

Abstract
Parathyroid hormone-related protein (PTHrP) is a critical regulator of bone resorption and augments osteolysis in skeletal malignancies. Here we report that the mature PTHrP1–36 hormone is processed by matrix metalloproteinases to yield a stable product, PTHrP1–17. PTHrP1–17 retains the ability to signal through PTH1R to induce calcium flux and ERK phosphorylation but not cyclic AMP production or CREB phosphorylation. Notably, PTHrP1–17 promotes osteoblast migration and mineralization in vitro, and systemic administration of PTHrP1–17 augments ectopic bone formation in vivo. Further, in contrast to PTHrP1–36, PTHrP1–17 does not affect osteoclast formation/function in vitro or in vivo. Finally, immunoprecipitation-mass spectrometry analyses using PTHrP1–17-specific antibodies establish that PTHrP1–17 is indeed generated by cancer cells. Thus, matrix metalloproteinase-directed processing of PTHrP disables the osteolytic functions of the mature hormone to promote osteogenesis, indicating important roles for this circuit in bone remodelling in normal and disease contexts.
INTRODUCTION
Parathyroid hormone-related protein (PTHrP), a member of the parathyroid hormone (PTH) family, has noted roles in skeletal development and cancer-induced bone destruction.1–3 PTHrP has three isoforms comprised of 139, 141 or 173 amino acids.4,5 In the Golgi, these preprohormones are processed by protein convertases (PCs) including PC1, 2, 4, 5 and furin.6 Further N-terminal processing in secretory vesicles yields a mature 36 amino acid form (PTHrP1–36) that, following release, can bind to the PTH1 receptor (PTH1R) and induce cyclic adenosine monophosphate (cAMP) production, calcium flux and activation of protein kinase A (PKA) and protein kinase C (PKC).6–8
PTH1R activation by PTH or PTHrP induces the expression of genes that direct bone remodelling, including receptor activator of nuclear kappa B ligand (RANKL), a critical mediator of osteoclast formation and bone resorption.9–11 Continuous administration of PTHrP1–36 has been shown to induce systemic osteolysis, yet intermittent application of the hormone promotes bone formation.1,12,13 The reason for these differential effects has been potentially ascribed to the labile nature of mature PTHrP.14
Multiple enzymatic cleavage sites exist within PTHrP1–36, suggesting that post-translational cleavage may control the powerful bone modelling activity of this hormone. To date, the extracellular proteases kallikrein-3/prostate serum antigen and the metalloproteinase, neprilysin, have been implicated in processing PTHrP, but the biological activity of any generated peptides remains undetermined.15–18 Further, it is also unknown if other proteases commonly found in skeletal tissues can process PTHrP1–36.
Matrix metalloproteinases (MMPs) are a family of 23 enzymes that collectively control processing and turnover of the extracellular matrix.19 Bone is collagen rich, and MMPs with collagenase activity, including MMP-1, −2, −3, −8, −13, −14 and −15, have reported effects on skeletal development and homeostasis.20,21 Further, MMPs function as key mediators of cell–cell communication given their ability to control the bioactivity and/or bioavailability of a wide array of growth factors and cytokines.22,23 This is especially true in the context of skeletal malignancies where there is heightened MMP expression at the tumour–bone interface.24–26
In the bone microenvironment, cancers provoke aberrant bone remodelling where lesions can be composed of extensive areas of bone resorption and/or bone formation.27 Primary and metastatic bone cancers have been shown to express PTHrP, which in turn induces RANKL expression in bone lining osteoblasts to trigger osteoclastogenesis.28 Osteoclasts then resorb the mineralized bone matrix, releasing bone sequestered growth factors such as transforming growth factor β that promote cancer cell survival.29 MMPs are key regulators of RANKL and transforming growth factor β bioavailability.24,30 Given the enzymatic susceptibility of PTHrP1–36, we hypothesized that PTHrP was a substrate of MMPs that are expressed in bone under normal and pathological conditions. Here we report that MMPs are capable of rapidly processing PTHrP1–36 to yield a unique PTHrP1–17 peptide. Moreover, PTHrP1–17 is stable and retains the ability to stimulate intracellular calcium flux via PTH1R but does not trigger cAMP production. Finally, PTHrP1–17 has robust biological activity, where it selectively directs osteoblast differentiation and osteogenesis without affecting osteoclastogenesis/bone resorption. Collectively these data suggest that MMPs are important regulators of PTHrP activity in the normal and pathological bone microenvironment.
RESULTS
PTHrP is an MMP substrate
To test if MMPs process PTHrP, we incubated recombinant PTHrP1–86 with MMP-3 and assessed immediate (1 h) cleavage products (Figures 1a and b). N-terminal amino acid sequencing identified that MMP-3 cleaved recombinant PTHrP to generate the mature form of the protein, PTHrP1–36 (Figure 1c). However, MALDI-TOF analysis demonstrated that PTHrP1–36 was further cleaved to distinct stable peptide products, including PTHrP1–17, 18–26 and 27–36 (Figure 1d). Kinetic analyses revealed that MMP-3 generated these main PTHrP products within 1 h (Supplementary Figures S1 and S2). We also examined the PTHrP processing activity of other MMPs present in the bone metastatic prostate cancer microenvironment and found that MMP-2, −7, −9 and −13 could generate PTHrP fragments and that all, with the exception of MMP-13, consistently generated PTHrP1–17 (Supplementary Table S1). Thus, PTHrP is an MMP substrate.
Figure 1.
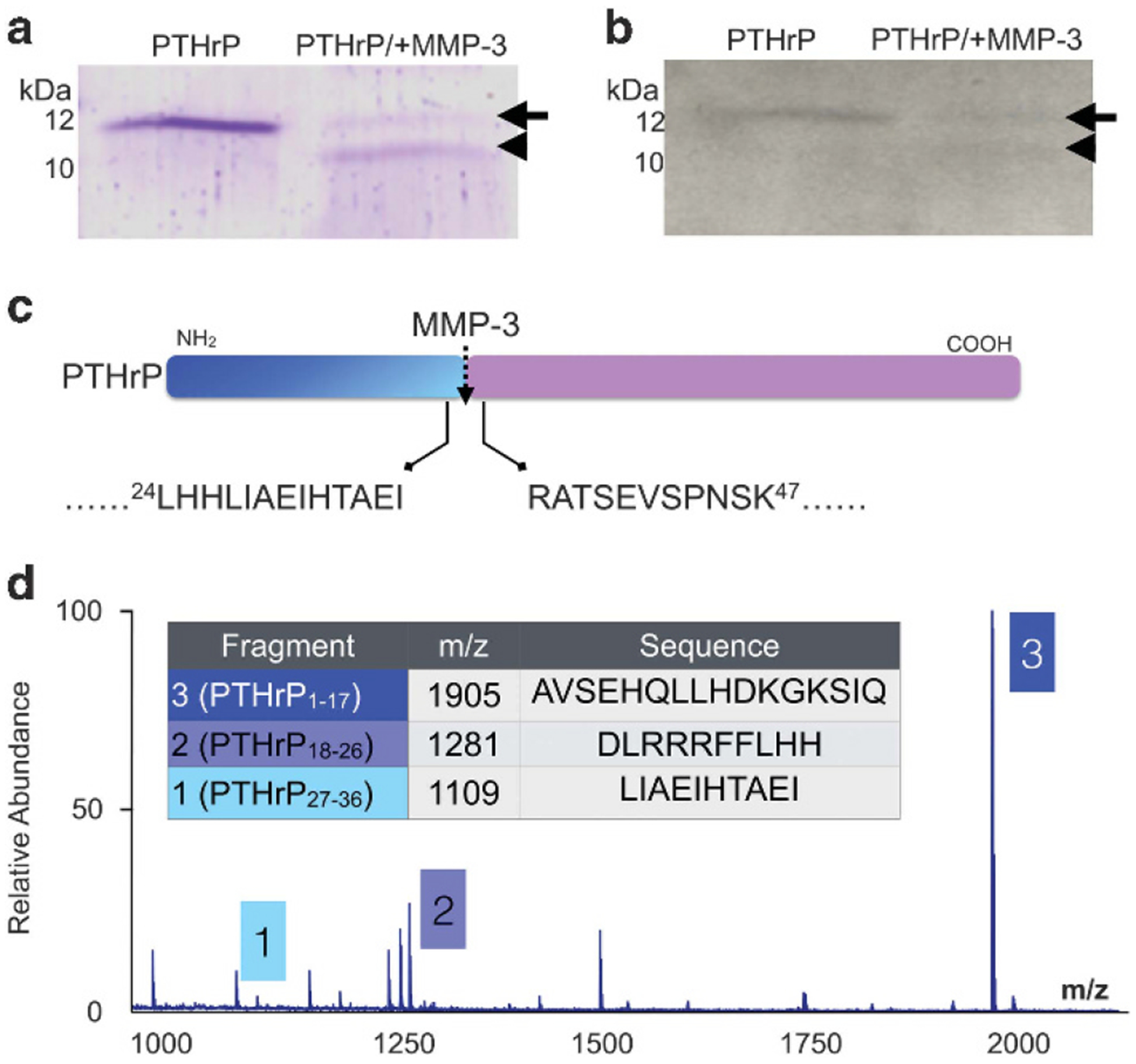
PTHrP is processed by MMPs. (a, b) Recombinant PTHrP1–86 (100 ng; arrow) was incubated for 1 h with active MMP-3 (100 ng) and products analysed by SDS-PAGE with Coomassie blue staining (a) and immunoblot analysis (b). Arrowhead indicates cleavage product. Molecular weight markers indicated in kilodaltons (kDa). (c) N-terminal amino acid sequencing revealed that MMP-3 cleaved (dashed arrow) PTHrP1–86 between amino acids 36 and 37. Arrows illustrate the amino acid sequence on either side of MMP-3 cleavage site. Amino acid position is indicated by numerical superscript. (d) MALDI TOF/MS analyses established that further incubation (1 h) of PTHrP with MMP-3 yields novel, stable PTHrP fragments, PTHrP1–17, PTHrP18–26 and PTHrP27–36.
MMP generated PTHrP1–17 has biological activity
To test if the MMP generated fragments of PTHrP retained biological activity, the major MMP generated PTHrP peptides, PTHrP1–17, 18–26 and 27–36 were synthesized and assessed for their biological effects on primary mouse mesenchymal stem cells (MSCs) and osteoblasts, which express and respond to signalling from the PTHrP receptor, PTH1R (Figure 2a).17 Low concentrations (10 nm) of PTHrP1–36 are sufficient to activate PTH1R and promote ERK phosphorylation in these cell types.31 Notably, treatment of MSCs and osteoblasts with 10 nm of PTHrP1–36 or PTHrP1–17 induced ERK phosphorylation within 5 min compared to control or scrambled peptide treated cells (Figure 2b). Increases in response to PTHrP18–26 and PTHrP27–36 were noted, but these increases were very subtle and variable in repeated experiments (data not shown). We next looked at CREB phosphorylation since it is another downstream target of PTH1R signalling.9 In contrast to our ERK analyses, we observed that phosphorylation of CREB was only induced by PTHrP1–36 (Figure 2c). PTH1R G-protein coupled receptor (GPCR) activation also induces rapid cAMP and calcium flux responses primarily via Gs and Gq signalling, respectively.6 Again, only the addition of PTHrP1–36 peptide induced cAMP (Figure 2d), whereas both PTHrP1–36 and PTHrP1–17 triggered increases in calcium flux (Figure 2e). No effects of the PTHrP18–26 or PTHrP27–36 MMP-generated peptides on signalling were noted. These differential effects for PTHrP1–17 on calcium flux versus cAMP production were recapitulated in HEK cells engineered to express the PTH1R receptor (Supplementary Figure S3). To determine if PTHrP1–17 effects were mediated via PTH1R, we generated multiple MC3T3 osteoblast shControl and shPTH1R clones (Figure 2f). The ability of PTHrP1–17 and PTHrP1–36 to induce ERK phosphorylation was abrogated in PTH1R knockdown cells versus control shRNA (Figure 2g). Further, calcium flux in response to PTHrP1–17 and PTHrP1–36 was significantly reduced in PTH1R knockdown cells (Figure 2h). These effects on ERK phosphorylation and calcium flux were validated with a separate shPTH1R clone (Supplementary Figure S4). Thus, our data indicate that PTHrP1–17 has biological activity and activates select arms of PTH1R-directed signalling circuits.
Figure 2.
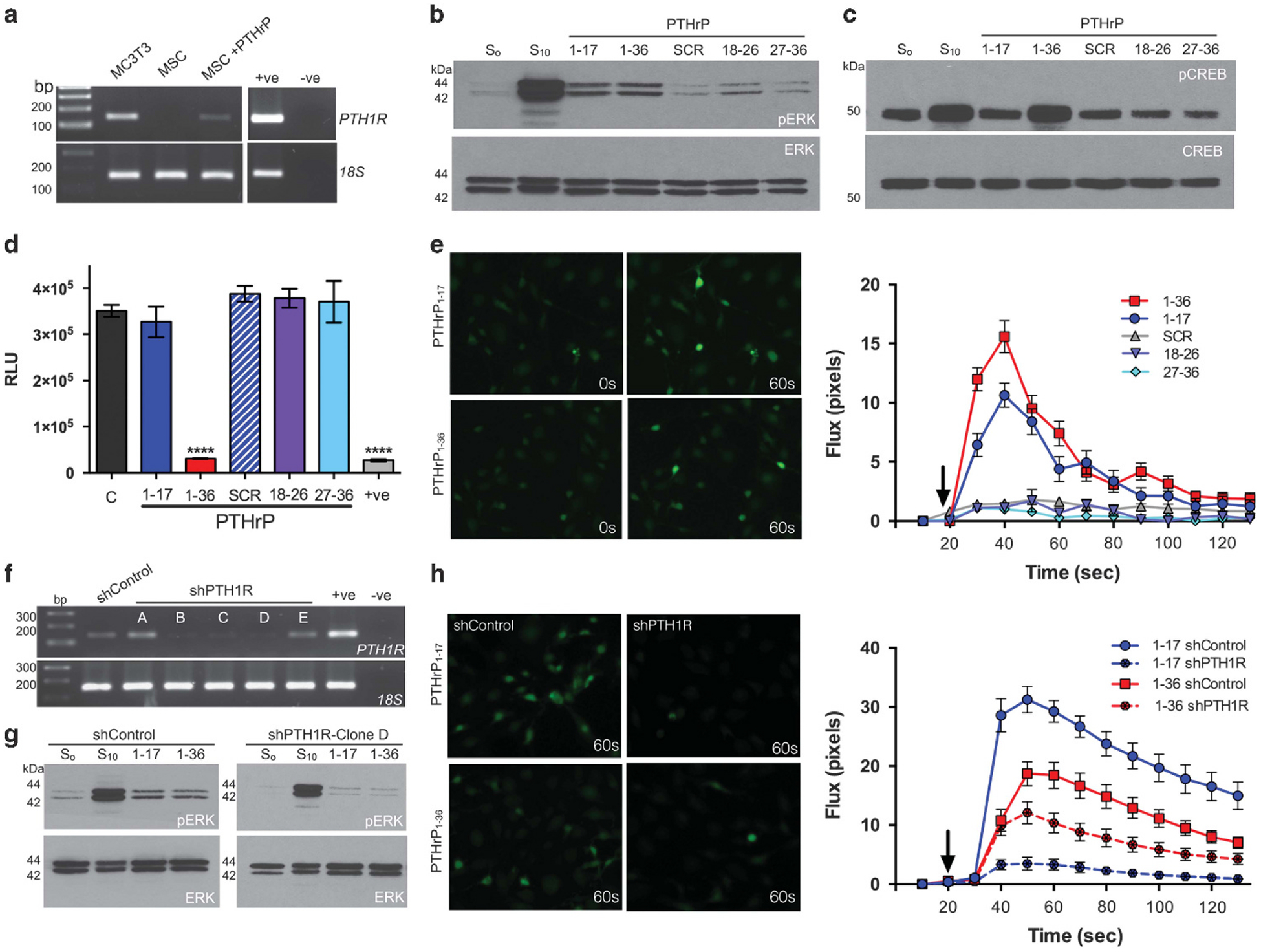
PTHrP1–17 has PTH1R-dependent signalling activity. (a) PTH1R expression in MC3T3 osteoblasts and primary MSCs treated in the absence or presence of PTHrP1–36 for 24 h. +ve indicates positive control (primary mouse osteoblasts) while –ve indicates negative non-template control. Molecular weight markers are illustrated in base pairs (bp). (b, c) ERK phosphorylation (pERK) and CREB phosphorylation (pCREB) in MC3T3 osteoblasts following treatment with PTHrP peptides (10 nm for 5 min in serum free media). S0 and S10 represent the addition of serum free and 10% serum, respectively. SCR is scrambled peptide control. (d) cAMP production in MC3T3 osteoblasts treated with PTHrP peptides (10 nm for 15 min). Asterisks denote statistical significance (*P < 0.05; ***P < 0.001). Forskolin (10 μm for 15 min) was used as a positive control (+ve). (e) Calcium flux analysis in MC3T3 osteoblasts after treatment with PTHrP peptides (10 nm). Left, representative images illustrate fluorescence activity prior to (0 s) and following treatment with PTHrP peptides (60 s). Graphs show increase in fluorescence measured in individual cells (n = 20/group) over time. Arrow on graph indicates the time point at which the PTHrP peptides were added. (f) Generation of PTH1R knockdown (shPTH1R) MC3T3 clones (a–e) via shRNA transduction. Scrambled control clones (shControl) were also selected for analysis. +ve indicates positive control (primary mouse osteoblasts) while –ve indicates negative non-template control. (g) ERK phosphorylation in shControl and shPTH1R cells (MC3T3 clone D) in response to PTHrP peptides (10 nm for 5 min). (h) Calcium flux assays were performed in shControl and shPTH1R clones after treatment with PTHrP1–36 and PTHrP1–17 (10 nm). Left, representative images illustrate fluorescence activity following addition of PTHrP peptides (60 s). Graphs show increase in fluorescence (RFU) measured in individual cells (n = 20/group) over time.
PTHrP1–17 promotes MSC/osteoblast cell migration
The biological effects of the MMP generated PTHrP fragments and PTHrP1–36 were assessed in primary MSCs, MC3T3 osteoblasts, an osteoclast precursor cell line (RAW 264.7), and cancer cell lines (PAIII, C4–2B, PC3–2M, SAOS-2). There were no overt effects of these four PTHrP peptides on cell growth (Supplementary Figures S5A–G), and treatment of osteoblasts with PTHrP1–17 or PTHrP1–36 did not prevent tumour necrosis factor-α-induced cell death (Supplementary Figure S5H).32 However, in assessing the effects of the PTHrP peptides on MSC and osteoblast proliferation, treated cells acquired a migratory phenotype, characterized by a more elongated shape (Figure 3a). PTHrP has been shown to contribute to the recruitment of osteoblasts in vivo.1 In keeping with this observation, both PTHrP1–36 and PTHrP1–17 significantly increased migration of MSCs and MC3T3 cells (Figure 3b). These effects of PTHrP1–17 are PTH1R-dependent, as knockdown of PTH1R abolished PTHrP1–17-induced osteoblast migration (Figure 3c).
Figure 3.
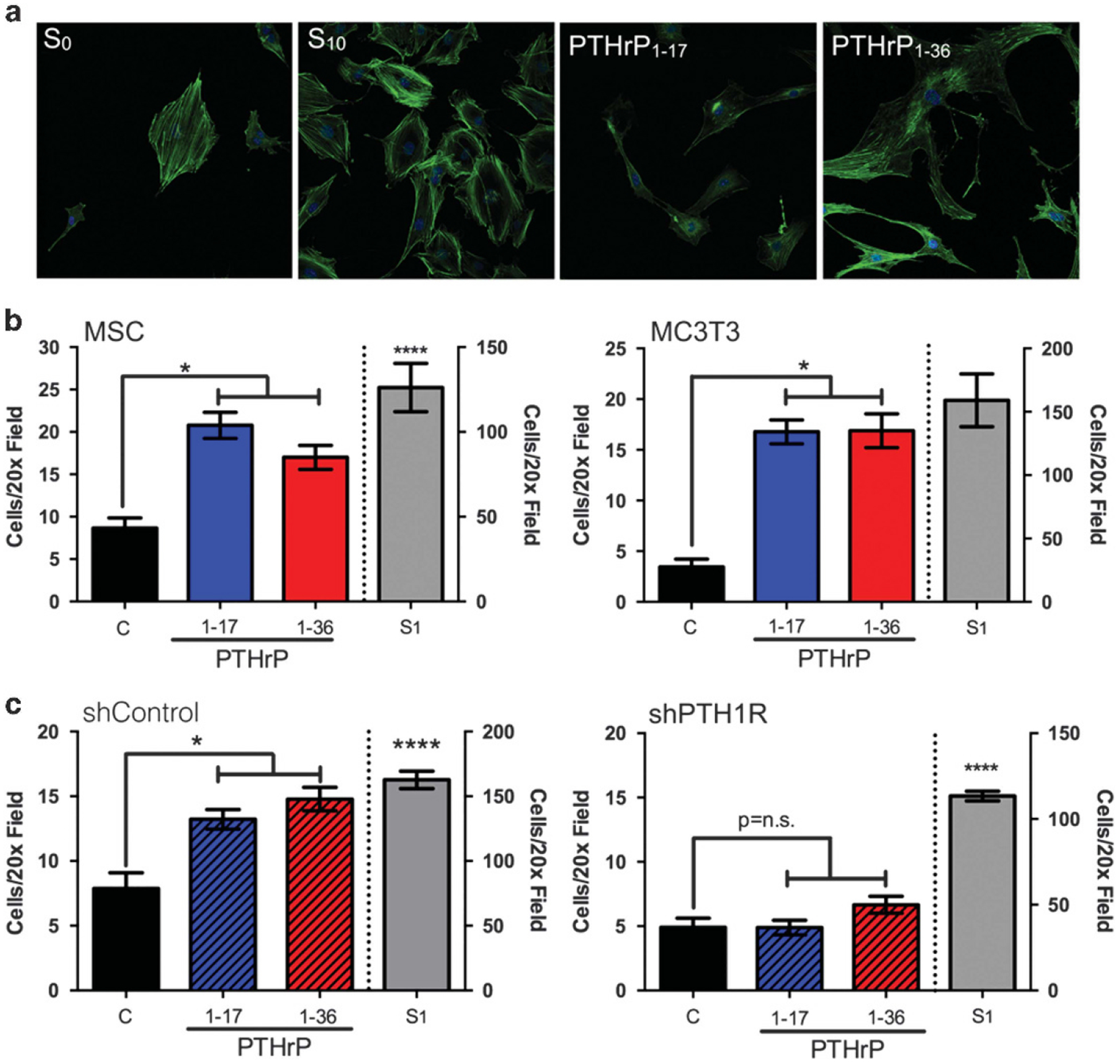
PTHrP1–17 promotes MSC and osteoblast migration via PTH1R. (a) Morphology of osteoblasts (MC3T3 cells) following treatment of PTHrP1–17 or PTHrP1–36 (10 nm for 1 hr) was determined by staining with anti-actin antibody and confocal fluorescence microscopy. (b) Migration of primary MSCs (left) and osteoblasts (MC3T3, right) treated with PTHrP1–17 versus PTHrP1–36 (10 nm for 6 h). (c) The migration of shControl (left) and PTH1R knockdown (right, shPTH1R) MC3T3 osteoblasts following treatment with PTHrP1–17 versus PTHrP1–36 (10 nm for 5 h). Cell number per × 20 field in five micrographs per condition were counted. Positive control for (b) and (c) was media containing 1% serum (S1). Asterisk denotes statistical significance (P < 0.05); n.s., non-significant differences.
PTHrP1–17 promotes MSC/osteoblast differentiation
PTHrP1–36 is a potent regulator of osteoclastogenesis and bone resorption, but intermittent treatment of osteoblasts can promote osteoblast differentiation and bone formation.33 To assess effects of PTHrP1–17 on osteoblast differentiation and mineralization, MSCs were treated for 16 days with PTHrP1–17 or PTHrP1–36 in the presence or absence of osteogenic media. Surprisingly, treatment with PTHrP1–17 alone was sufficient to promote osteoblast differentiation of MSCs and significantly enhanced the effects of the osteogenic media as determined by Alizarin red staining and colorimetric analysis (Figures 4a and b). Consistent with the ability of PTHrP1–17 to promote mineralization, we also observed that PTHrP1–17 could induce the expression of Type I Collagen, a major component of the bone extracellular matrix (Figure 4c). To test if these effects of PTHrP1 – 17 were also manifest in vivo, we used a murine model of ectopic bone formation.34 Primary MSCs were loaded onto Gelfoam scaffolds and implanted subcutaneously. Mice were treated daily with vehicle control, PTHrP1 – 17 or PTHrP1 – 36 (40 μg/kg/day; intermittent treatment regimen for 21 days35). High-resolution μCT scans of isolated ossicles revealed bone formation in the PTHrP1 – 17- and PTHrP1 – 36-treated animals (Figure 4d). Analysis of trichrome stained ossicle sections, which allows for the detection of collagen and bone (blue/green colour), supported μCT scans and demonstrated a significant amount of osteoid in both PTHrP1 – 17- and PTHrP1 – 36-treated cohorts (Figure 4e). Underscoring this observation, trabecular bone volume measurements of hind limbs revealed significantly more bone in the PTHrP1 – 17- and PTHrP1 – 36-treated mice compared to control (Supplementary Figures S6A and B).
Figure 4.
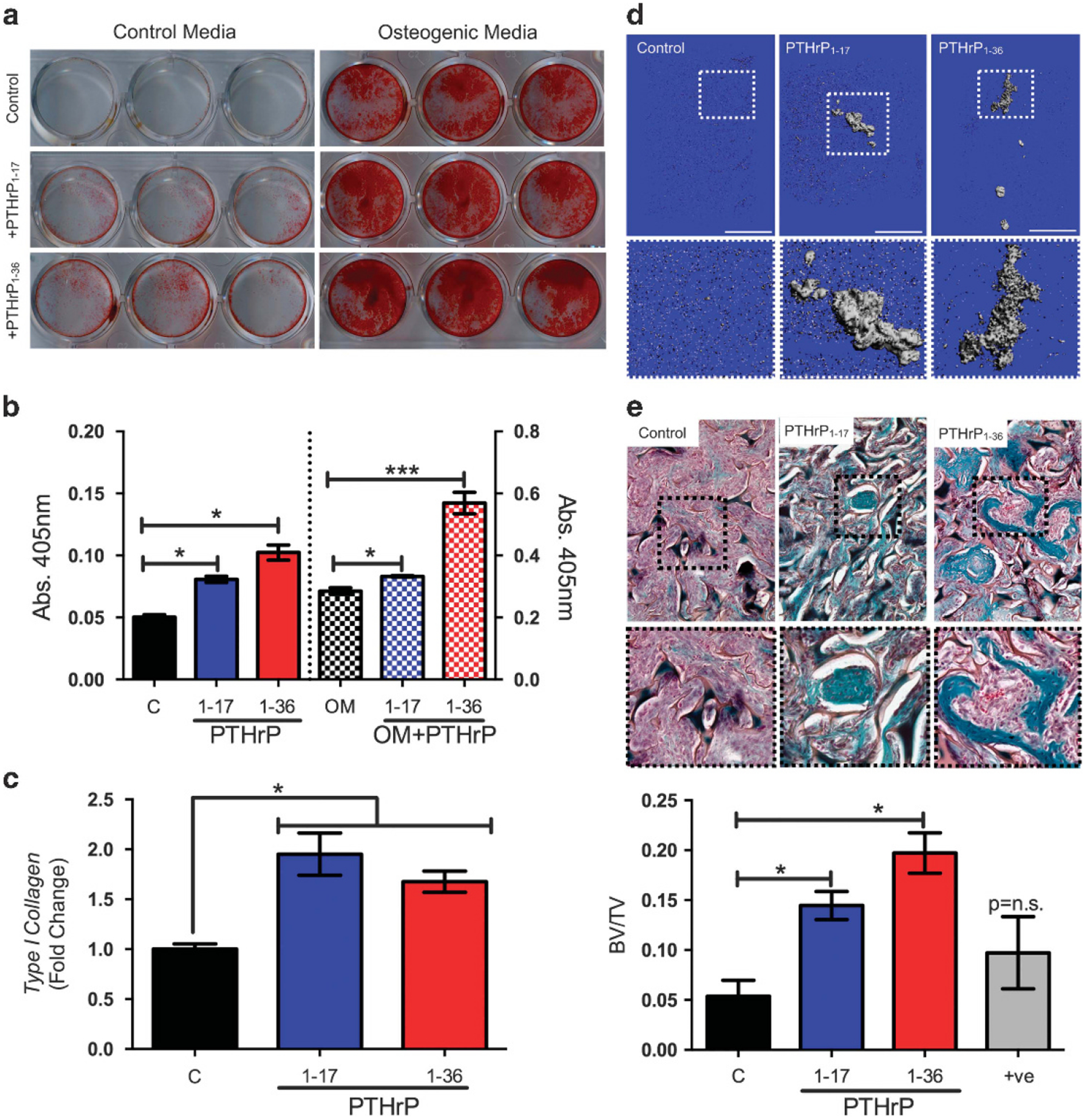
PTHrP1–17 promotes MSC and osteoblast differentiation. (a) Alizarin red staining of primary MSCs (n = 3) treated with PTHrP1–17 versus PTHrP1–36 (10 nm every other day for 16 days) in either normal media or in osteogenic media. (b) Quantitation of Alizarin red intensity in control and osteogenic media treated cells treated with the indicated PTHrP peptides. (c) Analysis of Type I Collagen expression in MC3T3 osteoblasts treated with PTHrP1–17 or PTHrP1–36 (10 nm for 48 h). RT qPCR was used to quantitate the relative fold change in expression. (d) Representative μCT scans of ectopic ossicles in control, PTHrP1–17 or PTHrP1–36 treated mice (n = 3/group). Scale bars are 1 mm. Dashed box represents area of magnification. (e) Trichrome-stained sections derived from control, PTHrP1–17 or PTHrP1–36 treated mice were quantitated for the amount of bone matrix (blue-green colour). Dashed box represents area of magnification. Asterisks denote significance (*P < 0.05; ***P < 0.001); n.s., non-significance.
PTHrP1–17 does not affect osteoclastogenesis/bone resorption
PTHrP1–36 promotes bone resorption by inducing the expression of factors such as RANKL.9,10 Treatment of whole bone marrow co-cultures with PTHrP1–36 revealed increased RANKL expression as expected, but this response was not observed following treatment with PTHrP1–17 (Figure 5a). We also noted that PTHrP1–36 appeared to suppress the expression of osteoprotegerin (OPG), which inhibits osteoclastogenesis (Figure 5a). Real-time PCR analyses confirmed these observations and demonstrated PTHrP1–36 significantly enhanced RANKL expression while suppressing OPG (Figure 5b and c). PTHrP1–17 had no effect on the expression of either of these genes. Taken together, the ratio of average RANKL:OPG transcripts was lower in PTHrP1–17 versus PTHrP1–36 treated cells (1.61 versus 15.03, respectively). These findings suggest that PTHrP1–17 does not contribute to osteoclastogenesis. To test this, we performed in vitro osteoclast formation assays using whole bone marrow co-cultures. As expected, PTHrP1–36 induced robust osteoclast formation, but PTHrP1–17 had no effect on osteoclast-togenesis (Figures 5d and e). To determine if this differential effect of PTHrP1–17 was also manifest in vivo, a calvarial injection assay was performed. In this model, repeated injections of PTHrP1–36 (every 6 h; continuous treatment regimen) over the calvaria promotes extensive osteolysis.36 Mice continuously treated with PTHrP1–36 displayed areas of extensive bone resorption while those injected with PTHrP1–17 did not (Figure 5f). TRAcP staining confirmed that there were significant increases in bone-lining osteoclasts in the PTHrP1–36-treated mice compared to PTHrP1–17 and control groups (Figure 5g). Finally, the differential effects of PTHrP1–17 and PTHrP1–36 on osteoclast activity were further supported by neonatal calvaria ex vivo assays. We found that calvaria treated with PTHrP1–36 displayed significant degradation of the calvaria, and that there was no evidence of bone formation (Supplementary Figure S6C). In contrast, PTHrP1–17 treatment significantly increased bone formation (Supplementary Figure S6D). Thus, PTHrP1–17 selectively promotes osteogenesis.
Figure 5.
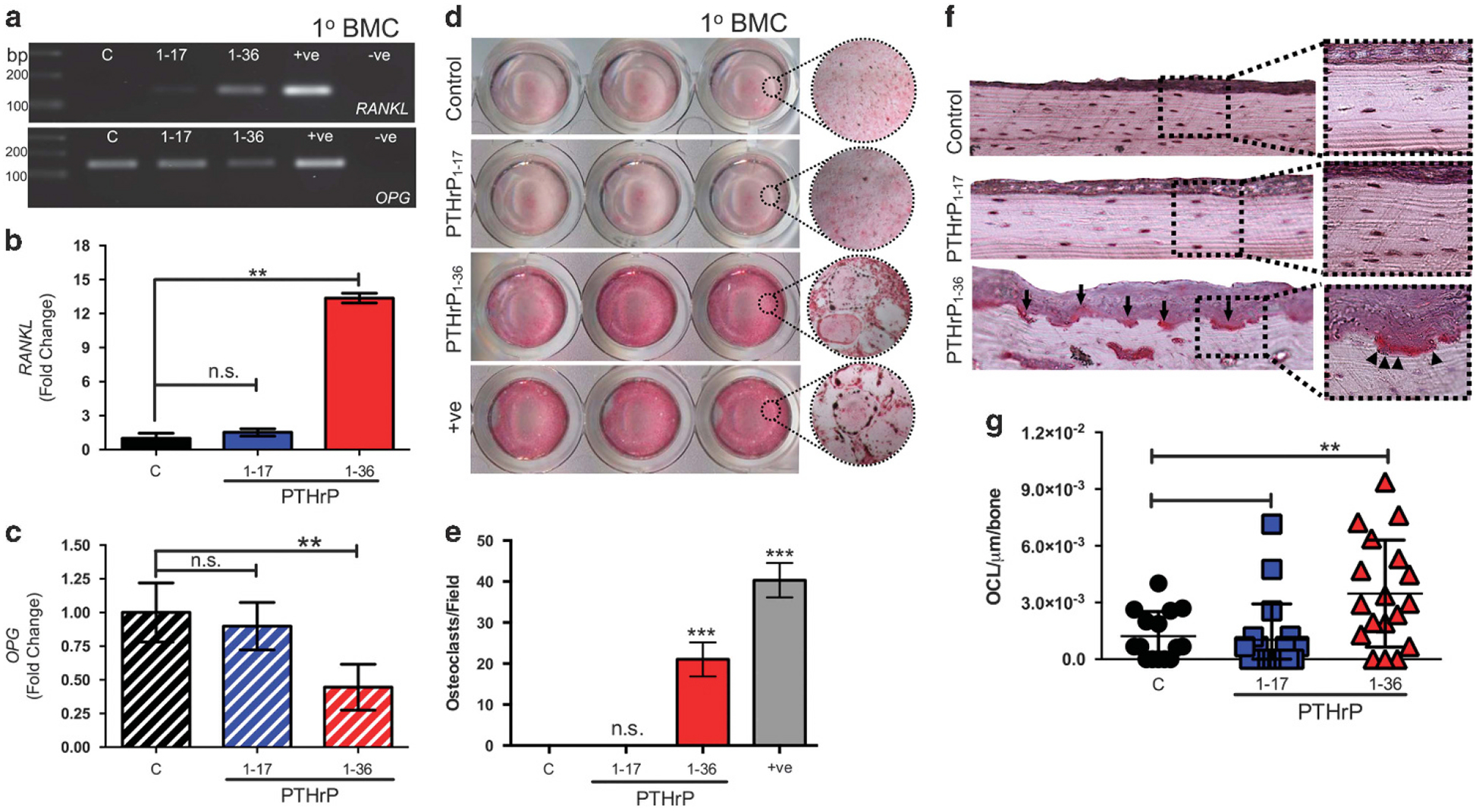
PTHrP1–17 does not stimulate osteoclastogenesis and bone resorption. (a) Expression of RANKL and OPG in response to PTHrP1–17 and PTHrP1–36 treatment (10 nm for 48 h) in primary bone marrow cultures (1°BMC). PTHrP1–36 stimulated MC3T3 osteoblasts were used as a positive control (+ve), while non-template was used as a negative control (−ve). (b, c) RT-qPCR analyses of effects of PTHrP1–17 or PTHrP1–36 on RANKL (B) and OPG (C) expression in bone marrow cultures (n = 3/group). (d, e) Bone marrow co-cultures were treated for 5 days with PTHrP1–17 or PTHrP1–36 (10 nm). Recombinant RANKL was used as a positive control (+ve). The number of TRAcP positive osteoclasts per field of view (d) were counted in each well (e). (f, g) The number of multinucleated osteoclasts/μm of bone (arrows, f) was determined in multiple tissue sections derived from animals in each group (n = 3/group) (g). Asterisks denote statistical significance (*P < 0.05; **P < 0.01); n.s., non-significant values.
PTHrP1–17 is produced by cancer cells
To address if the PTHrP1–17 peptide could be detected in biological samples, PTHrP1–17-specific antibodies were generated (Figure 6a). The lead antibody, clone 2D11 (CPTC-PTHrP-1), detects PTHrP1–17 and PTHrP1–36 at concentrations as low as 10 ng but did not cross-react with the PTHrP27–36 peptide (Figure 6b). Immuno-precipitation followed by mass spectrometry (IP-MS)37,38 allowed for the detection of, and delineation between, PTHrP1–17 and PTHrP1–36 in multiple PTHrP peptide mixtures (Figure 6c).
Figure 6.
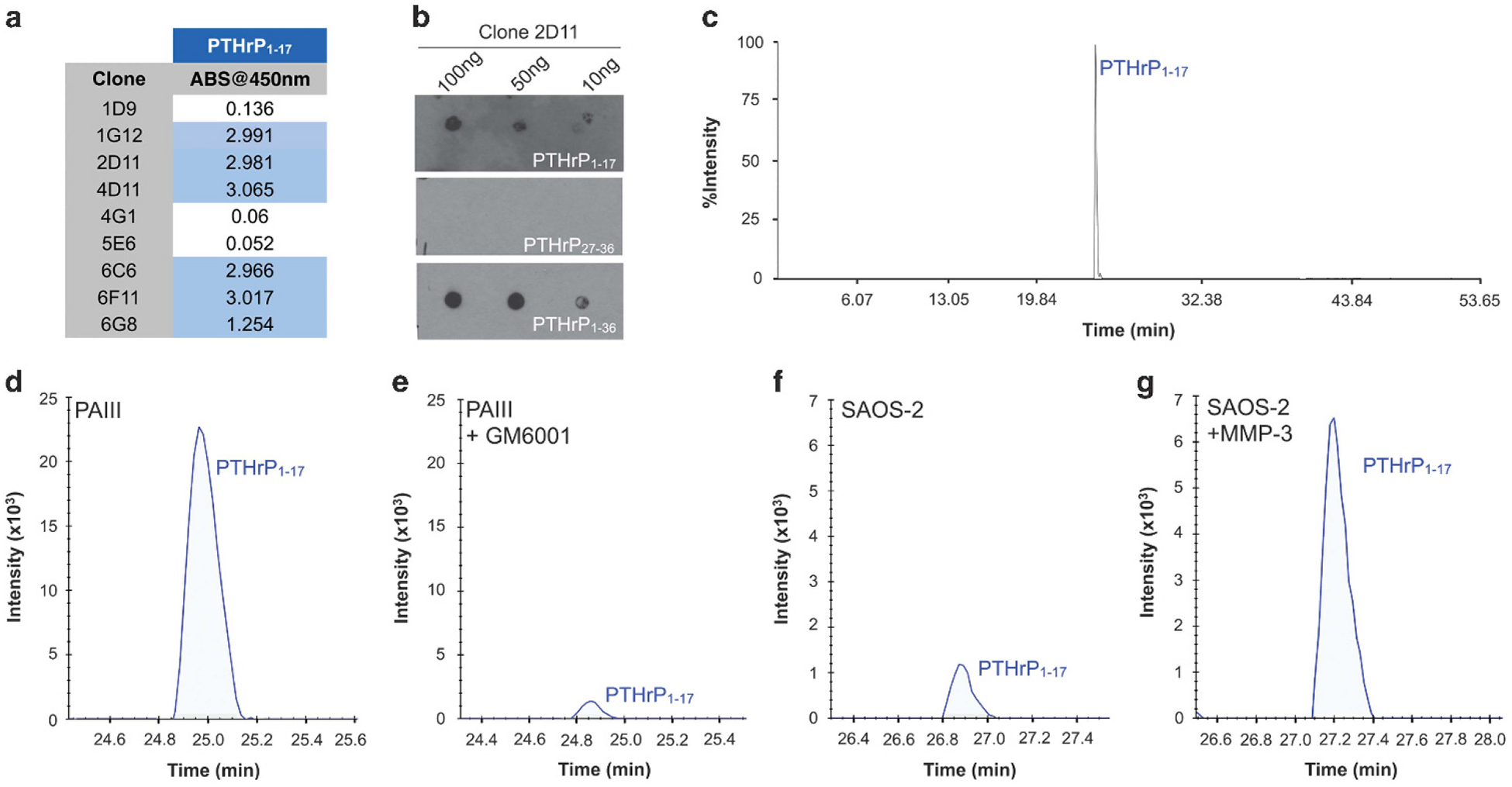
MMP generation of PTHrP1–17 in cancer cells. (a) Antibodies were raised against PTHrP1–17 and the ability of isolated clones to detect the peptide was measured by ELISA. (b) Dot blot titration of clone 2D11 against 100, 50 and 10 ng of PTHrP1–17, PTHrP27–36 and PTHrP1–36. (c) IP-MS detection of PTHrP1–17 after immunoprecipitation with 2D11 from an equimolar mixture of PTHrP1–17, PTHrP18–26, PTHrP27–36 and PTHrP1–36 peptides. The peak detected at 25 min corresponds to PTHrP1–17. (d, e) IP-MS of PTHrP1–17 from the conditioned media of the prostate cancer cell line, PAIII treated in the absence (d) or presence (e) of the broad spectrum MMP inhibitor GM6001. (f, g). IP-MS of PTHrP1–17 from the conditioned media of the human osteosarcoma cell line SAOS-2. SAOS-2 cells were treated in the absence (f) or presence of recombinant MMP-3 (g). The blue lines in (d–g) represent endogenous PTHrP1–17 at the +3 charge state.
PTHrP is expressed by a number of cancer cell lines, including those of a prostate and osteosarcoma origin (Supplementary Figure S7). We collected conditioned media from PTHrP expressing PAIII rat prostate adenocarcinoma cells incubated in the presence or absence of a broad-spectrum MMP inhibitor, GM6001. IP-MS of PAIII conditioned media clearly demonstrated the presence of PTHrP1–17 and that MMP inhibition reduced the amount of this product (Figures 6d and e). The incorporation of stable isotope-labelled standards (SIS1–17) allowed for the quantification of peak areas from IP-MS experiments and demonstrated GM6001 treatment reduced the amount of PTHrP1–17 by 77% (PaIII = 0.13, PaIII+GM6001 = 0.03; Supplementary Figure S7B and C). Conversely, despite the detection of PTHrP transcripts in SAOS-2 osteosarcoma cells, levels of PTHrP1–17 in SAOS-2 conditioned media were low compared to those in PAIII (Figure 6f). However, overnight incubation of SAOS-2 cells with recombinant exogenous MMP-3 resulted in the enhanced detection of the PTHrP1–17 peptide (Figure 6g). Use of SIS1–17 demonstrated that the addition of MMP-3 increased PTHrP1–17 levels by 400% (SAOS-2 = 0.004, SAOS-2+MMP-3 = 0.03, Supplementary Figures S6E and F). These data show that PTHrP1–17 can be biologically generated by cancer cells and in turn this novel MMP-generated product can selectively promote osteogenesis.
DISCUSSION
MMPs regulate bone matrix turnover as well as the bioactivity and bioavailability of non-matrix factors important in bone remodelling such as RANKL and transforming growth factor β. Here we have shown that MMPs also process PTHrP1–36 to yield a distinct, biologically active peptide, PTHrP1–17, which can be generated by cancer cells. Notably, PTHrP1–17 promotes osteogenesis yet has no effect on osteoclast formation and bone resorption. This suggests that MMP-directed cleavage of PTHrP1–36 is a new means for post-translationally regulating the potent osteolytic effects of this hormone, which has important implications for our understanding of bone remodelling and potentially for skeletal malignancies (Figure 7).
Figure 7.
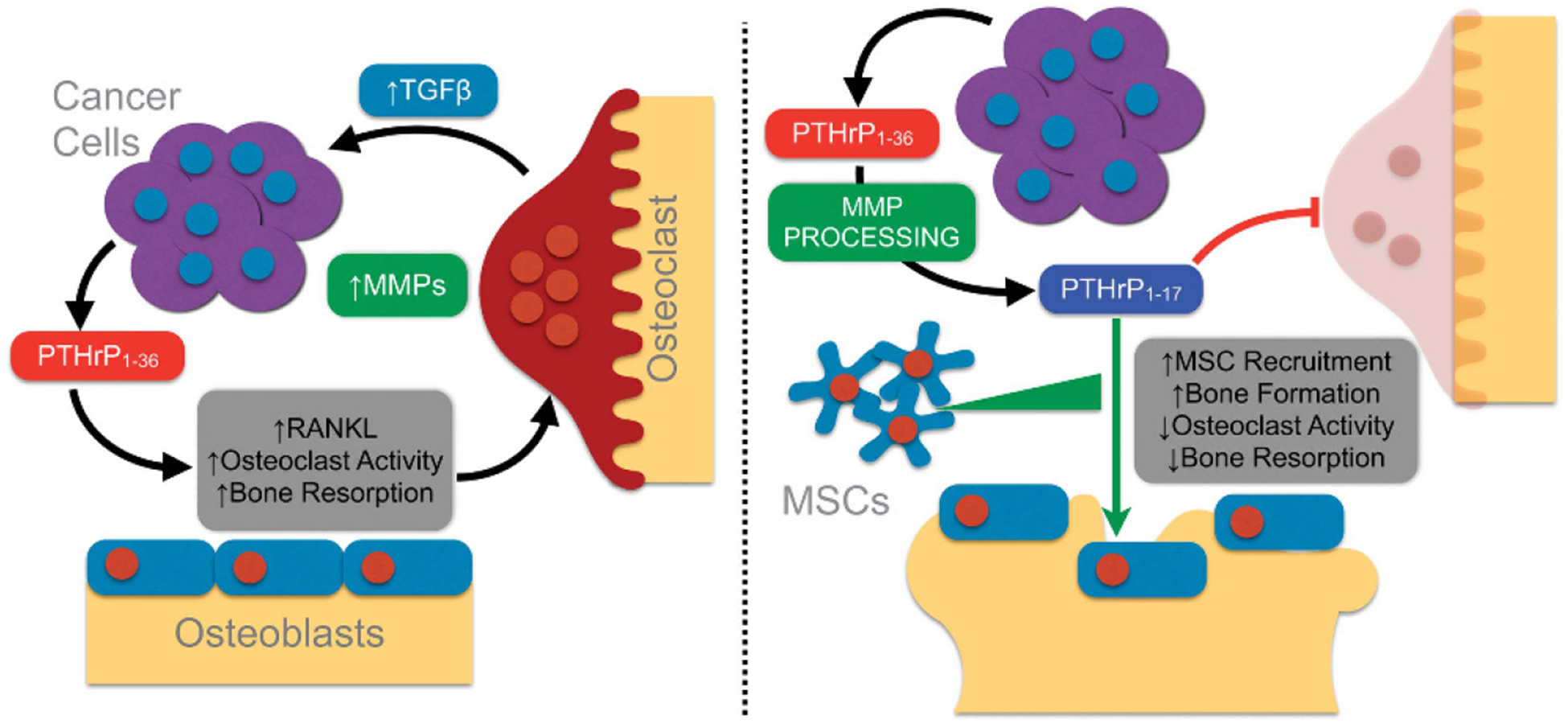
PTHrP1–17 working model in bone metastatic cancer. (a) The initiation of the vicious cycle involves the secretion of PTHrP1–36 from bone metastatic prostate cancer cells, which leads to the induction of RANKL, osteoclastogenesis and the release of growth factors from the bone matrix such as transforming growth factor β that enhance tumour survival. MMP expression is also heightened at the tumour bone interface. (b) Heightened MMP expression leads to the generation of PTHrP1–17 that in turn can promote osteogenesis while preventing osteoclastogenesis. Further, PTHrP1–17 can promote the recruitment of MSCs that can contribute to the osteogenic response.
Previous studies have shown that PTHrP1–36 is susceptible to proteolytic processing, but MMP-generated PTHrP1–17 appears to be a distinct product. Prostate serum antigen/kallikrein-3 and neprilysin have both been shown to generate PTHrP1–23.15,18 Our mass spectrometry data show that a 1–26 fragment can be generated by MMPs but that this species is rapidly reduced to PTHrP1–17. Comparative kinetic analyses between enzymes capable of processing PTHrP1–36 may reveal the dominant protease involved, but it is likely that spatial and temporal factors dictate which protease controls PTHrP1–36 cleavage. Further, serine proteases and MMPs may reciprocally activate each other. For example, prostate serum antigen can regulate MMP-2 activity while conversely, MMPs can activate kallikreins, suggesting that proteolytic cascades could converge to process PTHrP1–36.39,40 It is also possible that PTHrP1–36 can induce the expression of MMPs that in turn process the hormone. For example, PTHrP is known to induce the expression of MMP-2, −3 and −9 in growth plate chondrocytes;41 the induction of MMPs by PTHrP1–36 may result in a feedback loop that dampens osteolytic stimuli once bone resorption has been initiated. Further, PTHrP has also been shown to induce the expression of MMP-13,42 but interestingly, our data show MMP-13 does not yield a PTHrP1–17 fragment, again pointing to distinct roles for proteases in regulating PTHrP1–36 activity.
Adding further complexity to PTHrP regulation, a recent report has demonstrated that serum levels of PTHrP12–48 are a prognostic marker for bone metastatic breast cancer,43 indicating that a PTHrP1–11 fragment is also generated. Our mass spectrometry analyses show that MMPs do not reduce PTHrP further than PTHrP1–17, implying that other proteases must be involved in generating this species.3 Whether PTHrP1–11 retains biological activity is undetermined, but this is possible given the importance of the first two N-terminal amino acids in activating PTH1R.3
PTHrP1–36 is generated from a full-length form of PTHrP of up to 173 amino acids. We focused exclusively on peptides generated from PTHrP1–36; however, products generated from the remaining 37–173 sequence of PTHrP can impact bone remodelling.3,44 For example, osteostatin is generated via cleavage of PTHrP at amino acids 107–111/139 and is a potent inhibitor of osteoclastogenesis.45,46 The proteases responsible for generating this fragment have not been identified, but it is tempting to speculate that MMP generation of osteogenic PTHrP1–17 coupled with the generation of osteostatin would further promote the anabolic effects of PTHrP following the resorptive phase. It is also noteworthy that PTHrP87–107 contains a nuclear localization sequence that supports osteoblast survival and matrix mineralization.47 Whether this fragment is generated by MMPs remains to be explored. Understanding the precise temporal sequence of how PTHrP is cleaved is needed to define the complex roles it plays in regulating the catabolic and anabolic phases of bone remodelling.
PTHrP1–36 activation of PTH1R leads to cAMP generation and calcium flux.31 Our studies show that PTHrP1–17 rapidly induces calcium flux and ERK phosphorylation in osteoblasts but unlike PTHrP1–36, does not affect cAMP generation or CREB phosphorylation. Previous studies have shown that ERK phosphorylation is enhanced via the protein kinase C pathway and promotes osteogenic differentiation.48 Additionally, PTH1R-induced cAMP triggers CREB phosphorylation and the induction of RANKL.9 In contrast to PTHrP1–36, PTHrP1–17 has no effect on RANKL expression in osteoblasts. Thus, we posit that PTHrP1–17 activation of PTH1R leads to osteoblast differentiation and bone formation by promoting calcium flux and ERK phosphorylation. In accord with this notion, the N-terminal domain of PTHrP and PTH can stimulate calcium flux via PTH1R.49 In contrast, other studies have shown that N-terminal fragments of PTHrP and PTH can stimulate protein kinase A and cAMP activation,31,50 yet this effect is not observed in PTHrP1–17-treated primary bone cell cultures and osteoblast cell lines. In agreement with our findings, a recent study demonstrated that PTHrP1–16 does not result in cAMP production but interestingly also had no effect on calcium flux using PTH1R overexpressing CHO-K1 cells.31 This may indicate that either the glutamine at amino acid position 17 in PTHrP is an important mediator of calcium flux or that PTH1R activates different signalling effectors in osteoblasts. Based on PTH1R knockdown studies it is clear that the effects of PTHrP1–17 are dependent on PTH1R and not on another G-protein coupled receptor such as endothelin-A.51,52
Our discovery of MMP processing of PTHrP has potentially important clinical implications. For example, bone metastatic prostate cancer contains both areas of osteolysis and aberrant bone formation.53 Osteosarcoma and prostate cancer cells are now revealed to generate both PTHrP1–36 and PTHrP1–17, which could explain their divergent effects on osteogenesis rather than osteolysis. This is further supported by the ability of both PTHrP1–361,54 and PTHrP1–17 to recruit MSCs and osteoblast precursors (Figure 3). PTHrP1–36 expression is highly associated with osteolytic lesions such as bone metastatic breast cancer and multiple myeloma. While PTHrP1–17 may also be generated in these skeletal malignancies, the overall balance of osteolytic to osteogenic factors in these scenarios favours osteolysis. Our current research centres on the detection of PTHrP1–17 in the serum of prostate cancer patients with primary, castrate resistant and metastatic castrate resistant prostate cancer to determine whether PTHrP1–17 can be used as a potential readout for occult bone metastases or progression of bone metastatic disease. We are also using genetic approaches to eliminate MMPs in the host and cancer cell compartments to identify the key MMP responsible for the generation of PTHrP1–17 in vivo. Finally, the ratio of PTHrP1–36 to PTHrP1–17 has implications for other diseases such as osteoporosis, and may potentially explain the differential effects of chronic versus intermittent PTHrP administration on bone resorption versus formation.
MATERIALS AND METHODS
Cell lines, culture and animals
MC3T3-E1, HEK-293, RAW264.7 and SAOS2 cell lines were purchased from the ATCC and PC3–2M cells obtained from Perkin Elmer (Waltham, MA, USA). PAIII cells55 and C4–2B56 were kindly donated. All were periodically mycoplasma tested (#CUL001B, R&D Systems, Minneapolis, MN, USA) and short tandem repeat verified. Mouse bone marrow MSCs and co-cultures were isolated from C57BL/6 mice as described.57 For PTH1R knockdown studies, standard lentiviral transduction protocols were used. A pool of three different shRNA plasmids (Santa Cruz Biotechnology, Dallas, TX, USA; sc-40158-V) was transduced to generate stable knockdown clones: sc-40158-VA 5′-GATCCCACATGTTCCTGTCGTTTATTCAAGAGATAAACGACA GGAACATGTGTTTTT-3′. sc-40158-VB 5′-GATCCCATCTGTTGTGCTCAACT TTTCAAGAGAAAGTTGAGCACAACAGATGTTTTT-3′. sc-40158-VC 5′-GATCC CAAGCGTAAAGCACGAAGTTTCAAGAGAACTTCGTGCTTTACGCTTGTTTTT-3′. A scrambled sequence was used for shControl transductions. Transient transfection (Qiagen, Hilden, Germany; Superfect, 301305) for forced PTH1R expression studies (Origene #RG212841-Human, Rockville, MD, USA; MC201102-Mouse) was conducted per manufacturer’s instructions. For conditioned media collection, cells were incubated in serum free conditions for 24 h. The broad spectrum MMP inhibitor GM6001 (Millipore, #CC1010, 10 μm) or recombinant MMP-3 (Millipore, #444217, 100 ng/ml) were added during collection for MMP inhibition/treatment.
Mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). All animal experiments were performed with Institutional Animal Care and Use Committee (IACUC) approval (#IS000001283, CCL) from the University of South Florida.
Gene expression analyses
RNA was extracted with TRIzol (Invitrogen #15596, Carlsbad, CA, USA). Reverse transcription was performed using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA; #4368813). Primer sequences: Mouse PTH1R Forward 5′-AGCCAGACGATGTCTTTACCAA-3′; mouse PTH1R Reverse 5′-GATGCTG GCGTCCACCCTT-3′. Human PTH1R Forward 5′-AGAGAAGAAGTACCTGTGGGG-3′; human PTH1R Reverse 5′-GATGATCCACTTTTTGTTCCC-3′. PTHrP Forward 5′-GCAGTGGAGTGTCCT GGTATTC-3′; PTHrP Reverse 5′-TTGGATGGACTTGCCCTTGT-3′. RANKL Forward 5′-ACGCCAACATTTGCTTTCGG-3′; RANKL Reverse 5′-GACC AGTTT TTCGTGCTCCCT-3′. OPG Forward 5′-CCTTGCCCTGACCACTCTTA-3′; OPG Reverse 5′-CCTCACACTCACACACTCGGT-3′. Type I Collagen Forward 5′-ACAGACGAACAACCCAAACT-3′; Type I Collagen Reverse 5′-GGTT TTTGGTCACGTTCAGT-3′. 18S Forward 5′-GTAACCCGTTGAACCCCATT-3′; 18S Reverse 5′-CCATCCAATCGGTAGTAGCG-3′. GAPDH Forward 5′-CCT GCACCACCAACTGCTTA-3′; GAPDH Reverse 5′-CCACGATGCCAAAGTTGTCA −3′. All samples were run in triplicate and normalized to 18S or GAPDH.
MMP processing of PTHrP and identification of cleavage sites
Recombinant PTHrP1–86 (100 ng; Abcam, Cambridge, UK; ab50228) was incubated for 1 h in MMP digestion buffer (0.15 M NaCl, 50 mm Tris pH 7.6) in the presence of MMP-2, −3, −7, −9 or −13 (100 ng, Millipore) with products determined by SDS-PAGE/Coomassie staining and western blotting. N-terminal amino acid sequencing was performed using standard protocols (Pro-Seq, Boxford, MA, USA). Matrix-assisted laser desorption-time of flight (MALDI-TOF) was performed at the Moffitt Proteomics Core. Peptides were extracted using C18 ZipTips (Millipore ZTC18S096), dried, and were resuspended in 5 μl of aqueous 2% acetonitrile, 1% acetic acid plus 5 μl of α-cyano-4-hydoxycinnamic acid dissolved at 5 mg/ml in 50% H2O/50% acetonitrile.
PTHrP1–36 (ProImmune, Oxford, UK) and major MMP generated fragments (PTHrP1–17, PTHrP18–26 and PTHrP27–36) were synthesized via standard FMOC chemistry (Symphony, (manufactured by Gyros Protein Technologies) Tucson, AZ, USA; PTI) and characterized as described.58
Immunoblotting and immunoprecipitation
Cells were lysed with RIPA (150 mm NaCl, 1 mm EDTA, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 20 mm Tris, pH 8). Protein concentration was determined by BCA (Pierce, Waltham, MA, USA; #23225). Blots were blocked in 5% BSA for 1 h followed by primary antibody for phospho-ERK (Cell Signaling Technology #9101, Danvers, MA, USA; 1:1000), ERK (Cell Signaling Technology #4695; 1:1000), phospho-CREB (Cell Signaling Technology #9198, diluted 1:1000 in blocking solution +0.1% Tween-20), CREB (Cell Signaling Technology #9197, diluted 1:1000 in blocking solution +0.1% Tween-20) or PTHrP (Santa Cruz sc20728; 1:1000) overnight at 4 °C. Blots were washed then incubated with HRP-conjugated anti-species secondary (Cell Signaling Technology, Rabbit #7074/Mouse #7076, 1:1000). Actin (Santa Cruz sc-1615; 1:1000) was used as a loading control. All experiments were performed in triplicate.
Anti-PTHrP1–17 antibodies were developed by the NCI Office of Cancer Clinical Proteomics Research (https://antibodies.cancer.gov) and evaluated by ELISA and PTHrP peptide (1, 10, 50 and 100 ng) dot blotting. Candidates were evaluated by IP-MS. 1 μg of anti-PTHrP1–17 antibody was added to 1 ml conditioned media aliquots and incubated for 1 h at 4 °C at which point 15 μl of Protein G beads (Ultralink, Pierce) were added and incubated at 4 °C overnight. Beads were washed three times (100 mm NaCl 50 mm Tris HCl, 0.1% NP-40), followed by three washes with nanopure water (18 MΩ) and pooled. Peptide was eluted with 0.1% trifluoroacetic acid, dried, and resuspended in chromatography buffer containing 4 fmol/μl of stable isotope labeled standard (SIS) PTHrP peptides, which incorporate leucine. Samples were analysed using liquid chromatography-parallel reaction monitoring (LC-PRM; nanoRSLC and QExactive Plus, Thermo, Waltham, MA, USA59). Raw data were imported into Skyline (https://skyline.gs.washington.edu60) and quantified using selected transitions. Quantification of peak areas for specific fragment ions was used to determine the ratio of endogenous PTHrP1–17 to the PTHrP1–17 SIS.
PTH1R signalling assays
For cAMP analyses, MC3T3 and PTH1R-expressing HEK cells (2.5 × 104 cells/well, 384-well plate) were treated with PTHrP peptides (1–100 nm, 15 min, n = 5/group). cAMP activity was measured with the cAMP-Glo™ Assay (Promega, Madison, WI, USA; #V1501) as per manufacturer’s instructions. The forskolin analogue NKH477 (Tocris, Bristol, UK; 10 μm, 15 min) served as a positive control. Calcium flux in response to similar concentrations of PTHrP peptides was determined using Fluo-4 Direct™ calcium reagent (Invitrogen, #F10471, 1 × 105 cells, 48-well plate). Fluorescence intensity was measured by time-lapse microscopy (EVOS FL, Waltham, MA, USA; Invitrogen) and quantified from individual cells (n = 20/group) in three fields of view per condition (Definiens, Munich, Germany).
MTS proliferation assay
All cells were seeded at 5 × 104 cells/well in 96-well plate (n = 5/group) and treated for 24 h in 5% serum (MSC, Raw 264.7) or serum free (MC3T3, PAIII, PC3–2M, C4–2B, SAOS-2) containing media. CellTiter96 (Promega, #G5421) was used to determine cell number.
Morphology and migration assays
For immunofluorescence studies, MC3T3 (5 × 104 per well) were seeded in eight-well chamber slides and treated with PTHrP1–17 or PTHrP1–36 in serum-free media for 1 h (10 nm, n = 3/group). Actin filaments were stained using Alexa Fluor 488-Phalloidin (Invitrogen A12379, 1:1000). Osteoblast and MSC migration was assessed using modified Boyden chamber assay. Cells (5 × 105) were seeded in the upper chamber after 24-h serum starvation (n = 3/group). Migration to PTHrP peptides (10 nm in serum free media) was tested over a 5-h period at 37 °C. Serum free and 1% serum media were used as negative and positive controls, respectively.
In vitro osteoblast and osteoclast formation assays
For osteoblast differentiation studies, mouse MSCs (1.2 × 105 cells/well in 24-well plates, n = 3/group) were incubated for 21 days in the presence of PTHrP1–17 or PTHrP1–36 (10 nm, replenished every third day). Osteogenic media (R&D, CCM009) were used as a positive control. Subsequently, cells were fixed with 10% neutral buffered formalin, stained with Alizarin red (2%, pH 4.1–4.3), and quantified (absorbance at 405 nm). For osteoclast formation assays, adherent bone marrow macrophage precursors were cultured for 3 days with recombinant M-CSF (Preprotech, Rocky Hill, NJ, USA; 20 ng/ml) and then seeded into 48-well plates (30 000 cells/well). Cultures were expanded for two additional days then treated with PTHrP1–17 or PTHrP1–36 (100 nm, daily). Positive controls incorporated RANKL (Oriental Yeast Company, Tokyo, Japan; 100 ng/ml) plus M-CSF (25 ng/ml). After 7 days, cultures were stained for tartrate-resistant acid phosphatase (TRAcP) positivity as described below. Only multinucleated (43 nuclei per cell) TRAcP positive cells were counted as osteoclasts.
In vivo osteoclastogenesis assay
For in vivo calvarial injection assays, 2 μg of PTHrP1–17 or PTHrP1–36 were injected subcutaneously every 6 h for 3 days over the calvaria of randomized 4–6-week-old female SCID-Beige mice as reported (n = 3/group).36,61 Calvariae were harvested 10 h after the final injection, fixed and analysed by high-resolution μCT (SCANCO-μCT40) as described.62 Tissues were then decalcified (14% EDTA, pH 7.4, 3 days), processed and paraffin embedded. Sections (5 μm) were stained with hematoxylin and eosin or trichrome for histological analyses. For TRAcP staining and osteoclast measurements, slides were incubated in buffer (112 mm anhydrous sodium acetate, 49 mm dibasic dehydrate sodium tartrate, 0.28% glacial acetic acid) containing 1% naphthol-phosphate substrate (2% napthol AS-BI phosphate in 2-ethoxyethanol) for 1 h at 37 °C. Slides were then transferred to buffer containing 250 μl of 5% pararosaniline dye in 2 N HCl and 250 μl of 4% sodium nitrite at 37 °C and monitored for development of red stained osteoclasts. Slides were rinsed in H2O, counterstained with hematoxylin, and aqueously mounted. The number of bone-lining, multi-nucleated (43 nuclei per cell), TRAcP positive osteoclasts was quantified from multiple sections.
In vivo osteogenesis assay
For ex vivo calvarial organ cultures, calvariae were isolated from 4-day-old Rag2−/− neonates and cultured as described.63 Calvariae (n = 4/group) were treated with PTHrP1–17 or PTHrP1–36 (10 nm, 14 days). Ectopic ossicle formation assays were performed by subcutaneously implanting Gelfoam sponges loaded with 1 × 106 mouse MSCs into randomized 6-week-old male SCID-Beige mice (n = 3/group with two implants on each flank). After 1-week recovery, daily subcutaneous injections of PTHrP peptides were administered (40 μg/kg/day, 21 days). Ossicles were removed for μCT and histology as described.34,64
Statistical analyses
Sample sizes for in vitro and in vivo experiments were estimated based on empirical and published data for PTHrP1–36 effects on osteoblast/osteoclast function. For in vivo experiments, primary study exclusion criteria were death from PTHrP calvaria treatment or dehiscence of ectopic ossicles. No study mice met these exclusion criteria. For statistical analysis among groups, analysis of variance was performed followed by Tukey’s multiple comparison tests using Graph Pad Prism 6.0 (GraphPad Inc., La Jolla, CA, USA). Variance was further measured using the Brown–Forsythe test. P-value < 0.05 was considered statistically significant. Data are presented as s.e.m.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by R01-CA143094 (CCL), the State of Florida Bankhead Coley Program grant 5BC-O1 (CCL) and by a Miles for Moffitt Foundation Award. This work was also supported in part by the Core Facilities of the Moffitt Cancer Center Grant P30-CA076292. The authors thank Joe Johnson (Moffitt Analytical Microscopy), Robert Sprung (Moffitt Proteomics) and Gordon Whiteley (SAIC-Frederick, NCI-Frederick) for their helpful suggestions and expertise, and Drs John Cleveland and Srikumar Chellappan for critical review of this study.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary Information accompanies this paper on the Oncogene website (http://www.nature.com/onc)
REFERENCES
- 1.Miao D, He B, Jiang Y, Kobayashi T, Soroceanu MA, Zhao J et al. Osteoblast-derived PTHrP is a potent endogenous bone anabolic agent that modifies the therapeutic efficacy of administered PTH 1–34. J Clin Invest 2005; 115: 2402–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suva LJ, Winslow GA, Wettenhall RE, Hammonds RG, Moseley JM, Diefenbach-Jagger H et al. A parathyroid hormone-related protein implicated in malignant hypercalcemia: cloning and expression. Science 1987; 237: 893–896. [DOI] [PubMed] [Google Scholar]
- 3.Martin TJ. Parathyroid hormone-related protein, its regulation of cartilage and bone development, and role in treating bone diseases. Physiol Rev 2016; 96: 831–871. [DOI] [PubMed] [Google Scholar]
- 4.Guise TA, Yin JJ, Thomas RJ, Dallas M, Cui Y, Gillespie MT. Parathyroid hormone-related protein (PTHrP)-(1–139) isoform is efficiently secreted in vitro and enhances breast cancer metastasis to bone in vivo. Bone 2002; 30: 670–676. [DOI] [PubMed] [Google Scholar]
- 5.Philbrick W Parathyroid hormone-related protein: Gene structure, biosynthesis, metabolism, and regulation In: Bilezikian JP, Marcus R, Levine M (eds). The Parathyroids: Basic and Clinical Concepts. 2nd edn Academic Press: San Diego, CA, USA, 2001, p 881. [Google Scholar]
- 6.Bilezikian JP, Marcus R, Levine M, Marcocci C, Silverberg SJ, Potts J (eds). The Parathyroids: Basic and Clinical Concepts. 3rd edn Academic Press: San Diego, CA, USA, 2015, p 946. [Google Scholar]
- 7.Juppner H, Abou-Samra AB, Freeman M, Kong XF, Schipani E, Richards J et al. A G protein-linked receptor for parathyroid hormone and parathyroid hormone-related peptide. Science 1991; 254: 1024–1026. [DOI] [PubMed] [Google Scholar]
- 8.Orloff JJ, Reddy D, de Papp AE, Yang KH, Soifer NE, Stewart AF. Parathyroid hormone-related protein as a prohormone: posttranslational processing and receptor interactions. Endocr Rev 1994; 15: 40–60. [DOI] [PubMed] [Google Scholar]
- 9.Park HJ, Baek K, Baek JH, Kim HR. The cooperation of CREB and NFAT is required for PTHrP-induced RANKL expression in mouse osteoblastic cells. J Cell Physiol 2015; 230: 667–679. [DOI] [PubMed] [Google Scholar]
- 10.Fukushima H, Jimi E, Kajiya H, Motokawa W, Okabe K. Parathyroid-hormone-related protein induces expression of receptor activator of NF-{kappa}B ligand in human periodontal ligament cells via a cAMP/protein kinase A-independent pathway. J Dent Res 2005; 84: 329–334. [DOI] [PubMed] [Google Scholar]
- 11.Ma YL, Cain RL, Halladay DL, Yang X, Zeng Q, Miles RR et al. Catabolic effects of continuous human PTH (1–38) in vivo is associated with sustained stimulation of RANKL and inhibition of osteoprotegerin and gene-associated bone formation. Endocrinology 2001; 142: 4047–4054. [DOI] [PubMed] [Google Scholar]
- 12.Miao D, Li J, Xue Y, Su H, Karaplis AC, Goltzman D. Parathyroid hormone-related peptide is required for increased trabecular bone volume in parathyroid hormone-null mice. Endocrinology 2004; 145: 3554–3562. [DOI] [PubMed] [Google Scholar]
- 13.Stewart AF. PTHrP(1–36) as a skeletal anabolic agent for the treatment of osteoporosis. Bone 1996; 19: 303–306. [DOI] [PubMed] [Google Scholar]
- 14.McCauley LK, Martin TJ. Twenty-five years of PTHrP progress: from cancer hormone to multifunctional cytokine. J Bone Miner Res 2012; 27: 1231–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cramer SD, Chen Z, Peehl DM. Prostate specific antigen cleaves parathyroid hormone-related protein in the PTH-like domain: inactivation of PTHrP-stimulated cAMP accumulation in mouse osteoblasts. J Urol 1996; 156(2 Pt 1): 526–531. [DOI] [PubMed] [Google Scholar]
- 16.Philbrick WM, Wysolmerski JJ, Galbraith S, Holt E, Orloff JJ, Yang KH et al. Defining the roles of parathyroid hormone-related protein in normal physiology. Physiol Rev 1996; 76: 127–173. [DOI] [PubMed] [Google Scholar]
- 17.Martin TJ. Osteoblast-derived PTHrP is a physiological regulator of bone formation. J Clin Invest 2005; 115: 2322–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruchon AF, Marcinkiewicz M, Ellefsen K, Basak A, Aubin J, Crine P et al. Cellular localization of neprilysin in mouse bone tissue and putative role in hydrolysis of osteogenic peptides. J Bone Miner Res 2000; 15: 1266–1274. [DOI] [PubMed] [Google Scholar]
- 19.Lopez-Otin C, Matrisian LM. Emerging roles of proteases in tumour suppression. Nat Rev Cancer 2007; 7: 800–808. [DOI] [PubMed] [Google Scholar]
- 20.Krane SM, Inada M. Matrix metalloproteinases and bone. Bone 2008; 43: 7–18. [DOI] [PubMed] [Google Scholar]
- 21.Lynch CC. Matrix metalloproteinases as master regulators of the vicious cycle of bone metastasis. Bone 2010; 48: 44–53. [DOI] [PubMed] [Google Scholar]
- 22.Lopez-Otin C, Overall CM. Protease degradomics: a new challenge for proteomics. Nat Rev Mol Cell Biol 2002; 3: 509–519. [DOI] [PubMed] [Google Scholar]
- 23.Lynch CC. Matrix metalloproteinases as master regulators of the vicious cycle of bone metastasis. Bone 2011; 48: 44–53. [DOI] [PubMed] [Google Scholar]
- 24.Lynch CC, Hikosaka A, Acuff HB, Martin MD, Kawai N, Singh RK et al. MMP-7 promotes prostate cancer-induced osteolysis via the solubilization of RANKL. Cancer Cell 2005; 7: 485–496. [DOI] [PubMed] [Google Scholar]
- 25.Winding B, NicAmhlaoibh R, Misander H, Hoegh-Andersen P, Andersen TL, Holst-Hansen C et al. Synthetic matrix metalloproteinase inhibitors inhibit growth of established breast cancer osteolytic lesions and prolong survival in mice. Clin Cancer Res 2002; 8: 1932–1939. [PubMed] [Google Scholar]
- 26.Bonfil RD, Sabbota A, Nabha S, Bernardo MM, Dong Z, Meng H et al. Inhibition of human prostate cancer growth, osteolysis and angiogenesis in a bone metastasis model by a novel mechanism-based selective gelatinase inhibitor. Int J Cancer 2006; 118: 2721–2726. [DOI] [PubMed] [Google Scholar]
- 27.Croucher PI, McDonald MM, Martin TJ. Bone metastasis: the importance of the neighbourhood. Nat Rev Cancer 2016; 16: 373–386. [DOI] [PubMed] [Google Scholar]
- 28.Guise TA. Parathyroid hormone-related protein and bone metastases. Cancer 1997; 80(8 Suppl): 1572–1580. [DOI] [PubMed] [Google Scholar]
- 29.Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer 2002; 2: 584–593. [DOI] [PubMed] [Google Scholar]
- 30.Thiolloy S, Edwards JR, Fingleton B, Rifkin DB, Matrisian LM, Lynch CC. An osteoblast-derived proteinase controls tumor cell survival via TGF-beta activation in the bone microenvironment. PLoS One 2012; 7: e29862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cupp ME, Nayak SK, Adem AS, Thomsen WJ. Parathyroid hormone (PTH) and PTH-related peptide domains contributing to activation of different PTH receptor-mediated signaling pathways. J Pharmacol Exp Ther 2013; 345: 404–418. [DOI] [PubMed] [Google Scholar]
- 32.Jilka RL, Weinstein RS, Bellido T, Parfitt AM, Manolagas SC. Osteoblast programmed cell death (apoptosis): modulation by growth factors and cytokines. J Bone Miner Res 1998; 13: 793–802. [DOI] [PubMed] [Google Scholar]
- 33.Esbrit P, Alcaraz MJ. Current perspectives on parathyroid hormone (PTH) and PTH-related protein (PTHrP) as bone anabolic therapies. Biochem Pharmacol 2013; 85: 1417–1423. [DOI] [PubMed] [Google Scholar]
- 34.Holmbeck K, Bianco P, Caterina J, Yamada S, Kromer M, Kuznetsov SA et al. MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell 1999; 99: 81–92. [DOI] [PubMed] [Google Scholar]
- 35.Pettway GJ, Schneider A, Koh AJ, Widjaja E, Morris MD, Meganck JA et al. Anabolic actions of PTH (1–34): use of a novel tissue engineering model to investigate temporal effects on bone. Bone 2005; 36: 959–970. [DOI] [PubMed] [Google Scholar]
- 36.Yates AJ, Gutierrez GE, Smolens P, Travis PS, Katz MS, Aufdemorte TB et al. Effects of a synthetic peptide of a parathyroid hormone-related protein on calcium homeostasis, renal tubular calcium reabsorption, and bone metabolism in vivo and in vitro in rodents. J Clin Invest 1988; 81: 932–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anderson NL, Anderson NG, Haines LR, Hardie DB, Olafson RW, Pearson TW. Mass spectrometric quantitation of peptides and proteins using stable isotope standards and capture by anti-peptide antibodies (SISCAPA). J Proteome Res 2004; 3: 235–244. [DOI] [PubMed] [Google Scholar]
- 38.Katafuchi T, Esterhazy D, Lemoff A, Ding X, Sondhi V, Kliewer SA et al. Detection of FGF15 in plasma by stable isotope standards and capture by anti-peptide antibodies and targeted mass spectrometry. Cell Metab 2015; 21: 898–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoon H, Blaber SI, Li W, Scarisbrick IA, Blaber M. Activation profiles of human kallikrein-related peptidases by matrix metalloproteinases. Biol Chem 2013; 394: 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pezzato E, Sartor L, Dell’Aica I, Dittadi R, Gion M, Belluco C et al. Prostate carcinoma and green tea: PSA-triggered basement membrane degradation and MMP-2 activation are inhibited by (−)epigallocatechin-3-gallate. Int J Cancer 2004; 112: 787–792. [DOI] [PubMed] [Google Scholar]
- 41.Kawashima-Ohya Y, Satakeda H, Kuruta Y, Kawamoto T, Yan W, Akagawa Y et al. Effects of parathyroid hormone (PTH) and PTH-related peptide on expressions of matrix metalloproteinase-2, −3, and −9 in growth plate chondrocyte cultures. Endocrinology 1998; 139: 2120–2127. [DOI] [PubMed] [Google Scholar]
- 42.Ibaragi S, Shimo T, Iwamoto M, Hassan NM, Kodama S, Isowa S et al. Parathyroid hormone-related peptide regulates matrix metalloproteinase-13 gene expression in bone metastatic breast cancer cells. Anticancer Res 2010; 30: 5029–5036. [PubMed] [Google Scholar]
- 43.Washam CL, Byrum SD, Leitzel K, Ali SM, Tackett AJ, Gaddy D et al. Identification of PTHrP(12–48) as a plasma biomarker associated with breast cancer bone metastasis. Cancer Epidemiol Biomarkers Prev 2013; 22: 972–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Amizuka N, Henderson JE, White JH, Karaplis AC, Goltzman D, Sasaki T et al. Recent studies on the biological action of parathyroid hormone (PTH)-related peptide (PTHrP) and PTH/PTHrP receptor in cartilage and bone. Histol Histopathol 2000; 15: 957–970. [DOI] [PubMed] [Google Scholar]
- 45.Cuthbertson RM, Kemp BE, Barden JA. Structure study of osteostatin PTHrP [Thr107](107–139). Biochim Biophys Acta 1999; 1432: 64–72. [DOI] [PubMed] [Google Scholar]
- 46.Valin A, Garcia-Ocana A, De Miguel F, Sarasa JL, Esbrit P. Antiproliferative effect of the C-terminal fragments of parathyroid hormone-related protein, PTHrP-(107–111) and (107–139), on osteoblastic osteosarcoma cells. J Cell Physiol 1997; 170: 209–215. [DOI] [PubMed] [Google Scholar]
- 47.Garcia-Martin A, Ardura JA, Maycas M, Lozano D, Lopez-Herradon A, Portal-Nunez S et al. Functional roles of the nuclear localization signal of parathyroid hormone-related protein (PTHrP) in osteoblastic cells. Mol Endocrinol 2014; 28: 925–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lai CF, Chaudhary L, Fausto A, Halstead LR, Ory DS, Avioli LV et al. Erk is essential for growth, differentiation, integrin expression, and cell function in human osteoblastic cells. J Biol Chem 2001; 276: 14443–14450. [DOI] [PubMed] [Google Scholar]
- 49.Azarani A, Goltzman D, Orlowski J. Structurally diverse N-terminal peptides of parathyroid hormone (PTH) and PTH-related peptide (PTHRP) inhibit the Na+/H+ exchanger NHE3 isoform by binding to the PTH/PTHRP receptor type I and activating distinct signaling pathways. J Biol Chem 1996; 271: 14931–14936. [DOI] [PubMed] [Google Scholar]
- 50.Luck MD, Carter PH, Gardella TJ. The (1–14) fragment of parathyroid hormone (PTH) activates intact and amino-terminally truncated PTH-1 receptors. Mol Endocrinol 1999; 13: 670–680. [DOI] [PubMed] [Google Scholar]
- 51.Takuwa Y, Ohue Y, Takuwa N, Yamashita K. Endothelin-1 activates phospholipase C and mobilizes Ca2+ from extra- and intracellular pools in osteoblastic cells. Am J Physiol 1989; 257(6 Pt 1): E797–E803. [DOI] [PubMed] [Google Scholar]
- 52.Schluter KD, Katzer C, Piper HM. A N-terminal PTHrP peptide fragment void of a PTH/PTHrP-receptor binding domain activates cardiac ET(A) receptors. Br J Pharmacol 2001; 132: 427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frieling JS, Basanta D, Lynch CC. Current and emerging therapies for bone metastatic castration-resistant prostate cancer. Cancer Control: Journal of the Moffitt Cancer Center 2015; 22: 109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mak IW, Turcotte RE, Ghert M. Parathyroid hormone-related protein (PTHrP) modulates adhesion, migration and invasion in bone tumor cells. Bone 2013; 55: 198–207. [DOI] [PubMed] [Google Scholar]
- 55.Hodde JP, Suckow MA, Wolter WR, Hiles MC. Small intestinal submucosa does not promote PAIII tumor growth in Lobund-Wistar rats. J Surg Res 2004; 120: 189–194. [DOI] [PubMed] [Google Scholar]
- 56.Wu TT, Sikes RA, Cui Q, Thalmann GN, Kao C, Murphy CF et al. Establishing human prostate cancer cell xenografts in bone: induction of osteoblastic reaction by prostate-specific antigen-producing tumors in athymic and SCID/bg mice using LNCaP and lineage-derived metastatic sublines. Int J Cancer 1998; 77: 887–894. [DOI] [PubMed] [Google Scholar]
- 57.Helfrich MH, Ralston S. Bone Research Protocols. Humana Press: Totowa, NJ, 2003, pxiv, 448p. [Google Scholar]
- 58.Remily-Wood ER, Liu RZ, Xiang Y, Chen Y, Thomas CE, Rajyaguru N et al. A database of reaction monitoring mass spectrometry assays for elucidating therapeutic response in cancer. Proteomics Clin Appl 2011; 5: 383–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gallien S, Duriez E, Crone C, Kellmann M, Moehring T, Domon B. Targeted proteomic quantification on quadrupole-orbitrap mass spectrometer. Mol Cell Proteomics 2012; 11: 1709–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010; 26: 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao W, Byrne MH, Boyce BF, Krane SM. Bone resorption induced by parathyroid hormone is strikingly diminished in collagenase-resistant mutant mice. J Clin Invest 1999; 103: 517–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cook LM, Araujo A, Pow-Sang JM, Budzevich MM, Basanta D, Lynch CC. Predictive computational modeling to define effective treatment strategies for bone metastatic prostate cancer. Sci Rep 2016; 6: 29384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mohammad KS, Chirgwin JM, Guise TA. Assessing new bone formation in neonatal calvarial organ cultures. Methods Mol Biol 2008; 455: 37–50. [DOI] [PubMed] [Google Scholar]
- 64.Datta NS, Pettway GJ, Chen C, Koh AJ, McCauley LK. Cyclin D1 as a target for the proliferative effects of PTH and PTHrP in early osteoblastic cells. J Bone Miner Res 2007; 22: 951–964. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.