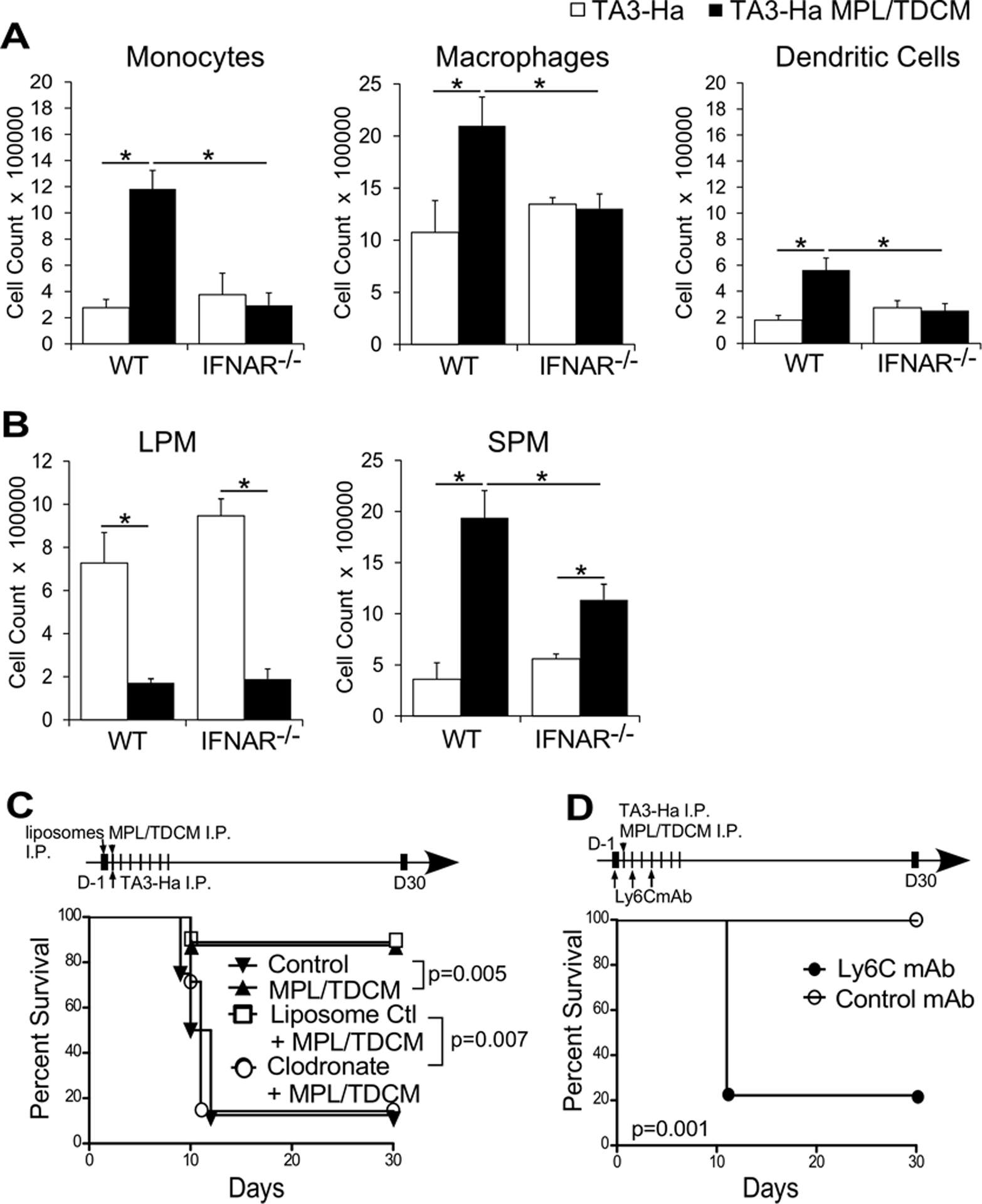
(A-B) WT and IFNAR−/− mice were challenged with 2.5×104 TA3-Ha cells ± MPL/TDCM on d0. On d3 (A) inflammatory monocytes, macrophages, CD11b+CD11c+ dendritic cells, and (B) SPM, and LPM were quantified in the peritoneal cavity as in Figure 2. Statistical analysis of one representative experiment using 5 mice per group is shown. In A-B, values represent means ± SEM. Asterisks (*) indicate significant differences (p<0.05). Similar results were obtained in 2 independent experiments. (C) Clodronate-containing liposomes or control liposomes were administered on day −1 of tumor challenge to WT mice, with survival monitored following TA3-Ha challenge (n=7–9/group; pooled results from two independent experiments). (D) Mice were treated with 200 μg anti-Ly6C or isotype control antibody on day −1, 1, and 3 of tumor challenge (i.p.) and monitored for survival (n=10–11/group; pooled results from two independent experiments). In C-D, p values indicate Log-rank results for analysis of pooled experimental survival results.
Given the striking decrease in monocyte and SPM numbers in MPL/TDCM-treated IFNAR−/− mice, we assessed the importance of phagocytic cells in the anti-tumor response stimulated by MPL/TDCM. One low intraperitoneal dose of clodronate-containing liposomes ablated the protective capacity of MPL/TDCM relative to control liposome-treated mice (Fig. 5C) but had no effect on MPL/TDCM-elicited tumor-reactive IgM production (Supplemental Fig. 9), indicating phagocytic cells are required for efficacy. Similarly, clodronate liposome treatment prevented MPL/TDCM-elicited protection against EL4 challenge (Supplemental Fig. 8B). Depletion of Ly6C+ cells also ablated MPL/TDCM-mediated protection against TA3-Ha challenge (Fig. 5D). Thus, type I IFN is critical for recruitment of Ly6Chi inflammatory monocytes and concomitant increases in SPM in response to MPL/TDCM treatment. Further, phagocytic cells and Ly6C+ cells and/or the cells they differentiate into play a critical role in MPL/TDCM-induced protection against TA3-Ha growth in the peritoneal cavity.
TNFα contributes to TA3-Ha killing, whereas nitric oxide contributes to EL4 killing
Ly6C+ inflammatory monocytes differentiate into TNFα- and iNOS-producing SPM and dendritic cells (23). TNFα and NO represent major tumor killing mechanisms employed by LPS-activated peritoneal macrophages (24). MPL and TDCM have also been shown to induce peritoneal macrophages to produce TNFα and NO (15,25,26). We therefore investigated the role that these factors play in MPL-induced anti-tumor protection. Inhibition of nitric oxide using aminoguanidine during TA3-Ha challenge (days 0–8) had no effect on MPL/TDCM-elicited protection against TA3-Ha growth (Fig. 6A). Daily N-acetylcysteine treatment, commonly used to inhibit reactive oxygen species, also had no effect (Supplemental Fig. 10).
Figure 6. MPL/TDCM-elicited anti-tumor responses depend on TNFα.
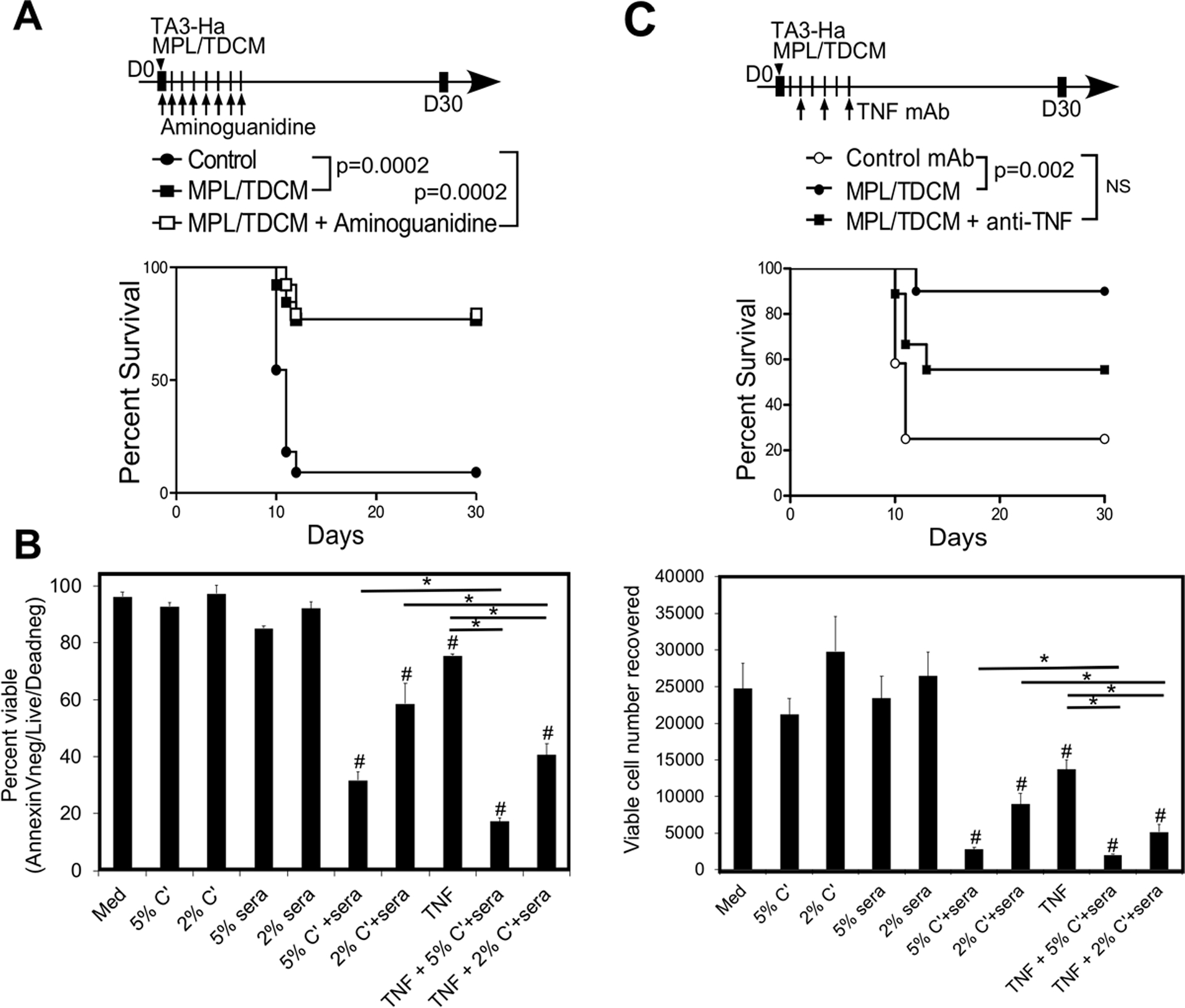
(A) Mice were challenged with 2.5 × 104 TA3-Ha cells ± MPL/TDCM i.p. on d0. Mice received aminoguanidine (400 mg/kg) i.p. daily from d0–8 (n=11–13/group; pooled from 2 independent experiments). (B) TA3-HA cells were cultured in medium alone, or in the presence rabbit complement (5 or 2%), d14 sera from MPL/TDCM-treated mice (5 or 2%), recombinant TNF-α (10 ng/ml), or a combination of these, as indicated, for 18 hrs. Cells were incubated with Live/Dead viability stain and Countbright beads, washed and stained with Annexin V, with the percentage (top panel) and number (lower panel) of live (A, AnnexinVnegLive/Deadneg) cells recovered. Hashtags (#) indicate significant differences from medium control, and asterisks (*) indicate differences among individual groups. Similar results obtained in 2 independent experiments. (C) Mice were challenged with 2.5 × 104 TA3-Ha cells ± MPL/TDCM on d0. Anti-TNFα antibody was given on days 2 (200 μg), 4 (200 μg), and 6 (100 μg) post-tumor challenge i.p., with survival assessed (n=9–11/group). Data pooled from two independent experiments, with similar results obtained in each. In A and C, p values indicate Log-rank results for pooled data plotted in survival curves.
TNFR1, the proapoptotic TNF receptor, is reported to be expressed by TA3-Ha cells (27). We therefore examined the effect of TNFα either alone or in combination with serum from MPL/TDCM-treated mice and rabbit complement in vitro. As shown in Fig. 6B, TNFα alone significantly reduced the percentage of AnnexinVnegLive/Deadneg cells (~20% reduction) and the number of viable cells recovered (55%) from 18 hr cultures. Moreover, addition of TNFα to either 5 or 2 % sera + complement cultures significantly reduced the percentage and number of viable cells recovered relative to either treatment alone. Enumeration of viable cells using trypan blue prior to staining revealed similar trends (Supplemental Fig. 11A). In contrast, EL4 cells, although very sensitive to complement-mediated killing, did not exhibit significant susceptibility to TNFα (Supplemental Fig. 11B–D). Nonetheless, we found that in contrast to what was observed with TA3-Ha cell challenge (Fig. 6A), MPL/TDCM-elicited protection against EL4 challenge was abrogated by aminoguanidine treatment (Supplementary Fig. 8C–D), consistent with the previously reported sensitivity of EL4 cells to NO killing in vivo (28). Given that TNFα and Ab-elicited complement-dependent killing pathways have additive effects on TA3-Ha cells in vitro, we tested the role of TNFα in vivo. Consistent with our in vitro findings supporting the sensitivity of TA3-Ha cells to TNFα, mice treated with an anti-TNFα neutralizing antibody beginning on d2 post TA3-Ha challenge were no longer afforded significant protection by MPL/TDCM treatment (Fig. 6C). Thus, TNFα cytotoxicity in combination with Ab-mediated complement killing contributes to MPL/TDCM anti-tumor efficacy in the TA3-Ha model whereas NO is involved in EL4 cytotoxicity.
Discussion
There are formidable barriers to stimulating immune responses against tumors. Promoting anti-tumor immune responses in the peritoneal cavity is particularly difficult, given the immunosuppressive environment along with an extensive surface area and volume available for tumor expansion. MPL/TDCM treatment is nonetheless highly effective at significantly decreasing tumor burden and ascites development resulting from aggressive TA3-Ha, as well as EL4, growth in the peritoneal cavity(10). Our previous study revealed a critical role for innate B-1a cell tumor-reactive IgM production and C4 in protection elicited by MPL/TDCM treatment(10). However, as we show in the current study, cancer cells may resist lysis by CDC such that accessory cells are required to assist in tumor cell killing. Our current study reveals an important role for phagocytes and Ly6C+ cells in supporting MPL/TDCM-elicited protection. MPL/TDCM treatment significantly increased the numbers of Ly6Chi monocytes and SPMs, which are well-known to produce TNFα and NO, in the peritoneal cavity. The increase in monocytes and SPM was dependent on type I interferon, which was rapidly and transiently induced in response to MPL/TDCM treatment. Depletion of either phagocytic cells or Ly6C+ cells abolished the anti-tumor effects of the combinational treatment, indicating their essential role in tumor cytotoxicity. Taken together, our results suggest that MPL/TDCM treatment stimulates high levels of B-1a cell tumor-reactive IgM production and subsequent classical complement deposition on tumor cells which in turn, elicits several downstream mechanisms of tumor cell killing, including CDC and possibly CDCC and TNF-α and NO cytotoxicity that may be highly dependent on Ly6Chi monocytes.
Ly6Chi monocytes derived from the bone marrow extravasate into various tissues and differentiate into macrophages or dendritic cells upon receiving appropriate chemokine and cytokine stimulation(18). In particular, inflammation within the peritoneal cavity has been reported to elicit local production of type I IFN, which in turn, stimulates CCL2 production (29). CCL2 is a potent chemokine that recruits CCR2-expressing Ly6Chi monocytes from the bone marrow and/or blood into the cavity. Based on our data, MPL/TDCM + squalene treatment recruits Ly6Chi monocytes through a similar IFN-dependent mechanism. The peritoneal cells that rapidly produce type I IFN in response to this treatment are not yet known. MPL is known to preferentially induce TRIF-dependent TLR4 signaling(30), which drives type I IFN production by multiple cell types, including peritoneal mesothelial cells(31). TDCM and squalene may also be involved. Despite the importance of B cells in driving the anti-tumor response, we did not find that their selective loss diminished monocyte recruitment, nor did loss of IFNAR expression on B cells affect tumor-reactive IgM production or MPL/TDCM anti-tumor efficacy. Future work is required to determine the critical players driving IFN I production and monocyte recruitment as well as factors which support further activation and differentiation of monocytes upon peritoneal cavity entry.
Given the concomitant increase in SPM following MPL/TDCM treatment, it is likely that the recruited monocytes differentiate into these cells upon entering the cavity. Depending on which interferon stimulated genes (ISGs) are triggered, type I interferon can influence macrophage polarization to either an M1 or M2 state(32). Additionally, cord factor analogs (ie., TDCM) promote conversion of an M2 to M1 or enhancement of an M1 phenotype (33). Consistent with the latter, the peritoneal macrophages elicited by MPL/TDCM treatment had markers indicative of an M1-like phenotype (MHCII+CD86+). Although it is not protective on its own, squalene may nonetheless also be playing a role in modulating these observed changes in myeloid cells. Synergistic activation of peritoneal macrophages with MPL + CpG or anti-CD40 also induces an M1 phenotype, along with high levels of TNFα and nitric oxide (NO) that are sufficient to kill tumor cells (34). Of note, a subset of inflammatory dendritic cells (TIP-DCs) also produces TNFα and NO in response to TLR stimulation (35), and could also contribute to TNFα-mediated cytotoxicity. Therefore, the costimulation of TDCM and type I interferon poses as a novel combination to promote a classically activated function of monocyte-derived phagocytes that are capable of eliciting anti-tumor responses. Importantly, there are possibly other mechanisms by which these cells or other cells may be promoting anti-tumor immunity in these tumor models which have not been ruled out by our study. Indeed, the recruited monocytes could in some way impact the recruitment and functionality of classical cytotoxic effectors (ie., CD8+ and CD4+ CTL and NK cells) as well as non-classical effectors which could further support clearance of tumor cells. Further study is needed to determine this.
Inflammatory monocytes have been shown to have a context-dependent role in tumorigenesis in the peritoneum. Studies have shown that peripheral monocytes and peritoneal macrophages from cancer patients can be activated with LPS to induce tumor cytotoxicity alone (36). Moreover, stimulation of monocytes ex vivo has been recently investigated as an adoptive immunotherapy for ovarian cancer patients (37). This work builds on earlier studies which used monocytes in conjunction with interferon-γ, or other stimuli, to stimulate anti-tumor responses directly in the cavity(38,39). Some studies have examined enhancing the cytotoxic activity of monocytes with co-administration of type I interferon(40). Type I interferons have been shown in a number of conditions and malignancies to promote antineoplastic effects(41). However, previous clinical trials have unveiled that type I interferon therapies given systemically often lead to autoimmunity(42,43). Administering type I interferon directly into the peritoneum is thought to ameliorate the adverse effects associated with giving systemic interferon while inducing the same level of cytotoxicity. Whether tumor-reactive IgM, classical complement, type I IFN, and activated inflammatory monocytes are sufficient to kill select peritoneal tumors remains to be determined. Future work in this area could potentially lead to novel combination therapies with increased efficacy and reduced patient toxicity.
We have elucidated that tumor-specific IgM and complement are critical players in MPL/TDCM-induced anti-tumor response(10). However, their ability to induce tumor cytotoxicity in either the presence of complement regulatory proteins or extensive surface mucin may be limited, as these factors may dampen membrane attack. The essential role of phagocytes and Ly6C+ cells in MPL/TDCM efficacy raises the possibility that CDCC may also be playing a critical role. This mechanism involves complement-coated tumor cell engagement of complement receptors expressed on monocytes/macrophages, which are then activated to kill tumor cells. CR1, CR3, CR4, and CrIg induce cytotoxicity through multiple mechanisms, including CDCC and complement-dependent cell-mediated phagocytosis (CDCP)(44,45). Mechanisms of tumor cell killing due to either CDCC and/or CDCP are not easily measured given the potential involvement of numerous receptors and the likelihood of both mechanisms occurring simultaneously(46). Our data suggests that tumor cytotoxicity depends in part, on TNFα production in the TA3-Ha model, and NO in the EL4 model. TNFα could directly kill tumor cells through either soluble or membrane forms, or alternatively, via supporting other killing mechanisms. Importantly, engagement of CR3 via complement complexes can induce iNOS protein and NO production in a dose dependent manner(47). In comparison, adequate macrophage TNFα secretion may require signals beyond CR3 such as ligation of Fcγ receptors(48). Indeed, TDCM, which activates Fc receptor common γ chain signaling via Mincle and MCL receptors, induces macrophages to produce both TNFα and NO(25). MPL additionally induces monocytes and macrophages to produce NO(49) and TNF-α(15). Thus, the robust anti-tumor response elicited by MPL/TDCM may rely on CR3 engagement by complement-coated tumor cells as well potentiation of TLR- and CLR-induced production of toxic intermediates (ie., TNFα, and NO) by resident and newly recruited monocytes/macrophages/Tip-DCs. The extent to which squalene may further enhance these activities remains unknown, but is certainly important to investigate. Future work will be required to gain an understanding of the nature and duration of these coordinated tumor-killing activities carried out within the unique environment of the peritoneal cavity.
In summary, our study highlights an exciting new combination of bacterial-derived agonists that has the capacity to elicit potent anti-tumor immunity by innate immune system components, namely B1 cells, natural IgM, complement and inflammatory monocytes/phagocytes. Further work is needed to translate our findings into effective treatments that are suitable for patients, beginning with identification and testing of agonists that may safely evoke similar responses in human patients. In addition, an understanding of the extent to which these responses direct adaptive anti-tumor immunity remains to be established, and may ultimately open up more possible combinations of immunotherapies that can be used to successfully treat patients.
Supplementary Material
Acknowledgements
This work was supported by DOD grant W81XWH-15-1-0585 and National Institute of Allergy and Infectious Diseases, National Institutes of Health grant R01AI18876 awarded to K. Haas. Shared resources support was provided by NCI-CCSG grant P30CA012197. A. Dyevoich was supported was in part by National Institutes of Health Training Grant AI007401. We thank Dr. Erik Barton for providing mice for these studies and Drs. Marlena Westcott and Douglas Lyles for assistance with the type I IFN bioassay.
Footnotes
The authors have no conflicts to disclose.
References
- 1.Sangisetty SL, Miner TJ. Malignant ascites: A review of prognostic factors, pathophysiology and therapeutic measures. World J Gastrointest Surg 2012;4:87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cavazzoni E, Bugiantella W, Graziosi L, Franceschini MS, Donini A. Malignant ascites: pathophysiology and treatment. Int J Clin Oncol 2013;18:1–9. [DOI] [PubMed] [Google Scholar]
- 3.Lambert LA, Hendrix RJ. Palliative Management of Advanced Peritoneal Carcinomatosis. Surg Oncol Clin N Am 2018;27:585–602. [DOI] [PubMed] [Google Scholar]
- 4.Sardi A, Jimenez WA, Nieroda C, Sittig M, Macdonald R, Gushchin V. Repeated cytoreductive surgery and hyperthermic intraperitoneal chemotherapy in peritoneal carcinomatosis from appendiceal cancer: analysis of survival outcomes. Eur J Surg Oncol 2013;39:1207–13. [DOI] [PubMed] [Google Scholar]
- 5.Grass F, Vuagniaux A, Teixeira-Farinha H, Lehmann K, Demartines N, Hubner M. Systematic review of pressurized intraperitoneal aerosol chemotherapy for the treatment of advanced peritoneal carcinomatosis. Br J Surg 2017;104:669–78. [DOI] [PubMed] [Google Scholar]
- 6.Capobianco A, Cottone L, Monno A, Manfredi AA, Rovere-Querini P. The peritoneum: healing, immunity, and diseases. J Pathol 2017;243:137–47. [DOI] [PubMed] [Google Scholar]
- 7.Stein SH, Phipps RP. Macrophage-secreted prostaglandin E2 potentiates immune complex-induced B cell unresponsiveness. Eur J Immunol 1990;20:403–7. [DOI] [PubMed] [Google Scholar]
- 8.Mikula-Pietrasik J, Uruski P, Tykarski A, Ksiazek K. The peritoneal “soil” for a cancerous “seed”: a comprehensive review of the pathogenesis of intraperitoneal cancer metastases. Cell Mol Life Sci 2018;75:509–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kipps E, Tan DS, Kaye SB. Meeting the challenge of ascites in ovarian cancer: new avenues for therapy and research. Nat Rev Cancer 2013;13:273–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haro MA, Dyevoich AM, Phipps JP, Haas KM. Activation of B-1 Cells Promotes Tumor Cell Killing in the Peritoneal Cavity. Cancer Res 2019;79:159–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haro MA, Littrell CA, Yin Z, Huang X, Haas KM. PD-1 Suppresses Development of Humoral Responses That Protect against Tn-Bearing Tumors. Cancer Immunol Res 2016;4:1027–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cain DW, O’Koren EG, Kan MJ, Womble M, Sempowski GD, Hopper K, et al. Identification of a tissue-specific, C/EBPbeta-dependent pathway of differentiation for murine peritoneal macrophages. J Immunol 2013;191:4665–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Westcott MM, Smedberg J, Jorgensen MJ, Puckett S, Lyles DS. Immunogenicity in African Green Monkeys of M Protein Mutant Vesicular Stomatitis Virus Vectors and Contribution of Vector-Encoded Flagellin. Vaccines (Basel) 2018;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fishelson Z, Attali G, Mevorach D. Complement and apoptosis. Mol Immunol 2001;38:207–19. [DOI] [PubMed] [Google Scholar]
- 15.Okemoto K, Kawasaki K, Hanada K, Miura M, Nishijima M. A potent adjuvant monophosphoryl lipid A triggers various immune responses, but not secretion of IL-1beta or activation of caspase-1. J Immunol 2006;176:1203–8. [DOI] [PubMed] [Google Scholar]
- 16.Martin M, Michalek SM, Katz J. Role of innate immune factors in the adjuvant activity of monophosphoryl lipid A. Infect Immun 2003;71:2498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Romero CD, Varma TK, Hobbs JB, Reyes A, Driver B, Sherwood ER. The Toll-like receptor 4 agonist monophosphoryl lipid a augments innate host resistance to systemic bacterial infection. Infect Immun 2011;79:3576–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity 2014;41:21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghosn EE, Cassado AA, Govoni GR, Fukuhara T, Yang Y, Monack DM, et al. Two physically, functionally, and developmentally distinct peritoneal macrophage subsets. Proc Natl Acad Sci U S A 2010;107:2568–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.del Fresno C, Soulat D, Roth S, Blazek K, Udalova I, Sancho D, et al. Interferon-beta production via Dectin-1-Syk-IRF5 signaling in dendritic cells is crucial for immunity to C. albicans. Immunity 2013;38:1176–86. [DOI] [PubMed] [Google Scholar]
- 21.Widman DG. Bioassay for the measurement of type-I interferon activity. Methods Mol Biol 2013;1031:91–6. [DOI] [PubMed] [Google Scholar]
- 22.Thomas KE, Galligan CL, Newman RD, Fish EN, Vogel SN. Contribution of interferon-beta to the murine macrophage response to the toll-like receptor 4 agonist, lipopolysaccharide. J Biol Chem 2006;281:31119–30. [DOI] [PubMed] [Google Scholar]
- 23.Cassado Ados A, D’Imperio Lima MR, Bortoluci KR. Revisiting mouse peritoneal macrophages: heterogeneity, development, and function. Front Immunol 2015;6:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Higuchi M, Higashi N, Taki H, Osawa T. Cytolytic mechanisms of activated macrophages. Tumor necrosis factor and L-arginine-dependent mechanisms act synergistically as the major cytolytic mechanisms of activated macrophages. J Immunol 1990;144:1425–31. [PubMed] [Google Scholar]
- 25.Ishikawa E, Ishikawa T, Morita YS, Toyonaga K, Yamada H, Takeuchi O, et al. Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin Mincle. J Exp Med 2009;206:2879–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Das A, Ali N. Combining cationic liposomal delivery with MPL-TDM for cysteine protease cocktail vaccination against Leishmania donovani: evidence for antigen synergy and protection. PLoS Negl Trop Dis 2014;8:e3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi KH, Choi HY, Ko JK, Park SS, Kim YN, Kim CW. Transcriptional regulation of TNF family receptors and Bcl-2 family by chemotherapeutic agents in murine CT26 cells. J Cell Biochem 2004;91:410–22. [DOI] [PubMed] [Google Scholar]
- 28.Komohara Y, Takemura K, Lei XF, Sakashita N, Harada M, Suzuki H, et al. Delayed growth of EL4 lymphoma in SR-A-deficient mice is due to upregulation of nitric oxide and interferon-gamma production by tumor-associated macrophages. Cancer Sci 2009;100:2160–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee PY, Li Y, Kumagai Y, Xu Y, Weinstein JS, Kellner ES, et al. Type I Interferon Modulates Monocyte Recruitment and Maturation in Chronic Inflammation. The American Journal of Pathology 2009;175:2023–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kolb JP, Casella CR, SenGupta S, Chilton PM, Mitchell TC. Type I interferon signaling contributes to the bias that Toll-like receptor 4 exhibits for signaling mediated by the adaptor protein TRIF. Sci Signal 2014;7:ra108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hwang EH, Kim TH, Oh SM, Lee KB, Yang SJ, Park JH. Toll/IL-1 domain-containing adaptor inducing IFN-beta (TRIF) mediates innate immune responses in murine peritoneal mesothelial cells through TLR3 and TLR4 stimulation. Cytokine 2016;77:127–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gunthner R, Anders HJ. Interferon-regulatory factors determine macrophage phenotype polarization. Mediators Inflamm 2013;2013:731023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kodar K, Harper JL, McConnell MJ, Timmer MSM, Stocker BL. The Mincle ligand trehalose dibehenate differentially modulates M1-like and M2-like macrophage phenotype and function via Syk signaling. Immun Inflamm Dis 2017;5:503–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi Y, Felder MA, Sondel PM, Rakhmilevich AL. Synergy of anti-CD40, CpG and MPL in activation of mouse macrophages. Mol Immunol 2015;66:208–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu Y, Zhan Y, Lew AM, Naik SH, Kershaw MH. Differential development of murine dendritic cells by GM-CSF versus Flt3 ligand has implications for inflammation and trafficking. J Immunol 2007;179:7577–84. [DOI] [PubMed] [Google Scholar]
- 36.Wilbanks GD, Ahn MC, Beck DA, Braun DP. Tumor cytotoxicity of peritoneal macrophages and peripheral blood monocytes from patients with ovarian, endometrial, and cervical cancer. Int J Gynecol Cancer 1999;9:427–32. [DOI] [PubMed] [Google Scholar]
- 37.Green DS, Nunes AT, David-Ocampo V, Ekwede IB, Houston ND, Highfill SL, et al. A Phase 1 trial of autologous monocytes stimulated ex vivo with Sylatron((R)) (Peginterferon alfa-2b) and Actimmune((R)) (Interferon gamma-1b) for intra-peritoneal administration in recurrent ovarian cancer. J Transl Med 2018;16:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stevenson HC, Keenan AM, Woodhouse C, Ottow RT, Miller P, Steller EP, et al. Fate of gamma-interferon-activated killer blood monocytes adoptively transferred into the abdominal cavity of patients with peritoneal carcinomatosis. Cancer Res 1987;47:6100–3. [PubMed] [Google Scholar]
- 39.Faradji A, Bohbot A, Frost H, Schmitt-Goguel M, Siffert JC, Dufour P, et al. Phase I study of liposomal MTP-PE-activated autologous monocytes administered intraperitoneally to patients with peritoneal carcinomatosis. J Clin Oncol 1991;9:1251–60. [DOI] [PubMed] [Google Scholar]
- 40.Green DS, Nunes AT, Annunziata CM, Zoon KC. Monocyte and interferon based therapy for the treatment of ovarian cancer. Cytokine Growth Factor Rev 2016;29:109–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bekisz J, Sato Y, Johnson C, Husain SR, Puri RK, Zoon KC. Immunomodulatory effects of interferons in malignancies. J Interferon Cytokine Res 2013;33:154–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tarhini AA, Shin D, Lee SJ, Stuckert J, Sander CA, Kirkwood JM. Serologic evidence of autoimmunity in E2696 and E1694 patients with high-risk melanoma treated with adjuvant interferon alfa. Melanoma Res 2014;24:150–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kretschmer S, Lee-Kirsch MA. Type I interferon-mediated autoinflammation and autoimmunity. Curr Opin Immunol 2017;49:96–102. [DOI] [PubMed] [Google Scholar]
- 44.Helmy KY, Katschke KJ Jr., Gorgani NN, Kljavin NM, Elliott JM, Diehl L, et al. CRIg: a macrophage complement receptor required for phagocytosis of circulating pathogens. Cell 2006;124:915–27. [DOI] [PubMed] [Google Scholar]
- 45.Weinstein JR, Quan Y, Hanson JF, Colonna L, Iorga M, Honda S, et al. IgM-Dependent Phagocytosis in Microglia Is Mediated by Complement Receptor 3, Not Fcalpha/mu Receptor. J Immunol 2015;195:5309–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee CH, Romain G, Yan W, Watanabe M, Charab W, Todorova B, et al. IgG Fc domains that bind C1q but not effector Fcgamma receptors delineate the importance of complement-mediated effector functions. Nat Immunol 2017;18:889–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matsuno R, Aramaki Y, Arima H, Adachi Y, Ohno N, Yadomae T, et al. Contribution of CR3 to nitric oxide production from macrophages stimulated with high-dose of LPS. Biochem Biophys Res Commun 1998;244:115–9. [DOI] [PubMed] [Google Scholar]
- 48.Stein M, Gordon S. Regulation of tumor necrosis factor (TNF) release by murine peritoneal macrophages: role of cell stimulation and specific phagocytic plasma membrane receptors. Eur J Immunol 1991;21:431–7. [DOI] [PubMed] [Google Scholar]
- 49.Salkowski CA, Detore GR, Vogel SN. Lipopolysaccharide and monophosphoryl lipid A differentially regulate interleukin-12, gamma interferon, and interleukin-10 mRNA production in murine macrophages. Infect Immun 1997;65:3239–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.