

Abstract
Inhibition of the bromodomain containing protein 9 (BRD9) by small molecules is an attractive strategy to target mutated SWI/SNF chromatin-remodeling complexes in cancer. However, reported BRD9 inhibitors also inhibit the closely related bromodomain-containing protein 7 (BRD7), which has different biological functions. The structural basis for differential potency and selectivity of BRD9 inhibitors is largely unknown because of the lack of structural information on BRD7. Here, we biochemically and structurally characterized diverse inhibitors with varying degrees of potency and selectivity for BRD9 over BRD7. Novel cocrystal structures of BRD7 liganded with new and previously reported inhibitors of five different chemical scaffolds were determined alongside BRD9 and BRD4. We also report the discovery of first-in-class dual bromodomain—kinase inhibitors outside the bromodomain and extraterminal family targeting BRD7 and BRD9. Combined, the data provide a new framework for the development of BRD7/9 inhibitors with improved selectivity or additional polypharmacologic properties.
Graphical Abstract
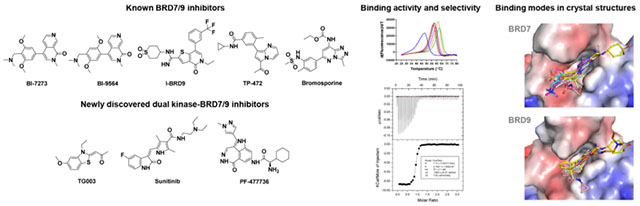
INTRODUCTION
Bromodomains (BRDs) are evolutionary conserved epigenetic reader modules that recognize N-acetylated lysine (KAc) residues on histones and other proteins.1,2 BRD-containing proteins have important functions in gene transcription and chromatin remodeling, gene splicing, protein scaffolding, and signal transduction.3 BRD-containing proteins, particularly those of the bromodomain and the extraterminal (BET) family, have been linked to tumorigenesis and inflammatory diseases, and several BET inhibitors are in clinical trials for oncology and nononcology indications.4 More recently, inhibitors targeting non-BET BRDs for which the physiological functions are less understood have been the subject of intense efforts in academia and pharmaceutical industry alike.5
The BRD-containing proteins BRD7 and BRD9 recognize acetylated lysines on histone H3 (H3Ac).1 Although they contain a structurally similar single BRD (62% sequence identity), the cellular functions of full-length BRD7 and BRD9 are remarkably different. BRD9 is a component of ncBAF (noncanonical BRG1/BRM-associated factor), a subtype of the BAF complex, while BRD7 is a component of the PBAF (polybromo-associated BAF) complex.6 Importantly, BRD9 has an essential role in a number of cancers such as acute myeloid leukemia, synovial sarcoma, and malignant rhabdoid tumor.7–9 In contrast, BRD7 is a potential tumor suppressor10 with critical functions in oligodendrocytes progenitor cell differentiation11 and spermatogenesis12.
BRD9 has attracted attention as a draggable subunit of mSWI/SNF chromatin-remodeling complexes, various subunits of which are mutated in nearly 20% of all human cancers.13 To target mSWI/SNF complexes in cancer, few small-molecule inhibitors of the acetyl-lysine binding site (KAc site) of BRD9 have been reported to date.14–18 These inhibitors showed varying degrees of selectivity across BRDs in general and within the BRD9/BRD7 subfamily in particular. As the role of BRD7 in human biology is not fully understood, off-target inhibition of BRD7 by nonselective BRD9 inhibitors bears the risk of unwanted pleiotropic effects. Recently, proteolysis-targeting chimeras (PROTACs), dBRD9 and VZ185, have been developed using the potent BRD7/9 inhibitor BI-7273 as a warhead. While dBRD9 induced the selective degradation of BRD9 over BRD7, VZ185 showed concomitant degradation of BRD9 and BRD7.19,20
An obstacle to the rational design of inhibitors selective for BRD9 is the limited structural information on BRD7. Specifying the binding interactions of an inhibitor in closely related proteins are crucial for the development of selective inhibitors. Here, we applied a multifaceted approach toward the characterization of diverse small-molecule inhibitors with varying degrees of potency and selectivity for BRD7 and BRD9. We report the discovery of first-in-class dual bromodomain–kinase inhibitors outside the BET family targeting BRD7 and BRD9. TG003, a Cdc2-like kinase (Clk2) inhibitor,21 sunitinib, a multitargeted tyrosine kinase inhibitor,22 and PF-477736, an inhibitor of cell cycle checkpoint kinase 1 (Chk1)23 were identified as moderately active BRD7/9 inhibitors with Kd values between 4 and 17 μM. Novel crystal structures of BRD7, BRD9 and BRD4 liganded with new and known inhibitors were determined, the information of which provides a structural basis for the differential potency and selectivity of BRD7/9 inhibitors. By combining, the data afford a new framework for the structure-based design of inhibitors with improved selectivity or additional polypharmacologic properties.
RESULTS
Structure–Activity Relationship of Reported BRD9 Inhibitors.
To address inhibitor selectivity over BRD7, structure–activity relationship (SAR) studies were performed with five previously reported BRD9 inhibitors (Figure 1A). Binding studies by differential scanning fluorimetry (DSF) and isothermal titration calorimetry (ITC) showed significant correlation between the data sets for both BRD7 and BRD9 (Figure 1B,C, Supporting Information Table S1, Figures S1, S2). For BRD9, the thermodynamic parameters of protein–inhibitor interactions were predominantly enthalpy driven and accompanied by entropic penalty (Figure 1D). By contrast, BRD7 showed neutral or favorable entropic contributions upon interaction with all inhibitors. The isomers BI-7273 and BI-9564 have been reported as potent BRD7/9 inhibitors, BI-7273 lacking selectivity and BI-9564 being 12 times more selective for BRD9.16,18 Our data using crystallization-grade proteins support a different notion with BI-7273 being the most potent (Kd = 9 nM) and selective inhibitor of BRD9 among the compounds tested (Figure 1C). This finding may explain the recent use of BI-7273 rather than BI-9564 as a warhead in the development of the BRD9-targeting PRO-TAC.19 BI-9564 shared with TP-472 similar activity and selectivity values; I-BRD9 and bromosporine were the least selective inhibitors.
Figure 1.
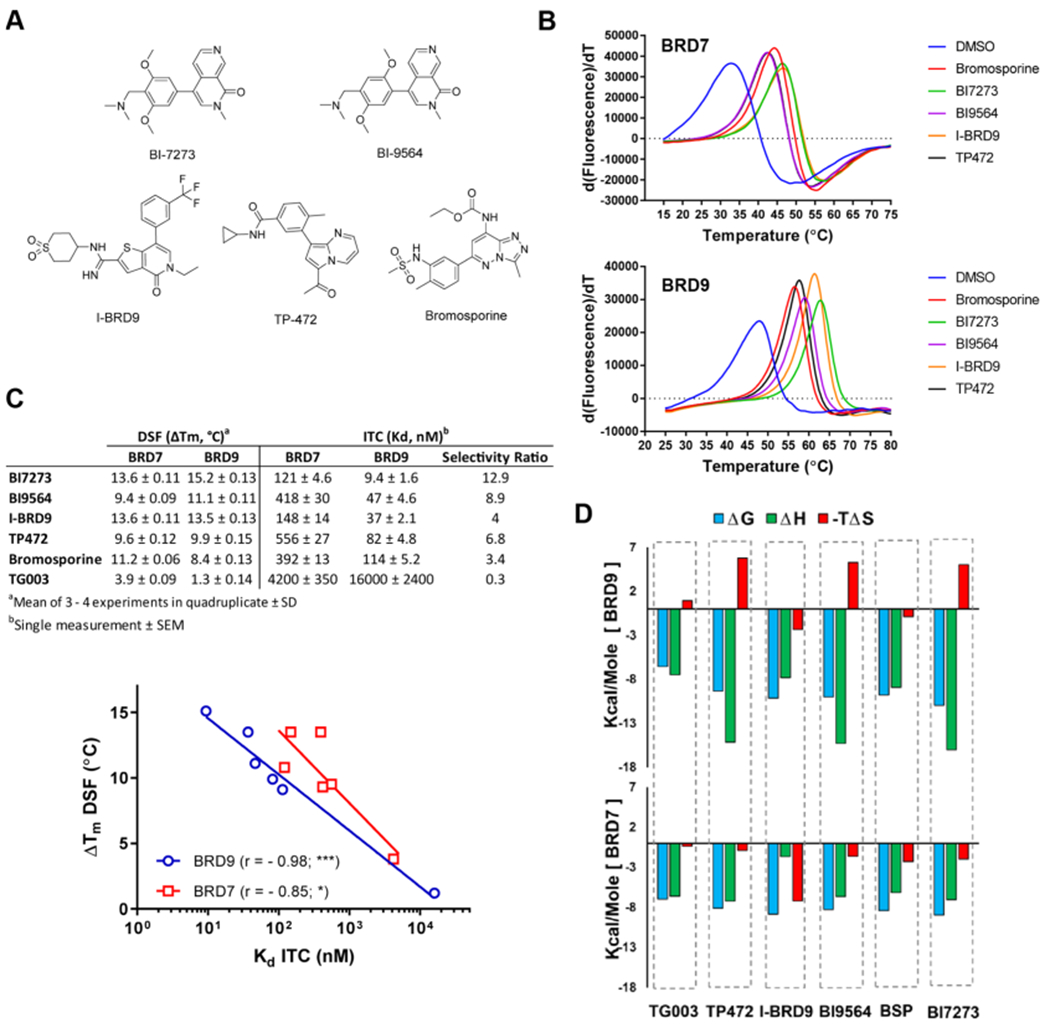
Binding potential of inhibitors for BRD7 and BRD9. (A) Chemical structures of reported BRD9 inhibitors studied herein. (B) Melting temperature shifts of BRD7 and BRD9 in the presence of the 100 μM inhibitor. (C) ΔTm values determined by DSF and Kd values determined by ITC and data correlation for BRD9 (blue) and BRD7 (red). For statistical analysis, r is Pearson’s correlation coefficient; asterisks indicate the significance of the two-tailed P value (α = 0.05). For graphical expression, data were fitted to semilogarithmic lines. (D) Thermodynamic parameters of inhibitor interactions with BRD9 (upper panel) and BRD7 (lower panel). ITC conditions and thermograms are shown in the Supporting Information Table S1 and Figures S1, S2.
While several co-crystal structures of BRD9 with inhibitors have been determined previously, the structure of BRD7 was described only in an unliganded state by NMR.24 Recently, the Structural Genomics Consortium deposited a crystal structure of BRD7 liganded with BI-9564 in the PDB (5MQ1), albeit without context. Although the BRDs of BRD7 and BRD9 share 62% sequence identity, BRD7 is temperature-sensitive with a melting temperature of Tm = 31 °C; whereas BRD9 shows the temperature stability typical of BET BRDs (Tm = 46 °C) (Figure 1B). The temperature sensitivity of BRD7 may explain the difficulties in obtaining X-ray crystals caused by protein denaturation during lengthy crystallization experiments. Therefore, purification of BRD7 was performed expeditiously, resulting in a highly homogeneous protein that readily formed X-ray grade crystals in unliganded and several liganded states under different crystallization conditions (Supporting Information Tables S2 and S3). The overall structures of the BRDs of BRD7 and BRD9 are highly similar (rmsd = 0.63 Å); residues within the KAc site are identical and six differing residues are located in the vicinity of the KAc site (Figure 2A). Distances between the opposing flanks of the KAc site are similar for both proteins (Figure 2B).
Figure 2.
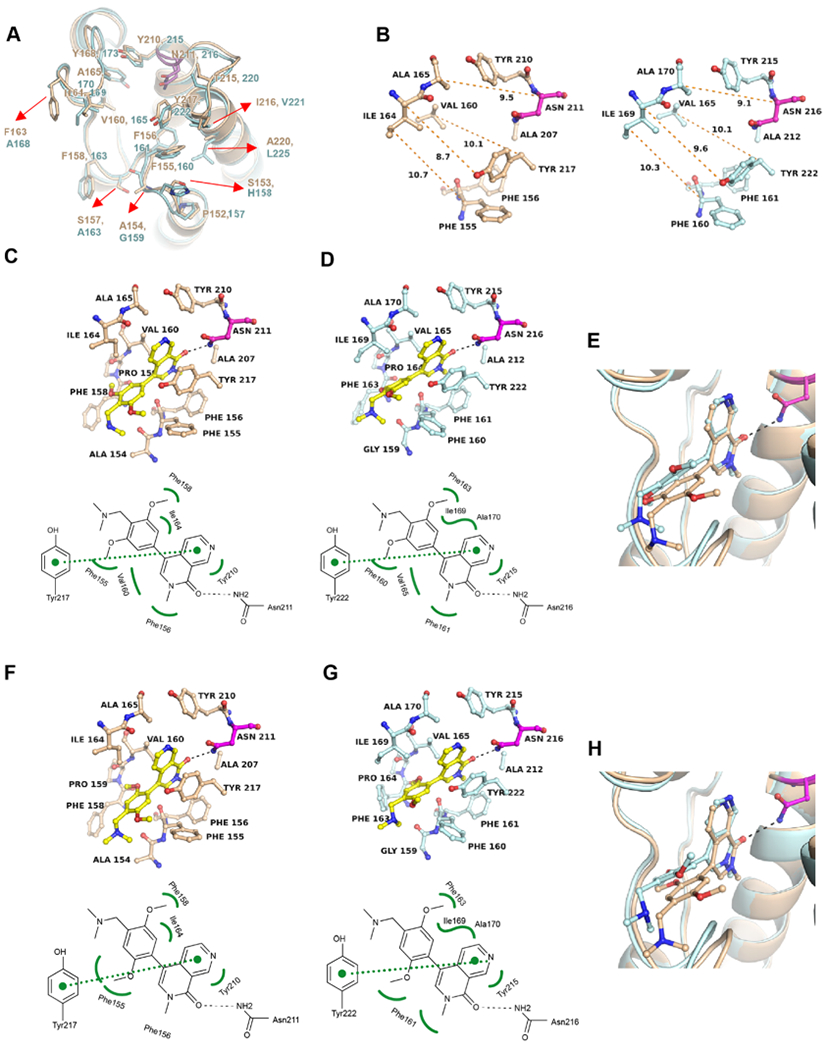
Differences in the overall structures of BRD7 and BRD9 affect the binding poses of BI-7273 and BI-9564. (A) Superposition of the crystal structures of BRD7 (beige) and BRD9 (cyan) in the unliganded state (PDB 6PPA, 6UZF). Differing residues (red arrows) are located outside the KAc site; the critical asparagine residue (BRD7N211 and BRD9N216) is drawn in magenta. (B) Comparison of the unliganded KAc sites of BRD7 and BRD9. Distances between the flanks of the binding site are indicated as orange-dotted lines (in Å). (C) Cocrystal structure of BRD7 with BI-7273 along with a schematic presentation of the binding interactions (PDB 6V1E). (D) Same as (C) for BI-7273 in BRD9 (PDB 5EU1). (E) Superposition of both structures. (F) Cocrystal structure of BRD7 with BI-9564 (PDB 6V1F) and (G) of BRD9 with BI-9564 (PDB 5F1H). (H) Superposition of both structures. BRD7 residues are shown in beige and BRD9 residues in cyan. H-bonding interactions are indicated as black-dotted lines, vdW hydrophobic interactions as green-curved lines and π-stacking interactions as green-dotted lines. Crystallization conditions, data collection and refinement statistics are shown in the Supporting Information Tables S2 and S3. 2Fo – Fc and Fo – Fc omit electron density maps are shown in the Supporting Information Figures S3 and S4.
Cocrystal structures were determined for BRD7 liganded with BI-7273 (Figure 2C) and BI-9564 (Figure 2F) showing the same principle binding mode as previously determined in BRD916 (Figure 2D,G). The naphthyridinone oxygen of both compounds establishes a single H-bond with BRD7N211 or BRD9N216. However, the dimethoxyphenyl moieties of BI-7273 and BI-9564, which establish multiple hydrophobic van der Waals (vdW) interactions with KAc site residues, adopt slightly different positions (Figure 2E,H). These differences appear to be caused by steric hindrance with residue F160 of BRD9. In BRD7, the main- and side-chain conformations of the equivalent residue F155 differ from BRD9 presumably due to alanine versus glycine as preceding residues (BRD7A154 and BRD9G159). In the absence of structural information on BRD7–inhibitor complexes, it was previously suggested that differences in the side-chain conformation of BRD9F163 and the equivalent residue BRD7F158 determine the potency and selectivity of BI-9564.16,18 However, comparison of BRD9 and BRD7 inhibitor complexes suggests that this residue adopts multiple conformations even in different monomers comprising the asymmetric unit. Additionally, the finding that BI-7273 is more potent and selective for BRD9 than BI-9564 (Figure 1C) suggests that the hydrophobic vdW interactions observed between BRD9F163 and BI-9564 are less significant.
Novel cocrystal structures were also obtained for BRD7 with I-BRD9 (Figure 3A), TP-472 (Figure 3D) and bromosporine (Figure 3G) as well as for BRD9 with TP-472 (Figure 3E). Crystal structures of BRD9 liganded with I-BRD9 and bromosporine have been determined previously,17,25 but we included a new data set of the BRD9–IBRD9 complex determined at higher resolution (1.5 Å) for structural comparison with BRD7 (Figure 3B). All three inhibitors showed the same principal interaction pattern but slightly different conformations and positioning in the respective KAc sites (Figure 3C,F,I). These differences were not as pronounced as with BI-7273 and BI-9564 (Figure 2E,F) and likely reflect the decreased selectivity of these compounds for BRD9 over BRD7 particularly of I-BRD9 and bromosporine (Figure 1C). TP-472 undergoes a significant shift toward the critical asparagine in the KAc site of BRD9, likely strengthening H-bonding activity and explaining the observed increased selectivity for BRD9.
Figure 3.
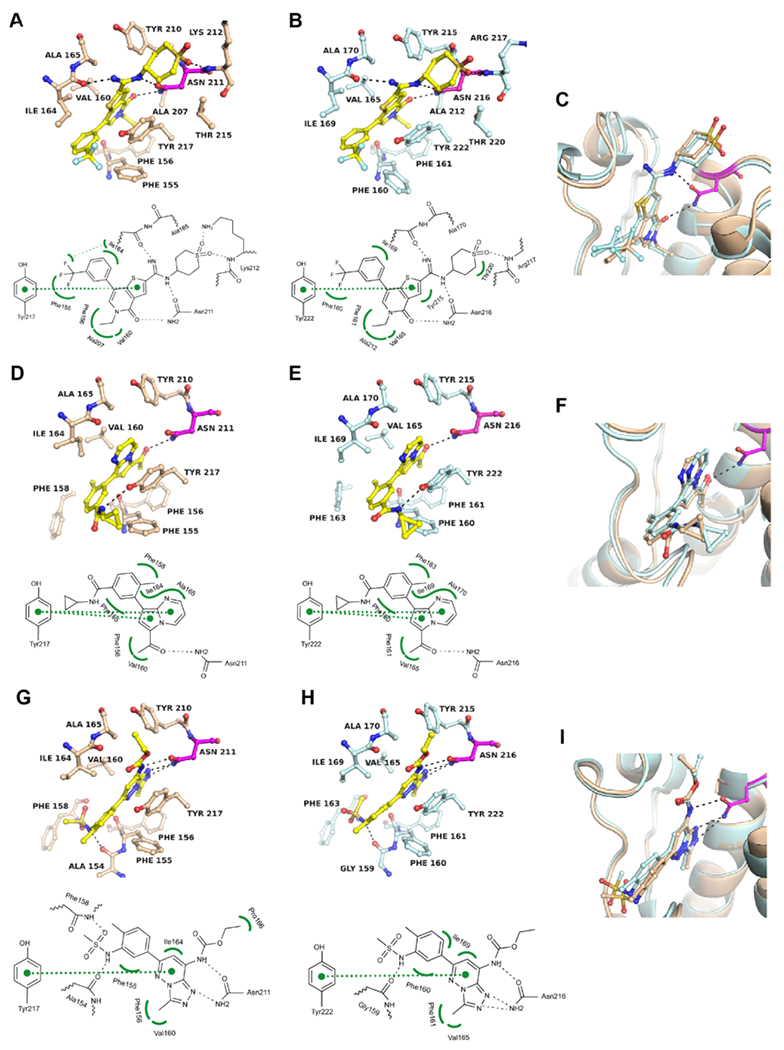
Binding modes of I-BRD9, TP-472, and bromosporine in BRD7 and BRD9. (A) Cocrystal structure of I-BRD9 in BRD7 (PDB 6V17) and (B) in BRD9 (PDB 6V1B). (C) Superposition of both structures. (D–F) Same as (A–C) for TP-472 (PDB 6V16 and 6V14). (G–I) Same as (A–C) for bromosporine (PDB 6V1H and 5IGM). Except for the BRD9-bromosporine complex, all structures were determined experimentally (Supporting Information Table S3). 2Fo – Fc and Fo – Fc electron density maps are shown in Figure S3 and S4.
To further evaluate the SAR of BRD9 inhibitors, we attempted cocrystal structure determination with the first BRD of BRD4, a member of the evolutionary distant BET family of BRD-containing proteins. BI-7273, BI-9564, and TP-472 showed weak binding potential for the first BRD of BRD4 (BRD4–1) by DSF corresponding to Kd values above 10 μM (data not shown). Novel cocrystal structures were determined for BRD4 liganded with these inhibitors showing the same principal interactions of the respective warheads with the critical asparagine side chain in the KAc site (BRD4N140) (Figure 4). However, all three inhibitors undergo significant conformational changes upon binding to BRD4, primarily caused by steric hindrance with the side chain of Trp81, which comprises the WPF shelf characteristic of all BET BRDs. The methoxyphenyl ring of BI-9564 rotates 180° to accommodate the ortho- and metamethoxy groups, while the double metaposition of the methoxy groups in BI-7273 forces the ring to rotate away from Trp81 because of steric hindrance (Figure 4A,B). TP-472 and bromosporine undergo the largest conformational changes upon binding to BRD4 versus BRD7 (Figure 4C,D). The conformational changes of BI-7273, BI-9564, and TP-472 suggest a loss of shape complementarity with the KAc site of BRD4, resulting in a weak binding potential. In contrast, the changed binding pose of bromosporine suggests a gain in shape complementarity, reflected by a substantial increase of binding potential for BRD4-1 (Kd = 42 nM25) over BRD7 (Kd = 390 nM).
Figure 4.
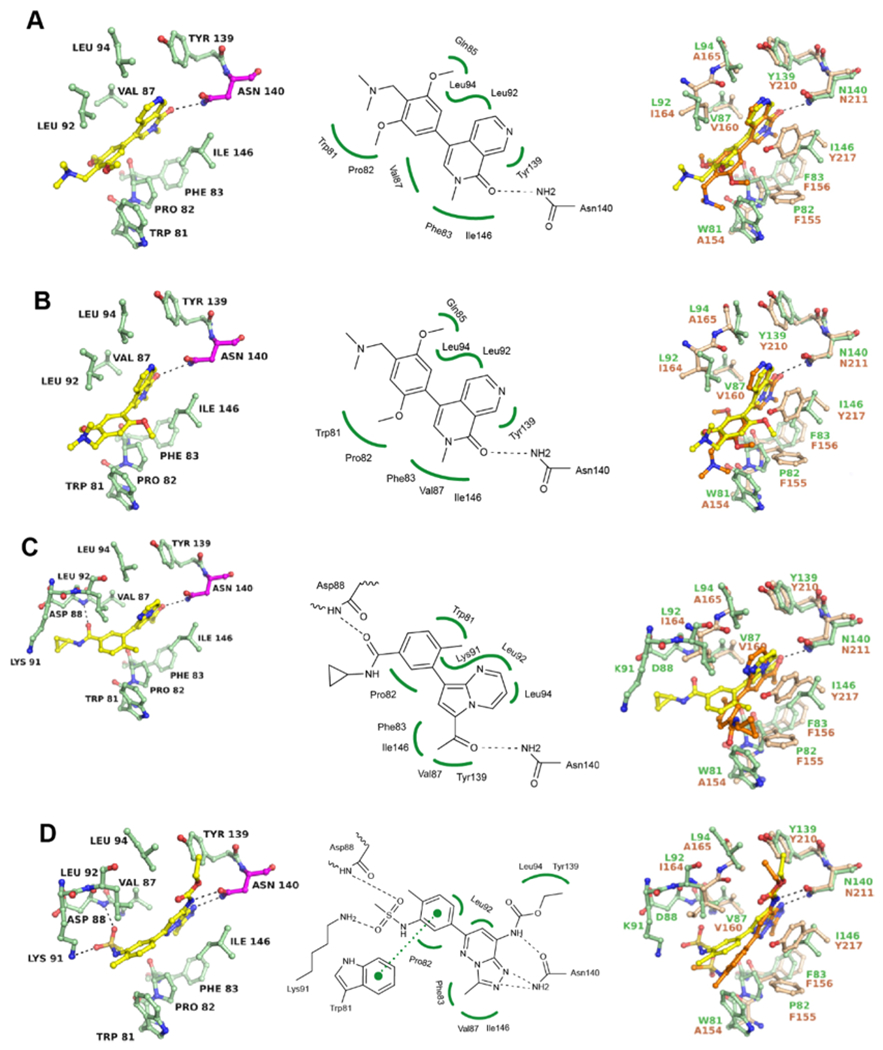
Binding modes of BRD7/9 inhibitors in BRD4. (A) Cocrystal structure of BI-7273 with BRD4-1 (PDB 6V1K) and superposition with the BRD7/BI-7273 complex (PDB 6V1E). (B) Same as (A) for BI-9564 (PDB 6V1L and 6V1F). (C) Same as (A) for TP-472 (PDB 6V1U and 6V16). (D) Same as (A) for bromosporine (PDB 6V0U and 6V1H). All structures were determined experimentally. Residues of BRD4 are shown in green and of BRD7 in beige. Inhibitor liganded with BRD4 is shown in yellow and liganded with BRD7 in orange. 2Fo – Fc and Fo – Fc electron density maps are shown in the Supporting Information Figure S5.
Discovery of Dual BRD7/9-Kinase Inhibitors.
Although BRD9 has an essential role in malignant transformation by modulating the functions of the BAF complex, it is not considered an oncogene. In cancer, altered BAF complexes complement driver oncogenes such as PIK3CA, KRAS, c-Myc, or ATR.8,15,26,27 While BRD9 inhibition alone may be insufficient to elicit robust anticancer activity, the concomitant inhibition of BRD9 and a kinase crucial for the survival of cancer cells may exert synergistic lethal effects. We explored the potential of kinase inhibitors to interact with BRD7 and BRD9 by screening a commercial library of 418 compounds against BRD9 using DSF. Three kinase inhibitors, TG003, a Clk2 inhibitor,21 sunitinib, a multitargeted tyrosine kinase inhibitor,22 and PF-477736, an inhibitor of cell cycle Chk123 were identified as potential hits (Figure 5A). These inhibitors showed small but reproducible temperature shifts against both BRD9 and BRD7 (Figure 5B). The highest temperature shift was 2.1 °C as compared to 13.6 °C for positive control BI-7273, suggesting relatively weak binding potential of kinase inhibitors for BRD9. ITC binding studies established that BRD7 and BRD9 interact with TG003 in the low micromolar range (Kd = 4 and 16 μM, respectively) (Figure 5C). Quantitative polymerase chain reaction (qPCR)-based competition assays confirmed that all three inhibitors bind to BRD7 and BRD9 with Kd values between 5 and 17 μM, except sunitinib which was significantly less active against BRD9 (Figure 5D).
Figure 5.
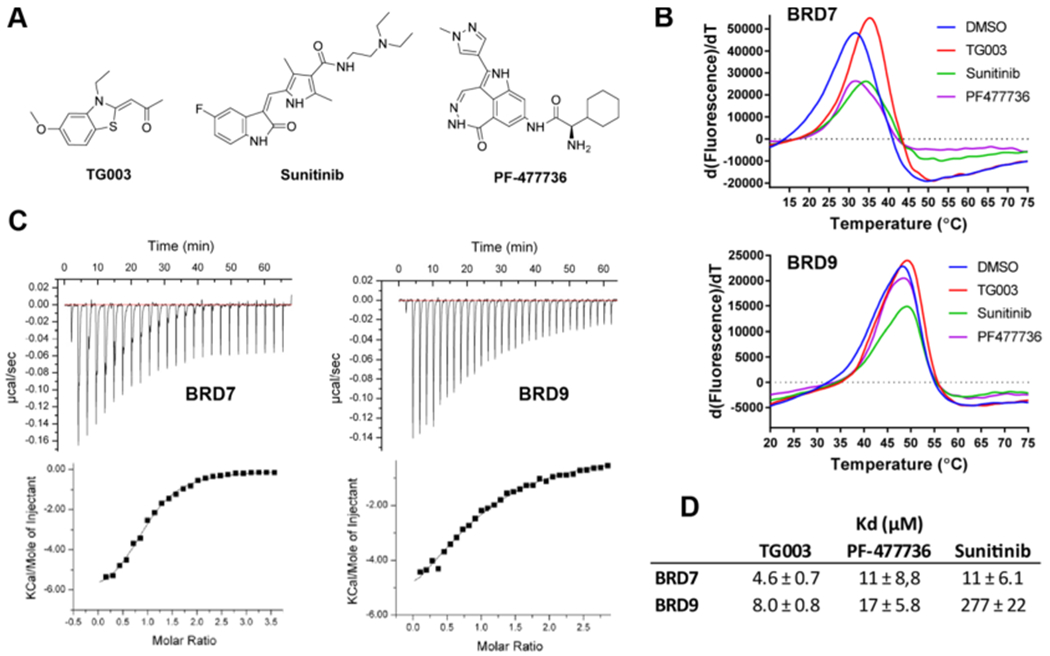
Discovery of BRD7/9-kinase inhibitors. (A) Chemical structures of kinase inhibitors identified as ligands of BRD7 and BRD9. (B) DSF derivative plots for BRD7 and BRD9 in the presence of 100 μM inhibitor. (C) ITC thermograms of TG003 interaction with BRD7 and BRD9. (D) Dissociation constants determined by the qPCR assay (performed by DiscoveRx) (N = 2).
Cocrystal structures of BRD7 and BRD9 liganded with TG003 show canonical H-bonding interactions of the propanone moiety with the conserved asparagine residue (BRD7N211 and BRD9N216) (Figure 6A,B). Several vdW interactions with residues of the flanks hold the inhibitor in place of which π-stacking interactions between the ring system of TG003 and the side chain of BRD7Y217 and BRD9Y222 are most prominent. The only difference between the KAc sites in the unliganded and liganded states is a slight movement of this tyrosine toward the inhibitor. Superposition of the two structures revealed almost identical positioning of the inhibitor in BRD7 and BRD9 (Figure 6C). A cocrystal structure was also obtained for BRD9 liganded with sunitinib, which binds to the KAc site in a noncanonical manner through H-bonding of the pyrrole amine group with the main chain carbonyl oxygen of F160 (Figure 6D). Thus, TG003 and sunitinib show significantly different binding modes in the KAc site of BRD9 (Figure 6E). Inhibitor PF-477736 resisted cocrystallization attempts because of low aqueous solubility.
Figure 6.
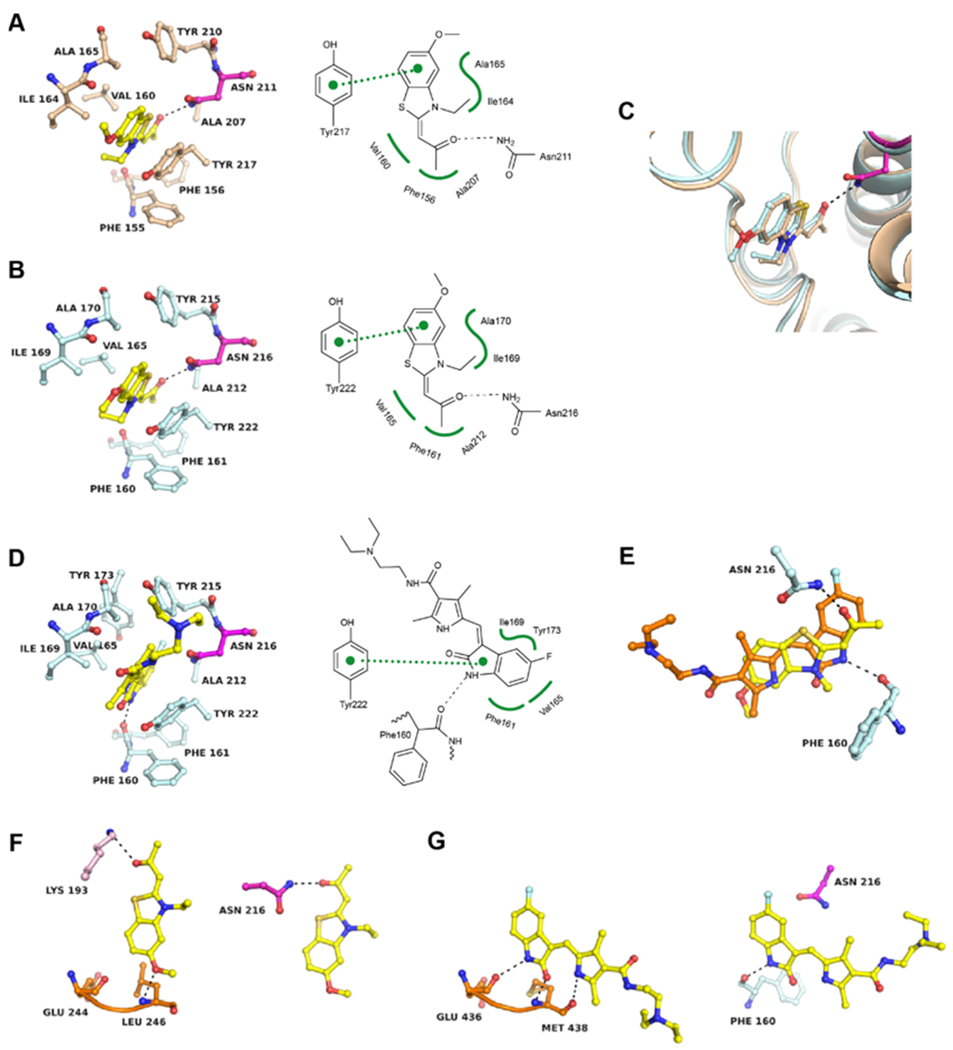
Structural basis of BRD7/9 inhibition by kinase inhibitors. (A) Cocrystal structure of TG003 in BRD7 (PDB 6V0Q) and (B) in BRD9 (PDB 6V0S). (C) Superposition of both structures. (D) Cocrystal structure of BRD9 liganded with sunitinib (PDB 6V0X). (E) Superposition of TG003 (yellow) and sunitinib (orange) bound to the KAc site of BRD9. (F) Comparison of the binding mode of TG003 in the ATP site of CLK2 (PDB 6FYI) (left panel) and the KAc site of BRD9 (right panel). The kinase hinge region is colored in orange and other kinase residues are colored in pink. (G) Comparison of the binding mode of sunitinib in the ATP site of ITK (PDB 3MIY) (left panel) and the KAc site of BRD9 (right panel). 2Fo – Fc and Fo – Fc electron density maps are shown in the Supporting Information Figures S3 and S4.
A comparative analysis of the binding interactions in kinases and BRDs was performed using the available cocrystal structures of TG003 in CLK228 and sunitinib in ITK.29 TG003 and sunitinib are type I inhibitors that bind to the ATP site of kinases through H-bonding interactions with main chain atoms of the hinge region. For TG003, methoxy oxygen is the hinge-binding group in CLK228 while it is solvent exposed in BRD9 (Figure 6F). The propanone group which is critical for binding to the KAc site in BRD7/9 is in H-bonding distance to a lysine residue in CLK2, but this interaction is probably less significant for the kinase inhibitory activity. Thus, the functional groups primarily responsible for binding to the ATP and KAc sites are different and positioned opposite to each other. Substitution of the methoxy group likely renders the TG003 pharmacophore less active against CLK2 while maintaining activity against BRD7/9. Similarly, modifications of the propanone moiety would reduce or eliminate the binding potential for BRD7/9 while maintaining activity against kinases. Sunitinib establishes multiple H-bonding interactions with the hinge region of ITK through its indolinone and pyrrole moieties (Figure 6G).29 As the pyrrole amine also establishes the only H-bond in the KAc site, the simultaneous inhibition of kinases and BRD7/9 is inherent to the sunitinib pharmacophore.
DISCUSSION
As with kinase inhibitors targeting the ATP site, a major challenge in the development of selective BRD inhibitors is the high similarity of the KAc site across BRD-containing proteins. Known BRD9 inhibitors also inhibit the closely related BRD7, an unwanted off-target because of its tumor suppressor properties.10,30,31 Previously, the structure-based design of the reported BRD9 inhibitors relied on the cocrystal structure information of BRD9 but lacked structural details on BRD7. We report the first crystal structures of BRD7 in unliganded and liganded states with inhibitors of five different chemical scaffolds. New cocrystal structures were also determined of BRD9 and BRD4. The data provide a comprehensive picture of the differences in binding interactions of the BRD9 inhibitors within and outside the BRD7/9 subfamily. Combined with biochemical data from direct binding studies, a structural basis for the potency and selectivity of diverse BRD7/9 inhibitors could be established. Although the KAc sites of BRD7 and BRD9 are composed of identical residues, small differences in the overall structures of the BRDs appear to affect the conformation and positioning of inhibitors upon binding. The observed differences in the binding pose are caused by steric hindrance with surrounding, mostly nonpolar residues. These changes may reflect loss or gain in shape complementarity of low energy conformations of an inhibitor with the KAc site, resulting in differential binding potency for BRD7 and BRD9.
Several kinase inhibitors that also inhibit BRDs have been identified for BRD4 and other BET BRDs.32–34 Our discovery of first-in-class dual BRD7/9-kinase inhibitors suggests that the biological activity of kinase inhibitors in development (TG003 and PF-477736) and in clinical use (sunitinib) may in part be caused by concomitant inhibition of BRD7 and BRD9. However, the bromodomain–kinase inhibitors identified in this work are significantly more potent against the respective kinase targets than against BRD7 or BRD9. TG003 inhibits Clk1 and Clk4 with IC50 values of 20 and 15 nM, respectively,21 about 250 times that of BRD7 (Figure 1). Sunitinib inhibits multiple tyrosine kinases with IC50 values below 10 nM,22 about 1,000 times that of BRD7, and PF-477736 inhibits Chk1 with a Ki of 0.49 nM,23 about 20,000 times that of BRD7. In order to exert a meaningful synergistic action of kinase and BRD inhibition in the cell, the binding affinity of these inhibitors for BRD7 and/or BRD9 is insufficient. The structural data of this work provide a new framework for the development of more potent dual kinase-BRD7/9 inhibitors based on the chemical scaffolds identified and beyond. Recent studies on potent BRD9 inhibitors against several cancer cell lines found only weak antiproliferative effects.7,15,16 While inhibition of BRD9 alone may lack efficacy, the simultaneous inhibition of kinases of specific signaling events could be beneficial in anticancer therapy.
EXPERIMENTAL SECTION
Protein Expression and Purification.
The expression plasmid for the BRD9 (Uniprot ID Q9H8M2) BRD (residues 134–239) was from Addgene (plasmid 39012). DNA sequences encoding the human full length BRD7 (Uniprot ID Q9NPI1) were custom synthesized by GeneArt (Thermo Fisher Scientific), and the BRD7 BRD (residues 129–250) were cloned in-frame of a modified pET15b vector providing an N-terminal hexa-histidine tag followed by a tobacco etch virus (TEV) cleavage site. The encoded BRD proteins were expressed in the BL21 (DE3) strain of Escherichia coli.
Plasmids were transformed into E. coli cells and grown at 37 °C in the LB medium (Fisher Scientific) containing carbenicillin (0.1 mg/mL). At OD600 of 0.6, the culture was cooled down to 18 °C and induced with 0.1 mM IPTG. After 18 h growth, the culture was harvested by centrifugation at 6000g for 25 min and stored at −80 °C. Harvested cell pellets were resuspended in 50 mM Na/K phosphate buffer (pH 7.4) containing 100 mM NaCl, 40 mM imidazole, 0.01% w/v lysozyme, and 0.01% v/v Triton X-100 at 4 °C for 1 h, subjected to sonication and the lysate was clarified by centrifugation (30,000g for 45 min at 4 °C). Proteins were purified by FPLC at 4 °C using columns and chromatography materials from GE Healthcare. The lysate was subjected to an immobilized Ni2+ affinity chromatography column equilibrated with 50 mM Na/K phosphate buffer (pH 7.4) containing 100 mM NaCl and 40 mM imidazole using a gradient from 40 to 500 mM of imidazole. Fractions containing the target protein were combined and incubated for 2–16 h with TEV protease at 4 °C, and the cleaved His6-tag was removed by a second Ni2+ affinity column. BRDs were purified to homogeneity by size exclusion chromatography using Superdex 75. The elution buffers were 50 mM HEPES/100 mM NaCl/1 mM DTT (pH 7.5) for BRD7 and 20 mM HEPES/150 mM NaCl/2 mM DTT (pH 7.5) for BRD9. BRDs eluted as monomeric proteins and were of crystallization grade quality (>95% purity as judged by SDS-PAGE). BRD-containing fractions were combined, concentrated to 10–15 mg/mL, and aliquots were flash-frozen in liquid N2 and stored at −80 °C.
Isothermal Titration Calorimetry.
All experiments were conducted using an ITC200 microcalorimeter from Malvern Panalytical (Spectris PLC). BRD7 and BRD9 were buffer exchanged (Supporting Information Table S1) using PD10 columns (GE life sciences) before the experiments and concentrated to 6–10 mg/mL. Experiments were carried out in the ITC buffer while stirring at 750 rpm, either in the normal titration method (BRD7) or reverse titration method (BRD9). The microsyringe (40 μL load volume) was loaded with a solution of the protein sample (150–300 μM protein in ITC buffer for BRD9) or compound sample (150–600 μM compound in ITC buffer for BRD7) and was inserted into the calorimetric cell (0.2 mL cell volume), which was filled with the solution of compounds (10–30 μM in ITC buffer) or proteins (15–50 μM in ITC buffer). All titrations were conducted using an initial control injection of 0.3 μL followed by 30 or 20 identical injections (1.58 or 2 μL per injection) with a duration of 3.16 or 4 s (per injection) and a spacing of 150 s between injections. The ratio of titrants in the titration experiments was optimized to ensure complete saturation of the titrant (protein or compounds) in the cell before the final injection thus facilitating the estimation of the baseline for each injection. The experimental data were corrected by subtracting the heats of dilution determined from independent titrations (the protein into the buffer for BRD9 or the compound into the buffer for BRD7). The collected data were analyzed using MicroCal Origin software provided with the ITC instrument to determine the enthalpies of binding (ΔH) and binding constants (KB) as previously described (Wiseman et al.).35 Thermodynamic parameters were calculated using the equation ΔG = ΔH − TΔS = −RT ln KB, where ΔG, ΔH, and ΔS are the changes in free energy, enthalpy, and entropy of binding, respectively. In all cases a single binding site model was used. Dissociation constants and thermodynamic parameters are shown in Figure 1C,D.
Differential Scanning Fluorimetry.
DSF experiments were carried out in Applied Biosystem QuantStudio 6 Flex (compound library screening) and a StepOnePlus (thermal-shift determination) real-time PCR system (Thermo Fisher Scientific) using sealed 384-well or 96-well format plates, assayed in quadruplicates. To obtain robust fluorescence signals, the assay was optimized regarding concentration of the protein (4 μM for screening, and 4.5 μM for thermal-shift determination) and the fluorescence dye SYPRO Orange (Invitrogen, Thermo Fisher Scientific) (5.5× for screening and 5× for thermal shift determination). For library screening, dilutions of the compound in the assay buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 2 mM DTT, 1% DMSO) were prepared using a Mosquito liquid dispenser (TTP Labtech Ltd). The protein in the assay buffer including the fluorescence dye was mixed with 100 μM compound and 2% DMSO in 20 μL reaction volumes. Reaction mixtures were heated from 25 to 95 °C at 1 °C/min with fluorescence readings every 0.5 °C at 610 nm. The observed thermal shift (ΔTm) was recorded as the difference between the Tm of the sample and DMSO reference wells.
Crystallization and X-ray Crystallography.
All crystallization experiments were performed at 18 °C. Aliquots of purified BRD7 and BRD9 were set up for crystallization using a mosquito crystallization robot (TTP Labtech). Initially, coarse screens were set up using Greiner 3-well plates at three different concentrations of the precipitant to the protein (200 + 400, 300 + 300, 400 + 200 nL) per condition. Conditions producing crystals were further optimized and scaled up for the manual set up of 2 μL drops. For cocrystallization, the compound was either premixed with protein on ice or added to the protein-reservoir solution drops to achieve final concentrations of the 1–2 mM compound and 5–10% DMSO (Supporting Information Table S2). Crystals were cryoprotected using the well solution supplemented with ethylene glycol (15–30%) and flash frozen in liquid nitrogen. X-ray diffraction data were collected at −180 °C at the beamlines 22-ID and 22-BM, SER-CAT, Advanced Photon Source, Argonne National Laboratories, and in the Moffitt Chemical Biology Core using Cu Kα X-rays generated by a Rigaku Micro-Max 007-HF X-ray generator, focused by mirror optics and equipped with a Rigaku CCD Saturn 944 system. Data were reduced and scaled with XDS36 or DIALS37 and Aimless.38 PHASER39 was employed for molecular replacement using the PDB entry 3HME as the search model for both BRD9 and BRD7. Refinement was carried out with PHENIX39 and model building was performed with Coot.40 Initial models for the small-molecule ligands were generated using MarvinSketch (ChemAxon, Cambridge, MA) with ligand restraints from the eLBOW of the PHENIX39 suite. All structures were validated by MolProbity. Figures were prepared using PyMOL (Schrödinger, LLC). Data collection and refinement statistics are shown in Supporting Information Table S3. The coordinates and structure factors have been deposited with the PDB.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Moffitt Chemical Biology Core for use of ITC and protein crystallography instruments (National Cancer Institute grant P30-CA076292) and the Southeast Regional Collaborative Access Team (SER-CAT, University of Georgia) for assistance with Synchrotron data collection. We thank Drs. Gunda I. Georg (University of Minnesota) and Min S. Lee (National Institute for Child Health & Human Development) for funds to initiate this project (grant HHSN275201300017C).
ABBREVIATIONS
- BET
bromodomain and extra-terminal domain protein
- BRD7
bromodomain-containing protein 7
- BRD9
bromodomain-containing protein 9
- Chk1
checkpoint kinase 1
- Clk2
Cdc2-like kinase
- DSF
differential scanning fluorimetry
- ITC
isothermal titration calorimetry
- ITK
IL2-inducible T-cell kinase
- KAc
acetylated lysine
- vdW
van der Waals
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01980.
Molecular formula strings (CSV)
Original ITC data, crystallization conditions for protein—inhibitor complexes, X-ray data collection, refinement statistics, and electron density maps (PDF)
Accession Codes
6PPA (apo), 6V1E (BI-7273), 6V1F (BI-9564), 6V1H (bromosporine), 6V17 (I-BRD9), 6V0Q (TG003) and 6V16 (TP-472) for BRD7. 6UZF (apo), 6V14 (TP-472), 6V0X (sunitinib), 6V1B (I-BRD9) and 6V0S (TG003) for BRD9. 6V1K (BI-7273), 6V1L (BI-9564), 6V0U (bromosporine), and 6V1U (TP-472) for BRD4-1.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.9b01980
The authors declare no competing financial interest.
Contributor Information
Rezaul Md Karim, Department of Drug Discovery, Moffitt Cancer Center, Tampa, Florida 33612, United States; Department of Molecular Medicine, USF Morsani College of Medicine, University of South Florida, Tampa, Florida 33612, United States.
Alice Chan, Department of Drug Discovery, Moffitt Cancer Center, Tampa, Florida 33612, United States.
Jin-Yi Zhu, Department of Drug Discovery, Moffitt Cancer Center, Tampa, Florida 33612, United States.
Ernst Schönbrunn, Department of Drug Discovery, Moffitt Cancer Center, Tampa, Florida 33612, United States; Department of Molecular Medicine, USF Morsani College of Medicine, University of South Florida, Tampa, Florida 33612, United States.
REFERENCES
- (1).Filippakopoulos P; Picaud S; Mangos M; Keates T; Lambert J-P; Barsyte-Lovejoy D; Felletar I; Volkmer R; Muller S; Pawson T; Gingras A-C; Arrowsmith CH; Knapp S Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell 2012, 149, 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Zou Z; Huang B; Wu X; Zhang H; Qi J; Bradner J; Nair S; Chen L-F Brd4 Maintains Constitutively Active Nf-Kappab in Cancer Cells by Binding to Acetylated Rela. Oncogene 2014, 33, 2395–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Fujisawa T; Filippakopoulos P Functions of Bromodomain-Containing Proteins and Their Roles in Homeostasis and Cancer. Nat. Rev. Mol. Cell Biol 2017, 18, 246–262. [DOI] [PubMed] [Google Scholar]
- (4).Xu Y; Vakoc CR Targeting Cancer Cells with Bet Bromodomain Inhibitors. Cold Spring Harbor Perspect. Med 2017, 7, a026674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Cochran AG; Conery AR; Sims RJ 3rd Bromodomains: A New Target Class for Drug Development. Nat. Rev. Drug Discovery 2019, 18, 609–628. [DOI] [PubMed] [Google Scholar]
- (6).Mashtalir N; D’Avino AR; Michel BC; Luo J; Pan J; Otto JE; Zullow HJ; McKenzie ZM; Kubiak RL; St Pierre R; Valencia AM; Poynter SJ; Cassel SH; Ranish JA; Kadoch C Modular Organization and Assembly of Swi/Snf Family Chromatin Remodeling Complexes. Cell 2018, 175, 1272–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Hohmann AF; Martin LJ; Minder JL; Roe J-S; Shi J; Steurer S; Bader G; McConnell D; Pearson M; Gerstberger T; Gottschamel T; Thompson D; Suzuki Y; Koegl M; Vakoc CR Sensitivity and Engineered Resistance of Myeloid Leukemia Cells to Brd9 Inhibition. Nat. Chem. Biol 2016, 12, 672–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Brien GL; Remillard D; Shi J; Hemming ML; Chabon J; Wynne K; Dillon ET; Cagney G; Van Mierlo G; Baltissen MP; Vermeulen M; Qi J; Frohling S; Gray NS; Bradner JE; Vakoc CR; Armstrong SA Targeted Degradation of Brd9 Reverses Oncogenic Gene Expression in Synovial Sarcoma. Elife 2018, 7, No. e41305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Krämer K; Moreno N; Frühwald M; Kerl K Brd9 Inhibition, Alone or in Combination with Cytostatic Compounds as a Therapeutic Approach in Rhabdoid Tumors. Int. J. Mol. Sci 2017, 18, 1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Drost J; Mantovani F; Tocco F; Elkon R; Comel A; Holstege H; Kerkhoven R; Jonkers J; Voorhoeve PM; Agami R; Del Sal G Brd7 Is a Candidate Tumour Suppressor Gene Required for P53 Function. Nat. Cell Biol 2010, 12, 380–389. [DOI] [PubMed] [Google Scholar]
- (11).Liu Z; Yan M; Liang Y; Liu M; Zhang K; Shao D; Jiang R; Li L; Wang C; Nussenzveig DR; Zhang K; Chen S; Zhong C; Mo W; Fontoura BMA; Zhang L Nucleoporin Seh1 Interacts with Olig2/Brd7 to Promote Oligodendrocyte Differentiation and Myelination. Neuron 2019, 102, 587–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wang H; Zhao R; Guo C; Jiang S; Yang J; Xu Y; Liu Y; Fan L; Xiong W; Ma J; Peng S; Zeng Z; Zhou Y; Li X; Li Z; Li X; Schmitt DC; Tan M; Li G; Zhou M Knockout of Brd7 Results in Impaired Spermatogenesis and Male Infertility. Sci. Rep 2016, 6, 21776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kadoch C; Hargreaves DC; Hodges C; Elias L; Ho L; Ranish J; Crabtree GR Proteomic and Bioinformatic Analysis of Mammalian Swi/Snf Complexes Identifies Extensive Roles in Human Malignancy. Nat. Genet 2013, 45, 592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Clark PGK; Vieira LCC; Tallant C; Fedorov O; Singleton DC; Rogers CM; Monteiro OP; Bennett JM; Baronio R; Müller S; Daniels DL; Méndez J; Knapp S; Brennan PE; Dixon DJ Lp99: Discovery and Synthesis of the First Selective Brd7/9 Bromodomain Inhibitor. Angew. Chem., Int. Ed 2015, 54, 6217–6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Crawford TD; Vartanian S; Côté A; Bellon S; Duplessis M; Flynn EM; Hewitt M; Huang H-R; Kiefer JR; Murray J; Nasveschuk CG; Pardo E; Romero FA; Sandy P; Tang Y; Taylor AM; Tsui V; Wang J; Wang S; Zawadzke L; Albrecht BK; Magnuson SR; Cochran AG; Stokoe D Inhibition of Bromodomain-Containing Protein 9 for the Prevention of Epigenetically-Defined Drug Resistance. Bioorg. Med. Chem. Lett 2017, 27, 3534–3541. [DOI] [PubMed] [Google Scholar]
- (16).Martin LJ; Koegl M; Bader G; Cockcroft X-L; Fedorov O; Fiegen D; Gerstberger T; Hofmann MH; Hohmann AF; Kessler D; Knapp S; Knesl P; Kornigg S; Müller S; Nar H; Rogers C; Rumpel K; Schaaf O; Steurer S; Tallant C; Vakoc CR; Zeeb M; Zoephel A; Pearson M; Boehmelt G; McConnell D Structure-Based Design of an in Vivo Active Selective Brd9 Inhibitor. J. Med. Chem 2016, 59, 4462–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Theodoulou NH; Bamborough P; Bannister AJ; Becher I; Bit RA; Che KH; Chung C-W; Dittmann A; Drewes G; Drewry DH; Gordon L; Grandi P; Leveridge M; Lindon M; Michon A-M; Molnar J; Robson SC; Tomkinson NCO; Kouzarides T; Prinjha RK; Humphreys PG Discovery of I-Brd9, a Selective Cell Active Chemical Probe for Bromodomain Containing Protein 9 Inhibition. J. Med. Chem 2016, 59, 1425–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Karim RM; Schönbrunn E An Advanced Tool to Interrogate Brd9. J. Med. Chem 2016, 59, 4459–4461. [DOI] [PubMed] [Google Scholar]
- (19).Remillard D; Buckley DL; Paulk J; Brien GL; Sonnett M; Seo H-S; Dastjerdi S; Wühr M; Dhe-Paganon S; Armstrong SA; Bradner JE Degradation of the Baf Complex Factor Brd9 by Heterobifunctional Ligands. Angew. Chem., Int. Ed 2017, 56, 5738–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zoppi V; Hughes SJ; Maniaci C; Testa A; Gmaschitz T; Wieshofer C; Koegl M; Riching KM; Daniels DL; Spallarossa A; Ciulli A Iterative Design and Optimization of Initially Inactive Proteolysis Targeting Chimeras (Protacs) Identify Vz185 as a Potent, Fast, and Selective Von Hippel-Lindau (Vhl) Based Dual Degrader Probe of Brd9 and Brd7. J. Med. Chem 2019, 62, 699–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Muraki M; Ohkawara B; Hosoya T; Onogi H; Koizumi J; Koizumi T; Sumi K; Yomoda J-I; Murray MV; Kimura H; Furuichi K; Shibuya H; Krainer AR; Suzuki M; Hagiwara M Manipulation of Alternative Splicing by a Newly Developed Inhibitor of Clks. J. Biol. Chem 2004, 279, 24246–24254. [DOI] [PubMed] [Google Scholar]
- (22).Sun L; Liang C; Shirazian S; Zhou Y; Miller T; Cui J; Fukuda JY; Chu J-Y; Nematalla A; Wang X; Chen H; Sistla A; Luu TC; Tang F; Wei J; Tang C Discovery of 5-[5-Fluoro-2-Oxo-1,2- Dihydroindol-(3z)-Ylidenemethyl]-2,4- Dimethyl-1h-Pyrrole-3-Carboxylic Acid (2-Diethylaminoethyl)Amide, a Novel Tyrosine Kinase Inhibitor Targeting Vascular Endothelial and Platelet-Derived Growth Factor Receptor Tyrosine Kinase. J. Med. Chem 2003, 46, 1116–1119. [DOI] [PubMed] [Google Scholar]
- (23).Blasina A; Hallin J; Chen E; Arango ME; Kraynov E; Register J; Grant S; Ninkovic S; Chen P; Nichols T; O’Connor P; Anderes K Breaching the DNA Damage Checkpoint Via Pf-00477736, a Novel Small-Molecule Inhibitor of Checkpoint Kinase 1. Mol. Cancer Ther 2008, 7, 2394–2404. [DOI] [PubMed] [Google Scholar]
- (24).Sun H; Liu J; Zhang J; Shen W; Huang H; Xu C; Dai H; Wu J; Shi Y Solution Structure of Brd7 Bromodomain and Its Interaction with Acetylated Peptides from Histone H3 and H4. Biochem. Biophys. Res. Commun 2007, 358, 435–441. [DOI] [PubMed] [Google Scholar]
- (25).Picaud S; Leonards K; Lambert J-P; Dovey O; Wells C; Fedorov O; Monteiro O; Fujisawa T; Wang C-Y; Lingard H; Tallant C; Nikbin N; Guetzoyan L; Ingham R; Ley SV; Brennan P; Muller S; Samsonova A; Gingras A-C; Schwaller J; Vassiliou G; Knapp S; Filippakopoulos P Promiscuous Targeting of Bromodomains by Bromosporine Identifies Bet Proteins as Master Regulators of Primary Transcription Response in Leukemia. Sci. Adv 2016, 2, No. e1600760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Chory EJ; Kirkland JG; Chang C-Y; D’Andrea VD; Gourinsankar S; Dykhuizen EC; Crabtree GR Inhibition of a Selective Swi/Snf Function Synergizes with Atr Inhibitors in Cancer Cell Killing. 2019, bioRxiv 660456. [Google Scholar]
- (27).Bell CM; Raffeiner P; Hart JR; Vogt PK Pik3ca Cooperates with Kras to Promote Myc Activity and Tumorigenesis Via the Bromodomain Protein Brd9. Cancers 2019, 11, 1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kallen J; Bergsdorf C; Arnaud B; Bernhard M; Brichet M; Cobos-Correa A; Elhajouji A; Freuler F; Galimberti I; Guibourdenche C; Haenni S; Holzinger S; Hunziker J; Izaac A; Kaufmann M; Leder L; Martus H-J; von Matt P; Polyakov V ; Roethlisberger P; Roma G; Stiefl N; Uteng M; Lerchner A X-Ray Structures and Feasibility Assessment of Clk2 Inhibitors for Phelan-Mcdermid Syndrome. ChemMedChem 2018, 13, 1997–2007. [DOI] [PubMed] [Google Scholar]
- (29).Kutach AK; Villasenor AG; Lam D; Belunis C; Janson C; Lok S; Hong L-N; Liu C-M; Deval J; Novak TJ; Barnett JW ; Chu W; Shaw D; Kuglstatter A Crystal Structures of Il-2-Inducible T Cell Kinase Complexed with Inhibitors: Insights into Rational Drug Design and Activity Regulation. Chem. Biol. Drug Des 2010, 76, 154–163. [DOI] [PubMed] [Google Scholar]
- (30).Chen CL; Wang Y; Pan QZ; Tang Y; Wang QJ; Pan K; Huang LX; He J; Zhao JJ; Jiang SS; Zhang XF; Zhang HX; Zhou ZQ; Weng de S; Xia JC Bromodomain-Containing Protein 7 (Brd7) as a Potential Tumor Suppressor in Hepatocellular Carcinoma. Oncotarget 2016, 7, 16248–16261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Park Y-A; Lee J-W; Kim H-S; Lee Y-Y; Kim T-J; Choi CH; Choi J-J; Jeon H-K; Cho YJ; Ryu JY; Kim B-G; Bae D-S . Tumor Suppressive Effects of Bromodomain-Containing Protein 7 (Brd7) in Epithelial Ovarian Carcinoma. Clin. Cancer Res 2014, 20, 565–575. [DOI] [PubMed] [Google Scholar]
- (32).Ciceri P; Müller S; O’Mahony A; Fedorov O; Filippakopoulos P; Hunt JP; Lasater EA; Pallares G; Picaud S; Wells C; Martin S; Wodicka LM; Shah NP; Treiber DK; Knapp S Dual Kinase-Bromodomain Inhibitors for Rationally Designed Polypharmacology. Nat. Chem. Biol 2014, 10, 305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Ember SWJ; Zhu J-Y; Olesen SH; Martin MP; Becker A; Berndt N; Georg GI; Schönbrunn E Acetyl-Lysine Binding Site of Bromodomain-Containing Protein 4 (Brd4) Interacts with Diverse Kinase Inhibitors. ACS Chem. Biol 2014, 9, 1160–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Carlino L; Rastelli G Dual Kinase-Bromodomain Inhibitors in Anticancer Drug Discovery: A Structural and Pharmacological Perspective. J. Med. Chem 2016, 59, 9305–9320. [DOI] [PubMed] [Google Scholar]
- (35).Wiseman T; Williston S; Brandts JF; Lin L-N Rapid Measurement of Binding Constants and Heats of Binding Using a New Titration Calorimeter. Anal. Biochem 1989, 179, 131–137. [DOI] [PubMed] [Google Scholar]
- (36).Kabsch W Integration, Scaling, Space-Group Assignment and Post-Refinement. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Winter G; Waterman DG; Parkhurst JM; Brewster AS; Gildea RJ; Gerstel M; Fuentes-Montero L; Vollmar M; Michels-Clark T; Young ID; Sauter NK; Evans G Dials: Implementation and Evaluation of a New Integration Package. Acta Crystallogr., Sect. D: Struct. Biol 2018, 74, 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Evans PR; Murshudov GN How Good Are My Data and What Is the Resolution? Acta Crystallogr., Sect. D: Biol. Crystallogr 2013, 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Afonine PV; Grosse-Kunstleve RW; Chen VB; Headd JJ; Moriarty NW; Richardson JS; Richardson DC; Urzhumtsev A; Zwart PH; Adams PD Phenix.Model_Vs_Data: A High-Level Tool for the Calculation of Crystallographic Model and Data Statistics. J. Appl. Crystallogr 2010, 43, 669–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and Development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.