

Summary
Background
Cemiplimab has shown substantial antitumour activity in patients with metastatic cutaneous squamous cell carcinoma. Patients with locally advanced cutaneous squamous cell carcinoma have poor prognosis with conventional systemic therapy. We present a primary analysis of the safety and antitumour activity of cemiplimab in patients with locally advanced cutaneous squamous cell carcinoma.
Methods
This pivotal open-label, phase 2, single-arm trial was done across 25 outpatient clinics, primarily at academic medical centres, in Australia, Germany, and the USA. Eligible patients (aged ≥18 years with histologically confirmed locally advanced cutaneous squamous cell carcinoma and an Eastern Cooperative Oncology Group performance status of 0–1) received cemiplimab 3 mg/kg intravenously over 30 min every 2 weeks for up to 96 weeks. Tumour measurements were done every 8 weeks. The primary endpoint was objective response, defined as the proportion of patients with complete or partial response, according to independent central review as per Response Evaluation Criteria in Solid Tumors version 1.1 for radiological scans and WHO criteria for medical photography. Data cutoff was Oct 10, 2018, when the fully enrolled cohort reached the prespecified timepoint for the primary analysis. Analyses were done as per the intention-to-treat principle. The safety analysis comprised all patients who received at least one dose of cemiplimab. This study is registered with ClinicalTrials.gov, number NCT02760498.
Findings
Between June 14, 2016, and April 25, 2018, 78 patients were enrolled and treated with cemiplimab. The median duration of study follow-up was 9·3 months (IQR 5·1–15·7) at the time of data cutoff. An objective response was observed in 34 (44%; 95% CI 32–55) of 78 patients. The best overall response was ten (13%) patients with a complete response and 24 (31%) with a partial response. Grade 3–4 treatment-emergent adverse events occurred in 34 (44%) of 78 patients; the most common were hypertension in six (8%) patients and pneumonia in four (5%). Serious treatment-emergent adverse events occurred in 23 (29%) of 78 patients. One treatment-related death was reported that occurred after onset of aspiration pneumonia.
Interpretation
Cemiplimab showed antitumour activity and an acceptable safety profile in patients with locally advanced cutaneous squamous cell carcinoma for whom there was no widely accepted standard of care.
Funding
Regeneron Pharmaceuticals and Sanofi.
Introduction
Advanced cutaneous squamous cell carcinoma comprises locally advanced and metastatic cutaneous squamous cell carcinoma not amenable to curative surgery or curative radiotherapy, or both. Patients with advanced cutaneous squamous cell carcinoma might respond to cytotoxic chemotherapy or epidermal growth factor receptor (EGFR) inhibitors.1–3 However, durable responses are uncommon with these treatments.4 In prospective studies of EGFR inhibition with antibodies or small molecules in patients with advanced cutaneous squamous cell carcinoma, an objective response was reported in 10–31% of patients and median overall survival times were 11–13 months.1,5–7 A phase 2 study of cetuximab reported an objective response of 28% and mean overall survival of 8·1 months.2
The low efficacy with cytotoxic chemotherapy or EGFR inhibitors has recently been confirmed in a retrospective study of 32 patients with advanced cutaneous squamous cell carcinoma by the Dermatologic Cooperative Oncology Group (DeCOG) of Germany and Austria.4 Similarly, a retrospective analysis of 82 patients with advanced cutaneous squamous cell carcinoma treated with conventional systemic therapy reported a median overall survival of 15 months from the start of first-line therapy.8 Therefore, advanced cutaneous squamous cell carcinoma is a life-threatening condition for patients who are treated with cytotoxic chemotherapy or EGFR inhibitors, and it is associated with substantial morbidity, impact on quality of life, and health-care burden.8–10 Patients older than 65 years are more likely than younger patients to require dose reductions in the first cycle of chemotherapy, underscoring the need for new treatment approaches for patients with advanced cutaneous squamous cell carcinoma, a predominantly elderly population.11,12
Because of ultraviolet-mediated mutagenesis, the median tumour mutational burden of cutaneous squamous cell carcinoma is approximately 45 mutations per megabase, three times higher than that of skin melanoma.13 High tumour mutational burden has been associated with efficacy of programmed cell death 1 (PD-1) checkpoint inhibition in various advanced solid tumour types.14 Additionally, the strong link between immunosuppression and risk of cutaneous squamous cell carcinoma15 indicates that natural immuno surveillance has an unusually strong role in controlling this tumour type, suggesting that approaches to enhance antitumour immune responses could be efficacious in treating this disease.
Cemiplimab is a high-affinity, highly potent, human, hinge-stabilised IgG4 monoclonal antibody to the PD-1 receptor.16 The primary analysis of the metastatic cutaneous squamous cell carcinoma cohort (group 1) of the phase 2 study of cemiplimab showed an objective response in 47% of patients as per independent central review, with emerging evidence of durable response and disease control.17 Here, we report the clinical activity of cemiplimab from the primary analysis and biomarker evaluation of patients with locally advanced cutaneous squamous cell carcinoma (group 2) from the phase 2 study (NCT02760498).
Methods
Study design and participants
Patients with locally advanced cutaneous squamous cell carcinoma (without nodal or distant metastasis) were enrolled in group 2 of the pivotal open-label, phase 2, single-arm trial assessing the clinical activity and safety of cemiplimab in patients with advanced cutaneous squamous cell carcinoma across 25 outpatient clinics in Australia, Germany, and the USA (appendix p 2). Eligible patients were aged 18 years or older, had histologically confirmed locally advanced cutaneous squamous cell carcinoma, Eastern Cooperative Oncology Group performance status of 0 or 1, adequate organ function, and at least one lesion measurable by digital medical photography according to modified WHO criteria or by radiological scans according to Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST 1.1).18
Clinical notes from the surgeon, radiation oncologist, or multidisciplinary tumour board were required for documentation that patients were not candidates for surgery or radiotherapy. Acceptable reasons for surgery to be considered inappropriate were cutaneous squamous cell carcinoma with substantial local invasion that precluded complete resection; cutaneous squamous cell carcinoma that was technically amenable to surgery, but clinically inappropriate because of the lesion being in an anatomically challenging location for which surgery might result in severe disfigurement or dysfunction; cutaneous squamous cell carcinoma that was technically amenable to surgery, but clinically inappropriate because of the lesion being in the same location after two or more surgical procedures and with curative resection deemed unlikely; or other conditions deemed contraindicating for surgery. Acceptable reasons for radiotherapy to be considered inappropriate were previous radiotherapy with further radiotherapy exceeding the threshold of an acceptable cumulative dose; judgment of the radiation oncologist that the tumour was unlikely to respond to radiotherapy; or a risk–benefit assessment that radio therapy was contraindicated for the patient.
During the screening period, baseline imaging requirements were standardised digital medical photography of externally visible cutaneous squamous cell carcinoma lesions and radiological imaging of all target lesions within 28 days of enrolment. A CT scan of the chest and MRI of the brain were also required during screening. Baseline laboratory tests to ascertain eligibility included complete blood cell count and comprehensive metabolic panel with serum creatinine, blood urea nitrogen, aspartate amino transferase, alanine aminotransferase, and total bilirubin (further details are in the protocol; appendix p 11–128).
Key exclusion criteria were ongoing or recent (within 5 years) autoimmune disease that required systemic immunosuppressive therapy, previous treatment with anti-PD-1 or anti-PD-L1 therapy, previous solid organ transplantation, or concurrent malignancies other than cutaneous squamous cell carcinoma (except for tumours with negligible risk of metastases or death such as adequately treated basal cell carcinoma of the skin, carcinoma in situ of cervix, ductal carcinoma in situ of the breast, or low-risk early-stage prostate adeno-carcinoma for which the management plan is active surveillance). Patients with haematological malignancies were not eligible.
The protocol and all amendments were approved by the institutional review boards at participating study sites. The study was done in accordance with the principles of the Declaration of Helsinki and the International Conference on Harmonisation Good Clinical Practice guidelines and overseen by a steering committee. All patients provided written, informed consent before enrolment.
Relevant protocol amendments included addition of interim analysis of data from patients with locally advanced cutaneous squamous cell carcinoma in response to health authority guidance (amendment date Sept 25, 2017) and provision of further justification for excluding patients with regional nodal metastasis from the locally advanced cutaneous squamous cell carcinoma group, who were instead enrolled in the metastatic cutaneous squamous cell carcinoma group (amendment date Jan 26, 2015). The full protocol amendment history is provided in the protocol.
Procedures
Patients received cemiplimab 3 mg/kg intravenously over 30 min every 2 weeks for up to 96 weeks. Tumour biopsy samples were taken at baseline and at day 29. Tumour response assessments were done every 8 weeks. Patients were recommended to undergo both digital medical photography and radiological imaging at each response assessment. In cases in which it was the opinion of the investigator that no substantial added information (beyond that provided by digital medical photography) was provided by baseline radio logical imaging of the lesion, the use of digital medical photography only (without radiological imaging) at post-baseline tumour assessments was allowed (or vice versa). All responses were confirmed with imaging at least 4 weeks after criteria for response were initially met; unconfirmed responses were considered to be stable disease for best overall response assessment. To establish complete response, biopsy of at least one site of a known externally visible target lesion was required to document the absence of residual malignant cells, otherwise the best response that could be assigned was partial response even with no externally visible cutaneous squamous cell carcinoma lesions on photo graphs. Treatment beyond progression was allowed if the patient tolerated treatment, after approval by the medical monitor.
Patients were allowed to withdraw consent from study participation at any time for any reason or at the discretion of the investigator if continued participation could put the patient at risk or if it was deemed in the best interest of the patient. Additionally, if patients developed a symptomatic lesion during the study for which palliative radiation was deemed appropriate by the investigator, these patients were removed from the study.
Treatment interruptions were allowed, following agreement between the investigator and study sponsor, if a patient had grade 3 or higher treatment-related adverse events, according to investigator assessment. Patients were considered for resumption of study treatment, at the discretion of the investigator, once the treatment-related adverse event resolved to grade 1 or baseline. Dose reductions were allowed. However, patients who required dose reduction below cemiplimab 0·3 mg/kg every 2 weeks were removed from the study.
All patients who received at least one dose of cemiplimab were assessed for safety. Assessments included reporting of treatment-emergent adverse events, laboratory monitoring, electrocardiograms, and measurement of vital signs. Safety assessment was done continuously from initiation of study treatment until 105 days after the last study treatment. Laboratory tests for blood chemistry and haematology were done on the same day as the administration of each study treatment and at the end of the study (30 days after the last dose of cemiplimab; see protocol for further details). The severity of treatment-emergent adverse events was graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.03).
The level of PD-L 1 expression was assessed by the PD-L1 immunohistochemistry 22C3 assay (Dako, Agilent, Santa Clara, CA, USA) in formalin-fixed paraffin embedded biopsy samples obtained before cemiplimab therapy. Expression level was quantified as the percentage of tumour cells with detectable PD-L1 membrane staining (tumour proportion score [TPS]). Tumour mutational burden was estimated in the DNA samples extracted from the formalin-fixed paraffin embedded tumour biopsies with the analytically validated TruSight Oncology 500 (Illumina, San Diego, CA, USA) to detect single nucleotide variants, insertions and deletions, copy number alterations in 500 genes, and a selected set of gene rearrangements. Tumour mutational burden was calculated as the total number of somatic single nucleotide variants and insertions and deletions in the coding regions of targeted genes per megabase of analysed genomic sequence. All somatic mutations were filtered to exclude the germline and oncogenic driver gene variants, according to the public database comparisons. The assay protocol included the addition of unique molecular identifier nucleotide barcodes during the sequencing library generation. The detection of unique molecular identifiers is used to identify sequence reads from complementary DNA strands in order to reduce the effect of formalin-fixed paraffin embedded DNA deamination artefacts on mutational variant calling.
Major deviations from the study protocol included enrolment error to the wrong treatment group, enrolment despite inclusion criteria not being met, failure to adhere to a procedure, and failure to report serious adverse events or adverse events of special interest within 24 h (appendix p 3).
Outcomes
The primary endpoint was objective response, defined as the proportion of patients with best overall response of complete or partial response, as assessed by independent central review. The composite independent central review committee was provided with the results of scan reviews by an independent radiological review committee as per RECIST 1.1 criteria18 and medical photography reviews by an independent photographic review committee as per WHO criteria.19 These assessments were integrated for the overall (composite) response as per prespecified criteria (appendix p 4). Secondary endpoints included objective response by investigator assessment; duration of response (by independent central review and investigator assessment), defined as the time between first measurement of complete or partial response and the first date of recurrent or progressive disease or death; progression-free survival (by independent central review and investigator assessment), defined as the time between start of treatment and the first date of recurrent or progressive disease or death from any cause; overall survival, defined as the time between the start of treatment and death from any cause; the proportion of patients achieving a best response of complete response after tumour biopsy confirmation; and safety and tolerability. Additional secondary outcomes were pharmacokinetics and immunogenicity, and patient-reported quality of life; analyses of these outcomes from the entire phase 2 study will be published when the analyses are completed. Protocol-defined exploratory outcomes included associations between PD-L1 immunohistochemistry and clinical activity of cemiplimab and between tumour mutational burden and clinical activity of cemiplimab.
Statistical analysis
The results are presented according to the intention-to-treat principle. According to the statistical plan, the prespecified timepoint for the primary efficacy analysis was approximately 6 months after the last patient started treatment with cemiplimab. The data cutoff date was Oct 10, 2018. The primary efficacy analysis was based on a single-stage exact binomial design, with a null hypothesis that an objective response would be observed in 25% of patients. We estimated that a sample size of 72 patients would give the study 90% power to reject the null hypothesis at a two-sided significance level of 5% if the true objective response was 44%. To account for premature withdrawal of patients from the study, we increased the sample size by 5%; therefore, the total planned sample size was 76 patients. The null hypothesis (objective response in 25% of patients) would be excluded with the lower limit of the 95% CI if an objective response was observed in 36% or more patients.
The proportion of patients achieving an objective response by investigator assessment was analysed in the same way as the primary efficacy endpoint (objective response by independent central review). Duration of response, progression-free survival, and overall survival were estimated by the Kaplan-Meier method. For duration of response, patients with complete or partial response without disease progression were censored at the time of their last valid tumour assessment. For progression-free survival, patients without disease recurrence or progression were censored at the time of their last valid tumour assessment. For overall survival, patients without a survival event were censored at the time of last known survival. The proportion of patients achieving a best response of complete response and safety analyses were summarised by descriptive statistics. Associations between PD-L1 status assessment at baseline and clinical activity of cemiplimab were summarised by descriptive statistics. Tumour mutational burden was summarised by descriptive statistics and displayed by box and scatter plots according to clinical activity of cemiplimab.
We also examined durable disease control, defined as the proportion of patients with response or stable disease for at least 105 days, in a post-hoc analysis. Additionally, as a post-hoc analysis, we examined clinical activity (objective response and disease control) by aforementioned reasons patients were not considered as candidates for curative surgery. The analysis of the proportion of patients achieving an objective response by these subgroups was done as per the primary efficacy endpoint. Disease control was defined as the proportion of patients with best overall response of complete response, partial response, or stable disease at the first response assessment, and was summarised descriptively.
An interim analysis was done when patients had been enrolled on the study for 9 months to assess the risk–benefit of cemiplimab in this patient group.
Statistical analyses were done with SAS (version 9.4).
This study is registered with ClinicalTrials.gov, (NCT02760498).
Role of the funding source
The study sponsors, in collaboration with the authors, were involved in the design of the study, data collection, data interpretation, data analysis, critical review and revision of drafts, and the decision to submit the Article for publication. The first draft of the manuscript was prepared by a medical writer funded by the study sponsors. The first draft was based on comments and revisions by authors on the manuscript outline, which was also prepared by the medical writer. The corresponding author had full access to all the data from the study and had final responsibility for the decision to submit for publication.
Results
Between June 14, 2016, and April 25, 2018, 78 patients with locally advanced cutaneous squamous cell carcinoma were enrolled, treated with cemiplimab, and included in the present analysis. Baseline characteristics, including reasons why patients were not considered candidates for surgery or radiotherapy according to the investigators, are shown in table 1. Data on the number of patients who were on treatment or off treatment (with reasons) as of the data cutoff are shown in the appendix (p 5). Data on exposure to cemiplimab are also shown in the appendix (p 6). At the time of data cutoff, the median duration of study follow-up was 9·3 months (IQR 5·1–15·7).
Table 1:
Patient demographics and baseline characteristics
| Patients (n=78) | |
|---|---|
| Median age, years (IQR) | 74 (65–81) |
| ≥65 years | 59 (76%) |
| Sex | |
| Female | 19 (24%) |
| Male | 59 (76%) |
| ECOG performance status | |
| 0 | 38 (49%) |
| 1 | 40 (51%) |
| Primary cutaneous squamous cell carcinoma site | |
| Head or neck* | 62 (79%) |
| Arm or leg | 14 (18%) |
| Trunk | 2 (3%) |
| Previous cancer-related systemic therapy† | 12 (15%) |
| Previous cancer-related radiotherapy | 43 (55%) |
| Reasons why patients were not considered candidates for surgery | |
| Lesion with substantial local invasion that precluded complete resection | 20 (26%) |
| Lesion in an anatomically challenging location for which surgery might result in severe disfigurement or dysfunction | 30 (38%) |
| Lesion in the same location after two or more surgical procedures and with curative resection deemed unlikely | 25 (32%) |
| Other conditions deemed contraindicating for surgery | 3 (4%) |
| Reasons why patients were not considered candidates for radiotherapy | |
| Previous radiotherapy with further radiotherapy exceeding the threshold of an acceptable cumulative dose | 10 (13%) |
| Judgment of the radiation oncologist that the tumour was unlikely to respond to radiotherapy | 17 (22%) |
| Risk-benefit assessment that radiotherapy was contraindicated for the patient | 38 (49%) |
| Other conditions deemed contraindicating for radiotherapy | 11 (14%) |
| Missing | 2 (3%) |
| Histological differentiation of tumour‡ | |
| Well differentiated | 26 (33%) |
| Moderately differentiated | 24 (31%) |
| Poorly differentiated | 22 (28%) |
| Undifferentiated | 1 (1%) |
| Unknown | 5 (6%) |
Data are n (%) unless otherwise specified. ECOG=European Cooperative Oncology Group.
Includes one patient with nodal metastasis who was incorrectly enrolled in the locally advanced group 2 (instead of a metastatic group) because of a protocol violation. Data for this patient were analysed in group 2 as per the intention-to-treat principle.
Ten patients had received one previous cancer-related systemic therapy and two had previously received two or more cancer-related systemic therapies.
Based on local pathology assessment of pre-treatment tumours.
By independent central review, an objective response was observed in 34 (44%; 95% CI 32–55) of 78 patients; best overall response was complete response in ten (13%) patients and partial response in 24 (31%). Table 2 summarises tumour response data. By investigator assessment, an objective response was observed in 41 (53%; 95% CI 41–64) of 78 patients; 13 patients had a complete response and 28 patients had a partial response.
Table 2:
Tumour response assessment as per independent central review
| Patients (n=78) | |
|---|---|
| Objective response* | 34 (44%; 32–55) |
| Best overall response | |
| Complete response | 10 (13%) |
| Partial response | 24 (31%) |
| Stable disease | 28 (36%) |
| Progressive disease | 9 (12%) |
| Not evaluable† | 7 (9%) |
| Disease control‡ | 62 (79%; 69–88) |
| Durable disease control‡ | 49 (63%; 51–74) |
| Median observed time to response, months (IQR)§ | 1.9 (1.9-3.7) |
| Median duration of response | Not reached |
| Observed duration of response ≥6 months§ | 23 (68%) |
| Estimated proportion of patients who remained in response at 12 months§ | 87.8% (66.7-95.9) |
Data are % (95% CI) or n (%), unless otherwise specified.
Not included among the responders are two patients who had progressive disease at initial response assessments as per independent central review, followed by subsequent responses (one partial response and one complete response, both ongoing at time of data cutoff).
Includes patients who did not undergo imaging studies after initiation of therapy or had imaging studies that were not evaluable.
Disease control was defined as the proportion of patients with best overall response of complete response, partial response, or stable disease at the first response assessment (after approximately 56 days on treatment) and durable disease control defined as the proportion of patients with best overall response of complete response, partial response, or stable disease for at least 105 days.
Data from patients with confirmed complete or partial responses.
By independent central review, two patients had progressive disease that was followed by subsequent response. Per protocol, these patients were classified as having disease progression, not responders, for the primary endpoint.
By independent central review, the median duration of response had not been reached at data cutoff. The longest duration of response at data cutoff was 24·2 months and is still ongoing. Characteristics of tumour responses are shown in figure 1.
Figure 1: Characteristics of tumour response to cemiplimab as per independent central review.
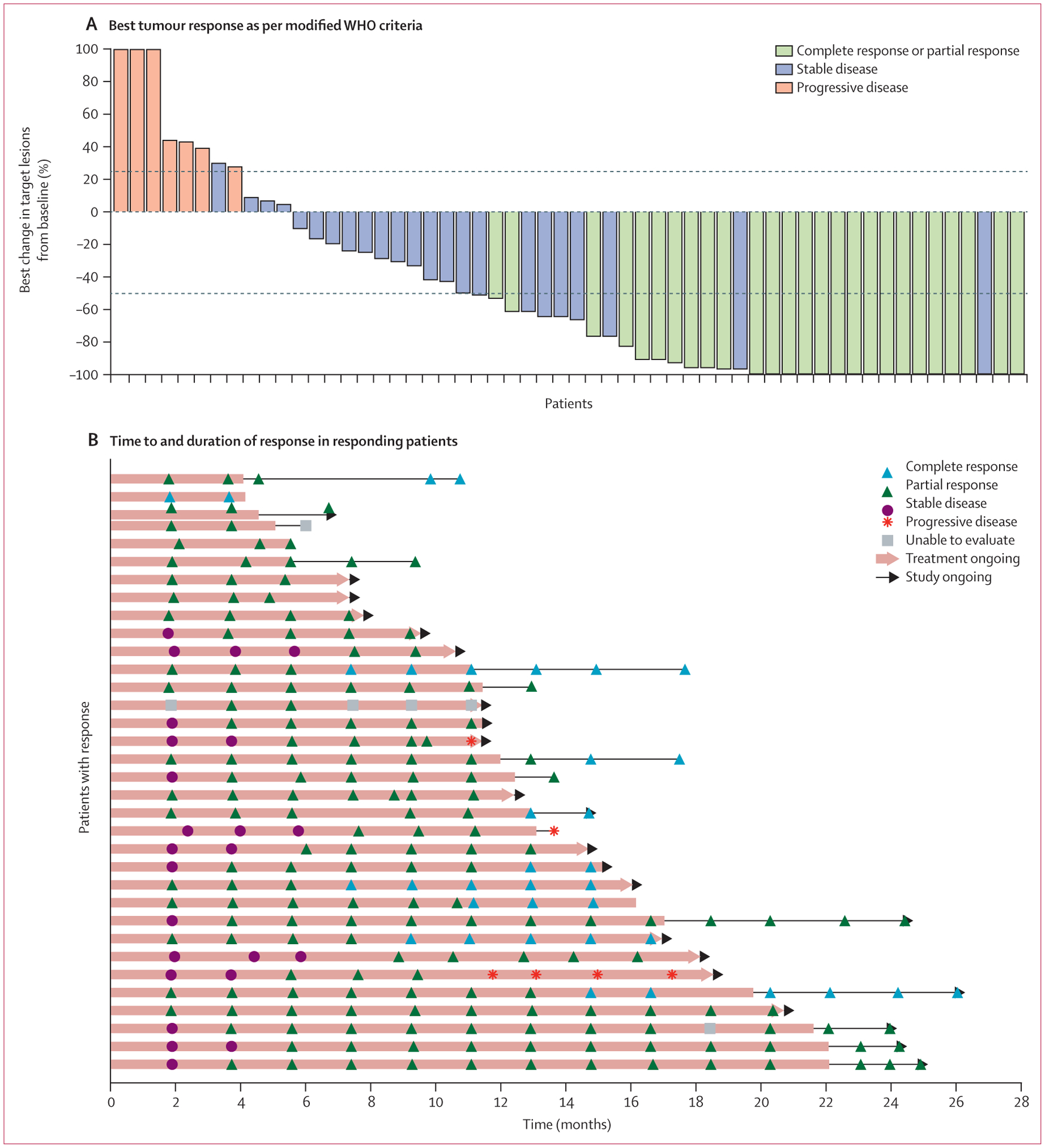
(A) Best change (percentage) in the sum of product (or products) of perpendicular longest dimensions of skin target lesion (or lesions) from baseline for 56 patients who had baseline skin target lesions and underwent at least one evaluable post-baseline medical photography evaluation as per modified WHO criteria by independent central review. Lesion measurements after progression were excluded. The dashed lines indicate WHO criteria for partial response (≥50% decrease in the sum of products of skin target lesion diameters) and progressive disease (≥25% increase in the sum of products of skin target lesion diameters). 22 patients who either did not have baseline skin target lesions or did not have evaluable post-baseline photography assessment are not included in the figure, but are included as non-responders in the overall response analysis as per the intention-to-treat principle. Eight patients had tumour reductions that met criteria for response on photographic measurements, but are classified as stable (blue bars >50% reduction in target lesions), either because there was no subsequent scan to confirm response (in seven patients) or because the composite response assessment was stable disease (in one patient). Eight of 34 patients with objective responses are not shown in this plot because the composite response assessments per independent central review included consideration of radiology results. (B) Time to response and duration of response in patients who responded to treatment. Each horizontal line represents one patient. Of the 34 responding patients, three had subsequent progressive disease. Among the remaining 31 patients who were still responding at the time of data cutoff, 12 were still on study treatment, nine were in post-treatment follow-up, and ten were off the study. One patient (sixth from bottom) had four progressive disease assessments because of discordance between investigator assessment and independent central review. Five patients with complete responses (top bar, and second, 12th, 17th, and 25th from top) are no longer on the study.
In a post-hoc subgroup analysis exploring the different reasons why patients were not considered candidates for curative surgery, clinical activity with cemiplimab was observed in all subgroups. An objective response was observed in ten (50%; 95% CI 27–73) and disease control in 16 (80%; 56–94) of the 20 patients with cutaneous squamous cell carcinoma lesions with substantial local invasion that precluded complete resection. An objective response was observed in 17 (57%; 95% CI 37–75) and disease control in 26 (87%; 69–96) of the 30 patients with cutaneous squamous cell carcinoma lesions in anatomically challenging locations for which surgery was anticipated to result in severe deformity or dysfunction. An objective response was observed in six (24%; 95% CI 9–45) and disease control in 17 (68%; 47–85) of the 25 patients with cutaneous squamous cell carcinoma lesions in the same location after two or more surgical procedures and with curative resection deemed unlikely.
Among all patients, median progression-free survival and median overall survival had not been reached at data cutoff. With regard to progression-free survival by independent central review, with 27 events (including 24 patients with progressive disease and three deaths) from the start of treatment until the data cutoff date of Oct 10, 2018, the estimated proportion of patients who were alive and had no disease progression at 12 months was 58% (95% CI 44–70). With regard to overall survival, with seven events from the start of treatment until the data cutoff date of Oct 10, 2018, the estimated proportion of patients who were alive at 12 months was 93% (95% CI 84–97; figure 2).
Figure 2: Kaplan-Meier curves for progression-free survival (as per independent central review; A) and overall survival (B).
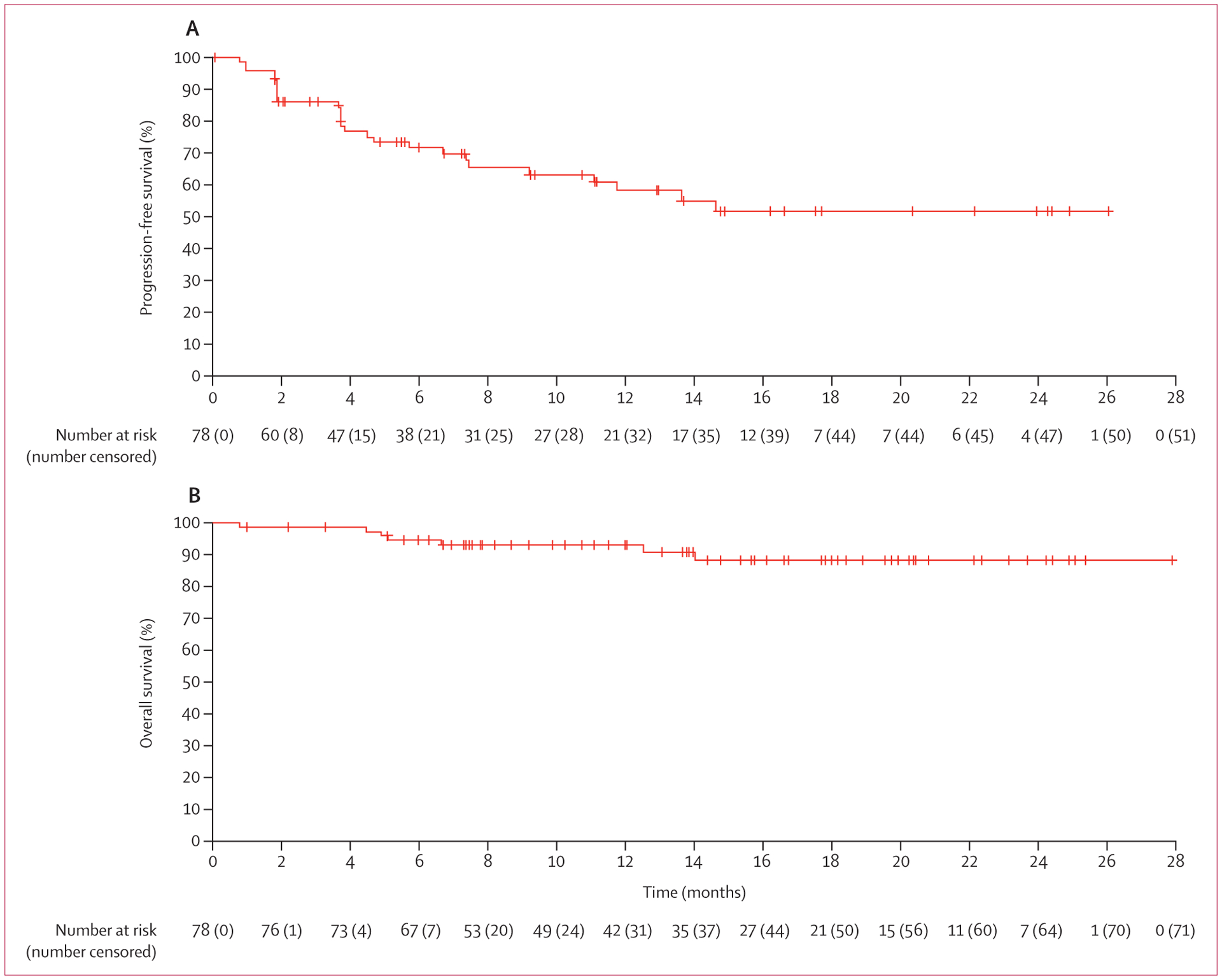
Median progression-free survival and median overall survival had not been reached at data cutoff.
48 (62%) of the 78 patients enrolled had samples available for tumour PD-L1 status assessment at baseline. An objective response was observed in six (35%; 95% CI 14–62) of the 17 patients with PD-L1 TPS of less than 1% and in 17 (55%; 36–73) of the 31 patients with PD-L1 TPS of 1% or higher. Objective responses were observed in patients regardless of baseline PD-L1 TPS (appendix p 7).
50 (64%) of the 78 patients enrolled had pre-treatment tumour samples available for the analysis of associations between tumour mutational burden and clinical activity of cemiplimab. Median tumour mutational burden was 74 (IQR 46–100) mutations per megabase among 21 responders and 29 (4–59) mutations per megabase among 29 non-responders (as per independent central review for both groups) (figure 3). Among 29 patients who achieved durable disease control, median tumour mutational burden was 65 (IQR 19–100) mutations per megabase and among 21 patients who did not, it was 31 (7–56) mutations per megabase, as per independent central review for both groups (figure 3). However, the differences in tumour mutational burden in survival analyses among patients who had at least 1 year of follow-up for progression or death did not show a clear separation (appendix p 9). Overall, among the activity endpoints studied, broad ranges in tumour mutational burden were seen among patients who had clinical benefit with cemiplimab and those who did not.
Figure 3: Clinical activity of cemiplimab and tumour mutational burden.
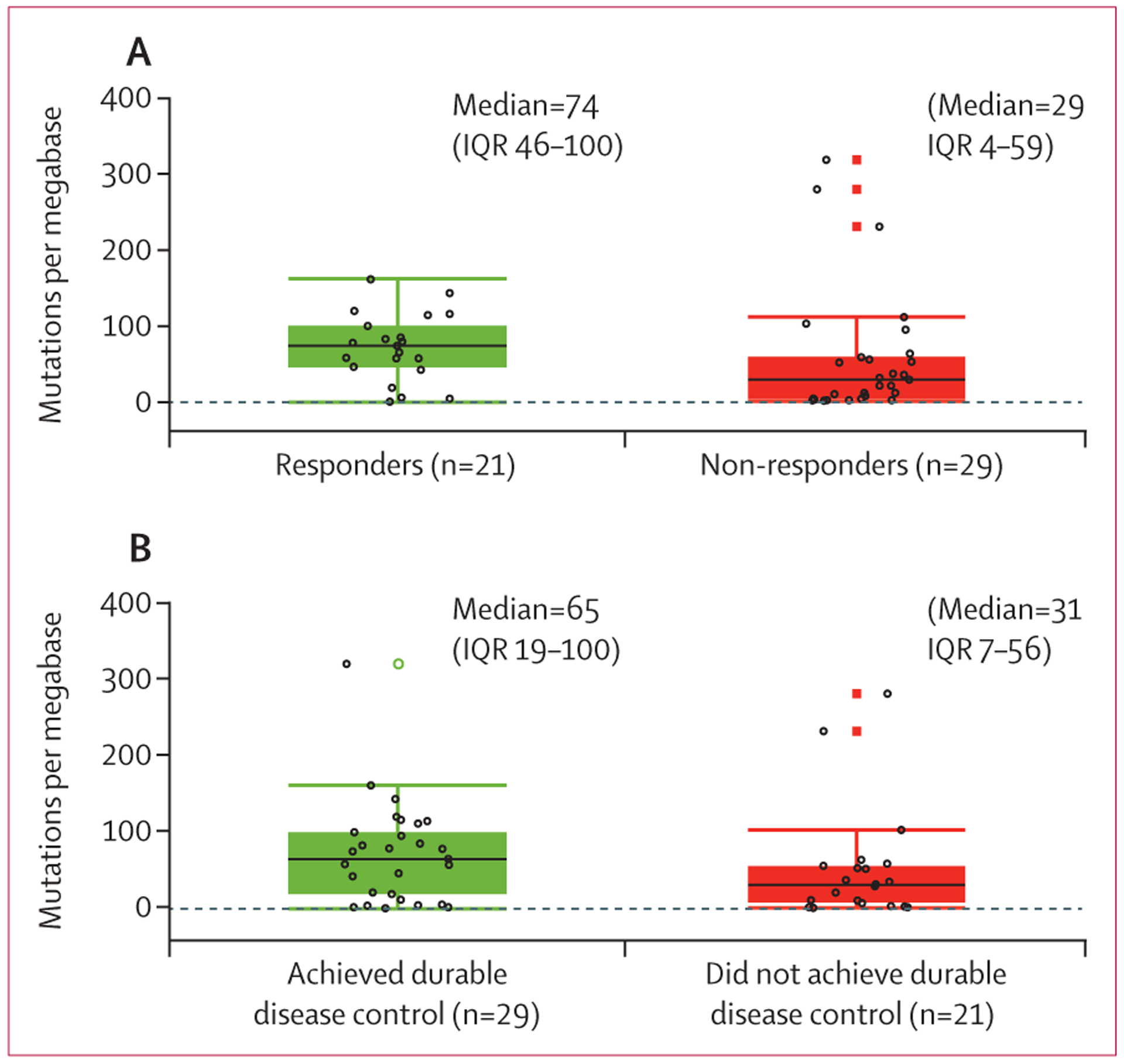
(A) Tumour mutational burden for responders (complete or partial response) versus non-responders (stable disease, progressive disease, or not evaluable) as per independent central review. (B) Tumour mutational burden for patients who achieved durable disease control (patients without progressive disease for at least 105 days) versus those who did not. Black lines in each box denote the median; lower and upper boundaries of box denote the IQR; and upper and lower whiskers indicate maximum and minimum values. Individual patients are indicated by open black circles. Open black circles beyond the whiskers are outliers. Open green circles and closed red boxes are duplicates of the outliers (the plots overlap box plots and scatter plots).
Grade 3–4 treatment-emergent adverse events occurred in 34 (44%) of 78 patients, with the most common being hypertension in six (8%) patients and pneumonia in four (5%; table 3). One (1%) of the 78 patients had a treatment dose reduction due to a treatment-emergent adverse event of grade 2 arthralgia that was considered related to study treatment. Six (8%) of the 78 patients discontinued treatment because of the following treatment-related adverse events: grade 4 pneumonia and grade 4 pneumonitis in one patient; grade 3 hepatitis, grade 3 increased alanine aminotransferase, grade 3 elevated aspartate aminotransferase, and grade 3 increased alkaline phosphatase in the second patient; and the following in each of the remaining four patients: grade 4 pneumonitis, grade 3 proctitis, grade 3 encephalitis, and grade 1 arthralgia. Serious treatment-emergent adverse events occurred in 23 (29%) of 78 patients, with seven (9%) considered treatment-related; the most common was pneumonitis in three (4%) patients. Treatment-related adverse events are summarised in the appendix (p 8). Grade 3 or higher immune-related adverse events, as per investigator attribution, occurred in eight (10%) of the 78 patients.
Table 3:
Treatment-emergent adverse events regardless of attribution
| Grade 1–2 | Grade 3 | Grade 4 | Grade 5 | |
|---|---|---|---|---|
| Any treatment-emergent adverse event | 77 (99%) | 33 (42%) | 4 (5%) | 2 (3%) |
| Fatigue | 32 (41%) | 1 (1%) | 0 | 0 |
| Diarrhoea | 21 (27%) | 0 | 0 | 0 |
| Pruritus | 21 (27%) | 0 | 0 | 0 |
| Nausea | 17 (22%) | 0 | 0 | 0 |
| Cough | 15 (19%) | 0 | 0 | 0 |
| Abdominal pain | 11 (14%) | 0 | 0 | 0 |
| Rash* | 10 (13%) | 0 | 0 | 0 |
| Vomiting | 9 (12%) | 1 (1%) | 0 | 0 |
| Actinic keratosis | 8 (10%) | 0 | 0 | 0 |
| Anaemia | 8 (10%) | 1 (1%) | 0 | 0 |
| Back pain | 8 (10%) | 0 | 0 | 0 |
| Constipation | 8 (10%) | 0 | 0 | 0 |
| Dry skin | 8 (10%) | 0 | 0 | 0 |
| Hypothyroidism | 8 (10%) | 0 | 0 | 0 |
| Maculopapular rash* | 8 (10%) | 0 | 0 | 0 |
| Arthralgia | 7 (9%) | 1 (1%) | 0 | 0 |
| Basal cell carcinoma | 7 (9%) | 1 (1%) | 0 | 0 |
| Dyspnoea | 6 (8%) | 1 (1%) | 0 | 0 |
| Hypokalaemia | 5 (6%) | 1 (1%) | 0 | 0 |
| Pain in extremity | 5 (6%) | 1 (1%) | 0 | 0 |
| Wound infection | 5 (6%) | 1 (1%) | 0 | 0 |
| Dizziness | 4 (5%) | 1 (1%) | 0 | 0 |
| Increased aspartate aminotransferase | 4 (5%) | 1 (1%) | 0 | 0 |
| Skin infection | 4 (5%) | 1 (1%) | 0 | 0 |
| Urinary tract infection | 4 (5%) | 2 (3%) | 0 | 0 |
| Fall | 3 (4%) | 2 (3%) | 0 | 0 |
| Hyperglycaemia | 3 (4%) | 3 (4%) | 0 | 0 |
| Neck pain | 3 (4%) | 1 (1%) | 0 | 0 |
| Pain | 3 (4%) | 1 (1%) | 0 | 0 |
| Pneumonitis | 3 (4%) | 1 (1%) | 2 (3%) | 0 |
| Squamous cell carcinoma of skin | 3 (4%) | 1 (1%) | 0 | 0 |
| Hypertension | 2 (3%) | 6 (8%) | 0 | 0 |
| Hyponatraemia | 2 (3%) | 2 (3%) | 0 | 0 |
| Hypotension | 2 (3%) | 1 (1%) | 0 | 0 |
| Muscular weakness | 2 (3%) | 2 (3%) | 0 | 0 |
| Musculoskeletal pain | 2 (3%) | 2 (3%) | 0 | 0 |
| Cellulitis | 1 (1%) | 3 (4%) | 0 | 0 |
| Hypophosphataemia | 1 (1%) | 1 (1%) | 0 | 0 |
| Influenza | 1 (1%) | 1 (1%) | 0 | 0 |
| Lymphopenia | 1 (1%) | 2 (3%) | 0 | 0 |
| Pneumonia | 1 (1%) | 3 (4%) | 1 (1%) | 1 (1%) |
| Soft tissue infection | 1 (1%) | 1 (1%) | 0 | 0 |
| Squamous cell carcinoma | 1 (1%) | 1 (1%) | 0 | 0 |
| Atopic dermatitis | 0 | 1 (1%) | 0 | 0 |
| Autoimmune hepatitis | 0 | 1 (1%) | 0 | 0 |
| Breast cancer | 0 | 2 (3%) | 0 | 0 |
| Cholecystitis | 0 | 1 (1%) | 0 | 0 |
| Clostridium difficile colitis | 0 | 1 (1%) | 0 | 0 |
| Death | 0 | 0 | 0 | 1 (1%) |
| Device-related infection | 0 | 1 (1%) | 0 | 0 |
| Encephalitis | 0 | 1 (1%) | 0 | 0 |
| Extradural abscess | 0 | 1 (1%) | 0 | 0 |
| Facial neuralgia | 0 | 1 (1%) | 0 | 0 |
| Hepatitis | 0 | 1 (1%) | 0 | 0 |
| Hip fracture | 0 | 1 (1%) | 0 | 0 |
| Increased international normalised ratio | 0 | 1 (1%) | 0 | 0 |
| Increased lipase | 0 | 1 (1%) | 0 | 0 |
| Increased weight | 0 | 1 (1%) | 0 | 0 |
| Myocardial infarction | 0 | 0 | 1 (1%) | 0 |
| Myocarditis | 0 | 1 (1%) | 0 | 0 |
| Nasal cavity cancer | 0 | 1 (1%) | 0 | 0 |
| Positive influenza A virus test | 0 | 1 (1%) | 0 | 0 |
| Pneumonia influenza | 0 | 1 (1%) | 0 | 0 |
| Proctitis | 0 | 1 (1%) | 0 | 0 |
| Psoas abscess | 0 | 1 (1%) | 0 | 0 |
| Radius fracture | 0 | 1 (1%) | 0 | 0 |
| Sepsis | 0 | 1 (1%) | 1 (1%) | 0 |
| Staphylococcal infection | 0 | 1 (1%) | 0 | 0 |
| Subdural haematoma | 0 | 1 (1%) | 0 | 0 |
| Syncope | 0 | 1 (1%) | 0 | 0 |
| Upper abdominal pain | 0 | 1 (1%) | 0 | 0 |
| Urinary tract obstruction | 0 | 1 (1%) | 0 | 0 |
| Wound complication | 0 | 1 (1%) | 0 | 0 |
Data are n (%) in all treated patients. Treatment-emergent adverse events, regardless of attribution, reported in at least 10% of patients (grades 1–2) or by any patient (grades 3–5) are shown.
Although rash and maculopapular rash might reflect the same condition, they were listed as two distinct events for the safety report of the study.
Two (3%) of 78 patients had treatment-emergent adverse events that resulted in death. One was due to infectious pneumonia that was considered unrelated to study treatment by the investigator. The other occurred 10 days after the onset of aspiration pneumonia and was considered related to study treatment.
Discussion
In this primary analysis, the clinical activity of cemiplimab in patients with locally advanced cutaneous squamous cell carcinoma was evident with an objective response observed in 34 (44%) patients by independent central review and in 41 (53%) patients by investigator assessment. Most responses were rapid, with median observed time to response corresponding to the time of first tumour assessment. Responses also show preliminary evidence of durability; longer follow-up analysis will better characterise durable responses. This analysis provides clinically meaningful data on disease control and the estimated proportion of patients who were alive at 12 months in a population of patients who, in the absence of effective treatment, often have poor prognosis. These data, combined with a previous report, suggest that cemiplimab is an active therapy in both locally advanced and metastatic cutaneous squamous cell carcinoma tumours.17
There were no new safety signals compared with previous reports of cemiplimab or other anti-PD-1 agents.17,20–22 Most of the treatment-related adverse events observed were grade 1 or 2, emphasising an acceptable safety profile of cemiplimab in this group of patients.
In addition to the primary endpoint, the clinical benefit of cemiplimab in advanced cutaneous squamous cell carcinoma could be further demonstrated by other endpoints and long term follow-up of these patients. For example, the results for durable disease control suggest that some patients who do not meet the criteria for response might have clinical benefit. Before our study, the largest prospective study in patients with advanced cutaneous squamous cell carcinoma reported a mean overall survival of 8·1 months (95% CI 6·9–9·3).2 Given the poor prognosis with previously available therapies, durable disease control of 105 days or longer in this study suggests a possible change in the natural history of this disease. Additionally, in this study, two patients had unconventional responses (progressive disease followed by subsequent response as per independent central review), an event called pseudoprogression that has been reported with cancer immunotherapy.23–25 RECIST criteria, rather than immune-related RECIST criteria, were used for radio logical assessments in this study to facilitate comparisons with previous studies of advanced cutaneous squamous cell carcinoma. For patients who are asymptomatic and do not have rapid disease progression, continued treatment with cemiplimab beyond an initial increase in tumour measurements might yield a clinically meaningful treatment effect. Furthermore, patients with durable disease control in this study are being followed up and, when clinically appropriate, biopsy samples might be obtained to establish whether a partial or complete response has been achieved histologically.
Immunohistochemistry analysis showed clinical activity with cemiplimab irrespective of baseline PD-L1 TPS. Analysis of the association between median tumour mutational burden and clinical activity of cemiplimab showed a wide range of tumour mutational burden in both responders and non-responders. Consequently, utilisation of PD-L1 TPS or tumour mutational burden, or both, to predict response or clinical benefit of cemiplimab in advanced cutaneous squamous cell carcinoma is not possible based on these analyses. Recent findings suggest that the characteristics of the mutations (eg, insertions and deletions vs substitutions) are an important determinant of the association between tumour mutational burden and responsiveness.26 New studies of cemiplimab in cutaneous squamous cell carcinoma are needed to evaluate other candidate biomarkers (alone or in combination with PD-L1 TPS) that might better predict clinical benefit from cemiplimab.
In a subgroup analysis, responses were observed regardless of the reason why patients were considered to be unsuitable candidates for surgery. However, the post-hoc subgroup analysis was not powered to support formal comparisons. Longer follow-up is ongoing to further describe the outcomes of patients in these subgroups. Management of patients with large or recurrent cutaneous squamous cell carcinoma lesions in aesthetically sensitive areas is complex and should be handled by multidisciplinary teams that include surgeons, medical oncologists, and radiation oncologists. The study definition of locally advanced cutaneous squamous cell carcinoma incorporates clinical judgment, similar to the established definition in previous studies of locally advanced basal cell carcinoma.27,28 In ongoing and future studies that integrate immunotherapy earlier into multidisciplinary management of cutaneous squamous cell carcinoma in both the preoperative (neoadjuvant) and postoperative (adjuvant) settings, standardised quantitative criteria will be used to define patient populations.
Limitations of this study include the single-arm design, small number of patients, and absence of long-term data. Longer-term follow-up data are being collected and will further characterise the clinical activity and durability of response to cemiplimab in patients with locally advanced cutaneous squamous cell carcinoma. Another limitation of this study is the use of a non-survival primary endpoint. However, objective response as per independent central review was a suitable primary endpoint when viewed in the context of the secondary endpoint of duration of response. Lastly, the sample sizes for the subgroup analyses of response were small. Nevertheless, objective responses were observed in all subgroups. Regardless of these limitations, cemiplimab showed clinical activity in a patient group who had poor prognosis before this study. Furthermore, to our knowledge, this is the largest prospective study of systemic therapy for patients with locally advanced cutaneous squamous cell carcinoma.
The data reported in this Article further support the approval of cemiplimab-rwlc by the US Food and Drug Administration (FDA) in September, 2018, and the conditional approval of cemiplimab by the European Commission in June, 2019, for the treatment of patients with metastatic cutaneous squamous cell carcinoma or locally advanced cutaneous squamous cell carcinoma who are not candidates for curative surgery or curative radiation.29,30 Notably, cemiplimab 3 mg/kg every 2 weeks was used in this study. The fixed-dose equivalent (cemiplimab 350 mg every 3 weeks) approved by the FDA and European Commission has been shown to have similar pharmacokinetics to weight-based dosing,29,30 and is being examined in a separate cohort of patients in the phase 2 study (NCT02760498).
In conclusion, these data show antitumour activity and an acceptable safety profile of cemiplimab in patients with locally advanced cutaneous squamous cell carcinoma.
Supplementary Material
Research in context.
Evidence before this study
Until recently, there was no approved systemic therapy for patients with advanced cutaneous squamous cell carcinoma. We previously reported substantial antitumour activity of cemiplimab with durable responses in patients in the metastatic and locally advanced cutaneous squamous cell carcinoma expansion cohorts in a phase 1 study and in a primary analysis of the metastatic cutaneous squamous cell carcinoma cohort (group 1) of a phase 2 study. We searched PubMed from Sept 1, 2010, to Sept 1, 2015, with the search terms “cutaneous squamous cell carcinoma OR squamous cell cancer of the skin AND treatment”, “immunotherapy AND cutaneous squamous cell carcinoma”, and “anti-PD-1 OR pembrolizumab OR nivolumab AND cutaneous squamous cell carcinoma”. This search was restricted to clinical trials. We found two studies that evaluated immunotherapy in cutaneous squamous cell carcinoma. In a phase 2 study, single-agent panitumumab showed an objective response in five (31%) of 16 patients with incurable cutaneous squamous cell carcinoma but the median duration of response was 6 months (range 5·0–17·5) and median overall survival was 11 months. In another relevant phase 2 study, cetuximab showed an objective response in ten (28%; 95% CI 14–45) of 36 patients with incurable cutaneous squamous cell carcinoma; the median duration of response was 6·8 months (95% CI 4·1–8·3) and mean overall survival was 8·1 months (95% CI 6·9–9·3). None of these immunotherapy agents was approved by regulatory bodies for the treatment of patients with cutaneous squamous cell carcinoma. A retrospective study confirmed low efficacy with chemotherapy or targeted therapy in this patient group.
Added value of this study
Findings from this study suggest that cemiplimab is an active treatment option for locally advanced cutaneous squamous cell carcinoma and has an acceptable safety profile. Associations between programmed cell death-ligand 1 (PD-L1) tumour proportion score and clinical activity of cemiplimab and between tumour mutational burden and clinical activity of cemiplimab were explored. Antitumour activity was observed regardless of PD-L1 status. A wide range of tumour mutational burden was found in both responders and non-responders.
Implications of all the available evidence
Our analysis shows meaningful clinical benefit in a patient population that previously had no widely accepted standard of care. It strengthens the previously available evidence that supported the US Food and Drug Administration’s approval of cemiplimab-rwlc for patients with metastatic cutaneous squamous cell carcinoma or locally advanced cutaneous squamous cell carcinoma who are not candidates for curative surgery or curative radiation, addressing a large unmet need. In exploratory correlative science objectives, immunohistochemistry results indicate that PD-L1 and tumour mutational burden are not predictive biomarkers in locally advanced cutaneous squamous cell carcinoma.
Acknowledgments
This study was funded by Regeneron Pharmaceuticals and Sanofi. We thank the patients, their families, all other investigators, and all investigational site members involved in this study. We also thank Q2 Solutions EA Genomics, Morrisville, NC, USA, for doing the tumour mutational burden analysis. Nikita Mehta and Robert Charnas from the study sponsors reviewed and provided editorial comments on the manuscript. Medical writing and editorial support under the direction of the authors was provided by Emmanuel Ogunnowo of Prime (Knutsford, UK) and funded by Regeneron Pharmaceuticals and Sanofi according to Good Publication Practice guidelines. Responsibility for all opinions, conclusions, and data interpretation lies with the authors.
Declaration of interests
MRM has received honoraria from Regeneron Pharmaceuticals, Sanofi, Novartis, Genentech, Eli Lilly, and Sun Pharma. NIK has received grants from Regeneron Pharmaceuticals; grants and advisory board fees from Bristol-Myers Squibb and HUYA Bioscience International; advisory board fees from EMD Serono, Regeneron Pharmaceuticals, Genentech, AstraZeneca (data safety monitoring committee), Merck, Array BioPharma, and Immunocore; grants from Merck, Novartis, GlaxoSmithKline, Cellgene, and Amgen; honorarium from Sanofi; and common stock ownership of Bellicum Pharmaceuticals, Mazor Robotics, Amarin, and Transenetrix. ALSC has received fees for being an expert panel member at Regeneron Pharmaceuticals and Merck. KDL has fulfilled consulting and advisory roles at Array BioPharma, Merck, Roche, and Regeneron Pharmaceuticals; received honoraria from Incyte and Array BioPharma, and research funding from Roche, Merck, Incyte, Array BioPharma, Nektar, Iovance Biotherapeutics, and Bristol-Myers Squibb. CDS is a steering committee member for Castle Biosciences; a steering committee member and consultant for Regeneron Pharmaceuticals; a consultant for Sanofi; has received research funding from Castle Biosciences, Regeneron Pharmaceuticals, Novartis, Genentech, and Merck; and is a chair for the National Comprehensive Cancer Network. LH-A has fulfilled consulting and advisory roles at Massive Bio; speakers’ bureau roles at Sanofi and Regeneron Pharmaceuticals, and received travel, accommodations, and expenses from Regeneron Pharmaceuticals, Sanofi, and Bristol-Myers Squibb; and research funding from Bristol-Myers Squibb, Regeneron Pharmaceuticals, Immunocore, Merck Sharp & Dohme, Polynoma, Corvus Pharmaceuticals, Roche, Merck Serono, Amgen, MedImmune, and Takeda. DS has received fees for consulting or advisory roles, honoraria, and travel, accommodation and expenses from Roche/Genentech and Merck Serono; fees for consulting or advisory roles, honoraria, speaker’s bureau, research funding, and travel, accommodation, and expenses from Novartis and Bristol-Myers Squibb; fees for consulting or advisory roles, honoraria, speaker’s bureau from Merck Sharp & Dohme, Incyte and Pierre Fabre; fees for consulting or advisory roles, honoraria, speaker’s bureau, and travel, accommodation and expenses from Amgen; fees for consulting or advisory roles and honoraria from Immunocore and 4SC; fees for consulting or advisory roles from Mologen, Sanofi, and Regeneron Pharmaceuticals; speaker’s bureau fees from Roche; travel, accommodation, and expenses from Merck; and honoraria from Sysmex, Grünenthal Group, Agenus, Array BioPharma, AstraZeneca, LEO Pharma, Pfizer, Philogen, Regeneron Pharmaceuticals, and Mologen. AG has received personal fees (advisory board and travel support) from Bristol-Myers Squibb, Sun Pharma, and Merck, and advisory boards fees from Eisai, Sanofi, Pfizer, and Roche. AH has received institutional grants, speaker’s honoraria, and consultancy fees from Amgen, Bristol-Myers Squibb, MSD/Merck, Pierre Fabre, Provectus, Sanofi Genzyme, Roche, Regeneron Pharmaceuticals, and Novartis, institutional grants and consultancy fees from Merck Serono and Philogen, and consultancy fees from Sun Pharma and OncoSec. DJW has received institutional research grant funding from Regeneron Pharmaceuticals, grants and personal fees from Bristol-Myers Squibb and Genentech, and grants from ARMO BioSciences, Merck Sharp & Dohme, Kura Oncology, AstraZeneca, and Regeneron Pharmaceuticals. GAD has received speaker fees from Regeneron Pharmaceuticals. CB has received an institutional research grant from Regeneron Pharmaceuticals; grants, personal fees, and non-financial support for consulting, lectures, and clinical trials from Amgen, Bristol-Myers Squibb, MSD, Novartis, and Roche; personal fees and non-financial support for consulting from Pierre Fabre, Sanofi Aventis, and Merck; a grant for a clinical trial from Array and personal fees (safety monitoring board) from Miltenyi. VJ, ES, JB, SL, DMW, IL, and MGF are employees and shareholders of Regeneron Pharmaceuticals. GDY is an employee, shareholder of, and member of Board of Directors at Regeneron Pharmaceuticals. DR has received an institutional research grant and funding from Regeneron Pharmaceuticals, Merck, Roche, GlaxoSmithKline, and Bristol-Myers Squibb, and uncompensated advisory board participation for Merck, Regeneron Pharmaceuticals, GlaxoSmithKline, Amgen, and Bristol-Myers Squibb; and travel support from Merck. FM declares no competing interests.
Footnotes
Data sharing
Qualified researchers can request access to study documents (including the clinical study report, study protocol with any amendments, blank case report form, and statistical analysis plan) that support the methods and findings reported in this report. Individual anonymised participant data will be made available once the indication has been approved by a regulatory body, if there is legal authority to share the data and there is not a reasonable likelihood of participant re-identification. Requests should be submitted on the Regeneron website.
To submit a request online see https://errs.regeneron.com/external
See Online for appendix
References
- 1.William WN Jr, Feng L, Ferrarotto R, et al. Gefitinib for patients with incurable cutaneous squamous cell carcinoma: a single-arm phase II clinical trial. J Am Acad Dermatol 2017; 77: 1110–13.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maubec E, Petrow P, Scheer-Senyarich I, et al. Phase II study of cetuximab as first-line single-drug therapy in patients with unresectable squamous cell carcinoma of the skin. J Clin Oncol 2011; 29: 3419–26. [DOI] [PubMed] [Google Scholar]
- 3.Shin DM, Glisson BS, Khuri FR, et al. Phase II and biologic study of interferon alfa, retinoic acid, and cisplatin in advanced squamous skin cancer. J Clin Oncol 2002; 20: 364–70. [DOI] [PubMed] [Google Scholar]
- 4.Hillen U, Leiter U, Haase S, et al. Advanced cutaneous squamous cell carcinoma: a retrospective analysis of patient profiles and treatment patterns—results of a non-interventional study of the DeCOG. Eur J Cancer 2018; 96: 34–43. [DOI] [PubMed] [Google Scholar]
- 5.Cavalieri S, Perrone F, Miceli R, et al. Efficacy and safety of single-agent pan-human epidermal growth factor receptor (HER) inhibitor dacomitinib in locally advanced unresectable or metastatic skin squamous cell cancer. Eur J Cancer 2018; 97: 7–15. [DOI] [PubMed] [Google Scholar]
- 6.Foote MC, McGrath M, Guminski A, et al. Phase II study of single-agent panitumumab in patients with incurable cutaneous squamous cell carcinoma. Ann Oncol 2014; 25: 2047–52. [DOI] [PubMed] [Google Scholar]
- 7.Gold KA, Kies MS, William WN Jr, Johnson FM, Lee JJ, Glisson BS. Erlotinib in the treatment of recurrent or metastatic cutaneous squamous cell carcinoma: a single-arm phase 2 clinical trial. Cancer 2018; 124: 2169–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cowey C, Robert N, Davies K, et al. Treatment patterns and outcomes among patients with advanced cutaneous squamous cell carcinoma (CSCC) in a US community oncology setting. Proc Am Soc Clin Oncol 2019; 37: e21033 (abstr). [Google Scholar]
- 9.Ruiz E, Chen CI, Deering K. Treatment patterns and costs in cutaneous squamous cell carcinoma (CSCC) patients with nodal dissection, chemotherapy, and/or radiation therapy. Proc Am Soc Clin Oncol 2018; 36: e18703 (abstr). [Google Scholar]
- 10.Group Work. Guidelines of care for the management of cutaneous squamous cell carcinoma. J Am Acad Dermatol 2018; 78: 560–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gajra A, Klepin HD, Feng T, et al. Predictors of chemotherapy dose reduction at first cycle in patients age 65 years and older with solid tumors. J Geriatr Oncol 2015; 6: 133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garcovich S, Colloca G, Sollena P, et al. Skin cancer epidemics in the elderly as an emerging issue in geriatric oncology. Aging Dis 2017; 8: 643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100 000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 2017; 9: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buttner R, Longshore JW, Lopez-Rios F, et al. Implementing TMB measurement in clinical practice: considerations on assay requirements. ESMO Open 2019; 4: e000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nehal KS, Bichakjian CK. Update on keratinocyte carcinomas. N Engl J Med 2018; 379: 363–74. [DOI] [PubMed] [Google Scholar]
- 16.Burova E, Hermann A, Waite J, et al. Characterization of the anti-PD-1 antibody REGN2810 and its antitumor activity in human PD-1 knock-in mice. Mol Cancer Ther 2017; 16: 861–70. [DOI] [PubMed] [Google Scholar]
- 17.Migden MR, Rischin D, Schmults CD, et al. PD-1 blockade with cemiplimab in advanced cutaneous squamous-cell carcinoma. N Engl J Med 2018; 379: 341–51. [DOI] [PubMed] [Google Scholar]
- 18.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009; 45: 228–47. [DOI] [PubMed] [Google Scholar]
- 19.World Health Organization. WHO handbook for reporting results of cancer treatment. WHO Offset Publication No. 48. 1979. https://apps.who.int/iris/bitstream/handle/10665/37200/WHO_OFFSET_48.pdf?sequence=1&isAllowed=y (accessed Sept 2, 2019).
- 20.Weber JS, Postow M, Lao CD, Schadendorf D. Management of adverse events following treatment with anti-programmed death-1 agents. Oncologist 2016; 21: 1230–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zimmer L, Goldinger SM, Hofmann L, et al. Neurological, respiratory, musculoskeletal, cardiac and ocular side-effects of anti-PD-1 therapy. Eur J Cancer 2016; 60: 210–25. [DOI] [PubMed] [Google Scholar]
- 22.Spain L, Diem S, Larkin J. Management of toxicities of immune checkpoint inhibitors. Cancer Treat Rev 2016; 44: 51–60. [DOI] [PubMed] [Google Scholar]
- 23.Katz SI, Hammer M, Bagley SJ, et al. Radiologic pseudoprogression during anti-PD-1 therapy for advanced non-small-cell lung cancer. J Thorac Oncol 2018; 13: 978–86. [DOI] [PubMed] [Google Scholar]
- 24.Seiwert TY, Burtness B, Mehra R, et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol 2016; 17: 956–65. [DOI] [PubMed] [Google Scholar]
- 25.Wolchok JD, Hoos A, O’Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res 2009; 15: 7412–20. [DOI] [PubMed] [Google Scholar]
- 26.Mandal R, Samstein RM, Lee KW, et al. Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science 2019; 364: 485–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med 2012; 366: 2171–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Migden MR, Guminski A, Gutzmer R, et al. Treatment with two different doses of sonidegib in patients with locally advanced or metastatic basal cell carcinoma (BOLT): a multicentre, randomised, double-blind phase 2 trial. Lancet Oncol 2015; 16: 716–28. [DOI] [PubMed] [Google Scholar]
- 29.Regeneron Pharmaceuticals. LIBTAYO [cemiplimab-rwlc] injection full US prescribing information. 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761097s000lbl.pdf (accessed Sept 9, 2019).
- 30.European Medicines Agency. LIBTAYO EPAR. 2019. https://www.ema.europa.eu/en/medicines/human/EPAR/libtayo (accessed Sept 9, 2019).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.