

Abstract
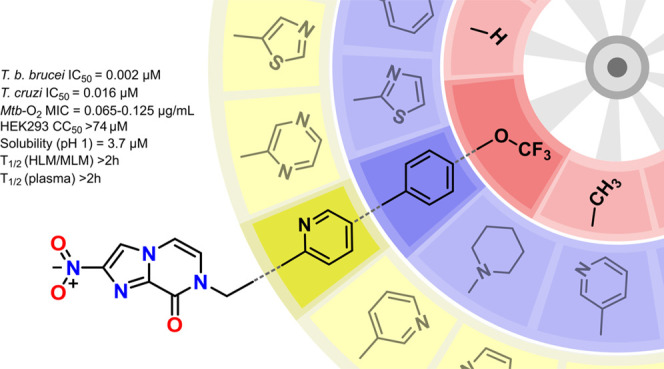
Following the approval of delamanid and pretomanid as new drugs to treat drug-resistant tuberculosis, there is now a renewed interest in bicyclic nitroimidazole scaffolds as a source of therapeutics against infectious diseases. We recently described a nitroimidazopyrazinone bicyclic subclass with promising antitubercular and antiparasitic activity, prompting additional efforts to generate analogs with improved solubility and enhanced potency. The key pendant aryl substituent was modified by (i) introducing polar functionality to the methylene linker, (ii) replacing the terminal phenyl group with less lipophilic heterocycles, or (iii) generating extended biaryl side chains. Improved antitubercular and antitrypanosomal activity was observed with the biaryl side chains, with most analogs achieved 2- to 175-fold higher activity than the monoaryl parent compounds, with encouraging improvements in solubility when pyridyl groups were incorporated. This study has contributed to understanding the existing structure–activity relationship (SAR) of the nitroimidazopyrazinone scaffold against a panel of disease-causing organisms to support future lead optimization.
Introduction
Diseases caused by protozoans and bacteria remain a major global health threat, especially in low- and medium-income countries where they affect millions of lives. Current treatment options are inadequate, attributed to issues such as toxicity, low efficacy, high cost, and rapid emergence of drug resistance.1,2 Therefore, new and effective therapeutics are urgently needed for the antibiotic and anti-protozoan drug discovery pipelines. Nitroimidazoles were discovered in the early 1950s.3 They have broad-spectrum activity across parasites, mycobacteria, and both Gram-positive and Gram-negative bacteria.3 Recently, there has been a great interest in developing nitroimidazoles based on new bicyclic core scaffolds, resulting in the success of a nitroimidazooxazole, delamanid 1 and a nitroimidazooxazine, pretomanid 2 (Figure 1) as approved drugs for treatment of tuberculosis (TB).4,5 Both 1 and 2 are pro-drugs that share an interesting dual mode of action. Under aerobic conditions, they have inhibitory activity against the biosynthesis of mycolic acid, a key component of the mycobacterial cell wall,6,7 while under hypoxic conditions, they release nitric oxide intracellularly, causing respiratory poisoning that kills the non-replicating bacteria.8 Nitroimidazole 1 was found to have excellent bactericidal and sterilizing activity, with MIC values of 0.006–0.024 μg/mL against both drug-susceptible and drug-resistant Mycobacterium tuberculosis under normoxic conditions.6 The compound was granted conditional approval by the European Medicines Agency (EMA) in 2014 for the treatment of multidrug-resistant TB.9 Nitroimidazole 2 was recently approved by the FDA in August 2019 as part of an all-oral combination regimen to treat drug-resistant TB, alongside bedaquiline and linezolid.10
Figure 1.

Bicyclic nitroimidazoles with antitubercular or antiparasitic activity.
Drug repurposing or repositioning programs have opened a new avenue for nitroimidazoles to target other neglected diseases where R&D funding is limited.11 Both enantiomers of 2 were shown to be active against Leishmania donovani, a causative agent of visceral leishmaniasis (VL).12 However the R enantiomer surpassed the activity of its S counterpart in leishmania-infected macrophages and murine models.12 A study of 1 in L. donovani revealed its superior activity against intracellular amastigotes, with >30-fold improvement of EC50 compared to the standard drug miltefosine.13 Another nitroimidazooxazole, DNDI-VL-2098 3 that was identified by the Drugs for Neglected Diseases initiative (DNDi), also demonstrated good efficacy in acute and chronic rodent models of VL after oral dosing.14 However, 3 was discontinued from further studies due to safety issues.15 Following the termination of 3, a novel 7-substituted nitroimidazooxazine, DNDI-0690 (4) was discovered within a backup program of 2 by the TB Alliance and the University of Auckland. This preclinical candidate was found to possess excellent in vitro activity against both L. donovani and Leishmania infantum as well as displaying a better safety profile than 3; hence, it was selected for further development.16 Another nitroimidazooxazine, DNDI-8219 (5) was discovered from a library of ∼900 pretomanid analogs, with broad-spectrum activity against a range of reference strains and clinical isolates of Leishmania while displaying favorable pharmacokinetic profile.17 Similar programs were expanded to other kinetoplastids, revealing a new thiazine oxide analog 6 that showed a complete cure in a mouse model of Trypanosoma brucei brucei infection, when dosed orally.18 A new nitroimidazolethiazole 7 with potent activity against Trypanosoma cruzi, the causative agent of Chagas disease, was also identified, although it was not active against L. infantum.(19) This suggests the utility of testing different subclasses of bicyclic nitroimidazoles for activity in various parasite species as well as the possibility to improve their selectivity against a specific organism.
The market approvals of nitroimidazoles are encouraging, but there is still room for improvement. Delamanid 1 suffers from poor water solubility and low absorption, requiring twice-daily dosing for the treatment of multidrug-resistant TB.20 Pretomanid 2 has better PK properties with daily dosing, but it is less potent compared to 1. A second-generation nitroimidazooxazine, TBA-354 8, was reported to have superior antitubercular activity than 2 and better metabolic stability than 1,21 but it was discontinued from phase I clinical trials due to neurotoxicity.22 To address some of these drawbacks, various strategies have been applied, mostly focusing on the modification of the aryl side chain of the imidazo-oxazole/oxazine ring.23−25 A library of new analogs of 2 with extended side chains and different biaryl linkers was synthesized by Palmer et al. to enhance the solubility and oral activity in chronic M. tuberculosis infection.23 Most of the lipophilic and polar linkers showed comparable or improved in vitro antitubercular activity, whereas the introduction of hydrophilic groups and replacement of phenyl with pyridine rings were beneficial in improving the aqueous solubility.23 Similar measures were also employed by Yempalla et al.24 and Thompson et al.25 to enhance the physicochemical/pharmacological properties of nitroimidazooxazole against M. tuberculosis and L. infantum, respectively.
Our previous work identified a new bicyclic nitroimidazole scaffold with promising antitubercular and antiparasitic activity, namely, nitroimidazopyrazinone 9.26 This scaffold was derived from the 4(5)-nitroimidazoles that were previously developed,27 taking inspiration from both metronidazole and 2. The O-alkylated nitroimidazopyrazine regioisomers of 9 were also isolated during the reaction, but they were inactive against M. tuberculosis, even though they retained antiparasitic activity against Giardia lamblia and T. b. brucei.26 This demonstrated the significance of the bicyclic core in determining their selectivity profile toward different organisms. Similar to other bicyclic nitroimidazoles such as 1 and 2, most of the potent nitroimidazopyrazinones displayed poor aqueous solubility, with a few exceptions for some of the actives against T. b. brucei.26 This led us to the current study, in which we have now designed a new series of analogs in an attempt to improve solubility without compromising the activity against a panel of disease-causing organisms. We investigated three approaches, all focused on modification of the substituent at R1 without altering the bicyclic imidazopyrazinone core (Figure 2). Modified compounds were tested for antibacterial, antiprotozoal, and antifungal activity in an effort to identify hit compounds against these organisms. We chose not to impart any substitutions at R2 and R3 as these modifications were previously shown to be less favorable for antitubercular activity, despite showing tolerable potency against T. b. brucei.(26) In the first approach, a small set of analogs was prepared where the linker was modified by introducing ketone, amide, and ether functionalities with varying lengths to the benzylic methylene moiety. In the second approach, less lipophilic heterocycles were used to replace the phenyl group. Lastly, extended side chains such as biphenyl analogs and their bioisosteres were synthesized, with and without the incorporation of less lipophilic heterocycles.
Figure 2.

Strategy to improve solubility of nitroimidazopyrazinones.
Chemistry
From our previous SAR findings, aromatic substituents at R1 were preferred over polar side chains and bulky aliphatic groups.26 Therefore in series 1, we sought an alternative approach to reduce the lipophilicity by varying the linker component without altering the active aromatic moiety. The key nitroimidazopyrazinone intermediate 16 was first synthesized in four steps as reported,26 starting from the commercially available imidazo-carboxylic acid 12 (Scheme 1). These involved nitration using concentrated H2SO4 and HNO3, formation of nitroimidazole carboxamide 14, alkylation with bromoacetaldehyde diethyl acetal under basic conditions, and finally cyclization between 1′ imidazole and 2′ free amide nitrogen to afford the bicyclic core 16.26 Different linker variants (17a–g) were synthesized from 16 through alkylation with alkyl halides using basic carbonate (Cs2CO3 or K2CO3) as a catalyst. Ketone-linked products (17a–c) were synthesized from bromoacetophenone derivatives at room temperature, though with poor yield (23–41%). For amide linker variants (17d–f), intermediate bromo-N-substituted acetamides (11a-c) were first synthesized via acylation of various amines and 2-bromoacetyl bromide in the presence of trimethylamine28 followed by N-alkylation under microwave irradiation. Hydroxyl derivative (17h) was prepared from 17g by deacetylation using K2CO3 in methanol, which was then coupled with the desired benzylic bromides in the presence of NaH to form the ether-linked analogs (17i–k).
Scheme 1. Synthesis of Nitroimidazopyrazinones 17a–k.

(i) Various amines, TEA, DCM, 0 °C → rt; (ii) H2SO4/HNO3, 60 °C; (iii) oxalyl chloride, catalytic DMF, DCM, 0 °C → rt, then concentrated NH4OH, 0 °C → rt; (iv) bromoacetaldehyde diethyl acetal, K2CO3, μW 180 °C; (v) 2 M HCl/1,4-dioxane, μW 120 °C; (vi) various alkyl bromides, Cs2CO3 or K2CO3, DMF, μW 80–100 °C; (vii) K2CO3, MeOH, rt; (viii) various benzyl bromides, NaH, DMF, 0 °C → rt. aReported in a previous study.26
For series 2, less lipophilic aromatic rings with hydrogen bond donors/acceptors, such as pyridine, thiazole, and imidazole were introduced. Two different routes, as described in Scheme 2, were utilized to synthesize 26a–k. In the first route, the chloride intermediates 18–20 were converted from the alcohol derivatives in thionyl chloride,29 whereas 21 was formed by bromination using phosphorus tribromide.30 These halide intermediates were used immediately in the following step due to their instability. Benzimidazoles 24 and 25 were made from 22 and 23 and chloroacetic acid by the Philip method,31 which were easily isolated in high purity by vacuum filtration after neutralizing with NaHCO3. Subsequently, N-alkylation was performed between 16 and the halide intermediates to afford the final products (26a, c–f, h) in 42–75% yield after purification. Alternatively, 26b, g, i–k were prepared via route 2, which involved the formation of 4(5)-nitroimidazole carboxamides 27b, g, i–k of different derivatives.27 Alkylation of 27b, g, i–k with bromoacetaldehyde diethyl acetal under microwave conditions provided 28b, g, i–k followed by cyclization in acid to give the bicyclic products in 13–83% yield.
Scheme 2. Synthesis of Nitroimidazopyrazinones 26a–k.

(i) SOCl2, DCM, 0 °C → rt; (ii) PBr3, DCM, 0 °C → rt; (iii) 4 N HCl, chloroacetic acid, reflux; (iv) 18–21, 24–25, K2CO3, DMF, μW 80–100 °C; (v) bromoacetaldehyde diethyl acetal, K2CO3, DMF, μW 150 °C; (vi) 1 M HCl, dioxane, μW 120 °C. Intermediates 19 and 20 were synthesized using the same method as 18.
For the third approach, a variety of biaryl side chain intermediates (33b–m) were synthesized to produce the series 3 analogs 34b–m, while biphenyl 34a was prepared from commercial source 33a (Scheme 3). Aldehydes 29a–c were first reacted with boronic acids 30a–e via palladium-catalyzed Suzuki coupling to form 31b–m,32 which were then purified by silica chromatography. Reduction of 31b–m using sodium borohydride produced alcohols 32b–m under mild conditions (room temperature) and good yield. Chlorination under treatment of thionyl chloride furnished 33b–m, which were used directly in the final step of alkylation. Synthesis of 34n,o was also accomplished using similar chemistry, starting from the reduction of aldehydes 31n,o. While the isolation yield of these extended biaryl analogs were varied (37–98%), they were synthesized rapidly (less than 1 h) under microwave conditions and less impurities were observed by LC-MS. Compound 34p was synthesized from 4(5)-nitroimidazole carboxamide 36 through acylation with (4-(piperidin-1-yl)phenyl)methanamine 35 followed by N-alkylation and cyclization under a similar reaction condition as 27b, g, i–k.
Scheme 3. Synthesis of Nitroimidazopyrazinones with Biaryl Side Chain 34a–p(32−34).

(i) Pd(PPh3)4, K2CO3, THF, rt → 80 °C, N2 ; (ii) NaBH4, EtOH, rt; (iii) thionyl chloride, DCM, 0 °C → rt; (iv) K2CO3, DMF, μW 80–100 °C; (v) nitroimidazole acid chlorideintermediate,27 TEA, DCM, 0 °C → rt; (vi) bromoacetaldehyde diethyl acetal, K2CO3, DMF, μW 150 °C; (vii) 1 M HCl, dioxane, μW 120 °C. Intermediate 33a was from a commercial source.
Results and Discussion
A total of 36 new nitroimidazopyrazinones (17a–f, 17i–k, 26a–k, and 34a–p) and 2 previously reported analogs (17g,h) were screened for in vitro activity against a wide range of disease-causing organisms. These include M. tuberculosis grown under normoxic and hypoxic conditions and T. b. brucei, a surrogate species for Human African trypanosomiasis (HAT). Following the potential of nitroimidazopyrazinones shown against T. b. brucei from our earlier investigation,26 we have also expanded our study to another pathogenic kinetoplastid, Trypanosoma cruzi, which causes Chagas disease. We hypothesized that these new nitroimidazopyrazinones might possess activity against Giardia lamblia and Entamoeaba histolytica as their first generation and the monocyclic intermediates, nitroimidazole carboxamides,27 were active against these intestinal parasites. Therefore, selected compounds from each series were screened for activity against these protozoans. Given other nitroimidazoles (e.g., metronidazole) have antibacterial activity beyond M. tuberculosis, we assayed their antibacterial properties against representative ESKAPE pathogens (Escherichia coli, Staphylococcus aureus, Klebsiella pneumoniae, Pseudomonas aeruginosa, and Actinetobacter baumannii) as well as their antifungal potential against Cryptoccoccus neoformans and Candida albicans were assessed. Mammalian cell viability was evaluated against human embryonic kidney (HEK293) cells in order to establish a preliminary selectivity index for antimicrobial activity versus mammalian toxicity. For compounds tested against T. cruzi, cytotoxicity against 3T3 host cells was also determined. Lipophilicities of these analogs were estimated using Pipeline Pilot (Accelrys, version 9.1.0.13). Solubility was measured in water and phosphate-buffered saline (PBS), pH 7.4 using HPLC with UV analysis. Representative compounds with weak base functionalities were also tested for their aqueous solubility in acetate buffer, pH 4.5 and 0.1 M HCl, pH 1. The low pH mimics the gastric fluid environment, which is beneficial to enhance dissolution of weakly basic drugs.35
Series 1 (Table 1)
For the first series 17a–k, insertion of ketone, amide, and ether groups to the methylene chain led to overall reduced lipophilicity based on Pipeline Pilot AlogP calculations (Table 1). The presence of oxygen in ether and ketone linked molecules functions as a strong hydrogen bond acceptor, whereas the amide linker serves as a strong hydrogen bond acceptor and as a modest hydrogen bond donor. Compared to our reported analog 16a with a MIC of 0.5–1 μg/mL against H37Rv strains under aerobic growth, 17a, 17d, 17e, and 17i were inactive (MICnormoxia ≥ 32 μg/mL), although 17i showed ∼11-fold of improvement in its solubility. Some activity was restored with the addition of an electron-donating 4-methyl group at the phenyl ring. However, this more polar series generally showed unfavorable antimycobacterial activity (Table 1). As many of the recently developed anti-TB agents are highly lipophilic, this suggests the importance of lipophilicity in a molecule to penetrate into the hydrophobic phase of mycobacteria cell wall.36
Table 1. Physicochemical Properties and Antimycobacterial and Antiparasitic Activities of 17a–k Compared to the Parent Analogs 16a–dg.
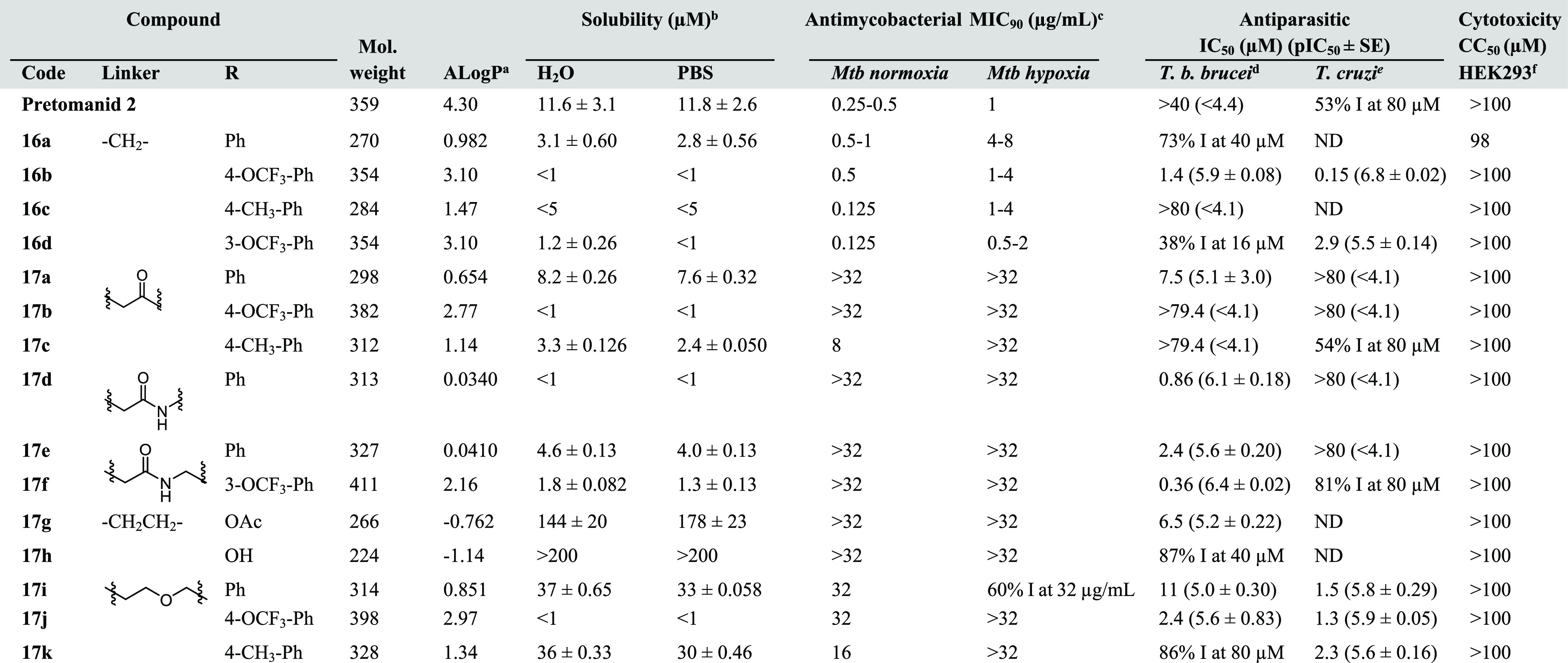
AlogP values were calculated using Pipeline Pilot (Accelrys, version 9.1.0.13).
Solubility in water and PBS (pH 7.4) were determined by LC-UV (254 nm).
H37RV, Mtb-O2 primary screen at 32 μg/mL n = 2, Mtb-hypoxia primary screen at 32 μg/mL n = 2, MIC-normoxia/hypoxia of active compounds n = 3. Isoniazid control Mtb-normoxia MIC = 0.04 μg/mL, Mtb-hypoxia MIC > 5 μg/mL.
Control pentamidine IC50 = 0.002 μM, diminazene aceturate IC50 = 0.062 μM, puromycin IC50 = 0.05 μM, n = 2.
Control nifurtimox IC50 = 1.4 μM, benznidazole IC50 = 5.2 μM, puromycin IC50 = 2.5 μM, posaconazole IC50 = 0.04 μM, n = 2.
ATCC CRL-1573, CC50n = 3. Additional cytotoxicity data are detailed in the Supporting Information, Table S2.
For compounds that did not completely inhibit growth at the concentration tested, the percentage inhibition (% I) is given. ND, not determined.
Determination of antitrypanosomal activity disclosed a different SAR. Amide linkers were overall well tolerated with respect to T. b. brucei activity, giving at least an 8-fold improvement of activity compared to the methylene linker (incomplete inhibition at 16 and 40 μM). Compound 17d (−CH2CONH−) with an amide bond directly linked to the phenyl side chain (IC50 = 0.86 μM) was ∼3-fold more potent than 17e (−CH2CONHCH2−) with an extra carbon length (IC50 = 2.4 μM). Addition of a 3-trifluoromethoxy group at the phenyl ring was also favorable (∼7-fold, IC50 = 0.36 μM). Though the presence of amide linker lowered the calculated lipophilicity (ΔAlogP ≈ −0.94 units), there was little or no improvement in their aqueous solubility. The introduction of a more soluble ether group linker displayed only a mild effect on T. b. brucei activity compared to the parent analogs (16a–c). These ether-linked derivatives 17i–k were the only actives in this series against T. cruzi, with IC50 values ranging from 1.3 to 2.3 μM, comparable activity to one of the standard drugs nifurtimox (IC50 = 1.4 μM) and more potent than benznidazole (IC50 = 5.2 μM). Though they were less active than the parent analog 16b, these series demonstrated improved solubility in some cases and had good selectivity indices of >43 to >77.
Series 2 (Table 2)
Overall, compounds with less lipophilic heterocycles replacing the aryl substituent displayed significant improvement of solubility compared to the phenyl substituent. However, in most cases, their antimycobacterial activity was compromised. For example, compound 26a containing a thiazol-5-yl substituent was >50-fold more soluble than 16a, though its activity against M. tuberculosis under normoxic conditions was reduced by 32-fold (Table 2). Replacement of the phenyl ring with a more soluble pyridine group was also detrimental to its activity. However, addition of a 4-CF3 electron-withdrawing group managed to restore some of the activity (MICnormoxia = 2 μg/mL cf. MICnormoxia of 16a = 0.5–1 μg/mL) and concurrently provided superior aqueous solubility (30 μM cf. 16a: 3.1 μM). It was found that compounds with negative AlogP values generally possessed poor MICnormoxia ≥ 16 μg/mL. Analogs with positive AlogP in this series remained active (MICnormoxia = 2–4 μg/mL), with the exception of compound 26f that displayed solubility problems even in the DMSO stock solution. None of the active analogs surpassed the activity of the parent compounds (for example, 16a–d), suggesting that an alternative strategy is required to achieve a desirable compromise between solubility and bioactivity.
Table 2. Physicochemical Properties and Antimycobacterial and Antiparasitic Activities of 26a–kg.
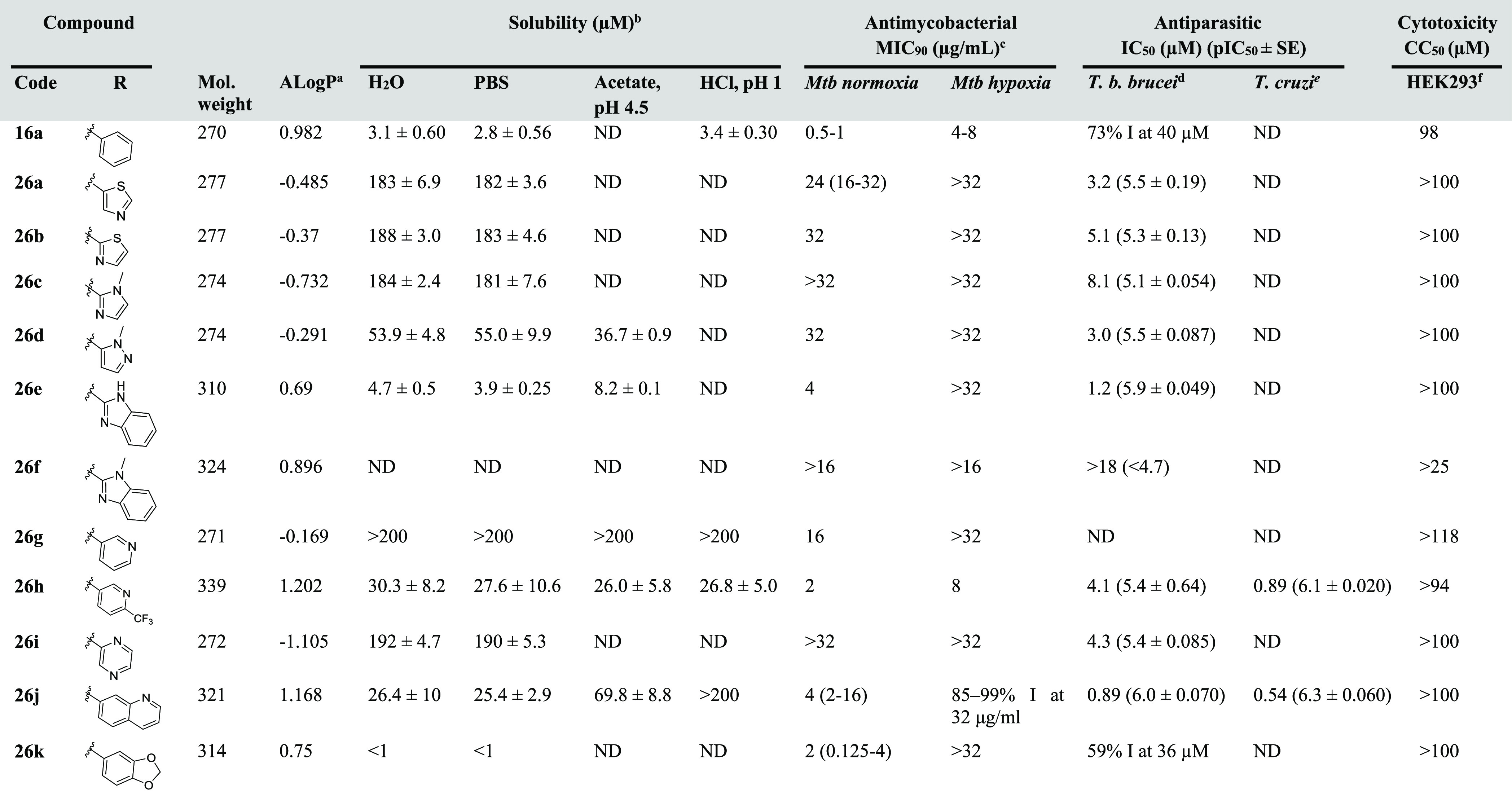
AlogP values were calculated using Pipeline Pilot (Accelrys, version 9.1.0.13).
Solubility were determined by LC-UV (254 nm).
H37RV, Mtb-O2 primary screen at 32 μg/mL n = 2, Mtb-hypoxia primary screen at 32 μg/mL n = 2, MIC-normoxia/hypoxia of active compounds n = 3–4. For compounds that gave varied MIC in different replicates (n = 3–6), a median value was reported with the MIC range indicated in parentheses. Isoniazid control Mtb-normoxia MIC = 0.04 μg/mL, Mtb-hypoxia MIC > 5 μg/mL.
Control pentamidine IC50 = 0.002 μM, diminazene aceturate IC50 = 0.062 μM, puromycin IC50 = 0.05 μM, n = 2.
Control nifurtimox IC50 = 1.4 μM, benznidazole IC50 = 5.2 μM, puromycin IC50 = 2.5 μM, posaconazole IC50 = 0.04 μM, n = 2.
ATCC CRL-1573, CC50n = 3. Additional cytotoxicity data are detailed in the Supporting Information, Table S2.
For compounds that did not completely inhibit growth at the concentration tested, the percentage inhibition (% I) is given. ND, not determined.
We next investigated the SAR of this series against T. b. brucei. As in series 1, in most cases, the less lipophilic heterocycles were well tolerated, with an IC50 of <10 μM. Compound 26j with a quinolone substituent was the most active in this series (IC50 = 0.89 μM) and gave a substantial enhancement of solubility (>8-fold) compared to 16a. The weak basic nature of 26j allowed for further improvement of solubility under acidic conditions (70 μM at pH 4.5 and > 200 μM at pH 1). It is noted that 26j was also active against T. cruzi at submicromolar concentrations (IC50 = 0.54 μM), and selectivity index of >136 to 3T3 host cells (Supporting Information, Table S2). As this analog was moderately active against M. tuberculosis (MICnormoxia = 4 μg/mL), this indicates the possibility to identify compounds with broad-spectrum activity and low toxicity against mammalian cells that could potentially treat both tuberculosis and kinetoplastid infections.
Series 3 (Table 3)
Following the precedent set by several new bicyclic nitroimidazoles with biaryl side chains such as 4 and 8 that are in pre-clinical development against leishmaniasis and tuberculosis, respectively,16,21 a similar strategy was employed in the third series. As suggested from the SAR for series 1 and 2, compounds with less lipophilicity had less favorable antimycobacterial activity. Therefore, extended and highly lipophilic biphenyl side chains were explored to understand whether this property is the main driver. It was also postulated that the extended side chains could interact more efficiently with the hydrophobic pocket in Ddn, the nitroreductase enzyme responsible for the bioactivation of bicyclic nitroimidazoles 1 and 2.37 To counter the solubility issue, bioisosteres were also introduced to replace one of the phenyl rings in this series. The effect of different biaryl geometries with 4′ and 3′-linked biaryls as well as the position of the substituents were investigated. Based on the results of the previously developed monoaryl series, we included the most active substituents, 4-trifluoromethoxy, 4-methyl, and 3-trifluoromethoxy, on the terminal ring.
Generally, the biphenyl analogs 34a–d from Table 3 show that these conformationally rigid extended side chains did not contribute to a better activity against M. tuberculosis. In fact, they were 16- to 64-fold less active than the monoaryl analogs 16a–d. This observation is different from what has been reported for the biphenyl structures of analogs of pretomanid 2, in which the para-linked compounds retained or improved their antitubercular activity compared to the monoaryl 2.38 This suggests a different binding mode of the nitroimidazopyrazinone scaffold 9 in the active site compared to 2, which is not unexpected given the planar nature of 9 compared to 2. There was a preference for substitution at the meta position over para at the terminal ring of the 4′ biphenyl analogs. A similar trend was observed for the less lipophilic phenylpyridine analogs (ΔAlogP ≈ −0.7 unit), where the 3-trifluoromethoxy congener 34i was 8-fold more active than its 4-trifluoromethoxy counterpart. Overall, the phenylpyridines 34f–i gave similar or 2-fold reduced activity than the 4′-biphenyl analogs 34a–d, though the solubility (at pH 1) was improved for some of these derivatives. The position of the nitrogen atom at the pyridine ring was also found to be important for its bioactivity. Replacement of the 3-pyridine ring in 34g by 2-pyridine (34j) substantially improved its antimycobacterial activity, from an MIC of 32 μg/mL to 0.0625–0.125 μg/mL against the replicating M. tuberculosis. However when M. tuberculosis was grown under hypoxic conditions (0.1% oxygen), 34j exhibited only marginal inhibitory activity, though still better than 34g. Under acidic conditions (pH 1), 34j possessed >3-fold better solubility than 34g.
Table 3. Physicochemical Properties and Antimycobacterial and Antiparasitic Activities of 34a–pg.
| compound |
solubility
(μM)b |
antimycobacterial
MIC90 (μg/mL)c |
antiparasitic
IC50 (μM) (pIC50 ± SE) |
cell viability CC50 (μM) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| code | R1 | link | R2 | mol. weight | ALogPa | H2O | PBS | acetate, pH 4.5 | HCl, pH 1 | Mtb normoxia | Mtb hypoxia | T. b. bruceid | T. cruzie | HEK293f |
| 34a | Ph | para | Ph | 346 | 2.50 | <1 | <1 | <1 | <1 | 16 | 79% I at 32 μg/mL | 53–55% I at 7.9 μM | 4.65 (5.3 ± 0.003) | >100 |
| 34b | 4-OCF3-Ph | 430 | 4.62 | <1 | <1 | <1 | <1 | 32 | >32 | 0.045 (7.4 ± 0.003) | 0.043 (7.4 ± 0.007) | >100 | ||
| 34c | 4-CH3-Ph | 360 | 2.97 | <1 | <1 | <1 | <1 | 6 (4–8) | 32 | 0.074 (7.13 ± 0.0003) | 0.077 (7.1 ± 0.013) | >50 | ||
| 34d | 3-OCF3-Ph | 430 | 4.62 | <1 | <1 | <1 | <1 | 2 (1–8) | 32 | 0.110 (7.0 ± 0.009) | 0.065 (7.2 ± 0.015) | >100 | ||
| 34e | Pyridin-3-yl | 347 | 1.35 | 3.9 ± 0.67 | 3.2 ± 1.0 | 19 ± 1.4 | >200 | 0.5 | 8 | 0.042 (7.4 ± 0.003) | 0.10 (7.0 ± 0.004) | >92 | ||
| 34f | pyridin-3-yl | Ph | 347 | 1.78 | <1 | <1 | <1 | 18 ± 0.3 | 32 | >32 | 0.067 (7.2 ± 0.003) | 0.073 (7.1 ± 0.005) | >50 | |
| 34g | 4-OCF3-Ph | 431 | 3.90 | <1 | <1 | <1 | <1 | 32 | >32 | 0.026 (7.6 ± 0.003) | 0.16 (6.8 ± 0.016) | >50 | ||
| 34h | 4-CH3-Ph | 361 | 2.26 | <1 | <1 | <1 | 7.8 ± 0.4 | 8 | 81% I at 32 μg/mL | 0.008 (8.1 ± 0.002) | 0.066 (7.2 ± 0.001) | >50 | ||
| 34i | 3-OCF3-Ph | 431 | 3.90 | <1 | <1 | <1 | <1 | 4 (2–16) | >32 | 0.021 (7.7 ± 0.0005) | 0.088 (7.0 ± 0.046) | >50 | ||
| 34j | pyridin-2-yl | 4-OCF3-Ph | 431 | 3.68 | <1 | <1 | <1 | 3.7 ± 0.69 | 0.0625 (0.0625–0.125) | 84% I at 8–32 μg/mL | 0.002 (8.6 ± 0.0001) | 0.016 (7.8 ± 0.0002) | >74 | |
| 34k | pyridin-3-yl | meta | 4-OCF3-Ph | 431 | 3.47 | <1 | <1 | <1 | 16 ± 0.5 | 0.25 (0.25–1) | 85% I at 4–32 μg/mL | 0.028 (7.6 ± 0.005) | 0.43 (6.4 ± 0.019) | >100 |
| 34l | 4-CH3-Ph | 361 | 1.84 | <1 | <1 | <1 | 27 ± 14 | 0.0625 (0.0625–0.125) | 84% I at 8–32 μg/mL | 0.067 (7.2 ± 0.018) | 0.12 (6.9 ± 0.002) | >94 | ||
| 34m | 3-OCF3-Ph | 431 | 3.47 | <1 | <1 | <1 | 6.6 ± 0.3 | 2 (0.125–4) | 84% I at 32 μg/mL | 0.093 (7.0 ± 0.024) | 0.23 (6.6 ± 0.014) | >100 | ||
| 34n | Ph | para | thiazol-2-yl | 353 | 1.36 | <1 | <1 | <1 | <1 | >32 | >32 | 0.53 (6.3 ± 0.051) | 0.21 (6.7 ± 0.005) | >50 |
| 34o | thiazol-5-yl | Ph | 353 | 1.44 | <1 | <1 | <1 | <1 | 0.5 (0.5–1) | >32 | 0.045 (7.3 ± 0.001) | 0.051 (7.3 ± 0.003) | >50 | |
| 34p | Ph | para | piperidinyl | 353 | 2.06 | 3.8 ± 0.5 | <1 | 2.7 ± 0.05 | >200 | 1.5 (1–2) | 8 | 39–67% I at 73.3 μM | 82–83% I at 36.6 μM | >100 |
ALogP values were calculated using Pipeline Pilot (Accelrys, version 9.1.0.13).
Solubility in water, PBS (pH 7.4), acetate buffer (pH 4.5), and 0.1 M HCl (pH 1 ) were determined by LC-UV (254 nm).
H37RV, Mtb-O2 primary screen at 32 μg/mL n = 2, Mtb-hypoxia primary screen at 32 μg/mL n = 2, MIC-normoxia/hypoxia of active compounds n = 3–4. For compounds that gave varied MIC in different replicates (n = 3–6), a median value was reported with the MIC range indicated in parentheses. Isoniazid control Mtb-normoxia MIC = 0.04 μg/mL, Mtb-hypoxia MIC > 5 μg/mL.
Control pentamidine IC50 = 0.002 μM, diminazene aceturate IC50 = 0.062 μM, puromycin IC50 = 0.05 μM, n = 2.
Control nifurtimox IC50 = 1.4 μM, benznidazole IC50 = 5.2 μM, puromycin IC50 = 2.5 μM, posaconazole IC50 = 0.04 μM, n = 2.
ATCC CRL-1573, CC50n = 3. Additional cytotoxicity data are detailed in the Supporting Information, Table S2.
For compounds that did not completely inhibit growth at the concentration tested, the percentage inhibition (% I) is given.
According to the general solubility equation developed by Jain and Yalkowsky, a compound can exhibit poor solubility due to high lipophilicity and high stability in the crystalline state. It is estimated that altering log P by 1 unit or melting point by 100 °C will change solubility by 10-fold.39 Hence, an alternative strategy was attempted by disrupting their molecular symmetry. The crystal packing of a molecule is influenced by its molecular symmetry and planarity, and improved solubility can be achieved by decreasing the efficiency of its crystal packing to lower the melting point.40 In the 3′-linked phenylpyridines 34k–m, we have observed at least 3- to >16-fold of improvement in solubility at pH 1 compared to 4′-linked phenylpyridines, with the 3′-substitution disrupting the potential for planar packing. Compounds 34k–m also showed 2- to 128-fold better antitubercular activity compared to their 4′-linked analogs. Among them, 34l displayed an MIC of 0.0625–0.125 μg/mL, which was equipotent with the parent analog 16c. As opposed to the 4′-linked biaryls, the para-substitution (34k) was preferred over its meta group (34m) in terms of potency and solubility.
Swapping the positions of the less lipophilic ring in the biaryl system led to substantial effects on potency and physicochemical properties (Figure 3). Biaryl compound 34e with a terminal 3-pyridine ring displayed 64-fold of improvement in antitubercular activity under aerobic conditions as compared to 34f with a terminal phenyl ring. Compound 34e was also more soluble than 34f in both water and PBS, pH 7.4, and its solubility was further improved at pH 4.5 (19 μM) and pH 1 (>200 μM). When pyridine was replaced by a thiazole group at the terminal ring (34n), there was a complete loss of activity, indicating that the antitubercular activity was strongly dependent on the nature of the heterocycles. In spite of having similar AlogP values, 34n was also at least 3-fold less soluble than 34e in water and PBS. When the position between thiazole and phenyl rings was inverted (34o), the antitubercular activity improved at least 32-fold, reaching an MICnormoxia of 0.5–1 μg/mL. Removing the aromaticity by replacing the pyridine ring with a piperidine ring (34p) resulted in 2- to 4-fold reduction of activity against replicating M. tuberculosis under aerobic conditions (MICnormoxia = 1–2 μg/mL) but retained similar activity against non-replicating M. tuberculosis (MIChypoxia = 8 μg/mL).
Figure 3.
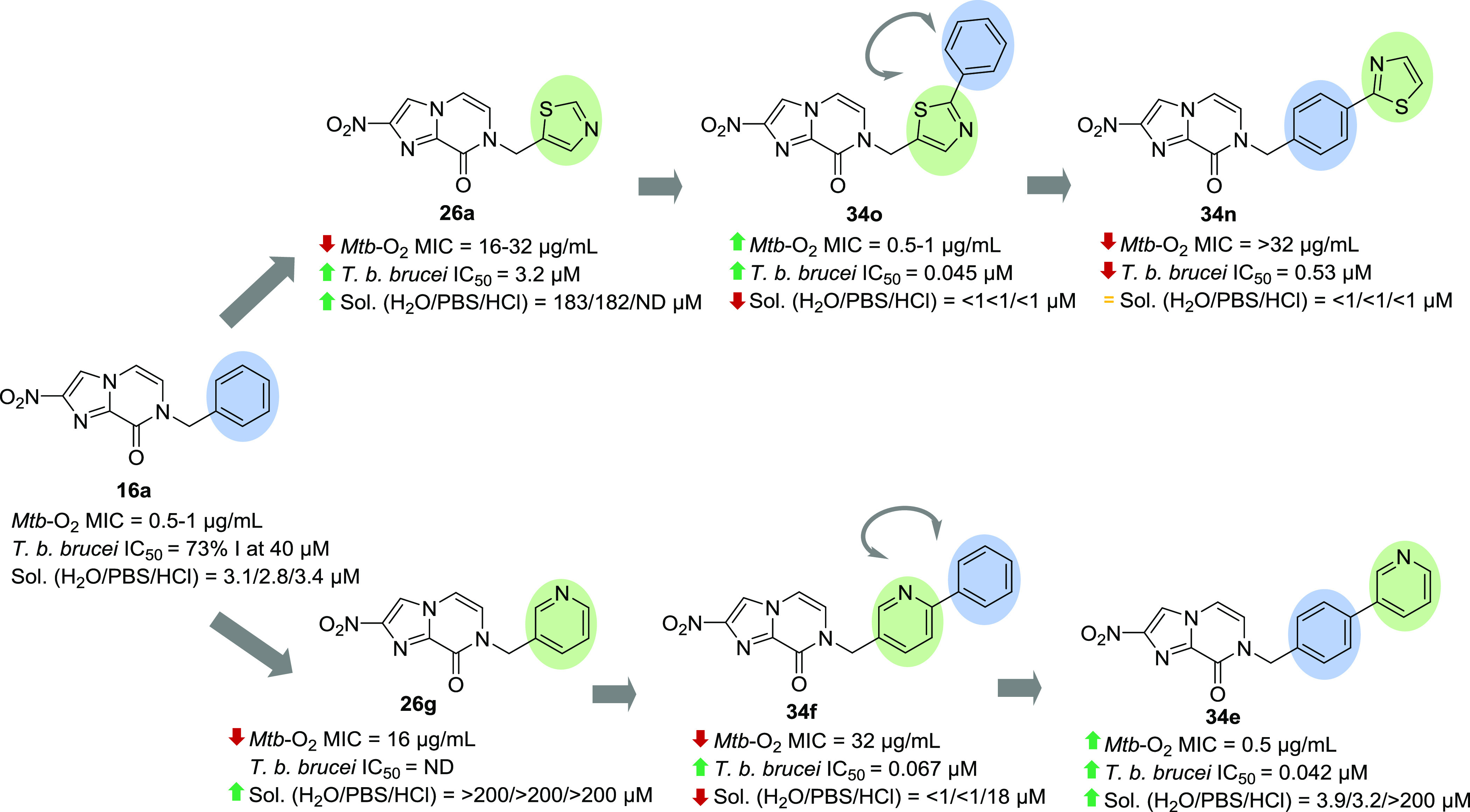
Influence of heterocyclic rings on the bioactivity and solubility of nitroimidazopyrazinones.
In comparison to the monoaryl compounds, the biaryl series demonstrated superior activity against both T. b. brucei and T. cruzi. Of a total of 16 biaryl analogs synthesized, 14 were identified as active hits, with mean IC50 values of <1 μM and selectivity indices of >10 for parasitic activity compared to mammalian cell cytotoxicity. More than half of these hits were able to achieve IC50 at ≤0.1 μM. Contrary to the SAR in mycobacteria, 4′-phenylpyridines 34f–i possessed better activity against T. b. brucei than the 4′-biphenyl side chains 34a–d. Compounds 34g-i were also ∼1.1- to 8.4-fold more active than 3′-linked phenylpyridines 34k–m, with an IC50 range of 0.008–0.026 μM. A similar SAR trend was observed in T. cruzi, though their activity was less compared to T. b. brucei in most of the cases. Compound 34j with 2-pyridine ring was identified as the most potent candidate, giving an impressive IC50 of 0.002 μM and 0.016 μM against T. b. brucei and T. cruzi, respectively, with 100% activity at Emax concentrations (maximal % inhibition plateau generated in dose–response curves). This compound also exhibited selectivity indices against HEK293 cells (20 h assay) of >37,000 and >4625, and selectivity of >2288 to 3T3 host cells (69 h assay, Supporting Information, Table S2). It should be noted that 34j was one of the most active antitubercular hits, suggesting a broad-spectrum activity. Altering the position of the thiazole ring from 34n to 34o enhanced the activity against both T. b. brucei and T. cruzi, whereas a marginal effect was observed when swapping pyridine from 34e to 34f. When the terminal 3-pyridine ring was replaced by a saturated heterocycle (piperidine), the antitrypanosomal activity of 34p was diminished.
Activity against Intestinal Parasites, ESKAPE Pathogens, and Fungi
Given the good activity shown by some of our previously reported nitroimidazopyrazinones against G. lamblia (IC50 = 1.6–3.5 μM),26 we first investigated compounds from Series 1 to examine the linker effect. Similar to the trends shown in M. tuberculosis, modification of the methylene chain resulted in drastic loss of activity (Table 4). Replacing the benzyl substituent with less lipophilic monoheterocycles (Series 2) also proved to be unfavorable. Fused rings such as quinolone (26j) and benzodioxole (26k) managed to restore some of the activity, with IC50 values of 7.1 and 2.4 μM, respectively. Most of the tested biaryl analogs remained active in the primary screen at 50 μM. However, some inconsistent SAR results were found when determining their IC50 values. The biphenyl compound 34a achieved 10-fold better potency than its monophenyl counterpart (IC50 = 0.5 μM cf. IC50 of 16a = 5 μM), whereas addition of a 3-OCF3 substituent (34d) reduced activity by >20-fold. Follow-up study is hence required in the future to determine the inhibitory concentration of more analogs from this series.
Table 4. Additional Activity Data for Selected Analogs from Each Series.
| antiparasitic
IC50 (μM) (pIC50 ± SE) |
antibacterial MIC (μg/mL) | |||
|---|---|---|---|---|
| compound | G. lambliaa | E. histolyticab | ESKAPEc | antifungald MIC (μg/mL) |
| pretomanid 2 | 3.0 (5.5 ± 0.02) | 9.3 (5.0 ± 0.03) | >32 | >32 |
| 16a | 5.0 (5.3 ± 0.05) | >50 (<4.3) | >32 | >32 |
| 16b | 3.5 (5.5 ± 0.01) | >50 (<4.3) | >32 | >32 |
| 16d | 1.7 (5.8 ± 0.03) | >50 (<4.3) | >32 | >32 |
| Series 1 | ||||
| 17a | 18 (4.7 ± 0.03) | >50 (<4.3) | >32 | >32 |
| 17d | >50 (<4.3) | >50 (<4.3) | >32 | >32 |
| 17e | >50 (<4.3) | >50 (<4.3) | >32 | >32 |
| 17i | >50 (<4.3) | >50 (<4.3) | >32 | >32 |
| 17k | 23 (4.6 ± 0.02) | >50 (<4.3) | >32 | >32 |
| Series 2 | ||||
| 26a | 40 (4.4 ± 0.04) | >50 (<4.3) | >32 | >32 |
| 26b | 19 (4.7 ± 0.04) | >50 (<4.3) | >32 | >32 |
| 26j | 7.1 (5.1 ± 0.02 | >50 (<4.3) | >32 | >32 |
| 26k | 2.4 (5.6 ± 0.04) | >50 (<4.3) | >32 | >32 |
| Series 3 | ||||
| 34a | 0.5 (6.3 ± 0.11) | >50 (<4.3) | >32 | >32 |
| 3d | 37 (4.4 ± 0.08) | >50 (<4.3) | >32 | >32 |
| 34k | 1.8 (5.7 ± 0.09) | >50 (<4.3) | >32 | >32 |
| 34p | 5 (5.3 ± 0.03) | >50 (<4.3) | >32 | >32 |
WB.
HM1:IMSS.
ESKAPE pathogens include Staphylococcus aureus (MRSA ATCC 43300), Escherichia coli (ATCC 25922), Klebsiella pneumoniae (ATCC 700603), Acinetobacter baumannii (ATCC 19606), and Pseudomonas aeruginosa (ATCC 27853).
Candida albicans (ATCC 90028) and Cryptococcus neoformans var. grubii H99 (ATCC 208821).
Besides antigiardial activity, selected analogs from each series were also screened against E. histolytica, ESKAPE bacteria strains and two fungal pathogens. Similar to our first generation of nitroimidazopyrazinones, none of the tested compounds were active against these organisms. We have also counter screened these compounds against mammalian cell line HEK293 and minimal reduction in cell viability was seen up to the highest tested concentrations (>50 or >100 μM, respectively, depending on the solubility in DMSO stock, Tables 1–3).
Plasma Protein Binding and Microsomal and Plasma Stability
To determine the drug-like properties of these new series, a small subset of compounds was evaluated in vitro to determine their plasma protein binding (PPB), microsomal stability, and plasma stability (Table 5). The more soluble analogs 17k and 26j demonstrated a reduced propensity to bind to plasma protein (72–79% PPB in human and mouse plasma) compared to other compounds. The biaryl series were generally highly bound to plasma, especially compounds 34g, 34j, and 34k that demonstrated >99% PPB due to their high lipophilicity (AlogP > 3). On the other hand, phenylpyridine 34e and phenylpiperidine 34p, with better solubility and lower lipophilicity, demonstrated a slightly lower PPB (93–99%). A good correlation between the compound lipophilicity and PPB was identified, suggesting the potential to utilize this parameter to modulate protein binding. However, it is also known that free drug concentration is not merely determined by PPB but also by hepatic intrinsic clearance after oral dosing.41 Thus, we investigated the metabolic stability of these analogs in human liver microsomes (HLM). The biaryl compounds tested were highly stable up to 2 h. Replacing the terminal aryl ring with piperidine (34p) reduced the microsomal stability from >95% to 37%. Similar to some of the reported pretomanid analogs,42 benzyl ether substituent (17k) was highly susceptible to metabolism, with 8.5% of intact compound remaining at 2 h. We have also expanded the metabolic assay to CD-1 mouse liver microsomes (MLM) for two hit compounds, 34e and 34j, prior to assessment in future in vivo studies against T. brucei and T. cruzi. The most potent compound 34j displayed excellent stability in both HLM and MLM. However, interspecies variation was observed for phenylpyridine 34e, with 20% of intact compound remaining at 2 h in MLM compared to 96% in HLM. Similar to the first generation of nitroimidazopyrazinones,26 these new series remained highly stable in plasma (>90% at 2 h) regardless of their side chains.
Table 5. In Vitro ADME Properties of Representative Compoundsa.
| PPB
(%) |
microsomal
stability (% remaining at 2 h) |
plasma
stability (% remaining at 2 h) |
||||
|---|---|---|---|---|---|---|
| compound | human | mouse | human | mouse | human | mouse |
| pretomanid 2 | 97 ± 1.1 | 91 ± 1.3 | 97 ± 5.0 | 92 ± 2.7 | 96 ± 6.9 | 96 ± 1.9 |
| 17k | 78 ± 4.1 | 74 ± 3.4 | 8.5 ± 0.73 | ND | 96 ± 6.8 | ND |
| 26j | 79 ± 0.44 | 72 ± 1.3 | 98 ± 2.5 | ND | 95 ± 4.3 | ND |
| 34e | 96 ± 0.88 | 93 ± 0.26 | 96 ± 7.7 | 20 ± 4.0 | 93 ± 2.6 | >99 |
| 34g | >99 | >99 | 98 ± 2.6 | ND | 98 ± 2.1 | ND |
| 34j | >99 | >99 | >99 | 98 ± 1.9 | >99 | >99 |
| 34k | >99 | >99 | >99 | ND | >99 | ND |
| 34p | 97 ± 0.30 | 99 ± 0.02 | 37 ± 1.7 | ND | >99 | ND |
Values are presented as mean of three replicates ± SD. Microsomal stability control, verapamil at 30 min = 9% (human), 2% (mouse). Plasma stability control, eucatropine at 2 h = 28% (human), 29% (mouse). % bound of PPB control, sulfamethoxazole = 57% (human), 9% (mouse). ND, not determined.
Conclusions
Current therapeutics against neglected tropical diseases are imperfect due to toxicity, inadequate efficacies against all subspecies/species, and prolonged regimens with poor patient compliance. M. tuberculosis has developed resistance rapidly, leading to the emergence of multidrug- and extensively drug-resistant TB with low cure rates. To improve treatment regimens in an effort to reduce poor patient compliance, low dosing frequency, reduced toxicity, and easy administration are required. Oral, affordable drugs are preferred as they are more convenient to be used, thus increasing compliance especially in developing countries where treatment is often far from the patient. However, many new chemical entities are poorly soluble, which makes them difficult to achieve desired therapeutic concentration after oral dosing.
In this study, we aimed to optimize the solubility and activity profiles of a new bicyclic nitroimidazole subclass, namely, nitroimidazopyrazinone, as an oral treatment against tuberculosis and parasitic infections. Three new exploratory series were synthesized, featuring a range of linker groups, heterocycles, and extended biaryl side chains. Modification of the methylene chain of 16 with polar linkers was not favorable for activity against M. tuberculosis, though some of them were active against the trypanosome species tested. Similarly, attempts to replace the phenyl substituent with less lipophilic heterocycles resulted in significant improvements of solubility but with compromised antitubercular activity. The series with biaryl extended side chains provided the most promising outcome in optimizing both the potency and solubility while retaining good selectivity index of >10 to mammalian cell lines. Several phenylpyridine analogs showed improved solubility at low pH conditions without compromising their activity against M. tuberculosis. It was also found that the meta-linked analogs were superior to the more linear, para-linked counterparts, in terms of their solubility and antimycobacterial activity. The biaryl compounds were highly active against both T. b. brucei and T. cruzi, with an increase in activity by 1–4 orders of magnitude in comparison to the monoaryl series. Compound 34e with a terminal pyridine ring combined both better solubility profile (>200 μM at pH 1) with good bioactivity against M. tuberculosis and trypanosomes but displayed a shorter half-life in mouse microsomes. The 2-pyridine 34j was identified as the most potent hit, demonstrating submicromolar to nanomolar inhibitory concentrations against M. tuberculosis, T. b. brucei, and T. cruzi while showing low cytotoxicity against HEK293 cells (CC50 > 74 μM, or selectivity indices of >510 to >37,000). Compound 34j also possessed good metabolic stability in both microsomes and plasma, despite having high PPB levels. As many clinical drugs are highly bound to plasma, it is not recommended to optimize this parameter in early-stage drug discovery.
This study highlights the potential of nitroimidazopyrazinone as a promising bicyclic subclass to be developed as anti-infective agents. It was found that some of the potent antitubercular biaryl compounds were highly active against both T. b. brucei and T. cruzi, unveiling their broad-spectrum activity for treating multiple infections including neglected tropical diseases. Several compounds were also moderately active against G. lamblia, albeit with no activity against E. histolytica. Similar to the activity profile of pretomanid 2, none of these nitroimidazopyrazinones was active against ESKAPE and fungal pathogens (MIC > 32 μg/mL). For further development of this series, it appears that both broad-spectrum (antitubercular and antiparasitic) and narrow-spectrum strategies are possible, but it is less clear which approach is most desirable. Co-infection with both mycobacteria and trypanosomes happens in some regions of the world, where a broad-spectrum treatment could be useful. However, this is offset by the possibility that widespread use against one organism could inadvertently lead to the development of resistance against the other. Since nitroimidazoles are pro-drugs that require bioactivation of their nitro group to exert the activity, resistance can arise when there is a change of activity or mutations of the reductive enzymes.3 In some cases, this resistance can be overcome by other nitroimidazoles with different mode of action. Further studies are needed to assess the cross-resistance profile of the nitroimidazopyrazinones. In vitro ADME and cytotoxicity studies have revealed favorable drug-like profiles for the hit compounds, which warrant advancement to in vivo testing to determine pharmacokinetic profiles and assess activity in both acute and chronic infection models of M. tuberculosis, T. b. brucei, and T. cruzi.
General Experimental Section
All the reagents and solvents were obtained from commercial sources and were used without further purification. Progression of reaction was monitored by TLC using Merck alumina sheets pre-coated with silica gel 60 F254 and was visualized using a UV lamp. Reactions requiring anhydrous conditions were performed under an inert atmosphere of nitrogen. Purification of compounds was done using a Biotage Isolera or Grace Reveleris chromatography system. NMR data were collected at 298 K on a Varian Unity 400 MHz or Bruker Advance 600 MHz spectrometer. Chemical shifts were measured relative to tetramethylsilane or the residual solvent signals (DMSO-d6) as the internal reference. Data are presented as follows: chemical shift (ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, bs = broad singlet) and coupling constant (Hz). All the peaks for final products were assigned based on one-dimensional and two-dimensional NMR analysis. Analytical LC-MS was performed on a Shimadzu LCMS-2020 using 0.05% formic acid in water (solvent A) and 0.05% formic acid in acetonitrile (solvent B) as the mobile phase. LC-MS condition A: column Zorbax Eclipse XDB-Phenyl, 3.0 × 100 mm, 3.5 μm; column temperature: 40 °C; flow: 1 mL/min; gradient timetable: 0.00 min, 5% B; 0.50 min, 5% B; 3.00 min, 100% B; 4.2 min, 100% B; 5.00 min, 5% B. LC-MS condition B: column: Waters Atlantis T3, 2.1 × 50 mm, 5 μm; column temperature: 40 °C; flow: 0.75 mL/min; gradient timetable: 0.00 min, 5% B; 0.50 min, 5% B; 3.30 min, 25%B; 3.50 min, 100% B; 4.00 min 100% B; 5.00 min, 0% B. All final products were >95% pure as determined by LC-MS using UV at 254 nm, ELSD, and APCI/ESI-MS. The purity percentage was indicated by Abs %.
Synthetic Procedures
General Procedure for the Preparation of 2-Bromoacetamides
Method A: Bromoacetyl bromide (1.0 equiv) was added dropwise to a stirring solution of amine (0.83 equiv) and triethylamine (1.0 equiv) in 20 vol of anhydrous dichloromethane at 0 °C under nitrogen. The reaction mixture was stirred at rt for 1–24 h and then concentrated under reduced pressure after completion of reaction.
General Procedure for Alkylation of Imidazopyrazinone
Method B: To a stirring suspension of imidazopyrazinone (1 equiv) in dry DMF (20 vol) was added 3 equiv of cesium carbonate or potassium carbonate, followed by addition of 1.2–1.5 equiv of benzyl bromide in dropwise. The reaction was either stirred at room temperature or microwaved at 80–100 °C.
Method C: To a stirring suspension of imidazopyrazinone (1 equiv) in dry DMF (20 vol) at 0 °C was added 1.5 equiv of 60% sodium hydride followed by addition of 1.2–1.5 equiv of benzyl bromide dropwise. The reaction was stirred at room temperature overnight under N2.
General Procedure for Chlorination of Alcohol
Method D: Alcohol (1 equiv) was added with DCM (20 vol) and was chilled at 0 °C. Thionyl chloride (2–3 equiv) was added dropwise, and the reaction was continued to stir at room temperature for 1–24 h. The mixture was then diluted with NaHCO3 (200 vol) and extracted with EtOAc (3x). The organic layer was collected and dried over MgSO4 before removing the volatiles in vacuo.
General Procedure for Amide Coupling
Method E: 4-Nitro-1H-imidazole-2-carboxylic acid (1 equiv) was added with anhydrous DCM (20 vol) and was chilled at 0 °C under N2. Oxalyl chloride (2 equiv) was added dropwise over 10 min followed by addition of catalytic DMF. The reaction was allowed to warm to room temperature and was continued to stir overnight. Additional oxalyl chloride and DMF was added to the reaction mixture when necessary. Volatiles were removed in vacuo, and the excess oxalyl chloride was removed by co-evaporation with toluene two times. The resulting acid chloride crude solid (1 equiv) was then added immediately to anhydrous DCM (15 vol) and triethylamine (2 equiv) in an ice bath. The blue-purple solution was then added dropwise to the respective amine (1.2 equiv) in 5 vol of anhydrous DCM at 0 °C under N2.
General Procedure for Alkylation of Imidazo Carboxamide
Method F: A mixture of imidazo carboxamide (1 equiv) and potassium carbonate (3 equiv) in dry DMF (20 vol) was reacted with bromoacetaldehyde diethyl acetal (1.5 equiv x 2) in a microwave reactor at 150 °C (15 min x 2). If necessary, the reaction was heated for another 15 min at 150 °C to consume the starting material.
General Procedure for Ring Closure to Form Imidazopyrazinones
Method G: To a stirring solution of imidazo carboxamide (1 equiv) in 1,4-dioxane (10 vol) was added 2 M aq. HCl (10 vol). The reaction was reacted at 120 °C under microwave for 20 min.
General Procedure for Suzuki Coupling Reaction
Method H: To a stirring mixture of 4-bromobenzaldehyde or bromonicotinaldehyde (1 equiv) in THF was added 3 equiv of potassium carbonate in distilled water followed by addition of 0.08 equiv of tetrakis(triphenylphosphine)palladium (0). The mixture was stirred at room temperature for 5 min. Boronic acid (1.1 equiv) was then added, and the reaction was heated at 80 °C for 18 h.
General Procedure for Reduction of Aldehyde to Alcohol
Method I: Sodium borohydride (1.3 equiv) was added in one portion to a stirring solution of aldehyde (1 equiv) in 20 vol of ethanol that was chilled to 0 °C. The mixture was continued to stir at room temperature for 30 min before the solvent was removed in vacuo. The crude product was diluted with 100 vol of distilled water and extracted with chloroform (3x). The organic layer was collected, washed with brine, and dried with MgSO4 before removing the volatiles in vacuo.
2-Bromo-N-phenylacetamide (11a)
2-Bromoacetyl bromide 10 (200 mg, 86 μL 0.991 mmol, 1 equiv) was reacted with aniline (76.9 mg, 78 μL, 0.825 mmol, 0.83 equiv) according to the general procedure method A. The residue was then purified over silica gel chromatography using 5–40% ethyl acetate in petroleum spirit to yield the desired product as a white powder (210 mg, 99%): LC-MS: Rt = 2.48 min, 99 Abs % @ 200 nm, [M + H]+ = 216.1; 1H NMR (600 MHz, DMSO-d6) δ 10.39 (s, 1H), 7.61–7.56 (m, 2H), 7.36–7.29 (m, 2H), 7.08 (tt, J = 7.4, 1.2 Hz, 1H), 4.04 (s, 2H).
N-Benzyl-2-bromoacetamide (11b)
2-Bromoacetyl bromide 10 (200 mg, 86 μL 0.991 mmol, 1 equiv) was reacted with benzylamine (88.5 mg, 90 μL, 0.825 mmol, 0.83 equiv) according to the general procedure method A. The residue was then purified over silica gel chromatography using 5–50% ethyl acetate in petroleum spirit to yield the desired product as a white powder (177 mg, 79%): LC-MS: Rt = 2.48 min, 99 Abs % @ 200 nm, [M + H]+ = 228.1; 1H NMR (600 MHz, DMSO-d6) δ 8.77 (t, J = 6.1 Hz, 1H), 7.37–7.31 (m, 2H), 7.30–7.23 (m, 2H), 4.29 (d, J = 5.9 Hz, 2H), 3.91 (s, 2H).
2-Bromo-N-(3-(trifluoromethoxy)benzyl)acetamide (11c)
2-Bromoacetyl bromide 10 (200 mg, 118 μL 0.991 mmol, 1 equiv) was reacted with 3-trifluoromethoxy benzylamine (157 mg, 129 μL, 0.825 mmol, 0.83 equiv) for 1 h according to the general procedure method A. The residue was then purified over silica gel chromatography using 0–4% methanol in dichloromethane to yield the desired product as a white powder (181 mg, 59%). LC-MS: Rt = 2.91 min, 99 Abs % @ 200 nm, [M + H]+ = 312.0; 1H NMR (600 MHz, DMSO-d6) δ 8.89–8.84 (m, 1H), 7.50–7.44 (m, 1H), 7.32–7.28 (m, 1H), 7.27–7.22 (m, 1H), 4.35 (d, J = 6.0 Hz, 2H), 3.92 (s, 2H).
2-Nitro-7-(2-oxo-2-phenylethyl)imidazo[1,2-a]pyrazin-8(7H)-one (17a)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (50 mg, 0.278 mmol, 1 equiv) was reacted with phenacyl bromide (82.9 mg, 0.416 mmol, 1.5 equiv) at room temperature for 30 min according to the general procedure method B. After the reaction was completed as analyzed by LC-MS, the mixture was diluted with distilled water (15 mL) and the precipitate was collected by filtration. The precipitate was then purified by recrystallization using hot ACN/MeOH to yield the desired product, 2-nitro-7-(2-oxo-2-phenylethyl)imidazo[1,2-a]pyrazin-8(7H)-one 17a, as a brown solid (30 mg, 36%): LC-MS: Rt = 2.60 min, 99 Abs % @ 254 nm, [M + H]+ = 299.1; 1H NMR (600 MHz, DMSO-d6) δ 8.89 (s, 1H), 8.12–8.06 (m, 2H), 7.75 (ddt, J = 7.8, 7.1, 1.3 Hz, 1H), 7.66 (d, J = 5.8 Hz, 1H), 7.65–7.59 (m, 2H), 7.34 (d, J = 5.8 Hz, 1H), 5.60 (s, 2H). 13C NMR (150 MHz, DMSO) δ 192.3, 152.7, 148.1, 134.7, 134.2, 134.2, 129.0, 128.1, 124.4, 116.9, 107.0, 54.2. HRMS (ESI): m/z calcd for C14H11N4O4 [M + H]+, 299.0775; found, 299.0785.
2-Nitro-7-(2-oxo-2-(4-(trifluoromethoxy)phenyl)ethyl)imidazo[1,2-a]pyrazin-8(7H)-one (17b)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (42 mg, 0.233 mmol, 1 equiv) was reacted with 2-bromo-1-(4-(trifluoromethoxy)phenyl)ethan-1-one (99 mg, 0.350 mmol, 1.5 equiv) at room temperature for 30 min according to the general procedure method B. After the reaction was completed as analyzed by LC-MS, the mixture was diluted with distilled water (15 mL) and the precipitate was collected by filtration. The precipitate was then purified by recrystallization using hot ACN/MeOH to yield the desired product, 2-nitro-7-(2-oxo-2-phenylethyl)imidazo[1,2-a]pyrazin-8(7H)-one 17b, as a yellow solid (37 mg, 41%): LC-MS: Rt = 2.91 min, 99 Abs % @ 254 nm, [M + H]+ = 383.1; 1H NMR (600 MHz, DMSO-d6) δ 8.89 (s, 1H), 8.26–8.20 (m, 2H), 7.66 (d, J = 5.8 Hz, 1H), 7.64–7.58 (m, 2H), 7.32 (d, J = 5.8 Hz, 1H), 5.61 (s, 2H). 13C NMR (150 MHz, DMSO) δ 191.3, 152.7, 152.1, 148.1, 134.7, 133.1, 130.7, 124.3, 121.1, 119.9 (q, J = 258.3 Hz), 116.9, 107.0, 54.2. HRMS (ESI): m/z calcd for C15H10 F3N4O5 [M + H]+, 383.0598; found, 383.0605.
2-Nitro-7-(2-oxo-2-(p-tolyl)ethyl)imidazo[1,2-a]pyrazin-8(7H)-one (17c)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (50 mg, 0.278 mmol, 1 equiv) was reacted with 2-bromo-4′-methylacetophenone (88.7 mg, 0.416 mmol, 1.5 equiv) at room temperature for 2 h according to the general procedure method B. After the reaction was completed as analyzed by LC-MS, the mixture was diluted with distilled water (15 mL) and the precipitate was collected by filtration. The precipitate was then purified by recrystallization using hot ACN/MeOH to yield the desired product, 2-nitro-7-(2-oxo-2-(p-tolyl)ethyl)imidazo[1,2-a]pyrazin-8(7H)-one 17c, as a red solid (20 mg, 23%): LC-MS: Rt = 2.74 min, 98 Abs % @ 254 nm, [M + H]+ = 313.1; 1H NMR (600 MHz, DMSO-d6) δ 8.88 (s, 1H), 8.01–7.96 (m, 2H), 7.65 (d, J = 5.8 Hz, 1H), 7.45–7.40 (m, 2H), 7.33 (d, J = 5.9 Hz, 1H), 5.56 (s, 2H), 2.43 (s, 3H); 13C NMR (150 MHz, DMSO) δ 191.8, 152.7, 148.1, 144.8, 134.7, 131.7, 129.5, 128.2, 124.4, 116.8, 106.9, 54.1, 21.3. HRMS (ESI): m/z calcd for C15H13N4O4 [M + H]+, 313.0931; found, 313.0941.
2-(2-Nitro-8-oxoimidazo[1,2-a]pyrazin-7(8H)-yl)-N-phenylacetamide (17d)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (75 mg, 0.416 mmol, 1 equiv) was reacted with 2-bromo-N-phenylacetamide 11a (107 mg, 0.500 mmol, 1.2 equiv) under microwave conditions at 90 °C for 15 min according to the general procedure method B. After the reaction was completed as analyzed by LC-MS, the mixture was diluted with distilled water (20 mL) and the precipitate was collected by filtration. The precipitate was then purified by recrystallization using hot ACN/MeOH to yield the desired product, 2-(2-nitro-8-oxoimidazo[1,2-a]pyrazin-7(8H)-yl)-N-phenylacetamide 17d, as a white solid (21 mg, 16%): LC-MS: Rt = 2.46 min, 99 Abs % @ 254 nm, [M + H]+ = 314.1; 1H NMR (600 MHz, DMSO-d6) δ 10.34 (s, 1H), 8.86 (s, 1H), 7.63 (d, J = 5.9 Hz, 1H), 7.60–7.55 (m, 2H), 7.38 (d, J = 5.8 Hz, 1H), 7.36–7.29 (m, 2H), 7.07 (tt, J = 7.3, 1.2 Hz, 1H), 4.80 (s, 2H). 13C NMR (150 MHz, DMSO) δ 165.0, 152.8, 148.0, 138.5, 134.8, 128.9, 124.8, 123.6, 119.1, 116.7, 106.6, 50.6. HRMS (ESI): m/z calcd for C14H12N5O4 [M + H]+, 314.0884; found, 314.0892.
N-Benzyl-2-(2-nitro-8-oxoimidazo[1,2-a]pyrazin-7(8H)-yl)acetamide (17e)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (70 mg, 0.389 mmol, 1 equiv) was reacted with N-benzyl-2-bromoacetamide 11b (106 mg, 0.466 mmol, 1.2 equiv) under microwave conditions at 90 °C for 15 min according to the general procedure method B. After the reaction was completed as analyzed by LC-MS, the mixture was diluted with distilled water (20 mL) and the precipitate was collected by filtration. The precipitate was then purified by recrystallization using hot ACN to yield the desired product, N-benzyl-2-(2-nitro-8-oxoimidazo[1,2-a]pyrazin-7(8H)-yl)acetamide 17e, as a white solid (93 mg, 73%): LC-MS: Rt = 2.46 min, 99 Abs % @ 254 nm, [M + H]+ = 328.1; 1H NMR (600 MHz, DMSO-d6) δ 8.84 (s, 1H), 8.69 (t, J = 6.0 Hz, 1H), 7.59 (d, J = 5.9 Hz, 1H), 7.37–7.30 (m, 3H), 7.30–7.22 (m, 3H), 4.64 (s, 2H), 4.32 (d, J = 6.0 Hz, 2H). 13C NMR (150 MHz, DMSO) δ 166.3, 152.9, 147.9, 138.9, 135.0, 128.3, 127.1, 126.8, 124.7, 116.6, 106.7, 50.2, 42.1. HRMS (ESI): m/z calcd for C15H14N5O4 [M + H]+, 328.1040; found, 328.1054.
2-(2-Nitro-8-oxoimidazo[1,2-a]pyrazin-7(8H)-yl)-N-(3-(trifluoromethoxy)benzyl)acetamide (17f)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (50 mg, 0.278 mmol, 1 equiv) was reacted with 2-bromo-N-(3-(trifluoromethoxy)benzyl)acetamide 11c (130 mg, 0.416 mmol, 1.5 equiv) under microwave conditions at 90 °C for 15 min according to the general procedure method B. After the reaction was completed as analyzed by LC-MS, the mixture was diluted with distilled water (15 mL) and the precipitate was collected by filtration. The precipitate was then purified by recrystallization using hot ACN to yield the desired product, 2-(2-nitro-8-oxoimidazo[1,2-a]pyrazin-7(8H)-yl)-N-(3-(trifluoromethoxy)benzyl)acetamide 17f, as a white solid (25 mg, 22%): LC-MS: Rt = 2.79 min, 99 Abs % @ 254 nm, [M + H]+ = 412.1; 1H NMR (600 MHz, DMSO-d6) δ 8.84 (s, 1H), 8.76 (t, J = 6.0 Hz, 1H), 7.60 (d, J = 5.9 Hz, 1H), 7.47 (t, J = 7.8 Hz, 1H), 7.37–7.29 (m, 2H), 7.27–7.22 (m, 2H), 4.65 (s, 2H), 4.37 (d, J = 6.0 Hz, 2H). 13C NMR (150 MHz, DMSO) δ 166.6, 152.0, 148.4, 147.9, 142.0, 135.0, 130.2, 126.1, 124.7, 120.1 (q, J = 256.7 Hz), 119.5, 119.3, 116.6, 106.7, 50.2, 41.6. HRMS (ESI): m/z calcd for C16H13F3N5O5 [M + H]+, 412.0863; found, 412.0856.
2-(2-Nitro-8-oxoimidazo[1,2-a]pyrazin-7(8H)-yl)ethyl Acetate (17g)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (130 mg, 0.722 mmol, 1 equiv) was reacted with 2-bromoethyl acetate (181 mg, 1.08 mmol, 1.5 equiv) under microwave conditions at 80 °C for 30 min according to the general procedure method B. After the reaction was completed as analyzed by LC-MS, the mixture was diluted with distilled water (15 mL) and the precipitate was collected by filtration to yield the desired product, 2-(2-nitro-8-oxoimidazo[1,2-a]pyrazin-7(8H)-yl)ethyl acetate 17g, as a cream solid (171 mg, 89%): LC-MS: Rt = 2.14 min, 99 Abs % @ 254 nm, [M + H]+ = 267.1; 1H NMR (600 MHz, DMSO-d6) δ 8.81 (s, 1H), 7.57 (d, J = 5.8 Hz, 1H), 7.35 (d, J = 5.8 Hz, 1H), 4.30 (t, J = 5.2 Hz, 2H), 4.16 (t, J = 5.2 Hz, 2H), 1.97 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 170.2, 152.8, 147.9, 134.9, 124.0, 116.5, 106.8, 61.3, 46.7, 20.5. HRMS (ESI): m/z calcd for C10H10N4NaO5 [M + Na]+, 289.0543; found, 289.0545.
7-(2-Hydroxyethyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one (17h)
To a stirring suspension of 2-(2-nitro-8-oxoimidazo[1,2-a]pyrazin-7(8H)-yl)ethyl acetate 17g (165 mg, 0.266 mmol, 1 equiv) in methanol (20 vol) was added 1.5 equiv of potassium carbonate (128 mg, 0.930 mmol). The reaction was stirred for 1 h at room temperature before removing the volatiles under N2. The solid was suspended with 5% TFA in methanol and purified by C18-reversed phase silica (Grace Reveleris X2, A: 0.1% TFA in water, B: 0.1% TFA in ACN, 0–50% B) to give final product, 7-(2-hydroxyethyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one 17h, as a light yellow solid (47 mg, 34%): LC-MS: Rt = 1.53 min, 99 Abs % @ 254 nm, [M + H]+ = 225.1; 1H NMR (600 MHz, DMSO-d6) δ 8.81 (s, 1H), 7.55 (d, J = 5.9 Hz, 1H), 7.29 (d, J = 5.8 Hz, 1H), 4.90 (t, J = 5.7 Hz, 1H), 3.96 (t, J = 5.5 Hz, 2H), 3.66 (q, J = 5.5 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 152.8, 147.9, 135.1, 124.8, 116.3, 106.3, 58.5, 50.3. HRMS (ESI): m/z calcd for C8H8N4NaO4 [M + Na]+, 247.0438; found, 247.0435.
7-(2-(Benzyloxy)ethyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one (17i)
7-(2-Hydroxyethyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one 17h (20 mg, 0.0892 mmol, 1 equiv) was reacted with benzyl bromide (18.3 mg, 12.7 μL, 0.107 mmol, 1.2 equiv) according to the general procedure method C. Additional 1.2 equiv of benzyl bromide and 1.5 equiv of sodium hydride were added to push the reaction. Once the reaction was completed as analyzed by LC-MS, the mixture was diluted with distilled water and extracted with EtOAc three times. The crude was then purified by C18-reversed phase silica (Grace Reveleris X2, A: 0.1% TFA in water, B: 0.1% TFA in ACN, 0–100% B) to give final product, 7-(2-(benzyloxy)ethyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one 17i, as a white solid (8.1 mg, 29%): LC-MS: Rt = 2.76 min, 99 A % @ 254 nm, [M + H]+ = 315.1; 1H NMR (600 MHz, DMSO-d6) δ 8.81 (s, 1H), 7.56 (d, J = 5.9 Hz, 1H), 7.33 (d, J = 5.9 Hz, 1H), 7.31–7.20 (m, 5H), 4.49 (s, 2H), 4.13 (t, J = 5.3 Hz, 2H), 3.71 (t, J = 5.3 Hz, 2H). 13C NMR (151 MHz, DMSO) δ 152.7, 147.9, 138.0, 134.9, 128.1, 127.4, 124.4, 116.4, 106.4, 71.8, 67.0, 47.4. HRMS (ESI): m/z calcd for C15H15N4O4 [M + H]+, 315.1088; found, 315.1100.
2-Nitro-7-(2-((4-(trifluoromethoxy)benzyl)oxy)ethyl)imidazo[1,2-a]pyrazin-8(7H)-one (17j)
7-(2-Hydroxyethyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one 17 h (57 mg, 0.254 mmol, 1 equiv) was reacted with 1-(bromomethyl)-4-(trifluoromethoxy)benzene (77.8 mg, 48.8 μL, 0.305 mmol, 1.2 equiv) according to the general procedure method C. After completion of reaction, the mixture was diluted with distilled water (12 mL) and extracted with EtOAc (3 × 12 mL). The organic layer was collected, washed with brine (12 mL), and dried with MgSO4. Volatiles were removed in vacuo, and the crude product was purified over silica gel by MPLC (Biotage Isolera, 0–8% DCM/MeOH), then re-purified by C18-reversed phase silica (Grace Reveleris X2, A: 0.1% TFA in water, B: 0.1% TFA in ACN, 0–100% B) to give final product, 2-nitro-7-(2-((4-(trifluoromethoxy)benzyl)oxy)ethyl)imidazo[1,2-a]pyrazin-8(7H)-one 17j, as a beige solid (16 mg, 16%): LC-MS: Rt = 3.00 min, 99 Abs % @ 254 nm, [M + H]+ = 399.1; 1H NMR (600 MHz, DMSO-d6) δ 8.80 (s, 1H), 7.56 (d, J = 5.9 Hz, 1H), 7.42–7.36 (m, 2H), 7.33 (d, J = 5.9 Hz, 1H), 7.29–7.24 (m, 2H), 4.51 (s, 2H), 4.13 (t, J = 5.2 Hz, 2H), 3.73 (t, J = 5.2 Hz, 2H). 13C NMR (150 MHz, DMSO) δ 152.7, 147.9, 147.5, 137.6, 134.9, 129.2, 124.4, 120.7, 120.1 (q, J = 256 Hz), 116.4, 106.5, 70.8, 67.2, 47.4. HRMS (ESI): m/z calcd for C16H14F3N4O5 [M + H]+, 399.0911; found, 399.0917.
7-(2-((4-Methylbenzyl)oxy)ethyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one (17k)
7-(2-Hydroxyethyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one 17h (55 mg, 0.245 mmol, 1 equiv) was reacted with 4-methylbenzyl bromide (54.5 mg, 34.2 μL, 0.294 mmol, 1.2 equiv) according to the general procedure method C. After completion of reaction, the mixture was diluted with distilled water (11 mL) and the precipitate was collected by filtration. The precipitate was then purified by recrystallization (slurry in hot ACN) to yield the desired product, 7-(2-((4-methylbenzyl)oxy)ethyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one 17k, as a beige solid (62 mg, 76%): LC-MS: Rt = 2.83 min, 99 Abs % @ 254 nm, [M + H]+ = 329.1; 1H NMR (600 MHz, DMSO-d6) δ 8.80 (d, J = 1.0 Hz, 1H), 7.55 (dd, J = 5.8, 0.9 Hz, 1H), 7.30 (dd, J = 5.9, 0.9 Hz, 1H), 7.12 (d, J = 7.7 Hz, 2H), 7.05 (d, J = 7.7 Hz, 2H), 4.42 (s, 2H), 4.09 (t, J = 5.2 Hz, 2H), 3.68 (t, J = 5.2 Hz, 2H), 2.22 (s, 3H).13C NMR (150 MHz, DMSO) δ 152.6, 147.9, 136.6, 134.9, 134.9, 128.6, 127.6, 124.5, 116.4, 106.4, 71.7, 66.7, 47.5, 20.6. HRMS (ESI): m/z calcd for C16H17N4O4 [M + H]+, 329.1244; found, 329.1258.
5-(Chloromethyl)thiazole (18)
Thiazol-5-ylmethanol (300 mg, 2.61 mmol, 1 equiv) and thionyl chloride (567 μL, 7.82 mmol, 3 equiv) were reacted and purified according to the general procedure method D. Volatiles were removed in vacuo to give 5-(chloromethyl)thiazole 18 (264 mg, 76%), which was used in the next step immediately: LC-MS: Rt = 2.23 min, 99 Abs % @ 254 nm, [M + H]+ = 134.1. This halide intermediate is unstable, and therefore NMR characterization data was not acquired.
2-(Chloromethyl)-1-methyl-1H-imidazole (19)
(1-Methyl-1H-imidazol-2-yl)methanol (300 mg, 2.68 mmol, 1 equiv) and thionyl chloride (388 μL, 5.35 mmol, 2 equiv) were reacted and purified according to the general procedure method D. Volatiles were then removed in vacuo a crude compound was then recrystallized from ethanol to give 2-(chloromethyl)-1-methyl-1H-imidazole 19 (220 mg, 63%) as a colorless crystal: LC-MS (Waters Atlantis T3): Rt = 0.29 min, 95 Abs % @ 210 nm, [M + H]+ = 131.0. 1H NMR (600 MHz, DMSO-d6) δ 7.76 (d, J = 1.9 Hz, 1H), 7.69 (d, J = 1.9 Hz, 1H), 5.18 (s, 2H), 3.87 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 141.5, 124.6, 119.4, 34.2, 31.7.
5-(Chloromethyl)-2-(trifluoromethyl)pyridine (20)
(6-(Trifluoromethyl)pyridin-3-yl)methanol (200 mg, 1.13 mmol, 1 equiv) and phosphoryl chloride (571 μL, 1.65 mmol, 3.3 equiv) were reacted and purified according to the general procedure method D. Volatiles were removed in vacuo to give 5-(chloromethyl)-2-(trifluoromethyl)pyridine 20 in a gel form, which was used in the next step immediately without further purification: LC-MS: Rt = 2.59 min, 96 Abs % @ 254 nm, [M + H]+ = 196.1. This halide intermediate is unstable, and therefore NMR characterization data was not acquired.
5-(Bromomethyl)-1-methyl-1H-pyrazole (21)
(1-Methyl-1H-pyrazol-5-yl)methanol (195 mg, 1.74 mmol, 1 equiv) was added with DCM (80 vol) and was chilled at 0 °C. Phosphorus tribromides (343 μL, 3.65 mmol, 2.1 equiv) was added dropwise, and the reaction was continued to stir at room temperature for 6 h. The mixture was then diluted with distilled water, adjusted to pH 8–9 using saturated NaHCO3, and extracted with DCM (3 x 40 mL). The organic layer was collected and washed with brine, and volatiles were removed in vacuo to give 5-(bromomethyl)-1-methyl-1H-pyrazole 21 (272 mg, 89%), which was used in the next step immediately without further purification: LC-MS: Rt = 2.28 min, 99 Abs % @ 254 nm, [M + H]+ = 177.1. This halide intermediate is unstable, and therefore NMR characterization data was not acquired.
2-(Chloromethyl)-1H-benzo[d]imidazole (24)
To a stirring solution of o-phenylenediamine 22 (500 mg, 4.62 mmol, 1 equiv) in 4 M HCl (12 vol) was added 1.2 equiv of chloroacetic acid (524 mg, 5.55 mmol, 1.2 equiv). The mixture was reflux overnight and then cooled to room temperature before neutralization with saturated NaHCO3. Precipitate formed was filtered under vacuum and washed with cold water to yield 2-(chloromethyl)-1H-benzo[d]imidazole 24 as a yellow solid (633 mg, 82%): LC-MS: Rt = 1.65 min, 98 Abs % @ 254 nm, [M + H]+ = 167.2; 1H NMR (600 MHz, DMSO-d6) δ 7.58–7.51 (m, 2H), 7.21 (dp, J = 7.1, 4.0 Hz, 2H), 4.92 (s, 2H).13C NMR (150 MHz, DMSO) δ 149.6, 122.3, 38.4.
2-(Chloromethyl)-1-methyl-1H-benzo[d]imidazole (25)
To a stirring solution of N-methyl-1,2-phenylenediamine 23 (500 mg, 4.09 mmol, 1 equiv) in 4 M HCl (12 vol) was added 1.2 equiv of chloroacetic acid (580 mg, 6.14 mmol, 1.5 equiv). The mixture was reflux overnight and then cooled to room temperature before neutralization with saturated NaHCO3. Precipitate formed was filtered under vacuum and washed with cold water to yield 2-(chloromethyl)-1-methyl-1H-benzo[d]imidazole 25 as a black solid (524 mg, 71%): LC-MS: Rt = 2.15 min, 99 Abs % @ 254 nm, [M + H]+ = 181.1; 1H NMR (600 MHz, DMSO-d6) δ 7.63 (dt, J = 8.0, 0.9 Hz, 1H), 7.58 (dt, J = 8.1, 1.0 Hz, 1H), 7.30 (ddd, J = 8.2, 7.1, 1.1 Hz, 1H), 7.23 (ddd, J = 8.2, 7.1, 1.2 Hz, 1H), 5.08 (s, 2H), 3.86 (s, 3H). 13C NMR (150 MHz, DMSO) δ 149.6, 141.7, 135.9, 123.0, 122.0, 119.3, 110.4, 37.1, 30.0.
2-Nitro-7-(thiazol-5-ylmethyl)imidazo[1,2-a]pyrazin-8(7H)-one (26a)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (55 mg, 0.305 mmol, 1 equiv) was reacted with 5-(chloromethyl)thiazole 18 (61.2 mg, 0.458 mmol, 1.5 equiv) under microwave conditions at 90 °C (20 min x 2) according to the general procedure method B. After the reaction was completed as analyzed by LC-MS, the mixture was diluted with distilled water (15 mL) and the precipitate was collected by filtration. The precipitate was then purified by recrystallization using DCM/MeOH (hot slurry) to yield the desired product, 2-nitro-7-(thiazol-5-ylmethyl)imidazo[1,2-a]pyrazin-8(7H)-one, 26a as a cream solid (54 mg, 62%): LC-MS: Rt = 2.23 min, 99 Abs % @ 254 nm, [M + H]+ = 278.1; 1H NMR (600 MHz, DMSO-d6) δ 9.05 (d, J = 0.8 Hz, 1H), 8.81 (s, 1H), 8.01 (d, J = 0.8 Hz, 1H), 7.61 (d, J = 5.9 Hz, 1H), 7.50 (d, J = 5.9 Hz, 1H), 5.36 (s, 2H); 13C NMR (150 MHz, DMSO-d6) δ 155.6, 152.5, 148.0, 143.3, 134.8, 132.8, 122.8, 116.8, 107.8, 42.8. HRMS (ESI): m/z calcd for C10H8N5O3S [M + H]+, 278.0342; found, 278.0348.
7-((1-Methyl-1H-imidazol-2-yl)methyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one (26c)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (50 mg, 0.278 mmol, 1 equiv) was reacted with 2-(chloromethyl)-1-methyl-1H-imidazole 19 (54.4 mg, 0.416 mmol, 1.5 equiv) under microwave conditions at 90 °C for 20 min according to the general procedure method B. After the reaction was completed as analyzed by LC-MS, the mixture was diluted with distilled water (15 mL) and the precipitate was collected by filtration. The precipitate was then purified by recrystallization using DCM/MeOH (hot slurry) to yield the desired product, 7-((1-methyl-1H-imidazol-2-yl)methyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one 26c, as a white solid (60 mg, 79%): LC-MS: Rt = 1.07 min, 99 Abs % @ 254 nm, [M + H]+ = 275.1; 1H NMR (600 MHz, DMSO-d6) δ 8.83 (s, 1H), 7.59 (d, J = 5.9 Hz, 1H), 7.31 (d, J = 5.9 Hz, 1H), 7.14 (d, J = 1.2 Hz, 1H), 6.81 (d, J = 1.2 Hz, 1H), 5.18 (s, 2H), 3.69 (s, 3H). 13C NMR (150 MHz, DMSO) δ 152.5, 148.0, 142.2, 134.9, 127.0, 123.4, 122.4, 116.7, 107.3, 42.1, 32.5. HRMS (ESI): m/z calcd for C11H11N6O3 [M + H]+, 275.0887; found, 275.0893.
7-((1-Methyl-1H-pyrazol-5-yl)methyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one (26d)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (60 mg, 0.333 mmol, 1 equiv) was reacted with 5-(bromomethyl)-1-methyl-1H-pyrazole 21 (87.5 mg, 0.500 mmol, 1.5 equiv) under microwave conditions at 90 °C for 20 min according to the general procedure method B. After the reaction was completed as analyzed by LC-MS, the mixture was diluted with distilled water (15 mL) and the precipitate was collected by filtration. The precipitate was then purified by recrystallization using DCM/MeOH (hot slurry) to yield the desired product, 7-((1-methyl-1H-pyrazol-5-yl)methyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one 26d, as a cream color solid (57 mg, 63%): LC-MS: Rt = 2.20 min, 98 Abs % @ 254 nm, [M + H]+ = 275.2; 1H NMR (600 MHz, DMSO-d6) δ 8.82 (s, 1H), 7.61 (d, J = 5.9 Hz, 1H), 7.36–7.32 (m, 2H), 6.27 (d, J = 1.9 Hz, 1H), 5.21 (s, 2H), 3.85 (s, 3H). 13C NMR (150 MHz, DMSO) δ 152.6, 148.0, 137.6, 137.2, 135.0, 122.9, 116.7, 107.7, 106.3, 41.1, 36.6. HRMS (ESI): m/z calcd for C11H11N6O3 [M + H]+, 275.0887; found, 275.0897.
7-((1H-Benzo[d]imidazol-2-yl)methyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one (26e)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (60 mg, 0.333 mmol, 1 equiv) was reacted with 2-(chloromethyl)-1H-benzo[d]imidazole 24 (83.2 mg, 0.500 mmol, 1.5 equiv) under microwave conditions at 90 °C (20 min x 2) according to the general procedure method B. After the reaction was completed as analyzed by LC-MS, the mixture was diluted with distilled water (15 mL) and the precipitate was collected by filtration. The crude product was purified by C18-reversed phase silica (Grace Reveleris X2, A: 0.1% TFA in water, B: 0.1% TFA in ACN, 0–100% B) to give a major product, 7-((1H-benzo[d]imidazol-2-yl)methyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one 26e, as a cream color solid (43 mg, 42%) and a minor product, 7-((1-((1H-benzo[d]imidazol-2-yl)methyl)-1H-benzo[d]imidazol-2-yl)methyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one as a cream color solid (35 mg, 24%). Major product LC-MS: Rt = 2.16 min, 99 Abs % @ 254 nm, [M + H]+ = 311.1; 1H NMR (600 MHz, DMSO-d6) 8.87 (s, 1H), 7.66 (d, J = 5.9 Hz, 1H), 7.51 (dt, J = 5.9, 3.5 Hz, 2H), 7.48 (d, J = 5.9 Hz, 1H), 7.20–7.14 (m, 2H), 5.39 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 152.9, 149.5, 148.0, 135.0, 124.2, 121.9, 116.7, 107.3, 45.2. HRMS (ESI): m/z calcd for C14H11N6O3 [M + H]+, 311.0887; found, 311.0900. Minor product LC-MS: Rt = 2.50 min, 99 Abs % @ 254 nm, [M + H]+ = 441.2.
7-((1-Methyl-1H-benzo[d]imidazol-2-yl)methyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one (26f)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (100 mg, 0.555 mmol, 1 equiv) was reacted with 2-(chloromethyl)-1-methyl-1H-benzo[d]imidazole 25 (150 mg, 0.833 mmol, 1.5 equiv) under microwave conditions at 90 °C for 20 min according to the general procedure method B. After the reaction was completed as analyzed by LC-MS, the mixture was diluted with distilled water (20 mL) and the precipitate was collected by filtration. The precipitate was then purified by recrystallization using DCM/MeOH (hot slurry) to yield the desired product, 7-((1-methyl-1H-benzo[d]imidazol-2-yl)methyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one 26f, as a light gray solid (135 mg, 75%): LC-MS: Rt = 2.30 min, 99 Abs % @ 254 nm, [M + H]+ = 325.1; 1H NMR (600 MHz, DMSO-d6) δ 8.87 (s, 1H), 7.65 (d, J = 5.9 Hz, 1H), 7.59–7.52 (m, 2H), 7.44 (d, J = 5.9 Hz, 1H), 7.25 (ddd, J = 8.2, 7.1, 1.2 Hz, 1H), 7.17 (ddd, J = 8.1, 7.1, 1.2 Hz, 1H), 5.47 (s, 2H), 3.88 (s, 3H. 13C NMR (150 MHz, DMSO-d6) δ 152.6, 149.7, 148.0, 141.8, 136.0, 134.8, 124.1, 122.3, 121.6, 118.8, 116.8, 110.1, 107.3, 43.4, 29.8. HRMS (ESI): m/z calcd for C15H13N6O3 [M + H]+, 325.1044; found, 325.1061.
2-Nitro-7-((6-(trifluoromethyl)pyridin-3-yl)methyl)imidazo[1,2-a]pyrazin-8(7H)-one (26h)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (50 mg, 0.278 mmol, 1 equiv) was reacted with 3-(4-(chloromethyl)-2-(trifluoromethyl)pyridine 20 (65 mg, 0.333 mmol, 1.2 equiv) under microwave conditions at 100 °C for 30 min according to the general procedure method B. After the reaction was completed as analyzed by LC-MS, the mixture was diluted with distilled water (20 mL) and the precipitate was collected by filtration. The precipitate was then purified by recrystallization using DCM/MeOH (hot slurry) to yield the desired product, 2-nitro-7-((6-(trifluoromethyl)pyridin-3-yl)methyl)imidazo[1,2-a]pyrazin-8(7H)-one 26h, as a white solid (45 mg, 48% yield). Filtrate from DCM/MeOH was collected and further purified over C18-reversed phase silica (Grace Reveleris X2, A: 0.1% TFA in water, B: 0.1% TFA in ACN, 5–100% B) to give additional product 26h (14 mg, 15%): LC-MS: Rt = 2.43 min, 99 Abs % @ 254 nm, [M + H]+ = 340.1; 1H NMR (600 MHz, DMSO-d6) δ 8.83 (s, 1H), 8.81 (d, J = 2.1 Hz, 1H), 8.07–8.02 (m, 1H), 7.90 (dd, J = 8.2, 0.8 Hz, 1H), 7.63 (d, J = 5.9 Hz, 1H), 7.51 (d, J = 5.9 Hz, 1H), 5.28 (s, 2H). 13C NMR (151 MHz, DMSO) δ 153.0, 149.6, 147.9, 145.6 (J = 33.9 Hz), 137.5, 136.2, 135.2, 123.4, 122.5, 121.6 (J = 273.3 Hz), 120.6 (J = 2.8 Hz), 107.7, 48.0. HRMS (ESI): m/z calcd for C13H9F3N5O3 [M + H]+, 340.0652; found, 340.0665.
4-Nitro-N-(thiazol-2-ylmethyl)-1H-imidazole-2-carboxamide (27b)
A mixture of acid chloride crude solid (150 mg, 0.855 mmol, 1 equiv) and triethylamine (238 μL, 1.71 mmol, 2 equiv) was reacted with thiazol-2-ylmethanamine (117 mg, 97 μL, 1.03 mmol, 1.2 equiv) according to general procedure method E. After completion of reaction, volatiles were removed in vacuo before purifying over C18-reversed phase silica (Grace Reveleris X2, A: 0.1% TFA in water, B: 0.1% TFA in ACN, 0–100% B) to give the final product, 4-nitro-N-(thiazol-2-ylmethyl)-1H-imidazole-2-carboxamide 27b, as a white solid (166 mg, 77%). LC-MS: Rt = 2.15 min, 99 Abs % @ 254 nm, [M + H]+ = 254.1; 1H NMR (600 MHz, DMSO-d6) δ 9.74 (t, J = 6.2 Hz, 1H), 8.49 (s, 1H), 7.73 (d, J = 3.2 Hz, 1H), 7.63 (d, J = 3.3 Hz, 1H), 4.72 (d, J = 6.1 Hz, 2H). 13C NMR (150 MHz, DMSO) δ 168.4, 157.5, 146.8, 142.2, 139.1, 121.7, 120.1, 40.6.
4-Nitro-N-(pyridin-3-ylmethyl)-1H-imidazole-2-carboxamide (27g)
A mixture of acid chloride crude solid (120 mg, 0.684 mmol, 1 equiv) and triethylamine (191 μL, 1.37 mmol, 2 equiv) was reacted with pyridin-3-ylmethanamine (88.8 mg, 0.820 mmol, 1.2 equiv) according to general procedure method E. After completion of reaction, volatiles were removed in vacuo before purifying over C18-reversed phase silica (Grace Reveleris X2, A: 0.1% TFA in water, B: 0.1% TFA in ACN, 0–50% B) to give the final product, 4-nitro-N-(pyridin-3-ylmethyl)-1H-imidazole-2-carboxamide 27g, as a white solid (128 mg, 76%). LC-MS: Rt = 1.21 min, 99% Abs @ 254 nm, [M + H]+ = 248.1; 1H NMR (600 MHz, DMSO-d6) δ 9.57 (t, J = 6.2 Hz, 1H), 8.73 (s, 1H), 8.65 (d, J = 4.8 Hz, 1H), 8.48 (s, 1H), 8.13 (dt, J = 8.1, 1.8 Hz, 1H), 7.70 (dd, J = 7.9, 5.2 Hz, 1H), 4.55 (d, J = 6.2 Hz, 2H). Triethylamine was presented as 5% when integrated by 1H NMR.
4-Nitro-N-(pyrazin-2-ylmethyl)-1H-iimidazole-2-carboxamide (27i)
A mixture of acid chloride crude solid (150 mg, 0.855 mmol, 1 equiv) and triethylamine (238 μL, 1.71 mmol, 2 equiv) was reacted with aminomethylpyrazine (112 mg, 1.03 mmol, 1.2 equiv) according to general procedure method E. After completion of reaction, volatiles were removed in vacuo before purifying over C18-reversed phase silica (Grace Reveleris X2, A: 0.1% TFA in water, B: 0.1% TFA in ACN, 0–100% B) to give the final product, 4-nitro-N-(pyrazin-2-ylmethyl)-1H-imidazole-2-carboxamide 27i, as a beige solid (121 mg, 57%). LC-MS: Rt = 1.99 min, 99 Abs % @ 254 nm, [M + H]+ = 249.1; 1H NMR (600 MHz, DMSO-d6) δ 9.47 (t, J = 6.1 Hz, 1H), 8.68–8.56 (m, 2H), 8.54 (d, J = 2.6 Hz, 1H), 8.48 (s, 1H), 4.62 (d, J = 6.1 Hz, 2H). 13C NMR (150 MHz, DMSO) δ 157.5, 153.5, 146.7, 143.9, 143.4, 143.2, 139.3, 121.6, 42.3.
4-Nitro-N-(quinolin-6-ylmethyl)-1H-imidazole-2-carboxamide (27j)
A mixture of acid chloride crude solid (310 mg, 1.77 mmol, 1 equiv) and triethylamine (492 μL, 3.53 mmol, 2 equiv) was reacted with 6-aminomethylquinoline (335 mg, 290 μL, 2.12 mmol, 1.2 equiv) according to general procedure method E. After completion of reaction, volatiles were removed in vacuo before purifying over C18-reversed phase silica (Grace Reveleris X2, A: 0.1% TFA in water, B: 0.1% TFA in ACN, 0–100% B) to give the final product 4-nitro-N-(quinolin-6-ylmethyl)-1H-imidazole-2-carboxamide 27j, as a white solid (612 mg, 91%, contained 0.7 equivalent of TFA). LC-MS: Rt = 2.15 min, 99 Abs % @ 254 nm, [M + H]+ = 298.1; 1H NMR (600 MHz, DMSO-d6) δ 9.63 (t, J = 6.3 Hz, 1H), 9.00 (dd, J = 4.5, 1.7 Hz, 1H), 8.62 (d, J = 8.3 Hz, 1H), 8.48 (d, J = 1.3 Hz, 1H), 8.07 (d, J = 8.7 Hz, 1H), 7.98 (d, J = 1.9 Hz, 1H), 7.87 (dd, J = 8.7, 2.0 Hz, 1H), 7.69 (dd, J = 8.3, 4.5 Hz, 1H), 4.67 (d, J = 6.2 Hz, 2H). 13C NMR (150 MHz, DMSO) δ 158.2, 158.0, 157.4, 148.7, 146.8, 139.5, 138.4, 130.9, 127.9, 126.6, 125.8, 121.8, 121.6, 42.20.
N-(Benzo[d][1,3]dioxol-5-ylmethyl)-4-nitro-1H-imidazole-2-carboxamide (27k)
A mixture of acid chloride crude solid (150 mg, 0.855 mmol, 1 equiv) and triethylamine (238 μL, 1.71 mmol, 2 equiv) was reacted with 1,3-benzodioxole-5-methanamine (155 mg, 128 μL, 1.03 mmol, 1.2 equiv) according to general procedure method E. After completion of reaction, volatiles were removed in vacuo before purifying over C18-reversed phase silica (Grace Reveleris X2, A: 0.1% TFA in water, B: 0.1% TFA in ACN, 0–100% B) to give the final product, N-(benzo[d][1,3]dioxol-5-ylmethyl)-4-nitro-1H-imidazole-2-carboxamide 27k, as a white solid (136 mg, 55%). LC-MS: Rt = 2.61 min, 98 Abs % @ 254 nm, [M – H]− = 288.9. 1H NMR (600 MHz, DMSO-d6) δ 9.37 (t, J = 6.4 Hz, 1H), 8.45 (s, 1H), 6.90 (d, J = 1.6 Hz, 1H), 6.84 (d, J = 7.9 Hz, 1H), 6.79 (dd, J = 7.9, 1.6 Hz, 1H), 5.97 (s, 2H), 4.33 (d, J = 6.3 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 157.0, 147.1, 146.7, 146.1, 139.6, 132.9, 121.5, 120.8, 108.2, 108.0, 100.8, 42.1.
1-(2,2-Diethoxyethyl)-4-nitro-N-(thiazol-2-ylmethyl)-1H-imidazole-2-carboxamide (28b)
4-Nitro-N-(thiazol-2-ylmethyl)-1H-imidazole-2-carboxamide (140 mg, 0.553 mmol, 1 equiv) was reacted with bromoacetaldehyde diethyl acetal (125 μL, 0.829 mmol, 1.5 equiv) according to the general procedure method F. After completion of reaction, the mixture was diluted with distilled water (28 mL) and extracted with EtOAc (3 × 28 mL). The organic layer was collected, washed with brine (28 mL), and dried with MgSO4. Volatiles were removed in vacuo and the crude product was purified over silica gel by MPLC (Biotage Isolera, 15–100% pet. spirit/EtOAc) to give 1-(2,2-diethoxyethyl)-4-nitro-N-(thiazol-2-ylmethyl)-1H-imidazole-2-carboxamide 28b as a beige solid (94 mg, 46%). LC-MS: Rt = 2.79 min, 93 Abs % @ 254 nm, [M + Na]+ = 370.1.
1-(2,2-Diethoxyethyl)-5-nitro-N-(pyridin-3-ylmethyl)-1H-imidazole-2-carboxamide (28g)
5-Nitro-N-(pyridin-3-ylmethyl)-1H-imidazole-2-carboxamide (120 mg, 0.485 mmol, 1 equiv) was reacted with bromoacetaldehyde diethyl acetal (110 μL, 0.728 mmol, 1.5 equiv) according to the general procedure method F. After completion of reaction, the mixture was diluted with distilled water (24 mL) and extracted with EtOAc (3 × 24 mL). The organic layer was collected, washed with brine (24 mL), and dried with MgSO4 before removing the volatiles in vacuo. The crude product was used in the next reaction without further purifying (175 mg, 99%). LC-MS: Rt = 2.43 min, 79 Abs % @ 254 nm, [M + H]+ = 364.2.
1-(2,2-Diethoxyethyl)-4-nitro-N-(pyrazin-2-ylmethyl)-1H-imidazole-2-carboxamide (28i)
4-Nitro-N-(pyrazin-2-ylmethyl)-1H-imidazole-2-carboxamide (137 mg, 0.552 mmol, 1 equiv) was reacted with bromoacetaldehyde diethyl acetal (125 μL, 0.828 mmol, 1.5 equiv) according to the general procedure method F. After completion of reaction, the mixture was diluted with distilled water (28 mL) and extracted with EtOAc (3 × 28 mL). The organic layer was collected, washed with brine (28 mL), and dried with MgSO4. Volatiles were removed in vacuo and the crude product was purified over silica gel by MPLC (Biotage Isolera, 15–100% pet. spirit/EtOAc) to give 1-(2,2-diethoxyethyl)-4-nitro-N-(pyrazin-2-ylmethyl)-1H-imidazole-2-carboxamide 28i as a light gray solid (38 mg, 19%). LC-MS: Rt = 2.72 min, 99 Abs % @ 254 nm, [M + Na]+ = 365.1. 1H NMR (600 MHz, CDCl3) δ 8.64 (d, J = 1.4 Hz, 1H), 8.57 (ddd, J = 6.9, 2.5, 1.5 Hz, 1H), 8.53 (d, J = 2.6 Hz, 1H), 8.16 (d, J = 5.9 Hz, 1H), 7.93 (s, 1H), 4.76 (d, J = 5.9 Hz, 2H), 4.73 (t, J = 4.9 Hz, 1H), 4.66 (d, J = 4.9 Hz, 2H), 3.75 (dq, J = 9.4, 7.1 Hz, 2H), 3.53 (dq, J = 9.4, 7.0 Hz, 2H), 1.18 (t, J = 7.0 Hz, 6H).
1-(2,2-Diethoxyethyl)-4-nitro-N-(quinolin-6-ylmethyl)-1H-imidazole-2-carboxamide (28j)
4-Nitro-N-(quinolin-6-ylmethyl)-1H-imidazole-2-carboxamide (180 mg, 0.606 mmol, 1 equiv) was reacted with bromoacetaldehyde diethyl acetal (137 μL, 0.908 mmol, 1.5 equiv) according to the general procedure method F. After completion of reaction, the mixture was diluted with distilled water (36 mL) and extracted with EtOAc (3 × 36 mL). The organic layer was collected, washed with brine (22 mL), and dried with MgSO4 before removing the volatiles in vacuo. The crude product looked clean when analyzed using LC-MS, and therefore was used in the next reaction without further purifying (204 mg, 81%). LC-MS: Rt = 2.72 min, 91 Abs % @ 254 nm, [M + Na]+ = 414.2. 1H NMR (600 MHz, CDCl3) δ 8.94 (dd, J = 4.2, 1.7 Hz, 1H), 8.20–8.08 (m, 2H), 7.99–7.87 (m, 2H), 7.83–7.75 (m, 1H), 7.70 (dd, J = 8.7, 2.0 Hz, 1H), 7.44 (dd, J = 8.3, 4.2 Hz, 1H), 4.86–4.76 (m, 3H), 4.71 (d, J = 4.8 Hz, 2H), 3.77 (dq, J = 9.4, 7.0 Hz, 2H), 3.56 (dq, J = 9.4, 7.1 Hz, 2H), 1.20 (t, J = 7.0 Hz, 6H).
N-(Benzo[d][1,3]dioxol-5-ylmethyl)-1-(2,2-diethoxyethyl)-4-nitro-1H-imidazole-2-carboxamide (28k)
N-(Benzo[d][1,3]dioxol-5-ylmethyl)-4-nitro-1H-imidazole-2-carboxamide (110 mg, 0.379 mmol, 1 equiv) was reacted with bromoacetaldehyde diethyl acetal (85.5 μL, 0.569 mmol, 1.5 equiv) according to the general procedure method F. After completion of reaction, the mixture was diluted with distilled water (22 mL) and extracted with EtOAc (3 × 22 mL). The organic layer was collected, washed with brine (22 mL), and dried with MgSO4 before removing the volatiles in vacuo. The crude product looked clean when analyzed using LC-MS, and therefore was used in the next reaction without further purifying. LC-MS: Rt = 2.95 min, 99 Abs % @ 254 nm, [M + Na]+ = 429.1. 1H NMR (600 MHz, DMSO-d6) δ 9.37 (t, J = 6.4 Hz, 1H), 8.48 (s, 1H), 6.90 (d, J = 1.6 Hz, 1H), 6.85 (d, J = 7.9 Hz, 1H), 6.79 (dd, J = 7.9, 1.6 Hz, 1H), 5.97 (s, 2H), 4.77 (t, J = 5.1 Hz, 1H), 4.60 (d, J = 5.1 Hz, 2H), 4.31 (d, J = 6.3 Hz, 2H), 3.60 (dq, J = 9.8, 7.0 Hz, 2H), 3.40 (dq, J = 9.8, 7.1 Hz, 2H), 1.01 (t, J = 7.0 Hz, 6H).
2-Nitro-7-(thiazol-2-ylmethyl)imidazo[1,2-a]pyrazin-8(7H)-one (26b)
1-(2,2-Diethoxyethyl)-4-nitro-N-(thiazol-2-ylmethyl)-1H-imidazole-2-carboxamide 28b (59 mg, 0.160 mmol, 1 equiv) was reacted according to the general procedure method G. After completion of reaction, the volatiles were evaporated in vacuo followed by recrystallization using DCM/MeOH (hot slurry) to yield the desired product, 2-nitro-7-(thiazol-2-ylmethyl)imidazo[1,2-a]pyrazin-8(7H)-one 26b, as a yellow solid (26 mg, 58%): LC-MS: Rt = 2.46 min, 99 Abs % @ 254 nm, [M + H]+ = 278.1; 1H NMR (600 MHz, DMSO-d6) δ 8.84 (s, 1H), 7.77 (d, J = 3.2 Hz, 1H), 7.74 (d, J = 3.3 Hz, 1H), 7.64 (d, J = 5.9 Hz, 1H), 7.50 (d, J = 5.9 Hz, 1H), 5.46 (s, 2H). 13C NMR (150 MHz, DMSO) δ 164.1, 152.5, 148.1, 142.4, 134.7, 123.7, 121.4, 116.9, 107.5, 48.0. HRMS (ESI): m/z calcd for C10H8N5O3S [M + H]+, 278.0342; found, 278.0356.
2-Nitro-7-(pyridin-3-ylmethyl)imidazo[1,2-a]pyrazin-8(7H)-one (26g)
1-(2,2-Diethoxyethyl)-5-nitro-N-(pyridin-3-ylmethyl)-1H-imidazole-2-carboxamide 28g (175 mg, 0.482 mmol, 1 equiv) was reacted according to the general procedure method G. Volatiles were removed in vacuo, and the crude product was purified by C18-reversed phase silica (Grace Reveleris X2, A: 0.1% TFA in water, B: 0.1% TFA in ACN, 0–100% B), then re-purified by HPLC (Gilson, A: 0.1% TFA in water, B: 0.1% TFA in ACN, 0–50% B) to give final product, 2-nitro-7-(pyridin-3-ylmethyl)imidazo[1,2-a]pyrazin-8(7H)-one 26g, as a light brown solid (17 mg, 13%). LC-MS: Rt = 1.61 min, 97 Abs % @ 254 nm, [M + H]+ = 272.1; 1H NMR (600 MHz, DMSO-d6) δ 8.82 (s, 1H), 8.62 (d, J = 2.2 Hz, 1H), 8.51 (dd, J = 4.8, 1.6 Hz, 1H), 7.77 (dt, J = 7.9, 2.0 Hz, 1H), 7.62 (d, J = 5.9 Hz, 1H), 7.49 (d, J = 5.9 Hz, 1H), 7.38 (dd, J = 7.9, 4.8 Hz, 1H), 5.17 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 152.9, 149.1, 149.0, 148.0, 135.6, 135.2, 132.2, 123.7, 123.5, 116.7, 107.6, 48.2. HRMS (ESI): m/z calcd for C12H10N5O3 [M + H]+, 272.0789; found, 272.0778.
2-Nitro-7-(pyrazin-2-ylmethyl)imidazo[1,2-a]pyrazin-8(7H)-one (26i)
1-(2,2-Diethoxyethyl)-4-nitro-N-(pyrazin-2-ylmethyl)-1H-imidazole-2-carboxamide 28i (37 mg, 0.102 mmol, 1 equiv) was reacted according to the general procedure method G. After completion of reaction, the volatiles were evaporated in vacuo followed by recrystallization using DCM/MeOH (hot slurry) to yield the desired product, 2-nitro-7-(pyrazin-2-ylmethyl)imidazo[1,2-a]pyrazin-8(7H)-one 26i, as a gray solid (10 mg, 35%): LC-MS: Rt = 2.19 min, 99 Abs % @ 254 nm, [M + H]+ = 273.1; 1H NMR (600 MHz, DMSO-d6) δ 8.84 (s, 1H), 8.74 (d, J = 1.4 Hz, 1H), 8.61–8.56 (m, 2H), 7.63 (d, J = 5.9 Hz, 1H), 7.48 (d, J = 5.8 Hz, 1H), 5.32 (s, 2H). 13C NMR (150 MHz, DMSO) δ 152.8, 151.2, 148.0, 144.1, 143.8, 135.0, 124.4, 116.7, 109.5, 107.1, 50.1. HRMS (ESI): m/z calcd for C11H9N6O3 [M + H]+, 273.0731; found, 273.0746.
2-Nitro-7-(quinolin-6-ylmethyl)imidazo[1,2-a]pyrazin-8(7H)-one (26j)
1-(2,2-Diethoxyethyl)-4-nitro-N-(quinolin-6-ylmethyl)-1H-imidazole-2-carboxamide 28j (200 mg, 0.484 mmol, 1 equiv) was reacted according to the general procedure method G. After completion of reaction, the volatiles were evaporated in vacuo. The crude product was then purified over silica gel by MPLC (Biotage Isolera, 1–25% DCM/MeOH), followed by recrystallization in DCM/MeOH (hot slurry) to give 2-nitro-7-(quinolin-6-ylmethyl)imidazo[1,2-a]pyrazin-8(7H)-one 26j as a light brown solid (129 mg, 83%): LC-MS: Rt = 2.21 min, 99 Abs % @ 254 nm, [M + H]+ = 322.1; 1H NMR (600 MHz, DMSO-d6) δ 8.89 (dd, J = 4.1, 1.7 Hz, 1H), 8.84 (s, 1H), 8.33 (ddd, J = 8.4, 1.8, 0.8 Hz, 1H), 8.02 (d, J = 8.7 Hz, 1H), 7.90 (d, J = 2.0 Hz, 1H), 7.75 (dd, J = 8.7, 2.1 Hz, 1H), 7.64 (d, J = 5.9 Hz, 1H), 7.53 (dd, J = 8.3, 4.1 Hz, 1H), 7.49 (d, J = 5.8 Hz, 1H), 5.34 (s, 2H). 13C NMR (150 MHz, DMSO) δ 152.9, 150.7, 148.0, 147.2, 135.9, 135.2, 134.7, 129.4, 129.2, 127.7, 126.3, 123.6, 121.8, 116.7, 107.5, 50.1. HRMS (ESI): m/z calcd for C16H12N5O3 [M + H]+, 322.0935; found, 322.0932.
7-(Benzo[d][1,3]dioxol-5-ylmethyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one (26k)
N-(Benzo[d][1,3]dioxol-5-ylmethyl)-1-(2,2-diethoxyethyl)-4-nitro-1H-imidazole-2-carboxamide 28k (150 mg, 0.369 mmol, 1 equiv) was reacted according to the general procedure method G. After completion of reaction, the volatiles were evaporated in vacuo followed by recrystallization using DCM/MeOH (hot slurry) to yield the desired product, 7-(benzo[d][1,3]dioxol-5-ylmethyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one 26k, as a yellow solid (71 mg, 61%): LC-MS: Rt = 2.66 min, 99 Abs % @ 254 nm, [M + H]+ = 315.1; 1H NMR (600 MHz, DMSO-d6) δ 8.80 (s, 1H), 7.58 (d, J = 5.9 Hz, 1H), 7.39 (d, J = 5.9 Hz, 1H), 6.96 (d, J = 1.4 Hz, 1H), 6.88 (d, J = 1.6 Hz, 2H), 5.99 (s, 2H), 5.02 (s, 2H). 13C NMR (150 MHz, DMSO) δ 152.7, 148.0, 147.4, 146.9, 135.1, 130.2, 123.3, 121.6, 116.6, 108.5, 108.3, 107.4, 101.1, 49.9. HRMS (ESI): m/z calcd for C14H11N4O5 [M + H]+, 315.0724; found, 315.0715.
4′-(Trifluoromethoxy)-[1,1′-biphenyl]-4-carbaldehyde (31b)
A mixture of 4-bromobenzaldehyde (1.10 g, 5.95 mmol), (4-(trifluoromethoxy)phenyl)boronic acid (1.1 equiv, 1.35 g, 6.54 mmol), K2CO3 (2.47 g, 17.8 mmol), and tetrakis(triphenylphosphine)palladium (0) (0.547 g, 0.476 mmol) in THF (16 mL) and distilled water (6 mL) was reacted according to the general procedure method H. After completion of reaction, the mixture was filtered through celite and the filtrate was evaporated in vacuo. The crude product was purified over silica gel by MPLC (Biotage Isolera, 2–20% pet. spirit/EtOAc) to give final product, 4′-(trifluoromethoxy)-[1,1′-biphenyl]-4-carbaldehyde 31b, as a white solid (1.31 g, 83% yield): LC-MS: Rt = 3.18 min, 99 Abs % @ 254 nm, [M + H]+ = 267.1; 1H NMR (600 MHz, DMSO-d6) δ 10.07 (s, 1H), 8.04–7.99 (m, 2H), 7.97–7.88 (m, 4H), 7.51 (dq, J = 7.7, 1.0 Hz, 2H). 13C NMR (150 MHz DMSO-d6) δ 192.7, 148.6, 144.3, 138.1, 135.3, 130.2, 129.2, 127.6, 121.6, 120.1 (J = 256.6 Hz).
4′-Methyl-[1,1′-biphenyl]-4-carbaldehyde (31c)
A mixture of 4-bromobenzaldehyde (550 mg, 2.97 mmol), p-tolylboronic acid (1.1 equiv, 445 mg, 3.27 mmol), K2CO3 (1.23 g, 8.92 mmol), and tetrakis(triphenylphosphine)palladium (0) (275 mg, 0.238 mmol) in THF (8 mL) and distilled water (3 mL) was reacted according to the general procedure method H. After completion of reaction, the mixture was diluted with distilled water and extracted with EtOAc (x 3). The combined organic layers were washed with brine, dried over MgSO4, and evaporated in vacuo. The crude product was purified over silica gel by MPLC (Biotage Isolera, 2–20% pet. spirit/EtOAc) to give final product, 4′-methyl-[1,1′-biphenyl]-4-carbaldehyde 31c as a white solid (355 mg, 61% yield): LC-MS: Rt = 3.07 min, 99 Abs % @ 254 nm, [M + H]+ = 197.2; 1H NMR (600 MHz, DMSO-d6) δ 10.04 (s, 1H), 7.98 (d, J = 8.0 Hz, 2H), 7.89 (d, J = 8.0 Hz, 2H), 7.67 (d, J = 7.9 Hz, 2H), 7.33 (d, J = 7.9 Hz, 2H), 2.36 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 192.7, 145.8, 138.2, 135.8, 134.8, 130.1, 129.7, 127.0, 126.9, 20.7.
3′-(Trifluoromethoxy)-[1,1′-biphenyl]-4-carbaldehyde (31d)
A mixture of 4-bromobenzaldehyde (550 mg, 2.97 mmol), (3-(trifluoromethoxy)phenyl)boronic acid (1.1 equiv, 673 mg, 3.27 mmol), K2CO3 (1.23 g, 8.92 mmol), and tetrakis(triphenylphosphine)palladium (0) (275 mg, 0.238 mmol) in THF (8 mL) and distilled water (3 mL) was reacted according to the general procedure method H. After completion of reaction, the mixture was filtered through celite and the filtrate was evaporated in vacuo. The crude product was purified over silica gel by MPLC (Biotage Isolera, 2–20% pet. spirit/EtOAc) to give final product, 3′-(trifluoromethoxy)-[1,1′-biphenyl]-4-carbaldehyde 31d, in a colorless gel form (710 mg, 90% yield): LC-MS: Rt = 3.18 min, 99 Abs % @ 254 nm, [M + H]+ = 267.1; 1H NMR (400 MHz, DMSO-d6) δ 10.08 (s, 1H), 8.06–7.93 (m, 4H), 7.83 (ddt, J = 7.8, 1.9, 0.9 Hz, 1H), 7.76 (s, 1H), 7.66 (t, J = 8.0 Hz, 1H), 7.50–7.42 (m, 1H).
6-Phenylnicotinaldehyde (31f)
A mixture of 6-bromonicotinaldehyde (550 mg, 2.96 mmol), phenylboronic acid (1.1 equiv, 397 mg, 3.25 mmol), K2CO3 (1.23 g, 8.87 mmol), tetrakis(triphenylphosphine)palladium (0) (273 mg, 0.237 mmol), THF (8 mL), and distilled water (3 mL) was reacted according to the general procedure method H. After completion of reaction, the mixture was diluted with distilled water and extracted with EtOAc (x 3). The combined organic layers were washed with brine, dried over MgSO4, and evaporated in vacuo. The crude product was purified over silica gel by MPLC (Biotage Isolera, 2–20% pet. spirit/EtOAc) to give the final product, 6-phenylnicotinaldehyde 31f, as a white solid (427 mg, 79% yield): LC-MS: Rt = 2.65 min, 99 Abs % @ 254 nm, [M + H]+ = 184.1; 1H NMR (600 MHz, DMSO-d6) δ 10.14 (s, 1H), 9.16 (p, J = 1.3 Hz, 1H), 8.35–8.26 (m, 1H), 8.23–8.18 (m, 3H), 7.54 (dtt, J = 8.6, 5.4, 1.7 Hz, 3H). 13C NMR (150 MHz DMSO-d6) δ 192.0, 160.4, 151.7, 137.4, 137.2, 130.4, 129.9, 129.0, 127.3, 120.5.
6-(4-(Trifluoromethoxy)phenyl)nicotinaldehyde (31g)
A mixture of 6-bromonicotinaldehyde (550 mg, 2.96 mmol), (4-(trifluoromethoxy)phenyl)boronic acid (1.1 equiv, 670 mg, 3.25 mmol), K2CO3 (1.23 g, 8.87 mmol), and tetrakis(triphenylphosphine)palladium (0) (273 mg, 0.237 mmol) in THF (8 mL) and distilled water (3 mL) was reacted according to the general procedure method H. After completion of reaction, the mixture was diluted with distilled water and extracted with EtOAc (x 3). The combined organic layers were washed with brine, dried over MgSO4, and evaporated in vacuo. The crude product was purified over silica gel by MPLC (Biotage Isolera, 2–20% pet. spirit/EtOAc) to give the final product, 6-(4-(trifluoromethoxy)phenyl)nicotinaldehyde 31g, as a white solid (560 mg, 71% yield): LC-MS: Rt = 2.99 min, 98 Abs % @ 254 nm, [M + H]+ = 268.1; 1H NMR (600 MHz, DMSO-d6) δ 1H NMR (600 MHz, DMSO-d6) δ 10.15 (s, 1H), 9.17 (dd, J = 2.2, 0.9 Hz, 1H), 8.37–8.29 (m, 3H), 8.25 (d, J = 8.2 Hz, 1H), 7.56–7.51 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 192.0, 158.9, 151.6, 149.8, 137.4, 136.6, 130.1, 129.4, 121.3, 120.8, 120.1 (J = 256.4 Hz).
6-(p-Tolyl)nicotinaldehyde (31h)
A mixture of 6-bromonicotinaldehyde (550 mg, 2.96 mmol), p-tolylboronic acid (1.1 equiv, 442 mg, 3.25 mmol), K2CO3 (1.23 g, 8.87 mmol), and tetrakis(triphenylphosphine)palladium (0) (273 mg, 0.237 mmol) in THF (8 mL) and distilled water (3 mL) was reacted according to the general procedure method H. After completion of reaction, the mixture was diluted with distilled water and extracted with EtOAc (x 3). The combined organic layers were washed with brine, dried over MgSO4, and evaporated in vacuo. The crude product was purified over silica gel by MPLC (Biotage Isolera, 2–20% pet. spirit/EtOAc) to give the final product, 6-(p-tolyl)nicotinaldehyde 31h, as a white solid (391 mg, 67% yield): LC-MS: Rt = 2.80 min, 99 Abs % @ 254 nm, [M + H]+ = 198.1; 1H NMR (600 MHz, DMSO-d6) δ 10.12 (s, 1H), 9.13 (dd, J = 2.2, 0.9 Hz, 1H), 8.29 (dd, J = 8.3, 2.2 Hz, 1H), 8.19–8.15 (m, 1H), 8.13–8.08 (m, 2H), 7.38–7.33 (m, 2H), 2.38 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 191.9, 160.4, 151.7, 140.3, 137.0, 134.7, 129.7, 129.6, 127.2, 120.1, 20.9.
6-(3-(Trifluoromethoxy)phenyl)nicotinaldehyde (31i)
A mixture of 6-bromonicotinaldehyde (550 mg, 2.96 mmol), (3-(trifluoromethoxy)phenyl)boronic acid (1.1 equiv, 670 mg, 3.25 mmol), K2CO3 (1.23 g, 8.87 mmol), and tetrakis(triphenylphosphine)palladium (0) (273 mg, 0.237 mmol) in THF (8 mL) and distilled water (3 mL) was reacted according to the general procedure method H. After completion of reaction, the mixture was diluted with distilled water and extracted with EtOAc (x 3). The combined organic layers were washed with brine, dried over MgSO4, and evaporated in vacuo. The crude product was purified over silica gel by MPLC (Biotage Isolera, 2–20% pet. spirit/EtOAc) to give the final product, 6-(3-(trifluoromethoxy)phenyl)nicotinaldehyde 31i, as a white solid (377 mg, 48% yield): LC-MS: Rt = 3.01 min, 99 Abs % @ 254 nm, [M + H]+ = 268.0; 1H NMR (600 MHz, DMSO-d6) δ 10.16 (s, 1H), 9.19 (d, J = 2.1 Hz, 1H), 8.36 (dd, J = 8.2, 2.1 Hz, 1H), 8.31 (d, J = 8.2 Hz, 1H), 8.25 (dt, J = 8.0, 1.1 Hz, 1H), 8.18 (d, J = 1.9 Hz, 1H), 7.70 (t, J = 8.0 Hz, 1H), 7.57–7.51 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 192.0, 158.4, 151.6, 149.0, 139.7, 137.5, 131.1, 130.5, 126.3, 122.7, 121.0, 120.2 (J = 256.6 Hz), 119.5.
5-(4-(Trifluoromethoxy)phenyl)picolinaldehyde (31j)
A mixture of 5-bromopicolinaldehyde (400 mg, 2.15 mmol), (4-(trifluoromethoxy)phenyl)boronic acid (1.1 equiv, 487 mg, 2.37 mmol), K2CO3 (892 mg, 6.45 mmol), tetrakis(triphenylphosphine)palladium (0) (199 mg, 0.172 mmol), THF (5.8 mL), and distilled water (2.2 mL) was reacted according to the general procedure method H. After completion of reaction, the mixture was diluted with distilled water and extracted with EtOAc (x 3). The combined organic layers were washed with brine, dried over MgSO4, and evaporated in vacuo. The crude product was purified over silica gel by MPLC (Biotage Isolera, 2–20% pet. spirit/EtOAc) to give the final product, 5-(4-(trifluoromethoxy)phenyl)picolinaldehyde 31j, as a white color solid (490 mg, 85% yield): LC-MS: Rt = 2.90 min, 98 Abs % @ 254 nm, [M + H]+ = 268.1; 1H NMR (600 MHz, DMSO-d6) δ 10.04 (d, J = 0.8 Hz, 1H), 9.18 (dd, J = 2.3, 0.9 Hz, 1H), 8.37 (ddd, J = 8.1, 2.3, 0.8 Hz, 1H), 8.02 (dd, J = 8.1, 0.9 Hz, 1H), 8.01–7.97 (m, 2H), 7.55 (dq, J = 7.8, 1.0 Hz, 2H). 13C NMR (150 MHz DMSO-d6) δ 193.2, 151.4, 149.0, 148.4, 138.1, 135.6, 135.2, 129.5, 121.9, 121.7, 120.0 (J = 256.3 Hz).
5-(4-(Trifluoromethoxy)phenyl)nicotinaldehyde (31k)
A mixture of 5-bromonicotinaldehyde (400 mg, 2.15 mmol), (4-(trifluoromethoxy)phenyl)boronic acid (1.1 equiv, 487 mg, 2.37 mmol), K2CO3 (892 mg, 6.45 mmol), tetrakis(triphenylphosphine)palladium (0) (199 mg, 0.172 mmol), THF (5.8 mL), and distilled water (2.2 mL) was reacted according to the general procedure method H. After completion of reaction, the mixture was diluted with distilled water and extracted with EtOAc (x 3). The combined organic layers were washed with brine, dried over MgSO4, and evaporated in vacuo. The crude product was purified over silica gel by MPLC (Biotage Isolera, 7–60% pet. spirit/EtOAc) to give the final product, 5-(4-(trifluoromethoxy)phenyl)nicotinaldehyde 31k, as a cream color solid (384 mg, 67% yield): LC-MS: Rt = 2.94 min, 98 Abs % @ 254 nm, [M + H]+ = 268.1; 1H NMR (600 MHz, DMSO-d6) δ 10.20 (s, 1H), 9.21 (dd, J = 2.4, 0.8 Hz, 1H), 9.11–9.07 (m, 1H), 8.56–8.52 (m, 1H), 8.00–7.94 (m, 2H), 7.54 (d, J = 8.3 Hz, 2H). 13C NMR (150 MHz DMSO-d6) δ 192.4, 152.5, 150.1, 148.7, 135.2, 134.6, 134.3, 131.2, 129.2, 121.7, 120.1 (J = 256.0 Hz).
5-(p-Tolyl)nicotinaldehyde (31l)
A mixture of 5-bromonicotinaldehyde (400 mg, 2.15 mmol), p-tolylboronic acid (1.1 equiv, 322 mg, 2.37 mmol), K2CO3 (892 mg, 6.45 smmol), tetrakis(triphenylphosphine)palladium (0) (199 mg, 0.172 mmol), THF (5.8 mL), and distilled water (2.2 mL) was reacted according to the general procedure method H. After completion of reaction, the mixture was diluted with distilled water and extracted with EtOAc (x 3). The combined organic layers were washed with brine, dried over MgSO4, and evaporated in vacuo. The crude product was purified over silica gel by MPLC (Biotage Isolera, 12–100% pet. spirit/EtOAc) to give the final product, 5-(p-tolyl)nicotinaldehyde 31l, as a yellow solid (240 mg, 57% yield): LC-MS: Rt = 2.71 min, 95 Abs % @ 254 nm, [M + H]+ = 198.2; 1H NMR (600 MHz, DMSO-d6) δ 10.19 (s, 1H), 9.16 (d, J = 2.4 Hz, 1H), 9.03 (d, J = 1.9 Hz, 1H), 8.48 (dd, J = 2.4, 1.9 Hz, 1H), 7.77–7.70 (m, 2H), 7.39–7.33 (m, 2H), 2.37 (s, 3H). 13C NMR (150 MHz DMSO-d6) δ 192.6, 152.2, 149.4, 138.3, 135.8, 133.7, 132.9, 131.3, 129.9, 126.9, 20.7.
5-(3-(Trifluoromethoxy)phenyl)nicotinaldehyde (31m)
A mixture of 5-bromonicotinaldehyde (400 mg, 2.15 mmol), (3-(trifluoromethoxy)phenyl)boronic acid (1.1 equiv, 487 mg, 2.37 mmol), K2CO3 (892 mg, 6.45 mmol), tetrakis(triphenylphosphine)palladium (0) (199 mg, 0.172 mmol), THF (5.8 mL), and distilled water (2.2 mL) was reacted according to the general procedure method H. After completion of reaction, the mixture was diluted with distilled water and extracted with EtOAc (x 3). The combined organic layers were washed with brine, dried over MgSO4, and evaporated in vacuo. The crude product was purified over silica gel by MPLC (Biotage Isolera, 7–60% pet. spirit/EtOAc) to give the final product, 5-(3-(trifluoromethoxy)phenyl)nicotinaldehyde 31m, as a cream color solid (331 mg, 58% yield): LC-MS: Rt = 2.94 min, 98 Abs % @ 254 nm, [M + H]+ = 268.1; 1H NMR (600 MHz, DMSO-d6) δ 10.20 (s, 1H), 9.23 (dd, J = 2.3, 0.9 Hz, 1H), 9.10 (d, J = 1.2 Hz, 1H), 8.58 (td, J = 2.2, 0.9 Hz, 1H), 7.92–7.86 (m, 2H), 7.72–7.65 (m, 1H), 7.49 (ddt, J = 8.2, 2.1, 1.1 Hz, 1H). 13C NMR (150 MHz DMSO-d6) δ 192.4, 152.6, 150.2, 149.1, 138.3, 134.7, 134.3, 131.2, 126.3, 121.1, 120.9, 120.1 (J = 256.4 Hz), 119.9.
(4′-(Trifluoromethoxy)-[1,1′-biphenyl]-4-yl)methanol (32b)
4′-(Trifluoromethoxy)-[1,1′-biphenyl]-4-carbaldehyde (500 mg, 1.88 mmol) and sodium borohydride (92.4 mg, 2.44 mmol) were reacted according to the general procedure method I. Product was obtained as a white solid (514 mg, quantitative yield): LC-MS: Rt = 3.00 min, 99 Abs % @ 254 nm, [M + H – H2O]+ = 251.1; 1H NMR (600 MHz, DMSO-d6) δ 7.83–7.75 (m, 2H), 7.69–7.62 (m, 2H), 7.43 (ddq, J = 11.9, 7.3, 0.9 Hz, 4H), 5.23 (t, J = 5.7 Hz, 1H), 4.54 (d, J = 5.7 Hz, 2H).13C NMR (150 MHz, DMSO-d6) δ 147.7, 142.3, 139.4, 137.0, 128.4, 127.0, 126.5, 121.4, 120.2 (J = 255.6 Hz), 62.5.
(4′-Methyl-[1,1′-biphenyl]-4-yl)methanol (32c)
4′-Methyl-[1,1′-biphenyl]-4-carbaldehyde (320 mg, 1.63 mmol) and sodium borohydride (80.2 mg, 2.12 mmol) were reacted according to the general procedure method I. Product was obtained as a white solid (350 mg, quantitative yield): LC-MS: Rt = 2.87 min, 98 Abs % @ 254 nm, [M – OH]+ = 181.2; 1H NMR (600 MHz, DMSO-d6) δ 7.61–7.57 (m, 2H), 7.57–7.52 (m, 2H), 7.38 (d, J = 8.1 Hz, 2H), 7.26 (d, J = 7.9 Hz, 2H), 5.20 (t, J = 5.7 Hz, 1H), 4.53 (d, J = 5.7 Hz, 2H), 2.34 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 141.4, 138.4, 137.2, 136.5, 129.5, 127.0, 126.3, 126.1, 62.6, 20.6.
(3′-(Trifluoromethoxy)-[1,1′-biphenyl]-4-yl)methanol (32d)
3′-(Trifluoromethoxy)-[1,1′-biphenyl]-4-carbaldehyde (500 mg, 1.88 mmol) and sodium borohydride (92.4 mg, 2.44 mmol) were reacted according to the general procedure method I. Product was obtained as a white solid (404 mg, 80% yield): LC-MS: Rt = 2.99 min, 99 Abs % @ 254 nm, [M + H – H2O]+ = 251.1; 1H NMR (600 MHz, DMSO-d6) δ 7.74–7.65 (m, 3H), 7.64–7.56 (m, 2H), 7.45–7.40 (m, 2H), 7.35 (ddt, J = 8.1, 2.3, 1.1 Hz, 1H), 5.25 (t, J = 5.7 Hz, 1H), 4.55 (d, J = 5.7 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 149.0, 142.7, 142.4, 136.7, 130.8, 127.0, 126.6, 125.6, 120.1 (J = 256.0 Hz), 119.5, 119.3, 62.5.
(4-(Pyridin-3-yl)phenyl)methanol (32e)
4-(Pyridin-3-yl)benzaldehyde (300 mg, 1.64 mmol) and sodium borohydride (80.5 mg, 2.13 mmol) were reacted according to the general procedure method I. Product was obtained as a cream color powder (195 mg, 65%): LC-MS: Rt = 1.39 min, 99 Abs % @ 254 nm, [M + H]+ = 186.2; 1H NMR (600 MHz, DMSO-d6) δ 8.89 (dd, J = 2.4, 0.9 Hz, 1H), 8.56 (dd, J = 4.7, 1.6 Hz, 1H), 8.06 (ddd, J = 7.9, 2.4, 1.6 Hz, 1H), 7.72–7.66 (m, 2H), 7.51–7.42 (m, 3H), 5.25 (td, J = 5.8, 0.7 Hz, 1H), 4.56 (d, J = 5.6 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 148.3, 147.5, 142.6, 135.5, 135.4, 133.9, 127.1, 126.6, 123.8, 62.5.
(6-Phenylpyridin-3-yl)methanol (32f)
6-Phenylnicotinaldehyde (310 mg, 1.69 mmol) and sodium borohydride (83.2 mg, 2.20 mmol) were reacted according to the general procedure method I. Product was obtained as a colorless gel (315 mg, quantitative yield): LC-MS: Rt = 1.87 min, 99 Abs % @ 254 nm, [M + H]+ = 186.2; 1H NMR (600 MHz, DMSO-d6) δ 1H NMR (600 MHz, DMSO-d6) δ 8.60 (dt, J = 2.2, 0.7 Hz, 1H), 8.11–8.05 (m, 2H), 7.93 (dd, J = 8.1, 0.8 Hz, 1H), 7.83–7.78 (m, 1H), 7.48 (dd, J = 8.2, 6.8 Hz, 2H), 7.45–7.39 (m, 1H), 5.34 (t, J = 5.7 Hz, 1H), 4.57 (d, J = 5.7 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 154.6, 148.0, 138.6, 136.4, 135.5, 128.8, 128.7, 126.4, 119.7, 60.5.
(6-(4-(Trifluoromethoxy)phenyl)pyridin-3-yl)methanol (32g)
6-(4-(Trifluoromethoxy)phenyl)nicotinaldehyde 31g (348 mg, 1.30 mmol) and sodium borohydride (64.0 mg, 1.69 mmol) were reacted according to the general procedure method I. The crude product was purified over C18-reversed phase silica (Grace Reveleris X2, A: H2O, B: ACN, 5–100% B) to give the final product as a white solid (207 mg, 59% yield): LC-MS: Rt = 2.61 min, 99 Abs % @ 254 nm, [M + H]+ = 270.1; 1H NMR (600 MHz, DMSO-d6) δ 8.62 (dd, J = 2.2, 0.9 Hz, 1H), 8.23–8.17 (m, 2H), 7.97 (dd, J = 8.1, 0.9 Hz, 1H), 7.83 (dd, J = 8.1, 2.2 Hz, 1H), 7.50–7.44 (m, 2H), 5.37 (t, J = 5.7 Hz, 1H), 4.58 (d, J = 5.5 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 153.2, 148.8, 148.1, 137.8, 136.9, 135.7, 128.3, 121.2, 120.1 (J = 256.8 Hz), 119.9, 60.4.
(6-(p-Tolyl)pyridin-3-yl)methanol (32h)
6-(p-Tolyl)nicotinaldehyde 31h (320 mg, 1.62 mmol) and sodium borohydride (79.8 mg, 2.11 mmol) were reacted according to the general procedure method I. The product was purified over silica gel by MPLC (Biotage Isolera, 1–20% pet. spirit/EtOAc) to give the final product as a white solid (250 mg, 77% yield): LC-MS: Rt = 2.08 min, 98 Abs % @ 254 nm, [M + H]+ = 200.2; 1H NMR (600 MHz, DMSO-d6) δ 8.57 (d, J = 2.2 Hz, 1H), 7.97 (d, J = 8.1 Hz, 2H), 7.89 (d, J = 8.1 Hz, 1H), 7.78 (dd, J = 8.2, 2.3 Hz, 1H), 7.29 (d, J = 7.7 Hz, 2H), 5.32 (t, J = 5.7 Hz, 1H), 4.56 (d, J = 5.6 Hz, 2H), 2.35 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 154.6, 148.0, 138.3, 136.0, 135.9, 135.5, 129.3, 126.3, 119.3, 60.5, 20.8.
(6-(3-(Trifluoromethoxy)phenyl)pyridin-3-yl)methanol (32i)
6-(3-(Trifluoromethoxy)phenyl)nicotinaldehyde 31i (330 mg, 1.23 mmol) and sodium borohydride (60.7 mg, 1.61 mmol) were reacted according to the general procedure method I. Product was obtained as a colorless gel in quant. yield: LC-MS: Rt = 2.67 min, 99 Abs % @ 254 nm, [M + H]+ = 270.1; 1H NMR (600 MHz, DMSO-d6) δ 8.63 (d, J = 2.1 Hz, 1H), 8.15–8.10 (m, 1H), 8.10–8.00 (m, 2H), 7.84 (dd, J = 8.1, 2.2 Hz, 1H), 7.63 (t, J = 8.0 Hz, 1H), 7.45–7.40 (m, 1H), 5.38 (t, J = 5.6 Hz, 1H), 4.59 (d, J = 5.1 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 152.7, 149.0, 148.1, 140.9, 137.3, 135.7, 130.8, 125.3, 121.2, 120.2 (J = 255.8 Hz), 120.1, 118.6, 60.4.
(5-(4-(Trifluoromethoxy)phenyl)pyridin-2-yl)methanol (32j)
5-(4-(Trifluoromethoxy)phenyl)picolinaldehyde 31j (490 mg, 1.83 mmol) and sodium borohydride (90.2 mg, 2.38 mmol) were reacted according to the general procedure method I. Product was obtained as a colorless gel (454 mg, 92% yield): LC-MS: Rt = 2.42 min, 98 Abs % @ 254 nm, [M + H]+ = 270.1; 1H NMR (600 MHz, DMSO-d6) δ 8.81 (dd, J = 2.4, 0.8 Hz, 1H), 8.11 (dd, J = 8.2, 2.4 Hz, 1H), 7.89–7.82 (m, 2H), 7.59–7.54 (m, 1H), 7.49 (dq, J = 7.8, 1.0 Hz, 2H), 5.47 (t, J = 5.8 Hz, 1H), 4.61 (d, J = 5.9 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 161.4, 148.2, 146.6, 136.5, 134.8, 132.3, 128.7, 121.6, 120.2, 120.1 (J = 256.0 Hz), 64.0.
(5-(4-(Trifluoromethoxy)phenyl)pyridin-3-yl)methanol (32k)
5-(4-Trifluoromethoxy)phenyl)nicotinaldehyde 31k (285 mg, 1.07 mmol) and sodium borohydride (52.5 mg, 1.39 mmol) were reacted according to the general procedure method I. Product was then purified over C18 silica gel (Grace Reveleris X2, A: H2O, B: ACN, 0–100% B) to yield a colorless gel (285 mg, quant. yield): LC-MS: Rt = 2.46 min, 97 Abs % @ 254 nm, [M + H]+ = 270.1; 1H NMR (600 MHz, DMSO-d6) δ 8.79 (d, J = 2.3 Hz, 1H), 8.57–8.53 (m, 1H), 8.00 (s, 1H), 7.89–7.83 (m, 2H), 7.50 (dq, J = 7.6, 1.0 Hz, 2H), 5.39 (t, J = 5.7 Hz, 1H), 4.62 (dt, J = 5.8, 0.7 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 148.3, 147.4, 146.2, 137.8, 136.5, 133.7, 132.5, 128.8, 121.6, 120.1 (J = 256.4 Hz), 60.5.
(5-(p-Tolyl)pyridin-3-yl)methanol (32l)
5-(p-Tolyl)nicotinaldehyde 31l (230 mg, 1.17 mmol) and sodium borohydride (57.3 mg, 1.52 mmol) were reacted according to the general procedure method I. Product was obtained as a colorless gel (229 mg, quant. yield): LC-MS: Rt = 2.46 min, 94 Abs % @ 254 nm, [M + H]+ = 270.1; 1H NMR (600 MHz, DMSO-d6) δ 8.74 (d, J = 2.3 Hz, 1H), 8.49 (d, J = 2.0 Hz, 1H), 7.95 (t, J = 2.2 Hz, 1H), 7.66–7.59 (m, 2H), 7.36–7.29 (m, 2H), 5.37 (td, J = 5.7, 0.7 Hz, 1H), 4.60 (dd, J = 5.7, 0.7 Hz, 2H), 2.36 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 146.7, 145.9, 137.6, 137.5, 134.9, 134.2, 132.0, 129.7, 126.6, 60.6, 20.7.
(5-(3-(Trifluoromethoxy)pphenyl)pyridin-3-yl)mmethanol (32m)
5-(3-Trifluoromethoxy)phenyl)nicotinaldehyde 31m (250 mg, 0.936 mmol) and sodium borohydride (46.0 mg, 1.22 mmol) were reacted according to the general procedure method I. Product was obtained as a colorless gel (283 mg, quant. yield): LC-MS: Rt = 2.67 min, 98 Abs % @ 254 nm, [M + H]+ = 270.1; 1H NMR (600 MHz, DMSO-d6) δ 8.82 (d, J = 2.3 Hz, 1H), 8.57 (d, J = 1.9 Hz, 1H), 8.04 (d, J = 2.2 Hz, 1H), 7.79 (ddd, J = 7.8, 1.7, 0.9 Hz, 1H), 7.73 (d, J = 2.2 Hz, 1H), 7.65 (t, J = 8.0 Hz, 1H), 7.43 (ddt, J = 8.2, 2.3, 1.1 Hz, 1H), 5.39 (t, J = 5.7 Hz, 1H), 4.62 (d, J = 5.7 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 149.0, 147.7, 146.2, 139.5, 137.8, 133.4, 132.6, 131.1, 126.0, 120.5, 120.1 (J = 256.6 Hz), 119.5, 60.5.
(4-(Thiazol-2-yl)phenyl)methanol (32n)
4-Thiazol-2-yl-benzaldehyde 31n (200 mg, 1.06 mmol) and sodium borohydride (52.0 mg, 1.37 mmol) were reacted according to the general procedure method I. Product was obtained as a white solid (203 mg, quantitative yield): LC-MS: Rt = 2.32 min, 99 Abs % @ 254 nm, [M + H]+ = 192.1; 1H NMR (600 MHz, DMSO-d6) δ 7.96–7.90 (m, 3H), 7.77 (d, J = 3.1 Hz, 1H), 7.45 (d, J = 7.9 Hz, 2H), 5.31 (t, J = 5.7 Hz, 1H), 4.56 (d, J = 5.7 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 167.1, 144.8, 143.7, 131.6, 127.0, 126.0, 120.1, 62.4.
(2-Phenylthiazol-5-yl)methanol (32o)
2-Phenylthiazole-5-carbaldehyde 31o (200 mg, 1.06 mmol) and sodium borohydride (52.0 mg, 1.37 mmol) were reacted according to the general procedure method I. Product was then purified over C18 silica gel (Grace Reveleris X2, A: H2O, B: ACN, 5–100% B) to yield a white solid (190 mg, 94%): LC-MS: Rt = 2.84 min, 99 Abs % @ 254 nm, [M + H]+ = 192.1; 1H NMR (600 MHz, DMSO-d6) δ 7.94–7.89 (m, 2H), 7.74 (s, 1H), 7.53–7.44 (m, 3H), 5.63 (t, J = 5.7 Hz, 1H), 4.71 (d, J = 5.6 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 166.4, 141.1, 140.5, 133.3, 130.0, 129.2, 125.9, 55.8.
4-(Chloromethyl)-4′-(trifluoromethoxy)-1,1′-biphenyl (33b)
4′-(Trifluoromethoxy)-[1,1′-biphenyl]-4-yl)methanol 32b (350 mg, 1.30 mmol) and thionyl chloride (189 μL, 2.61 mmol) were reacted according to the general procedure method D. Product was obtained as a white solid (330 mg, 88% yield): LC-MS: Rt = 3.39 min, 98 Abs % @ 254 nm, [M + Na]+ = 310, [M – Cl]+ = 251.2; 1H NMR (600 MHz, DMSO-d6) δ 7.85–7.77 (m, 2H), 7.73–7.66 (m, 2H), 7.54 (d, J = 8.4 Hz, 2H), 7.46 (dq, J = 7.6, 1.0 Hz, 2H), 4.82 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 147.9, 138.9, 138.6, 137.3, 129.5, 128.6, 127.1, 121.5, 120.1 (J = 255.8 Hz), 45.8.
4-(Chloromethyl)-4′-methyl-1,1′-biphenyl (33c)
(4′-Methyl-[1,1′-biphenyl]-4-yl)methanol 32c (215 mg, 1.08 mmol) and thionyl chloride (157 μL, 2.17 mmol) were reacted according to the general procedure method D. Product was obtained as a white solid (247 mg, 94% yield): LC-MS: Rt = 3.32 min, 92 Abs % @ 254 nm, [M – Cl]+ = 181.2; 1H NMR (600 MHz, DMSO-d6) δ 7.68–7.63 (m, 2H), 7.60–7.55 (m, 2H), 7.54–7.48 (m, 2H), 7.28 (d, J = 8.0 Hz, 2H), 4.80 (s, 2H), 2.34 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 140.0, 137.0, 136.6, 136.4, 129.5, 129.4, 126.6, 126.5, 46.0, 20.7.
4-(Chloromethyl)-3′-(trifluoromethoxy)-1,1′-biphenyl (33d)
3′-(Trifluoromethoxy)-[1,1′-biphenyl]-4-yl)methanol 32d (330 mg, 1.23 mmol) and thionyl chloride (178 μL, 2.46 mmol) were reacted according to the general procedure method D. Product was obtained as a white solid (369 mg, quant. yield): LC-MS: Rt = 3.37 min, 95 Abs % @ 254 nm, [M + Na]+ = 310, [M – Cl]+ = 251.1; 1H NMR (600 MHz, DMSO-d6) δ 7.77–7.70 (m, 3H), 7.66 (d, J = 2.3 Hz, 1H), 7.61 (t, J = 8.0 Hz, 1H), 7.55 (d, J = 8.0 Hz, 2H), 7.41–7.35 (m, 1H), 4.83 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 148.9, 141.9, 138.3, 137.7, 130.9, 129.6, 127.2, 125.9, 120.1 (J = 256.8 Hz), 120.0, 119.3, 45.2.
3-(4-(Chloromethyl)phenyl)pyridine (33e)
(4-(Pyridin-3-yl)phenyl)methanol 32e (190 mg, 1.03 mmol) and thionyl chloride (149 μL, 2.05 mmol) were reacted according to the general procedure method D. Product was obtained as a gel form in quant. yield: LC-MS: Rt = 2.80 min, 99 Abs % @ 254 nm, [M + H]+ = 204.1; 1H NMR (600 MHz, DMSO-d6) δ 8.73 (d, J = 2.2 Hz, 1H), 8.12–8.06 (m, 2H), 8.02–7.93 (m, 2H), 7.53–7.48 (m, 2H), 7.48–7.43 (m, 1H), 4.87 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 155.9, 149.7, 138.1, 137.7, 132.3, 129.3, 128.8, 126.6, 120.1, 43.1.
5-(Chloromethyl)-2-phenylpyridine (33f)
(6-Phenylpyridin-3-yl)methanol 32f (200 mg, 1.08 mmol) and thionyl chloride (157 μL, 2.16 mmol) were reacted according to the general procedure method D. Product was obtained as a white solid (185 mg, 84% yield): LC-MS: Rt = 2.80 min, 99 Abs % @ 254 nm, [M + H]+ = 204.1; 1H NMR (600 MHz, DMSO-d6) δ 8.73 (d, J = 2.2 Hz, 1H), 8.12–8.06 (m, 2H), 8.02–7.93 (m, 2H), 7.53–7.48 (m, 2H), 7.48–7.43 (m, 1H), 4.87 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 155.9, 149.7, 138.1, 137.7, 132.3, 129.3, 128.8, 126.6, 120.1, 43.1.
5-(Chloromethyl)-2-(4-(trifluoromethoxy)phenyl)pyridine (33g)
(6-(4-(Trifluoromethoxy)phenyl)pyridin-3-yl)methanol 32g (105 mg, 0.390 mmol) and thionyl chloride (56.6 μL, 0.780 mmol) were reacted according to the general procedure method D. Product was obtained as a white solid (95 mg, 84% yield): LC-MS: Rt = 3.14 min, 99 Abs % @ 254 nm, [M + H]+ = 288.0; 1H NMR (600 MHz, DMSO-d6) δ 8.74 (d, J = 2.1 Hz, 1H), 8.25–8.20 (m, 2H), 8.06–8.01 (m, 1H), 7.98 (dd, J = 8.2, 2.2 Hz, 1H), 7.51–7.47 (m, 2H), 4.88 (d, J = 1.3 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 154.4, 149.8, 149.1, 137.9, 137.3, 132.7, 128.6, 121.2, 120.3, 120.1 (J = 256.8 Hz), 43.0.
5-(Chloromethyl)-2-(p-tolyl)pyridine (33h)
(6-(p-Tolyl)pyridin-3-yl)methanol 32h (240 mg, 1.20 mmol) and thionyl chloride (175 μL, 2.41 mmol) were reacted according to the general procedure method D. Product was obtained as a white solid (247 mg, 94% yield): LC-MS: Rt = 2.90 min, 98 Abs % @ 254 nm, [M + H]+ = 218.1; 1H NMR (600 MHz, DMSO-d6) δ 8.69 (dd, J = 2.2, 0.9 Hz, 1H), 8.02–7.97 (m, 2H), 7.96 (dd, J = 8.2, 1.0 Hz, 1H), 7.92 (dd, J = 8.2, 2.3 Hz, 1H), 7.30 (d, J = 7.7 Hz, 2H), 4.86 (s, 2H), 2.36 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 155.9, 149.6, 138.9, 137.6, 135.3, 131.9, 129.4, 126.5, 119.7, 43.2, 20.8.
5-(Chloromethyl)-2-(3-(trifluoromethoxy)phenyl)pyridine (33i)
(6-(3-(Trifluoromethoxy)phenyl)pyridin-3-yl)methanol 32i (330 mg, 1.23 mmol) and thionyl chloride (178 μL, 2.45 mmol) were reacted according to the general procedure method D. Product was obtained as a colorless gel (368 mg, quant. yield): LC-MS: Rt = 3.17 min, 99 Abs % @ 254 nm, [M + H]+ = 288.0; 1H NMR (600 MHz, DMSO-d6) δ 8.76 (d, J = 1.6 Hz, 1H), 8.14 (ddd, J = 7.9, 1.7, 0.9 Hz, 1H), 8.11–8.05 (m, 2H), 8.00 (dd, J = 8.2, 2.3 Hz, 1H), 7.64 (t, J = 8.0 Hz, 1H), 7.46 (ddt, J = 8.2, 2.4, 1.0 Hz, 1H), 4.88 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 154.0, 149.8, 149.0, 140.4, 137.9, 133.2, 130.9, 125.5, 121.7, 121.3 (J = 255.7 Hz), 120.5, 118.8, 42.9.
2-(Chloromethyl)-5-(4-(trifluoromethoxy)phenyl)pyridine (33j)
(5-(4-(Trifluoromethoxy)phenyl)pyridin-2-yl)methanol 32j (450 mg, 1.67 mmol) and thionyl chloride (398 μL, 3.34 mmol) were reacted according to the general procedure method D. Product was obtained as a light brown solid (95 mg, 84% yield): LC-MS: Rt = 3.04 min, 98 Abs % @ 254 nm, [M + H]+ = 288.1; 1H NMR (600 MHz, DMSO-d6) δ 8.90 (dd, J = 2.4, 0.8 Hz, 1H), 8.16 (dd, J = 8.1, 2.4 Hz, 1H), 7.94–7.83 (m, 2H), 7.66 (dd, J = 8.1, 0.8 Hz, 1H), 7.51 (dq, J = 7.8, 1.0 Hz, 2H), 4.84 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 155.7, 148.4, 147.5, 135.9, 135.4, 133.7, 129.0, 123.4, 121.7, 120.1 (J = 255.2 Hz), 46.6.
3-(Chloromethyl)-5-(4-(trifluoromethoxy)phenyl)pyridine (33k)
(5-(4-(Trifluoromethoxy)phenyl)pyridin-3-yl)methanol 32k (205 mg, 0.761 mmol) and thionyl chloride (110 μL, 1.52 mmol) were reacted according to the general procedure method D. Product was obtained as a light yellow solid (215 mg, 98% yield): LC-MS: Rt = 3.06 min, 99 Abs % @ 254 nm, [M + H]+ = 288.1; 1H NMR (600 MHz, DMSO-d6) δ 8.89 (d, J = 2.2 Hz, 1H), 8.68 (d, J = 2.0 Hz, 1H), 8.20 (d, J = 2.3 Hz, 1H), 7.91–7.86 (m, 2H), 7.52 (d, J = 8.3 Hz, 2H), 4.89 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 148.9, 148.5, 147.5, 135.8, 134.7, 134.1, 133.8, 129.0, 121.7, 120.1 (J = 256.2 Hz), 43.0.
3-(Chloromethyl)-5-(p-tolyl)pyridine (33l)
(5-(p-Tolyl)pyridin-3-yl)methanol 32l (200 mg, 1.00 mmol) and thionyl chloride (146 μL, 2.01 mmol) were reacted according to the general procedure method D. Product was obtained as an orange solid (210 mg, 96% yield): LC-MS: Rt = 2.79 min, 95 Abs % @ 254 nm, [M + H]+ = 218.1; 1H NMR (600 MHz, DMSO-d6) δ 8.84 (d, J = 2.2 Hz, 1H), 8.62 (d, J = 2.0 Hz, 1H), 8.13 (t, J = 2.2 Hz, 1H), 7.67–7.60 (m, 2H), 7.36–7.29 (m, 2H), 4.88 (s, 2H), 2.36 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 148.3, 147.2, 137.9, 135.3, 134.1, 133.7, 133.5, 129.8, 126.7, 43.2, 20.7.
3-(Chloromethyl)-5-(3-(trifluoromethoxy)phenyl)pyridine (33m)
(5-(3-(Trifluoromethoxy)phenyl)pyridin-3-yl)methanol 32m (205 mg, 0.761 mmol) and thionyl chloride (110 μL, 1.52 mmol) were reacted according to the general procedure method D. Product was obtained as a cream color solid in quant. yield: LC-MS: Rt = 3.06 min, 99 Abs % @ 254 nm, [M + H]+ = 288.1; 1H NMR (600 MHz, DMSO-d6) δ 8.92 (d, J = 2.3 Hz, 1H), 8.69 (d, J = 2.0 Hz, 1H), 8.24 (t, J = 2.2 Hz, 1H), 7.82 (ddd, J = 7.8, 1.7, 0.9 Hz, 1H), 7.77 (d, J = 2.2 Hz, 1H), 7.66 (t, J = 8.0 Hz, 1H), 7.46 (ddt, J = 8.2, 2.3, 1.1 Hz, 1H), 4.89 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 149.3, 149.0, 147.6, 138.9, 134.8, 133.8, 133.8, 131.2, 126.1, 120.7, 120.1 (J = 256.3 Hz), 119.7, 43.0.
2-(4-(Chloromethyl)phenyl)thiazole (33n)
(4-(Thiazol-2-yl)phenyl)methanol 32n (180 mg, 0.941 mmol) and thionyl chloride (137 μL, 1.88 mmol) were reacted according to the general procedure method D. Product was obtained as a white solid (124 mg, 80% yield): LC-MS: Rt = 2.92 min, 99 Abs % @ 254 nm, [M + H]+ = 210.1; 1H NMR (600 MHz, DMSO-d6) δ 7.98–7.95 (m, 2H), 7.94 (d, J = 3.3 Hz, 1H), 7.81 (d, J = 3.2 Hz, 1H), 7.61–7.54 (m, 2H), 4.82 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 166.5, 143.9, 139.5, 132.9, 129.7, 126.4, 120.8, 45.6.
5-(Chloromethyl)-2-phenylthiazole (33o)
(2-Phenylthiazol-5-yl)methanol 32o (140 mg, 0.732 mmol) and thionyl chloride (106 μL, 1.46 mmol) were reacted according to the general procedure method D. Product was obtained as a beige solid (171 mg, 87% yield): LC-MS: Rt = 2.95 min, 81 Abs % @ 254 nm, [M + H]+ = 210.1; 1H NMR (600 MHz, DMSO-d6) δ 7.97–7.90 (m, 3H), 7.51 (dd, J = 4.9, 1.9 Hz, 3H), 5.16 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 168.6, 143.8, 135.9, 132.8, 130.6, 129.3, 126.1, 38.0.
7-([1,1′-Biphenyl]-4-ylmethyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one (34a)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (60 mg, 0.333 mmol, 1 equiv) was reacted with 4-(bromomethyl)-1,1′-biphenyl 33a (98.8 mg, 0.400 mmol, 1.2 equiv) under microwave conditions at 90 °C for 15 min according to the general procedure method B. After the reaction was completed as analyzed by LC-MS, the mixture was diluted with distilled water (15 mL) and the precipitate was collected by filtration. The precipitate was then purified by recrystallization using DCM/MeOH (hot slurry) to yield the desired product, 7-([1,1′-biphenyl]-4-ylmethyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one 34a, as a white solid (113 mg, 98%): LC-MS: Rt = 3.04 min, 99 Abs % @ 254 nm, [M + H]+ = 347.1; 1H NMR (600 MHz, DMSO-d6) δ 8.82 (s, 1H), 7.68–7.60 (m, 5H), 7.50–7.42 (m, 5H), 7.36 (ddt, J = 7.9, 6.8, 1.2 Hz, 1H), 5.18 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 152.8, 148.0, 139.7, 139.7, 135.7, 135.1, 128.9, 128.4, 127.5, 126.9, 126.6, 123.6, 116.6, 107.4, 50.0. HRMS (ESI): m/z calcd for C19H15N4O3 [M + H]+, 347.1139; found, 347.1146.
2-Nitro-7-((4′-(trifluoromethoxy)-[1,1′-biphenyl]-4-yl)methyl)imidazo[1,2-a]pyrazin-8(7H)-one (34b)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (50 mg, 0.278 mmol, 1 equiv) was reacted with 4-(chloromethyl)-4′-(trifluoromethoxy)-1,1′-biphenyl 33b (95.5 mg, 0.333 mmol, 1.2 equiv) under microwave conditions at 90 °C for 20 min according to the general procedure method B. After the reaction was completed as analyzed by LC-MS, the mixture was diluted with distilled water (10 mL) and the precipitate was collected by filtration. The precipitate was then purified by recrystallization using MeOH/acetone (hot slurry) to yield the desired product, 2-nitro-7-((4′-(trifluoromethoxy)-[1,1′-biphenyl]-4-yl)methyl)imidazo[1,2-a]pyrazin-8(7H)-one 34b, as a white solid (57 mg, 48%): LC-MS: Rt = 3.13 min, 99 Abs % @ 254 nm, [M]+ = 431.1; 1H NMR (600 MHz, DMSO-d6) δ 8.82 (s, 1H), 7.81–7.74 (m, 2H), 7.70–7.64 (m, 2H), 7.62 (dd, J = 5.9, 0.5 Hz, 1H), 7.51–7.42 (m, 5H), 5.18 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 152.8, 148.0, 147.9, 139.0, 138.2, 136.2, 135.1, 128.6, 128.4, 127.1, 123.6, 121.5, 120.1 (J = 256.6 Hz), 116.6, 107.4, 50.0. HRMS (ESI): m/z calcd for C20H14F3N4O4 [M + H]+, 431.0962; found, 431.0965.
7-((4′-Methyl-[1,1′-biphenyl]-4-yl)methyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one (34c)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (60 mg, 0.333 mmol, 1 equiv) was reacted with 4-(chloromethyl)-4′-methyl-1,1′-biphenyl 33c (79.4 mg, 0.366 mmol, 1.1 equiv) under microwave conditions at 90 °C for 20 min according to the general procedure method B. After the reaction was completed as analyzed by LC-MS, the mixture was diluted with distilled water (12 mL) and the precipitate was collected by filtration. The precipitate was then purified by recrystallization (hot slurry) in DCM/MeOH to yield the desired product, 7-((4′-methyl-[1,1′-biphenyl]-4-yl)methyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one 34c, as a white solid (73 mg, 61%): LC-MS: Rt = 3.13 min, 99 Abs % @ 254 nm, [M + H]+ = 431.1; 1H NMR (600 MHz, DMSO-d6) δ 8.82 (s, 1H), 7.65–7.59 (m, 3H), 7.56–7.51 (m, 2H), 7.49–7.41 (m, 3H), 7.26 (d, J = 7.9 Hz, 2H), 5.16 (s, 2H), 2.33 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 152.8, 148.0, 139.6, 136.8, 136.7, 135.3, 135.1, 129.5, 128.4, 126.6, 126.4, 123.5, 116.6, 107.4, 50.0, 20.6. HRMS (ESI): m/z calcd for C20H17N4O3 [M + H]+, 361.1295; found, 361.1295.
2-Nitro-7-((3′-(trifluoromethoxy)-[1,1′-biphenyl]-4-yl)methyl)imidazo[1,2-a]pyrazin-8(7H)-one (34d)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (55 mg, 0.305 mmol, 1 equiv) was reacted with 4-(chloromethyl)-3′-(trifluoromethoxy)-1,1′-biphenyl 33d (105 mg, 0.366 mmol, 1.2 equiv) under microwave conditions at 90 °C for 20 min according to the general procedure method B. After the reaction was completed as analyzed by LC-MS, the mixture was diluted with distilled water (11 mL) and the precipitate was collected by filtration. The precipitate was then purified by recrystallization using DCM/MeOH (hot slurry) to yield the desired product, 2-nitro-7-((3′-(trifluoromethoxy)-[1,1′-biphenyl]-4-yl)methyl)imidazo[1,2-a]pyrazin-8(7H)-on 34d, as a white solid (109 mg, 83%): LC-MS: Rt = 3.13 min, 99 Abs % @ 254 nm, [M + H]+ = 431.1;1H NMR (600 MHz, DMSO-d6) δ 8.82 (s, 1H), 7.71 (dd, J = 8.6, 2.3 Hz, 3H), 7.64–7.56 (m, 3H), 7.50–7.45 (m, 3H), 7.36 (ddt, J = 8.1, 2.3, 1.1 Hz, 1H), 5.19 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 13C NMR (151 MHz, DMSO) δ 152.8, 148.9, 148.0, 142.0, 137.9, 136.5, 135.1, 130.9, 128.5, 127.2, 125.8, 123.5, 120.1 (J = 255.6 Hz), 119.8, 119.2, 116.7, 107.4, 49.9. HRMS (ESI): m/z calcd for C20H14F3N4O4 [M + H]+, 431.0962; found, 431.0967.
2-Nitro-7-(4-(pyridin-3-yl)benzyl)imidazo[1,2-a]pyrazin-8(7H)-one (34e)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (50 mg, 0.278 mmol, 1 equiv) was reacted with 3-(4-chloromethyl)phenyl)pyridine 33e (67.8 mg, 0.333 mmol, 1.2 equiv) under microwave conditions at 100 °C for 45 min according to the general procedure method B. After the reaction was completed as analyzed by LC-MS, the mixture was diluted with distilled water (10 mL) and the precipitate was collected by filtration. The precipitate was then purified by recrystallization in DCM/MeOH to yield the desired product, 2-nitro-7-(4-(pyridin-3-yl)benzyl)imidazo[1,2-a]pyrazin-8(7H)-one 34e, as an orange solid (36 mg, 37%): LC-MS: Rt = 2.19 min, 99 Abs % @ 254 nm, [M + H]+ = 348.1; 1H NMR (600 MHz, DMSO-d6) δ 8.87 (d, J = 2.4 Hz, 1H), 8.82 (d, J = 0.6 Hz, 1H), 8.56 (dd, J = 4.7, 1.6 Hz, 1H), 8.05 (dddd, J = 7.9, 2.2, 1.6, 0.5 Hz, 1H), 7.77–7.70 (m, 2H), 7.62 (dd, J = 5.9, 0.6 Hz, 1H), 7.52–7.45 (m, 4H), 5.19 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 152.8, 148.5, 148.0, 147.6, 136.5, 136.5, 135.1, 135.1, 134.1, 128.5, 127.2, 123.8, 123.5, 116.7, 107.5, 50.0. HRMS (ESI): m/z calcd for C18H14N5O3 [M + H]+, 348.1091; found, 348.1107.
2-Nitro-7-((6-phenylpyridin-3-yl)methyl)imidazo[1,2-a]pyrazin-8(7H)-one (34f)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (55 mg, 0.305 mmol, 1 equiv) was reacted with 5-(chloromethyl)-2-phenylpyridine 33f (74.6 mg, 0.366 mmol, 1.2 equiv) under microwave conditions at 100 °C for 20 min according to the general procedure method B. After the reaction was completed as analyzed by LC-MS, the mixture was diluted with distilled water (11 mL) and the precipitate was collected by filtration. The precipitate was then purified by recrystallization (hot slurry) in DCM/MeOH to yield the desired product, 2-nitro-7-((6-phenylpyridin-3-yl)methyl)imidazo[1,2-a]pyrazin-8(7H)-one 34f, as a white solid (90 mg, 85%): LC-MS: Rt = 2.60 min, 99 Abs % @ 254 nm, [M + H]+ = 348.1; 1H NMR (600 MHz, DMSO-d6) δ 8.82 (s, 1H), 8.71 (d, J = 2.2 Hz, 1H), 8.06 (dd, J = 7.3, 1.7 Hz, 2H), 7.96 (d, J = 8.3 Hz, 1H), 7.87 (dd, J = 8.2, 2.3 Hz, 1H), 7.63 (d, J = 5.9 Hz, 1H), 7.53 (d, J = 5.9 Hz, 1H), 7.49 (t, J = 7.5 Hz, 2H), 7.43 (t, J = 7.4 Hz, 1H), 5.21 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 155.5, 152.9, 149.1, 148.0, 138.2, 136.8, 135.2, 130.9, 129.1, 128.8, 126.5, 123.4, 120.1, 116.6, 107.6, 48.0. HRMS (ESI): m/z calcd for C18H14N5O3 [M + H]+, 348.1091; found, 348.1094.
2-Nitro-7-((6-(4-(trifluoromethoxy)phenyl)pyridin-3-yl)methyl)imidazo[1,2-a]pyrazin-8(7H)-one (34g)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (45 mg, 0.250 mmol, 1 equiv) was reacted with 5-(chloromethyl)-2-(4-(trifluoromethoxy)phenyl)pyridine 33g (86.2 mg, 0.300 mmol, 1.2 equiv) under microwave conditions at 100 °C for 20 min according to the general procedure method B. After the reaction was completed as analyzed by LC-MS, the mixture was diluted with distilled water (9 mL) and the precipitate was collected by filtration. The precipitate was then purified by recrystallization (hot slurry) in DCM/MeOH to yield the desired product, 2-nitro-7-((6-(4-(trifluoromethoxy)phenyl)pyridin-3-yl)methyl)imidazo[1,2-a]pyrazin-8(7H)-one 34g, as a white solid (82 mg, 76%): LC-MS: Rt = 2.94 min, 99 Abs % @ 254 nm, [M + H]+ = 432.1; 1H NMR (600 MHz, DMSO-d6) δ 8.82 (s, 1H), 8.73 (d, J = 2.2 Hz, 1H), 8.22–8.16 (m, 2H), 8.00 (d, J = 8.2 Hz, 1H), 7.89 (dd, J = 8.2, 2.3 Hz, 1H), 7.63 (d, J = 5.9 Hz, 1H), 7.53 (d, J = 5.9 Hz, 1H), 7.47 (d, J = 8.4 Hz, 2H), 5.21 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 154.0, 152.9, 149.2, 149.0, 148.0, 137.4, 136.9, 135.2, 131.3, 128.5, 123.4, 121.2, 120.3, 120.1 (J = 256.5 Hz), 116.6, 107.6, 48.0. HRMS (ESI): m/z calcd for C19H13F3N5O4 [M + H]+, 432.0914; found, 432.0917.
2-Nitro-7-((6-(p-tolyl)pyridin-3-yl)methyl)imidazo[1,2-a]pyrazin-8(7H)-one (34h)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (60 mg, 0.333 mmol, 1 equiv) was reacted with 5-(chloromethyl)-2-(p-tolyl)pyridine 33h (87.02 mg, 0.400 mmol, 1.2 equiv) under microwave conditions at 90 °C for 20 min according to the general procedure method B. After the reaction was completed as analyzed by LC-MS, the mixture was diluted with distilled water (12 mL) and the precipitate was collected by filtration. The precipitate was then purified by recrystallization (hot slurry) in MeOH/DCM to yield the desired product, 2-nitro-7-((6-(p-tolyl)pyridin-3-yl)methyl)imidazo[1,2-a]pyrazin-8(7H)-one 34h, as a white solid (88 mg, 63%): LC-MS: Rt = 2.69 min, 99 Abs % @ 254 nm, [M + H]+ = 362.1; 1H NMR (600 MHz, DMSO-d6) δ 8.82 (s, 1H), 8.68 (d, J = 2.2 Hz, 1H), 7.96 (d, J = 8.1 Hz, 2H), 7.91 (d, J = 8.2 Hz, 1H), 7.83 (dd, J = 8.3, 2.4 Hz, 1H), 7.62 (d, J = 5.8 Hz, 1H), 7.52 (d, J = 5.9 Hz, 1H), 7.29 (d, J = 7.8 Hz, 2H), 5.19 (s, 2H), 2.35 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 155.5, 152.9, 149.1, 148.0, 138.7, 136.7, 135.5, 135.1, 130.5, 129.4, 126.4, 123.4, 119.6, 116.6, 107.6, 48.0, 20.8. HRMS (ESI): m/z calcd for C19H16N5O3 [M + H]+, 362.1248; found, 362.1253.
2-Nitro-7-((6-(3-(trifluoromethoxy)phenyl)pyridin-3-yl)methyl)imidazo[1,2-a]pyrazin-8(7H)-one (34i)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (60 mg, 0.333 mmol, 1 equiv) was reacted with 5-(chloromethyl)-2-(3-(trifluoromethoxy)phenyl)pyridine 33i (138.1 mg, 0.400 mmol, 1.2 equiv) under microwave conditions at 90 °C for 20 min according to the general procedure method B. After the reaction was completed as analyzed by LC–MS, the mixture was diluted with distilled water (12 mL) and the precipitate was collected by filtration. The precipitate was then purified by recrystallization (hot slurry) in MeOH/DCM to yield the desired product, 2-nitro-7-((6-(3-(trifluoromethoxy)phenyl)pyridin-3-yl)methyl)imidazo[1,2-a]pyrazin-8(7H)-one 34i as a white solid (109 mg, 76%): LC-MS: Rt = 2.95 min, 99 Abs % @ 254 nm, [M + H]+ = 432.1; 1H NMR (600 MHz, DMSO-d6) δ 8.82 (s, 1H), 8.74 (d, J = 2.2 Hz, 1H), 8.11 (dt, J = 8.0, 1.1 Hz, 1H), 8.08–8.02 (m, 2H), 7.90 (dd, J = 8.2, 2.3 Hz, 1H), 7.66–7.60 (m, 2H), 7.53 (d, J = 5.9 Hz, 1H), 7.44 (ddd, J = 8.1, 2.3, 1.1 Hz, 1H), 5.22 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 153.6, 152.9, 149.2, 149.0, 148.0, 140.5, 137.0, 135.2, 131.8, 130.9, 125.4, 123.4, 121.5, 120.5, 120.1 (J = 256.7 Hz), 118.7, 116.6, 107.6, 48.0. HRMS (ESI): m/z calcd for C19H13F3N5O4 [M + H]+, 432.0914; found, 432.0924.
2-Nitro-7-((5-(4-(trifluoromethoxy)phenyl)pyridin-2-yl)methyl)imidazo[1,2-a]pyrazin-8(7H)-one (34j)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (50 mg, 0.278 mmol, 1 equiv) was reacted with 2-(chloromethyl)-5-(4-(trifluoromethoxy)phenyl)pyridine 33j (95.8 mg, 0.333 mmol, 1.2 equiv) under microwave conditions at 100 °C for 15 min according to the general procedure method B. After the reaction was completed as analyzed by LC-MS, the mixture was diluted with distilled water (10 mL) and the precipitate was collected by filtration. The precipitate was then purified by recrystallization (hot slurry) in MeOH/DCM to yield the desired product, 2-nitro-7-((5-(4-(trifluoromethoxy)phenyl)pyridin-2-yl)methyl)imidazo[1,2-a]pyrazin-8(7H)-one 34j, as an off-white solid (88 mg, 74%): LC-MS: Rt = 2.90 min, 99 Abs % @ 254 nm, [M + H]+ = 432.1; 1H NMR (600 MHz, DMSO-d6) δ 8.85 (s, 1H), 8.83 (dd, J = 2.4, 0.8 Hz, 1H), 8.11 (dd, J = 8.1, 2.4 Hz, 1H), 7.88–7.81 (m, 2H), 7.64 (d, J = 5.9 Hz, 1H), 7.53–7.46 (m, 4H), 5.30 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 154.8, 152.8, 148.3 (J = 2.3 Hz), 148.0, 147.3, 136.1, 135.2, 135.1, 133.2, 128.9, 124.6, 121.9, 121.6, 120.1 (J = 256.4 Hz), 116.6, 107.0, 51.9. HRMS (ESI): m/z calcd for C19H13F3N5O4 [M + H]+ 432.0914; found, 432.0924.
2-Nitro-7-((5-(4-(trifluoromethoxy)phenyl)pyridin-3-yl)methyl)imidazo[1,2-a]pyrazin-8(7H)-one (34k)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (55 mg, 0.305 mmol, 1 equiv) was reacted with 3-(chloromethyl)-5-(4-(trifluoromethoxy)phenyl)pyridine 33k (105.4 mg, 0.366 mmol, 1.2 equiv) under microwave conditions at 100 °C for 40 min according to the general procedure method B. After the reaction was completed as analyzed by LC-MS, the mixture was diluted with distilled water (11 mL) and the precipitate was collected by filtration. The precipitate was then purified by recrystallization in DCM/MeOH to yield the desired product, 2-nitro-7-((5-(4-(trifluoromethoxy)phenyl)pyridin-3-yl)methyl)imidazo[1,2-a]pyrazin-8(7H)-one 34k, as an orange solid (50 mg, 38%): LC-MS: Rt = 2.87 min, 99 Abs % @ 254 nm, [M + H]+ = 432.1; 1H NMR (600 MHz, DMSO-d6) δ 8.85 (d, J = 1.6 Hz, 1H), 8.83 (s, 1H), 8.81 (s, 1H), 8.66–8.63 (m, 1H), 8.08 (d, J = 2.3 Hz, 1H), 7.86–7.80 (m, 2H), 7.61 (dd, J = 5.9, 0.7 Hz, 1H), 7.55–7.47 (m, 3H), 5.24 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 153.0, 148.4, 148.4, 147.9, 147.1, 136.1, 135.3, 134.1, 133.9, 132.3, 129.0, 123.4, 121.7, 120.1 (J = 256.5 Hz), 116.6, 107.6, 48.1. HRMS (ESI): m/z calcd for C19H13F3N5O4 [M + H]+, 432.0914; found, 432.0912.
2-Nitro-7-((5-(p-tolyl)pyridin-3-yl)methyl)imidazo[1,2-a]pyrazin-8(7H)-one (34l)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (50 mg, 0.278 mmol, 1 equiv) was reacted with 3-(chloromethyl)-5-(p-tolyl)pyridine 33l (72.5 mg, 0.333 mmol, 1.2 equiv) under microwave conditions at 110 °C for 20 min according to the general procedure method B. After the reaction was completed as analyzed by LC-MS, the mixture was diluted with distilled water (10 mL) and the precipitate was collected by filtration. The precipitate was then purified by recrystallization using DCM/MeOH (hot slurry) to yield the desired product, 2-nitro-7-((5-(p-tolyl)pyridin-3-yl)methyl)imidazo[1,2-a]pyrazin-8(7H)-one 34l, as a white solid (46 mg, 46%): LC-MS: Rt = 2.61 min, 99 Abs % @ 254 nm, [M]+ = 362.1; 1H NMR (600 MHz, DMSO-d6) δ 8.81 (s, 1H), 8.81 (d, J = 2.2 Hz, 1H), 8.58 (d, J = 2.1 Hz, 1H), 8.04 (t, J = 2.2 Hz, 1H), 7.64–7.58 (m, 3H), 7.53 (d, J = 5.9 Hz, 1H), 7.34–7.28 (m, 2H), 5.22 (s, 2H), 2.35 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 153.0, 147.9, 147.7, 146.8, 137.8, 135.3, 135.2, 133.7, 133.4, 132.2, 129.7, 126.8, 123.4, 116.6, 107.6, 48.1, 20.7. HRMS (ESI): m/z calcd for C19H16N5O3 [M + H]+, 362.1248; found, 362.1253.
2-Nitro-7-((5-(3-(trifluoromethoxy)phenyl)pyridin-3-yl)methyl)imidazo[1,2-a]pyrazin-8(7H)-one (34m)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (37 mg, 0.205 mmol, 1 equiv) was reacted with 3-(chloromethyl)-5-(3-(trifluoromethoxy)phenyl)pyridine 33m (70.9 mg, 0.247 mmol, 1.2 equiv) under microwave conditions at 100 °C for 20 min according to the general procedure method B. After the reaction was completed as analyzed by LC-MS, the mixture was diluted with distilled water (8 mL) and the precipitate was collected by filtration. The precipitate was then purified by recrystallization in hot DCM/MeOH to yield the desired product, 2-nitro-7-((5-(3-(trifluoromethoxy)phenyl)pyridin-3-yl)methyl)imidazo[1,2-a]pyrazin-8(7H)-one 34m, as a white solid (63 mg, 71%): LC-MS: Rt = 2.87 min, 99 Abs % @ 254 nm, [M + H]+ = 432.1; 1H NMR (600 MHz, DMSO-d6) δ 8.88 (d, J = 2.2 Hz, 1H), 8.81 (s, 1H), 8.65 (d, J = 2.0 Hz, 1H), 8.14 (t, J = 2.2 Hz, 1H), 7.77 (ddd, J = 7.8, 1.8, 0.9 Hz, 1H), 7.73 (d, J = 2.1 Hz, 1H), 7.65 (t, J = 8.0 Hz, 1H), 7.61 (d, J = 6.0 Hz, 1H), 7.53 (d, J = 6.0 Hz, 1H), 7.44 (ddt, J = 8.2, 2.4, 1.1 Hz, 1H), 5.24 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 153.0, 149.0, 148.7, 147.9, 147.2, 139.1, 135.3, 134.2, 133.8, 132.3, 131.1, 126.2, 123.4, 120.6, 120.1 (J = 256.4 Hz), 119.7, 116.6, 107.6, 48.1. HRMS (ESI): m/z calcd for C19H13F3N5O4 [M + H]+, 432.0914; found, 432.0915.
2-Nitro-7-(4-(thiazol-2-yl)benzyl)imidazo[1,2-a]pyrazin-8(7H)-one (34n)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (60 mg, 0.333 mmol, 1 equiv) was reacted with 2-(4-(chloromethyl)phenyl)thiazole 33n (83.8 mg, 0.400 mmol) under microwave conditions at 100 °C for 20 min according to the general procedure method B. After the reaction was completed as analyzed by LC-MS, the mixture was diluted with distilled water (12 mL) and the precipitate was collected by filtration. The precipitate was then purified by recrystallization (hot slurry) in DCM/MeOH to yield the desired product, 2-nitro-7-(4-(thiazol-2-yl)benzyl)imidazo[1,2-a]pyrazin-8(7H)-one 34n, as a yellow solid (78 mg, 67%): LC-MS: Rt = 2.66 min, 99 Abs % @ 254 nm, [M + H]+ = 354.1; 1H NMR (600 MHz, DMSO-d6) δ 8.83 (s, 1H), 7.95–7.92 (m, 2H), 7.92 (d, J = 3.2 Hz, 1H), 7.79 (d, J = 3.2 Hz, 1H), 7.63 (d, J = 5.9 Hz, 1H), 7.47 (dd, J = 8.7, 7.1 Hz, 3H), 5.19 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 166.6, 152.8, 148.0, 143.8, 138.5, 135.1, 132.5, 128.5, 126.4, 123.5, 120.6, 116.6, 107.5, 50.0. HRMS (ESI): m/z calcd for C16H12N5O3S [M + H]+, 354.0655; found, 354.0654.
2-Nitro-7-((2-phenylthiazol-5-yl)methyl)imidazo[1,2-a]pyrazin-8(7H)-one (34o)
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one 16 (55 mg, 0.305 mmol, 1 equiv) was reacted with 5-(chloromethyl)-2-phenylthiazole 33o (76.8 mg, 0.366 mmol, 1.2 equiv) under microwave conditions at 90 °C for 20 min according to the general procedure method B. Additional 5-(chloromethyl)-2-phenylthiazole (0.25 equiv) was added and reacted at 100 °C for 20 min to push the reaction to completion. The mixture was diluted with distilled water (11 mL), and the precipitate was collected by filtration. The precipitate was then purified by recrystallization (hot slurry) in DCM/MeOH to yield the desired product, 2-nitro-7-((2-phenylthiazol-5-yl)methyl)imidazo[1,2-a]pyrazin-8(7H)-one 34o as a cream color solid (84 mg, 78%): LC-MS: Rt = 2.73 min, 99 Abs % @ 254 nm, [M + H]+ = 354.1; 1H NMR (600 MHz, DMSO-d6) δ 8.82 (s, 1H), 8.00 (t, J = 0.8 Hz, 1H), 7.93–7.87 (m, 2H), 7.63 (d, J = 5.9 Hz, 1H), 7.54 (d, J = 5.9 Hz, 1H), 7.52–7.44 (m, 3H), 5.38 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 168.2, 152.5, 148.0, 143.9, 134.8, 133.1, 132.9, 130.4, 129.3, 126.0, 122.8, 116.8, 107.8, 43.1. HRMS (ESI): m/z calcd for C16H12N5O3S [M + H]+, 354.0655; found, 354.0656.
4-Nitro-N-(4-(piperidin-1-yl)benzyl)-1H-imidazole-2-carboxamide (36)
A mixture of acid chloride crude solid (300 mg, 1.71 mmol, 1 equiv) and triethylamine (476 μL, 3.42 mmol, 2 equiv) was reacted with (4-(piperidin-1-yl)phenyl)methanamine 35 (390 mg, 2.05 mmol, 1.2 equiv) according to general procedure method E. After completion of reaction, volatiles were removed in vacuo before purifying over C18-reversed phase silica (Grace Reveleris X2, A: 0.1% TFA in water, B: 0.1% TFA in ACN, 0–100% B) to give the final product 4-nitro-N-(4-(piperidin-1-yl)benzyl)-1H-imidazole-2-carboxamide 36 as a pink solid (494 mg, 88%). LC-MS: Rt = 2.23 min, 99 Abs % @ 254 nm, [M + H]+ = 330.2; 1H NMR (600 MHz, DMSO-d6) δ 9.43 (s, 1H), 8.46 (s, 1H), 7.37 (s, 4H), 4.41 (d, J = 6.2 Hz, 2H), 3.34 (s, 4H), 1.78 (s, 4H), 1.60 (s, 2H). 13C NMR (150 MHz, DMSO) δ 158.2, 158.0, 157.1, 146.7, 139.6, 130.0, 128.7, 121.5, 45.7, 41.7, 24.1, 8.6.
1-(2,2-Diethoxyethyl)-4-nitro-N-(4-(piperidin-1-yl)benzyl)-1H-imidazole-2-carboxamide (37)
4-Nitro-N-(4-(piperidin-1-yl)benzyl)-1H-imidazole-2-carboxamide 36 (200 mg, 0.607 mmol, 1 equiv) was reacted with bromoacetaldehyde diethyl acetal (137 μL, 0.911 mmol, 1.5 equiv) according to the general procedure method F. After completion of reaction, the mixture was diluted with distilled water (40 mL) and extracted with EtOAc (3 × 40 mL). The organic layer was collected, washed with brine, and dried with MgSO4 before removing the volatiles in vacuo. The crude product was purified over silica gel by MPLC (Biotage Isolera, 7–60% pet. spirit/EtOAc) to give the desired final product (95 mg, 35%): LC-MS: Rt = 2.73 min, 99 Abs % @ 254 nm, [M + Na]+ = 446.2.
2-Nitro-7-(4-(piperidin-1-yl)benzyl)imidazo[1,2-a]pyrazin-8(7H)-one (34p)
1-(2,2-Diethoxyethyl)-4-nitro-N-(4-(piperidin-1-yl)benzyl)-1H-imidazole-2-carboxamide 37 (95 mg, 0.213 mmol, 1 equiv) was reacted according to the general procedure method G. After completion of reaction, the volatiles were evaporated in vacuo followed by recrystallization using DCM/MeOH (hot slurry) to yield the desired product, 2-nitro-7-(4-(piperidin-1-yl)benzyl)imidazo[1,2-a]pyrazin-8(7H)-one 34p, as a light brown solid (47 mg, 63%): LC-MS: Rt = 2.28 min, 99 Abs % @ 254 nm, [M + H]+ = 354.1; 1H NMR (600 MHz, DMSO-d6) δ 8.83 (s, 1H), 7.62 (d, J = 5.9 Hz, 2H), 7.44 (d, J = 5.9 Hz, 4H), 5.13 (s, 2H), 3.39 (s, 4H), 1.84 (s, 4H), 1.60 (s, 2H). 13C NMR (150 MHz, DMSO) δ 152.8, 148.0, 135.1, 129.1, 123.5, 116.6, 107.4, 49.8, 23.6. HRMS (ESI): m/z calcd for C18H20N5O3 [M + H]+, 354.1561; found, 354.1565.
Minimum Inhibitory Concentration Assay: M. tuberculosis H37Rv
The minimum inhibitory concentration (MIC) study was performed using a resazurin reduction microplate assay as previously described.26 For normoxic conditions, the plates were incubated for 5 days at 37 °C in a humidified incubator prior to the addition of 30 μL of a 0.02% resazurin solution and 12.5 μL of 20% Tween-80 to each well. After 24 h incubation (37 °C), sample fluorescence was measured on a FLUOstar Omega fluorescent plate reader (BMG LABTECH) with an excitation wavelength of 530 nm and emission read at 590 nm. For hypoxic assays, the same method was used except assay plates were incubated for 5 days at 0.1% oxygen, and after addition of the resazurin solution, the fluorescence was measured after a prolonged incubation time of 48 h. Percent fluorescence relative to the positive control wells (H37Rv without compound) minus the negative control wells (without H37Rv) was plotted for the determination of the MIC (≤90% reduction in growth). The assays were performed in replicate on independent occasions (n = 3–6).
Antiparasitic Assay: T. b. brucei
Compounds were screened for activity against T. b. brucei (strain 427, BS427) using an established 384-well resazurin viability assay, as previously described.26,43 Serial compound concentrations were prepared in 100% DMSO and diluted 1:21 in DMEM. Five microliters of these dilutions were subsequently added to assay plates to give final compound concentrations ranging from a top final assay concentration of 80 or 40 μM to 4 × 10–3 or 2 × 10–3 μM, respectively. Plates were incubated for 48 h before the addition of a final concentration of 70 μM resazurin. The IC50 value was determined for compounds that exhibited a plateau of inhibition and were calculated from two independent experiments. IC50 values were calculated in GraphPad Prism 5 (GraphPad Software), using a sigmoidal dose–response analysis, with variable slope.
Antiparasitic Assay: T. cruzi
Compounds were screened for antitrypanosomal activity against T. cruzi using an established image-based assay, as previously described.44 Briefly, 3T3 fibroblasts (ATCC CCL92) were added at a concentration of 1 × 103 cells/well to 384-well Collagen coated plates (PerkinElmer). Following 24 h of incubation at 37 °C and 5% CO2, 10 μL of T. cruzi Tulahuen strain parasites at a multiplicity of 5:1 were added to wells. After 24 h incubation, non-infected parasites were washed from wells with a Bravo liquid handling device (Agilent Technologies). Compounds, pre diluted in milli-Q H2O, were added to wells to give final assay concentrations ranging in serial log dilutions from 73.3 or 36.6 μM to 1.8 × 10–4 or 9 × 10–5 μM, respectively. Plates were incubated for 48 h before fixing infected cells with paraformaldehyde and staining with Hoechst 3348 and HCS CellMask green (ThermoFisher Scientific). Plates were imaged on an Opera confocal imager (PerkinElmer) and analyzed to determine the number of host cells and infected cells using the assay language interface on the Opera in a developed image-based script.44 The control compounds were nifurtimox, benznidazole, puromycin, and posaconazole. The positive control for the parasite was 12 μM nifurtimox and for 3T3 host cells was 30 μM puromycin. A final concentration of 0.37% DMSO was the negative control. The activity of compounds was determined over two biological replicates. IC50 values were calculated in GraphPad Prism 5 (GraphPad Software), using a sigmoidal dose–response analysis, with variable slope.
Antiparasitic Assay: G. lamblia and E. histolytica
Compounds were screened for antiparasitic activity using an ATP-bioluminescence based assay, as previously described.45 All experiments were performed using trophozoites harvested during the logarithmic phase of growth. Compounds (0.5 μL) were added into 96-well microtiter plates followed by addition of 99.5 μL of trophozoites (5000 parasites). Final assay concentrations of compounds were in the range of 0.39–50 or 0.78–100 or 0.003–100 μM. Assay plates were incubated for 48 h at 37 °C in the GasPak EZ Anaerobe Gas Generating Pouch Systems (VWR) to maintain anaerobic conditions. Viable cell numbers were determined in triplicate using the CellTiter-Glo Luminescent Cell Viability Assay (Promega). Dose–response curves including IC50 calculation were processed using GraphPad Prism 5 (GraphPad Software).
Mammalian Cell Viability Assay
Similar to cytotoxicity assays previously described,46−48 human HEK293 cells (20 μL) were seeded at 5000 cells per well in 384-well plates. Cells were cultured in DMEM, supplemented with 10% fetal bovine serum, and 50 U/mL penicillin, and 50 mg/mL streptomycin, for 24 h at 37 °C, 5% CO2. Compounds prepared in DMSO were diluted a 1-in-2 series for eight points in DMEM, to a highest concentration of 100 μM, and were added (20 μL) to the cells. A tamoxifen dilution series, with a highest concentration of 200 μM, was used as a plate control for cell viability inhibition. The final concentration of DMSO in culture media was 0.5%, which showed no effect on cell growth. After 20 h incubation with the compounds, resazurin (11 μM final) was added into each well and then incubated at 37 °C, 5% CO2 for 3 h. The fluorescence intensity (FI) was read using an Infinite M1000 PRO (TECAN) with excitation/emission of 560/590 nm. Cell viability as a percent fluorescence relative to the positive control wells (wells with untreated cells) minus the negative control wells (wells without cells) was calculated. Data were analyzed with GraphPad Prism 6 software (GraphPad Software) to determine CC50 (concentration at 50% cell viability) using sigmoidal dose–response analysis, with variable fitting for top, bottom, and slope. Assays were performed in replicate on three independent occasions (n = 3). To determine the selectivity indices of active compounds, additional HEK293 cytotoxicity assay was carried out independently at a longer incubation time over 3 days (Supporting Information, S3).
Microsome Stability
Metabolic stability was determined using pooled human (HMMC-PL, Thermo Fisher Scientific) and mouse (CD-1) (MCMCPL, Thermo Fisher Scientific) liver microsomes. Test compound (3 μM, final DMSO concentration 0.2%) and liver microsomes (1 mg/mL) were mixed in 100 mM potassium phosphate buffer, pH 7.4 preincubated at 37 °C. The reaction was initiated by addition of NADPH solution (cofactor) in 0.1 M potassium phosphate buffer at a final NADPH concentration of 1 mM. The reaction was incubated in a shaking incubator at 37 °C, 150 rpm. Aliquots from the reaction mixture were withdrawn (t = 0, 10, 30, 60, and 120 min) and quenched by adding 3 X volume of ice-cold precipitating solution (270 μL) comprising a 0.5 μM carbutamide internal standard in acetonitrile:methanol:formic acid (1:1:0.001 v/v). Reaction samples were incubated at 4 °C for 30 min and centrifuged at 14,000g for 8 min, and the clear supernatant was analyzed by LC-MS/MS. The percentage of compound remaining at different times was calculated by comparing the peak area ratio of the parent compound (compound peak area/internal standard peak area) at the start of incubation (t = 0 min sample). All samples were tested in triplicate except for the control samples (without NADPH), matrix blank, and verapamil standard (time points = 0, 10, and 30 min). LC-MS/MS parameters are detailed in the Supporting Information.
Plasma Stability
Plasma stability studies were performed using human (HMPLNAHP, BioReclamationIVT) and mouse (CD-1) plasma (MSEPLNAHP, BioReclamationIVT) at five different time points. A solution of plasma and phosphate-buffered saline (PBS), pH 7.4 (50:50; v/v) were pre-heated at 37 °C for 30 min. The reaction was initiated by addition of the test compounds (3 μM, final DMSO concentration 1%), and the reaction was incubated in a shaking incubator at 37 °C, 150 rpm. Aliquots from the reaction mixture were withdrawn and processed as described for microsome stability assay. All samples were tested in triplicate, and eucatropine was used as a positive control.
Plasma Protein Binding
Plasma protein binding (PPB) was performed using an ultrafiltration method.58,59 Both human and mouse plasma were obtained commercially from BioReclamationIVT. Test compounds (5 μM) were incubated in 100% human plasma at 37 °C for 30 min. For unfiltered samples, an aliquot (50 μL) was removed, diluted with PBS (50 μL), and quenched with ice-cold precipitating solution comprising a 0.5 μM carbutamide MS internal standard in acetonitrile:methanol:formic acid (1:1:0.001). Samples were incubated at 4 °C for 30 min, then centrifuged at 14,000 × g for 8 min before the clear supernatant was transferred to a vial for LC–MS/MS analysis. For filtered samples, the plasma sample (250 μL) was filtered using Amicon Ultra-0.5 Centrifugal Filter Devices 30K NMWL (Merck Millipore) at 14,000g for 7 min, and then an aliquot (50 μL) was processed as described for unfiltered samples. The fraction of unbound compound was calculated by determining the concentration of the filtered sample and the concentration of unfiltered sample. All samples were tested in triplicate with sulfamethoxazole as a control.
Solubility Determination
Stock compound solution (20 mM in DMSO) was aliquoted into water, phosphate-buffered saline (PBS), pH 7.4 and 0.1 M HCl (pH 1), respectively, to a final concentration of 200 μM, 1% DMSO. After 24 h of incubation in a shaking incubator at room temperature, 130 rpm, samples were filtered using centrifuge filter tubes (Corning Costar Spin-X centrifuge tube filters, CLS8169) at 8000 rpm for 1 min. The filtrates were further diluted with acetonitrile (1:1, v/v) prior to analysis using LC-UV as detailed in the General Experimental Section. The solubility was determined based on the peak area at a UV absorbance of 254 nm, with reference to the standard calibration curve prepared from 20 mM DMSO stock. Compounds and standards (caffeine and pretomanid) were prepared in duplicate, and each sample was analyzed in duplicate by LC-UV.
Acknowledgments
We thank the Community for Open Antimicrobial Drug Discovery (CO-ADD)49 for performing MIC assays against the ESKAPE bacteria and fungal pathogens. The antimicrobial screening performed by CO-ADD was funded by the Wellcome Trust (UK; Strategic Funding Award: 104797/Z/14/Z) and The University of Queensland (Australia; Strategic Funding Award). We thank Emily Kennedy for assisting with the T. cruzi in vitro assays and Elouise Gaylord for assisting with the T. b. brucei in vitro assays. We also thank Angelo Frei for his help in graphic design. C.W.A. was supported by an Australian Government Research Training Program scholarship. M.L.S. was the recipient of a Griffith University Post-doctoral Fellowship (GUPF) award. A.D. was supported by the grants 1KL2TR001444, R21AI141210, R21AI133394, and R21AI146460 from the NIH. M.A.B. was supported in part by Wellcome Trust Strategic Award 104797/Z/14/Z. M.A.C. is a NHMRC Principal Research Fellow (APP1059354) and holds a fractional professorial research fellow appointment at The University of Queensland, with his remaining time as the CEO of Inflazome Ltd., a company developing drugs to address clinical unmet needs in inflammatory disease. Cell lines (bacteria, fungi, and mammalian) were sourced from the American Type Culture Collection (ATCC).
Glossary
Abbreviations Used
- SAR
structure–activity relationship
- TB
tuberculosis
- VL
visceral leishmaniasis
- PBS
phosphate-buffered saline
- MIC
minimum inhibitory concentration
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01372.
Author Present Address
# Present address: School of Science, Monash University Malaysia, 47500 Subang Jaya, Selangor, Malaysia (C.W.A.).
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Mackey T. K.; Liang B. A.; Cuomo R.; Hafen R.; Brouwer K. C.; Lee D. E. Emerging and reemerging neglected tropical diseases: a review of key characteristics, risk factors, and the policy and innovation environment. Clin. Microbiol. Rev. 2014, 27, 949–979. 10.1128/CMR.00045-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norrby S. R.; Nord C. E.; Finch R.; Lack of development of new antimicrobial drugs: a potential serious threat to public health. Lancet Infect. Dis. 2005, 5, 115–119. 10.1016/S1473-3099(05)70086-4. [DOI] [PubMed] [Google Scholar]
- Ang C. W.; Jarrad A. M.; Cooper M. A.; Blaskovich M. A. T. Nitroimidazoles: molecular fireworks that combat a broad spectrum of infectious diseases. J. Med. Chem. 2017, 60, 7636–7657. 10.1021/acs.jmedchem.7b00143. [DOI] [PubMed] [Google Scholar]
- Ryan N. J.; Lo J. H. Delamanid: first global approval. Drugs 2014, 74, 1041–1045. 10.1007/s40265-014-0241-5. [DOI] [PubMed] [Google Scholar]
- Keam S. J. Pretomanid: first approval. Drugs 2019, 79, 1797–1803. 10.1007/s40265-019-01207-9. [DOI] [PubMed] [Google Scholar]
- Matsumoto M.; Hashizume H.; Tomishige T.; Kawasaki M.; Tsubouchi H.; Sasaki H.; Shimokawa Y.; Komatsu M. OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med. 2006, 3, e466 10.1371/journal.pmed.0030466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover C. K.; Warrener P.; VanDevanter D. R.; Sherman D. R.; Arain T. M.; Langhorne M. H.; Anderson S. W.; Towell J. A.; Yuan Y.; McMurray D. N.; Kreiswirth B. N.; Barry C. E.; Baker W. R. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 2000, 405, 962–966. 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
- Singh R.; Manjunatha U.; Boshoff H. I. M.; Ha Y. H.; Niyomrattanakit P.; Ledwidge R.; Dowd C. S.; Lee I. Y.; Kim P.; Zhang L.; Kang S.; Keller T. H.; Jiricek J.; Barry C. E. III PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science 2008, 322, 1392–1395. 10.1126/science.1164571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xavier A. S.; Lakshmanan M. Delamanid: A new armor in combating drug-resistant tuberculosis. J. Pharmacol. Pharmacother. 2014, 5, 222–224. 10.4103/0976-500X.136121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://www.fda.gov/news-events/press-announcements/fda-approves-new-drug-treatment-resistant-forms-tuberculosis-affects-lungs; (Accessed September 29, 2019)
- Klug D. M.; Gelb M. H.; Pollastri M. P. Repurposing strategies for tropical disease drug discovery. Bioorg. Med. Chem. Lett. 2016, 26, 2569–2576. 10.1016/j.bmcl.2016.03.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson S.; Wyllie S.; Stojanovski L.; Perry M. R.; Simeons F. R. C.; Norval S.; Osuna-Cabello M.; De Rycker M.; Read K. D.; Fairlamb A. H. The R enantiomer of the antitubercular drug PA-824 as a potential oral treatment for visceral leishmaniasis. Antimicrob. Agents Chemother. 2013, 57, 4699–4706. 10.1128/AAC.00722-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson S.; Wyllie S.; Norval S.; Stojanovski L.; Simeons F. R. C.; Auer J. L.; Osuna-Cabello M.; Read K. D.; Fairlamb A. H. The anti-tubercular drug delamanid as a potential oral treatment for visceral leishmaniasis. Elife 2016, 5, e09744 10.7554/eLife.09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S.; Yardley V.; Vishwakarma P.; Shivahare R.; Sharma B.; Launay D.; Martin D.; Puri S. K. Nitroimidazo-oxazole compound DNDI-VL-2098: an orally effective preclinical drug candidate for the treatment of visceral leishmaniasis. J. Antimicrob. Chemother. 2015, 70, 518–527. 10.1093/jac/dku422. [DOI] [PubMed] [Google Scholar]
- http://www.dndi.org/diseases-projects/portfolio/completed-projects/vl-2098/; (Accessed December 14, 2016)
- Thompson A. M.; O’Connor P. D.; Marshall A. J.; Yardley V.; Maes L.; Gupta S.; Launay D.; Braillard S.; Chatelain E.; Franzblau S. G.; Wan B.; Wang Y.; Ma Z.; Cooper C. B.; Denny W. A. 7-Substituted 2-nitro-5,6-dihydroimidazo[2,1-b][1,3]oxazines: novel antitubercular agents lead to a new preclinical candidate for visceral leishmaniasis. J. Med. Chem. 2017, 60, 4212–4233. 10.1021/acs.jmedchem.7b00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A. M.; O’Connor P. D.; Marshall A. J.; Blaser A.; Yardley V.; Maes L.; Gupta S.; Launay D.; Braillard S.; Chatelain E.; Wan B.; Franzblau S. G.; Ma Z.; Cooper C. B.; Denny W. A. Development of (6R)-2-nitro-6-[4-(trifluoromethoxy)phenoxy]-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (DNDI-8219): a new lead for visceral leishmaniasis. J. Med. Chem. 2018, 61, 2329–2352. 10.1021/acs.jmedchem.7b01581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A. M.; Marshall A. J.; Maes L.; Yarlett N.; Bacchi C. J.; Gaukel E.; Wring S. A.; Launay D.; Braillard S.; Chatelain E.; Mowbray C. E.; Denny W. A. Assessment of a pretomanid analogue library for African trypanosomiasis: Hit-to-lead studies on 6-substituted 2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]thiazine 8-oxides. Bioorg. Med. Chem. Lett. 2018, 28, 207–213. 10.1016/j.bmcl.2017.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A. M.; Blaser A.; Palmer B. D.; Anderson R. F.; Shinde S. S.; Launay D.; Chatelain E.; Maes L.; Franzblau S. G.; Wan B.; Wang Y.; Ma Z.; Denny W. A. 6-Nitro-2,3-dihydroimidazo[2,1-b][1,3]thiazoles: facile synthesis and comparative appraisal against tuberculosis and neglected tropical diseases. Bioorg. Med. Chem. Lett. 2017, 27, 2583–2589. 10.1016/j.bmcl.2017.03.069. [DOI] [PubMed] [Google Scholar]
- Szumowski J. D.; Lynch J. B. Profile of delamanid for the treatment of multidrug-resistant tuberculosis. Drug Des., Dev. Ther. 2015, 9, 677–682. 10.2147/DDDT.S60923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton A. M.; Cho S.; Yang T. J.; Kim Y.; Wang Y.; Lu Y.; Wang B.; Xu J.; Mdluli K.; Ma Z.; Franzblau S. G. In vitro and in vivo activities of the nitroimidazole TBA-354 against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 136–144. 10.1128/AAC.03823-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessem E.The tuberculosis treatment pipeline: activity; but no answers http://pipelinereport.org/2016/tb-treatment.; (Accessed November 23, 2016)
- Palmer B. D.; Sutherland H. S.; Blaser A.; Kmentova I.; Franzblau S. G.; Wan B.; Wang Y.; Ma Z.; Denny W. A.; Thompson A. M. Synthesis and structure–activity relationships for extended side chain analogues of the antitubercular drug (6S)-2-nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (PA-824). J. Med. Chem. 2015, 58, 3036–3059. 10.1021/jm501608q. [DOI] [PubMed] [Google Scholar]
- Yempalla K. R.; Munagala G.; Singh S.; Kour G.; Sharma S.; Chib R.; Kumar S.; Wazir P.; Singh G. D.; Raina S.; Bharate S. S.; Khan I. A.; Vishwakarma R. A.; Singh P. P. Synthesis and biological evaluation of polar functionalities containing nitrodihydroimidazooxazoles as anti-TB agents. ACS Med. Chem. Lett. 2015, 6, 1059–1064. 10.1021/acsmedchemlett.5b00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A. M.; O’Connor P. D.; Blaser A.; Yardley V.; Maes L.; Gupta S.; Launay D.; Martin D.; Franzblau S. G.; Wan B.; Wang Y.; Ma Z.; Denny W. A. Repositioning antitubercular 6-nitro-2,3-dihydroimidazo[2,1-b][1,3]oxazoles for neglected tropical diseases: structure–activity studies on a preclinical candidate for visceral leishmaniasis. J. Med. Chem. 2016, 59, 2530–2550. 10.1021/acs.jmedchem.5b01699. [DOI] [PubMed] [Google Scholar]
- Jarrad A. M.; Ang C. W.; Debnath A.; Hahn H. J.; Woods K.; Tan L.; Sykes M. L.; Jones A. J.; Pelingon R.; Butler M. S.; Avery V. M.; West N. P.; Karoli T.; Blaskovich M. A. T.; Cooper M. A. Design, synthesis, and biological evaluation of 2-nitroimidazopyrazin-one/–es with antitubercular and antiparasitic activity. J. Med. Chem. 2018, 61, 11349–11371. 10.1021/acs.jmedchem.8b01578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrad A. M.; Debnath A.; Miyamoto Y.; Hansford K. A.; Pelingon R.; Butler M. S.; Bains T.; Karoli T.; Blaskovich M. A. T.; Eckmann L.; Cooper M. A. Nitroimidazole carboxamides as antiparasitic agents targeting Giardia lamblia, Entamoeba histolytica and Trichomonas vaginalis. Eur. J. Med. Chem. 2016, 120, 353–362. 10.1016/j.ejmech.2016.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Więckowska A.; Kołaczkowski M.; Bucki A.; Godyń J.; Marcinkowska M.; Więckowski K.; Zaręba P.; Siwek A.; Kazek G.; Głuch-Lutwin M.; Mierzejewski P.; Bienkowski P.; Sienkiewicz-Jarosz H.; Knez D.; Wichur T.; Gobec S.; Malawska B. Novel multi-target-directed ligands for Alzheimer’s disease: combining cholinesterase inhibitors and 5-HT6 receptor antagonists. Design, synthesis and biological evaluation. Eur. J. Med. Chem. 2016, 124, 63–81. 10.1016/j.ejmech.2016.08.016. [DOI] [PubMed] [Google Scholar]
- Hasvold L.; Hexamer L.; Li G.; Lin N.-H.; Sham H.; Sullivan G.; Wang L.; Xia P.. Heterocyclic kinase inhibitors. US20,040,254,159A1, 2004.
- Albrecht B. K.; Audia J. E.; Cook A. S.; Dakin L. A.; Duplessis M.; Gehling V. S.; Harmange J.-C.; Nasveschuk C. G.; Vaswani R. G.. Modulators of methyl modifying enzymes, compositions and uses thereof. US9,085,583B2, 2014.
- Bi F.; Guo L.; Wang Y.; Venter H.; Semple S. J.; Liu F.; Ma S. Design, synthesis and biological activity evaluation of novel 2,6-difluorobenzamide derivatives through FtsZ inhibition. Bioorg. Med. Chem. Lett. 2017, 27, 958–962. 10.1016/j.bmcl.2016.12.081. [DOI] [PubMed] [Google Scholar]
- Leung S. C.; Gibbons P.; Amewu R.; Nixon G. L.; Pidathala C.; Hong W. D.; Pacorel B.; Berry N. G.; Sharma R.; Stocks P. A.; Srivastava A.; Shone A. E.; Charoensutthivarakul S.; Taylor L.; Berger O.; Mbekeani A.; Hill A.; Fisher N. E.; Warman A. J.; Biagini G. A.; Ward S. A.; O’Neill P. M. Identification, design and biological evaluation of heterocyclic quinolones targeting Plasmodium falciparum type II NADH:quinone oxidoreductase (PfNDH2). J. Med. Chem. 2012, 55, 1844–1857. 10.1021/jm201184h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaser A.; Palmer B. D.; Sutherland H. S.; Kmentova I.; Franzblau S. G.; Wan B.; Wang Y.; Ma Z.; Thompson A. M.; Denny W. A. Structure–activity relationships for amide-, carbamate-, and urea-linked analogues of the tuberculosis drug (6S)-2-nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (PA-824). J. Med. Chem. 2012, 55, 312–326. 10.1021/jm2012276. [DOI] [PubMed] [Google Scholar]
- Lu X.; Tang J.; Cui S.; Wan B.; Franzblauc S. G.; Zhang T.; Zhang X.; Ding K. Pyrazolo[1,5-a]pyridine-3-carboxamide hybrids: design, synthesis and evaluation of anti-tubercular activity. Eur. J. Med. Chem. 2017, 125, 41–48. 10.1016/j.ejmech.2016.09.030. [DOI] [PubMed] [Google Scholar]
- Kataoka M.; Fukahori M.; Ikemura A.; Kubota A.; Higashino H.; Sakuma S.; Yamashita S. Effects of gastric pH on oral drug absorption: in vitro assessment using a dissolution/permeation system reflecting the gastric dissolution process. Eur. J. Pharm. Biopharm. 2016, 101, 103–111. 10.1016/j.ejpb.2016.02.002. [DOI] [PubMed] [Google Scholar]
- Piccaro G.; Poce G.; Biava M.; Giannoni F.; Fattorini L. Activity of lipophilic and hydrophilic drugs against dormant and replicating Mycobacterium tuberculosis. J. Antibiot. 2015, 68, 711–714. 10.1038/ja.2015.52. [DOI] [PubMed] [Google Scholar]
- Cellitti S. E.; Shaffer J.; Jones D. H.; Mukherjee T.; Gurumurthy M.; Bursulaya B.; Boshoff H. I.; Choi I.; Nayyar A.; Lee Y. S.; Cherian J.; Niyomrattanakit P.; Dick T.; Manjunatha U. H.; Barry C. E. III; Spraggon G.; Geierstanger B. H. Structure of Ddn, the deazaflavin-dependent nitroreductase from Mycobacterium tuberculosis involved in bioreductive activation of PA-824. Structure 2012, 20, 101–112. 10.1016/j.str.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer B. D.; Thompson A. M.; Sutherland H. S.; Blaser A.; Kmentova I.; Franzblau S. G.; Wan B.; Wang Y.; Ma Z.; Denny W. A. Synthesis and structure–activity studies of biphenyl analogues of the tuberculosis drug (6S)-2-nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (PA-824). J. Med. Chem. 2010, 53, 282–294. 10.1021/jm901207n. [DOI] [PubMed] [Google Scholar]
- Walker M. A. Novel tactics for designing water-soluble molecules in drug discovery. Expert Opin. Drug Discovery 2014, 9, 1421–1433. 10.1517/17460441.2014.960839. [DOI] [PubMed] [Google Scholar]
- Ishikawa M.; Hashimoto Y. Improvement in aqueous solubility in small molecule drug discovery programs by disruption of molecular planarity and symmetry. J. Med. Chem. 2011, 54, 1539–1554. 10.1021/jm101356p. [DOI] [PubMed] [Google Scholar]
- Liu X.; Wright M.; Hop C. E. C. A. Rational use of plasma protein and tissue binding data in drug design. J. Med. Chem. 2014, 57, 8238–8248. 10.1021/jm5007935. [DOI] [PubMed] [Google Scholar]
- Thompson A. M.; Sutherland H. S.; Palmer B. D.; Kmentova I.; Blaser A.; Franzblau S. G.; Wan B.; Wang Y.; Ma Z.; Denny W. A. Synthesis and structure-activity relationships of varied ether linker analogues of the antitubercular drug (6S)-2-nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1, 3]oxazine (PA-824). J. Med. Chem. 2011, 54, 6563–6585. 10.1021/jm200377r. [DOI] [PubMed] [Google Scholar]
- Sykes M. L.; Avery V. M. Development of an Alamar BlueTM viability assay in 384-well format for high throughput whole cell screening of Trypanosoma brucei brucei bloodstream form strain 427. Am. J. Trop. Med. Hyg. 2009, 81, 665–674. 10.4269/ajtmh.2009.09-0015. [DOI] [PubMed] [Google Scholar]
- Sykes M. L.; Avery V. M. Development and application of a sensitive, phenotypic, high-throughput image-based assay to identify compound activity against Trypanosoma cruzi amastigotes. Int. J. Parasitol. 2015, 5, 215–228. 10.1016/j.ijpddr.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrad A. M.; Karoli T.; Debnath A.; Tay C. Y.; Huang J. X.; Kaeslin G.; Elliott A. G.; Miyamoto Y.; Ramu S.; Kavanagh A. M.; Zuegg J.; Eckmann L.; Blaskovich M. A. T.; Cooper M. A. Metronidazole-triazole conjugates: activity against Clostridium difficile and parasites. Eur. J. Med. Chem. 2015, 101, 96–102. 10.1016/j.ejmech.2015.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien J.; Wilson I.; Orton T.; Pognan F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. 10.1046/j.1432-1327.2000.01606.x. [DOI] [PubMed] [Google Scholar]
- McMillian M. K.; Li L.; Parker J. B.; Patel L.; Zhong Z.; Gunnett J. W.; Powers W. J.; Johnson M. D. An improved resazurin-based cytotoxicity assay for hepatic cells. Cell Biol. Toxicol. 2002, 18, 157–173. 10.1023/A:1015559603643. [DOI] [PubMed] [Google Scholar]
- Grace J. L.; Amado M.; Reid J. C.; Elliott A. G.; Landersdorfer C. B.; Truong N. P.; Kempe K.; Cooper M. A.; Davis T. P.; Montembault V.; Pascual S.; Fontaine L.; Velkov T.; Quinn J. F.; Whittaker M. R. An optimised Cu(0)-RDRP approach for the synthesis of lipidated oligomeric vinyl azlactone: toward a versatile antimicrobial materials screening platform. J. Mater. Chem. B 2019, 7, 6796–6809. 10.1039/C9TB01624D. [DOI] [PubMed] [Google Scholar]
- Blaskovich M. A. T.; Zuegg J.; Elliott A. G.; Cooper M. A. Helping chemists discover new antibiotics. ACS Infect. Dis. 2015, 1, 285–287. 10.1021/acsinfecdis.5b00044. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.