

Abstract
Coronavirus disease 2019 (COVID-19) is a global health crisis caused by the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), and there is a critical need to produce large quantities of high-quality SARS-CoV-2 Spike (S) protein for use in both clinical and basic science settings. To address this need, we have evaluated the expression and purification of two previously reported S protein constructs in Expi293F and ExpiCHO-S cells, two different cell lines selected for increased protein expression. We show that ExpiCHO-S cells produce enhanced yields of both SARS-CoV-2 S proteins. Biochemical, biophysical, and structural (cryo-EM) characterizations of the SARS-CoV-2 S proteins produced in both cell lines demonstrate that the reported purification strategy yields high-quality S protein (nonaggregated, uniform material with appropriate biochemical and biophysical properties), and analysis of 20 deposited S protein cryo-EM structures reveals conformation plasticity in the region composed of amino acids 614–642 and 828–854. Importantly, we show that multiple preparations of these two recombinant S proteins from either cell line exhibit identical behavior in two different serology assays. We also evaluate the specificity of S protein-mediated host cell binding by examining interactions with proposed binding partners in the human secretome and report no novel binding partners and notably fail to validate the Spike:CD147 interaction. In addition, the antigenicity of these proteins is demonstrated by standard ELISAs and in a flexible protein microarray format. Collectively, we establish an array of metrics for ensuring the production of high-quality S protein to support clinical, biological, biochemical, structural, and mechanistic studies to combat the global pandemic caused by SARS-CoV-2.
Introduction
Most human coronavirus infections are associated with mild symptoms, but in the last two decades, three beta coronaviruses, SARS-CoV, MERS, and SARS-CoV-2, have emerged that are able to infect humans and cause severe disease.1,2 The current pandemic of coronavirus disease 19 (COVID-19) is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2),3 an enveloped virus from the Coronaviridae family with a single positively stranded RNA genome.3 This RNA virus, which likely originated in bats, has several structural components, including Spike (S), Envelope (E), Membrane (M), and Nucleocapsid (N) proteins.2
The S protein is a class I viral fusion protein, which consists of two subunits (S1 and S2) and forms a trimer on the viral membrane.4 The S1 subunit contains the receptor binding domain (RBD), which is responsible for host cell receptor binding, while the S2 subunit facilitates membrane fusion between the viral and host cell membranes.4−7 Host cell proteases are essential for activating the S protein for Coronaviridae cellular entry.8 The S protein in many Coronaviridae, including SARS-CoV-2, is cleaved between the S1 and S2 subunits (S1/S2 cleavage site) and at an additional site present in S2 (S2’ cleavage site).8−10 Similar to SARS-CoV, the SARS-CoV-2 trimeric S glycoprotein mediates viral entry into the cell by utilizing angiotensin converting enzyme 2 (ACE2) as a human cell surface entry receptor.8 Processing of both SARS-CoV and SARS-CoV-2 S proteins is dependent on the endosomal cysteine proteases cathepsin B and cathepsin L and the serine protease TMPRSS2.8 In many coronaviruses, these events lead to conformational rearrangements in S2, which ultimately result in the fusion of the host and viral cell membranes, and delivery of the viral genome into the newly infected cell.11,12
Due to the global COVID-19 pandemic, the SARS-CoV-2 S ectodomain protein has become an important target for clinical, biological, and structural investigations, and future studies will require the efficient and streamlined production of this protein. Clinically, the production of large amounts of S ectodomain protein enables testing of individuals for SARS-CoV-2 seropositivity. Serological testing is important for determining individuals who have been exposed to the virus, and the resulting antibody titer can facilitate identification of potential donors of convalescent plasma.13 Additionally, the S protein ectodomain could be used to identify therapeutic monoclonal antibodies (mAbs) through single B cell cloning from convalescent patients. Furthermore, the development of small molecules and protein therapeutics designed to inhibit viral infection by targeting the S protein needs to be tested and biochemically characterized. Biologically, the complete mechanisms of viral host cell fusion and replication remain to be completely understood. Structural studies will continue to support these ongoing clinical, therapeutic, and biological investigations.
To provide support for these investigations, we examined the expression and purification of two recently reported recombinant versions of the S protein (here termed OptSpike1 and OptSpike2, see Figure 1A). OptSpike1 was initially reported by McLellan and co-workers and used to determine the cryo-EM structure of the Spike protein in the prefusion conformation.7 This construct has additionally been utilized as an antigen for clinical ELISAs at Montefiore Medical Center (Bronx, NY).14 OptSpike2 was described by Krammer and co-workers and has been successfully used to conduct serum ELISAs to test patients for the presence of anti-S antibodies at Mount Sinai Hospital (New York, NY).15 The successful use of both of these recombinant forms of the S protein in clinical (OptSpike1 and OptSpike2) and structural (OptSpike1) applications has made them attractive targets for future COVID-19 studies. Both constructs are cloned into the mammalian expression vector pCAGGS and include either the majority of the ectodomain (OptSpike1: AAs 1–1208) or the full-length ectodomain (OptSpike2: 1–1213) of the SARS-CoV-2 S protein (based on the Wuhan-Hu-1 sequence).16 Both constructs include the K986P and K987P stabilizing mutations and use a T4 Foldon motif (T4) to enhance trimerization.17,18 Both constructs lack the furin cleavage site: OptSpike1 contains the mutation RRAR:GSAS, while OptSpike2 contains the mutation RRAR:A.19 OptSpike1 contains the T4-HRV3C protease cleavage sequence-8x HisTag and a TwinStrepTag at the C-terminus, while OptSpike2 contains a Thrombin cleavage sequence-T4-6x HisTag at the C-terminus.
Figure 1.

Expression and purification of the SARS-CoV-2 S protein. (A) Schematic showing the characteristics of OptSpike1 (upper) and OptSpike2 (lower). (B) Average yields of OptSpike1 and OptSpike2 produced in either Expi293FTM or ExpiCHO-STM cells harvested on either day 6 (Expi293FTM) or day 12 (ExpiCHO-STM) and purified using nickel affinity chromatography and size exclusion chromatography. (C) SDS-PAGE showing apparent molecular mass and purity of OptSpike1 and OptSpike2 purifications. Lanes 1–4: 3 μg of OptSpike1-ExpiCHO-STM, OptSpike1-Expi293FTM, OptSpike2-ExpiCHO-STM, OptSpike2-Expi293FTM in the presence of 100 μM DTT, Lanes 5–8: same as 1–4 without DTT. (D,E) Representative HiLoadTM 16/600 SuperdexTM 200 purification of (D) OptSpike1-Expi293FTM and (E) OptSpike2-Expi293FTM. (F) OptSpike1-ExpiCHO-STM and (G) OptSpike2-CHO after nickel affinity purification (the second peak represents imidazole). (H) Yield from 1 mL crude nickel purification of OptSpike1 or OptSpike2 on indicated day post transfection in Expi293F cells. (I) Yield from 1 mL crude nickel purification of OptSpike1 or OptSpike2 on indicated day post transfection in ExpiCHO-STM cells.
Here, we examine the expression of OptSpike1 and OptSpike2 in Expi293F and ExpiCHO-S expression systems20 and provide detailed protein production protocols, including a Standard Operating Procedure (SOP) in the Supporting Information Appendix 1. We then evaluate the biochemical and biophysical properties of these proteins to define standards of protein quality and evaluate the recognition specificity of the S protein within the human secretome. Furthermore, we demonstrate the functional reproducibility of recombinant S protein in serum ELISAs and develop a multiantigen COVID-19 protein microarray that can simultaneously test for antibodies against multiple antigens, including the full-length S protein, the receptor binding domain (RBD) of the S protein, and the full-length N protein. Finally, we determine the 3.22 Å cryo-EM structure of OptSpike1 produced in ExpiCHO-S cells and the 3.44 Å cryo-EM structure of OptSpike1 produced in Expi293F cells, which are nearly indistinguishable and in agreement with previously described structures.7,21
This work provides a comprehensive strategy, with a wide range of guiding standards and metrics, for production of large quantities of high-quality recombinant S protein for use in future clinical, diagnostic, biological, biochemical, structural, and mechanistic studies that will be needed to combat the global pandemic caused by SARS-CoV-2.
Results
Enhanced Expression and Purification of SARS-CoV-2 S Proteins
To determine conditions for the enhanced expression and purification of the SARS-CoV-2 S protein, we tested the expression of two recently reported recombinant variants (OptSpike1 and OptSpike2, Figure 1A) in both the Expi293F and ExpiCHO-S cells.20 The ExpiCHO-S expression system reproducibly afforded the highest yield per liter (∼28 mg/L) of transfected cells for both versions of the S protein (Figure 1B). OptSpike1 and OptSpike2 were readily produced at a high level of purity, via transient transfection of both Expi293F and ExpiCHO-S cells, followed by purification with nickel affinity and size exclusion chromatography (SEC) (Figure 1C). SEC revealed that both S proteins ran at the appropriate size on a HiLoad 16/600 Superdex 200 gel filtration column (Figure 1D–G), without the presence of significant aggregates. To identify optimal culture growth times, a time course was performed for both constructs in both expression systems, and the optimal day for harvest was found to be day 6 in Expi293F cells and day 12 in ExpiCHO-S cells (Figure 1H,I). Additionally, the ExpiCHO-S standard titer and high titer protocols (Figure S1H) were compared. Collectively, these data reveal that the ExpiCHO-S expression system is more efficient than the Expi293F expression system for the production of recombinant Spike protein. To provide further information about the stability, quality, and aggregation state of OptSpike1 and OptSpike2, their biophysical properties were characterized by a number of approaches.
PNGase F Digestion of Recombinant S Protein Produced in Expi293F or ExpiCHO-S Cells
OptSpike1 and OptSpike2 proteins migrated as slightly larger species on SDS-PAGE when expressed in the Expi293F cells than in the ExpiCHO-S system (Figure 2A). We hypothesized that this difference in size of the proteins was due to differential N-linked glycosylation associated with Expi293F and ExpiCHO-S cells. Heat-denatured OptSpike1 was digested with PNGase F, an amidase that cleaves between the innermost GlcNAc and asparagine residues in N-linked glycoproteins, and analyzed by SDS-PAGE. While undigested OptSpike1 produced in Expi293F cells runs larger than OptSpike1 produced in ExpiCHO-S, the difference in apparent molecular weight is abolished after PNGase F treatment (Figure 2A). It is known that recombinant S proteins are heavily glycosylated,22 and these results confirm that these proteins exist as distinct N-linked glycoforms when expressed in different mammalian expression systems, as has been previously reported with a number of other glycoproteins.23−25 Importantly, these differences in glycosylation had no effect on reactivity with sera from convalescent patients (see Figure 4B).
Figure 2.

Biophysical characterization of recombinant SARS-CoV-2 S protein indicates that it is stable, uniform, and has appropriate molecular mass. (A) SDS-PAGE analysis of OptSpike1-Expi293F (lane 1) or OptSpike1-ExpiCHO-S (lane 2) compared to the same protein denatured and treated with PNGase F for 60 min (lanes 3 and 4, respectively), the lower MW band is PNGase F. (B) Representative SEC traces of OptSpike1-Expi293F analyzed on a Yarra 3 μm SEC-4000 LC column on day 1 post purification (black – left Y axis) and day 14 post purification (blue – right Y axis). (C) Representative SEC traces of OptSpike1-ExpiCHO-S analyzed on a Yarra 3 μm SEC-4000 LC column on day 1 post purification (black – left Y axis) and day 14 post purification (blue – right Y axis). (D) Representative MALS analysis of OptSpike1-ExpiCHO-S day 1 (red curve: light scattering, green curve: UV280, blue curve: refractive index, black line: Mw). (E) Representative MALS analysis of OptSpike1- ExpiCHO-S day 14. (F) Molecular mass of OptSpike1-ExpiCHO-S and PDI (Mw:Mn) determined by MALS (the right peak in panel (E)). (G) Results of SYPRO Orange thermal denaturation of OptSpike1-ExpiCHO-S and OptSpike2-ExpiCHO-S showing a graph of first derivative vs temperature and a table showing Tm1 (left peak) and Tm2 (right peak) for each, all experiments are representative of three individual replicates. (H) The c(s) distribution obtained by AUC analysis of OptSpike1-ExpiCHO-S at 4.8 μM total protein concentration. The asterisk denotes the S protein trimeric species. The best fit molecular mass resolved for the trimer is 455 kDa. The best fit ratio of f/fo is 1.63.
Figure 4.
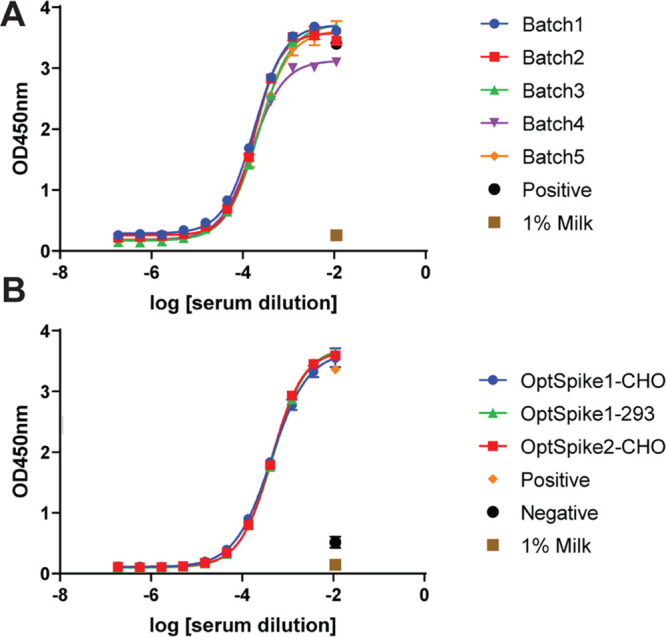
Highly reproducible data in COVID-19 convalescent serum ELISA experiments using OptSpike1 and OptSpike2. (A) Five different production batches of OptSpike1-ExpiCHO-STM were tested by ELISA for the detection of anti-S IgG antibodies from convalescent COVID-19 patients. Low batch-to-batch variability in OptSpike1 was observed, with no statistically significant differences in EC50 values between batches (P > .9999). (B) Different S protein constructs OptSpike1-ExpiCHO-STM, OptSpike1Expi293FTM, and OptSpike2-ExpiCHO-STM were tested by ELISA for the detection of anti-S IgG antibodies from confirmed COVID-19 convalescent patients. Very little variability was seen between different constructs and expression cell lines used, with no statistically significant differences in EC50 values (P > .9999).
Size Exclusion Chromatography and Multiangle Light Scattering (SEC-MALS) Analysis of OptSpike1 and OptSpike2
The purified, concentrated OptSpike1 and OptSpike2 proteins were analyzed by FPLC on a Superose 6 Increase 10/300 GL column (Figure S1A–D) and by HPLC on a Yarra 3 μm SEC-4000 LC column (Figure 2B,C, Figure S1E,F), both of which are appropriate for resolving proteins in the range of, and greater than, the predicted molecular mass (based on the amino acid sequence) of ∼420 kDa of trimeric OptSpike1 and OptSpike2. On the Superose 6 Increase 10/300 GL column, OptSpike1 and Optspike2 eluted as a single peak within the included volume, with an apparent molecular mass of ∼670 kDa, and did not form aggregates (Figure S1A–D). Interestingly, analysis on the Yarra 3 μm SEC-4000 LC column showed that both OptSpike1 and OptSpike2, expressed in either Expi293F or ExpiCHO-S cells, eluted as two partially overlapping peaks (elution times of 10.35 and 11.00 mL) (Figure 2B,C, Figure S1E,F), which could not be resolved on the Superose 6 Increase 10/300 GL column (Figure S1A–D). Furthermore, after storage at 4 °C for 14 days, the distribution of the elution volume shifted entirely to the faster migrating peak at 10.35 mL (Figure 2B,C). The OptSpike1 produced in ExpiCHO-S cells was analyzed with multiangle light scattering (MALS) 1 day after purification, and the polydispersity index (PDI) for species present across both peaks was 1.006, indicating that the species across both peaks are uniform with respect to molecular mass (Figure 2D,F). The molecular mass of this species was calculated to be 486 kDa ± 2.1% using MALS. This measurement is larger than the predicated molecular mass of 420 kDa (based on the amino acid sequence of trimeric OptSpike1 and not accounting for glycans). The PDI of OptSpike1 on day 14 (which ran as one peak) was determined to be 1.005, which also indicated that this peak is uniform, and the molecular mass from MALS was 493 kDa ± 1.08% (Figure 2E,F). OptSpike-2-Expi293F and OptSpike2-ExpiCHO-S exhibited a similar two-peak profile when analyzed on the Yarra 3 μm SEC-4000 LC column (Figure S1E,F) Together, these data indicate the existence of closely related species that likely arise from conformational conversion and not from changes in the oligomeric state (see below).
Thermal Denaturation of OptSpike1 and OptSpike2
Differential scanning fluorimetry (DSF) was used to assess the relative thermal stabilities of OptSpike1 and OptSpike2 produced in ExpiCHO-S cells. Analysis of DSF data revealed no discernible difference in melting behavior, with both OptSpike1 and 2 exhibiting two similar melting transitions (OptSpike1 Tm1 = 49.2 ° C, Tm2 = 63.4 ° C and OptSpike2 Tm1 = 48.7 ° C, Tm2 = 63.9 ° C) (Figure 2G). This consistent behavior suggests that differences in expression yields of OptSpike1 and Optspike2 are likely not due to inherent differences in protein stability.
Analytical Ultracentrifugation (AUC) Analysis of OptSpike1
To confirm that purified OptSpike1-ExpiCHO-S is a stable trimer and further evaluate the molecular mass, we conducted a series of sedimentation velocity experiments as a function of protein concentration. Six concentrations of S protein were centrifuged spanning a range from 136 nM to 4.8 μM. The distribution of sedimentation coefficients g(s*) and in particular the maxima of the main peak are invariant over the concentration range examined (Figure S1H). This behavior demonstrates that the protein molecules present in the preparation are stable noninteracting particles. The distribution of species present in a solution of noninteracting particles can be deconvolved using a continuous distribution, c(s), analysis (Figure 2H, Figure S1H). The dominant peak (68%) in the c(s) distribution has a mass of 455 kDa, consistent with the S protein trimer being the dominant protein present in the solution. This calculated molecular mass is comparable to both the predicted molecular mass based on the amino acid sequence (420 kDa) and the molecular mass determined by MALS (490 kDa). The best fit ratio of f/fo is 1.63, indicative of significant geometric asymmetry. These data demonstrate that OptSpike1 is a stable trimer in solution from nM to μM protein concentrations.
Biochemical Characterization of OptSpike1 Interaction with Human ACE2
Purified OptSpike1-ExpiCHO-S protein activity was demonstrated by binding to human ACE2 (hACE2) but not mouse ACE2 (mACE2) or human CD26 (hCD26), the entry receptor for MERS. A 500 nM OptSpike1 trimer was incubated with FreeStyle 293-F cells expressing hACE2, mACE2, or hCD26—all C-terminally tagged with GFP—and binding was detected by flow cytometry with an anti-His PE labeled antibody recognizing the His8 tag on the S protein (see Figure S3A). The expression and cell surface localization of hACE2, mACE2, and hCD26 were confirmed by antibody staining (Figure S3E,F,K). Strong binding to cells expressing hACE2 (Figure 3A,C) was observed, but not to cells expressing mACE2 or hCD26 (Figure 3B,C), confirming that the S protein does not bind to CD26 (in agreement with a previous report21) and confirming specificity of the S protein for hACE2. Titration of hACE2-expression FreeStyle 293-F cells with recombinant OptSpike1 (0.0025–400 nM) yielded an EC50 of 2.0 ± 0.6 nM (Figure 3D).
Figure 3.

SARS-CoV-2 S protein binds to human ACE2 but not mouse ACE2 or human CD26. (A) Representative flow plot of data quantified in panel (C) showing that OptSpike1-ExpiCHOSTM binds to FreeStyleTM 293-F cells expressing hACE2, binding was detected using an antibody against the 8x-HIS tag on OptSpike1. (B) Representative flow plot of data quantified in panel (C) showing that OptSpike1-CHO does not bind to FreeStyleTM 293-F cells expressing mACE2, binding was detected using an antibody against the 8x-HIS tag on OptSpike1. (C) 500 nM OptSpike1-CHO was incubated with cells expressing human ACE2, mouse ACE2, or human CD26, binding was detected with an anti-HIS antibody, and data was acquired by flow cytometry, n = 4. (D) OptSpike1-ExpiCHO-STM was titrated on FreeStyleTM 293-F cells expressing human ACE2 from .0025–.400 nM, binding was quantified with an anti-HIS antibody using flow cytometry, and a binding curve was fit in GraphPad using the equation Y=Bmax*Xh/(Kdh+Xh), n = 4.
Screening OptSpike1 for Binding to Members of the Human Secretome
To further evaluate the specificity of the SARS-CoV-2 S protein, OptSpike1-ExpiCHO-S was screened for binding to 900 members (around ∼20%) of the human secretome (from the Ig superfamily, TNFR superfamily, Integrin family, chemokine family, and GPCR family – see Table S1). Each member of this library (tagged with cytosolic GFP to confirm expression – see Table S1 and Figure S3) was individually transfected into FreeStyle 293-F cells. Individual transfections were then incubated with 200 nM OptSpike1 in the 96-well plate format, and binding was analyzed by flow cytometry with an anti-HIS PE labeled antibody as above. hACE2-GFP expressing cells were included as a positive control on each plate. Integrins were screened both as individual transfections and as alpha-beta pairs. While strong binding to each replicate of hACE2 was detected, we did not detect binding to any other members of the library (Figure S3, Table S1), including proteins suggested to be targets of S protein binding, including CD14726,27 and Siglec 9 and 10,28 as well as the host cell receptors for other coronaviruses, including Ceacam1, which is the receptor for Murine Coronavirus29 and CD26 and Ceacam5, which are both receptors for MERS.30 Expression and cell surface presentation of these putative receptors were validated using monoclonal antibodies (Figure S3I–L).
OptSpike1 and OptSpike2 Can Be Used Reproducibly and Interchangeably in COVID-19 Convalescent Serum ELISA
Enzyme-linked immunosorbent assays (ELISAs) are commonly used in clinical settings to detect the presence of viral antibodies. Therefore, we assessed the reactivity of serum antibodies from one COVID-19 convalescent patient toward multiple independent preparations of the S ectodomain proteins (Figure 4A). We analyzed the EC50 and standard error by one-way ANOVA and found that there is no statistical significance in EC50 values when different production batches of OptSpike1 were used as the target (Figure 4A, p > .9999) or when the different S protein constructs OptSpike1-ExpiCHO-S, OptSpike2-ExpiCHO-S, and OptSpike1-Expi293F were used as the target (Figure 4B, p > .9999). These data demonstrate that the expression and purification protocols reported herein consistently yield OptSpike1 with reproducible behavior in ELISAs detecting anti-S IgG antibodies. Additionally, we have shown that reactivity of serum antibodies from convalescent patients toward OptSpike1 and OptSpike2 is not distinguishable.
Development of a COVID-19 Multiantigen Protein Array
Protein microarray technology allows for the high-throughput, multiplexed screening of numerous parameters within a single experiment.31 To simultaneously and rapidly screen the serum from convalescent COVID-19 patients against multiple SARS-CoV-2 antigens at once, we developed a COVID-19 multiantigen protein array presenting purified S protein, the RBD of the S protein (Figure S2C), and Nucleocapsid protein (Figure S2A,B,D) of SARS CoV-2 (printed with a Marathon Argus piezoelectric printer from Arrayjet). This multiantigen array was challenged with either convalescent sera from COVID-19 patients that had been previously diagnosed using RT-PCR and ELISA tests or serum from control individuals that had been collected prior to the SARS-CoV-2 outbreak in November 2019. Seropositivity of COVID-19 patients was confirmed by ELISA (Figure S5). In total, serum from eight individuals was screened. Individuals 2, 3, 5, 6, and 7 are confirmed SARS-CoV-2-positive. Individuals 1, 4, and 8 had serum collected before SARS-CoV-2 was reported (Figure 5 and Figure S4).
Figure 5.

SARS-CoV-2 multiantigen protein array. (A) Representative image of antigen detection from SARS-CoV-2 positive serum 2. Serum 2 detects all of the SARS-CoV-2 antigens printed on the protein array: S protein (OptSpike1 and 2), receptor binding domain of S protein (RBD), and Nucleocapsid protein (N). (B) Representative image of screening with SARS-CoV2 negative serum 4. Serum 4 is negative for all SARS-CoV-2 antigens printed and only positive control, human IgG1 (hIgG1) is detected. (C) Representative image of antigen detection from SARS-CoV-2 positive serum 5. Serum 5 does not detect all of the SARS-CoV-2 antigens printed on the protein array and only detects S protein and N proteins. All protein arrays contain negative buffer and 1× PBS controls as well as negative protein controls (huAche). (E) Quantifications of serums 2, 4, and 5 results with titrations of OptSpike1 produced in ExpiCHO-S cells from 200 to 25 pg. (F) Quantifications of serums 2, 4, and 5 results with titrations of RBD from 200 to 25 pg. (G) Quantifications of serums 2, 4, and 5 results with titrations of N protein from 200 to 25 pg.
Each array was printed with 16 identical subarrays. A range of target protein concentrations was spotted (25, 50, 100, and 200 pg per spot), and each serum was screened in duplicate (Figure 5A–C, Figure S4A,B). Recombinant human IgG1, which is recognized by the anti-human-647 labeled secondary antibody, was included in each array as a positive control for protein printing. Negative controls (human acetyl cholinesterase (huAche), printing buffer, and PBS) were used to determine nonspecific binding and background correction for data analysis. All confirmed COVID-19 patients had strong antibody responses to the S protein in our multiantigen protein array, which was in good agreement with the S protein ELISAs (Figure S4D–F, Figure S5). Additionally, all confirmed COVID-19 patient sera tested positive for N protein in our protein multiantigen array (Figure S4H). Antibodies against the RBD were detected in serums 2, 3, 6, and 7. However, antibodies against the RBD from patients 5 (which additionally had relatively lower detectable levels of antibodies against the S and N proteins) could not be detected in this assay (Figure 5D–G, Figure S4C–H). These data demonstrate the ability of the COVID-19 multiantigen protein array to simultaneously analyze antibody responses to multiple antigens in a high-throughput format and again validate the use of our recombinant S protein in an antigen detection platform.
Understanding variable antibody responses to the spectrum of SARS-CoV-2 antigens will substantially impact our understanding of population-wide immune responses to COVID-19.
Cryo-EM Structure of the SARS-CoV-2 S Protein Produced in ExpiCHO-S Cells
Cryogenic electron microscopy (cryo-EM) single particle reconstruction was conducted to assess the tertiary and quaternary arrangements of the S protein expressed in the Expi293F and ExpiCHO-S systems (Table S2). Aliquots of purified OptSpike1 protein from each expression host (1 mg/mL in 50 mM Tris and 250 mM NaCl, pH 8.0) were applied to cryo-EM grids, blotted, and plunge-frozen in liquid ethane at LN2 temperature. Micrographs collected on a Titan Krios at 300 kV, using a K3 camera, were aligned and motion-corrected followed by CTF estimation and 2D classification as described in the Materials and Methods. Particle picking, 3D refinement, and reconstruction yielded high-quality maps for samples arising from both Expi293F and ExpiCHO-S productions (Figure S6, Table S2). Using the OptSpike1-ExpiCHO-S material, we obtained a 3.22 Å resolution reconstruction of the symmetrical trimer in the closed conformation (all three RBDs in the down position) (Figure 6A,B, Figure S6, Table S2). Similarly, the OptSpike1-Expi293F material yielded a 3.44 Å reconstruction of the symmetrical trimer in the closed conformation (Figure S6, Table S2). Rigid body refinement and manual adjustment in COOT followed by RSR refinement against the reconstructed maps with PHENIX32 yielded models with good stereochemistry and validation metrics (Table S2, Figure 6, Figure S6A–F). Comparison of the molecular envelopes from the current work with the recently deposited cryo-EM structure of the closed state of the SARS-CoV-2 SPIKE protein (PDB: 6VXX) showed excellent agreement. For example, structural alignment of the trimeric ExpiCHO-produced S protein coordinates with those from 6VXX resulted in a core RMSD of 0.63 Å for 2889 aligned Cα pairs. Likewise, comparison of the coordinates for the Expi293F and ExpiCHO-S coordinates from the current work aligned with an RMSD of 0.60 Å over 2696 aligned Cα pairs. This is consistent with proper tertiary folding and trimeric quaternary organization of both samples.21
Figure 6.

Cryo-EM structure of OptSpike1-ExpiCHO-STM in the closed state. (A) Side view of the SARS-CoV-2 OptSpike1 trimer in the prefusion conformation. (B) Top view of the SARS-CoV-2 OptSpike1 trimer in the prefusion conformation. Two protomers are displayed with the cryo-EM density maps (dark gray and light gray), and the third protomer is displayed as a ribbon structure (magenta) with glycans represented on the ribbon structure (cyan).
Analysis of Cryo-EM Structures of the SARS-CoV-2 S Protein Reveals Two Distinct Conformations at Residues 614–642
We next compared all available structures of the SARS-CoV-2 S protein in the closed (all three RBDs pointing down, no additional binding partners) conformation (Figure 7A,B). The structures were aligned pairwise to Cα atoms in chain A of PDB 6VXX using PyMol, revealing general tertiary and quaternary agreement among all models. It is apparent that the N-terminal domain and the RBD of S1 show greater positional variability among the structures than do the S2 domains (Figure 7A,B). One region, amino acid segment 614–642, adopted two distinct conformations across different models (Figure 7C–F). In nine structures that align well in this region (conformation 1: 6VXX, 6X29, 6X2C, 6X79, 6ZOX, 6ZOY, 6ZP0, 6ZP1, 6ZWV), the amino acid segment 621–640 was not modeled, presumably due to disorder (Figure 7C). Seven structures (conformation 2: 6XR8, 6ZGE, 6ZGH, 6ZGI, 6ZP2, 7JJI, 7JJJ) show a different chain trace in this region (first reported elsewhere33) and are missing amino acids 619–631, except for 7JJJ, which exhibits a continuous trace in this region (shown in green – Figure 7D) by virtue of stabilization due to packing against the N-terminal domain of a second trimer. Additionally, these conformation structures contain an ordered segment spanning amino acids 828–854 (colored blue, Figure 7D) not observed in the above structures characterized by conformation 1 (Figure 7C).
Figure 7.
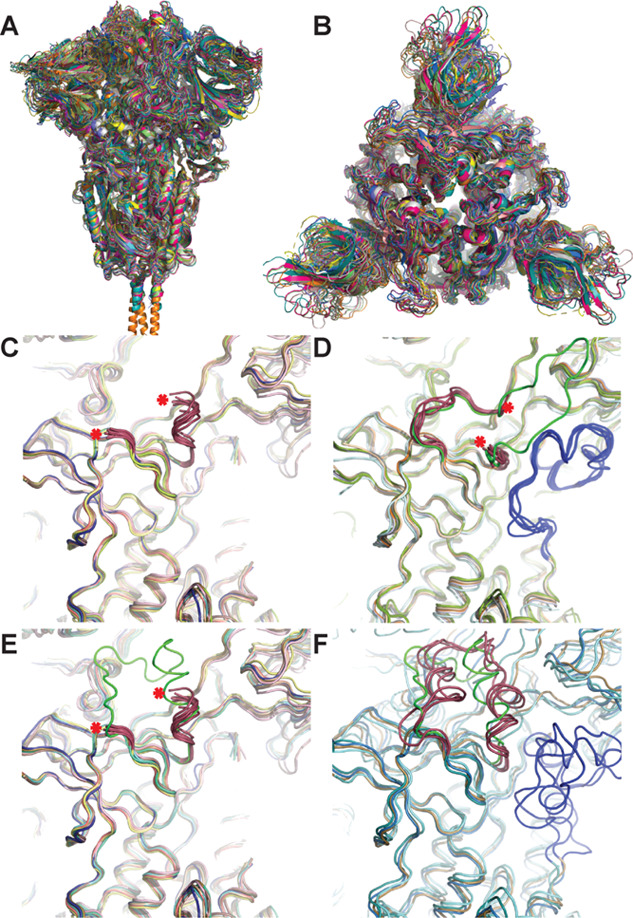
Comparison of 20 S protein structures in the RBD down conformation reveals two distinct structural variations at amino acids 614–642. (A) Side and (B) top views of superposition of 19 trimeric S structures with RBDs in the down conformation (6VXX, 6X29, 6X2C, 6X6P, 6X79, 6XLU, 6XM5, 6XR8, 6ZGE, 6ZGH, 6ZGI, 6ZOX, 6ZOY, 6ZOZ, 6ZP0, 6ZP1, 6ZP2, 6ZWV, 7JJI) aligned pairwise to Cα atoms in chain A of 6VXX using Pymol. (C–F) Examination of the two regions of S protein structures exhibiting structural variability at amino acid segments 614–642 (colored magenta or green in all panels, red asterisk denotes the end of the trace) and 828–854 (shown in blue): (C) nine structures (6VXX, 6X29, 6X2C, 6X79, 6ZOX, 6ZOY, 6ZP0, 6ZP1, 6ZWV) displaying conformation 1 between the region 614–642 and the amino acid segment 621–640 was not modeled; (D) seven structures (6XR8, 6ZGE, 6ZGH, 6ZGI, 6ZP2, 7JJI, 7JJJ) displaying conformation 2 in this region and missing amino acids 619–631 (except 7JJJ, shown in green) and which also have an ordered segment spanning amino acids 828–854 (colored blue) not observed in structures in panel (C); (E) the structure from this work (6X6P, in green) overlaid on the structures from panel (C) showing general structural agreement; (F) five structures (6X6P, 6XLU, 6XM5, 6ZOZ with 6X6P in green for 621–640) for which amino acids from 621 to 640 in conformation 1 were modeled, amino acids 828–854 are shown in blue.
The structure from the present work (6X6P) displays conformation 1 and is overlaid (in green) on the structures in Figure 7C, which reveals closely matched organization for the amino acid segments that are mutually present (614–620 and 641–642) (Figure 7E).We next overlaid all structures displaying conformation 1, which also exhibited a complete trace in this region (residues 614–642) (6X6P, 6XLU, 6XM5, 6ZOZ in magenta; 6X6P in green Figure 7F). These structures are not fully conserved structurally in segment 614–642 but generally follow a similar chain path distinct from conformation 2. Three of these four conformation 1 structures also have features modeled for amino acids 828–854 (colored blue Figure 7F), although these segments do not overlay as well as those present in the conformation 2 structures (colored blue, Figure 7D). Based on this analysis, it is likely that at least two discrete structural states for the amino acid segment spanning residues 614–642 exist, which may influence the conformation of residues 828–854.
Discussion
To effectively address the COVID-19 global health crisis, reproducibility in biological and clinical studies will be critical. Efficient and uniform production of key biological reagents is essential for the generation of reliable data in the coming months and years. Herein, we describe our protocols (including the SOP) for the optimized production of two recently reported recombinant forms of the SARS-CoV-2 S protein. We show that using the ExpiCHO-S expression system, recombinant S protein can be reproducibly produced in both high quantities and high quality. Our reported yields of 28 mg/L for OptSpike1 and 18 mg/L for OptSpike2 exceed those previously reported (OptSpike1: .5 mg/L in FreeStyle 293-F cells,7 OptSpike2: 5 mg/L in Expi293F cells, and .5 mg/L in insect cells15). To validate protein quality, we employed analytical size exclusion chromatography, multiangle light scattering, thermal denaturation, and analytical ultracentrifugation and demonstrated that these recombinant S proteins have similar biophysical properties and antigenicity.
Interestingly, we found that all preparations consistently exhibited different behaviors when examined on two different analytical size exclusion columns. These proteins eluted as a single peak on the Superose 6 Increase 10/300 GL column (agarose-based resin) but eluted as two overlapping peaks on the Yarra 3 μm SEC-4000 (silica-based resin). We also noted a time-dependent evolution as the distribution of peaks on the Yarra 3 μm SEC-4000 moved entirely to the faster migrating species. Furthermore, MALS analysis demonstrated that there was no mass difference between the two overlapping peaks and AUC analysis of OptSpike1-ExpiCHO-S confirmed that the predominant species in solution was a stable trimer. We propose a modest time-dependent structural alteration of the S protein, involving two states with distinct physicochemical properties, resulting in differential interactions with the silica-based Yarra 3 μm SEC-4000 resin. This proposal is consistent with the reported cryo-EM structures displaying the trimeric S protein existing in both a closed conformation with all RBDs pointing downward (PDB: 6VXX) and an open conformation with one RBD pointing upward (PDB: 6VSB and 6VYB).7,21 Future work will be required to fully understand the nature and functional consequences of the multiple solution species observed for the S protein (Figure 2B).
We demonstrate that the protein purified here is well suited for structural studies by determining the cryo-EM structures of OptSpike1-Expi293F at 3.44 Å resolution and OptSpike1-ExpiCHO-S at 3.22 Å. These structures are nearly identical to each other and are in good agreement with the closed form observed in previously reported structures of the S protein (which were produced in FreeStyle 293-F cells).7,21 Interestingly, the structures determined from protein expressed in mammalian cells differ from a recently reported cryo-EM structure of wild-type S protein produced from High Five insect cells. This structure appeared to be more compact overall, and the model could not accommodate the presence of the SD1 domain.34 Whether these disparities are the consequence of differences in expression hosts (e.g., glycosylation or other processing functions) or the lack of stabilizing mutations remains an open question, although RBD recognition and antigenicity were preserved.
We compared 20 deposited cryoEM structures of the S protein with the RBDs in the down conformation. We found that, though there was generally good overall structural agreement, one region (amino acid segment 614–642) was modeled as one of the two distinct paths in each structure (Figure 7). This behavior indicates that this region of the S protein can adopt at least two discrete conformations (conformation 1: Figure 7C,E,F; conformation 2: Figure 7D), which may influence neighboring segments of the S protein (notably amino acids 828–854). Interestingly, this region is proximal to both S cleavage sites, and residue 614 (the start of the 614–642 site) is often mutated in SARS-CoV-2 (i.e., D614G, which is associated with increased infectivity).35 A recent structural study of the D614G variant reported a model in which neither residues 621–639 nor the RBD was included (PDB: 6XS6),36 and the trace modeled for residues 614–620 follows the path observed in Figure 7C,E,F (conformation 1). The D614G structure did contain several differences in the S protein conformation, including an increased distance between protomers and an increase in the fraction of S protein exhibiting two or three RBDs in the open (up) conformation compared to D614.35 However, it has been recently reported that the structure of the S protein and the distribution of RBD conformations are affected by changes in pH,37 and yet, another group reported temperature sensitivity of the S protein structure.38 Thus, the apparent propensity for the immediate physicochemical environment of the S protein to alter conformation makes it difficult to compare distributions of structures when samples were not prepared identically.
One structure (PDB: 7JJJ – the only structure in the “down” conformation to fully model every amino acid in conformation 2- Figure 7D)39 formed a “dimer of trimers” in which the region spanning amino acids 619–631 extends away from one trimer and interacts with the NTD of a second trimer. While the biological significance of this conformation and interaction is unclear, the authors proposed that “inter-spike interactions may play a role in viral viability”, which is consistent with a mutagenesis experiment they conducted.39
Thus, the determinants of different observed S protein conformations remain unclear, and future studies are needed to fully define the structural and dynamic characteristics of the S protein and to determine how these properties contribute to SARS-CoV-2 infectivity.
Both OptSpike1 and OptSpike2 proteins exhibit comparable thermal denaturation profiles with two similar melting transitions (Figure 2G). This finding is consistent with the thermal denaturation profile of wild-type S protein purified from High Five insect cells despite the overall structural differences.34 Additional work will be required to assign these transitions to specific domains or modules within the full-length COVID-19 spike protein.
We examined the receptor-binding specificity of the SARS-CoV-2 S protein. We demonstrate strong binding between the S protein and hACE2, but not mACE2, as has been previously reported.8,40 Furthermore, we screened recombinant OptSpike1 against a sublibrary of 900 members of the human secretome that were expressed on FreeStyle 293-F cells and did not identify any additional interactions (Figure S3C). This library included many proteins that others have suggested may bind to the SARS-CoV-2 S protein, including CD147, Siglec 9, Siglec 10, and proteins that other coronaviruses use for viral entry, including CD26 (MERS), Ceacam5 (MERS), and Ceacam1 (Murine Coronavirus).28,29,41,42 CD147 is of particular interest because of the ongoing clinical trial that aims to treat SARS-CoV-2 with Meplazumab (a humanized anti-CD147 antibody).26 Importantly, for this subset of proteins, we conducted antibody staining to explicitly evaluate cell surface localization (Figure S3I–L). While all members of this subset could be detected by antibody staining and are therefore properly localize to the cell surface, we were not able to detect binding to the S protein. It is important to note that the assay we employed represents only one possible format for detecting protein–protein interactions (recombinant S protein binding to the cell surface displayed receptor: Figure S3A). Because we did not directly recapitulate the experiments that originally detected interactions between the S protein and CD147 (SPR, ELISA, Co-IP27) or Siglec 9 and 10 (ELISA28), it is possible that the S protein binds to these receptors in a way that is undetectable in this assay or that there will be cell-specific differences in the presentation of the putative receptors (i.e., the requirement an unknown co-receptor not expressed on FreeStyle 293-F cells, etc.). It is important to note that anti-CD147 Abs were reported to block COVID-19 infection in Vero E6 cells, which also express ACE2,43 highlighting the potential importance of cell-specific differences in receptor presentation and recognition. Further investigations are warranted to evaluate these potential interactions. Finally, as our screening efforts only included ∼20% of the human secretome, it is important to continue to examine the remaining 80% for alternate S protein interactions, which might reveal additional mechanisms of viral entry for SARS-CoV-2.
We additionally demonstrate that these recombinant S proteins can be used interchangeably and reproducibly in two different assays screening for a human serum response to S protein (ELISA: see Figure 4, and protein microarray: see Figure 5, Figure S4, and below). Validating that both S proteins can be utilized in a serum ELISA screen is important because both OptSpike1 and OptSpike2 are being widely utilized for serology testing at different clinical sites. These data collectively confirm that the strategy for antigen preparation described here can be used reliably and that the antigenicity of OptSpike1 and OptSpike2 is comparable regardless of the cell line used (Expi293F vs ExpiCHO-S: Figure 5B). These data are critical for interpreting clinical results from various institutions, which are using a wide array of different serological tests to detect anti-S antibodies (for a complete list of antibody tests approved for use by the FDA, please see their website44). Consequently, additional versions of the S protein that are being used clinically should be analyzed similarly, so as to further ensure clinical reproducibility and compatibility of different antibody serology tests.
We developed a protein microarray platform to simultaneously analyze serum for antibodies against the S protein, as well as recombinant RBD and N protein. Microarray results from a small cohort of serum from five seroreactive SARS-CoV-2 patients and serum from three individuals collected before November 2019 revealed that detectable anti-S antibody titer correlated well with results of serological ELISAs (Figures S4 and S5) and additionally demonstrated the simultaneous detection of antibodies against the S protein, the N protein, and the RBD of the S protein. It is important to note the limitations of the current platform. In particular, we stress that at present, this is a qualitative approach as a number of variables can impact the ability to detect antibody reactivity, including patient titers for specific antigens and the relative affinities of an antigen-specific pool. Furthermore, it should be appreciated that the current platform is programmed to detect the capture of IgG antibodies. Thus, the resultant IgG signal (or the lack thereof) for a particular antigen could be the consequence of different prevalences between different isotypes (IgA, IgD, IgE, IgG, IgM), which are known to evolve during the course of infection and subsequent resolution. Although currently focused on three antigens, this platform can be readily expanded to study differential antibody responses to different SARS-CoV-2 antigens and subdomains of those antigens among individuals, which are actively being investigated by others.45−48 We are currently working to include not only other antigens from SARS-CoV-2 (E protein, M protein, etc.) but also antigens from other coronaviruses that may be cross reactive with SARS-CoV-2 antibodies. The analysis of antibody reactivity to multiple SARS-CoV-2 and related antigens will provide broad insight into the humoral immune response to SARS-CoV-2.
Collectively, this work provides an array of standards and metrics for high-quality protein reagents that can yield comparable clinical, biological, and structural data as we continue to combat the global health crisis caused by SARS-CoV-2.
Materials and Methods
Recombinant Spike Protein Production
An SOP for the production of high-quality recombinant S antigen is provided in the Supporting Information Appendix 1. Both variants of the recombinant SARS-CoV-2 S protein (OptSpike1 and OptSpike2) were expressed in Expi293F and ExpiCHO-S cells according to the manufacturer’s instructions (Thermo Fisher Scientific). For Expi293F (Thermo Fisher Cat # A14527) purifications, cells were grown in sterile TC flasks, vented, with a baffled bottom (example: Fisher Scientific Cat # BBV12-5) in a Climo Shaker ISF4-X at 110 rpm (an orbital diameter of 50 mm) at 37 °C and 8% CO2. On the day of transfection, Expi293F cells can reach a density between 4.5−5.5 × 106 cells/mL. The Expi293F cells are diluted to 3 × 106 cells/mL in the appropriate volume and shaker flask. For example, for a transfection volume of 30 mL, 75 × 106 cells were diluted in 25 mL of fresh media in a 125 mL shaker flask. Cells were then transfected by diluting 30 μg of plasmid DNA in Opti-MEM I Reduced Serum Medium (Cat # 31985-062) to a total volume of 1.5 mL in a 15 mL conical tube and then mixing briefly by inverting the tube. 80 μL of ExpiFectamine 293 Reagent was then diluted in Opti-MEM I medium to a total volume of 1.5 mL in a separate 15 mL conical tube. Both reactions were incubated for 5 min. The transfection reagent mixture was then carefully pipetted into the DNA mixture and mixed by inverting the 15 mL conical tube three times. The combined DNA and transfection reagent mixture was incubated at room temperature for 25 min, after which the complexes were added to the cell culture in a dropwise fashion while swirling the cell culture flask to ensure uniform distribution of the DNA complexes. 16 h post-transfection, 150 μL of ExpiFectamine 293 Transfection Enhancer 1 and 1.5 mL of ExpiFectamine 293 Transfection Enhancer 2 were added to the transfected cells, bringing the total volume of the transfection to 30 mL. Expi293F transfections were harvested on day 6 post transfection unless otherwise specified. This protocol can be scaled linearly for larger Expi293F cultures, according to the manufacturer’s protocol, for both OptSpike1 and OptSpike2.
For ExpiCHO-S transfections, we obtained the ExpiCHO-S (Thermo Fisher Cat # A29127) cells and ExpiCHO Expression System kit (Cat # A29133) from Thermo Fisher Scientific. ExpiCHO-S cells were grown in sterile Erlenmeyer-shaped flasks with plain bottoms with vented screw caps (appropriate flasks for cell culture volume should be used; see, for example, Fisher brand PBV125) in a Climo Shaker ISF4-X at 110 rpm (an orbital diameter of 50 mm) at 37 °C and 8% CO2. ExpiCHO-S cells were passaged and split every 2–3 days when cell densities reached 0.3 × 106 to 6 × 106 viable cells per mL. ExpiCHO-S cells are allowed to reach a density of 7–10 × 106 cells per mL on the day of transfection, with a viability of at least 95%. ExpiCHO-S cells are diluted to a final density of 6 × 106 cells per mL in a plain bottom Erlenmeyer flask. For spike protein transfections, the manufacturer’s ExpiCHO expression system manual was followed closely. For example, for a 25 mL transfection, 25 μg of filtered DNA was diluted into 1 mL of OptiPRO SFM (Cat # 12309019), and 80 μL of ExpiFectamine CHO reagent was added to 920 μL of OptiPRO SFM. Dilutions were mixed by inversion, and then diluted DNA was mixed with diluted ExpiFectamine CHO reagent and mixed by inversion. ExpiFectamine CHO and DNA complexes were incubated at room temperature for 5 min and slowly transferred to a 125 mL plain bottom flask containing 25 mL of ExpiCHO-S cells at a cell density of 6 × 106 cells per mL. For all spike transfections, we followed the max titer protocol (unless otherwise stated), and thus, on day 1 post transfection, we added 150 μL of ExpiCHO-S Enhancer and 4 mL of ExpiCHO-S Feed. On day 1 post transfection, the cells were shifted to another Climo Shaker ISF4-X at 110 rpm (an orbital diameter of 50 mm) with temperature set at 32 °C and 5% CO2. On day 5 post transfection, another 4 mL of ExpiCHO-S Feed was added and the cells were set back at 32 °C. ExpiCHO-S transfections were harvested on day 12 post transfection unless otherwise specified.
S Protein Purification
For purifications of the SARS-CoV-2 S protein (OptSpike1 and OptSpike2), on the indicated day post transfection (optimally day 6 for Expi293F and day 12 for ExpiCHO-S), the media was harvested by centrifuging cells at 500g for 10 min, removing the supernatant, and centrifuging the supernatant at 2000g for 10 min. For ExpiCHO-S, the supernatant was clarified by filtering through a rapid flow filter from Thermo Fisher (example: 524-0020). The supernatant was then adjusted to 50 mM ArgCl using a 2 M stock solution of ArgCl at pH 6.5 to increase stability of the protein during purification. OptSpike1 and OptSpike2 were subsequently purified by His60 Ni2+ superflow resin (Takara Cat: 635664) using a batch binding method: .6 mL of resin was added for every 10 mL of supernatant, and supernatants were incubated with resin for 2 h at 4 °C with shaking or rotating. Batch adsorption was followed by gravity flow over a column. The Ni2+ His60 resin was washed with 3 column volumes of wash buffer (50 mM Tris pH 8.0, 100 mM ArgCl, 5 mM imidazole, 150 mM NaCl, 10% glycerol), and the bound protein was eluted with 5 mL of the same buffer containing 500 mM imidazole. Elution from nickel resin was concentrated by centrifugation in a 100 K concentrator (Thermo Fisher Cat # 88533) by spinning at 1100g for intervals of 8 min (if necessary) and further purified by gel filtration on a HiLoad 16/600 Superdex 200 column (GE) equilibrated with 50 mM Tris, 100 mM ArgCl, 150 mM NaCl, 10% glycerol, pH 8.0. For use in cryo-EM experiments, nickel eluates were further purified by gel filtration on a HiLoad 16/600 Superdex 200 (GE) equilibrated with 50 mM Tris, 150 mM NaCl, pH 8.0. For the protein used in serum ELISA or protein microarray experiments, 30 mL of nickel eluates was dialyzed against 5 L of 50 mM Tris, 150 mM NaCl, pH 8.0 at 4 °C for 16 h. Generally, the SEC step did not affect the serum ELISA experiments but was important for all other experiments. Protein concentration was determined using an extinction coefficient (146,850 M–1 cm–1) estimated from the amino acid sequence by the Expasy online ProtParam tool.49
SARS-CoV-2 RBD Expression and Purification
The pCAGGS50 SARS-CoV-2 RBD plasmid (provided by Florian Krammer) was used for recombinant RBD expression as previously described. FreeStyle 293-F cells (Thermo Fisher, R79007) were transiently transfected with a mixture of plasmid DNA diluted in PBS (0.67 μg of total plasmid DNA per mL of culture) and polyethylenimine (PEI) (Polysciences, Inc., 23,966) at a DNA-to-PEI ratio of 1:3. At 6 days post-transfection, cultures were harvested by centrifugation at 4000g for 20 min, and the supernatant was incubated with Ni-NTA resin (Goldbio) for 2 h at 4 °C with gentle stirring. The resin was collected in columns by gravity flow, washed with 16 column volumes of wash buffer (50 mM Tris HCl pH 8.0, 250 mM NaCl, 20 mM imidazole), and eluted in 12 mL of elution buffer (50 mM Tris HCl pH 8.0, 250 mM NaCl, 250 mM imidazole). Eluates were concentrated and exchanged into storage buffer (50 mM Tris HCl pH 8.0, 250 mM NaCl) using an Amicon centrifugal units (EMD Millipore). Protein concentration was determined using an extinction coefficient (33,350 M–1 cm–1), estimated from the amino acid sequence by Expasy online ProtParam, and was further analyzed by SDS-PAGE.
Recombinant Expression and Purification of SARS-CoV-2 N Protein
The nucleocapsid sequence was PCR-amplified from a diagnostic test positive control plasmid obtained from IDTDNA (cat # 10006625 GenBank: NC_045512.2) and InFusion-cloned into a derivative of the NYSGRC pSGC-HIS vector. 50 ng of plasmid was used to transform 20 μL of the BL21 DE3 strain of E. coli. Cultures were then grown overnight (16 h) in LB at 37 °C and used to inoculate either LB media the next day (1:100× dilution of overnight culture). Inoculated cultures were grown at 37 °C until they reached OD600 of 0.7, at which point they were induced using 500 μM IPTG. Upon induction of LB media, the temperature of the cultures was immediately lowered to 25 °C for 16 h.
To harvest protein, cells were lysed by sonic disruption using a 550 sonic dismembrator from Fisher Scientific. Every 5 g was resuspended in 30 mL of a lysis buffer consisting of 50 mM HEPES, 250 mM KCl, 10% glycerol, 10 mM BME, 0.1% Igepal CA-630 (Sigma Aldrich), pH 7.5 and 1/2 protease inhibitor tablets (Roche). After lysis, samples were cleared by centrifugation at 20,000 rpm. The resulting supernatant was purified on an AKTA FPLC (GE Biosciences). Supernatants were loaded onto fast flow HisTrap columns and washed with 20 column volumes of lysis buffer and eluted with 2 column volumes of Buffer B (Buffer A + 500 mM imidazole, pH 7.5). The resulting eluent with high OD280 absorbance was collected and loaded onto a HiPrep 16/60 S-200 size exclusion column equilibrated with 50 mM HEPES, 250 mM KCl, 10% glycerol, 5 mM DTT, pH 7.5. Protein concentrations of fractions were approximated using an extinction coefficient of 43,890 M–1 cm–1, and a molecular mass of 45.62570 kDa was estimated from the amino acid sequence by the Expasy online ProtParam tool.49
Analytical Size Exclusion Chromatography
After nickel elutes were concentrated and purified by gel filtration on a HiLoad 16/600 Superdex 200 column and concentrated, the protein aggregation state was assessed by analytical gel filtration on a Superose 6 Increase 10/300 GL column. The void for this column runs at 8.5 mL. The aggregation state was monitored over time and after freeze–thaw cycles on this column.
Molecular Mass Determination Using Multiangle Light Scattering (MALS)
30 μL of OptSpike1, Optspike2, or Nucleocapsid was run over a Yarra 3 μm SEC-4000 LC column using an Agilent Technologies 1260 Infinity instrument, equipped with an autoinjector. 10 μL samples were injected onto the column in 50 mM Tris, 150 mM NaCl, 100 mM ArgCl, 10% glycerol, pH 8.0 at a flow rate of 0.25 mL/min. MALS analysis was performed using a miniDAWN Treos MALS detector (Wyatt) and Optilab T-rEX and analyzed using the associated Astra software. Baselines were determined automatically; peaks were manually delineated.
OptSpike1 Protein Melting Curve
A 5000× stock of SYPRO dye (Thermo Fisher Scientific) was diluted to 200× in Tris HCl (pH 7.5), 100 mM KCl buffer, the same buffer as OptSpike1 and OptSpike2 that were diluted in for the assay. We prepared a mixture with a final protein concentration of 1.58 μM and a final SYPRO orange concentration of 20×.The samples were split into three technical triplicates (one in each well of a 384-well plate), and the reactions were placed into a 7900 HT Fast Real Time PCR System. The PCR machine was programmed to monitor the fluorescence of the dye over a temperature gradient spanning 25 to 99 °C. The protocol was designed to hold the samples at 25 °C for 2 min and then ramp the temperature. Background fluorescence was measured using a 20× dye, and all measurements were taken in technical triplicate.
Data was exported in Excel and then analyzed and graphed by GraphPad Prism. To discern the melting temperature (Tm) of the protein, two methods were used as outlined in (1) fitting the Boltzmann equation to the nonlinear raw data and (2) detection of local maximum values in the 1st derivative of melting curve. The curve had two apparent melting transitions, one between 42 and 55 °C and the other between 55 and 70 °C. The curves in these two temperature ranges were fitted with Boltzmann equations (listed below). The function fit to data in each of the temperature ranges contained an approximate value for each Tm. In order to determine the local maximum values, the 1st and 2nd derivatives were graphed (the 2nd derivative should equal 0 at the local maxima of the 1st derivative, which can be visually verified by the graph of the 1st derivative). Both methods produced comparable melting temperature values: OptSpike1 Tm1 = 49.04 ± 0.1699 °C and OptSpike2 Tm1 = 49.36 ± 0.1652 °C; OptSpike1 Tm2 = 63.04 ± 0.1659 °C and OptSpike2 Tm2 = 63.31 ± 0.09236 °C. The melting temperature can also be approximated by determining local maxima of the 1st derivative of the fluorescence vs temperature curve. There was not a significant difference in either Tm1 and Tm2 between the OptSpike1 and OptSpike2 constructs.
Analytical Ultracentrifugation of OptSpike1
Analytical ultracentrifugation (AUC) studies were conducted with OptSpike1 in 250 mM NaCl, 50 mM Tris, pH 8.0 in a Beckman XL-AUC using the absorption optics to scan cells assembled with a double sector charcoal EPON centerpiece and sapphire glass windows inserted into an AN-60 Ti rotor. Centrifuge runs were conducted at 20 °C and 30,000 rpm. The first run was scanned at 230 nm. A second run was scanned at 280 nm. This protocol allowed protein concentrations ranging from 136 nM to 4.8 μM to be analyzed. A minimum of 50 scans was acquired for each sample. An overview of the sedimentation behavior of each sample was obtained by time-derivative g(s*) analysis conducted using either the program SedAnal51,52 or the program DCDT+ by John Philo v2.5.1.53,54 Component analysis was conducted using the program Sedfit55,56 to deconvolute the species present in a solution. The program Prism v8 was used to generate the AUC figures shown in the text and the Supporting Information.
Convalescent Serum and Plasma Samples
Serum and plasma samples were collected from healthy adult volunteers residing in Westchester County, NY, who had recovered from COVID-19 in April 2020. Patients had reported a positive nasopharyngeal swab by qPCR for SARS-CoV-2 during illness and had been asymptomatic for at least 14 days prior to sample collection. After obtaining informed consent, serum was obtained by venipuncture (BD Vacutainer, serum), centrifuged, aliquoted, and stored at −80 °C prior to use. The sera were heat-inactivated at 56 °C for 30 min and stored at 4 °C prior to analysis. Protocol approval was obtained from the Institutional Review Board (protocol IRB# 2016-6137) of the Albert Einstein College of Medicine.
Protein Microarray Production and Processing
The COVID-19 protein array included OptSpike1, OptSpike2, receptor binding domain (RBD) of Spike protein, and nucleocapsid proteins, along with positive (human IgG) and negative (human acetylcholinesterase, PBS, or printing buffer) controls. Arrays were generated by printing purified proteins onto aminosilane-coated slides with a printing buffer containing an amine-to-amine homo-bifunctional cross-linker (bissulfosuccinimidyl suberate, BS3, Thermo Scientific Pierce Cat # A39266) and glycerol. BS3 was used for covalent immobilization of the proteins to the slide, and glycerol was used to keep them hydrated at all times. Protein concentrations were adjusted to 25, 50, 100, and 200 pg per spot (2-fold dilutions between 25 and 200 pg per spot). The array layout was designed with 16 identical subarrays. Each sample was spotted in duplicate with a total drop deposition volume of 800 pL (8 drops of 100 pL) per spot using a Marathon Argus piezoelectric printer (Arrayjet, Edinburgh, UK) at 50–55% humidity. The slides were incubated in a humidity-controlled (50%) enclosure for 45 min and blocked by incubation with Superblock blocking buffer (Thermo Fisher Scientific, Rockford, IL) for 45 min at room temperature with shaking at 60 rpm.
After blocking, a multiwell chamber (ProPlate Multi-Well Chambers, Grace Bio-labs, Bend, OR) was used to screen multiple serum samples with subarrays on the same slide. As negative control for screening, we used three serum samples (1, 4, and 8), which were received from the New York Blood Center, which were collected before the 2019 SARS-CoV-2 outbreak and stored as aliquots at −80 °C. COVID-19 convalescence sera samples (2, 3, 5, 6, 7) were previously validated to be negative with RT-PCR for SARS-CoV-2 and positive ELISA for antibodies against SARS-CoV-2.
Each subarray was challenged with 250 μL of 1:100 patient sera diluted in 5% (w/v) milk, PBS, and 0.2% (v/v) Tween-20 (5% milk-PBST) overnight at 4 °C with gentle shaking on a plate shaker. The arrays were rinsed with 5% milk-PBST briefly and incubated for 1 h at room temperature with a fluorescently labeled secondary antibody (AlexaFluor 647 labeled goat anti-human IgG (H + L) cross-adsorbed secondary antibody, Thermo Fisher Scientific, Cat # A21445) diluted 1:150 in 5% milk-PBST. After washing three times with PBST, the slides were rinsed with water and dried in falcon tubes by centrifugation at 1000g for 3 min. The slides were scanned with a GenePix 4400A microarray scanner (Molecular Devices), and data were analyzed with GenePix Pro 7 software. Mean fluorescence intensities (MFI) of every spot were quantified after background correction was performed for each serum sample by subtracting the MFI of huAche, which was printed as negative control within the subarray. Duplicate spot measurements were averaged. The results were normalized relative to the corresponding signal of hIgG1 and multiplied by a hundred for presentation as percentage.
Recombinant Protein ELISAs
Recombinant SARS-CoV-2 Spike protein was coated onto high-binding 96-well plates (Corning, 3690) at 2 μg/mL, overnight at 4 °C. Wash steps were done using 1× PBS containing 0.05% Tween-20 (Sigma, P1379), and all incubations were done at 25 °C. Plates were blocked with 3% nonfat dry milk (Bio-Rad, P1379) in 1× PBS for 1 h. Serum from COVID-19 convalescent patients was serially diluted in 1× PBS, containing 1% nonfat dry milk and 0.1% Tween-20, and then added to plates and incubated for 2 h. Following plate washing, serum reactivity to the Spike protein was measured using an HRP-conjugated goat anti-human IgG (H + L) (Invitrogen, 31410) at a 1:3000 dilution for 1 h. After a final wash, the Ultra-TMB substrate (Thermo Fisher, 34029) was added to the plates and incubated for 5 min followed by quenching of the reaction by addition of an equal volume of 0.5 M H2SO4. Absorption was measured at OD450 using a Synergy4 plate reader (Biotek), and data was analyzed using GraphPad Prism7.0 to calculate IC50 values.
PNGase F Digestion of Spike Protein
For PNGase F digestion, purified Spike protein expressed from Expi293F or ExpiCHO-S cells was digested under denaturing reaction conditions. 5 μg of protein, 1 μL of glycoprotein denaturing buffer (NEB Cat # B0701S, 10X), and 4 μL of water were mixed to a total of 10 μL. The spike glycoproteins were denatured at 100 ° C for 10 min, and the denatured proteins were incubated on ice for 5 min and centrifuged for 10 s at max speed on a microcentrifuge. Next, 2 μL of GlycoBuffer 2 (NEB Cat # B0701S, 10X), 2 μL of 10% NP-40 (NEB Cat # B0701S), and 6 μL of water were mixed with 10 μL of denatured spike glycoprotein. Finally, 1 μL of PNGase F (NEB Cat # P0704S) was added to the reaction, and the digestion was allowed to proceed at 37 ° C for 1 h. Glycoproteins were analyzed by SDS-PAGE to observe loss of N-linked glycans compared to undigested glycoproteins.
FreeStyle 293-F Transient Transfections and Culture
FreeStyle 293-F suspension cells were cultured in FreeStyle 293 Expression Media (Invitrogen) at 37 °C in a humidified shaking platform incubator (Kuhner) with 5% CO2. For transfection, cells were pelleted at 500g and resuspended in fresh media. For small-scale (1 mL of cells at 1 × 106/mL) transient transfections performed in 24-well nontreated tissue culture plates, 2 μg of polyethylenimine (PEI) was added to 0.5 μg of diluted plasmid DNA in a final volume of 100 μL. For small-scale (.25 mL of cells at 1 × 106/mL) transient transfections performed in 48-well nontreated tissue culture plates, .5 μg of polyethylenimine (PEI) was added to 0.125 μg of diluted plasmid DNA in a final volume of 25 μL. DNA–PEI complexes were incubated at room temperature for 10 min and then added directly to cells in 48-well plates. FreeStyle 293-F cells were utilized to screen for spike protein interactions, so genes that were transfected into FreeStyle 293-F cells included human ACE2 and CD26, mouse ACE2, the secretome library, and GFP controls.
Flow Cytometry Titration Experiment
Flow cytometry titration assays were performed with the OptSpike1 and OptSpike2 proteins described above. FreeStyle 293-F suspension cells were transfected with DNA encoding human ACE2, mouse ACE2, human CD26, or GFP. Both ACE2 variants were expressed with GFP tags on their cytosolic C term, while CD26 was expressed in an IRES vector expressing GFP. Two days post transfection, cells were counted and diluted to 1 × 106 cells/mL in 1× PBS, .2% BSA. OptSpike1 and OptSpike2 proteins were serially diluted to a range of concentrations from 1 nM to 10 μM. Subsequently, 10 μL of diluted Spike protein was added to 90 μL of diluted cells in wells of a 96-well plate (90,000 cells per well). Binding was performed at room temperature for 1 h with shaking at 900 rpm, after which the cells were washed twice with 1× PBS, .2% BSA by centrifugation. Cells were then incubated with a PE labeled anti-6x His tag antibody (Abcam Cat # ab72467) to detect Spike protein binding. Antibody binding was performed for 30 min at room temperature with shaking, after which the cells were washed twice with 1× PBS, .2% BSA by centrifugation. Cells were analyzed by flow cytometry/spectral analysis on a Sony Spectral Analyzer. Gated live cells were subgated for GFP, and GFP-positive cells were subgated for PE-positive events. Data points represent the average of three independent experiments fit to the single site binding equation: Y=Bmax*Xh/(Kdh+Xh).
Generation of a Human Plasma Membrane Protein Library for Mammalian Expression
The human genome was analyzed for all transcripts that contain at least one transmembrane domain (TMHMM Server 2.0) and a predicted secretion signal peptide sequence.57−59 The resulting list of 14,028 transcripts was manually purged of mitochondrial, nuclear, and ER membrane proteins, resulting in a final target set of 9065 potential human plasma membrane transcripts representing a set of 4860 genes. All of the full-length cDNAs available from GeneCopoeia were purchased as covalent C-terminal GFP fusions in a CMV mammalian expression plasmid (3926 total). To enhance library coverage of proteins missing from the GeneCopoeia library, a separate set of synthetic transcripts was ordered from Gen9 (1282 total). This set was selected by identifying the largest transcript for each of the missing proteins with the limitation that they be under 4 kb in length (the limit for high-throughput synthesis). Each full-length cDNA was codon-optimized, synthesized, and subcloned into the Clonetech pEGFP N1 vector as C-terminal GFP fusions. Working libraries of Ig superfamily proteins (IGSF), TNF receptor superfamily proteins (TNFRSF), G-Protein coupled receptor proteins (GPCR), Integrins, and chemokines were identified for each subfamily based on lists generated using the HUGO Gene Nomenclature Committee (HGNC) gene family resource.60 Each set was re-arrayed manually from glycerol stocks into liquid 2xYT media in 96-well deep-well blocks. Overnight cultures were mini-prepped in 96-well plates (Macherey Nagel kits) for use in downstream high-throughput mammalian cell transfections.
High-Throughput Screening of the Human Plasma Membrane Protein Library
The human plasma membrane protein library59,61 was transfected into FreeStyle 293-F cells in the 48-well format. Two days post-transfection, cells were diluted to 1 × 106 cells/mL in .2% BSA. Binding reactions were set up in 96-well V-bottom plates by incubating 100 μL of cells with 200 nM OptSpike1; each plate also contained hACE2-GFP expressing cells as an internal positive control. After 45 min, cells were pelleted and washed twice and then incubated with an anti-HIS PE antibody, washed twice, and analyzed on a Sony Spectral Analyzer. The percent bound was calculated as the number of double-positive events (GFP and mCherry) divided by the total number of GFP positive cells. Expressions of hACE2, mACE2, CD147, CD26, Siglec 9, Siglec 10, Ceacam1, and Ceacam5 were confirmed by antibody staining of transfected FreeStyle 293-F cells. Binding was conducted as described for S protein binding. Antibodies used were as follows: hACE2 and mACE2 (R&D Cat # AF933-SP), CD147 (Biolegend Clone HIM6), CD26 (Biolegend Clone BA5b), Siglec 9 (Biolegend Clone K8), Siglec 10 (Biolegend Clone FG6), Ceacam1, and Ceacam5 (Biolegend Clone ASL-32).
CryoEM
Grid Preparation
3 μL of protein (1 mg/mL in 50 mM Tris, 250 mM NaCl, pH 8.0) was applied to plasma-cleaned C-flat 1.2/1.3400 mesh Cu holey carbon grids (Protochips, Raleigh, NC) or 1.2/1.3300 mesh UltrAuFoil gold holey gold grids (Quantifoil Micro Tools GmbH, Großlöbichau, Germany), blotted for 2.5 s after a 30 s wait time, and then plunge frozen in liquid ethane, cooled by liquid nitrogen, using the EM GP2 (Leica Microsystems, Inc., Buffalo Grove, IL) or Vitrobot Mark IV (Thermo Fisher Scientific, Hillsboro, Oregon).
Microscopy
A Thermo Fisher Titan Krios operated at 300 kV, Gatan GIF-Bioquantum with a 20 eV slit, and K3 camera were used with 100 μm C2 aperture, 100 μm objective aperture, and 1.058 Å calibrated pixel size.
Imaging
Movies were collected in counting mode using Leginon62 at a dose rate of 26.6 e–/Å2/s with a total exposure time of 2.5 s, for an accumulated dose of 66.5 e–/Å2. Intermediate frames were recorded every 50 ms for a total of 50 frames per micrograph. Defocus values ranged from approximately 0.8 to 2.5 μm.
Image Processing
Movies recorded on the K3 were aligned using Appion63 and MotionCor264 and CTF estimated and 2D classified in cryoSPARC v2.14.2.65 Particle picking was done with TOPAZ66 as implemented in cryoSPARC. A Topaz picking model was trained using frame-summed micrographs of 4000 particles manually curated from 100 micrographs initially picked from 7500 blob picks in cryoSPARC. For the final reconstruction, particles were selected and subjected to 3D refinement in cryoSPARC with a final box size of 384 × 384pixels. The ExpiCHO expressed dataset (n20apr21a) processing used 1131 micrographs and 75,582 particles that was curated to 54,395 particles. The Expi293F expressed dataset (n20apr22) processing used 1694 micrographs and 99,154 particles that was curated to 54,066 particles.
Acknowledgments
This work was supported by the NIH (R01 CA198095 to S.C.A., R01-AI125462 to J.R.L., 5R01-GM129350 to M.B., U19AI142777 and R01AI132633 to K.C., R01AI143453 and R01AI123654 to L.P., and R21AI141367 to J.P.D.). Some of this work was performed at the Simons Electron Microscopy Center and National Resource for Automated Molecular Microscopy located at the New York Structural Biology Center, supported by grants from the Simons Foundation (SF349247), NYSTAR, and the NIH National Institute of General Medical Sciences (GM103310) with additional support from the National Center for CryoEM Access and Training (NCCAT) supported by the NIH Common Fund Transformative High Resolution Cryo-Electron Microscopy program (U24 GM129539) and NY State Assembly Majority. A.J.N. was supported by a grant from the NIH National Institute of General Medical Sciences (NIGMS) (F32GM128303). M.E.D. is a Latin American Fellow in the Biomedical Sciences, supported by the Pew Charitable Trusts. J.H.L., R.H.B.III., and R.J.M. were partially supported by the NIH training grant 2T32GM007288-45 (Medical Scientist Training Program) at Albert Einstein College of Medicine. L.P. was additionally supported by a grant from the Mathers Foundation. We thank Jason McLellan for providing the stabilized spike plasmid (OptSpike1) and Arvin Dar, Sarah Pegno, and Zaigham Khan for sharing the OptSpike2 plasmid. We also thank Daija Bobe at SEMC and Al Tucker at Einstein for technical support and Phaneendra Duddempudi for useful discussion. We also thank Bridget Carragher and Clint Potter for assistance and input in cryo-EM work. The TOC graphic was generated using BioRender.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c03512.
S, N, and RBD protein quality control data. Screen of S protein against 20% of the human secretome Additional data on the SARS-CoV-2 multiantigen protein array. ELISA confirming S protein reactivity to COVID-19 positive serum. Additional Cryo-EM data. Table of Cryo-EM data collection, refinement, and validation statistics. Appendix 1: Standard Operating Procedure detailing our SARS CoV-2 Spike protein expression and purification protocol (PDF)
Additional information showing the results of the S protein screen to 20% of the human secretome (XLSX)
Author Contributions
+ N.G.H., N.C.M., and A.C. are equally contributing authors. N.G.H., N.C.M., A.C., S.J.G., J.R.L., and S.C.A. conceived the project. N.G.H., N.C.M., A.C., J.H.L., S.J.G., G.I.G., R.J.M., K.T., O.V., L.Y.Y., M.B., and E.T.E. conducted experiments. N.G.H., N.C.M., A.C., J.H.L., G.I.G., R.J.M., K.T., O.V., L.Y.Y., A.J.N., M.K., J.B.B., D.B.H., S.J.G., E.T.E., J.R.L, and S.C.A. analyzed data. R.H.B., A.S.W., C.F., E.L., D.H., J.M.S., M.U.D., R.K.J., J.B., and K.C. processed and curated samples and developed the ELISA assay. A.M., D.K., J.P.D., and L.P. recruited donors and collected and curated samples. A.B.M. and S.C.G. designed key aspects of the project. J.B., J.P.D., L.P., S.J.G., E.T.E., J.R.L., and S.C.A. supervised research. N.G.H. and N.C.M. generated the initial manuscript. N.G.H., N.C.M., and S.C.A. generated the final manuscript with comments and edits from A.C., G.I.G., R.J.M, J.H.L., O.V., J.B.B., D.B.H., M.B., S.J.G., E.T.E., and J.R.L. All authors approved the final manuscript.
The authors declare no competing financial interest.
Notes
The accession numbers for the EM maps, models, and raw movie stack for the SPIKE structure from protein produced in ExpiCHO-S cells reported in this paper are EMD-22078, PDB Entry 6X6P, and EMPIAR-10433.
Supplementary Material
References
- Su S.; Wong G.; Shi W.; Liu J.; Lai A. C. K.; Zhou J.; Liu W.; Bi Y.; Gao G. F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. 10.1016/j.tim.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R.; Zhao X.; Li J.; Niu P.; Yang B.; Wu H.; Wang W.; Song H.; Huang B.; Zhu N.; Bi Y.; Ma X.; Zhan F.; Wang L.; Hu T.; Zhou H.; Hu Z.; Zhou W.; Zhao L.; Chen J.; Meng Y.; Wang J.; Lin Y.; Yuan J.; Xie Z.; Ma J.; Liu W. J.; Wang D.; Xu W.; Holmes E. C.; Gao G. F.; Wu G.; Chen W.; Shi W.; Tan W. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5 (), 536–544, 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millet J. K.; Whittaker G. R. Physiological and molecular triggers for SARS-CoV membrane fusion and entry into host cells. Virology 2018, 517, 3–8. 10.1016/j.virol.2017.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan J.; Ge J.; Yu J.; Shan S.; Zhou H.; Fan S.; Zhang Q.; Shi X.; Wang Q.; Zhang L.; Wang X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 215. 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- Li F.; Li W.; Farzan M.; Harrison S. C. Structure of SARS Coronavirus Spike Receptor-Binding Domain Complexed with Receptor. Science 2005, 309, 1864–1868. 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- Wrapp D.; Wang N.; Corbett K. S.; Goldsmith J. A.; Hsieh C. L.; Abiona O.; Graham B. S.; McLellan J. S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M.; Kleine-Weber H.; Schroeder S.; Krüger N.; Herrler T.; Erichsen S.; Schiergens T. S.; Herrler G.; Wu N. H.; Nitsche A.; Müller M. A.; Drosten C.; Pöhlmann S. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottier P. J. M., The Coronavirus Membrane Glycoprotein. In The Coronaviridae; Siddell S. G., Ed. Springer US: Boston, MA, 1995; pp. 115–139. [Google Scholar]
- Hoffmann M.; Kleine-Weber H.; Pöhlmann S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 779. 10.1016/j.molcel.2020.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A. C.; Tortorici M. A.; Snijder J.; Xiong X.; Bosch B.-J.; Rey F. A.; Veesler D. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 11157. 10.1073/pnas.1708727114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X.; Cao D.; Kong L.; Zhang X. Cryo-EM analysis of the post-fusion structure of the SARS-CoV spike glycoprotein. Nat. Commun. 2020, 11, 3618. 10.1038/s41467-020-17371-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadevall A.; Pirofski L. A. The convalescent sera option for containing COVID-19. J. Clin. Invest. 2020, 130, 1545–1548. 10.1172/JCI138003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortz R. H.; Florez C.; Laudermilch E.; Wirchnianski A. S.; Lasso G.; Malonis R. J.; Georgiev G. I.; Vergnolle O.; Herrera N. G.; Morano N. C.; Campbell S. T.; Orner E. P.; Mengotto A.; Dieterle M. E.; Fels J. M.; Haslwanter D.; Jangra R.; Celikgil A.; Kimmel D.; Lee J. H.; Mariano M.; Nakouzi A.; Quiroz J.; Rivera J.; Szymczak W. A.; Tong K.; Barnhill J.; Forsell M. N. E.; Ahlm C.; Stein D. T.; Pirofski L.-a.; Goldstein D. Y.; Garforth S. J.; Almo S. C.; Daily J. P.; Prystowsky M. B.; Faix J. D.; Fox A. S.; Weiss L. M.; Lai J. R.; Chandran K. Development, clinical translation, and utility of a COVID-19 antibody test with qualitative and quantitative readouts. medRxiv 2020, 10.1101/2020.09.10.20192187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amanat F.; Stadlbauer D.; Strohmeier S.; Nguyen T.; Chromikova V.; McMahon M.; Jiang K.; Asthagiri-Arunkumar G.; Jurczyszak D.; Polanco J.; Bermudez-Gonzalez M.; Kleiner G.; Aydillo T.; Miorin L.; Fierer D.; Lugo L. A.; Milunka Kojic E.; Stoever J.; Liu S. T. H.; Cunningham-Rundles C.; Felgner P. L.; Caplivski D.; Garcia-Sastre A.; Cheng A.; Kedzierska K.; Vapalahti O.; Hepojoki J.; Simon V.; Krammer F.; Moran T. A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat. Med. 2020, 26, 1033–1036. 10.1101/2020.03.17.20037713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F.; Zhao S.; Yu B.; Chen Y. M.; Wang W.; Song Z. G.; Hu Y.; Tao Z. W.; Tian J. H.; Pei Y. Y.; Yuan M. L.; Zhang Y. L.; Dai F. H.; Liu Y.; Wang Q. M.; Zheng J. J.; Xu L.; Holmes E. C.; Zhang Y. Z. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier S.; Güthe S.; Kiefhaber T.; Grzesiek S. Foldon, the natural trimerization domain of T4 fibritin, dissociates into a monomeric A-state form containing a stable beta-hairpin: atomic details of trimer dissociation and local beta-hairpin stability from residual dipolar couplings. J. Mol. Biol. 2004, 344, 1051–1069. 10.1016/j.jmb.2004.09.079. [DOI] [PubMed] [Google Scholar]
- Kirchdoerfer R. N.; Wang N.; Pallesen J.; Wrapp D.; Turner H. L.; Cottrell C. A.; Corbett K. S.; Graham B. S.; McLellan J. S.; Ward A. B. Stabilized coronavirus spikes are resistant to conformational changes induced by receptor recognition or proteolysis. Sci. Rep. 2018, 8, 15701. 10.1038/s41598-018-34171-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.; Qiu Y.; Li J. Y.; Zhou Z. J.; Liao C. H.; Ge X. Y. A Unique Protease Cleavage Site Predicted in the Spike Protein of the Novel Pneumonia Coronavirus (2019-nCoV) Potentially Related to Viral Transmissibility. Virol. Sin. 2020, 337. 10.1007/s12250-020-00212-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain N. K.; Barkowski-Clark S.; Altman R.; Johnson K.; Sun F.; Zmuda J.; Liu C. Y.; Kita A.; Schulz R.; Neill A.; Ballinger R.; Patel R.; Liu J.; Mpanda A.; Huta B.; Chiou H.; Voegtli W.; Panavas T. A high density CHO-S transient transfection system: Comparison of ExpiCHO and Expi293. Protein Expression Purif. 2017, 134, 38–46. 10.1016/j.pep.2017.03.018. [DOI] [PubMed] [Google Scholar]
- Walls A. C.; Park Y. J.; Tortorici M. A.; Wall A.; McGuire A. T.; Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acharya P.; Williams W.; Henderson R.; Janowska K.; Manne K.; Parks R.; Deyton M.; Sprenz J.; Stalls V.; Kopp M.; Mansouri K.; Edwards R. J.; Meyerhoff R. R.; Oguin T.; Sempowski G.; Saunders K.; Haynes B. F. A glycan cluster on the SARS-CoV-2 spike ectodomain is recognized by Fab-dimerized glycan-reactive antibodies. bioRxiv 2020, 10.1101/2020.06.30.178897. [DOI] [Google Scholar]
- Goh J. B.; Ng S. K. Impact of host cell line choice on glycan profile. Crit. Rev. Biotechnol. 2018, 38, 851–867. 10.1080/07388551.2017.1416577. [DOI] [PubMed] [Google Scholar]
- Watanabe Y.; Allen J. D.; Wrapp D.; McLellan J. S.; Crispin M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020, 369, 330. 10.1126/science.abb9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shajahan A.; Supekar N. T.; Gleinich A. S.; Azadi P. Deducing the N- and O- glycosylation profile of the spike protein of novel coronavirus SARS-CoV-2. Glycobiology 2020, 10.1093/glycob/cwaa042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich H.; Pillat M. M. CD147 as a Target for COVID-19 Treatment: Suggested Effects of Azithromycin and Stem Cell Engagement. Stem. Cell Rev. Rep. 2020, 16, 1–440. 10.1007/s12015-020-09976-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K.; Chen W.; Zhou Y.-S.; Lian J.-Q.; Zhang Z.; Du P.; Gong L.; Zhang Y.; Cui H.-Y.; Geng J.-J.; Wang B.; Sun X.-X.; Wang C.-F.; Yang X.; Lin P.; Deng Y.-Q.; Wei D.; Yang X.-M.; Zhu Y.-M.; Zhang K.; Zheng Z.-H.; Miao J.-L.; Guo T.; Shi Y.; Zhang J.; Fu L.; Wang Q.-Y.; Bian H.; Zhu P.; Chen Z.-N. SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. bioRxiv 2020, 10.1101/2020.03.14.988345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiodo F.; Bruijns S. C. M.; Rodriguez E.; Li R. J. E.; Molinaro A.; Silipo A.; Di Lorenzo F.; Garcia-Rivera D.; Valdes-Balbin Y.; Verez-Bencomo V.; van Kooyk Y. Novel ACE2-Independent Carbohydrate-Binding of SARS-CoV-2 Spike Protein to Host Lectins and Lung Microbiota. bioRxiv 2020, 10.1101/2020.05.13.092478. [DOI] [Google Scholar]
- Miura H. S.; Nakagaki K.; Taguchi F. N-terminal domain of the murine coronavirus receptor CEACAM1 is responsible for fusogenic activation and conformational changes of the spike protein. J. Virol. 2004, 78, 216–223. 10.1128/JVI.78.1.216-223.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C.-M.; Chu H.; Wang Y.; Wong B. H.-Y.; Zhao X.; Zhou J.; Yang D.; Leung S. P.; Chan J. F.-W.; Yeung M.-L.; Yan J.; Lu G.; Gao G. F.; Yuen K.-Y. Carcinoembryonic Antigen-Related Cell Adhesion Molecule 5 Is an Important Surface Attachment Factor That Facilitates Entry of Middle East Respiratory Syndrome Coronavirus. J. Virol. 2016, 90, 9114–9127. 10.1128/JVI.01133-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Templin M. F.; Stoll D.; Schrenk M.; Traub P. C.; Vöhringer C. F.; Joos T. O. Protein microarray technology. Drug Discovery Today 2002, 7, 815–822. 10.1016/S1359-6446(00)01910-2. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Echols N.; Headd J. J.; Hung L.-W.; Jain S.; Kapral G. J.; Grosse Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R. D.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. The Phenix software for automated determination of macromolecular structures. Methods 2011, 55, 94–106. 10.1016/j.ymeth.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y.; Zhang J.; Xiao T.; Peng H.; Sterling S. M.; Walsh R. M. Jr.; Rawson S.; Rits-Volloch S.; Chen B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, eabd4251 10.1126/science.abd4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T.; Zheng Q.; Yu H.; Wu D.; Xue W.; Zhang Y.; Huang X.; Zhou L.; Zhang Z.; Zha Z.; Chen T.; Wang Z.; Chen J.; Sun H.; Deng T.; Wang Y.; Chen Y.; Zhao Q.; Zhang J.; Gu Y.; Li S.; Xia N. Characterization of the SARS-CoV-2 Spike in an Early Prefusion Conformation. bioRxiv 2020, 10.1101/2020.03.16.994152. [DOI] [Google Scholar]
- Zhang L.; Jackson C. B.; Mou H.; Ojha A.; Rangarajan E. S.; Izard T.; Farzan M.; Choe H. The D614G mutation in the SARS-CoV-2 spike protein reduces S1 shedding and increases infectivity. bioRxiv 2020, 10.1101/2020.06.12.148726. [DOI] [Google Scholar]
- Yurkovetskiy L.; Wang X.; Pascal K. E.; Tomkins-Tinch C.; Nyalile T.; Wang Y.; Baum A.; Diehl W. E.; Dauphin A.; Carbone C.; Veinotte K.; Egri S. B.; Schaffner S. F.; Lemieux J. E.; Munro J.; Rafique A.; Barve A.; Sabeti P. C.; Kyratsous C. A.; Dudkina N.; Shen K.; Luban J. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell 2020, 739. 10.1016/j.cell.2020.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T.; Tsybovsky Y.; Olia A. S.; Gorman J.; Rapp M.; Cerutti G.; Chuang G.-Y.; Katsamba P. S.; Nazzari A.; Sampson J. M.; Schön A.; Wang P.; Bimela J.; Shi W.; Teng I. T.; Zhang B.; Boyington J. C.; Sastry M.; Stephens T.; Stuckey J.; Wang S.; Friesner R. A.; Ho D. D.; Mascola J. R.; Shapiro L.; Kwong P. D.. Cryo-EM Structures Delineate a pH-Dependent Switch that Mediates Endosomal Positioning of SARS-CoV-2 Spike Receptor-Binding Domains. Cell Host Microbe 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards R. J.; Mansouri K.; Stalls V.; Manne K.; Watts B.; Parks R.; Gobeil S. M. C.; Janowska K.; Li D.; Lu X.; Deyton M.; Sprenz J.; Williams W.; Saunders K.; Sempowski G. D.; Henderson R.; Alam M.; Haynes B. F.; Acharya P.. Cold sensitivity of the SARS-CoV-2 spike ectodomain. bioRxiv 20202020.07.12.199588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangaru S.; Ozorowski G.; Turner H. L.; Antanasijevic A.; Huang D.; Wang X.; Torres J. L.; Diedrich J. K.; Tian J. H.; Portnoff A. D.; Patel N.; Massare M. J.; Yates J. R.; Nemazee D.; Paulson J. C.; Glenn G.; Smith G.; Ward A. B.. Structural analysis of full-length SARS-CoV-2 spike protein from an advanced vaccine candidate. Science 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao L.; Deng W.; Huang B.; Gao H.; Liu J.; Ren L.; Wei Q.; Yu P.; Xu Y.; Qi F.; Qu Y.; Li F.; Lv Q.; Wang W.; Xue J.; Gong S.; Liu M.; Wang G.; Wang S.; Song Z.; Zhao L.; Liu P.; Ye F.; Wang H.; Zhou W.; Zhu N.; Zhen W.; Yu H.; Zhang X.; Guo L.; Chen L.; Wang C.; Wang Y.; Wang X.; Xiao Y.; Sun Q.; Liu H.; Zhu F.; Ma C.; Yan L.; Yang M.; Han J.; Xu W.; Tan W.; Peng X.; Jin Q.; Wu G.; Qin C. The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature 2020, 830. 10.1038/s41586-020-2312-y. [DOI] [PubMed] [Google Scholar]
- Sigrist C. J.; Bridge A.; Le Mercier P. A potential role for integrins in host cell entry by SARS-CoV-2. Antiviral Res. 2020, 177, 104759–104759. 10.1016/j.antiviral.2020.104759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vankadari N.; Wilce J. A. Emerging COVID-19 coronavirus: glycan shield and structure prediction of spike glycoprotein and its interaction with human CD26. Emerging Microbes Infect. 2020, 9, 601–604. 10.1080/22221751.2020.1739565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Moore M. J.; Vasilieva N.; Sui J.; Wong S. K.; Berne M. A.; Somasundaran M.; Sullivan J. L.; Luzuriaga K.; Greenough T. C.; Choe H.; Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Administration U. S. F. D.EUA Authorized Serology Test Performance; https://www.fda.gov/medical-devices/emergency-situations-medical-devices/eua-authorized-serology-test-performance. (accessed June 10th)
- Palm K.; Jaago M.; Rahni A.; Pupina N.; Pihlak A.; Sadam H.; Avarlaid A.; Planken A.; Planken M.; Haring L.; Vasar E.; Bacevic M.; Lambert F.; Kalso E.; Pussinen P.; Tienari P.; Vaheri A.; Lindholm D.; Timmusk T.; Ghaemmaghami A. M. High seroreactivity against SARS-CoV-2 Spike epitopes in a pre SARS-CoV-2 cohort: implications for antibody testing and vaccine design. medRxiv 2020, 10.1101/2020.05.18.20105189. [DOI] [Google Scholar]
- de Assis R. R.; Jain A.; Nakajima R.; Jasinskas A.; Felgner J.; Obiero J. M.; Adenaiye O.; Tai S.; Hong F.; Norris P. J.; Stone M.; Simmons G.; Bagri A.; Schreiber M.; Buser A.; Holbro A.; Battegay M.; Hosimer P.; Noesen C.; Milton D. K.; Davies D. H.; Contestable P.; Corash L. M.; Busch M. P.; Felgner P. L.; Khan S. Analysis of SARS-CoV-2 Antibodies in COVID-19 Convalescent Blood using a Coronavirus Antigen Microarray. bioRxiv 2020, 10.1101/2020.04.15.043364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Annessa I.; Marchetti F.; Colombo G. Differential Antibody Recognition by Novel SARS-CoV-2 and SARS-CoV Spike Protein Receptor Binding Domains: Mechanistic Insights and Implications for the Design of Diagnostics and Therapeutics. bioRxiv 2020, 10.1101/2020.03.13.990267. [DOI] [Google Scholar]
- Ravichandran S.; Coyle E. M.; Klenow L.; Tang J.; Grubbs G.; Liu S.; Wang T.; Golding H.; Khurana S. Antibody repertoire induced by SARS-CoV-2 spike protein immunogens. bioRxiv 2020, 10.1101/2020.05.12.091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins M. R.; Gasteiger E.; Bairoch A.; Sanchez J. C.; Williams K. L.; Appel R. D.; Hochstrasser D. F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1999, 112, 531–552. 10.1385/1-59259-584-7:531. [DOI] [PubMed] [Google Scholar]
- Hitoshi N.; Ken-ichi Y.; Jun-ichi M. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 1991, 108, 193–199. 10.1016/0378-1119(91)90434-D. [DOI] [PubMed] [Google Scholar]
- Sontag C. A.; Stafford W. F.; Correia J. J. A comparison of weight average and direct boundary fitting of sedimentation velocity data for indefinite polymerizing systems. Biophys. Chem. 2004, 108, 215–230. 10.1016/j.bpc.2003.10.029. [DOI] [PubMed] [Google Scholar]
- Stafford W. F.; Sherwood P. J. Analysis of heterologous interacting systems by sedimentation velocity: curve fitting algorithms for estimation of sedimentation coefficients, equilibrium and kinetic constants. Biophys. Chem. 2004, 108, 231–243. 10.1016/j.bpc.2003.10.028. [DOI] [PubMed] [Google Scholar]
- Philo J. S. Improved methods for fitting sedimentation coefficient distributions derived by time-derivative techniques. Anal. Biochem. 2006, 354, 238–246. 10.1016/j.ab.2006.04.053. [DOI] [PubMed] [Google Scholar]
- Stafford W. F. 3rd Boundary analysis in sedimentation transport experiments: a procedure for obtaining sedimentation coefficient distributions using the time derivative of the concentration profile. Anal. Biochem. 1992, 203, 295–301. 10.1016/0003-2697(92)90316-Y. [DOI] [PubMed] [Google Scholar]
- Brown P. H.; Schuck P. Macromolecular size-and-shape distributions by sedimentation velocity analytical ultracentrifugation. Biophys. J. 2006, 90, 4651–4661. 10.1529/biophysj.106.081372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 2000, 78, 1606–1619. 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen H.; Engelbrecht J.; Brunak S.; von Heijne G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1997, 10, 1–6. 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- Krogh A.; Larsson B.; von Heijne G.; Sonnhammer E. L. L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Garrett-Thomson S. C.; Massimi A.; Fedorov E. V.; Bonanno J. B.; Scandiuzzi L.; Hillerich B.; Seidel R. D. 3rd; Love J. D.; Garforth S. J.; Guha C.; Almo S. C. Mechanistic dissection of the PD-L1:B7-1 co-inhibitory immune complex. PLoS One 2020, 15, e0233578 10.1371/journal.pone.0233578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray K. A.; Seal R. L.; Tweedie S.; Wright M. W.; Bruford E. A. A review of the new HGNC gene family resource. Hum. Genomics 2016, 10, 6. 10.1186/s40246-016-0062-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E. L.; Harrington K.; Staehr M.; Masakayan R.; Jones J.; Long T. J.; Ng K. Y.; Ghoddusi M.; Purdon T. J.; Wang X.; Do T.; Pham M. T.; Brown J. M.; De Larrea C. F.; Olson E.; Peguero E.; Wang P.; Liu H.; Xu Y.; Garrett-Thomson S. C.; Almo S. C.; Wendel H. G.; Riviere I.; Liu C.; Sather B.; Brentjens R. J.. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci. Transl. Med. 2019, 11 (). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suloway C.; Pulokas J.; Fellmann D.; Cheng A.; Guerra F.; Quispe J.; Stagg S.; Potter C. S.; Carragher B. Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 2005, 151, 41–60. 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Lander G. C.; Stagg S. M.; Voss N. R.; Cheng A.; Fellmann D.; Pulokas J.; Yoshioka C.; Irving C.; Mulder A.; Lau P. W.; Lyumkis D.; Potter C. S.; Carragher B. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J. Struct. Biol. 2009, 166, 95–102. 10.1016/j.jsb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S. Q.; Palovcak E.; Armache J. P.; Verba K. A.; Cheng Y.; Agard D. A. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 2017, 14, 331–332. 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punjani A.; Rubinstein J. L.; Fleet D. J.; Brubaker M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 2017, 14, 290–296. 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- Bepler T.; Morin A.; Rapp M.; Brasch J.; Shapiro L.; Noble A. J.; Berger B. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 2019, 16, 1153–1160. 10.1038/s41592-019-0575-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.