

Abstract
Manganese (Mn) is a trace element essential for normal human development and is required for the proper functioning of a variety of physiological processes. Chronic exposure to Mn can cause manganism, a neurodegenerative disorder resembling idiopathic Parkinson’s disease (PD). Mn(II) neurotoxicity is characterized by astrocytic impairment both in the expression and activity of glutamine (Gln) transporters. Because protein kinase C (PKC) activation leads to the downregulation of a number of neurotransmitter transporters and Mn(II) increases PKC activity, we hypothesized that the PKC signaling pathway contributes to the Mn(II)-mediated disruption of Gln turnover. Our results have shown that Mn exposure increases the phosphorylation of both the PKCα and PKCδ isoforms. PKC activity was also shown to be increased in response to Mn(II) treatment. Corroborating our earlier observations, Mn(II) also caused a decrease in Gln uptake. This effect was blocked by PKC inhibitors. Notably, PKC activation caused a decrease in Gln uptake mediated by systems ASC and N, but had no effect on the activities of systems A and L. Exposure to α-phorbol 12-myristate 13-acetate significantly decreased SNAT3 (system N) and ASCT2 (system ASC) protein levels. Additionally, a co-immunoprecipitation study demonstrated the association of SNAT3 and ASCT2 with the PKCδ isoform, and Western blotting revealed the Mn(II)-mediated activation of PKCδ by proteolytic cleavage. PKC activation was also found to increase SNAT3 and ubiquitin ligase Nedd4-2 binding and to induce hyperubiquitination. Taken together, these findings demonstrate that the Mn(II)-induced dysregulation of Gln homeostasis in astrocytes involves PKCδ signaling accompanied by an increase in ubiquitin-mediated proteolysis.
Keywords: glutamine transporter, manganese, protein kinase C, ubiquitination
INTRODUCTION
l-Glutamine (Gln) serves a multitude of roles in the central nervous system (CNS); it is a precursor of glutamic acid (Glu) and γ-aminobutyric acid (GABA) neurotransmitters and an essential component of intermediary metabolism (Hamberger et al., 1979; Norenberg and Martinez-Hernandez, 1979; Sonnewald et al., 1993). The passage of Gln across astrocytic and neuronal plasma membranes is mediated by specific transporting proteins and is a key factor in the Glu-Gln-GABA cycle (Daikhin and Yudkoff, 2000). The bi-directionally acting systems N and ASC transporters are expressed in astrocytes, whereas the Gln uptake-promoting system A transporter, SNAT2, and system L transporter, LAT2, are expressed both in neurons and astrocytes, with the higher expression levels in neurons when compared with astrocytes (Bröer and Brookes, 2001; Mackenzie and Erickson, 2003). In astrocytes, at control conditions, Gln uptake is mediated mainly by systems ASC and N, in 50% and 30%, respectively, with a lesser contribution by systems A and L (for both systems in 10%) (Sidoryk-Wegrzynowicz et al., 2009). Gln transport in the CNS is essential for the Glu-Gln-GABA cycle, and the deregulation of this cycle has been implicated in response to manganese (Mn) exposure (Lee et al., 2009; Milatovic et al., 2007; Sidoryk-Wegrzynowicz et al., 2009, 2010). Primary cultures of astrocytes treated with Mn(II) displayed a significant reduction in the protein levels of SNAT3 (system N), SNAT2 (system A), LAT2 (system L), and ASCT2 (system ASC). The Mn(II)-induced disruption of Gln transporter expression was associated with Gln transport alteration (Sidoryk-Wegrzynowicz et al., 2009) . Recently, we also showed that the Mn-induced downregulation of the astroglial Gln transporter, SNAT3, involves the ubiquitin-mediated proteolytic system (Sidoryk-Wegrzynowicz et al., 2010); however, the source of initiation and the specific regulatory mechanism(s) of this process have yet to be fully elucidated. It is well known that Mn(II) at excessive levels is toxic to the CNS. Pathological Mn(II) concentrations lead to a neurological disorder, called manganism, which is characterized by early psychotic symptoms, and frequently followed by chronic symptoms similar to idiopathic Parkinson’s disease (PD) (Barbeau, 1984; Mena et al., 1967; Racette et al., 2001). At the histopathological level, manganism results in neurodegeneration and gliosis in the substantia nigra, the globus pallidus, and the striatum (Erikson and Aschner, 2003; Pal et al., 1999). Mn exposure causes membrane translocation and the subsequent activation of protein kinase C (PKC) (Liao et al., 2007), resulting in the downregulation of a number neurotransmitter transporters, including those for dopamine, norepinephrine, serotonin, GABA, glycine, and glutamate (Apparsundaram et al., 1998; Beckman et al., 1999; Fang et al., 2002; Gomeza et al., 1995; Melikian and Buckley, 1999; Zhang et al., 1997). Accordingly, we hypothesized that PKC signaling contributes to the Mn(II)-mediated disruption of astrocytic Gln carrier expression and function.
PKC isozymes participate in diverse signaling pathways that play critical roles in regulating brain function (Battaini, 2001; Govoni et al., 2010). The PKC family of serine/threonine kinases consists of at least 10 isoforms classified by their activation requirements (Newton, 2001, 2003). Although many neurotransmitters and amino acid transporters have been documented to be controlled by PKC signaling (Balkrishna et al., 2010; Bode et al., 1998; Foster et al., 2008), the specific isoform(s) that is involved in this process has not yet been identified.
The present study was designed to address the following objectives: (1) to determine which of the different PKC isoforms is activated after Mn(II) exposure; (2) to identify the role of PKC in the Mn(II)-mediated disruption of Gln transporter expression and function; and (3) to examine the involvement of PKC signaling in the ubiquitin-mediated proteolytic system.
MATERIALS AND METHODS
Chemicals, Reagents, Cell-Culture Supplies, and Antibodies
Manganese chloride (MnCl2) and the PKC inhibitor bisindolylmaleimide II (BisII) and activator α-phorbol 12-myristate 13-acetate (PMA), amino acids: Gln, histidine (His), threonine (Thr), 2-methylaminoisobutyric acid (MeAIB) leucine (Leu) were purchased from Sigma Chemical Co. (St Louis, MO). Minimal essential medium (MEM) with Earle’s salts, heat-inactivated horse serum, penicillin, and streptomycin were purchased from Invitrogen (Carlsbad, CA). Primary antibodies against the following proteins were used: SNAT3, SNAT2, ASCT2, LAT2 (Santa Cruz Biotechnology, Santa Cruz, CA); Nedd4-2, conjugated ubiquitin, (Abcam, Cambridge, MA), phospho-PKCβ (Thr 642), phosho-PKCα (Thr 638), phospho-PKCδ (Thr 505) (Invitrogen, Carlsbad, CA), PKCβ, PKCα, PKCδ (Abcam), and β-actin (Sigma). Secondary, HRP-conjugated antibodies were purchased from Santa Cruz Biotechnology: against goat, mouse, and rabbit IgGs, and from Sigma: against mouse IgG.
Primary Cultures of Astrocytes
Primary cultures of astrocytes were prepared according to previously established protocols (Aschner et al., 1992). One-day-old Sprague–Dawley rats were decapitated under halothane anesthesia, and the cerebral cortices were dissected out and digested with bacterial neutral protease (Dispase, Invitrogen, Eugene, OR). Astrocytes were recovered by the repeated removal of dissociated cells and plated at a density of 1 × 105 cells/mL. Twenty-four hours after the initial plating, the medium was changed to preserve the adhering astrocytes and to remove neurons and oligodendrocytes. The cultures were maintained at 37°C in a 95% air/5% CO2 incubator for 3 weeks in MEM with Earle’s salts supplemented with 10% fetal bovine serum, 100 U/mL of penicillin, and 100 μg/mL of streptomycin. The medium was replaced twice per week. The surface-adhering monolayer cultures were >95% positive for the astrocytic marker, glial fibrillary acidic protein. All experiments were performed 3 weeks postisolation, when the cultures reached confluency.
Experimental Treatments
The chosen MnCl2 concentrations were based on estimates from the literature. The physiological range of Mn is 75–100 μM; clinical signs increase in frequency and severity above this level. Cells were exposured to 100 (physiological range) and to 500 μM or 1 mM MnCl2, representing a range of Mn(II) concentrations considered to be toxic in cultured astrocytes (Milatovic et al., 2007) as well as in the in vivo brain in animals (Suzuki et al.,1975) and humans models (Tracqui et al., 1995). For the assessment of the involvement of PKC in Mn(II)-induced Gln transporters downregulation, cells were treated with specific activator of PKCs: α, βI, βII, γ, δ, , η, θ-PMA (100 nM), and inhibitor of PKCs-Bis II (10 μM).
RNA Isolation and RT-PCR
For real time polymerized chain reaction (RT-PCR) analysis, total RNA was isolated with the RNeasy kit (Qiagen, Valencia, CA) from primary cultures of astrocytes followed by reverse-transcription with a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s protocols. RT-PCR analyses were carried out using the following TaqMan Gene Expression: GAPDH-Rn99999916_s1; PKC α-Rn01496144_m1; PKCbeta-Rb01275210_m1 PKC delta Rn01450219_g1 (Applied Biosystems). The reaction mixtures contained Taqman Universal PCR Mastermix, TaqMan® Gene Expression and 1 μl cDNA in a total volume of 5 μl. Reactions were performed in 384-well optical reaction plates using an Applied Biosystems 7500 Sequence Detection System. Expression values obtained from triplicate runs of each cDNA were calculated relative to the triplicate value for the GAPDH from the same cDNA preparation. Relative quantifications of mRNA in the samples were performed with the postrun data analysis software (SDS 2.3) provided by the Cycler system from Applied Biosystems.
Western Blot Analysis
Western blot analyses with anti-SNAT3 (1:400) anti-ASCT2 (1:500), anti-SNAT2 (1:500), anti-LAT2 (1:500), anti-p-PKCα (1:1,000), anti-p-PKCδ (1:1,000), anti-p-PKCβ (1:1,000), anti-PKCα (1:1,000), anti-PKCδ (1:1,000), anti-PKCβ (1:1,000), anticonjugated Ub (1:1,000), antibeta actin (1:3,000) and secondary antibody anti-rabbit IgG (1:5,000), anti-goat IgG (1:1,000), or anti-mouse IgG (1:3,000) were performed as described (Sidoryk-Wegrzynowicz et al., 2010).
Biotinylation of Cell Surface Protein
Primary astrocyte cultures were treated with PMA (100 nM) for 4, 6, 8, and 24 h, and the cell surface biotinylation assay was performed using the cell surface protein isolation kit (Pierce, Rockford, IL), following the manufacturer’s protocol. In brief, astrocytes were washed twice with ice-cold PBS and biotinylated using EZ-Link Sulfo-NHS-SS biotin in PBS for 30 min at 4°C on an orbital shaker. The reaction was stopped by adding a quenching solution (TBS, 25 mM Tris, and 0.15 M sodium chloride; pH 7.2), and cells were scraped and centrifuged at 500g for 3 min. The pellets were washed with TBS, centrifuged at 500g for 3 min, and lysed and centrifuged at 10,000g for 2 min at 4°C. The supernatant was added to a column containing NeutrAvidin™ gel and incubated for 60 min at room temperature with end-over-end mixing using a rotator. The column was washed three times with TBS, and the biotinylated proteins adherent to NeutrAvidin were suspended in SDS buffer (62.5 mM Tris–HCl pH 6.8, 1% SDS, and 10% glycerol containing 50 mM dithiothreitol), heated in a heat block for 5 min at 95°C, and eluted by centrifugation at 1,000g for 2 min. Proteins in each sample were measured by the BCA method (Pierce), and equal amount of protein was used for Western blots. Beta-actin, which is not expressed in cell surface membranes, was also evaluated to assure the purity of the isolated cell surface proteins.
Co-Immunoprecipitation
Co-immunoprecipitation study for SNAT3, ASCT2, SNAT2, LAT2, Nedd4-2, and PKC δ was performed as described (Sidoryk-Wegrzynowicz et al., 2010).
PKC Activity Assays
Primary astrocyte cultures were treated with 100 μM, 500 μM, and 1 mM Mn for 4, 8, and 24 h and lysed with RIPA buffer-containing protease and phosphatase inhibitors. PKC activity was measured with a PKC assay kit (Millipore, Temecula, CA) following the manufacturer’s protocol. In brief, lysates containing 50 μg of protein were mixed with the PKC substrate cocktail, assay dilution buffer, PKC lipid activator, and Mg2+/ATP cocktail with 100 μCi of the (γ32P)ATP (Perkin Elmer, Waltham, CA). After incubation for 10 min at 30°C, the radiolabeled substrate was transferred onto the center of a phosphocellulose paper square and immersed into a 50-mL conical tube containing 40 mL 0.75% phosphoric acid. The assay squares were gently shaken for 5 min on a rotator, washed with acetone for 5 min, and drained and transferred to the scintillation vial with the LSC cocktail (Fisher Scientific, Pittsburgh, PA). Radioactivity was measured with a scintillation counter (LS 6500 Beckman), and PKC activity was expressed as a counts per minute/milligram protein.
Analysis of 3H-Gln Uptake
Glutamine (Gln) uptake was measured after incubating the astrocytes for 5 min with 0.25 μCi of l-(G-3H)-Gln per well (specific activity: 49.0 Ci/mmol; Amersham Biosciences, Piscataway, NJ). Cells grown in 24-well plates were washed three times with 2 mL of fresh sodium-HEPES buffer consisting of the following: 122 mM NaCl, 3.3 mM KCl, 0.4 mM MgSO4, 1.3 mM CaCl2, 1.2 mM KH2PO4, 10 mM glucose, and 25 mM HEPES adjusted to a pH of 7.4 with 10 M NaOH and were preincubated for 0.5, 1, 2, 4, 6, 8, and 24 h with this buffer alone or containing 100 nM PMA at 37°C and 95% air/5% CO2. For other uptake analyses, cells were treated for 4, 8, and 24 h with 500 μM or 1 mM MnCl2 in the presence of 100 nM PMA or 10 μM Bis II. The role of PKC in regulating the activity of various Gln transporting systems was analyzed in the presence of system-specific competing amino acids in the presence or absence of PMA for 4, 6, and 24 h. Competitors included histidine (His) for the suppression of system N; threonine (Thr) for the suppression of system ASC; MeAIB for the suppression of system A; and Leu for the suppression of system L. The concentration of unlabeled Gln was 0.158 mM; competing amino acids used to differentiate between the transport systems were added at 32-fold excess (to Gln). The reaction was stopped after 5 min with ice-cold sodium-HEPES buffer, and cells were lysed by incubation with 1 M NaOH for 30 min at 37°C. An aliquot of the lysate was transferred to a scintillation vial, LSC cocktail (Fisher Scientific) was added, and radioactivity was measured in a scintillation counter (LS 6500 Beckman). The remainder of the lysate was used for protein determination by the bicinchoninic acid protein assay (Pierce). Uptake of Gln was expressed as nanomole per minute per milligram of protein.
Statistical Analysis
Results are expressed as the mean ± SD from a minimum of three independent astrocytic cultures. Statistical analysis was carried out with one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test. All analyses were performed with GraphPad Prism 4.02 for Windows (GraphPad Software, San Diego, CA).
RESULTS
Mn(II) Exposure Increases PKC Activity in Primary Cultures of Astrocytes
To test the hypothesis that astrocytic PKC activity is induced in response to Mn(II) exposure, mRNA and protein levels of the classical isoforms, PKCα and PKCβ, as well as the novel isoform, PKCδ, were measured. The quantitative RT-PCR analysis showed no effect on astrocytic PKCα, PKCβ, PKCδ mRNA levels after exposure to Mn(II) (100 μM, 500 μM, and 1 mM) for 4, 8, and 24 h (Fig. 1A,C,E). Treatment with 1 mM Mn(II) for 8 h or treatment with 500 μM and 1 mM Mn(II) for 24 h significantly increased pPKCα protein levels (Fig. 1B). Exposure to 500 μM and 1 mM Mn(II) for 8 and 24 h significantly increased pPKCδ protein levels (Fig. 1D), while phosphorylation levels of PKCβ remained unchanged under all experimental conditions (Fig. 1F). PKC activity assays revealed a significant increase in PKC activity after 4 h (2.25 ± 0.7-fold 500 μM Mn(II) relative to the control; 3.78 ± 0.8-fold 1 mM Mn(II) relative to the control), 8 h (2.19 ± 0.6-fold 500 μM Mn(II) relative to the control; 3.19 ± 0.9-fold 1 mM Mn(II) relative to the control), and 24 h (3.48 ± 0.8-fold 500 μM Mn(II) relative to the control; 3.84 ± 0.8-fold 1 mM Mn(II) relative to the control) (see Fig. 2). Both the Western blot and PKC assay results strongly support the hypothesis that Mn upregulates PKC activity.
Fig. 1.
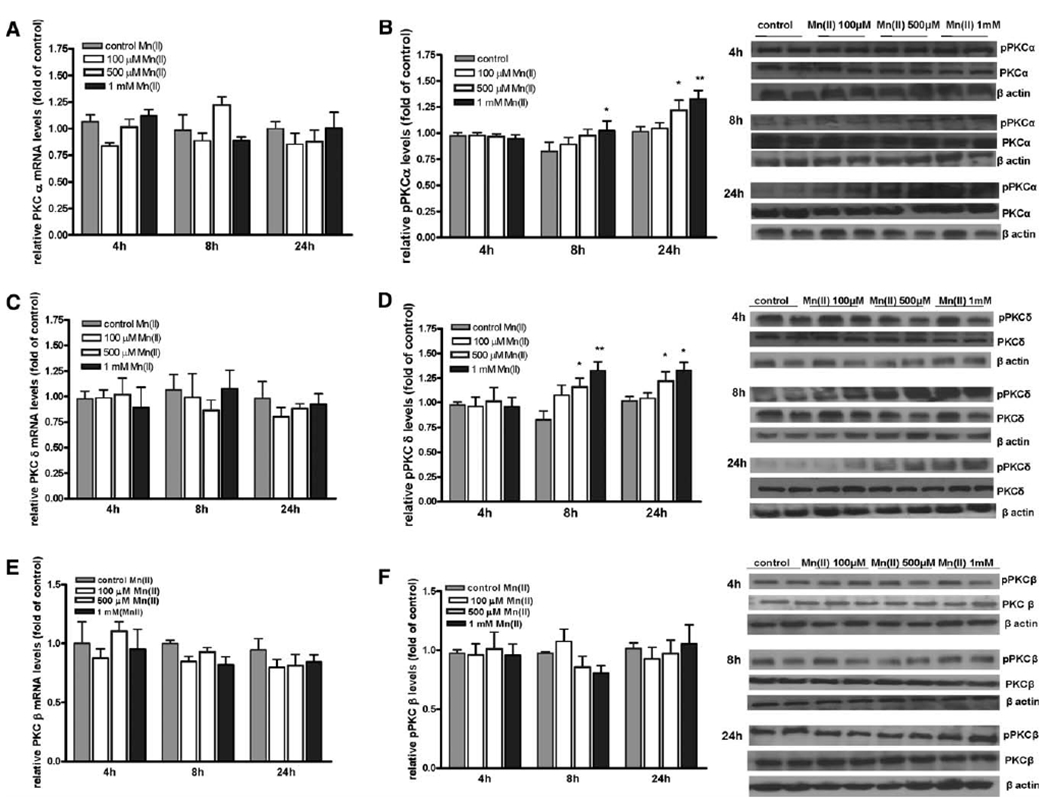
Effect of Mn on astrocytic PKC isozymes mRNA and protein phosphorylation. Levels of PKCs mRNA and protein phosphorylation were determined in primary cultures of astrocytes treated with 100 μM, 500 μM, or 1 mM Mn(II). Expression of PKCα (A), PKCδ (C), and PKCβ (E) mRNA after 4, 8, or 24 h exposure to Mn(II) was measured by quantitative real-time PCR and normalized to levels of GAPDH mRNA. Levels of phosphorylated protein PKCα (B), PKCδ (D), and PKCβ (F) after 4, 8, and 24 h exposure to Mn(II) were determined by Western blotting and normalized to the levels of total protein PKCα, PKCβ, and PKCδ. Data represent the mean ± SD from three to four independent sets of cultures.
Fig. 2.
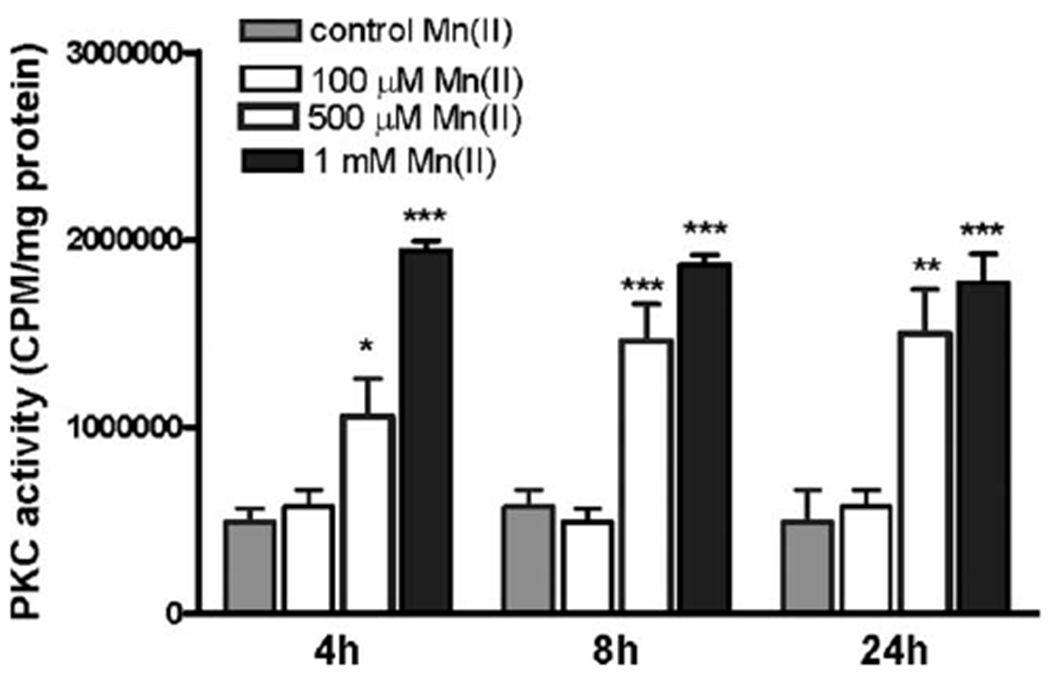
Effects of Mn on astrocytic PKC activity. PKC activity was determined by measuring phosphorylation of substrate peptide with radiolabeled ATP. Confluent cultures of astrocytes were treated with 100 μM, 500 μM, or 1 mM Mn(II) for 4, 8, and 24 h. Lysates containing 1000 μg of proteins were subjected for PKC activity measurement. Data represent the mean ± SD from three independent sets of cultures; *P < 0.05, **P < 0.01, and ***P < 0.001 control versus Mn(II)-exposed cells.
Activation of PKC Attenuates Gln Uptake in Primary Cultures of Astrocytes
Because Mn(II) increases PKC activity (see Fig. 2), we next sought to determine whether the modulation of PKC activity with the PKC-activating phorbol 12-myristate 13-acetate, PMA (100 nM), interferes with the total astrocytic Gln uptake. As shown in Figure 3A, PKC stimulation for 0.5, 1, and 2 h did not significantly affect astrocytic Gln uptake. Continued incubation (≥4 h) with PMA decreased the total astrocytic Gln uptake with a maximal effect after 8 h (49% ± 8% of control), which persisted through 24 h (52% ± 9% of control) post exposure (Fig. 3A).
Fig. 3.
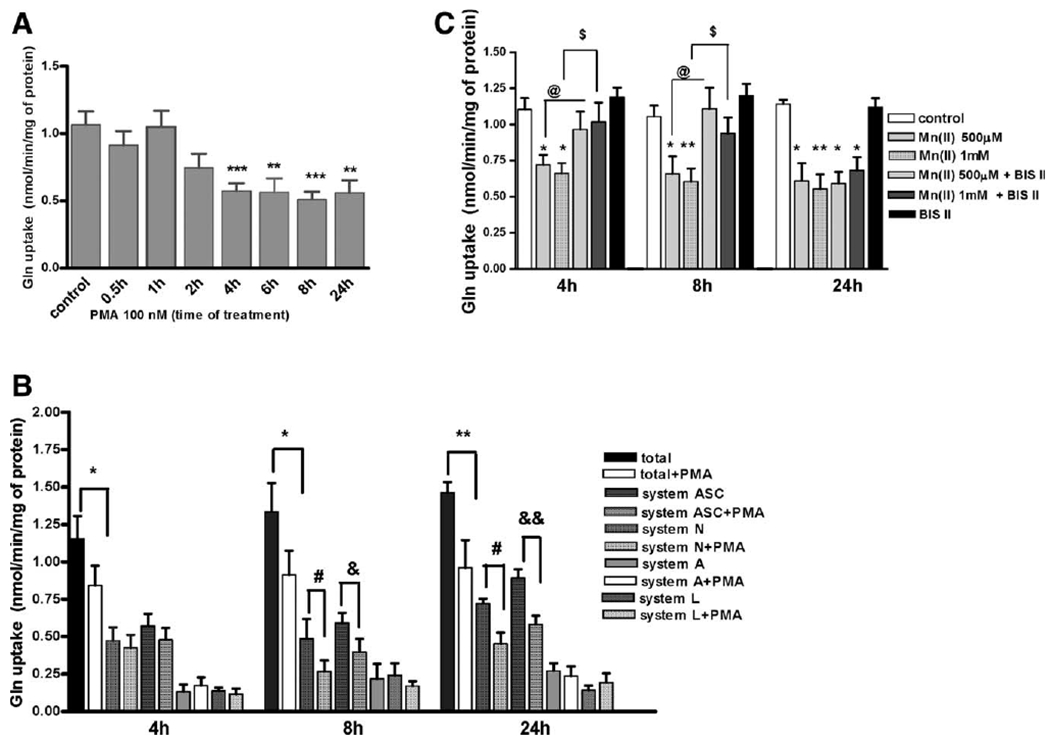
PKC downregulates astrocytic glutamine uptake. Total L-(G-3H)-Gln uptake was measured in control and 100 nM PMA treated for 1, 2, 4, 6, 8, and 24 h primary cultures of astrocytes. Data represent the mean ± SD from three independent sets of cultures each performed in triplicate; **P < 0.01, ***P < 0.001 control versus PMA-exposed cells (A). For competition analysis, cells were incubated for 4, 8, and 24 h with or without 100 nM PMA. Results are mean ± SD of three to four independent experiments each performed in triplicate; *P < 0.05 total versus total PMA-exposed cells, **P < 0.01 total versus total PMA-exposed cells, #P < 0.05 system N versus system N PMA-exposed cells, &P < 0.05 system ASC versus system ASC PMA-exposed cells, and &&P < 0.01 system ASC versus system ASC PMA-exposed cells following (B). Cells were treated with 500 μM and 1 mM Mn(II) in the presence or absence of 10 μM BIS II for 4, 8, and 24 h. Results are mean ± SD of three to four independent experiments each performed in triplicate; *P < 0.05 control versus Mn(II)-exposed cells, **P < 0.01 control versus Mn(II)-exposed cells, @P < 0.05 500 μM Mn(II) versus 500 μM Mn(II) and BIS II-exposed cells, and $P < 0.05 1 mM Mn(II) versus 1 mM Mn(II) and BIS II-exposed cells.
To asses which Gln transporting system(s) is most affected by PKC activation, competition analyses for the A, N, ASC, and L transport systems were performed. Shorter exposure to PMA (4 h) caused a small and statistically insignificant decrease in Gln uptake mediated by the principle Gln transporting systems N and ASC, but not by systems A and L (Fig. 3B). Longer (>4 h) PKC stimulation caused a significant Gln transport inhibition; in the case of system N, after exposure to PMA for 8 h, Gln uptake was 57% ± 8% of control, and after exposure for 24 h, Gln uptake was 63% ± 5% of control. Uptake mediated by system ASC decreased to 67% ± 8% of control after exposure for 8 h and to 62% ± 6% after exposure for 24 h.
Notably, our previous study showed a Mn-induced decrease in Gln uptake, which was mediated by systems N and ASC (Sidoryk-Wegrzynowicz et al., 2009). Accordingly, we next sought to determine whether PKC inhibition can block the Mn-mediated disruption of Gln transport. Astrocytic Gln transport was not affected by preincubation with 10 μM of the general PKC inhibitor, Bis II (Fig. 3C). Interestingly, the inhibition of Gln uptake after exposure to 500 μM and 1 mM Mn(II) for 4 and 8 h was abrogated by Bis II. These results suggest that the Mn-induced downregulation of Gln transport is mediated by PKC-dependent signaling. Bis II failed to reverse the astrocytic inhibition of Gln uptake after 24 h of exposure to Mn(II) (Fig. 3C).
PKC Activation Decreases Gln Transporter Protein Levels in Primary Cultures of Astrocytes
Our previous study showed that Mn(II) decreases SNAT3, SNAT2, ASCT2, and LAT1 transporter protein levels (Sidoryk-Wegrzynowicz et al., 2009). In the current study, we examined whether PKC signaling influences the expression of Gln transporters. As shown in Figure 4A,B, PKC activation by the PMA (100 nM) did not alter the protein levels of SNAT2 and LAT2, but significantly decreased SNAT3 protein levels after 8 h (67% ± 4% of control) and 24 h (58% ± 8% of control), and ASCT2 after 6 h (49% ± 5% of control), 8 h (67% ± 7% of control), and 24 h (61% ± 5% of control) (Fig. 4C,D). Additionally, a surface biotinylation study showed decreased of SNAT3 in the plasma membrane after PMA exposure for 4 h (15% ± 0.9% of control), 6 h (11% ± 0.7% of control), 8 h (17% ± 0.6% of control), and 24 h (21% ± 0.9% of control), decreased of ASCT2 after PMA exposure for 6 h (48% ± 5% of control), 8 h (26% ± 4% of control), and 24 h (28% ± 6% of control), while had no effect on SNAT2 and LAT2 levels (Fig. 5A–D).
Fig. 4.
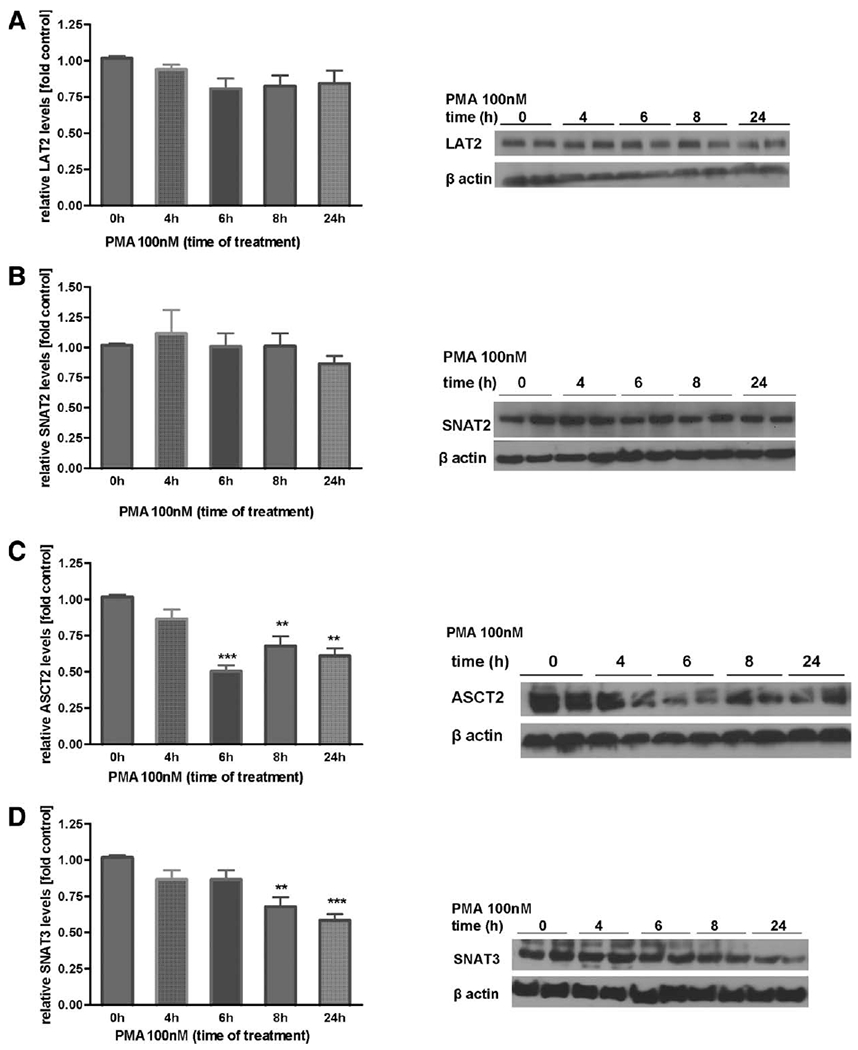
Effect of PKC activation on protein levels of Gln transporters in whole astrocyte lysates. Levels of LAT2 (A), SNAT2 (B), ASCT2 (C), and SNAT3 (D) in primary cultures of astrocytes treated with PMA (100 nM) for 4, 6, 8, and 24 h in whole cell lysates. Shown are results from typical Western blots and immunostaining images. For each lane, protein levels were adjusted to the levels of β-actin. Results are mean ± SD from three to five independent experiments each performed in triplicate; **P < 0.01 control and ***P < 0.01 control (0 h) versus PMA-exposed cells.
Fig. 5.
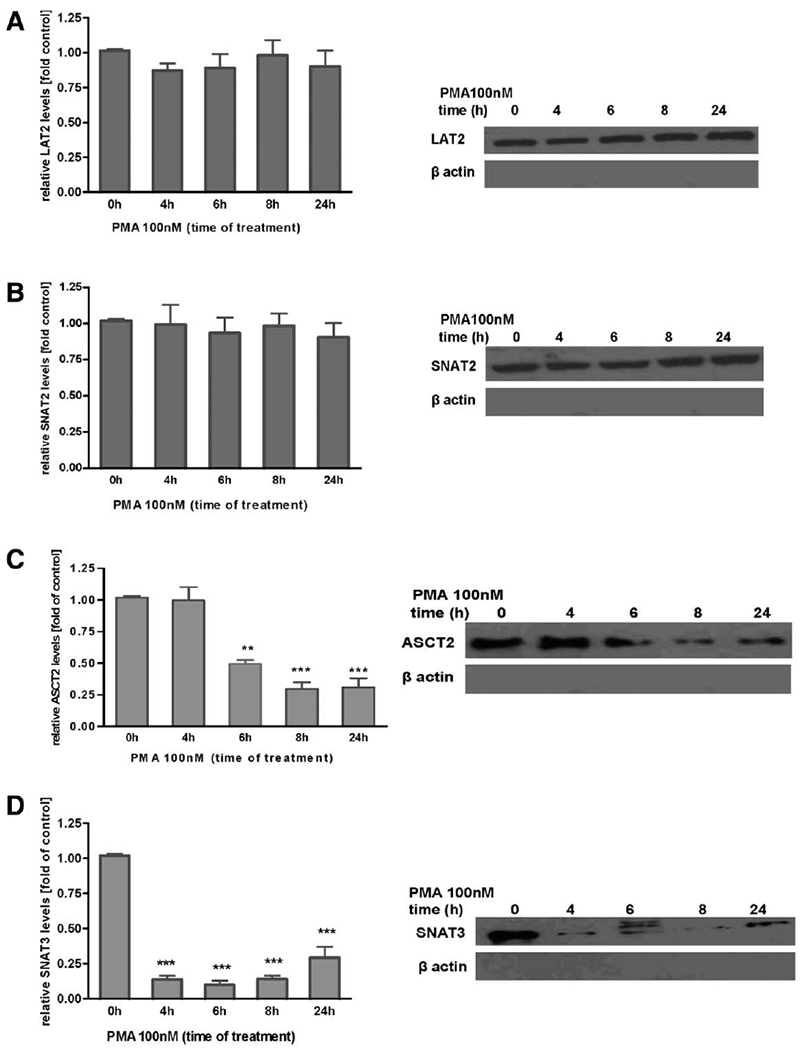
Effect of PKC activation on protein levels of astrocytic Gln transporters in plasma membranes. Levels of LAT2 (A), SNAT2 (B), ASCT2 (C), and SNAT3 (D) were determined upon PKC activation by 100 nM PMA for 4, 6, 8, and 24 h. All samples were adjusted to contain the same amount of proteins prior to loading on the gel. β-Actin was evaluated to assure the purity of cell surface proteins. Results are mean ± SD from three independent experiments each performed in triplicate; **P < 0.01 and ***P < 0.01 control (0 h) versus PMA-exposed cells.
PKCδ Associates with Gln Transporters in Primary Cultures of Astrocytes
Next, we performed a series of experiments to examine the possible conjugation/complexation of Gln transporters with different PKC isoforms. We also sought to determine whether Mn(II) exposure can affect this interaction. Cell lysates were subjected to immunoprecipitation using antibodies against several Gln transporters: SNAT3, SNAT2, ASCT2, and LAT2. We found that the PKCδ isozyme is present in SNAT3 and ASCT2, but not in SNAT2 and LAT2 immunoprecipitates. Exposure to 500 μM Mn(II) increased the interaction of PKCδ interaction with ASCT2 with the strongest signal observed at 4 h (5.34 ± 0.7-fold relative to the control) (Fig. 6A). The interaction of PKCδ and SNAT3 was observed after 2 h of Mn(II) exposure, but not after longer periods of Mn(II) exposure (Fig. 6B). Furthermore, no interaction was observed between PKCδ and SNAT2 and LAT2 (Fig. 6C,D). Notably, the co-localization of SNAT3 and ASCT2 transporters with PKCδ was specific, and there was no interaction between the transporters and the others investigated, namely the PKCα and PKCβ isoforms (data not shown).
Fig. 6.
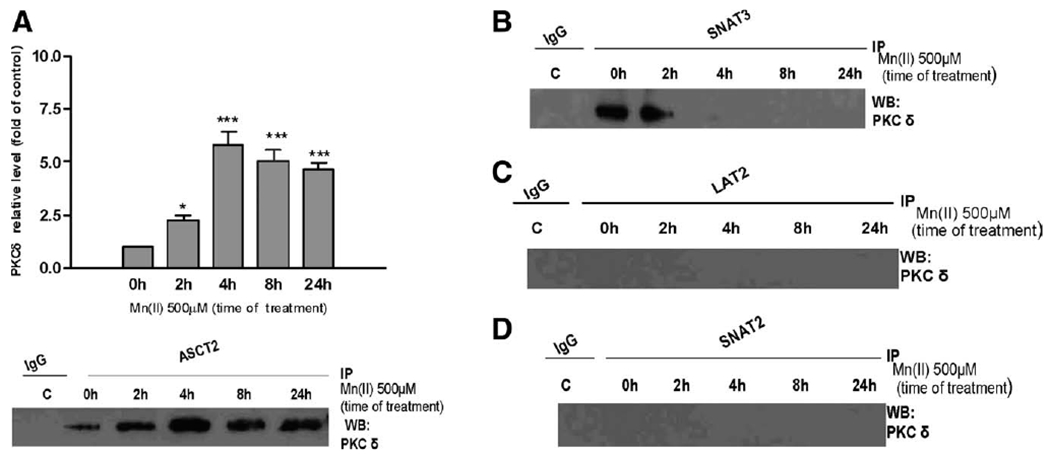
PKCδ interacts with ASCT2 and SNAT3. Cells were exposed to 500 μM Mn(II) for 2, 4, 6, 8, and 24 h then lysed and subjected to immunoprecipitation with antibodies against ASCT2 (A), SNAT3 (B), LAT2 (C), or SNAT2 (D) and probed for PKCδ. In each experiment, an antibody against control IgG was additionally used for immunoprecipitation as the control for nonspecific binding. Similar results were obtained in three independent sets of cultures; *P < 0.05 and ***P < 0.001 control (0 h) versus Mn(II)-exposed.
Mn has been postulated to activate PKCδ by caspase-3-dependent proteolytic cleavage in dopaminergic neurons (Latchoumycandane et al., 2005). Because PKCδ, among all investigated isoforms, was the most affected by Mn exposure (Fig. 1D), we examined PKCδ proteolytic cleavege followed by Mn(II) exposure. Western blot data revealed that the exposure of astrocytes to 1 mM Mn(II) for 4 h (5.25 ± 0.6-fold relative to the control), 8 h (1.51 ± 0.3-fold 500 μM Mn(II) relative to the control; 2.61 ± 0.4-fold 1 mM Mn(II) relative to the control), and 24 h (1.24 ± 0.4-fold 500 μM Mn(II) relative to the control; 5.12 ± 0.6-fold 1 mM Mn(II) relative to the control) caused a dramatic increase in cleaved bands staining positive for PKCδ, suggesting that Mn(II) induces proteolytic cleavage-dependent PKC activation (see Fig. 7).
Fig. 7.
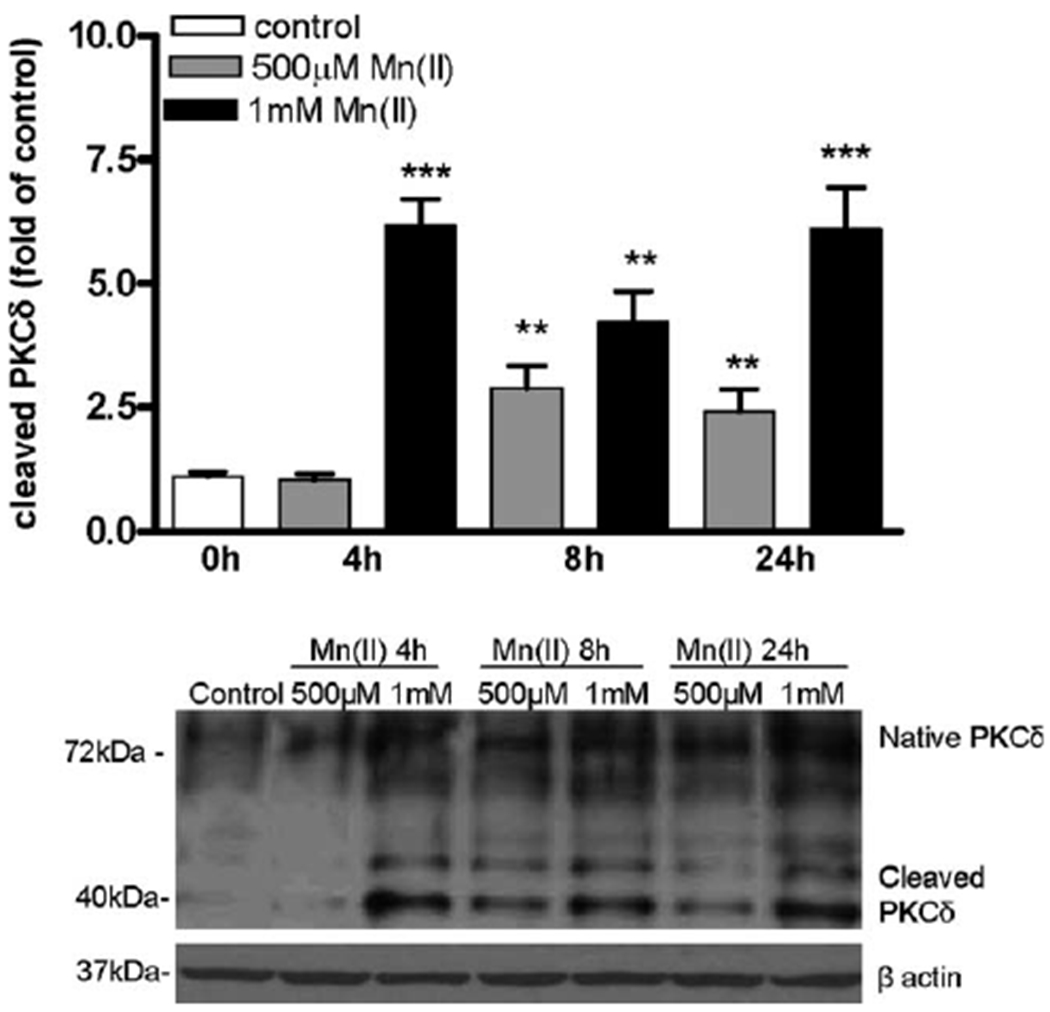
Manganese mediates proteolytic cleavage of astrocytic PKCδ. Cells were exposed to 1 mM and 500 μM Mn(II) for 4, 8, and 24 h. Both native (72 kDa) and cleaved (40 kDa) PKCδ bands were determined by Western blotting. The blot is representative of three independent experiments; **P < 0.01 and ***P < 0.001 control (0 h) versus Mn(II)-exposed cells.
PKC Activation Increases the SNAT3 and Nedd4-2 Interaction in Primary Cultures of Astrocytes
Our previous study (Sidoryk-Wegrzynowicz et al.,2010) demonstrated that, among all investigated Gln transporters, only SNAT3 interacts with the ubiquitin ligase, Nedd4-2 (neural precursor cells expressed developmentally downregulated 4–2). To determine whether PKC activation can affect this protein interaction, astrocytes were exposed to PMA for 1, 2, 4, 6, 8, and 24 h and subjected to co-immunoprecipitation. As shown in Figure 8, the PMA-dependent activation of PKC increased the interaction of SNAT3 with Nedd4-2 with the strongest signal observed at 8 h (4.32 ± 0.8-fold relative to the control). Moreover, PKC stimulation failed to induce the interaction of Nedd4-2 with ASCT2, SNAT2, and LAT2 transporters (data not shown).
Fig. 8.
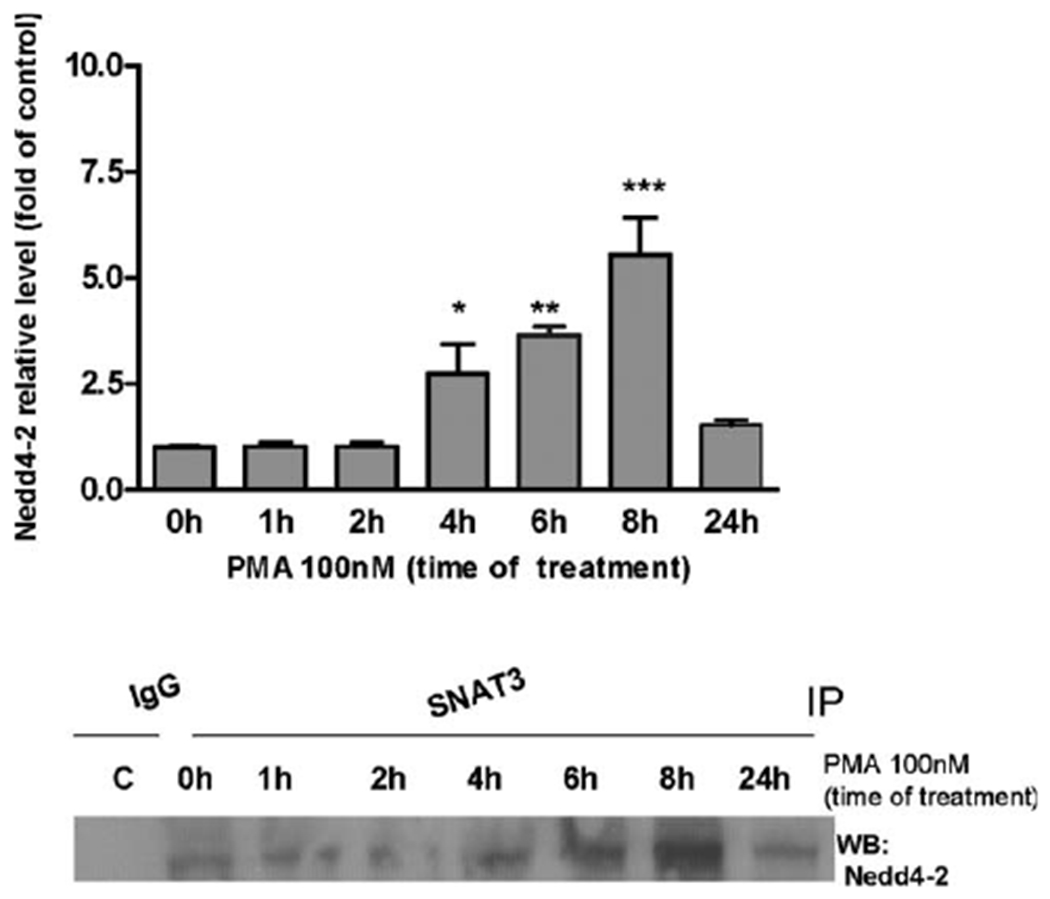
PKC stimulation influences astrocytic SNAT3 and Nedd4-2 interaction. Cells were exposure to 100 nM PMA for 1, 2, 6, 8, and 24 h and subjected to immunoprecipitation using an antibody against SNAT3 and were then probed for Nedd4-2. In each experiment, an antibody against control IgG was additionally used for immunoprecipitation as the control for nonspecific binding. Similar results were obtained in three to four independent sets of cultures; *P < 0.05, **P < 0.01, and ***P < 0.001 control (0 h) versus Mn(II)-exposed cells.
PKC Involvement in the Ubiquitin-Mediated Proteolytic System in Primary Cultures of Astrocytes
Our previous study (Sidoryk-Wegrzynowicz et al., 2010) showed that Mn(II) exposure elevated free ubiquitin (UB) protein levels and total protein ubiquitination. We have previously posited that the Mn(II)-mediated downregulation of Gln transporters involves the ubiquitin-mediated proteolytic system (Sidoryk-Wegrzynowicz et al., 2010). Accordingly, in the current study, we sought to determine whether PKCδ, as a possible downstream enzyme for Mn(II) action, can mediate ubiquitination. We assessed overall levels of protein ubiquitination by Western blotting using an antibody that targets poly- and monoubiqui-tinated proteins, and we measured the total intensity of the smear produced. Results from these studies revealed that activating PKC with PMA for 4 h (6.72 ± 0.8-fold relative to the control), 6 h (6.64 ± 0.6-fold relative to the control), 8 h (5.75 ± 0.9-fold relative to the control), and 24 h (5.66 ± 0.7-fold relative to the control) significantly increased ubiquitination (see Fig. 9). These findings are consistent with the Mn(II)-mediated downregulation of Gln transport via PKC activation and the subsequent stimulation of the ubiquitin-dependent proteolytic system.
Fig. 9.
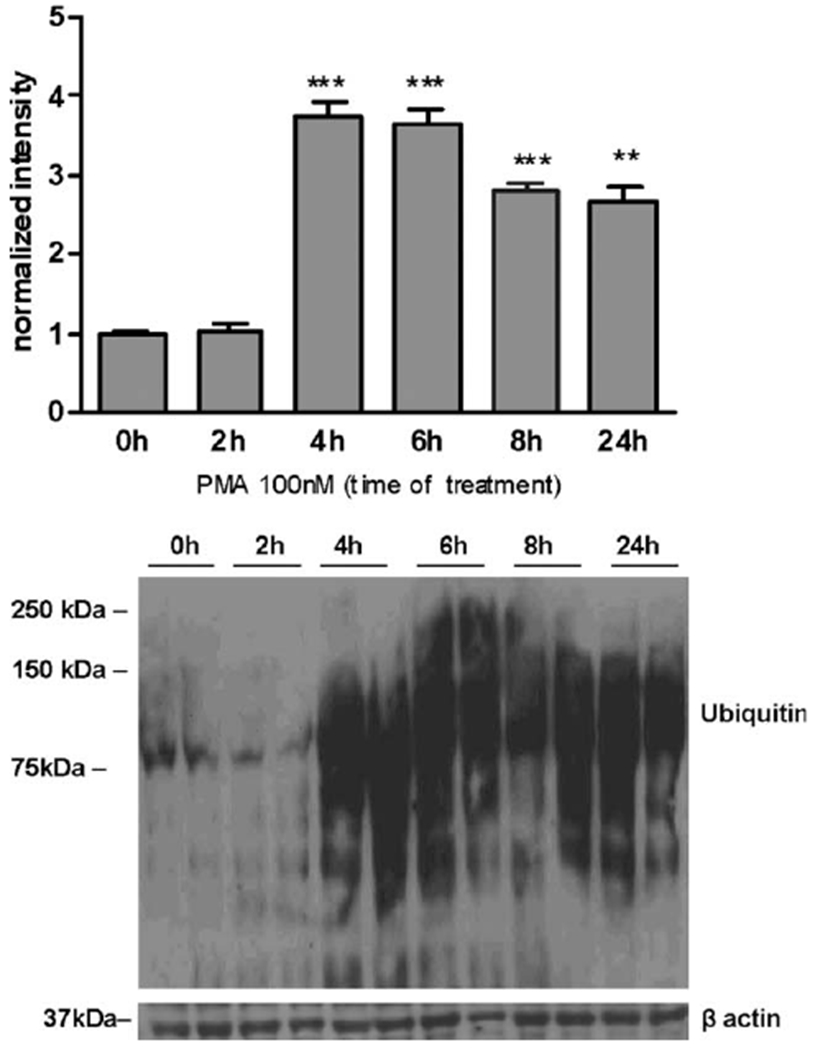
PMA exposure increases ubiquitination in astrocytes. Levels of protein ubiquitination after exposure to 100 nM PMA for 2, 4, 6, 8, and 24 h were determined by Western blotting and normalized to the levels of β-actin protein using an antibody that targets mono- and poly-ubiquitinated proteins. Data represent the mean ± SD from three independent sets of cultures each performed in triplicate; **P < 0.01 and ***P < 0.001 versus control (0 h) versus PMA-exposed cells.
DISCUSSION
In the current studies, we have examined whether PKC signaling is involved in the Mn(II)-mediated disruption of Gln transporter expression and function. We have demonstrated, for the first time, that the phosphorylation levels of both the PKCα and PKCδ isozymes (Fig. 1B,D) as well as PKC activity are significantly increased in Mn-treated astrocytes (see Fig. 2). These findings are consistent with earlier observations of Mn(II)-induced PKC membrane translocation and activation (Liao et al., 2007).
More recent findings have revealed that PKC activation by phorbol esters for a short period of time (1 h) failed to influence Gln uptake in primary astrocytes (Balkrishna et al., 2010); this finding is consistent with our observation (Fig. 3A). Here, we report that the total Gln uptake was downregulated upon 4, 8, and 24 h of exposure with the specific PKC activator PMA. Our observations suggest that prolonged (>4 h) PKC stimulation dysregulates ASC and N system specific-mediated Gln uptake (Fig. 3B). Notably, the general PKC inhibitor, BisII, blocked the Mn(II)-dependent decrease in Gln uptake (Fig. 3C). Accordingly, the Mn(II)-mediated disruption of astrocytic Gln transport, at least in part, was mediated by the dysregulation of PKC signaling.
Additional studies demonstrated that PKC activation decreases ASCT2 (system ASC) and SNAT3 (system N) protein levels in whole astrocytic cell lysates and in plasma membranes, but has no effect on SNAT2 and LAT2 protein levels (Figs. 4 and 5). It has been previously shown that several PKC-regulated transporters are phosphorylated by this kinase (Foster et al., 2008; Jayanthi et al., 2004). Both SNAT3 and ASCT2 contain putative PKC phosphorylation sites that are conserved in the human, rat, and mouse (Balkrishna et al., 2010; Pawlik et al., 2000). SNAT3 was found to be downregulated by PKC, not by the direct phosphorylation of the transporter, but in a caveolin-dependent manner (Balkrishna et al., 2010). On the other hand, recent in situ study showed that PKC activation induces phosphorylation and internalization of SNAT3 (Nissen-Meyer et al., 2011) . The exact mechanism(s) responsible for mediating the downregulation of ASCT2 and SNAT3 by PKC in the primary cultures of astrocytes has not yet been determined.
Notably, our findings showing that PKC activation downregulates SNAT3 and ASCT2 expression (Fig. 4C,D) are consistent with the uptake study results, where PKC stimulation inhibited system N- and system ASC-mediated Gln influx (Fig. 3B). PMA is a general PKC activator; therefore, additional studies with specific inhibitors or gene knockdown will be necessary to ascertain which of the PKC isoforms is specifically involved in the response PKC signaling to Mn.
The negative effect of PKC activation on Gln transporters has been demonstrated for rat SNAT3 in a Xenopus laevis oocyte expression system (Balkrishna et al., 2010) and for ASCT2 in human colon carcinoma cells (Pawlik et al., 2000). Additionally, other transporting proteins, such as the dopamine transporter (DAT) and the glutamate transporter, GLT1, are downregulated by PKC (Foster et al., 2008; Gonzalez et al., 2005). Contrary to these findings, a PKC-activating effect on Gln uptake has been demonstrated in Caco-2 cells (Pan et al., 2002). These observations underscore the complex nature of the regulatory role of PKC in transmembrane transport, with differing functional effects in various cell types.
In addition, we demonstrated that only PKCδ associates with the Gln transporters, SNAT3 and ASCT2, in the primary cultures of astrocytes (see Fig. 6). SNAT3 failed to interact with PKCδ after 4 h of Mn(II) exposure (Fig. 6B). As previously shown, the SNAT3 protein was completely degraded upon Mn(II) treatment (Sidoryk-Wegrzynowicz et al., 2009), which may account for the lack of interaction between SNAT3 and PKCδ under this experimental condition. Notably, exposure to Mn increased PKCδ and ASCT2 binding (Fig. 6A). These observations may suggest that ASCT2 is a major PKCδ target among Gln transporters after Mn exposure. PKC signaling was found to be involved in downregulation of neurotransmiter transporters via different mechanisms, including induction of endocytosis or inhibition of redistribution of the transporters from a subcellular compartment to the plasma membrane as well as disruption of transcription and posttranslational modification (Foster et al., 2008; Gonzalez et al., 2005, Robinson 2002). Further investigations of PKCδ-mediated ASCT2 modification will have to be reconciled with the complex nature of the PKC pathway.
In contrast to many other PKC isozymes, which are considered to be prosurvival mediators, PKCδ is a ubiquitously expressed isozyme, which has a proapoptotic isoform (Reyland, 2009). PKCδ is activated by a variety of apoptotic stimuli. A putative mechanism of PKCδ activation is proteolytic cleavage, which is mediated by caspase-3. Proteolytically activated PKC translocates to the nucleus where it mediates DNA fragmentation. Additionally, the catalytically active PKCδ fragment activates caspase-3 via a feedback activation loop, causing the effective amplification of apoptosis (Kanthasamy et al., 2003). Notably, PKCδ expression increases in the aged brain (Goldberg and Steinberg, 1996), and Parkinsonian neurodegeneration, which shares multiple symptoms with manganism, is strongly associated with PKCδ signaling (Hanrott et al., 2008). In a model of PD, caspase-3 proteolytic cleavage and PKCδ activity were found to be increased (Hanrott et al., 2006). PKCδ was also shown to be activated by Mn(II) in dopaminergic neurons via proteolytic cleavage (Latchoumycandane et al., 2005) Here, we have reported a similar process in cultured primary astrocytes (see Fig. 7). Therefore, Mn(II) exposure increases PKCδ activity via two different mechanisms, namely, phosphorylation and proteolytic cleavage. There is no information in the literature about correlation of these two mechanisms in PKCδ activation. It is possible that those mechanisms are not related, because they are cell compartment-specific: PKCδ activated by phosphorylation is present in cytoplasm or in plasma membrane, while localization of PKCδ activated by proteolytic cleavage is restricted to the nuclear fraction (DeVries-Seimon et al., 2002; Wang et al., 2010). Therefore, it is possible that PKC activated by phosphorylation may disrupt Gln transport in different way than PKC activated by proteolysis, or only one pool of PKCs may affect Gln turnover.
We recently established that the Mn(II)-mediated downregulation of the astroglial transporter, SNAT3, involves the ubiquitin-mediated proteolytic system (Sidoryk-Wegrzynowicz et al., 2010). Ubiquitination precedes the degradation of many receptors, transporters, and channels (Kamsteeg et al., 2006; Miranda et al., 2005, 2007; Miranda and Sorkin, 2007). Therefore, we hypothesized that PKCδ plays a role in the Mn-induced activation of the ubiquitin system. Our results have established that PKC activation causes significant hyperubiquitination (see Fig. 9) along with reduced Gln uptake (see Fig. 3), consistent with parallel observations on the increased ubiquitination, decreased expression, and reduced functionality of GLT1-mediated astrocytic Glu transport (Sheldon et al., 2008). Moreover, it has been shown that PKC activation results in the ubiquitination of the DAT, leading to its internalization and degradation (Miranda et al., 2007; Sorkina et al., 2006). These results clearly demonstrate that PKC activation is involved in the downregulation of multiple neurotransmitter transporters in an ubiquitin-dependent manner.
The ubiquitin ligase Nedd4-2 belongs to the family of E3 ubiquitin ligases and is involved in the regulation of numerous receptor and transporter protein levels (Kabra et al., 2008). Notably, the knockdown of Nedd4-2 results in the dramatic reduction of the PKC-dependent ubiquitination of the DAT (Sorkina et al., 2006). Our previous study showed that Mn(II) exposure increases Nedd4-2 expression and that Nedd4-2 specifically associates with SNAT3 in astrocytes (Sidoryk-Wegrzynowicz et al., 2010). SNAT3 has been also reported as a target of Nedd4-2 in a Xenopus laevis oocyte expression system (Boehmer et al., 2003). Here, we have demonstrated that PKC activation increases the interaction of SNAT3 with Nedd4-2 (see Fig. 8), but does not induce the interaction of Nedd4-2 with other Gln transporters (data not shown). We demonstrated that PMA exposure leads to the hyperubiquitinization of proteins in general; therefore, more specific study will have to be reconciled for investigations of PKC-mediated single transporter modification. However, our findings showing PKC-mediated downregulaltion of SNAT3 in plasma membranes and whole cell lysates as well ability of PKC to promote the targeting of SNAT3 to Nedd4-2 prompted us to hypothesize that PKC(s) signaling may lead to the SNAT3 ubiquitinization. In contrast to the SNAT3 proteolysis pathway, Nedd4-2 is not involved in PKC-mediated ASCT2 downregulation.
In summary, the present studies provide direct evidence that Mn(II) exposure activates PKC. Our results also indicate that PKC inhibition can block the Mn(II)-mediated deregulation of Gln turnover. Specific interactions of PKCδ with SNAT3 and ASCT2, as well as the contrasting role of this isoform in cell survival (comparing to other PKC isoforms), establish, for the first time, a critical role for PKCδ in mediating the Mn-dependent disruption of Gln transporter expression and function.
Acknowledgments
Grant sponsor: National Institute of Environmental Health Sciences; Grant number: R01 ES10563.
Abbreviations
- Bis II
bisindolylmaleimide II
- NEDD4-2
neuronal precursor cell expressed, developmentally downregulated 4-2
- PMA
phorbol 12-myristate 13-acetate
REFERENCES
- Apparsundaram S, Schroeter S, Giovanetti E, Blakely RD. 1998. Acute regulation of norepinephrine transport. II. PKC-modulated surface expression of human norepinephrine transporter proteins. J Pharmacol Exp Ther 287:744–751. [PubMed] [Google Scholar]
- Aschner M, Gannon M, Kimelberg HK. 1992. Manganese uptake and efflux in cultured rat astrocytes. J Neurochem 58:730–735. [DOI] [PubMed] [Google Scholar]
- Balkrishna S, Bröer A, Kingsland A, Bröer S. 2010. Rapid downregulation of the rat glutamine transporter SNAT3 by a caveolin-dependent trafficking mechanism in Xenopus laevis oocytes. Am J Physiol Cell Physiol 299:C1047–C1057. [DOI] [PubMed] [Google Scholar]
- Barbeau A 1984. Manganese and extrapyramidal disorders (a critical review and tribute to Dr. George C. Cotzias). Neurotoxicology 5:13–35. [PubMed] [Google Scholar]
- Battaini F 2001. Protein kinase C isoforms as therapeutic targets in nervous system disease states. Pharmacol Res 44:353–361. [DOI] [PubMed] [Google Scholar]
- Beckman ML, Bernstein EM, Quick MW. 1999. Multiple G protein-coupled receptors initiate protein kinase C redistribution of GABA transporters in hippocampal neurons. J Neurosci 19:RC9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehmer C, Okur F, Setiawan I, Bröer S, Lang F. 2003. Properties and regulation of glutamine transporter SN1 by protein kinases SGK, PKB. Biochem Biophys Res Commun 306:156–162. [DOI] [PubMed] [Google Scholar]
- Bode BP, Reuter N, Conroy JL, Souba WW. 1998. Protein kinase C regulates nutrient uptake and growth in hepatoma cells. Surgery 124:260–268. [PubMed] [Google Scholar]
- Bröer S, Brookes N. 2001. Transfer of glutamine between astrocytes and neurons. J Neurochem 77:705–719. [DOI] [PubMed] [Google Scholar]
- DeVries TA, Neville MC, Reyland ME. 2002. Nuclear import of PKCδ is required for apoptosis: identification of a novel nuclear import sequence. EMBO J 21:6050–6060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daikhin Y, Yudkoff M. 2000. Compartmentation of brain glutamate metabolism in neurons and glia. J Nutr 130:1026S–1031S. [DOI] [PubMed] [Google Scholar]
- Erikson KM, Aschner M. 2003. Manganese neurotoxicity and glutamate-GABA interaction. Neurochem Int 43:475–480. [DOI] [PubMed] [Google Scholar]
- Fang H, Huang Y, Zuo Z. 2002. The different responses of rat glutamate transporter type 2 and its mutant (tyrosine 403 to histidine) activity to volatile anesthetics and activation of protein kinase C. Brain Res 953:255–264. [DOI] [PubMed] [Google Scholar]
- Foster JD, Adkins SD, Lever JR, Vaughan RA. 2008. Phorbol ester induced trafficking-independent regulation and enhanced phosphorylation of the dopamine transporter associated with membrane rafts and cholesterol. J Neurochem 105:1683–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg M, Steinberg SF. 1996. Tissue-specific developmental regulation of protein kinase C isoforms. Biochem Pharmacol 51:1089–1093. [DOI] [PubMed] [Google Scholar]
- Gomeza J, Zafra F, Olivares L, Gimenez C, Aragon C. 1995. Regulation by phorbol esters of the glycine transporter (GLYT1) in glioblastoma cells. Biochim Biophys Acta 1233:41–46. [DOI] [PubMed] [Google Scholar]
- Gonzalez MI, Susarla BT, Robinson MB. 2005. Evidence that protein kinase C alpha interacts with and regulates the glial glutamate transporter GLT-1. J Neurochem 94:1180–1188. [DOI] [PubMed] [Google Scholar]
- Govoni S, Amadio M, Battaini F, Pascale A. 2010. Senescence of the brain: Focus on cognitive kinases. Curr Pharm Des 16:660–671. [DOI] [PubMed] [Google Scholar]
- Hamberger A, Chiang GH, Nylén G, Scheff SW, Cotman CW. 1979. Glutamate as a CNS transmitter. Evaluation of glucose and glutamine as precursors for the synthesis of preferentially released glutamate. Brain Res 168:513–530. [DOI] [PubMed] [Google Scholar]
- Hanrott K, Gudmunsen L, O’Neill MJ, Wonnacott S. 2006. 6-Hydroxydopamine-induced apoptosis is mediated via extracellular auto-oxidation and caspase 3-dependent activation of protein kinase C delta. J Biol Chem 281:5373–5382. [DOI] [PubMed] [Google Scholar]
- Hanrott K, Murray TK, Orfali Z, Ward M, Finlay C, O’Neill MJ, Wonnacott S. 2008. Differential activation of PKCδ in the substantia nigra of rats following striatal or nigral 6-hydroxydopamine lesions. Eur J Neurosci 27:1086–1096. [DOI] [PubMed] [Google Scholar]
- Jayanthi LD, Samuvel DJ, Ramamoorthy S. 2004. Regulated internalization and phosphorylation of the native norepinephrine transporter in response to phorbol esters. Evidence for localization in lipid rafts and lipid raft-mediated internalization. J Biol Chem 279:19315–19326. [DOI] [PubMed] [Google Scholar]
- Kabra R, Knight KK, Zhou R, Snyder PM. 2008. Nedd4–2 induces endocytosis and degradation of proteolytically cleaved epithelial Na+ channels. J Biol Chem 283:6033–6039. [DOI] [PubMed] [Google Scholar]
- Kamsteeg EJ, Hendriks G, Boone M, Konings IB, Oorschot V, van der Sluijs P, Klumperman J, Deen PM. 2006. Short-chain ubiquitination mediates the regulated endocytosis of the aquaporin-2 water channel. Proc Natl Acad Sci USA 103:18344–18349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanthasamy AG, Kitazawa M, Kanthasamy A, Anantharam V. 2003. Role of proteolytic activation of protein kinase Cdelta in oxidative stress-induced apoptosis. Antioxid Redox Signal 5:609–620. [DOI] [PubMed] [Google Scholar]
- Latchoumycandane C, Anantharam V, Kitazawa M, Yang Y, Kanthasamy A, Kanthasamy AG. 2005. Protein kinase C delta is a key downstream mediator of manganese-induced apoptosis in dopaminergic neuronal cells. J Pharmacol Exp Ther 313:46–55. [DOI] [PubMed] [Google Scholar]
- Lee ES, Sidoryk M, Jiang H, Yin Z, Aschner M. 2009. Estrogen and tamoxifen reverse manganese-induced glutamate transporter impairment in astrocytes. J Neurochem 110:530–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao SL, Ou YC, Chen SY, Chiang AN, Chen CJ. 2007. Induction of cyclooxygenase-2 expression by manganese in cultured astrocytes. Neurochem Int 50:905–915. [DOI] [PubMed] [Google Scholar]
- Mackenzie B, Erickson JD. 2003. Sodium-coupled neutral amino acid (System N/A) transporters of the SLC38 gene family. Pflugers Arch 447:784–795. [DOI] [PubMed] [Google Scholar]
- Melikian HE, Buckley KM. 1999. Membrane trafficking regulates the activity of the human dopamine transporter. J Neurosci 19:7699–7710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mena I, Marin O, Fuenzalida S, Cotzias GC. 1967. Chronic manganese poisoning. Clinical picture and manganese turnover. Neurology 17:128–136. [DOI] [PubMed] [Google Scholar]
- Milatovic D, Yin Z, Gupta RC, Sidoryk M, Albrecht J, Aschner JL, Aschner M. 2007. Manganese induces oxidative impairment in cultured rat astrocytes. Toxicol Sci 98:198–205. [DOI] [PubMed] [Google Scholar]
- Miranda M, Dionne KR, Sorkina T, Sorkin A. 2007. Three ubiquitin conjugation sites in the amino terminus of the dopamine transporter mediate protein kinase C-dependent endocytosis of the transporter. Mol Biol Cell 18:313–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda M, Sorkin A. 2007. Regulation of receptors and transporters by ubiquitination: New insights into surprisingly similar mechanisms. Mol Interv 7:157–167. [DOI] [PubMed] [Google Scholar]
- Miranda M, Wu CC, Sorkina T, Korstjens DR, Sorkin A. 2005. Enhanced ubiquitylation and accelerated degradation of the dopamine transporter mediated by protein kinase C. J Biol Chem 280:35617–35624. [DOI] [PubMed] [Google Scholar]
- Newton AC. 2001. Protein kinase C: Structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem Rev 101:2352–2364. [DOI] [PubMed] [Google Scholar]
- Newton AC. 2003. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem J 370:361–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen-Meyer LS, Popescu MC, Hamdani el H, Chaudhry FA. 2011. Protein kinase C-mediated phosphorylation of a single serine residue on the rat glial glutamine transporter SN1 governs its membrane trafficking. J Neurosci 31:6565–6575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norenberg MD, Martinez-Hernandez A. 1979. Fine structural localization of glutamine synthetase in astrocytes of rat brain. Brain Res 161:303–310. [DOI] [PubMed] [Google Scholar]
- Pal PK, Samii A, Calne DB. 1999. Manganese neurotoxicity: A review of clinical features, imaging and pathology. Neurotoxicology 20:227–238. [PubMed] [Google Scholar]
- Pan M, Wolfgang CA, Karinch AM, Lin C, Meng Q, Vary TC, Souba WW. 2002. Protein kinase C activation of intestinal glutamine transport is mediated by mitogen-activated protein kinases. J Surg Res 106:137–144. [DOI] [PubMed] [Google Scholar]
- Pawlik TM, Souba WW, Sweeney TJ, Bode BP. 2000. Phorbol esters rapidly attenuate glutamine uptake and growth in human colon carcinoma cells. J Surg Res 15:149–155. [DOI] [PubMed] [Google Scholar]
- Robinson MB. 2002. Regulated trafficking of neurotransmitter transporters: Common notes but different melodies. J Neurochem 80:1–11. [DOI] [PubMed] [Google Scholar]
- Racette L, McGee-Minnich SM, Moerlein JW Mink JW, Videen TO, Perlmutter JS. 2001. Videen and J.S. Perlmutter, Welding-related parkinsonism: Clinical features, treatment, and pathophysiology. Neurology 56:8–13. [DOI] [PubMed] [Google Scholar]
- Reyland ME. 2009. Protein kinase C isoforms: Multi-functional regulators of cell life and death. Front Biosci 14:2386–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon AL, Gonzalez MI, Krizman-Genda EN, Susarla BT, Robinson MB. 2008. Ubiquitination-mediated internalization and degradation of the astroglial glutamate transporter, GLT-1. Neurochem Int 53:296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoryk-Wegrzynowicz M, Lee E, Albrecht J, Aschner M. 2009. Manganese disrupts astrocyte glutamine transporter expression and function. J Neurochem 110:822–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoryk-Wegrzynowicz M, Lee ES, Ni M, Aschner M. 2010. Manganese-induced downregulation of astroglial glutamine transporter SNAT3 involves ubiquitin-mediated proteolytic system. Glia 58:1905–19012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnewald U, Westergaard N, Schousboe A, Svendes JS, Unsgard G, Petersen SB. 1993. Direct demonstration by [13C] NMR spectroscopy that glutamine from astrocytes is a precursor for GABA synthesis in neurons. Neurochem Int 22:19–29. [DOI] [PubMed] [Google Scholar]
- Sorkina T, Miranda M, Dionne KR, Hoover BR, Zahniser NR, Sorkin A. 2006. RNA interference screen reveals an essential role of Nedd4–2 in dopamine transporter ubiquitination and endocytosis. J Neurosci 26:8195–8205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Mouri T, Nishiyama K, Fujii N. 1975. Study of subacute toxicity of manganese dioxide in monkeys. Tokushima J Exp Med 22:5–10. [PubMed] [Google Scholar]
- Tracqui A, Tayot J, Kintz P, Alves G, Bosque MA, Mangin P. 1995. Determination of manganese in human brain samples. Foren Sci Int 76:199–203. [DOI] [PubMed] [Google Scholar]
- Wang L, Wu B, Sun Y, Xu T, Zhang X, Zhou M, Jiang W. 2010. Translocation of protein kinase C isoforms is involved in propofol-induced endothelial nitric oxide synthase activation. Br J Anaesth 104:606–612. [DOI] [PubMed] [Google Scholar]
- Zhang L, Coffey LL, Reith ME. 1997. Regulation of the functional activity of the human dopamine transporter by protein kinase C. Biochem Pharmacol 53:677–688. [DOI] [PubMed] [Google Scholar]