

Abstract

Lythrum salicaria herb (LSH) was applied in diarrhea therapy since ancient times. Despite empirically referenced therapeutic effects, the bioactivity mechanisms and chemical constituents responsible for pharmacological activity remain not fully resolved. Taking into consideration the historical use of LSH in treatment of diarrhea in humans and farm animals, the aim of the study was to examine in vitro the influence of LSH and its C-glycosylic ellagitannins on processes associated with maintaining intestinal epithelium integrity and enteropathogenic Escherichia coli (EPEC) growth and adhesion. LSH was not only inhibiting EPEC growth in a concentration dependent manner but also its adhesion to IPEC-J2 intestinal epithelial cell monolayers. Inhibitory activity toward EPEC growth was additionally confirmed ex vivo in distal colon samples of postweaning piglets. LSH and its dominating C-glycosylic ellagitannins, castalagin (1), vescalagin (2), and salicarinins A (3) and B (4) were stimulating IPEC-J2 monolayer formation by enhancing claudin 4 production. Parallelly tested gut microbiota metabolites of LSH ellagitannins, urolithin C (5), urolithin A (6), and its glucuronides (7) were inactive. The activities of LSH and the isolated ellagitannins support its purported antidiarrheal properties and indicate potential mechanisms responsible for its beneficial influence on the intestinal epithelium.
Lythraceae, Lythrum salicaria L. (purple loosestrife), is a medicinal plant that was traditionally applied in therapy of diarrhea and dysentery since ancient times until the early 20th century. It was used both in traditional and official medical treatment of diarrhea in humans and farm animals.1,2 Its antibacterial activities were examined in the mid 20th century, long before the etiology and pathophysiology of intestinal infections were fully recognized.3,4 Its effectiveness may be validated from historical sources reporting its use during dysentery epidemies in the 18th and 19th centuries and in military hospitals during World War I.5−8 Despite empirical therapeutic effects, the mechanisms of action and chemical constituents responsible for pharmacological activity remain not fully resolved. The main specialized metabolites of Lythrum salicaria herb belong to two groups: C-glycosylic ellagitannins (CGEs) (vescalagin, castalagin, and characteristic for L. salicaria, dimeric salicarinins A, B, C) and the C-glycosylic flavonoids (vitexin, orientin, and isoorientin).9 The high performance liquid chromatography with charged aerosol detection (UPLC–CAD) analysis have shown that the former group significantly dominates in polar extracts.10
Diarrhea among farm animals is one of the most important health conditions, where new preventive and therapeutic strategies are urgently needed. Postweaning diarrhea in piglets is a disease which strongly affects pig production. Numerous stress factors associated with weaning, including social, environmental, and dietary changes, alter the homeostasis of the intestinal epithelium and microbiota, rendering young piglets more inclined to gastrointestinal tract infections. These changes include disrupted intestinal structure, reduced digestive and absorptive capacity, damaged intestinal barrier, loss of microbial diversity, and unbalanced intestinal immune homeostasis.11 The etiology of postweaning diarrhea is multifactorial, although it is commonly associated with the infection with enteropathogenic Escherichia coli (EPEC). Piglets ingest EPEC found in their environment, which originates from the gut of animals with EPEC diarrhea, or subclinical carrier farm animals.12 EPEC adhere to the piglet’s small intestinal epithelium using fimbriae and pili and colonize the surface of the intestinal tract. The release of enterotoxins causes an increase of water and electrolyte secretion into the intestinal lumen and alters the functions of enterocytes by increasing secretion of water and reducing absorption of small intestinal epithelial cells.13
The rising incidence of antibiotic-resistant bacterial infections prompted the European Union in 2006 to introduce a ban on growth-promoting antimicrobials (Regulation 1831/2003/EC). Nevertheless, the overall use of therapeutic antimicrobials to treat infections in farm livestock worldwide remained high. Owing to limitations on antimicrobial use in veterinary medicine and serious concerns of antibiotic resistance development and its transfer to human pathogens, antidiarrheal agents based on novel mechanistic concepts should be developed. Recent advances in understanding EPEC microbiology, pathogenesis, ecology, and interactions with the host become essential for development of novel preventive and/or therapeutic approaches to fight EPEC-induced diarrhea.14 The alternative approaches that could contribute to optimal gut health are a prerequisite to minimize the prophylactic use of antimicrobials and could lead to improved productivity of the health and welfare of pigs and sustainability of pig production.
In recent years, much research was done on alternative agents and their potential use in the treatment of diarrhea in piglets.15,16 However, the important aspect associated with the wide application of natural products in livestock as feed additives is the cost of its production and availability, which should be suitable to cover the demands of farm production. L. salicaria, which widely grows in its natural state in Europe and is recognized as an invasive plant in North America17 fulfills these criteria of availability, which makes its eventual reintroduction as feed additive feasible and associated with additional economic and ecological benefits.
Taking into consideration the historical use of L. salicaria herb in treatment of diarrhea in humans and farm animals, this plant material can be considered as a potential candidate for development of a new feed additive, which could be applied in prevention or therapy of diarrhea in postweaning piglets. Thus, the aim of the study was to examine in vitro the influence of L. salicaria extract and dominating CGEs on processes associated with maintaining intestinal epithelium integrity as well as EPEC growth and adhesion to epithelial cells. Because our previous study has shown metabolism of L. salicaria CGEs to urolithins by distal colon microbiota of postweaning piglets,18 these metabolites were included in the studied biological activity studies.
Results and Discussion
Infection with EPEC takes place in the small intestine.19 In our previous studies conducted on gut microbiota of healthy postweaning piglets, we have shown that ellagitannins present in L. salicaria extract (LSH) are stable in the gut microbiota of the jejunum and ileum and that their metabolism begins in the cecum and continues to the distal colon.18 Thus, it can be foreseen that, following the oral ingestion of LSH, the ellagitannins will not be decomposed at this level of the gastrointestinal tract and will potentially interact with the small intestine epithelium as well as with pathogenic bacteria. However, it has to be considered that the ellagitannins of LSH were shown to be metabolized to urolithins by distal colon microbiota of piglets. Previous studies on pigs and humans indicate that urolithins are bioavailable but undergo intensive enterohepatic circulation, being released with bile, which results in their presence in the small intestine in the free and conjugated form.20,21 Taking into consideration the metabolic fate of LSH, we have included in the study not only LSH and known ellagitannins castalagin (1), vescalagin (2), salicarinin A (3), and salicarinin B (4) but also the representative gut microbiota metabolites, urolithin C (5) and urolithin A (6) together with a mixture of its phase II conjugates, urolithins A 3-O-glucuronide and 8-O-glucuronide (7). Experiments using the different extraction procedures indicated that only the aqueous extract prepared at 40 °C contained 1, 2, 3, and 4, while extraction at 80 °C led to degradation of dimeric ellagitannins 3 and 4. Owing to the high polarity of the ellagitannins, they were present only in small amounts in the 70% EtOH extract (Figure S7, Supporting Information).
Inhibition of E. coli growth by LSH was determined for two strains, the enteropathogenic strain E. coli Abbotstown O147:K89:K88 (ECA) and an E. coli DSM 2840 control strain (DSM). The growth inhibition of both strains, seen as an increase in lag time and an increase in doubling time, were observed (Table 1, Figure S3A, Supporting Information). The inhibition was concentration-dependent at a range of 20–2000 μg/mL. There were some differences in the level of inhibition between the two E. coli strains at certain concentrations, but for both strains, the minimal inhibitory concentration (MIC) was determined at the level of 2000 μg/mL. Similar results of inhibition growth were observed for 1 (Table 1, Figure S3A, Supporting Information) with an MIC value of 500 μM associated with high turbidity of tested samples. To determine whether in samples of 2000 μg/mL of LSH and 500 μM of 1E. coli growth was observed, after a 24 h measurement, 100 μL of well content was seeded on a brain heart infusion (BHI) agar plate and incubated at 37 °C for 48 h. No growth of E. coli colonies was observed. The experiments conducted for gut microbiota metabolites 5, 6, and 7 showed no inhibition of bacterial growth at concentration range 20–500 μM (Figure S3B, Supporting Information). In contrast to tested LSH and 1, significant differences in susceptibility between ECA and DSM strains toward the positive control (ciprofloxacin) were observed (Figure S3C, Supporting Information), the former being far more resistant to chemotherapeutics with an MIC of 100 ng/mL.
Table 1. Growth Parameters: Doubling Time (G) and Lag Time (λ) for E. coli Growth Curvesa.
| G [min] | SD | μmax | λ [min] | d | SD | ||
|---|---|---|---|---|---|---|---|
| E. coli IMT 0147:K89:K88 | H2O | 29.2 | 0.6 | 0.023 72 | 302 | 0 | 3 |
| LSH 20 μg/mL | 33.8 | 0.6 | 0.020 52 | 297 | –5 | 6 | |
| LSH 50 μg/mL | 40.7 | 0.5 | 0.017 02 | 333 | 32 | 3 | |
| LSH 100 μg/mL | 42.8 | 1.8 | 0.016 21 | 400 | 98 | 9 | |
| LSH 200 μg/mL | 41.1 | 0.6 | 0.016 86 | 552 | 250 | 13 | |
| LSH 500 μg/mL | 37.0 | 1.3 | 0.018 74 | 717 | 415 | 10 | |
| LSH 1000 μg/mL | 40.8 | 1.6 | 0.017 00 | 808 | 507 | 8 | |
| LSH 2000 μg/mL | >1440 | >1440 | |||||
| DMSO 0.5% | 34.1 | 0.5 | 0.020 34 | 300 | 0 | 0 | |
| 1 5 μM | 36.8 | 0.4 | 0.018 86 | 317 | 17 | 3 | |
| 1 10 μM | 34.3 | 1.0 | 0.020 23 | 310 | 10 | 9 | |
| 1 20 μM | 45.6 | 2.0 | 0.015 21 | 380 | 80 | 10 | |
| 1 50 μM | 45.3 | 2.2 | 0.015 29 | 502 | 202 | 8 | |
| 1 100 μM | 36.6 | 1.5 | 0.018 92 | 647 | 347 | 33 | |
| 1 200 μM | 35.9 | 1.5 | 0.019 33 | 722 | 422 | 13 | |
| 1 500 μM | >1440 | >1440 | |||||
| E. coli DSM 2840 | H2O | 32.2 | 1.7 | 0.021 55 | 320 | 0 | 0 |
| LSH 20 μg/mL | 34.6 | 2.5 | 0.020 05 | 312 | –8 | 8 | |
| LSH 50 μg/mL | 37.3 | 1.2 | 0.018 60 | 332 | 12 | 3 | |
| LSH 100 μg/mL | 38.9 | 1.4 | 0.017 82 | 373 | 53 | 6 | |
| LSH 200 μg/mL | 40.3 | 0.6 | 0.017 19 | 447 | 127 | 8 | |
| LSH 500 μg/mL | 54.1 | 3.5 | 0.012 80 | 593 | 273 | 8 | |
| LSH 1000 μg/mL | 59.9 | 3.5 | 0.011 58 | 890 | 570 | 40 | |
| LSH 2000 μg/mL | >1440 | >1440 | |||||
| DMSO 0.5% | 31.6 | 0.8 | 0.021 90 | 312 | 0 | 3 | |
| 1 5 μM | 33.4 | 1.3 | 0.020 73 | 322 | 10 | 3 | |
| 1 10 μM | 34.9 | 1.5 | 0.019 87 | 320 | 8 | 5 | |
| 1 20 μM | 38.1 | 1.1 | 0.018 19 | 347 | 35 | 6 | |
| 1 50 μM | 43.0 | 1.3 | 0.016 11 | 422 | 110 | 8 | |
| 1 100 μM | 42.9 | 0.4 | 0.016 16 | 515 | 203 | 5 | |
| 1 200 μM | 56.8 | 11.8 | 0.012 20 | 870 | 558 | 50 | |
| 1 500 μM | >1440 | >1440 |
IMT O147:K89:K88 (Abbottstown) and control strain DSM 2840 were incubated with LSH or castalagin (1). The data are representative for three independent experiments assayed in triplicate. Growth curves are provided in Figure S2A, Supporting Information.
EPEC spreads among farm animals via fecal–oral transmission.22,23 To check if LSH remains active against EPEC growth in the presence of gut microbiota, a model of ex vivo distal colon microbiota infection with the ECA strain was developed. In order to selectively monitor the strain growth, the DNA coding fae fimbriae was quantified using qPCR at the chosen time points. First, the model was developed and validated (Figure S4, Supporting Information). It was shown that the growth curves of ECA are dependent on the size of initial inoculum and no bacteria carrying fae gene are initially present in piglet feces. For further studies, the 104 CFU/mL inoculum was chosen. The studies on pure ECA cultures confirmed the concentration-dependent inhibition of bacteria growth observed in an experiment using the turbidimetric method; however, at 2000 μg/mL LSH, although significantly attenuated, growth was still observed (Figure 1). As expected, the ECA growth in fecal samples was significantly weakened; at the same time, the inhibitory activity of LSH was stronger, and at 2000 μg/mL, no fae gene was detected. This implicates the complete eradication of the inoculated E. coli strain from fecal samples (Figure 1).
Figure 1.

Growth curves of enteropathogenic E. coli strain, IMT O147:K89:K88, incubated with LSH in piglet fecal samples monitored by qPCR determination of fimbrial fae gene copies. pc = pure E. coli culture (BHI), and M = fecal sample. The data are representative for two independent experiments assayed in duplicate.
The cytotoxicity tests of LSH and 1 showed no negative influence on viability of IPEC-J2 (Figure S5, Supporting Information). The cell monolayers were incubated with LSH or 1 for 24 h and washed prior to infection with the ECA strain, to avoid direct impact of tested principles on bacterial growth. Treatment of IPEC-J2 cells with LSH at the concentration of 100 μg/mL resulted in statistically significant inhibition of EPEC adhesion by 20.4 ± 6.3%. A similar effect was observed for 1, which at a concentration of 100 μM inhibited EPEC adhesion by 37.7 ± 2.4% (Figure 2). Metabolites 5, 6, and 7 were inactive (data not shown).
Figure 2.

Influence of LSH and castalagin (1) on the adhesion of the enteropathogenic E. coli strain O147:K89:K88 to IPEC-J2 cell monolayers. Data are expressed as mean ± SD of three separate experiments conducted in triplicate. Statistical significance *p < 0.05, **p < 0.01 decrease versus PBS control (Dunnett’s posthoc test).
Transepithelial electrical resistance (TEER) measurement is used to assess the barrier function of epithelial cell monolayers cultured on inserts. TEER measurement is applied to determine permeability changes, being a consequence of tight junction protein production/degradation dynamics and the membrane physical perturbations induced by pharmacological or mechanistic agents.24 The IPEC-J2 cell monolayers were treated with LSH or pure compounds when the TEER values reached 1 kΩ cm2. The addition of culture medium containing LSH resulted in significant stimulation of monolayer formation determined as increased TEER during subsequent 6 day treatment including 2 times medium exchange (Figure 3A). Similar effects were observed for isolated ellagitannins, monomeric 1 and 2 and dimeric 3 and 4, at a concentration of 20 μM (Figures 3B,C). At the end of the experiment, the cells were lysed and the amount of TJ proteins was determined using Western blot. An increase in claudin 4 and ZO-1 protein production was observed for LSH, while only dimeric ellagitannins 3 and 4 showed activity toward claudin 4 induction (Figures 3D–F, Table S1, Supporting Information). Moreover, Spearman correlation analysis performed for each examined well indicated statistically significant correlation between determined TEER values and claudin 4 levels (Table S2, Supporting Information). The qPCR analysis conducted for cells treated with LSH at 500 μg/mL also indicated a significant increase of mRNA expression for claudin 4 and ZO-1 (Figure 4). Metabolites 5, 6, and 7 had no effect on IPEC-J2 cell monolayer development (Figure S6, Supporting Information).
Figure 3.
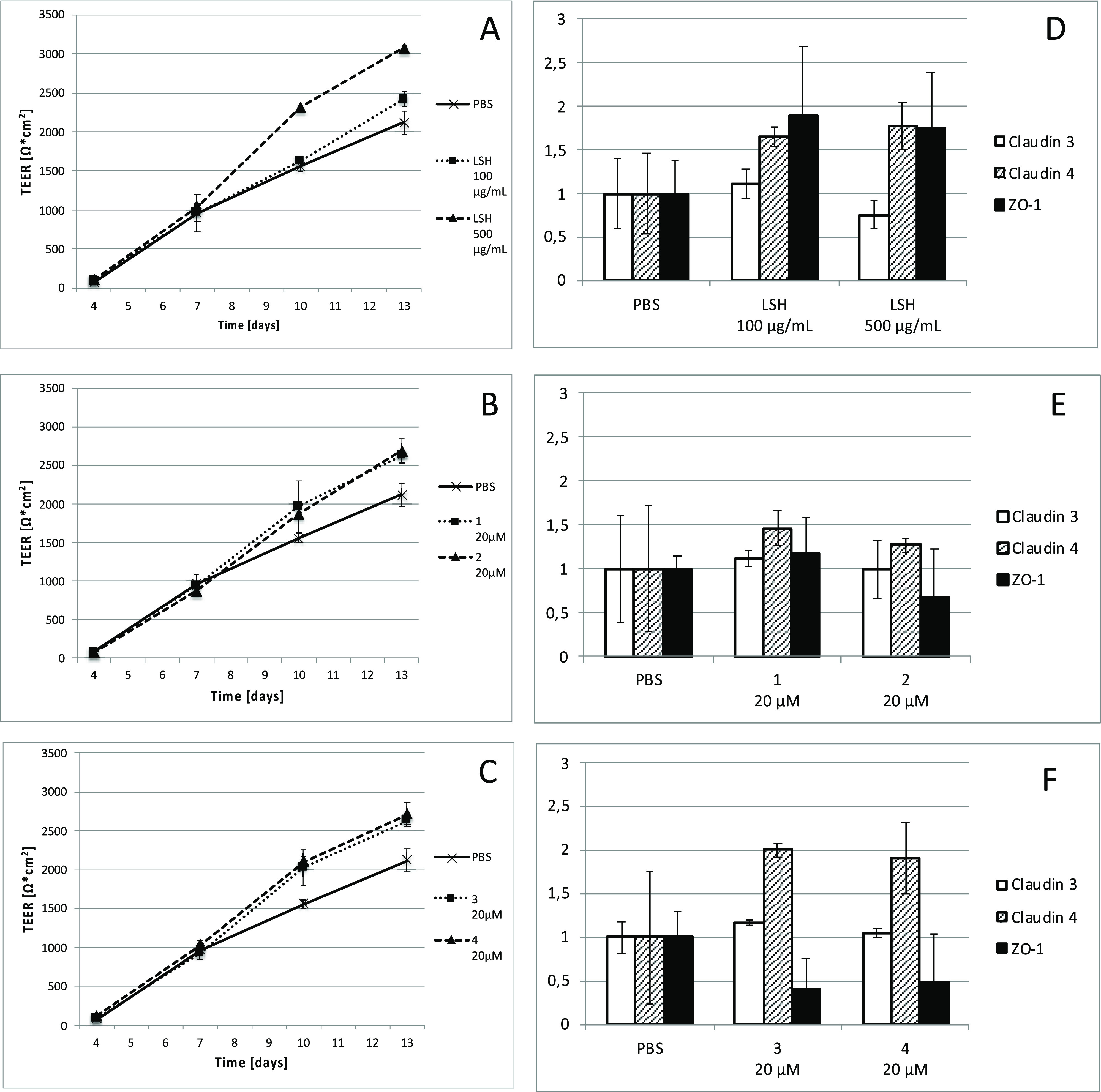
(A–C) Changes in TEER of IPEC-J2 cell monolayers incubated from day 7 with medium containing LSH at concentrations of 100 and 500 μg/mL, castalagin (1), vescalagin (2), salicarinin A (3), or salicarinin B (4) at a 20 μM concentration. The data are representative for three independent experiments assayed in triplicate (LSH and 1) or one experiment assayed in triplicate (2, 3, and 4). (D–F) Respective changes in tight junction protein (TJ) expression using Western blot performed at day 13. Western blots and calculations are provided in Table S1 (Supporting Information). Spearman correlation analysis between TEER and TJ levels are provided in Table S2 (Supporting Information).
Figure 4.
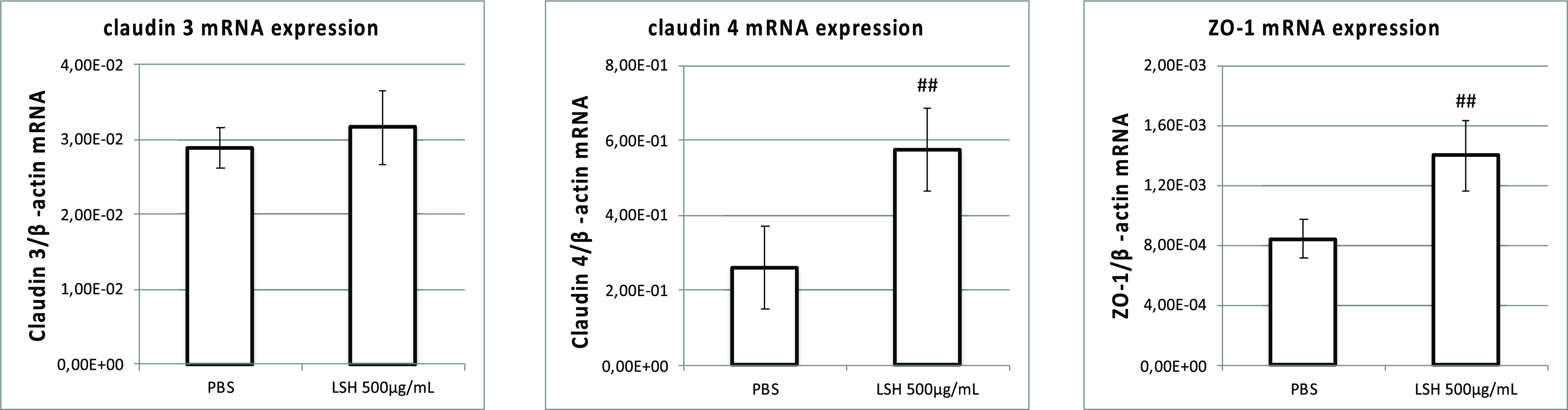
Expression of mRNA at day 13 for claudin 3 and 4 and zona occludens-1 in IPEC-J2 cell monolayers incubated from day 7 with LSH at 500 μg/mL. Data are expressed as mean ± SD. Statistical significance #p < 0.05, ##p < 0.01 increase versus PBS control (Mann–Whitney test).
Postweaning diarrhea in piglets is often associated with proliferation of one or more strains of enteropathogenic EPEC in the gastrointestinal tract. EPEC colonizes the small intestine by binding to the apical side of enterocytes via F4 (or K88) fimbriae which interact with glycoprotein receptors. The attached bacteria then secrete different enterotoxins such as the heat-labile toxin (LT) or heat-stable toxins a and b (STa and STb). These toxins trigger an intracellular signaling cascade resulting in inflammation and the hypersecretion of electrolytes and water into the intestinal lumen, leading to watery diarrhea.25
LSH clearly inhibited the growth of two different E. coli strains in a concentration dependent manner, reaching an MIC at 2000 μg/mL. There were however only minor differences in activity between ECA and control DSM strains, which indicates a lack of strain-specificity in the antimicrobial activity of the extracts. The experiments conducted on the C-glycosylic ellagitannin 1 confirmed that this compound contributes to the inhibition of bacterial growth. Previous studies on LSH regarding its antibacterial activity toward E. coli have shown no effects to moderate effects. The studies showing no activity of LSH were conducted for MeOH extracts26 or EtOH(aq) extracts prepared using a Soxhlet apparatus.27 The determined activity of 1 indicates that ellagitannins are the compounds responsible for inhibition of E. coli growth by LSH. They are highly polar and heat liable compounds, especially dimeric 2 and 3, which could explain the lack of activity determined during previous examinations. The aqueous extract was prepared at temperatures not exceeding 40 °C. This prevented ellagitannin hydrolysis and resulted in inhibitory activity toward E. coli growth. The results obtained for 1 are congruent with previous studies, which showed its exceptional activity among other examined ellagitannins and condensed tannins toward growth of different E. coli strains (including clinical EPEC isolates) with a mean MIC of 705 ± 169 μM.28 The broader studies including a more diverse array of polyphenols and different bacteria groups showed structure dependent activities of polyphenols and that the Enterobacteriaceae is the least susceptible group of bacteria. However, 1 was shown to be exceptionally active in E. coli growth inhibition with an MIC value of 533 μM.29,30 Structure–activity studies showed that the pyrogallol-type structural element is crucial for the antimicrobial activity of tannins; however, the exact mechanism responsible for growth inhibition has not yet been determined.29,31 In the case of E. coli, it can be hypothesized that the iron chelating properties of ellagitannins may be responsible for growth inhibition. The C-glycosylic ellagitannins were previously shown to form navy complexes with Fe3+ ions.9 Ferric ions are necessary for E. coli growth, and their chelation is a known mechanism of antibacterial activity of lactoferrin present in human milk or released by infiltrating leucocytes.31 This hypothesis requires further mechanistic studies to be fully verified.
The antimicrobial activity of LSH was further confirmed in an ex vivo model with distal colon samples inoculated with the ECA strain. The addition of LSH at a concentration of 2000 μg/mL resulted in complete inhibition of ECA growth in distal colon samples. This observation reveals that the presence of the microbiota in colon samples does not affect the inhibitory properties of LSH by inactivation of its active principles under the conditions of the ex vivo assay. After we take into consideration that E. coli spreads among piglets via fecal–oral transmission, the potential activity of LSH to decrease EPEC fecal shedding can be considered to be of epidemiological significance.22,23
Adhesion to the intestinal epithelium is a prerequisite step for many pathogenic bacteria, and thus, the influence of LSH on this process was examined. The pretreatment of IPEC-J2 monolayers with LSH and 1 resulted in a significant inhibition of bacterial adhesion despite the fact that the tested principles were removed from the culture medium before infection. Based on the commonly attributed nonspecific tannin–protein interactions, it could be hypothesized that the interaction of E. coli with epithelial cells was prevented by the tannin molecules randomly attached to the cell surface proteins. However, a study conducted by Tang et al. (2003)32 clearly showed that nonspecific protein binding in the case of CGEs is not strongly marked, in contrast to other ellagitannins, condensed tannins, and gallotannins. In CGEs, the intergalloyl linkages severely restrict the flexibility of galloyl groups and reduce the hydrophobic interactions. In addition, the specificity of the biological activities of CGEs were confirmed not only in enzymatic studies, but also in in vitro experiments on cell models.33−36 The characteristic feature of LSH is that, in comparison to other ellagitannin-containing plant materials with potential application in postweaning diarrhea (Castanea sativa, Quercus sp., Rhus sp.), it strictly contains only ellagitannnins of C-glycosylic nature.9,37−39 After we take into consideration the crucial role of actin polymerization beneath the attached bacteria in the adhesion of EPEC to epithelial cells,40 it can be hypothesized that previously determined specific activity of CGEs to bind actin filaments without any perturbation of the microtubule network41 may contribute to the observed inhibition of bacterial adhesion of LSH and 1. The proposed mechanism, however, requires further verification. Until now, only a few feed additives such as extracts from wheat bran, casein glycomacropeptide, mannan-oligosaccharides, locust bean extract, and Aspergillus oryzae fermentation product or autolyzed yeast product were shown to reduce EPEC attachment to IPEC-J2 cells.42,43 However, due to a high composition heterogeneity, none of them could serve as a referenced positive control in the adhesion experiments.
To evaluate the potential long-term impact of LSH on the epithelial cell monolayer condition, cultures of IPEC-J2 cells were grown on inserts. The addition of LSH to the apical side stimulated monolayer formation (determined as increase in TEER), while experiments conducted on isolated CGEs confirmed their contribution to the augmented monolayer development. The analysis of TJ protein levels at the end of conducted experiments indicated that the stimulation of claudin 4 mRNA expression and protein production contributes to the observed increase in TEER values. Although all tested ellagitannins stimulated monolayer formation, dimeric 3 and 4 enhanced only the production of claudin 4. Elevated levels of ZO-1 mRNA expression and protein production were observed for LSH, but this effect was less significant and could not be attributed to any of the tested ellagitannins. These observations indicate that other compounds present in the extract could contribute to LSH activity toward IPEC-J2 monolayers, such as C-glycosylic flavonoids, anthocyanins, or polysaccharides,1 especially in terms of ZO-1 stimulation. This is the first description of compounds that stimulate IPEC-J2 monolayer formation.
The metabolites formed from ellagitannins in the distal colon by the gut microbiota in post weaning piglets18 (potentially present in the small intestine due to enterohepatic circulation20) do not show inhibitory activity neither toward E.coli growth nor toward its adhesion to the epithelial cell surface. Although 6 was previously shown in vivo and in vitro to significantly enhance gut barrier function and inhibit unwarranted inflammation44 as well as restore LPS-triggered decrease in ZO-1 production in Caco-2 cells,45 in the current study no activity toward stimulation of IPEC-J2 cell monolayer formation was observed (Figure S6, Supporting Information). These data permit us to hypothesize that the beneficial activity of urolithins toward the intestinal barrier could be of significance mostly due to their anti-inflammatory properties, due to the lack of immune responsiveness of IPEC-J2 cells to LPS (neither decrease in TEER nor induction of TNF-α mRNA expression was observed for cells stimulated with 1 μg/mL LPS, data not shown).
Until now, studies of L. salicaria herb on antidiarrheal activity referred to its impact in vivo and ex vivo on intestine contractility gave contradictory results.3,46−48 Experiments conducted on weaned rabbits fed with a diet supplemented with 0.2 and 0.4% of ground herb indicated that LSH does not cause significant adverse effects in terms of performance, blood parameters, and nutrient digestibility as well as gut microbiota community composition and metabolism.49 Despite the antimicrobial activity for LSH and 1 toward E. coli, our previous study conducted ex vivo on gut microbiota of healthy postweaning piglets indicated no negative impact on the cecal or distal colon microbiota diversity and metabolism.18
The observed activities of LSH and isolated ellagitannins support its traditionally attributed antidiarrheal properties and indicate potential mechanisms responsible for its beneficial influence on the condition of the intestinal epithelium. It was clearly shown that LSH inhibits EPEC growth not only in pure cultures but also ex vivo in fecal samples that could be of significance in terms of prevention of fecal shedding of infecting bacteria. The extract also prevented the adhesion of the E. coli enteropathogenic strain and stimulated the formation of cell monolayers by enhancement of TJ protein production. The experiments conducted for the isolated compounds permit attribution of these effects to the presence of CGEs. The high polarity and susceptibility to decomposition at high temperatures of these compounds implicate the use of polar solvents and low temperatures for obtaining preparations applied in further in vitro and in vivo studies. Comparative studies conducted for urolithins, the gut microbiota metabolites of L. salicaria ellagitannins, indicate that these compounds do not influence the applied processes. However, they do not exclude their anti-inflammatory activities, which could be of significance in infection-triggered intestinal inflammation. After we take into consideration the complexity of the biological effects depending on extract composition, metabolism, and distribution of its constituents, it is necessary to confirm the observed effects in further in vivo studies.
Experimental Section
Plant Material Collection
Lythrum salicaria L. herb was collected in August 2016 in Umer, świętokrzyskie region, Poland (50°59′16.7″N 20°35′37.5″E), from its natural habitat. The plant was identified by Dr. Jakub Piwowarski in the Department of Pharmacognosy and Molecular Basis of Phytotherapy, Medical University of Warsaw, based on morphological characters according to Rutkowski, 1998 (klucz), where a voucher specimen LSH2016U is deposited.
Extract and Compounds
The LSH was prepared and standardized as previously described.9,10 Plant material was dried in the shade and cut into pieces. A portion (100 g) was with distilled water (3 × 1L, 30 min each) at 40 °C using an ultrasonic bath. The extract was filtered, combined, and freeze-dried. The amount of crude dry extract was 21.0 g. The ellagitannins, castalagin (1), vescalagin (2), salicarinin A (3), and salicarinin B (4), were isolated from L. salicaria aqueous extract.9 The content of compounds in the extract was determined using a published method.10 LSH contained 33.1 ± 1.5 mg/g of 1, 47.5 ± 2.4 mg/g of 2, 52.1 ± 2.5 mg/g of 3, and 45.0 ± 2.3 mg/g of 4. Urolithins C and A (5 and 6, respectively) were synthesized according to Bialonska et al.;50 phase II conjugate mixture of urolithin A 3-O- and 8-O-glucuronides (7) was isolated from human urine according to Piwowarski et al.51 The ultra high performance liquid chromatography–diode array detector–tandem mass spectrometry (UPLC–DAD–MS) chromatogram of LSH and structures of tested compounds are provided in Figures S1 and S2 (Supporting Information).
Examination of the Stability of Tannins Stability
Plant material (10 g) was extracted once with 10 mL of distilled water or 70% EtOH/water (v/v) at 40 °C for 30 min using an ultrasonic bath or at 80 °C for 24 h. The extracts were filtered and analyzed using UPLC–DAD.
Inhibition of E. coli Growth
The enterotoxigenic E. coli strain Abbotstown, serotype O147:K89:K88 (ECA), was used throughout the experiments as representative strain for enteropathogenic E. coli (EPEC); E. coli DSM 2840 strain (DSM) (DSMZ, Braunschweig, Germany) was used as a nonpathogenic, nonfimbriated control strain. Bacteria were obtained from cryopreservation and grown in BHI medium (Roth, Karlsruhe, Germany) under constant shaking at 37 °C, starting 2 days prior to experimentation. Twice a day, 10 μL of bacterial solution was transferred to 10 mL of new, sterile BHI medium. Double concentrated BHI medium was dispensed into microtiter plates at 100 μL per well. LSH stock solution was prepared in deionized water, 1, 5, 6, and 7 stock solutions were prepared in DMSO. The solutions were sterilized by filtration through 0.22 μm PVDF syringe filters. Ciprofloxacin (Polpharma, Warsaw, Poland) was applied as a positive control. Appropriately diluted extract/compound stock solution (20 μL) was added to each well to achieve the designated concentration. The E. coli strains ECA and DSM were added to each well yielding a final concentration of 104E. coli CFU/mL. Plates were sealed with adhesive film to provide anaerobic conditions and cultured at 37 °C in a microplate reader (Tecan Infinite200Pro, Germany). The turbidity (OD690 nm) was recorded every 5 min for 24 h. For determination of lag time (λ [min]), the lag-exponential (λL) method was used.52
Inhibition of E. coli Growth in Fecal Microbiota Cultures
Feces obtained from 6–8-week-old piglets by rectal stimulation were immediately transported to an anaerobic chamber (BACTRON anaerobic chamber, model BACTRON300-2 6.0A, Sheldon Manufacturing, Inc., Cornelius, Oregon, USA) and mixed with PBS 1:5 (w/v) to create a fecal slurry (FS). LSH was dissolved in deionized water and sterilized by filtration through 0.22 μm PVDF syringe filters. FS (4.5 mL) was mixed with 5 mL of BHI, and 0.5 mL of LSH solution was added to achieve the appropriate concentration. For determination of EPEC growth in microbiota cultures, E. coli IMT 0147:K89:K88 (ECA) strain was added to cultures to achieve concentrations of 106, 105, 104, and 103 CFU/mL. For determination of EPEC growth in the presence of LSH, a concentration of 104 CFU/mL of the ECA strain was chosen. To exclude the presence of EPEC in feces, FS cultures without inoculation with ECA strain were performed. For comparison, the influence of LSH on ECA growth in pure cultures without FS was tested in parallel. Cultures were performed in an anaerobic chamber incubator at 37 °C. A sample (1 mL) for each time point (0, 1, 2, 4, 8, and 24 h) was taken and immediately frozen at −30 °C for further analysis. After they were thawed, samples were centrifuged at 4 °C for 10 min at 21 000 g, the supernatant was removed, and the pellet was subjected to DNA isolation. DNA from culture pellets was extracted using the QIAamp DNA Stool Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. The amount of DNA was quantified using NanoDrop fluorospectrometer ND-3300 (Peqlab, Erlangen, Germany).
Real Time qPCR
The growth of the ECA strain in microbiota cultures was monitored through determination of K88 (fae) fimbrial operon copies. The DNA was quantified by Brilliant II SYBR Green QPCR Kit (Agilent Technologies, Palo Alto, CA, USA) using Aria Mx Real-Time PCR system (Agilent Technologies). For the qPCR experiments, primers against the sequence coding K88 fimbriae were used (sense = 5′-GTTGGTACAGGTCTTAATGG-3′, antisense = 5′-GAATCTGTCCGAGAATATCA-3′). The cycling conditions were 95 °C for 10 min followed by 40 cycles of denaturing at 95 °C for 15 s and annealing/extension for 1 min at 55 °C. The ECA strain growth was expressed as the number of fae copies/ng DNA against calibration samples with known copy numbers.
IPEC-J2 Cell Culture
The IPEC-J2 cell line (ACC 701) was obtained from DSMZ (Braunschweig, Germany) and grown in Dulbecco’s MEM/Ham’s F-12 (DMEM, Biochrom GmbH, Berlin, Germany) medium supplemented with 10% FBS, with addition of penicillin (100 IU/mL) and streptomycin 100 mg/mL (Biochrom GmbH, Berlin, Germany), at 37 °C in 5% CO2. The cells from passages 42–60 were used for the experiments. The cell culture was routinely tested to be free of Mycoplasma spp. contamination.
IPEC-J2 Cell Viability
The IPEC-J2 cells were seeded in 24-well plates at a density of 105 cells/well and maintained in the culture plates for 7 days at 37 °C with 5% CO2, with a medium change every second day. To assess the impact on viability, cells were incubated with increasing concentrations of LSH (20, 100, and 500 μg/mL) and 1 (5, 20, and 100 μM). After 24 h of incubation, the cells were washed twice with PBS, trypsinized, stained with propidium iodide (PI), and analyzed using FACS (MACSQuant Analyze, Miltenyi Biotec, Bergisch Gladbach, Germany). The viability was assessed as the percentage of PI(−) cells.
IPEC-J2 Adhesion Studies
The IPEC-J2 cells were seeded in 24-well plates at a density of 105 cells/well and maintained in the cell culture plates during 7 days at 37 °C with 5% CO2 and a medium change every second day. On day 8, the medium was removed, cells were washed twice with warm PBS, and medium containing LSH or tested compounds at designated concentration was added to each well. On day 9, the medium was removed, the cells were washed twice with warm PBS to remove LSH, 1, and antibiotics, and fresh antibiotic-free medium was added. The cells were infected with the ECA strain using a multiplicity of infection (MOI) of 100 as described previously,53 equivalent to 1 × 107 bacteria per well. A nonfimbriated E. coli strain DSM 2840 (DSM) was used as a negative, nonadherent control. Staining and detection of E. coli strains were performed using fluorescent stain 5,6-carboxymethyl fluorescein diacetate succinimidyl ester (CFDA-SE) (Sigma-Aldrich, Steinheim, Germany). Bacteria were incubated with 1 mM CFDA-SE at 37 °C for 120 min and then centrifuged (13 000 g, 4 min, 4 °C), resuspended in PBS, counted, and used for infection. After 45 min of incubation with stained bacteria, the cells were washed with PBS (three times) to remove nonadherent bacteria, trypsinized with trypsin/EDTA (Biochrom AG, Berlin, Germany), suspended in 1 mL of cell medium, and centrifuged at 4000 g for 5 min at 4 °C. The pellet was resuspended in 300 μL of antibiotic free medium. Flow cytometric measurement was carried out using MACSQuant Analyze (Miltenyi Biotec, Bergisch Gladbach, Germany). The mean fluorescence intensity (MFI) in the gated cell population was measured (10 000 events per sample).
TEER Measurement of IPEC-J2 Monolayers
For the TEER assay, IPEC-J2 cells (5 × 105 cells/well) were seeded to ThinCertTM cell culture inserts (polyethylene terephthalate capillary pore membranes; 0.4 μm pore size; Greiner BioOne, Frickenhausen, Germany) compatible with 6-well plates. Cells were cultured for 7 days with medium exchange in the apical and basolateral side every third day. The TEER was measured using an epithelial voltammeter EVOM2 with a chopstick electrode STX2 (World Precision Instruments Inc., Sarasota, FL, USA) and used as indicator for confluency.54 When TEER of the cell monolayer reached values over 1 kΩ cm2 (day 7), the medium in the apical compartment was changed to medium containing tested compounds or LSH at selected concentrations or PBS as a control. At day 10, the TEER was measured, and medium was once again exchanged to fresh medium containing tested compounds or LSH. The medium in the basolateral side was changed in parallel. At day 13, the TEER was measured, and cells were washed twice with PBS and harvested to RNAlater (Qiagen GmbH, Hilden, Germany) for qPCR analysis or lysed with cOmplete Lysis-M (Roche, Basel, Switzerland) containing protease and phosphatase inhibitor cocktail (Roche, Basel, Switzerland) for Western blot analysis.
Western Blot Analysis
The cell lysate was centrifuged at 14 000 g for 15 min at 4 °C. The total protein concentration of the cell and nuclear lysates were quantified with the BCA protein assay kit (Bio-Rad, Hercules, CA, USA) using BSA as a standard. After equilibration of protein concentration between the samples, the lysates were boiled with 4xLaemmli Sample Buffer (Bio-Rad) and stored at −20 °C. The protein lysates were separated by 10 or 12.5% SDS–PAGE and transferred to nitrocellulose membrane Amersham Protran (Sigma-Aldrich). The membrane was blocked with 5% skimmed milk powder (Sigma-Aldrich) in Tris-buffered saline (Roth GmbH, Karlsruhe, Germany) containing 0.1% (v/v) Tween 20 (SERVA) and then incubated overnight at 4 °C with primary rabbit monoclonal antibodies, anti-Claudin 3, polyclonal (Thermo-Fisher, Waltham, MA, USA), anti-ZO-1, polyclonal (Bioss Antibodies, Woburn, MA, USA), antibeta Actin, polyclonal (Abcam, Cambridge, UK), and mouse anti-Claudin 4, monoclonal (Thermo-Fisher). The membranes were washed (three times) and incubated with the secondary antibody at a dilution of 1:3000 (v/v). HRP-conjugated goat antirabbit IgG antibody (Abcam) or antimouse IgG, HRP-linked antibody (Cell Signaling Technology, Denver, MA, USA) was used as a secondary antibody. The protein bands were detected with SignalFire ECL Reagent (Cell Signaling Technology) and visualized using PXi 4 Touch (SYNGENE, Frederick, MD, USA). The β-actin was used as a loading control. The protein level was determined by normalization to that of β-actin. Relative expression was determined as the ratio of the intensity of the protein band to that of the corresponding control band and finally represented as the fold increase over the control group.
RNA Extraction and Reverse Transcription
The total cellular RNA was extracted using NucleoSpin RNA Clean-up (Macherey-Nagel, Düren, Germany) according to the manufacturer’s instructions. RNA samples were treated with PureLink DNase (Invitrogen, Carlsbad, CA, USA). The quality and quantity of isolated RNA was evaluated using 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). Total RNA was reversely transcribed to cDNA using Superscript III reverse Transcriptase kit (Life Technologies, Carlsbad, CA, USA) according to the manufacturers protocol.
Real Time qPCR Analysis
The levels of mRNA expression for Claudin 3 (Cldn3), Claudin 4 (Cldn4), and Zona occludens-1 (ZO-1) were quantified by Brilliant II SYBR Green QPCR Kit (Agilent Technologies, Palo Alto, CA, USA) using Aria Mx Real-Time PCR system (Agilent Technologies). For the qPCR experiments, the following are primers against the sequences of porcine: Cldn3 (sense = 5′-GCCAAGATCCTCTACTCCGC-3′, antisense = 5′-ATACGTAGTCCTTGCGGTCG-3′), Cldn4 (sense = 5′-CAACTGCGTGGATGATGAGA-3′, antisense = 5′- CCAGGGGATTGTAGAAGTCG-3′), ZO-1 (sense = 5′-ACAGTGCCCAGAGACCAAGA-3′, antisense = 5′-CATTTCCTCGGGGTAGGGGT-3′) and the housekeeping genes β-actin (ACTB) (sense = 5′- CACCGCAAATGCTTCTAGGC-3′, antisense = 5′-TGCGCAAGTTAGGTTTTGTCA-3′), succinate dehydrogenase subunit A (SDHA) (sense = 5′- CAAACTCGCTCCTGGACCTC-3′, antisense = 5′-CCGGAGGATCTTCTCACAGC-3′), and β2-microglobulin (β2-glob) (sense = 5′-CCCCGAAGGTTCAGGTTTAC-3′, antisense = 5′-CGGCAGCTATACTGATCCAC-3′). The cycling conditions were 95 °C for 10 min followed by 40 cycles of denaturing at 95 °C for 15 s and annealing/extension for 1 min at 60 °C. The amount of cDNA content of each gene in the tested sample was calculated based on the respective calibration curve and normalized to β-actin cDNA content.
Statistical Analysis
The data were analyzed using Statistica 13 software (StatSoft, Tulsa, OK, USA) or GraphPad Prism. For samples with normal distribution, one-way analysis of variance followed by the Tukey’s or Dunnet’s posthoc test was conducted. To compare samples with distributions other than normal, nonparametric Kruskal–Wallis one-way analysis of variance or nonparametric Mann–Whitney test was performed. Associations between the TEER and TJ protein production were determined using the Spearman’s rank-order correlation analysis.
Acknowledgments
The project was financially supported by Alexander von Humboldt Foundation Research Fellowship for Postdoctoral Researchers awarded to J.P.P. The authors would like to thank Petra Huck and Marita Eitinger for assistance in laboratory works. This project was partially carried out with the use of CePT infrastructure financed by the European Regional Development Found within the Operational Programme “Innovative economy” for 2007–2013.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jnatprod.0c00776.
UPLC–DAD–MS chromatograms, structures of tested gut microbiota metabolites, growth curves, influence of LSH and castalagin (1) on IPEC-J2 cell viability, changes in TEER of IPEC-J2 cell monolayers, and UPLC–DAD–MS analysis (PDF)
Changes in tight junction protein (TJ) expression determined using Western blot of proteins from each well at the end of conducted experiment (PDF)
Spearman correlation analysis between TEER and TJ levels in IPEC-J2 monolayers (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Piwowarski J. P.; Granica S.; Kiss A. K. J. Ethnopharmacol. 2015, 170, 226–50. 10.1016/j.jep.2015.05.017. [DOI] [PubMed] [Google Scholar]
- Akerreta S.; Calvo M.I.; Cavero R.Y. J. Ethnopharmacol. 2010, 130, 369–378. 10.1016/j.jep.2010.05.023. [DOI] [PubMed] [Google Scholar]
- Vincent D. S. G. Acta Phytotherapeutica 1954, 1, 1–12. [Google Scholar]
- Sartory A.; Quevauviller A.; Richard P. C. R. Hebd. Seances Acad. Sci. 1949, 228, 782–784. [PubMed] [Google Scholar]
- Gougeon M. M.; Laumonier J. L’Union Pharm. 1918, 59, 147–149. [Google Scholar]
- Murray J. A.Apparatus Medicaminum tam Simplicium quam Praeparatorum et Compositorum; J.C. Dieterich: Goettingae, 1793. [Google Scholar]
- Cazin F. J.Traité Pratique et Raisonné des Plantes Médicinales Indigènes; Paris, Labé, 1858. [Google Scholar]
- Gardane J. J. Gaz. Sante 1773, 16, 65–66. [Google Scholar]
- Piwowarski J. P.; Kiss A. K. Phytochem. Anal. 2013, 24, 336–348. 10.1002/pca.2415. [DOI] [PubMed] [Google Scholar]
- Granica S.; Piwowarski J. P.; Kiss A. K. Phytochem. Anal. 2014, 25, 201–206. 10.1002/pca.2492. [DOI] [PubMed] [Google Scholar]
- Xiong X.; Tan B.; Song M.; Ji P.; Kim K.; Yin Y.; Liu Y. Front. Vet. Sci. 2019, 6, 46. 10.3389/fvets.2019.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyles C. L.; Prescott J. F.; Songer J. G.; Thoen C. O.. Pathogenesis of Bacterial Infections in Animals, 4th ed.; Blackwell Publishing, 2010. [Google Scholar]
- Nagy B.; Fekete P. Z. Int. J. Med. Microbiol. 2005, 295, 443–454. 10.1016/j.ijmm.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Croxen M. A.; Law R. J.; Scholz R.; Keeney K. M.; Wlodarska M.; Finlay B. B. Clin. Microbiol. Rev. 2013, 26, 822–880. 10.1128/CMR.00022-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Espinosa C. D.; Abelilla J. J.; Casas G. A.; Lagos L. V.; Lee S. A.; Kwon W. B.; Mathai J. K.; Navarro D.; Jaworski N. W.; Stein H. H. Anim. Nutr. 2018, 4, 113–125. 10.1016/j.aninu.2018.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard M.; Bee G. Animal 2020, 14, 95–107. 10.1017/S1751731119002143. [DOI] [PubMed] [Google Scholar]
- Lavoie C. Biol. Invasions 2010, 12, 1967–1999. 10.1007/s10530-009-9600-7. [DOI] [Google Scholar]
- Dadi T. H.; Vahjen W.; Zentek J.; Melzig M. F.; Granica S.; Piwowarski J. P. J. Ethnopharmacol. 2020, 261, 113073. 10.1016/j.jep.2020.113073. [DOI] [PubMed] [Google Scholar]
- Nisa S.; Scanlon K. M.; Donnenberg M. S.. Escherichia coli (Second ed.). Pathotypes and Principles of Pathogenesis; Academic Press: Cambridge, MA, USA, 2013. [Google Scholar]
- Espin J. C.; Gonzalez-Barrio R.; Cerda B.; Lopez-Bote C.; Rey A. I.; Tomas-Barberan F. A. J. Agric. Food Chem. 2007, 55, 10476–10485. 10.1021/jf0723864. [DOI] [PubMed] [Google Scholar]
- Andreux P. A.; Blanco-Bose W.; Ryu D.; Burdet F.; Ibberson M.; Aebischer P.; Auwerx J.; Singh A.; Rinsch C. Nat. Metab. 2019, 1, 595–603. 10.1038/s42255-019-0073-4. [DOI] [PubMed] [Google Scholar]
- Cornick N. A.; Helgerson A. F. Appl. Environ. Microbiol. 2004, 70, 5331–5335. 10.1128/AEM.70.9.5331-5335.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luise D.; Lauridsen C.; Bosi P.; Trevisi P. J. Anim. Sci. Biotechnol. 2019, 10, 53. 10.1186/s40104-019-0352-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Einspanier R.; Schoen J. Histochem. Cell Biol. 2015, 144, 509–515. 10.1007/s00418-015-1351-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo J. M.; Opapeju F. O.; Pluske J. R.; Kim J. C.; Hampson D. J.; Nyachoti C. M. J. Anim. Physiol. Anim. Nutr. 2013, 97, 207–237. 10.1111/j.1439-0396.2012.01284.x. [DOI] [PubMed] [Google Scholar]
- Becker H.; Scher J. M.; Speakman J. B.; Zapp J. Fitoterapia 2005, 76, 580–584. 10.1016/j.fitote.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Dulger B.; Gonuz A. Asian J. Plant Sci. 2004, 3, 104–107. 10.3923/ajps.2004.104.107. [DOI] [Google Scholar]
- Taguri T.; Tanaka T.; Kouno I. Biol. Pharm. Bull. 2004, 27, 1965–1969. 10.1248/bpb.27.1965. [DOI] [PubMed] [Google Scholar]
- Taguri T.; Tanaka T.; Kouno I. Biol. Pharm. Bull. 2006, 29, 2226–2235. 10.1248/bpb.29.2226. [DOI] [PubMed] [Google Scholar]
- Buzzini P.; Arapitsas P.; Goretti M.; Branda E.; Turchetti B.; Pinelli P.; Ieri F.; Romani A. Mini-Rev. Med. Chem. 2008, 8, 1179–87. 10.2174/138955708786140990. [DOI] [PubMed] [Google Scholar]
- Scalbert A. Phytochemistry 1991, 30, 3875–3883. 10.1016/0031-9422(91)83426-L. [DOI] [Google Scholar]
- Tang H. R.; Covington A. D.; Hancock R. A. Biopolymers 2003, 70, 403–413. 10.1002/bip.10499. [DOI] [PubMed] [Google Scholar]
- Piwowarski J. P.; Kiss A. K.; Kozlowska-Wojciechowska M. J. Ethnopharmacol. 2011, 137, 937–941. 10.1016/j.jep.2011.05.039. [DOI] [PubMed] [Google Scholar]
- Auzanneau C.; Montaudon D.; Jacquet R.; Puyo S.; Pouysegu L.; Deffieux D.; Elkaoukabi-Chaibi A.; De Giorgi F.; Ichas F.; Quideau S.; Pourquier P. Mol. Pharmacol. 2012, 82, 134–141. 10.1124/mol.111.077537. [DOI] [PubMed] [Google Scholar]
- Douat-Casassus C.; Chassaing S.; Di Primo C.; Quideau S. ChemBioChem 2009, 10, 2321–2324. 10.1002/cbic.200900287. [DOI] [PubMed] [Google Scholar]
- Richard T.; Lefeuvre D.; Descendit A.; Quideau S.; Monti J. P. Biochim. Biophys. Acta, Gen. Subj. 2006, 1760, 951–958. 10.1016/j.bbagen.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Sanz M.; Cadahia E.; Esteruelas E.; Munoz A. M.; Fernandez de Simon B.; Hernandez T.; Estrella I. J. Agric. Food Chem. 2010, 58, 9631–9640. 10.1021/jf102718t. [DOI] [PubMed] [Google Scholar]
- Gallardo A.; Morcuende D.; Solla A.; Moreno G.; Pulido F.; Quesada A. Physiol. Plant. 2019, 165, 319–329. 10.1111/ppl.12848. [DOI] [PubMed] [Google Scholar]
- Chen H.; Liu J.; Cui K.; Lu Q.; Wang C.; Wu H.; Yang Z.; Ding W.; Shao S.; Wang H.; Ling X.; King-Jones K.; Chen X. Sci. Rep. 2018, 8, 9841. 10.1038/s41598-018-28153-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro-Garcia F.; Serapio-Palacios A.; Ugalde-Silva P.; Tapia-Pastrana G.; Chavez-Duenas L. BioMed Res. Int. 2013, 2013, 374395. 10.1155/2013/374395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quideau S.; Douat-Casassus C.; Delannoy Lopez D. M.; Di Primo C.; Chassaing S.; Jacquet R.; Saltel F.; Genot E. Angew. Chem., Int. Ed. 2011, 50, 5099–50104. 10.1002/anie.201006712. [DOI] [PubMed] [Google Scholar]
- Hermes R. G.; Manzanilla E. G.; Martin-Orue S. M.; Perez J. F.; Klasing K. C. Comp. Immunol. Microbiol. Infect. Dis. 2011, 34, 479–488. 10.1016/j.cimid.2011.08.006. [DOI] [PubMed] [Google Scholar]
- Spitzer F.; Speiser S.; Vahjen W.; Zentek J. Cytotechnology 2016, 68, 1463–1471. 10.1007/s10616-015-9905-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R.; Chandrashekharappa S.; Bodduluri S. R.; Baby B. V.; Hegde B.; Kotla N. G.; Hiwale A. A.; Saiyed T.; Patel P.; Vijay-Kumar M.; Langille M. G. I.; Douglas G. M.; Cheng X.; Rouchka E. C.; Waigel S. J.; Dryden G. W.; Alatassi H.; Zhang H. G.; Haribabu B.; Vemula P. K.; Jala V. R. Nat. Commun. 2019, 10, 89. 10.1038/s41467-018-07859-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R.; Long X.; Yang J.; Du L.; Zhang X.; Li J.; Hou C. Food Funct. 2019, 10, 8273–8285. 10.1039/C9FO02077B. [DOI] [PubMed] [Google Scholar]
- Bencsik T.; Bartho L.; Sandor V.; Papp N.; Benko R.; Felinger A.; Kilar F.; Horvath G. Nat. Prod. Commun. 2013, 8, 1247–1250. [PubMed] [Google Scholar]
- Brun Y.; Wang X. P.; Willemot J.; Sevenet T.; Demenge P. Fundam. Clin. Pharmacol. 1998, 12, 30–36. 10.1111/j.1472-8206.1998.tb00920.x. [DOI] [PubMed] [Google Scholar]
- Torrent Marti M. T. Circ. Farm. 1975, 33, 265–307. [Google Scholar]
- Kovitvadhi A.; Gasco L.; Ferrocino I.; Rotolo L.; Dabbou S.; Malfatto V.; Gai F.; Peiretti P. G.; Falzone M.; Vignolini C.; Cocolin L.; Zoccarato I. Animal 2016, 10, 10–18. 10.1017/S1751731115001822. [DOI] [PubMed] [Google Scholar]
- Bialonska D.; Kasimsetty S. G.; Khan S. I.; Ferreira D. J. Agric. Food Chem. 2009, 57, 10181–10186. 10.1021/jf9025794. [DOI] [PubMed] [Google Scholar]
- Piwowarski J. P.; Stanislawska I.; Granica S.; Stefanska J.; Kiss A. K. Drug Metab. Dispos. 2017, 45, 657–665. 10.1124/dmd.117.075200. [DOI] [PubMed] [Google Scholar]
- Baty F.; Delignette-Muller M. L. Int. J. Food Microbiol. 2004, 91, 261–277. 10.1016/j.ijfoodmicro.2003.07.002. [DOI] [PubMed] [Google Scholar]
- Sargeant H. R.; Miller H. M.; Shaw M. A. Mol. Immunol. 2011, 48, 2113–2121. 10.1016/j.molimm.2011.07.002. [DOI] [PubMed] [Google Scholar]
- Schierack P.; Nordhoff M.; Pollmann M.; Weyrauch K. D.; Amasheh S.; Lodemann U.; Jores J.; Tachu B.; Kleta S.; Blikslager A.; Tedin K.; Wieler L. H. Histochem. Cell Biol. 2006, 125, 293–305. 10.1007/s00418-005-0067-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.