

Abstract
Three physiologically mineralizing tissues — teeth, cartilage and bone — have critical common elements and important evolutionary relationships. Phylogenetically the most ancient densely mineralized tissue is teeth. In jawless fishes without skeletons, tooth formation included epithelial transport of phosphates, a process echoed later in bone physiology. Cartilage and mineralized cartilage are skeletal elements separate from bone, but with metabolic features common to bone. Cartilage mineralization is coordinated with high expression of tissue nonspecific alkaline phosphatase and PHOSPHO1 to harvest available phosphate esters and support mineralization of collagen secreted locally. Mineralization in true bone results from stochastic nucleation of hydroxyapatite crystals within the cross-linked collagen fibrils. Mineral accumulation in dense collagen is, at least in major part, mediated by amorphous aggregates — often called Posner clusters — of calcium and phosphate that are small enough to diffuse into collagen fibrils. Mineral accumulation in membrane vesicles is widely suggested, but does not correlate with a definitive stage of mineralization. Conversely mineral deposition at non-physiologic sites where calcium and phosphate are adequate has been shown to be regulated in large part by pyrophosphate. All of these elements are present in vertebrate bone metabolism. A key biological element of bone formation is an epithelial-like cellular organization which allows control of phosphate, calcium and pH during mineralization.
Keywords: Bone mineral, Hydroxyapatite, Osteoblast, Calcium transport, Phosphate transport, Acid transport
1. The first densely mineralized tissue, teeth
To develop a context and perspective for later discussion it is helpful and important to consider evolution and development of teeth. Teeth arise in epithelial tissues and require membrane transport to form calcium-phosphate mineral. This process depends on genes occurring in tooth development and prominent in bone of modern vertebrates, including mammals. The data we present are a consensus view, emphasizing the chemical basis for the appearance of densely mineralized tissue. That said, this is not the place for a comprehensive review of tooth development and we apologize for important contributions not included.
Teeth appeared in early jawless fishes (thelodonts or ostracoderms) as appendages to skin epithelia, at the endoderm-ectoderm interface, preceding the evolution of the mineralized skeleton [1]. The hard matrix of teeth depends on calcium phosphate salts, mainly hydroxyapatite, but phosphate is not present in sea water in concentrations needed for precipitation of calcium salts [2]. Phosphate is accumulated by the cells of these organisms and permits the making of teeth. Tooth formation occurs in epithelia internal to the animal; teeth then are moved to the surface of the integument. This reflects that calcium is abundant in sea water (or plasma), but phosphate is present in sea water at nanomolar concentrations [2]. Phosphate for teeth therefore must be concentrated locally by cells. Hence, teeth develop behind epithelia internal to the animal (Fig. 1B–C), later moving to the surface of the integument. Two epithelial layers function in tooth formation: one forms enamel; an epithelial root sheath generates bone-like dentin [3]. Because of the calcium and phosphate mineral components, enamel and dentin require mineral transport and deposition. Mineral transport depends on membrane potential, pH and ion gradients. This is a defining function of epithelia. In most cases, work in the area of tooth development has focused on inductive signals in the dental epithelium [4]. However, disruption of phosphate transport affects dentin mineralization [5], supporting the concept. The major phosphate transporters appear to be sodium-dependent [6], which later will be discussed in presenting bone mineral transport by osteoblast epithelial-like cell layers.
Fig. 1.
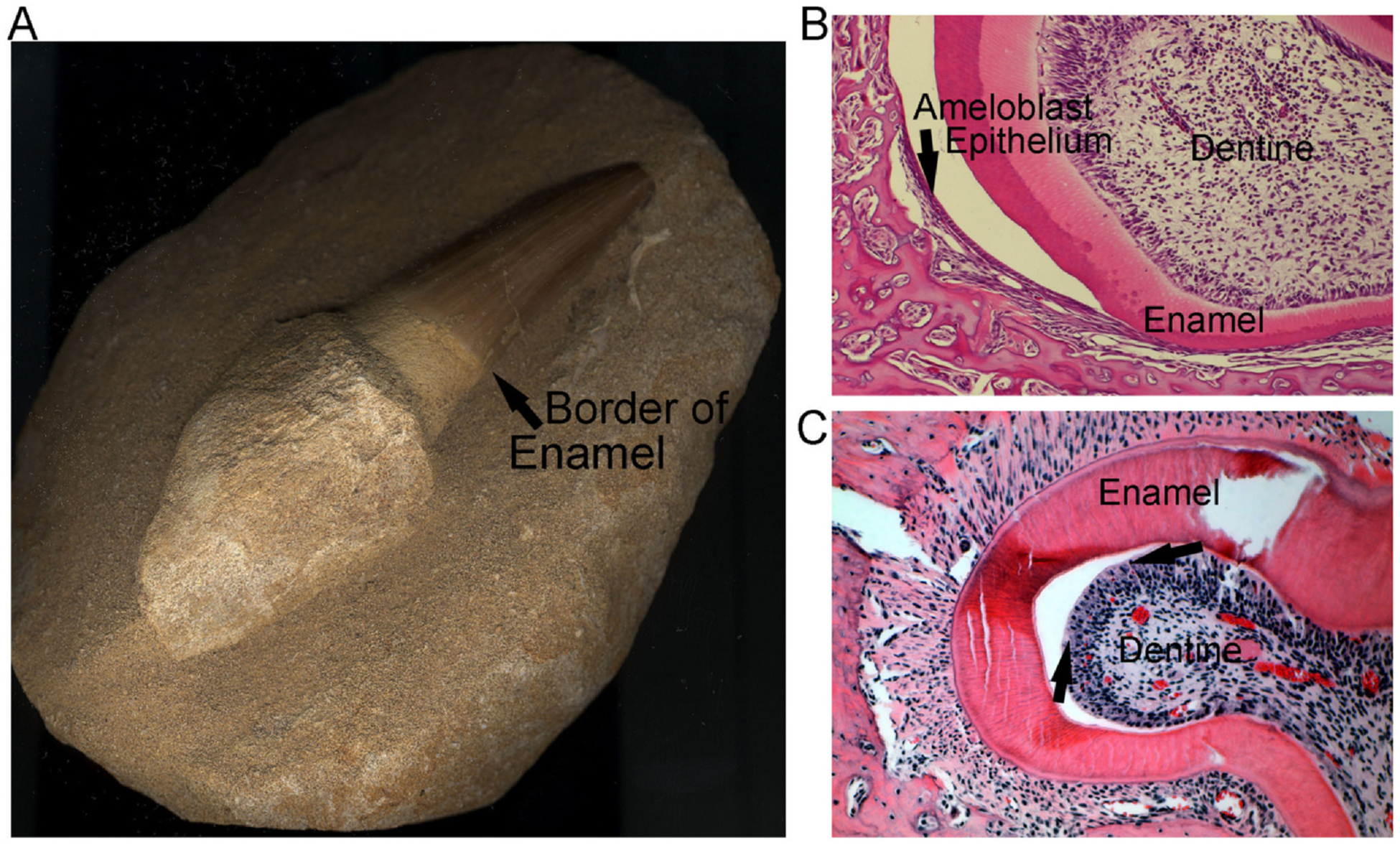
Key features of teeth in higher vertebrates. Unpublished photographs; the mosasaur tooth, (A), was a kind gift of Dr. Mary Sorrells, Pittsburgh PA. Developing mouse teeth in (B,C) are unpublished sections from [11].
A. A fossil tooth from a mosasaur, a Cretaceous period marine dinosaur-related reptile, ~100–66 million years ago. Note that the enamel, even in the fossil, is clearly seen above the arrow. The tooth, from root to tip, is 5 cm long.
B. Developing tooth from a wild type C57Bl/6 mouse showing the ameloblast layer (arrow); this epithelial layer is difficult to see in intact sections; where the tooth is separated by an artifact of cutting it is clearly seen. Section 500 μm across.
C. Developing tooth from a wild type C57Bl/6 mouse showing the odontoblast layer (example between the two arrows) at a site where the overlapping enamel is separated during cutting of the specimen. The section is tangential, making the odontoblast layer appear more than one cell layer thick. Section 500 μm across.
Teeth were retained, or perhaps reappeared, as the skeleton evolved in bony fishes and cartilaginous fishes [7]. The cartilaginous fishes, rays and sharks, go back ~450 million years but employ genes in tooth expression found in the jawless fishes [8]. Modern sharks do not have bone but exhibit well developed teeth that are structurally and evolutionarily homologous to the teeth of air-breathing vertebrates. This co-evolution of teeth that are structurally and genetically similar demonstrates the developmental and functional pressure characterizing the appearance of teeth independent of the bony skeleton. Data include considerable information on growth factors and differentiation of dental epithelium showing that hard tissues are from neural crest cells. These differentiation patterns are conserved in animals, including vertebrates with teeth [1,8].
Shark teeth are composed of dentin of several types, and heavily mineralized enamel [9]. The enamel is generated first, involving amyloid or a closely related ancestral protein. The dentin is not well characterized but contains exclusively type I collagen [10]. All of the higher vertebrates have teeth that retain this pattern (Fig. 1A). In the mammal, there are separate epithelial layers for the ameloblasts and odontoblasts (Fig. 1B–C). We hypothesize that chondrichthyes teeth are closely related to those structures of higher phyla, and that the tooth epithelia form enamel and bone-like dentin layers of teeth in vertebrates, representing a precursor of bone.
In the molecular era, the genetic relationships of bone and teeth are seen [12]. Differences include that only teeth make enamel [13] and only bone is remodeled. Genes expressed in bone and teeth are very similar: tooth or bone stem cells produce bone [14]. Both dentin and bone are composed of type I collagen and express strongly osteocalcin, osteopontin, and alkaline phosphatase [15–18]. Many consider this the consequence of convergent evolution, but identical genes are expressed, so the functional relationship is clear. Specifically there is evidence that some enamel proteins might function in bone [19]. Our experience with cRNA screens for human osteoblasts suggests that, if enamel genes are expressed, they occur only at low levels (not shown). A further important fact is that, while bone-like dentin is composed of proteins and mineral found in bone, teeth do not remodel. Teeth are formed and lost, but once formed they are not significantly modified.
2. Cartilage and mineralized cartilage
This discussion is based on cartilage and its transition to bone via mineralized cartilage in mammals; homology in the lower classes of animals is interesting, but too complex and variable for inclusion. However, the discussion here applies to all mammals with little variation.
Developmentally, the skeleton is formed mainly as a cartilage template that is converted to bone. Exceptions are membranous or dermal bones, the skull and clavicles, which are not discussed. Cartilage is a matrix produced by embedded chondrocytes, mainly type II collagen held to an stiff elastic consistency by hydrophilic proteoglycans. Chondrocytes are cells interspersed in this matrix without cell-cell communications in a anisotropic distribution. Unlike bone, cartilage is avascular. Cartilage is expanded by mitosis of chondrocytes, which redistribute within the cartilage [20]; the mechanism for chondrocyte migration is not well studied. In the region of chondrocyte proliferation, type II collagen and collagenase synthesis are documented [21], presumably allowing new matrix to be formed between cells and limited matrix remodeling.
Mineralization of cartilage occurs at points where bone is to be formed. Preceding cartilage mineralization, chondrocytes align in rows (Fig. 2B, C). There is a complex series of events including expression of Indian hedgehog and the PTH-related protein receptor [22]. The cells then expand with gradual elimination of organelles leaving only vesicles in the cells [23]; at this point they, traditionally, are named “hypertrophic”. In the opinion of the authors, mineralizing chondrocytes is a better term; the cells do not make cartilage and are, effectively, pre-apoptotic. The chondrocytes are in the process of matrix mineralization coordinated with death and degradation. On the other hand, while apoptosis promotes mineralization it might not be essential for mineralization [24]. The apoptotic fate of chondrocytes is currently unclear although macrophages, along with blood vessels invade the mineralizing region and form osteoclasts [25].
Fig. 2.
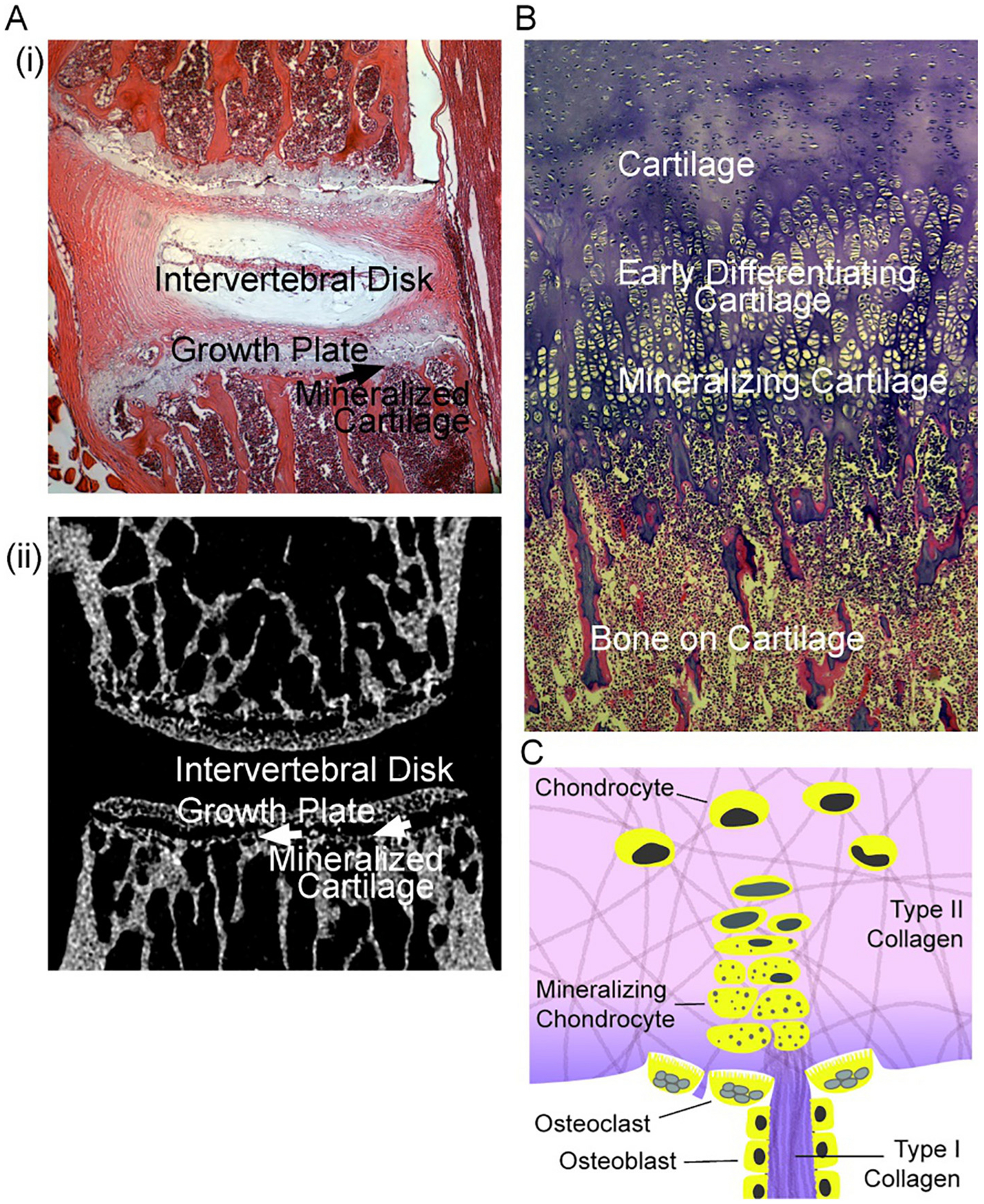
Characteristics of mineralizing cartilage. Mouse bone section photographs are unpublished frames of wild type mice [11]; the section of term human bone is an unpublished frame from [30].
A. Left. (i) At the top, a section of mouse vertebrae L2 and L3, showing the disk and adjacent growth plates. (ii) At the bottom, a similar section by micro computed tomography showing the mineralized cartilage at the transition of growth plates to bone, and the adjacent trabecular bone. Each section is 2 mm across.
B. Right. Morphology of cartilage to bone transition in a human term fetus, with cartilage and its transition to bone labeled. This is a section of rib at the transition to bone; in the rib, a broad section of cartilage is involved; narrower regions of growing cartilage occur in vertebral growth plates as in (A) or diagrammed in (C). The region “mineralizing cartilage” is often called “hypertrophic”, see text. The section is 1 mm across and ~2 mm vertically.
D. Diagram of typical structure of a growth plate. Shown are cartilage from the growing plate (top) composed of chondrocytes in mainly type II collagen, and “hypertrophic” chondrocytes that in part lose nuclei and retain vesicles [24] with mineral producing enzymes [31]. At the transition to mineralized cartilage, some type I collagen is present, with trans-differentiation of chondrocytes [32]. Mineralized cartilage (darker color) is degraded mainly by chondroclasts (osteoclasts) and osteoblasts form on this matrix. The authors hypothesize that calcium-phosphate complexes are important in mineralizing collagen, including type I collagen produced in mineralizing cartilage or associated osteoblast-like cells [33], although in early mineralization mineral in vesicles might be important; see text.
Early mineralization of cartilage [26] is associated with expression of collagen X and matrix metalloproteinase-13 [22]. As the chondrocytes lose organelles vesicles are formed that exhibit high levels of tissue-nonspecific alkaline phosphatase and PHOSPHO1 [27]. PHOSPHO1 is a phosphatase that removes phosphate from phosphocholine and phosphoethanolamine [28] in intact membranes [29]. Phospholipase C uniquely provides this substrate releasing phosphocholine and phosphoethanolamine, although the same molecules are generated by kinases in vertebrate cells. How these pathways influence mineralization is not clear. Indeed, expression of phospholipases in mineralizing cartilage (and osteoblasts) is not well studied, and PHOSPHO1 is recently discovered; its distribution is uncertain, although it clearly is important along with alkaline phosphatase in bone and cartilage [21]. At present we view alkaline phosphatase, organic phosphates, and membrane lipids as sources of phosphate for mineral formation (Fig. 2). Additional sources include Na-dependent phosphate transport in vesicles and osteoblasts, with the pyrophosphatase (ENPP1), producing a high ratio of Pi to PPi [27]. Cartilage is not a vascular tissue and chondrocytes are not connected by tight junctions, so substrates and products of mineralization would diffuse, but perhaps not as effectively as in hierarchical bone, see below.
The morphology of the cartilage-bone transformation has long been studied by light and electron microscopy. From these studies it was concluded include that the cells often lose organelles and nuclei, becoming collections of vesicles in otherwise empty lacunae [24]. The mechanisms by which these transformations occur is unclear. A key issue is how bone mineral deposition occurs in matrix. As reported by standard fixation for electron microscopy (that is, with calcium in solutions) with vacuum processing, hydroxyapatite crystals appear inside vesicles. This was studied by Anderson [34] and later by Glimcher in a comprehensive review of mineralization [35]: while early cartilage mineralization is consistent with retention of large crystals in vesicles, later cartilage mineralization produces smaller, collagen associated crystals that are distinct from vesicle crystals. We hypothesize that the large crystals are, in part, artifacts of sample processing for electron microscopy and offer an alternative suggestions for cartilage mineralization and endochondral bone formation. During mineralization in living tissue and conversion of cartilage to bone, calcium phosphate aggregates (Posner clusters) and amorphous calcium aggregates of 2–5 nm form [36], small enough to diffuse into dense collagen in mineralizing cartilage and in compact bone matrix of osteoblasts [37]. There is, however, convincing characterization of matrix vesicle mineral deposition, including by atomic force microscopy [38]. The problem is that calcium crystals larger than a few nm cannot diffuse into dense collagen, and crystals of ~100 nm cannot transit vesicle membranes, but might contribute to the non-hierarchical cartilage matrix.
Type I collagen in bone is a material into which mineral crystals are added in a defined arrangement; in the type II collagen of cartilage mineral packing is not well understood. While the issue is controversial, mineral deposition in cartilage might involve a mixture of type II, type X [33], and type I collagen in mineralizing cartilage. In this regard, there is convincing data indicating that chondrocytes, during epiphyseal bone formation, can produce bone proteins including type I collagen, and might trans-differentiate to form bone [32]. That said, a satisfactory overall mechanism for the transition from apatite crystals with matrix vesicles to mineralized cartilage matrix is not available. In any case, mineralization of avascular cartilage reaches ~half the density achieved in bone (Fig. 3), where an epithelial-like transport system allows packing inward of phosphate and proton removal [40]: see Mineralization of True Bone, below. Mineralized cartilage has been studied in detail, pointing, in addition to lower density than bone, to a more variable mineral composition [39].
Fig. 3.
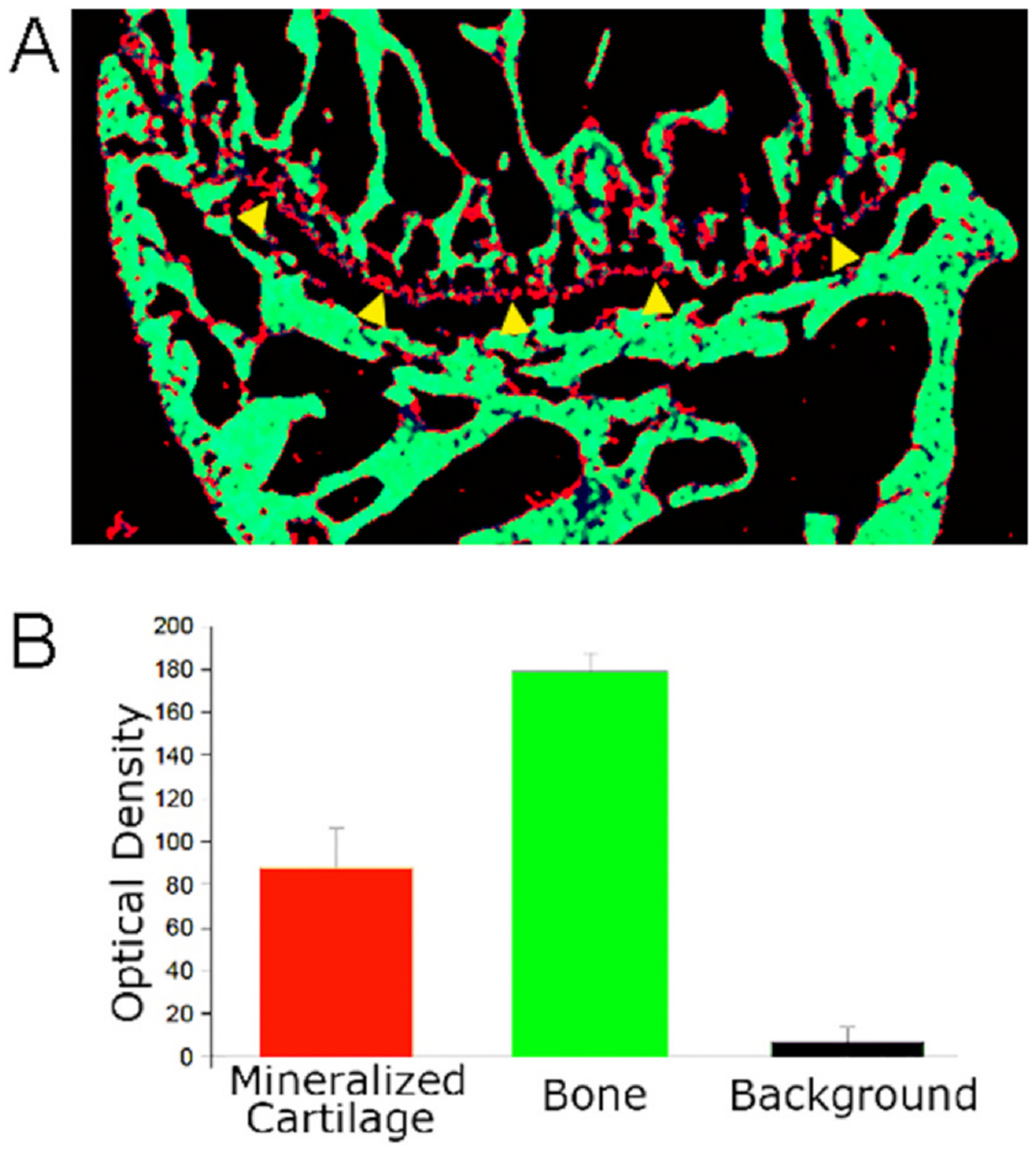
Comparison of Cartilage and Bone Mineral Density. Unpublished photographs from [11]; mouse sections and density are of wild type mice, as in Fig. 1.
A. Micro-computed tomography of a mouse femur tibial end. The pixels are color coded for density of < 10, black, 10–100, red, and > 100, green. Note the mineralized cartilage is seen as a red line (marked by yellow arrowheads at its bottom; compare with Fig. 2A, lower panel).
B. Density of mineralized cartilage, bone, and background from micro computed tomography as in (A). N = 4, mean ± SD. The cartilage has approximately half the density of bone; the difference is significant, p < 0.001. This difference in cartilage and bone mineral density reflects, in major part, that cartilage is not vascular, and in addition to having a lower mineral density than bone, cartilage has more variable mineral composition [39], see text.
3. Regulation of mineralization in normally non-mineralized tissues
Orderly mineralization of bone is essential for the development of higher organisms. However, absence of mineralization in soft tissue is equally critical. The calcium and phosphorous in circulation is adequate for mineral formation in the absence of a preventative mechanism [41]. In this context, we are not discussing ectopic bone, but mineralization of tissues that normally do not mineralize. There are many examples of ectopic mineralization of varying severity, sometimes associated with trauma, e.g. [42–44]. However, an important mechanism of potentially fatal ectopic mineralization results from inability to form low levels of pyrophosphate (PPi) in normally non-mineralized tissues. We will concentrate on syndromes related to this, which are studied at the molecular level. Pyrophosphate is long recognized as an inhibitor of mineralization, including in bone [45,46], but its importance in non-bone tissues has only recently been established.
Generalized arterial calcification of infancy (GACI) is a rare syndrome in which afflicted patients calcify their hearts, aorta, and large arteries during the third trimester of pregnancy [47,48]. If the fetus survives to term, newborns present before 4 months, half in the first week of life, in respiratory or cardiac distress. Half of affected infants will not survive to 6 months of age despite supportive care. Those that survive undergo a physiologically adaptive response with elevated FGF23 and phosphate wasting [49]. Vascular and cardiac calcifications stabilize or may even regresses [50,51], but stabilization of ectopic calcification is at the expense of a rachitic skeletal phenotype, autosomal recessive hypophosphatemic rickets Type 2 [52,53].
There are two genetic causes of GACI. One is homozygous loss of function mutations in the enzyme ectonucleotide pyrophosphate/phosphodiesterase-1 (ENPP1), seen in 75% of patients [54,55]. Remaining patients with GACI have bi-allelic loss of function mutations in the transporter ABCC6 [56,57], which regulates extracellular ATP. Mutations of ABCC6 cause soft tissue calcification disorders overlapping with GACI, also called pseudoxanthoma elasticum (PXE) [58]. Severely affected PXE presents as GACI. ENPP1 is the only enzyme in humans for synthesis of extracellular pyrophosphate (PPi) [54]. Both ENPP1 and ABCC6 deficiency cause decreased plasma [PPi] [59–61] accounting for the overlapping clinical phenotype.
ENPP1 generates PPi by cleaving NTPs to generate PPi (Fig. 4). Extracellular PPi is never generated unless ENPP1 is present. However, when ENPP1 is present, PPi is produced at a rate of approximately 3.5 molecules per second, an acceleration of 1027 fold above the non-catalyzed rate.
Fig. 4.
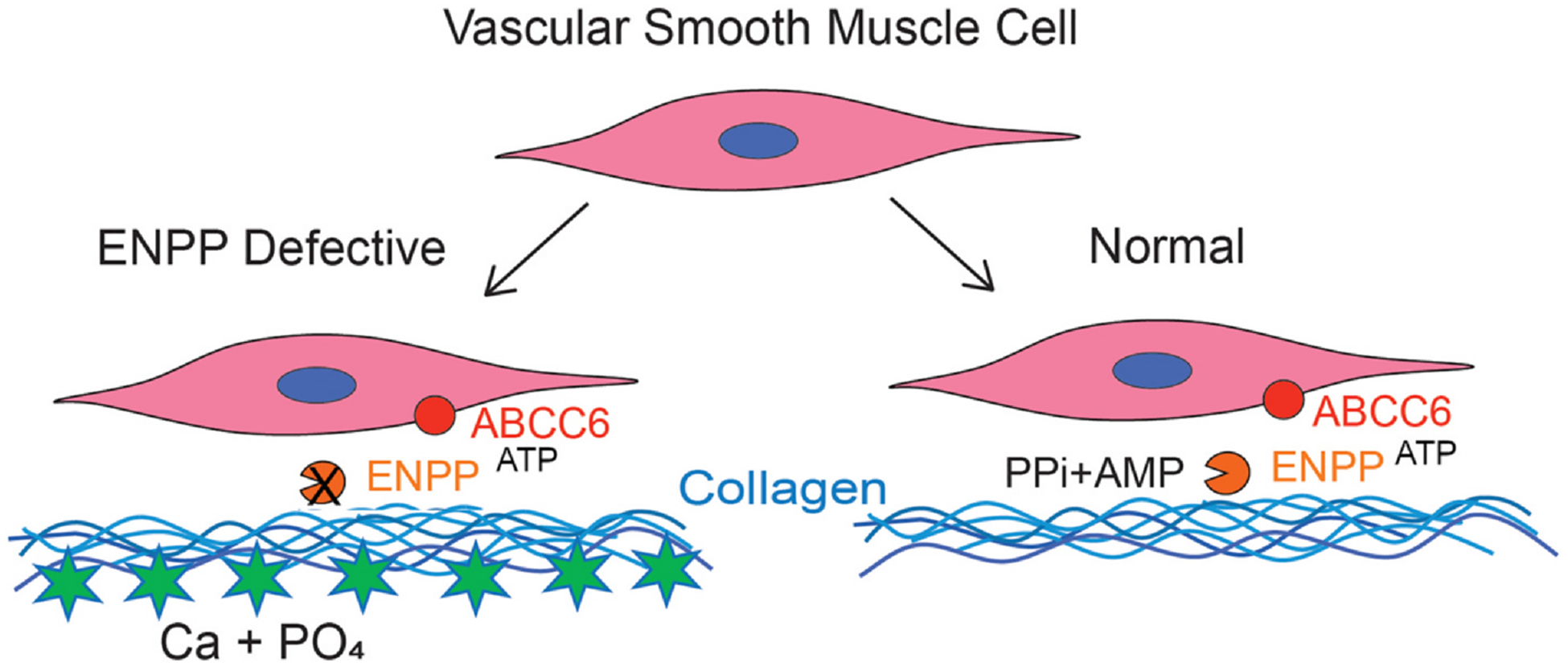
Mineral formation in normally non-mineralized tissues occurs when pyrophosphate cannot be produced.
The model shows arterial tissue including type I collagen and arterial smooth muscle cells. Normally, the transporter ABCC6 produces small amounts of extracellular ATP, converted to pyrophosphate and AMP by ENPP1 (right). If ENPP1 or ABCC6 are absent (left) calcium and phosphorus in the serum are adequate to cause extracellular calcification.
Extracellular [PPi] is ~2.5 μM in patients without loss of function mutations in either ENPP1 or ABCC6. Bi-allelic ENPP1 mutations lead to extracellular PPi < 10% of WT [59,62]. Patients with bi-allelic ABCC6 mutations have PPi ~30% of WT (~0.75–1.0 μM) [61]. Lower limits of ‘normal’ plasma PPi have not been established, but in chronic kidney disease vascular calcifications occur at plasma [PPi] below 1.5 μM [63]. Patients with heterozygous ENPP1 mutations have plasma PPi ~ half of normal [59,64]. Patients with heterozygous loss of function mutations in ENPP1 are may develop early onset osteoporosis [59], believed to be a consequence of adaptive response as discussed above.
3.1. Modeling of GACI
The best current model is the Enpp1asj/asj mouse [65], progeny of an ENU-mutagenized C57BL/6 mouse. ‘Asj’ signifies ‘age with stiffened joints’ due to a V246D point mutation in ENPP1 [65], near the catalytic threonine at AA 238 in mouse ENPP1. This attenuates the enzymatic activity ~75%. Enpp1asj/asj mice have accelerated mortality, pervasive arterial calcifications, and cardiac death on a special diet to accelerate calcification [65,66]. These mice have been used to verify the mechanism by ENPP1 enzyme replacement [67]. To convert the membrane-bound enzyme to a soluble, stable, and bioavailable protein, the signal sequence of Enpp2 was used to release Enpp1 from the membrane surface. The protein was fused to the Fc domain of murine IgG1 to increase the bioavailability and stability. The resulting solubilized Enpp2 has a half-life of ~6–8 h. In initial dosing experiments, there was a dramatic clinical response to ~8–10 mg/kg daily of replacement enzyme. Treated Enpp1asj/asj mice normalized their weights compared to WT, normalized plasma PPi levels, and eliminated vascular and cardiac calcifications [67].
4. Mineralization of true bone
We focus on the air breathing vertebrates, emphasizing mammals. Lower orders are useful in study of specific genetic defects, such as type 1 collagen defects in zebrafish [68], but differences in hard tissue structure and regulation detract from the utility of fish models to inform the study of bone in mammals.
4.1. The bone composite
Over a half century ago it was postulated that the addition of mineral to the bone organic matrix was critical in achieving the necessary mechanical properties [69]. Decades of sophisticated study concluded that bone represents a composite mainly of hydroxyapatite (HA) and collagen [70]. This suggested to many that the mechanism of bone formation to be the stochastic nucleation of HA crystals around the cross linked collagen fibrils. Over time, it has been agreed that in order to achieve the observed and required changes in properties, HA crystallization must occur in association with collagen molecules and not as appendages of collagen fibrils [71,72]. A hierarchical and defined structure of this sort allows the two major components to share resistance to stress and provide the elastic properties that are present in cortical bone. In human cortical bone, synchrotron X-ray diffraction and scattering revealed the intimacy of the mineral–organic interaction [73]. In bone tissue, the maximum displacement achieved in response to stress forces increases to twice the estimated fracture tolerance of bulk apatite. This indicates a bidirectional relationship between the organic and mineral components of the bone composite [73]. Recent studies using cryo-transmission EM and transmission EM indicate that crystalline structures infiltrate the individual fibrils, even when solution crystallization is suppressed with inhibitors [74]. This was extended in vitro to confirm that intra-fibrillar mineralization occurs in the presence of crystallization inhibitors, even though they can be covalently linked to collagen, suppressing inter-fibrillar crystallization but not mineralization and its effects on bone quality [75]. Most importantly, mineralization improves the collagen Young’s modulus to values similar to that in quality bone. These observations lead us to conclude that the local environment in the collagen fibril of the osteoid matrix is critical in fostering effective bone mineralization that provides the strength and elasticity of quality bone. Furthermore the intra-fibrillar mineralization may not be the result of stochastic crystallization, but rather result from specific chemistry between calcium-phosphates and the collagen.
4.2. Key elements of bone formation
Osteoblasts perform the key biological functions during bone formation. Osteoblasts form an epithelium-like layer over the osteon allowing the development of an osteogenic environment (Fig. 5). Osteoblasts synthesize Type I collagen in massive quantities, which is heavily crosslinked and produced in lamina with alternating orientation along and orthogonal to the axis of bone growth [18]. Osteoblasts make small quantities of other bone proteins including osteopontin and bone sialoprotein. Osteoblasts convey phosphate and calcium into the osteogenic matrix [76], and remove acid produced by mineral deposition [18,40]. The mechanism linking mineral transport to dense mineral formation remains controversial; calcium phosphate complexes (Posner complexes) diffuse to the site of dense mineralization. A small fraction of apatite in bone is substituted by carbonate or ionized phosphates [77–79], but hydroxyapatite constitutes the vast majority of bone mineral.
Fig. 5.
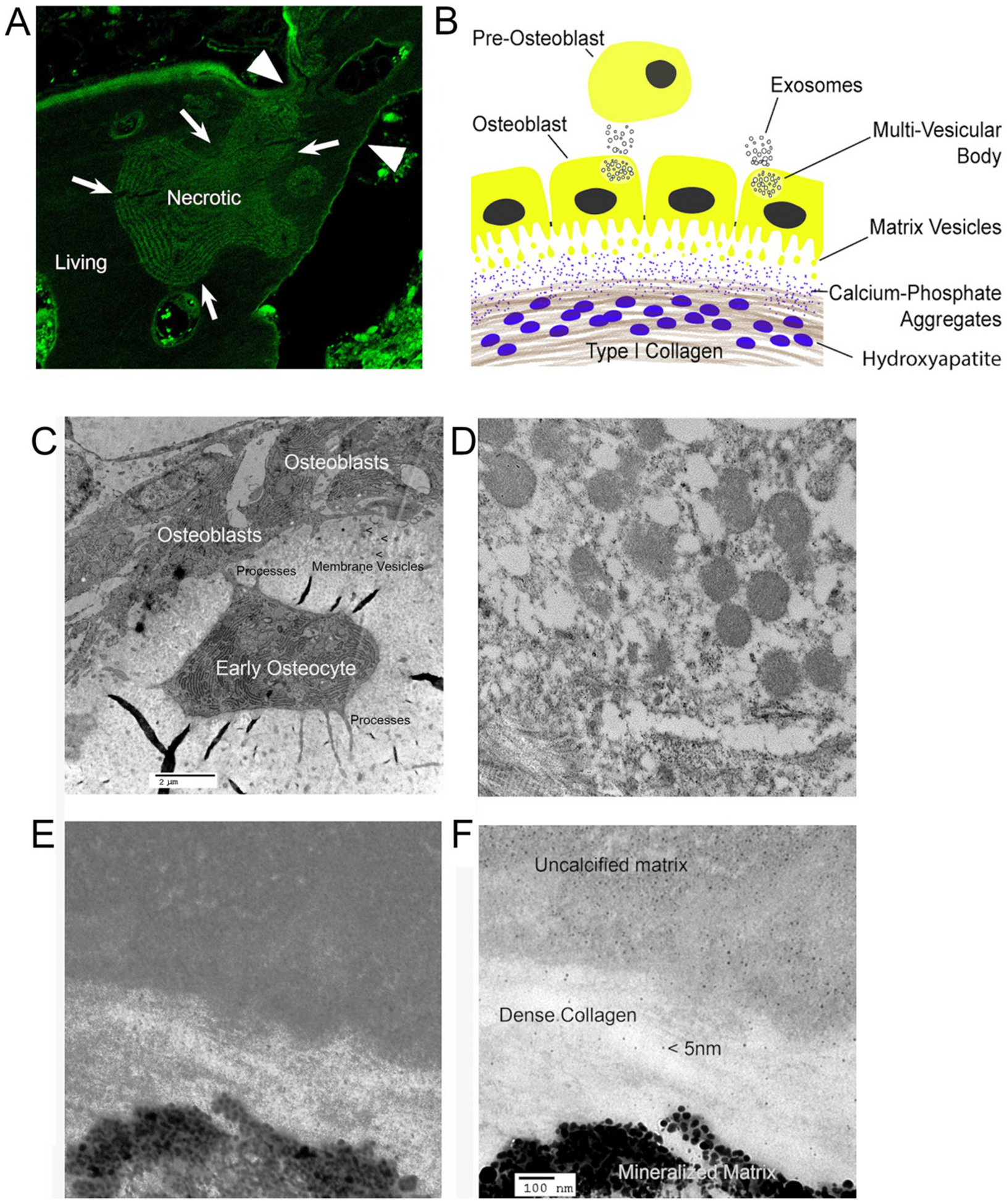
Membranes, vesicles, and the apical surface of the bone epithelioid layer. (A) is previously published in [79]; (C–F) are unpublished frames from [40], except (E), from that work, used with permission.
A. Bone is separated from general extracellular fluid by an epithelial-like cell layer. The photograph shows tetracycline labeling of bone formation (top of section) but that tetracycline otherwise is excluded from living (appears dark), but not necrotic (shows labeling) bone; bone is impervious to water and most small ions, but not calcium. For details, see [79].
B. Diagram of bone formation with two types of vesicles. Vesicles, at least in part from multivesicular bodies, are produced on the basolateral surface and serve, at least mainly, to communicate with other cells. At the apical surface, matrix vesicles [80] as in (C) and (D) increase surface area of the cells at the site of mineralization and express phosphate producing enzymes also found in the osteoblast. Matrix vesicles are excluded from dense collagen. Small calcium-phosphate complexes diffuse into dense matrix and form mineral on collagen strands.
C. Plastic embedded thin section of mouse bone in EM (~20 μm across) [40] showing membrane vesicles at the osteoblast apical membrane, the bone secreting side. To retain calcified material, 2 mm thick calvarial bone was fixed 5 min in 4% paraformaldehyde in phosphate buffer at 4 °C and moved directly to dehydration for plastic embedding. An early osteocyte, recently separated from the osteoblast layer, is seen with processes connecting the cell to osteoblasts, and below the osteocyte connecting to deeper osteocytes (not seen here). Irregular black areas are tears in the section.
D. The appearance of this section of the apical surface at higher power. Many vesicles from the membrane are seen. Average vesicle size is ~300 nm. Collagen fibers with crosslinking are seen below the membrane vesicles. Vesicles are excluded from dense collagen fibers, where mineral accumulates (see text).
E. Early mineralized bone on EM of a similar section with dense collagen. Hydroxyapatite crystals ~30 nm associated with collagen are seen. No bilayer vesicles occur in dense crosslinked collagen.
F. The section (E) at higher power. Amorphous calcium phosphates (based on Posner complexes) of small size, ~5 nm, occur in the non-mineralized and dense collagen layer, hypothetical diffusible calcium phosphate aggregates [36,37].
4.3. The tight epithelium-like cell layer of bone
A comprehensive understanding of bone remodeling requires that one appreciate that skeletal formation is locally bounded by a tight epithelium-like layer that has operational homology to dental epithelium, Fig. 1. The osteon surface layer of epithelioid osteoblasts displays functional and histological characteristics of a tight cell layer with regulated permeability. Living bone is impermeable to most anions. Dead bone is generally diffusely permeable to these ions, [79], (Fig. 5A). The rest of the time osteoblasts form a tight cell layer excluding small molecules including anionic fluorescent indicators such as tetracycline. The impermeability is related to tight junctions in the surface cells, which have long been known [81]. Primary osteoblasts form epithelial layers with significant trans-epithelial resistance of ≈150 Ohm/cm2 [82]. In addition to tight junctions, gap junctions have long been recognized [83], which mostly are connexin45 and connexin43 based, and link both surface osteoblasts including with calcium signaling, along with deeper layers of osteocytes [84,85]. During matrix synthesis tetracyclines and other fluorescent anions are rapidly taken up and deposited in the osteogenic compartment. This activity is short lived but is responsible for a widely used experimental and clinical assay of bone growth [79], (Fig. 5A). It is also clear that bone regulates the water content of matrix [86], which, again, reflects isolation of the matrix from general extracellular space. The mechanism for regulation of bone matrix water is not established.
The osteoblast cell layer supports transport of phosphate [76] and calcium (discussed below) into the osteogenic compartment and the removal out of that same compartment of acid produced during sustained mineralization [40]. As previously reviewed, the osteon is a bone forming unit that occurs between the osteoblast epithelial-like layer and the cement lines [87]. This compartment is morphologically complex but possible to identify in rapidly fixed semi-thin sections and in culture [88]. At the macroscopic level this barrier has been used for decades to evaluate bone growth; a range of calcium-binding fluorescent anions are taken up by new bone specifically in narrow layers only during bone synthesis. These are useful as probes for live-cell microscopic studies in vitro, and have long been used to mark bone synthesis in vivo [89]. Similar tetracycline deposition occurs in teeth, and highlights the known dependence of tooth development on the dental epithelium for mineralization [90].
4.4. pH and transport
There is limited data on the pH of mineralizing bone matrix, but studies on the chemistry and structure of calcium phosphate precipitation provides solid support for the importance of pH in bone mineralization. Over 60 years ago a monograph characterizing the chemistry of bone mineral as dynamic and uniquely pH sensitive was published [91]. This assertion was confirmed by showing that the calcium phosphate solubility increased dramatically as pH went from 7.4 to 6.8 [92]. It was further noted that even in buffered solutions, as precipitation proceeded, the pH decreased, indicating that protons were generated by precipitation of hydroxyapatite. Finally, it was observed that soluble collagen promoted precipitation and the pH change, while gelatin did not. Later it was shown that under conditions when pH was held constant at a slightly alkaline value (for example 7.4) that HPO4 −2 was the dominant phosphate species (> 60%) entering the apatite and that each phosphate entering the crystal generated two protons [93]. This allows the formulation of a balanced equation for a solution of calcium phosphate becoming apatite crystals:
| (1) |
Thus predicting that bone mineralization will produce substantial protons, in the osteon, that must be removed.
In a direct study of pH in healing bone, Chakkalakal et al. [94] found that in early healing, tissue pH is lower than serum pH, consistent with accumulation of acidic metabolites. Subsequently, pH increased to alkaline levels with rapid mineralization, in keeping with removal of acid produced. The results of these studies support observations suggesting that the pH of tissue plays a regulatory role in mineralization of bone. Considering in vitro studies where hydroxyapatite precipitation generates protons ([92] and Eq. (1)) the observed alkalinization is consistent with high activity of Na+/H+ exchangers in active osteoblasts [40,95].
4.5. Calcium transport into bone
Bone and teeth constitute a major reservoir of calcium. Specific calcium transport mechanisms are largely undefined in osteoblasts, but it appears that calcium is not actively transported. In other sites including the gut and kidney, para-cellular transport is of key importance, and regulated [96]. Calcium transport involves transient receptor potential vanilloid 6 (Trpv6) calcium channels mediating calcium transfer across the intestinal apical membrane [97], but Trpv6 is expressed only at low levels in osteoblasts [98], so this mechanism is unimportant in bone. Osteoblasts express several claudins [82] that might be involved in similar transport in bone, but calcium paracellular transport in osteoblasts is not specifically studied. These include claudins 2 and 12 [82], critical for paracellular calcium absorption in enterocytes and renal tubular cells [99]. Significant active calcium transport by osteoblasts is unlikely: Osteoblasts have Na+/Ca2+ exchangers that are negatively regulated by dihydroxyvitamin D and parathyroid hormone [100]. Osteoblasts express a calmodulin-activated plasma membrane Ca2+-ATPase, but not in quantities expected for mineralization transport of calcium [101,102]. Osteoblasts and osteocytes contain the calcium binding protein calbindin, which is upregulated by dihydroxyvitamin D [103]. In teeth, calcium transport is independent of calbindin [104], suggesting independence of bulk calcium transport from an intracellular calcium pool. On the other hand, sodium-calcium exchangers are expressed in quantities consistent with transport at the osteoblast-matrix (apical) membrane [105], so it cannot be excluded that some calcium transcytosis occurs in osteoblasts.
4.6. Phosphate transport to form bone mineral
Phosphate metabolism is a universal aspect of eukaryotic biology but, especially in vertebrates, is important in bone and teeth. It has been studied extensively, but it is complex, and major conceptual and experimental gaps remain. One major issue the contribution of organic phosphate esters to the transcellular movement of phosphate for collagen mineralization. Massive osteoblast expression of alkaline phosphatase was reported 100 years ago, as phosphate ester hydrolysis [106] by the orthophosphoric-monoester phosphohydrolase, now generally called alkaline phosphatase, reflecting to the enzyme’s alkaline pH optimum [107], in contrast to the osteoclast’s acid phosphatase. Twenty-five years later hypophosphatasia was designated an inborn error of metabolism with severe skeletal consequences [108]. Subsequently hypophosphatasia has been studied in illuminating detail, contributing to our understanding of collagen mineralization [109]. The bone isoform of the enzyme is an unusual ecto-enzyme, that is, attached (at least mostly) to the matrix side of the osteoblast apical surface [110]. This supports the hypothesis that bone alkaline phosphatase isoenzyme (also called tissue nonspecific alkaline phosphatase or liver/bone/kidney alkaline phosphatase – transcribed from the same gene) supplies phosphate for bone mineral. It is clear that phosphate is trafficked into the osteogenic matrix by a transcellular pathway and is presented for mineralization extracellularly as summarized below and illustrated in Fig. 6. The alkaline phosphatase degrades pyrophosphate, which accumulates in its absence [46], and all organic phosphates as well. One key organic phosphate that accumulates in its absence is pyridoxal phosphate [111,112], although it is not known whether pyridoxal phosphate is a major part of organic phosphate transmitted by osteoblasts.
Fig. 6.
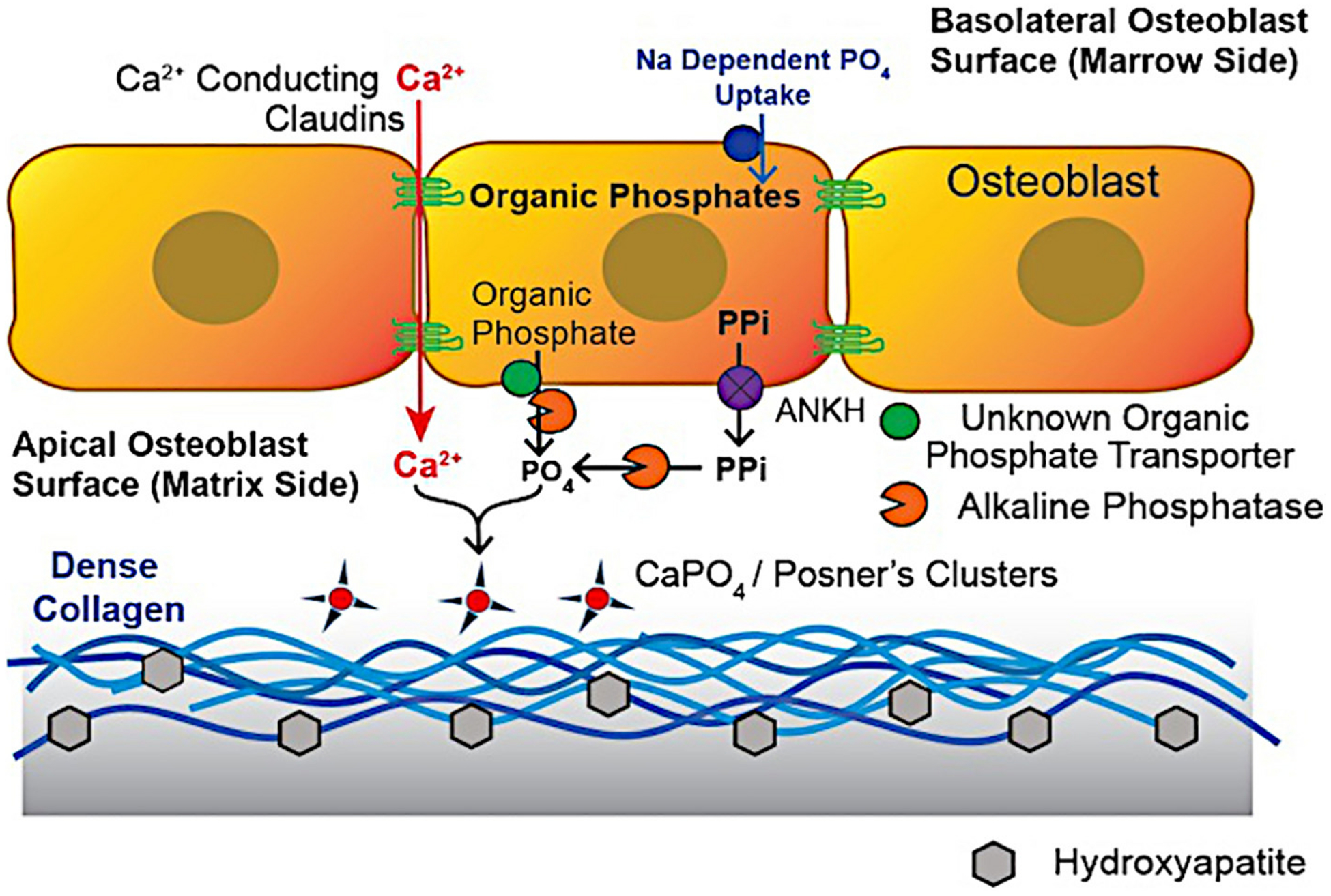
Major mineral transport mechanisms in bone. For Ca2+, transport is largely trans-cellular [82]. Phosphate from sodium-dependent uptake at the basolateral surface [76] is converted to organic phosphate [119] and exported from the apical surface by unknown transport mechanisms. This is followed by release of phosphate by an ecto-enzyme (extracellular membrane attached) alkaline phosphatase [110]. The osteoblast also massively expresses the pyrophosphate transport regulator ANKH [121], possibly capturing PPi produced during collagen synthesis: one pyrophosphate is produced per amino acid added. In the absence of alkaline phosphatase, accumulation of organic phosphates including pyridoxal phosphate [110] and of pyrophosphate [112] occurs, both related to attenuation of mineralization.
4.7. Phosphate transport overall and in bone
Phosphate is transported in major quantities in the gut and in the kidney, as well as in bone. In all cases described to date, phosphate movement into cells is mediated by sodium-phosphate co-transporters, a subject not specific to bone and reviewed recently [113]. Phosphate entry in osteoblasts is demonstrated to occur by sodium-dependent neutral phosphate transport [76]. Neutral phosphate transport in osteoblasts is, in part, regulated by the sodium hydrogen exchange regulator factor-1 (NHERF1) [95]. Known sodium-linked transport mechanisms in apical membranes of epithelial cells are SLC34 and SLC20 [114]. SLC34 isoforms are found in osteogenic cells and during tooth mineralization; this transporter is specific for the phosphate di-anion and might to lessen acid load during mineralization [115]. Neutral phosphate transporters SLC20A1 and A2 are active in bone [76]. Selective genetic suppression of sodium-dependent phosphate transport produces strong phenotypes with the absence of SLC34A2 is an embryonic lethal condition in mice [116]. Knockdown or mutation of SLC34A1 [117] or A3 [118] in mice are survivable, with disorders of mineral metabolism, the portion due to osteoblast effects being unknown.
4.8. The distribution of phosphate after uptake in osteoblasts
It was conclusively demonstrated that phosphate entering bone in the presence of sodium is rapidly, and essentially quantitatively, converted into organic phosphates. Specifically, by 32P analysis, 87% of phosphate entering osteoblasts is found in the organic fraction after 5 min [119,120] (Fig. 6).
4.9. Export of phosphate from cells
The delivery of phosphate from cells is much less fully understood (Fig. 6). Only one transporter is known to deliver phosphate from cells on the side opposite sodium-dependent phosphate uptake, that being, in kidney, the xenotropic and polytropic retrovirus receptor 1 (XPR1 or SLC53A1), believed to liberate free phosphate [122,123], although Na+ dependency of transport, possibly related to secondary factors, is reported [124]. Sodium dependency of transport by a closely related protein, GLVR1, was reported but would not favor Pi release into the mineralizing matrix [125]. Deletion of XPR1 in the renal proximal tubule causes hypophosphatemic rickets [126]. In the intestine, although transport into the body is well known, the mechanism phosphate export is not studied, but is hypothesized to use XPR1 [113].
In osteoblasts specific phosphate export mechanisms into the osteon matrix are not described (Fig. 6). Expression libraries from osteoblasts show XPR1 is present only at low levels (not shown). Furthermore, when two homologs of XPR1 were genetically eliminated in zebrafish, bone formation was not impaired [127]. On the other hand, organic phosphates transported to the apical membrane are degraded to produce free phosphate [112,128] by the osteoblast external membrane-bound alkaline phosphatase [110]. Among the interesting and yet not fully studied possibilities include that, in hypophosphatasia, pyridoxal phosphate accumulates in serum [111], suggesting that pyridoxal phosphate is a substrate for the alkaline phosphatase ectoenzyme and might contribute directly to phosphate transport in osteoblasts. This has only been studied indirectly [129]. Pyridoxal kinase is one of several mechanisms to mediate rapid conversion of osteoblast intracellular phosphate [76,114] to organic forms [119]. Other substrates for alkaline phosphatase include pyrophosphate, which is a second barrier to mineral formation in hypophosphatasia [46]. This is particularly, if still hypothetically (Figs. 4, 6), important in bone because osteoblasts express highly the pyrophosphate transport-regulating protein ANKH [121], thus, potentially, capturing, by pyrophosphate export, the large amount of pyrophosphate produced during collagen synthesis in osteoblasts. However, pyrophophosphate exported must be completely degraded to avoid preventing hydroxyapatite formation. The role of pyrophosphate inhibiting mineralization discussed in “Regulation of mineralization in normally non mineralized tissues”, above. Free phosphate and calcium near the apical membrane of osteoblasts produce small calcium phosphate aggregates that are consistent with the small hydroxyapatite crystals that develop in dense mineral within type I collagen subjacent to the cells [35]; (see Fig. 5).
4.10. Initialization of mineralization
No-one disputes that hydroxyapatite is the dominant bone mineral. Similarly, the organic matrix is overwhelmingly Type I collagen. Controversy occurs when considering the transformation from soluble mineral components and collagen gel to the bone composite, Earlier in this review we present a speculation on the formation of the hydroxyapatite-collagen composite that is catalyzed by Type I collagen substrate and strongly promoted by pH. Here we present our understanding of several alternate pathways through early mineralization.
There are many parallels between bone mineralization and cartilage mineralization, including matrix vesicles [130] at mineralization sites, and high levels of alkaline phosphatase and PHOSPHO1 [131,132]. In cartilage the majority of the mineral is located in the organic matrix, not matrix vesicles [35]. However, the mineral density and the material properties of cartilage never reach those of bone (see Fig. 3). We propose that this is primarily because cartilage is not mineralized behind an epithelial-like cell layer (osteoblasts) controlling the local environment [133]. Thus, deposition of mineral in cartilage is driven by exceeding the mineral solubility product, and not influenced by fine pH control and a uniform dense Type I collagen substrate.
4.11. Intra-vesicular and intra-cellular crystallization
A confounding thread of speculation follows observations of crystals in vacuoles in cartilage [34] and bone [134]. This hypothesis is that mineral formation occurs within osteoblasts or osteoblast matrix vesicles [130]. These crystals are proposed to be transferred by unknown mechanisms to form, or catalyze, the formation of dense and smaller hydroxyapatite crystals in cross-linked collagen fibrils. Indeed, it has been hypothesized that there is no direct transport of mineral into bone matrix and mineralization occurs by the formation of amorphous crystals in ~0.2 to 0.3 μm cell vesicles that are released into the extracellular matrix, where they can accrete more mineral [135]. This is often called the “crystal nucleation hypothesis.” This hypothesis is attractive for its simplicity, but it supposes the spontaneous and coordinated occurrence of complex events and in the opinion of the authors needs significant modification. Specifically, it does not include phosphate (or calcium) transport into the matrix or account for export of acid produced in formation of hydroxyapatite [40]. It does not account for the isolation of bone matrix from the general extracellular fluid [79], or for why the activity of alkaline phosphatase resides on the matrix side of the osteoblast cell membrane. As discussed above, calcium transcytosis is believed to be a minor part of calcium transport for bone formation, so calcium is less available for intra-cellular or intravesicular crystal formation. It should be noted that for crystal formation there is always crystal nucleation. We assert that kilograms of mineral in the human skeleton are accreted mainly by regulated transport of calcium, phosphate, and removal of acid released from formation of hydroxyapatite. Further in this regard, Landis et al. pointed out that extracellular vesicles are not uniformly present during mineralization [136]. We know of no proposal defining how the crystalline structures are transferred out of the extracellular vesicles into crosslinked collagen fibrils to assemble the bone composite. Finally, bone mineralization is exquisitely controlled, and yields an atomistically integrated composite with physical properties distinct from separate collagen and hydroxyapatite components [73].
4.12. The fate of osteoblast membrane vesicles
In the mineralized matrix the only membrane bilayer structures are associated with osteocytes, indeed vesicles are excluded once dense cartilage develops and the only exceptions are canaliculi connecting osteocytes and osteoblasts (Fig. 5D–E). This raises an important question as to where vesicles produced by mineralizing osteoblasts are destined. Since there are no macrophages in bone matrix, the vesicles are not degraded by phagocytic cells; other possibilities include that the vesicles are taken back up by osteoblasts, or that the membranes are completely degraded. While vesicles bear the alkaline phosphatase ectoenzyme, and the phosphates from the membranes are removed by PHOSPHO1, and this phosphate probably contributes to initial mineralization, there are no precedents for membrane bilayer dissolution, although some reports point to penetration and release of crystals from vesicles [137]. For formation of vesicles, there are also multiple hypotheses including ectosomes from vesicle fusion and exosomes from multi-vesicular bodies [80]. We hypothesize that membrane vesicles in developing bone matrix fuse back to osteoblasts, since bone matrix does not contain significant membrane lipid, bone matrix is not patrolled by macrophages, and matrix vesicles are excluded from dense type I collagen (Fig. 5F). Autophagy in osteoblasts [138,139] is in keeping with recycling of membrane vesicles.
Apical membrane vesicles (Fig. 5C–D) are proposed to participate in the production of new mineral. However the importance of this could be judged better if their mechanism of formation was understood, budding verses multi-vesicular-body exocytosis. The enzymes for phosphate production, PHOSPHO1 and alkaline phosphatase, are present, although the amount of phosphate associated with vesicles is by itself insufficient for the mass of dense mineral formation in bone. Osteogenesis is known to require membrane transport of phosphate in, and of acid out, by an epithelial (or epithelioid) osteoblast cell layer [40,140]. Alkaline phosphatase and PHOSPHO1 can support initial mineralization, for which there is a large literature [141]. However, the connection of this to collagen-bound hydroxyapatite, and the development from initial mineral to dense hydroxy-apatite collagen layers, is not worked out. Aside from initial mineralization and matrix vesicles, bulk phosphate transport through the osteoblast surface layer, and removal of acid produced by hydroxyapatite deposition, is at a further step critical to the formation of dense mineral in bone [18].
Osteoblasts also shed vesicles from their basolateral surface (Fig. 5B) [142]. These may be generated from either plasma membrane budding (ectosomes) or fusion of multi-vesicular bodies with the plasma membranes. While the fate and function of these vesicles is also not established, potential roles in RNA and protein transport to modify differentiation of developing new osteoblasts and related cells have been described [142]. Signaling by cell surface glycosaminoglycans promoting osteoblast formation is described [143].
5. Reprise: support of mineralization by the surface layer of bone forming cells
The osteoblast surface cells are an epithelial-like layer that isolates the forming bone matrix from surrounding extracellular fluid (Fig. 5A) [79]. As shown in Eq. (1), formation of hydroxyapatite from the soluble calcium and phosphate ions at physiologic pH generates protons. The number of protons depends upon the phosphate species that participates in crystallization. The removal of these protons is accomplished in the same layer of surface osteoblasts that define the mineralization frontier. Along their basolateral border these cells express massive sodium‑hydrogen exchange capacity that can transport acid out of the osteon [40,144]. This transport capacity is under the control of the sodium-hydrogen exchange regulator-1 [95], which is expressed on surface epithelial-like osteoblasts, but not significantly on the deeper osteocytes derived from these cells in bone formation. Actual H+ transport is a mixture of NHE1 and NHE6 activity, a likely reason for survivability of NHE1 knockout [145]. NHE activity removes H+ from the osteoblast using the Na+ gradient maintained in all mammalian cells. Antibody localization of NHE-1 in the basolateral membrane of osteoblasts [144] is consistent with this, although how NHE activity is regulated is unclear.
It is not clear how protons from mineral formation enter through the osteoblast apical membrane [18]. In expression libraries of bone-producing osteoblasts the levels of mRNA for the chloride-proton anti-porters, ClC-3 and ClC-5 are elevated to unprecedented levels, and the proteins are, surprisingly, histologically associated with the apical membranes of osteogenic osteoblasts [140]. Further, surface membranes from mineralizing osteoblasts in culture, reconstituted into membrane vesicles, show that chloride-dependent proton transport is present, but is blocked if Cl− inside vesicles is replaced by gluconate. These same preparations do not exhibit ATP-dependent acidification are therefore not detectably contaminated by endosome, lysosome or secretory vesicle membrane, the usual location of ClC-3 and 5 [40]. However endosome and secretory vesicle membranes cycle through the plasma membrane and it is known that ClC-3 cell surface isoforms depend on an N-terminal retention signal [146].
6. Conclusion
Physiologic mineralization of tissues is essential in mammals. Non-physiological mineralization is disease producing, often fatal. Although complex, key aspects of mineralization are conserved in phylogeny and are important to consider in medicine. Included are the occurrence of mineral behind an epithelial barrier and selective transport of Ca2+, phosphate, and H+. Important gaps in understanding of mineralization remain, but we are confident the phylogeny and ontology of mineralization are constrained by physics, chemistry [147], reflect ancestral mechanisms and parallel transport mechanisms in other organs.
Grant support
Supported in part by grant 1 R01 AR076146-01 from the National Institutes of Health, USA, and by BX002490-06A1 from the Department of Veteran’s Affairs, USA.
References
- [1].McCollum M, Sharpe PT, Evolution and development of teeth, J. Anat 199 (2001) 153–159, 10.1046/j.1469-7580.2001.19910153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Menzel DW, Corwin N, The measurement of total phosphorus in seawater based on liberation of organically bound fractions by persulfate oxidation, Limnol. Oceanogr 10 (1965) 280–282, 10.4319/lo.1965.10.2.0280. [DOI] [Google Scholar]
- [3].Yang Y, Ge Y, Chen G, Yan Z, Yu M, Feng L, Jiang Z, Guo W, Tian W, Hertwig’s epithelial root sheath cells regulate osteogenic differentiation of dental follicle cells through the Wnt pathway, Bone 63 (2014) 158–165, 10.1016/j.bone.2014.03.006. [DOI] [PubMed] [Google Scholar]
- [4].Balic A, Thesleff I, Tissue interactions regulating tooth development and renewal, Curr. Top. Dev. Biol 115 (2015) 157–186, 10.1016/bs.ctdb.2015.07.006. [DOI] [PubMed] [Google Scholar]
- [5].Merametdjian L, Beck-Cormier S, Bon N, Couasnay G, Sourice S, Guicheux J,Gaucher C, Beck L, Expression of phosphate transporters during dental mineralization, J. Dent. Res 97 (2018) 209–217, 10.1177/0022034517729811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lundquist P, Odontoblast phosphate and calcium transport in dentinogenesis, Swed. Dent. J suppl, 154 (2002) 1–52. doi not available. [PubMed] [Google Scholar]
- [7].Smith MM, Johanson Z, Separate evolutionary origins of teeth from evidence in fossil jawed vertebrates, Science 299 (2003) 1235–1236, 10.1126/science.1079623. [DOI] [PubMed] [Google Scholar]
- [8].Rasch LJ, Martin KJ, Cooper RL, Metscher BD, Underwood CJ, Fraser GJ, An ancient dental gene set governs development and continuous regeneration of teeth in sharks, Dev. Biol, 415 (2016) 347–370. doi: 10.1016/j.ydbio.2016.01.038. [DOI] [PubMed] [Google Scholar]
- [9].Whitenack LB, Simkins DC Jr., Motta PJ, Hirai M, Kumar A, Young’s modulus and hardness of shark tooth biomaterials, Arch. Oral Biol 55 (2010) 203–209, 10.1016/j.archoralbio.2010.01.001. [DOI] [PubMed] [Google Scholar]
- [10].Ciena AP, de S Rangel B, Bruno CE, Miglino MA, de Amorim AF, Rici RE, Watanabe I, Morphological Aspects of Oral Denticles in the Sharpnose Shark Rhizoprionodon lalandii (Müller and Henle, 1839) (Elasmobranchii, Carcharhinidae), Anat Histol Embryol, 45 (2016) 109–114. doi: 10.1111/ahe.12178. [DOI] [PubMed] [Google Scholar]
- [11].Robinson LJ, Mancarella S, Songsawad D, Tourkova IL, Barnett JB, Gill DL,Soboloff J, Blair HC, Gene disruption of the calcium channel Orai1 results in inhibition of osteoclast and osteoblast differentiation and impairs skeletal development, Lab. Investig 92 (2012) 1071–1083, 10.1038/labinvest.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chinsembu KC, Teeth are bones: signature genes and molecules that underwrite odontogenesis, J. Med.Genet. Genomics 4 (2012) 13–24, 10.5897/JMGG11.022. [DOI] [Google Scholar]
- [13].Smith CEL, Poulter JA, Antanaviciute A, Kirkham J, Brookes SJ,Inglehearn CF, Mighell AJ, Amelogenesis imperfecta; genes, proteins, and pathways, Front. Physiol 8 (2017) 435, 10.3389/fphys.2017.00435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Winning IA Karim El, Lundy FT. A, Comparative Analysis of the Osteogenic Potential of Dental Mesenchymal Stem Cells, Stem Cells Dev, 28 (2019) 1050–1058. doi: 10.1089/scd.2019.0023. [DOI] [PubMed] [Google Scholar]
- [15].Bidder M, Latifi T, Towler DA, Reciprocal temporospatial patterns of Msx2 and osteocalcin gene expression during murine odontogenesis, J. Bone Miner. Res 13 (1998) 609–619, 10.1359/jbmr.1998.13.4.609. [DOI] [PubMed] [Google Scholar]
- [16].Saito K, Nakatomi M, Ida-Yonemochi H, Ohshima H, Osteopontin is essential for type I collagen secretion in reparative dentin, J. Dent. Res 95 (2016) 1034–1041, 10.1177/0022034516645333. [DOI] [PubMed] [Google Scholar]
- [17].Ching HS, Luddin N, Rahman IA, Ponnuraj KT, Expression of odontogenic and osteogenic markers in DPSCs and SHED, Curr. Stem Cell Res. Ther 12 (2017) 71–79, . [DOI] [PubMed] [Google Scholar]
- [18].Blair HC, Larrouture QC, Li Y, Lin H, Beer-Stoltz D, Liu L, Tuan RS,Robinson LJ, Schlesinger PH, Nelson DJ, Osteoblast differentiation and bone matrix formation in vivo and in vitro, Tissue Eng Part B Rev 22 (2017) 268–280, 10.1089/ten.TEB.2016.0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lu X, Fukumoto S, Yamada Y, Evans CA, Diekwisch TG, Luan X, Ameloblastin, an extracellular matrix protein, affects long bone growth and mineralization, J. Bone Miner. Res 31 (2016) 1235–1246, 10.1002/jbmr.2788. [DOI] [PubMed] [Google Scholar]
- [20].Chang C, Lauffenburger DA, Morales TI, Motile chondrocytes from newborn calf: migration properties and synthesis of collagen II, Osteoarthr. Cartil 11 (2003) 603–612, 10.1016/s1063-4584(03)00087-6. [DOI] [PubMed] [Google Scholar]
- [21].Tchetina EV, Mwale F, Poole AR, Changes in gene expression associated with matrix turnover, chondrocyte proliferation and hypertrophy in the bovine growth plate, Acta Naturae, 6 (2014) 89–97. doi not available. [PMC free article] [PubMed] [Google Scholar]
- [22].Hata K, Takahata Y, Murakami T, Nishimura R, Transcriptional Network Controlling Endochondral Ossification, J. Bone Metab, 24 (2017) 75–82. doi: 10.11005/jbm.2017.24.2.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Roy R, Kudryashov V, Binderman I, Boskey AL, The role of apoptosis in mineralizing murine versus avian micromass culture systems, J. Cell. Biochem 111 (2010) 653–658, 10.1002/jcb.22748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Gibson GJ, Kohler WJ, Schaffler MB, Chondrocyte apoptosis in endochondral ossification of chick sterna, Dev. Dyn 203 (1995) 468–476, 10.1002/aja.1002030409. [DOI] [PubMed] [Google Scholar]
- [25].Engsig MT, Chen QJ, Vu TH, Pedersen AC, Therkidsen B, Lund LR,Henriksen K, Lenhard T, Foged NT, Werb Z, Delaissé JM, Matrix metalloproteinase 9 and vascular endothelial growth factor are essential for osteoclast recruitment into developing long bones, J. Cell Biol 151 (2000) 879–889, 10.1083/jcb.151.4.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Nishimura R, Hata K, Takahata Y, Murakami T, Nakamura E, Yagi H, Regulation of Cartilage Development and Diseases by Transcription Factors, J. Bone Metab, 24 (2017) 147–153. doi: 10.11005/jbm.2017.24.3.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bolean M, Simão AMS, Barioni MB, Favarin BZ, Sebinelli HG, Veschi EA, Janku TAB, Bottini M, Hoylaerts MF, Itri R, Millán JL, Ciancaglini P, Biophysical aspects of biomineralization, Biophys. Rev, 9 (2017) 747–760. doi: 10.1007/s12551-017-0315-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Roberts SJ, Stewart AJ, Sadler PJ, Farquharson C, Human PHOSPHO1 exhibits high specific phosphoethanolamine and phosphocholine phosphatase activities, Biochem. J 382 (2004) 59–65, 10.1042/BJ20040511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mebarek S, Abousalham A, Magne D, Dole D, Bandorowicz-Pikula J, S. Pikula S, Buchet R, Phospholipases of mineralization competent cells and matrix vesicles: roles in physiological and pathological mineralizations, Int. J. Mol. Sci, 14 (2013) 5036–5129. doi: 10.3390/ijms14035036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Blair HC, Zaidi M, Schlesinger PH, Mechanisms balancing skeletal matrix synthesis and degradation, Biochem. J 364 (2002) 329–341, 10.1042/BJ20020165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Huesa C, Yadav MC, Finnilä MA, Goodyear SR, Robins SP, Tanner KE, Aspden RM, Millán JL, Farquharson C, PHOSPHO1 is essential for mechanically competent mineralization and the avoidance of spontaneous fractures, Bone 48 (2011) 1066–1074, 10.1016/j.bone.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Aghajanian P, Xing W, Cheng S, Mohan S, Epiphyseal bone formation occurs via thyroid hormone regulation of chondrocyte to osteoblast transdifferentiation, Sci. Rep 7 (2017) 10432, 10.1038/s41598-017-11050-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Garimella R, Bi X, Camacho N, Sipe JB, Anderson HC, Primary culture of rat growth plate chondrocytes: an in vitro model of growth plate histotype, matrix vesicle biogenesis and mineralization, Bone 34 (2004) 961–970, 10.1016/j.bone.2004.02.010. [DOI] [PubMed] [Google Scholar]
- [34].Anderson HC, Electron microscopic studies of induced cartilage development and calcification, The J. Cell Biol 35 (1967) 81–101, 10.1083/jcb.35.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Glimcher MJ, Bone: Nature of the Calcium Phosphate Crystals and Cellular, Structural, and Physical Chemical Mechanisms in Their Formation, Rev. Mineralogy Geochem, 64 (2006) 223–282. doi:0.2138/rmg.2006.64.8. [Google Scholar]
- [36].Posner AS, Crystal chemistry of bone mineral, Physiol. Rev 49 (1969) 760–792, 10.1152/physrev.1969.49.4.760. [DOI] [PubMed] [Google Scholar]
- [37].Lutsko JF, How crystals form: A theory of nucleation pathways, Sci. Adv, 5 (2019) eaav7399. doi: 10.1126/sciadv.aav7399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Plaut JS, Strzelecka-Kiliszek A, Bozycki L, Pikula S, Buchet R, Mebarek S,Chadli M, Bolean M, Simao AMS, Ciancaglini P, Magrini A, Rosato N,Magne D, Girard-Egrot A, Farquharson C, Esener SC, Millan JL, Bottini M, Quantitative atomic force microscopy provides new insight into matrix vesicle mineralization, Arch. Biochem. Biophys 667 (2019) 14–21, 10.3390/10.1016/j.abb.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Rey C, Beshah K, Griffin R, Glimcher MJ, Structural studies of the mineral phase of calcifying cartilage, J. Bone Miner. Res 6 (1991) 515–525, 10.1002/jbmr.5650060514. [DOI] [PubMed] [Google Scholar]
- [40].Blair HC, Larrouture QC, Tourkova IL, Liu L, Bian JH, Stolz DB, Nelson DJ, Schlesinger PH, Support of bone mineral deposition by regulation of pH, Am. J. Physiol. Cell Physiol 315 (2018) C587–C597, 10.1152/ajpcell.00056.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Tian E, Watanabe F, Martin B, Zangari M, Innate Biomineralization, International J. Mol. Sci 21 (2020) E4820, , 10.3390/ijms21144820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Fournier DE, Kiser PK, Beach RJ, Dixon SJ, Séguin CA, Dystrophic calcification and heterotopic ossification in fibrocartilaginous tissues of the spine in diffuse idiopathic skeletal hyperostosis (DISH), Bone Res 8 (2020) 16, 10.1038/s41413-020-0091-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ranganathan K, Loder S, Agarwal S, Wong VW, Forsberg J, Davis T, Wang S, Aaron J, Levi B, Benjamin, Heterotopic ossification: basic-science principles and clinical correlates, Bone Joint Surg Am, 97 (2015) 1101–11. doi: 10.2106/JBJS.N.01056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Pignolo RJ, Ramaswamy G, Fong JT, Shore E,M, Kaplan FS, Progressive osseous heteroplasia: diagnosis, treatment, and prognosis. Appl Clin Genet. 8 (2015) 37–48. doi: 10.2147/TACG.S51064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Meyer JL, Can biological calcification occur in the presence of pyrophosphate? Arch. Biochem. Biophys 231 (1984) 1–8, 10.1016/0003-9861(84)90356-4. [DOI] [PubMed] [Google Scholar]
- [46].Whyte MP, Hypophosphatasia: an overview for 2017, Bone 102 (2017) 15–25, 10.1016/j.bone.2017.02.011. [DOI] [PubMed] [Google Scholar]
- [47].Bryant J, White W, A case of calcification of the arteries and obliterative endarteritis, associated with hydronephrosis, in a child aged six months, Guy’s Hos. Rep, 55 (1901) 17–28. doi not available. [Google Scholar]
- [48].Ferreira C, Ziegler S, Gahl WA, Generalized Arterial Calcification of Infancy, in: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A (Eds.), GeneReviews, University of Washington, Seattle, 1993–2020, 2014. November 13 [eBook]. [PubMed] [Google Scholar]
- [49].Rutsch F, Boyer P, Nitschke Y, Ruf N, Lorenz-Depierieux B, Wittkampf T,Weissen-Plenz G, Fischer RJ, Mughal Z, Gregory JW, Davies JH, Loirat C, Strom TM, Schnabel D, Nurnberg P, Terkeltaub R, Group GS, Hypophosphatemia, hyperphosphaturia, and bisphosphonate treatment are associated with survival beyond infancy in generalized arterial calcification of infancy, Circ. Cardiovasc. Genet 1 (2008) 133–140, 10.1161/CIRCGENETICS.108.797704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Marrott PK, Newcombe ND, Becroft DM, Friedlander DH, Idiopathic infantile arterial calcification with survival to adult life, Pediatr. Cardiol 5 (1984) 119–122, 10.1007/BF02424963. [DOI] [PubMed] [Google Scholar]
- [51].Sholler GF, Yu JS, Bale PM, Hawker RE, Celermajer JM, Kozlowski K, Generalized arterial calcification of infancy: three case reports, including spontaneous regression with long-term survival, J. Pediatr 105 (1984) 257–260, 10.1016/s0022-3476(84)80123-7. [DOI] [PubMed] [Google Scholar]
- [52].Levy-Litan V, Hershkovitz E, Avizov L, Leventhal N, Bercovich D, Chalifa-Caspi V, Manor E, Buriakovsky S, Hadad Y, Goding J, Parvari R, Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene, Am. J. Hum. Genet 86 (2010) 273–278, 10.1016/j.ajhg.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lorenz-Depiereux B, Schnabel D, Tiosano D, Hausler G, Strom TM, Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets, Am. J. Hum. Genet 86 (2010) 267–272, 10.1016/j.ajhg.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Rutsch F, Vaingankar S, Johnson K, Goldfine I, Maddux B, Schauerte P,Kalhoff H, Sano K, Boisvert WA, Superti-Furga A, Terkeltaub R, PC-1 nucleoside triphosphate pyrophosphohydrolase deficiency in idiopathic infantile arterial calcification, Am. J. Pathol 158 (2001) 543–554, 10.1016/S0002-9440(10)63996-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Rutsch F, Ruf N, Vaingankar S, Toliat MR, Suk A, Hohne W, Schauer G,Lehmann M, Roscioli T, Schnabel D, Epplen JT, Knisely A, Superti-Furga A,McGill J, Filippone M, Sinaiko AR, Vallance H, Hinrichs B, Smith W, Ferre M,Terkeltaub R, Nurnberg P, Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification, Nat. Genet 34 (2003) 379–381, 10.1038/ng1221. [DOI] [PubMed] [Google Scholar]
- [56].Nitschke Y, Baujat G, Botschen U, Wittkampf T, du Moulin M, Stella J, Le Merrer M, Guest G, Lambot K, Tazarourte-Pinturier MF, Chassaing N, Roche O, Feenstra I, Loechner K, Deshpande C, Garber SJ, Chikarmane R, Steinmann B, Shahinyan T, Martorell L, Davies J, Smith WE, Kahler SG, McCulloch M, Wraige E, Loidi L, Hohne W, Martin L, Hadj-Rabia S, Terkeltaub R, Rutsch F, Generalized arterial calcification of infancy and pseudoxanthoma elasticum can be caused by mutations in either ENPP1 or ABCC6, Am. J. Hum. Genet, 90 (2012) 25–39. doi: 10.1016/j.ajhg.2011.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Li Q, Brodsky JL, Conlin LK, Pawel B, Glatz AC, Gafni RI, Schurgers L,Uitto J, Hakonarson H, Deardorff MA, Levine MA, Mutations in the ABCC6 gene as a cause of generalized arterial calcification of infancy: genotypic overlap with pseudoxanthoma elasticum, J. Invest. Dermatol 134 (2014) 658–665, 10.1038/jid.2013.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Nitschke Y, Rutsch F, Generalized arterial calcification of infancy and pseudoxanthoma elasticum: two sides of the same coin, Front. Genet 3 (2012) 302, 10.3389/fgene.2012.00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Oheim R, Zimmerman K, Maulding ND, Sturznickel J, von Kroge S,Kavanagh D, Stabach PR, Kornak U, Tommasini SM, Horowitz MC,Amling M, Thompson D, Schinke T, Busse B, Carpenter TO, Braddock DT, Human heterozygous ENPP1 deficiency is associated With early onset osteoporosis, a phenotype recapitulated in a mouse model of Enpp1 deficiency, J. Bone Miner. Res 35 (2020) 528–539, 10.1002/jbmr.3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Jansen RS, Kucukosmanoglu A, de Haas M, Sapthu S, Otero JA, Hegman IE, Bergen AA, Gorgels TG, Borst P, van de Wetering K, ABCC6 prevents ectopic mineralization seen in pseudoxanthoma elasticum by inducing cellular nucleotide release, Proc. Nat. Acad. Sci. U.S.A, 110 (2013) 20206–20211. doi: 10.1073/pnas.1319582110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Jansen RS, Duijst S, Mahakena S, Sommer D, Szeri F, Varadi A, Plomp A, Bergen AA, Oude Elferink RP, Borst P, van de Wetering K, ABCC6-mediated ATP secretion by the liver is the main source of the mineralization inhibitor inorganic pyrophosphate in the systemic circulation-brief report, Arterioscler. Thromb. Vasc. Biol 34 (2014) 1985–1989, 10.1161/ATVBAHA.114.304017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Stella J, Buers I, van de Wetering K, Hohne W, Rutsch F, Nitschke Y, Effects of different variants in the ENPP1 gene on the functional properties of ectonucleotide pyrophosphatase/phosphodiesterase family member 1, Hum. Mutat 37 (2016) 1190–1201, 10.1002/humu.23057. [DOI] [PubMed] [Google Scholar]
- [63].O’Neill WC, Sigrist MK, McIntyre CW, Plasma pyrophosphate and vascular calcification in chronic kidney disease, Nephrol. Dial. Transplant 25 (2010) 187–191, 10.1093/ndt/gfp362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Kotwal A, Ferrer A, Kumar R, Singh RJ, Murthy V, Schultz-Rogers L,Zimmermann M, Lanpher B, Zimmerman K, Stabach PR, Klee E,Braddock DT, Wermers RA, Clinical and biochemical phenotypes in a family With ENPP1 mutations, J. Bone Min. Res 35 (2020) 662–670, 10.1002/jbmr.3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Li Q, Guo H, Chou DW, Berndt A, Sundberg JP, Uitto J, Mutant Enpp1asj mice as a model for generalized arterial calcification of infancy, Dis. Models Mech 6 (2013) 1227–1235, 10.1242/dmm.012765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Jiang Q, Uitto J, Restricting dietary magnesium accelerates ectopic connective tissue mineralization in a mouse model of pseudoxanthoma elasticum (Abcc6−/−), Exp. Dermatol 21 (2012) 694–969, 10.1111/j.1600-0625.2012.01553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Albright RA, Stabach P, Cao W, Kavanagh D, Mullen I, Braddock AA, Covo MS, Tehan M, Yang G, Cheng Z, Bouchard K, Yu ZX, Thorn S, Wang X, Folta-Stogniew EJ, Negrete A, Sinusas AJ, Shiloach J, Zubal G, Madri JA, De La Cruz EM, Braddock DT, ENPP1-Fc prevents mortality and vascular calcifications in rodent model of generalized arterial calcification of infancy, Nat. Commun 6 (2015) 10006, 10.1038/ncomms10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Gistelinck C, Kwon RY, Malfait F, Symoens S, P Harris M, Henke K, Hawkins MB, Fisher S, Sips P, Guillemyn B, Bek JW, Vermassen P, De Saffel H, Witten PE, Weis M, De Paepe A, Eyre DR, Willaert A, Coucke PJ, Zebrafish type I collagen mutants faithfully recapitulate human type I collagenopathies, Proc. Natl. Acad. Sci. U.S.A, 115 (2018) E8037–E8046. doi: 10.1073/pnas.1722200115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Currey JD, Strength of bone, Nature 195 (1962) 513–514, 10.1038/195513a0. [DOI] [Google Scholar]
- [70].Weiner S, Wagner HD, The material bone: structure-mechanical function relations, Ann. Rev. Materials Sci 28 (1998) 271–298, 10.1146/annurev.matsci.28.1.271. [DOI] [Google Scholar]
- [71].Olszta MJ, Cheng X, Jee SS, Kumar R, Kim Y-Y, Kaufman MJ, Douglas EP,Gower LB, Bone structure and formation: a new perspective, Mater. Sci. Eng. Rep 58 (2007) 77–116, 10.1016/j.mser.2007.05.001. [DOI] [Google Scholar]
- [72].Fratzl P, Fratzl-Zelman N, Klaushofer K, Vogl G, Koller K, Nucleation and growth of mineral crystals in bone studied by small-angle X-ray scattering, Calcif. Tiss. Int 48 (1991) 407–413, 10.1007/BF02556454. [DOI] [PubMed] [Google Scholar]
- [73].Gupta HS, Seto J, Wagermaier W, Zaslansky P, Boesecke P, Fratzl P, Cooperative deformation of mineral and collagen in bone at the nanoscale, Proc. Nat. Acad. Sci. U.S.A, 103 (2006) 17741–17746. doi: 10.1073/pnas.0604237103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Nudelman F, Pieterse K, George A, Bomans PHH, Friedrich H, Brylka LJ, Hilbers PA, de With G, Sommerdijk NAJM, The role of collagen in bone apatite formation in the presence of hydroxyapatite nucleation inhibitors, Nat. Mater 9 (2010) 1004–1009, 10.1038/nmat2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Song Q, Jiao K, Tonggu L, Wang LG, Zhang SL, Yang YD, Zhang L, Bian JH, Hao DX, Wang CY, Ma YX, Arola DD, Breschi L, Chen JH, Tay FR, Niu LN, Contribution of biomimetic collagen-ligand interaction to intrafibrillar mineralization, Sci. Adv, 5 (2019) eaav9075. doi: 10.1126/sciadv.aav9075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Wang B, Yang Y, Liu L, Blair HC, Friedman PA, NHERF1 regulation of PTH-dependent bimodal Pi transport in osteoblasts, Bone 52 (2013) 268–277, 10.1016/j.bone.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Paschalis EP, DiCarlo E, Betts F, Sherman P, Mendelsohn R, Boskey AL, FTIR microspectroscopic analysis of human osteonal bone, Calcif. Tiss. Int 59 (1996) 480–487, 10.1007/bf00369214. [DOI] [PubMed] [Google Scholar]
- [78].Von Euw S, Wang Y, Laurent G, Drouet C, Babonneau F, Nassif N, Azaïs T, Bone mineral: new insights into its chemical composition, Sci. Rep 9 (2019) 8456, 10.1038/s41598-019-44620-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Eberhardt AW, Yeager-Jones A, Blair HC, Regional trabecular bone matrix degeneration and osteocyte death in femora of glucocorticoid treated rabbits, Endocrinology 142 (2001) 1333–1340, 10.1210/endo.142.3.8048. [DOI] [PubMed] [Google Scholar]
- [80].Shapiro IM, Landis WJ, Risbud MV, Matrix vesicles: are they anchored exosomes? Bone 79 (2015) 29–36, 10.1016/j.bone.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Weinger JM, Holtrop ME, An ultrastructural study of bone cells: the occurrence of microtubules, microfilaments and tight junctions, Calcif. Tissue Res 14 (1974) 15–29, 10.1007/bf02060280. [DOI] [PubMed] [Google Scholar]
- [82].Wongdee K, Pandaranandaka J, Teerapornpuntakit J, Tudpor K, Thongbunchoo J, Thongon N, Jantarajit W, Krishnamra N, Charoenphandhu N, Osteoblasts express claudins and tight junction-associated proteins, Histochem. Cell Biol 130 (2008) 79–90, 10.1007/s00418-008-0419-6. [DOI] [PubMed] [Google Scholar]
- [83].Arana-Chavez VE, Soares AM, Katchburian E, Junctions between early developing osteoblasts of rat calvaria as revealed by freeze-fracture and ultrathin section electron microscopy, Arch. Histol. Cytol 58 (1995) 285–292, 10.1679/aohc.58.285. [DOI] [PubMed] [Google Scholar]
- [84].Jorgensen NR, Geist ST, Civitelli R, Steinberg TH, ATP- and gap junction-dependent intercellular calcium signaling in osteoblastic cells, J. Cell Biol 139 (1997) 497–506, 10.1083/jcb.139.2.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Moorer MC, Stains JP, Connexin43 and the intercellular signaling network regulating skeletal remodeling, Curr Osteoporos Rep 15 (2017) 24–31, 10.1007/s11914-017-0345-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Granke M, Does MD, Nyman JS, The role of water compartments in the material properties of cortical bone, Calcif. Tissue Int 97 (2015) 292–307, 10.1007/s00223-015-9977-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Schlesinger PH, Blair HC, Beer Stolz D, Riazanski V, Ray EC, Tourkova IL, Nelson DJ, Cellular and extracellular matrix of bone, with principles of synthesis and dependency of mineral deposition on cell membrane transport, Am. J. Physiol. Cell Physiol 318 (1) (2020) C111–C124, 10.1152/ajpcell.00120.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Raman A, Appositional growth rate in rat bones using the tetracycline labelling method, Acta Orthop. Scand 40 (2) (1969) 193–197, 10.3109/17453676908989498. [DOI] [PubMed] [Google Scholar]
- [89].Frost HM, Tetracycline bone labeling in anatomy, Am. J. Phys. Anthropol 29 (2) (1968) 183–195, 10.1002/ajpa.1330290212. [DOI] [PubMed] [Google Scholar]
- [90].Nalbandian J, Hagopian M, Patters M, The microscopic distribution of tetracycline in human teeth, J. Biol. Buccale, 10(4) (1982) 271–279. doi not available. [PubMed] [Google Scholar]
- [91].Neuman WF, Neuman MW, The Chemical Dynamics of Bone Mineral, The University of Chicago Press, Chicago IL, USA, 1958. [Google Scholar]
- [92].Strates BS, Neuman WF, Levinskas GJ, The solubility of bone mineral. II. Precipitation of near-neutral solutions of calcium and phosphate, J. Phys. Chem 61 (1957) 279–282, 10.1021/j150549a005. [DOI] [Google Scholar]
- [93].Inskeep WP, Silvertooth JC, Kinetics of hydroxyapatite precipitation at pH 7.4 to 8.4, Geochimica Cosmochimica Acta 52 (1988) 1883–1893, 10.1016/0016-7037(88)90012-9. [DOI] [Google Scholar]
- [94].Chakkalakal DA, Mashoof AA, Novak J, Strates BS, McGuire MH, Mineralization and pH relationships in healing skeletal defects grafted with demineralized bone matrix, J. Biomed. Mater. Res 28 (1994) 1439–1443, 10.1002/jbm.820281209. [DOI] [PubMed] [Google Scholar]
- [95].Liu L, Alonso V, Guo L, Tourkova I, Henderson SE, Almarza AJ,Friedman PA, Blair HC, Na+/H+ exchanger regulatory factor 1 (NHERF1) directly regulates osteogenesis, J. Biol. Chem 287 (2012) 43312–43321, 10.1074/jbc.M112.422766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Alexander RT, Rievaj J, Dimke H, Paracellular calcium transport across renal and intestinal epithelia, Biochem. Cell Biol 92 (2014) 467–480, 10.1139/bcb-2014-0061. [DOI] [PubMed] [Google Scholar]
- [97].Lieben L, Benn BS, Ajibade D, Stockmans I, Moermans K, Hediger MA, Peng JB, Christakos S, Bouillon R, Carmeliet G, Trpv6 mediates intestinal calcium absorption during calcium restriction and contributes to bone homeostasis, Bone 47 (2010) 301–308, 10.1016/j.bone.2010.04.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Little R, Muimo R, Robson L, Harris K, Grabowski PS, The transient receptor potential ion channel TRPV6 is expressed at low levels in osteoblasts and has little role in osteoblast calcium uptake, PLoS One 6 (2011) e28166, , 10.1371/journal.pone.0028166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Fujita H, Sugimoto K, Inatomi S, Maeda T, Osanai M, Uchiyama Y,Yamamoto Y, Wada T, Kojima T, Yokozaki H, Yamashita T, Kato S, Sawada N, Chiba H, Tight junction proteins claudin-2 and −12 are critical for vitamin D-dependent Ca2+ absorption between enterocytes, Mol. Biol. Cell 19 (2008) 1912–1921, 10.1091/mbc.E07-09-0973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Short CL, Monk RD, Bushinsky DA, Krieger NS, Hormonal regulation of Na+-Ca2+ exchange in osteoblast-like cells, J. Bone Miner. Res 9 (1994) 1159–1166, 10.1002/jbmr.5650090805. [DOI] [PubMed] [Google Scholar]
- [101].Gay CV, Lloyd QP, Characterization of calcium efflux by osteoblasts derived from long bone periosteum, Comp. Biochem. Physiol. A Physiol 111 (1995) 257–261, 10.1016/0300-9629(95)00004-q. [DOI] [PubMed] [Google Scholar]
- [102].Lloyd QP, Kuhn MA, Gay CV, Characterization of calcium translocation across the plasma membrane of primary osteoblasts using a lipophilic calcium-sensitive fluorescent dye, calcium green c18, J. Biol. Chem 270 (1996) 22445–22451, 10.1074/jbc.270.38.22445. [DOI] [PubMed] [Google Scholar]
- [103].Balmain N, Berdal A, Hotton D, Cuisinier-Gleizes P, Mathieu H, Calbindin-D9K immunolocalization and vitamin D-dependence in the bone of growing and adult rats, Histochemistry 92 (1989) 359–365, 10.1007/BF00492492. [DOI] [PubMed] [Google Scholar]
- [104].Turnbull CI, Looi K, Mangum JE, Meyer M, Sayer RJ, Hubbard MJ, Calbindin independence of calcium transport in developing teeth contradicts the calcium ferry dogma, J. Biol. Chem 279 (2004) 55850–55854, 10.1074/jbc.M409299200. [DOI] [PubMed] [Google Scholar]
- [105].Stains JP, Weber JA, Gay CV, Expression of Na+/Ca2+ exchanger isoforms (NCX1 and NCX3) and plasma membrane Ca2+ ATPase during osteoblast differentiation, J. Cell. Biochem 84 (2002) 625–635, 10.1002/jcb.10050.abs. [DOI] [PubMed] [Google Scholar]
- [106].Robison R, The possible significance of hexosephosphoric esters in ossification, Biochem. J 17 (1923) 286–293, 10.1042/bj0170286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Skillen AW, Harrison J, Serum alkaline phosphatases. Effect of pH and buffer on optimum substrate concentration, Clin. Chim. Acta, 45 (1973) 287–291. doi: 10.1016/0009-8981(73)90440-3. [DOI] [PubMed] [Google Scholar]
- [108].Rathbun JC, Hypophosphatasia: a new developmental anomaly, Am. J. Dis. Child 75 (1948) 822–831, 10.1001/archpedi.1948.02030020840003. [DOI] [PubMed] [Google Scholar]
- [109].P Whyte M, Hypophosphatasia: Nature’s window on alkaline phosphatase function in humans, In : Bilezikian JP, Raisz LG, and Rodan GA, Eds., Principles of Bone Biology, 2ed, pp. 1569–1599. Academic Press, New York, 2002. doi: 10.1016/B978-012098652-1.50172-4. [DOI] [Google Scholar]
- [110].Fedde KN, Lane CC, Whyte MP, Alkaline phosphatase is an ectoenzyme that acts on micromolar concentrations of natural substrates at physiologic pH in human osteosarcoma (SAOS-2) cells, Arch. Biochem. Biophys 264 (1988) 400–409, 10.1016/0003-9861(88)90305-0. [DOI] [PubMed] [Google Scholar]
- [111].Whyte MP, Mahuren JD, Vrabel LA, Coburn SP, Markedly increased circulating pyridoxal-5′-phosphate levels in hypophosphatasia. Alkaline phosphatase acts in vitamin B6 metabolism, J. Clin. Invest 76 (1985) 752–756, 10.1172/JCI112031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Whyte MP, Landt M, Ryan LM, Mulivor RA, Henthorn PS, Fedde KN, Mahuren JD, Coburn SP, Alkaline phosphatase: placental and tissue-nonspecific isoenzymes hydrolyze phosphoethanolamine, inorganic pyrophosphate, and pyridoxal 5′ phosphate. Substrate accumulation in carriers of hypophosphatasia corrects corrects during pregnancy, J. Clin. Invest, 95 (1995) 1440–1445. doi: 10.1172/JCI117814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Hernando N, Gagnon KB, Lederer ED, Phosphate transport in epithelial and nonepithelial tissue, Physiol. Rev (2020. April 30), 10.1152/physrev.00008.2019 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- [114].Forster IC, The molecular mechanism of slc34 proteins: insights from two decades of transport assays and structure-function studies, Pflugers Arch. 471 (2019) 15–42, 10.1007/s00424-018-2207-z. [DOI] [PubMed] [Google Scholar]
- [115].Beck L, Expression and function of slc34 sodium-phosphate co-transporters in skeleton and teeth, Pflügers Archiv, 471 (2019)175–184. doi: 10.1007/s00424-018-2240-y. [DOI] [PubMed] [Google Scholar]
- [116].Shibasaki Y, Etoh N, Hayasaka M, Takahashi MO, Kakitani M, Yamashita T,Tomizuka K, Hanaoka K, Targeted deletion of the tybe IIb Na+-dependent Pi-co-transporter, NaPi-IIb, results in early embryonic lethality, Biochem. Biophys. Res. Commun 381 (2009) 482–486, 10.1016/j.bbrc.2009.02.067. [DOI] [PubMed] [Google Scholar]
- [117].Beck L, Karaplis AC, Amizuka N, Hewson AS, Ozawa H, Tenenhouse HS, Targeted inactivation of Npt2 in mice leads to severe renal phosphate wasting, hypercalciuria, and skeletal abnormalities, Proc. Natl. Acad. Sci. U. S. A, 95 (1998) 5372–5377. doi: 10.1073/pnas.95.9.5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Segawa H, Onitsuka A, Kuwahata M, Hanabusa E E, Furutani J, Kaneko I, Tomoe Y, Aranami F, Matsumoto N, Ito M, Matsumoto M. Li N,. Amizuka N, Miyamoto K, Type IIc sodium-dependent phosphate transporter regulates calcium metabolism, J. Am. Soc. Nephrol, 20 (2009) 104–113. doi: 10.1681/ASN.2008020177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Kemp GJ, Khouja HI, Ahmado A, Graham R, Russell G, Bevington A, Regulation of the phosphate (Pi) concentration in UMR 106 osteoblast-like cells: effect of Pi, Na+ and K+, Cell Biochem. Funct 11 (1993) 13–23, 10.1002/cbf.290110103. [DOI] [PubMed] [Google Scholar]
- [120].Kemp GJ, Bevington A, Khodja D, Challa A, Russell RG, 32P-labelling anomalies in human erythrocytes. Is there more than one pool of cellular Pi? Biochem. J, 264 (1989) 729–736. doi: 10.1042/bj2640729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Moochhala SH, Extracellular pyrophosphate in the kidney: how does it get there and what does it do? Nephron. Physiol 120 (2012) 33–38, 10.1159/000341597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Giovannini D, Touhami J, Charnet P, Sitbon M, Battini JL, Inorganic phosphate export by the retrovirus receptor XPR1 in metazoans, Cell Rep. 3 (2013) 1866–1873, 10.1016/j.celrep.2013.05.035. [DOI] [PubMed] [Google Scholar]
- [123].Legati A, Giovannini D, Nicolas G, López-Sánchez U, Quintáns B, Oliveira JR, Sears RL, Ramos EM, Spiteri E, Sobrido MJ, Carracedo A, Castro-Fernández C, Cubizolle S, Fogel BL, Goizet C, Jen JC, Kirdlarp S,Lang AE, Miedzybrodzka Z, Mitarnun, Coppola G, Mutations in XPR1 cause primary familial brain calcification associated with altered phosphate export, Nat. Genet, 47 (2015) 579–581. doi: 10.1038/ng.3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].López-Sánchez U, Tury S, Nicolas G, Wilson MS, Jurici S, Ayrignac X, Courgnaud V, Saiardi A, Sitbon M, Battini JL, Interplay between PFBC-associated SLC20A2 and XPR1 phosphate transporters requires inositol polyphosphates for control of cellular phosphate homeostasis, J. Biol. Chem, 2020. May 11 pii: jbc. RA119.011376. doi: 10.1074/jbc.RA119.011376. (Epub ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Olah Z, Lehel C, Anderson WB, Eiden MV, Wilson CA, The cellular receptor for gibbon ape leukemia virus is a novel high affinity sodium-dependent phosphate transporter, J. Biol. Chem, 269 (1994) 25426–25431. doi not available. [PubMed] [Google Scholar]
- [126].Balani S, Perwad F, Fibroblast growth factor 23 and phosphate homeostasis, Curr. Opin. Nephrol. Hypertens (2019) 465–473, 10.1097/MNH.0000000000000526. [DOI] [PubMed] [Google Scholar]
- [127].Meireles AM, Shiau CE, Guenther CA, Sidik H, Kingsley DM, Talbot WS WS, The phosphate exporter xpr1b is required for differentiation of tissue-resident macrophages, Cell Rep., 8 (2014) 1659–1667. doi: 10.1016/j.celrep.2014.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Millan JL, The role of phosphatases in the initiation of skeletal mineralization, Calcif. Tissue Int 93 (2013) 299–306, 10.1007/s00223-012-9672-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Narisawa S, Wennberg C, Millán JL, Abnormal vitamin B6 metabolism in alkaline phosphatase knock-out mice causes multiple abnormalities, but not the impaired bone mineralization, J. Pathol 2001 (193) (2001) 125–133, . [DOI] [PubMed] [Google Scholar]
- [130].Anderson HC, Matrix vesicles and calcification, Curr. Rheumatol. Rep 5 (2003) 222–226, 10.1007/s11926-003-0071-z. [DOI] [PubMed] [Google Scholar]
- [131].Orimo H, The mechanism of mineralization and the role of alkaline phosphatase in health and disease, J. Nippon Med. Sch 77 (2010) 4–12, 10.1272/jnms.77.4. [DOI] [PubMed] [Google Scholar]
- [132].Yadav MC, Bottini M, Cory E, Bhattacharya K, Kuss P, Narisawa S, Sah RL,Beck L, Fadeel B, Farquharson C, Millán JL, Skeletal mineralization deficits and impaired biogenesis and function of chondrocyte-derived matrix vesicles in phospho1−/− and phospho1/pi t1 double-knockout mice, J. Bone Miner. Res 31 (2016) 1275–1286, 10.1002/jbmr.2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Maroudas A, Distribution and diffusion of solutes in articular cartilage, Biophys.J 10 (1970) 365–379, 10.1016/S0006-3495(70)86307-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Anderson HC, Normal and abnormal mineralization in mammals, Trans. Am. Soc. Artif. Intern. Organs, 27 (1981) 702–708. doi not available. [PubMed] [Google Scholar]
- [135].Peacock M, Phosphate metabolism in health and disease, Calcif. Tissue Int (2020. April 7), 10.1007/s00223-020-00686-3 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- [136].Landis WJ, Pains MC, Hodgens KJ, Glimcher MJ, Matrix vesicles in embryonic chick bone: considerations of their identification, number, distribution, and possible effects on calcification of extracellular matrices, J. Ultrastruct. Mol. Struct. Res 95 (1986) 142–163, 10.1016/0889-1605(86)90037-6. [DOI] [PubMed] [Google Scholar]
- [137].Hasegawa T, Ultrastructure and biological function of matrix vesicles in bone mineralization, Histochem. Cell Biol 149 (2018) 289–304, 10.1007/s00418-018-1646-0. [DOI] [PubMed] [Google Scholar]
- [138].Li H, Li D, Ma Z, Qian Z, Kang X, Jin X, Li F, Wang X, Chen Q, Sun H, Wu S, Defective autophagy in osteoblasts induces endoplasmic reticulum stress and causes remarkable bone loss, Autophagy 14 (2018) 1726–1741, 10.1080/15548627.2018.1483807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Nollet M, Santucci-Darmanin S, Breuil V, Al-Sahlanee R, Cros C, Topi M, Momier D, Samson M, Pagnotta S, Cailleteau L, Battaglia S, Farlay D, Dacquin R,Barois N, Jurdic P, Boivin G, Heymann D, Lafont F, Lu SS, Dempster DW, Carle GF, Pierrefite-Carle V, Autophagy in osteoblasts is involved in mineralization and bone homeostasis, Autophagy, 10 (2014) 1965–1677. doi: 10.4161/auto.36182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Larrouture QC, Nelson DJ, Robinson LJ, Liu L, Tourkova I, Schlesinger PH, Blair HC. Chloride-hydrogen antiporters ClC-3 and ClC-5 drive osteoblast mineralization and regulate fine-structure bone patterning in vitro. Physiol Rep. 3 (2015) pii: e12607. doi: 10.14814/phy2.12607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Wuthier RE, Lipscomb GF, Matrix vesicles: structure, composition, formation and function in calcification, Front. Biosci 16 (2011) 2812–2902, 10.2741/3887. [DOI] [PubMed] [Google Scholar]
- [142].Cappariello A, Loftus A, Muraca M, Maurizi A, Rucci N, Teti A, Osteoblast-derived extracellular vesicles are biological tools for the delivery of active molecules to bone, J. Bone Miner. Res 33 (2018) 517–533, 10.1002/jbmr.3332. [DOI] [PubMed] [Google Scholar]
- [143].Schmidt JR, Kliemt S, Preissler C, Moeller S, von Bergen M, Hempel U,Kalkhof S, Osteoblast-released matrix vesicles, regulation of activity and composition by sulfated and non-sulfated glycosaminoglycans, Mol. Cell. Proteomics 15 (2016) 558–572, 10.1074/mcp.M115.049718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Liu L, Schlesinger PH, Slack NM, Friedman PA, Blair HC, High capacity Na+/H+ exchange activity in mineralizing osteoblasts, J. Cell. Physiol 226 (2011) 1702–1712, 10.1002/jcp.22501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Bell SM, Schreiner CM, Schultheis PJ, Miller ML, Evans RL, Vorhees CV, Shull GE, Scott WJ, Targeted disruption of the murine Nhe1 locus induces ataxia, growth retardation, and seizures, Am. J. Phys 276 (1999) C788–C795, 10.1152/ajpcell.1999.276.4.C788. [DOI] [PubMed] [Google Scholar]
- [146].Guzman RE, Miranda-Laferte E, Franzen A, Fahlke C, Neuronal ClC-3 splice variants differ in subcellular localizations, but mediate identical transport functions, J. Biol. Chem 290 (2015) 25851–25862, 10.1074/jbc.M115.668186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Gould SJ, Ontogeny and Phylogeny, Belknap Press, Cambridge, MA, 1977. [Google Scholar]