

Abstract
Curative therapy for metastatic cancers is equivalent to causing extinction of a large, heterogeneous, and geographically dispersed population. Although eradication of dinosaurs is a dramatic example of extinction dynamics, similar application of massive eco-evolutionary force in cancer treatment is typically limited by host toxicity. Here, we investigate the evolutionary dynamics of Anthropocene species extinctions as an alternative model for curative cancer therapy. Human activities can produce extinctions of large, diverse, and geographically distributed populations. The extinction of a species typically follows a pattern in which initial demographic and ecological insults reduce the size and heterogeneity of the population. The surviving individuals, with decreased genetic diversity and often fragmented ecology, are then vulnerable to small stochastic perturbations that further reduce the population until extinction is inevitable. We hypothesize large, diverse, and disseminated cancer populations can be eradicated using similar evolutionary dynamics. Initial therapy is applied to reduce population size and diversity and followed by new treatments to exploit the eco-evolutionary vulnerability of small and/or declining populations. Mathematical models and computer simulations demonstrate initial reductive treatment followed immediately by demographic and ecological perturbations, similar to the empirically derived treatment of pediatric acute lymphocytic leukemia, can consistently achieve curative outcomes in nonpediatric cancers.
Introduction
The ideal outcome of cancer therapy is eradication of the malignant population without significant harm to the patient. This is achieved in localized cancers by surgical removal or focused therapy such as radiation. A curative outcome becomes more difficult and much less likely in a metastatic setting. The growth of tumors in multiple locations requires systemic treatment. Walther and colleagues and (1) and Korolev and colleagues (2) have noted the similarities between eradicating a cancer and the extinction of natural populations. Here we propose that the eradication of a disseminated cancer population is analogous to “Anthropocene extinctions,” the intentional or unintentional eradication of species due to human activities.
Prior theoretical (3–5), experimental (5, 6), and clinical studies (7) have demonstrated how integrating evolutionary dynamics (Fig. 1) into cancer therapy can prolong tumor control for incurable metastatic cancers. Here, we take these studies a step further. We propose integrating the ecological and evolutionary dynamics of extinction into treatment protocols with curative intent. Specifically, we focus on metastatic cancers that initially show robust responses to therapy, yet almost always recur and progress as the cancer cells evolve resistance and ecologically recover. We hypothesize that the probability of cure in these clinical settings may be increased with existing drugs using a treatment strategy that integrates the eco-evolutionary dynamics of anthropogenic extinctions observed in nature.
Figure 1.
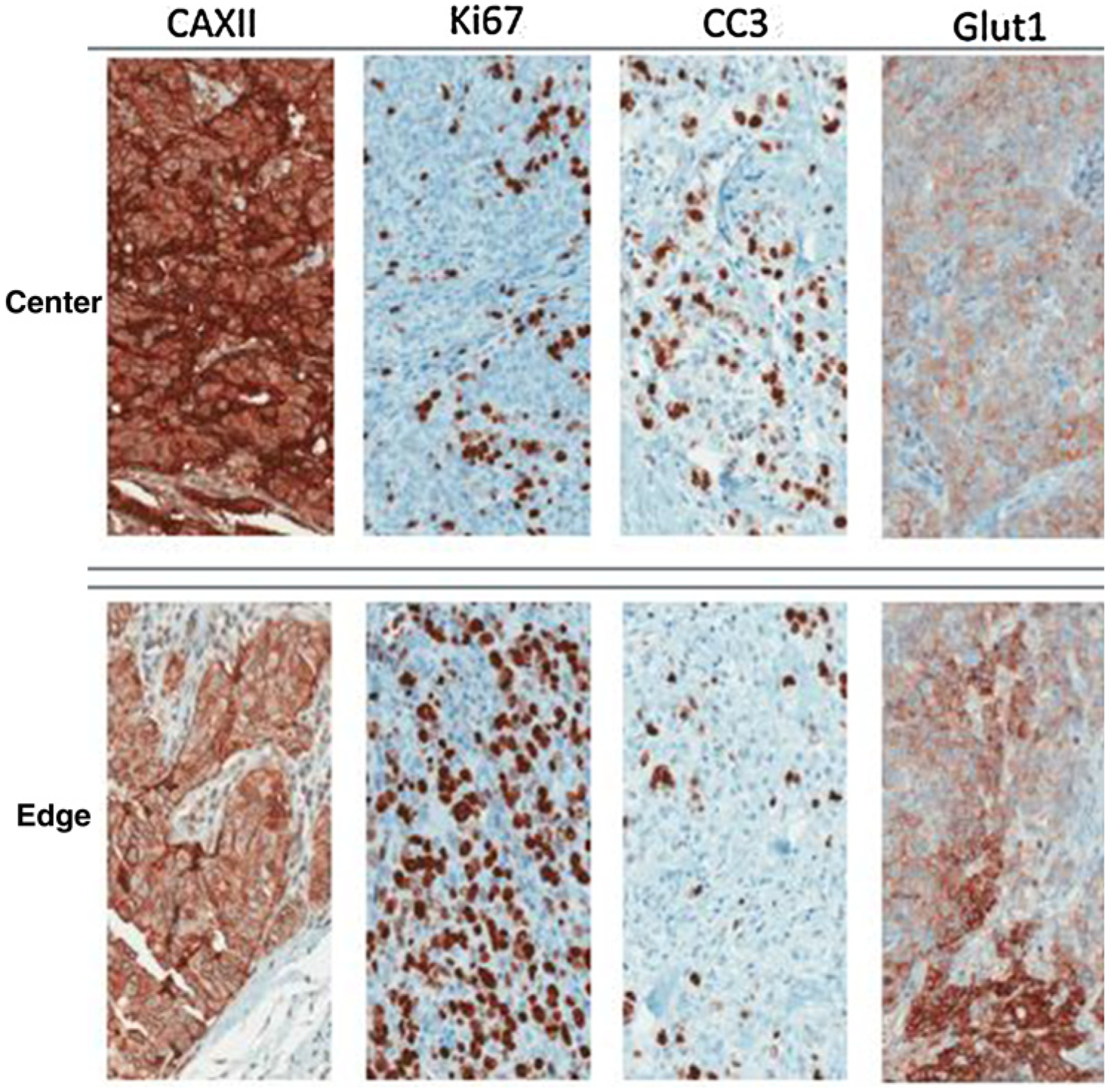
IHC stains of two regions from a clinical invasive ductal breast cancer. The Ki67 stain identifies cells that are proliferating, while the cleaved caspase-3 (CC3) stain shows cell undergoing apoptosis. These spatial variations are governed by variations in the environmental selection forces (note associated difference in GLUT1 and CAIX expression). This continuous but variable birth and death rate permits evolution and can result in both tumor growth and tumor extinction. Note that if the birth/death dynamics remained unchanged, the tumor population in the top panels would become extinct, while the population in the bottom panels would grow rapidly. In small populations, stochastic variations in birth and death rates have a disproportionately large population effect and can result in extinction (adapted from Lloyd et al.; ref. 36).
In general, cancer treatment research, influenced in many ways by Ehrlich’s magic bullet paradigm and the remarkable success of antibiotics in eliminating infectious diseases, has focused on drug development. This approach has been highly successful. The continuous emergence of new agents has generally increased overall survival in patients with metastatic disease. However, despite the introduction of many highly effective treatment agents, most common metastatic cancers (e.g., breast, prostate, colon, and lung) remain fatal because cancer cells have a remarkable capacity to evolve resistance strategies.
Thus, while searching for cancer magic bullets is certainly appropriate, it is possible that the size, phenotypic diversity, and ecological heterogeneity of widely disseminated cancers may preclude consistent eradication by the continuous application of a single drug or combination of drugs. However, disseminated cancers may yet be curable with these same drugs. Here we hypothesize that the limitation of cancer treatment in some clinical settings is not the efficacy of available drugs, but rather the efficacy of current treatment tactics.
Most drugs used in cancer therapy are applied at MTDs until progression (8). The intuitive appeal of this strategy has similarities to the extinction of dinosaurs following the massive ecological and evolutionary forces unleashed by a meteor impact at the Cretaceous-Tertiary boundary (KT impact). While the application of overwhelming force to eradicate a target population may at times be highly effective, it is also highly indiscriminate. The KT impact not only eliminated the entire dinosaur clade it also drove 1 in 3 of all other species to extinction. Furthermore, extinction events in the distant past were likely more complex than is apparent in the fossil record. Thus, we propose it is more useful to examine well-studied recent extinction events.
Here, we examine cancer therapy in the context of well-studied extinctions caused by humans during the Anthropocene era. Although far less dramatic than mass extinctions, the subtle and complex dynamics that produce the extinction of a single species may be informative particularly when the species originally had a large, highly diverse, and geographically dispersed population similar to a metastatic cancer population.
A number of well-studied, unintended Anthropocene era extinctions, such as the passenger pigeon (9) and heath hen (10), have been closely investigated. In particular, the recent intentional anthropogenic extinction of goats on the Galápagos Islands provides a close analogy to cancer therapy. Beginning in the 18th century, sailors deposited domesticated animals, including goats, on the Galápagos Islands to provide food for future visits. The feral goat population remained stable until the late 20th century when it rapidly expanded to approximately 140,000 to 250,000 individuals spread across the Isa-bela, Santiago, and Pinta Islands causing significant environmental damage and threatening native species. Using a grant from the Global Environment Facility, an eradication effort commenced using sharpshooters riding trucks and helicopters. These tactics were highly successful, eradicating about 90% of the feral goat population. However, small groups of survivors became uncatchable, perhaps alert to the noise made by helicopters they learned to escape into the forests. These “resistant” goats began to breed. Because the initial strategy was no longer effective, the rangers deployed a new approach, the “Judas goats.” These females were sterilized, coated with hormones, and wore a radio-tracking device. When released, they attracted and mingled with the small surviving groups allowing hunters to locate and kill them finally eliminating the population in 2005.
The Galápagos goat extinction illustrates two-step dynamics typically found in other observed extinction events including the passenger pigeon and the heath hen. A population’s slide to extinction begins with an initial perturbation or series of perturbations that greatly reduces the size, spatial distribution, and diversity of an initially large heterogeneous population. In the case of the Galápagos goats, the initial perturbation was hunting. However, this “first strike” did not cause extinction because of “evolutionary rescue.” That is, a population can survive and ultimately recover from an abrupt and massive perturbation via high genetic, phenotypic or behavioral diversity. Large populations have the time and diversity to allow for resistant variants that rescue the population from extinction (11, 12). Furthermore, once evolutionary rescue has produced a resistant population, continued application of the initial perturbation is futile. However, extinction of the population remains possible because the surviving population is small, fragmented, and lacks the numbers, genetic diversity and spatial dispersion of the original population. Such a damaged population has only limited capacity to withstand additional perturbations especially those that differ from the first strike. For example, continued hunting of the Galápagos goats by helicopter became futile as the goats became fearful to the danger. But then, the release of a small number of Judas goats was highly effective. Such “Judas goats” would have been impractical and less effective when the goat populations were still large and dispersed.
An important concept in preventing Anthropocene extinctions is the minimum viable population (MVP). The MVP estimates the minimum number of individuals necessary to save a population threatened with extinction. Small populations are extremely vulnerable to stochastic demographic and environmental perturbations. Below an MVP, extinction becomes highly probable (13). These dynamics have been extensively investigated in conservation studies (14) using population viability analysis (PVA; refs. 15, 16). PVAs estimate the likelihood of extinction based on the time frame and given some existing number of individuals. Although cancer cells differ in important ways (e.g., they reproduce asexually so that finding a mate is not limiting) from most threatened populations in nature, many PVA principles can be applied to cancer treatment but with the somewhat ironic goal of increasing the probability of extinction.
Materials and Methods
Mathematical model and simulation
The ability to describe and predict tumor growth and decline is essential for developing strategies to eradicate cancer cell populations. The kinetics of tumor growth can change in different environments and as a function of cancer cell population numbers and their underlying population genetics. Hence, consideration of the kinetics associated with both low and high cell population numbers is of importance, because it is processes taking place at the interface of these regimens that ultimately govern the observed patterns associated tumor initiation, treatment response and tumor relapse, as well as cancer evolutionary trajectories, which can change in response to shifts in selection forces acting on the cancer population in different regimes. Applying ecological and evolutionary principles at the stage where there is a fast transition from large tumor sizes to small ones due to chemotherapy, therefore, could help improve therapeutic strategies, lowering the probability of relapse and promoting full tumor eradication. Another aspect that is important to consider is stochastic factors that determine extinction risk (i.e., the likelihood of successfully eradicating a tumor and avoiding relapse; ref. 17). Of these, there are three important factors for cancer treatment: (i) demographic stochasticity (which arises from the probabilistic nature of birth and death at the level of individual cells); (ii) population heterogeneity (which is related to variation in vital rates among individual cells); and (iii) the Allee or “aggregation” effects (which are the benefits each cancer cells gains during therapy because it is a member of a group; refs. 18–22). How and to what extent each of these factors shape extinction risk will likely change during the course of treatment. Thus, small stochastic fluctuations in the birth and death rate that would cause no discernible change in the original metastatic population may have catastrophic effects on small isolated tumor populations following initial treatment. Each isolated group of cells may randomly fall below its extinction threshold, which acts as an “absorbing boundary.” Furthermore, adding even small changes to this underlying demographic stochasticity through administration of therapy further increases the probability of extinction. Similarly, decreased cellular heterogeneity and Allee protective effects in the surviving populations also increase the potential effects of any given therapy.
Deterministic population model
The two aspects of tumor kinetics discussed above, that is, those governing large tumor population dynamics and those governing small ones, are addressed separately in most cancer models. We therefore begin by defining a population level deterministic model of tumor dynamics that incorporates both regimes. This mean-field model is then used as a basis for developing a stochastic individual-based model for studying the system. The model is based on a simple ordinary differential equation describing birth death processes at the population level, with N(t)representing the number of cancer cells and g(t) the per capita growth rate of the population:
| (A) |
In the simplest scenario, the intrinsic tumor growth rate is proportional to the number of cells present, and can be captured by a single per capita constant representing the difference between cells added by birth and those removed by death: , where and are respectively mean per capita cancer cell proliferation and death rates assumed to be independent of population size. Hence, at intermediate tumor sizes cancer cell numbers usually increase exponentially when g > 0. In contrast, at low and high tumor cell densities, cell proliferation may be influenced by other factors with growth rates deviating from exponential. As tumors become larger, growth rate usually slows due to limitation of resources [e.g., from diminished blood flow (Fig. 1)]. In these large tumor regimes, the per capita growth rate can be described as , where K represents a carrying capacity. Here, the closer the cancer population size is to K, the slower the per capita growth rate. At the other extreme, when tumor size becomes small, its growth rate can also slow but due to different factors. Here, the per capita growth rate can be described as (19), where A is an “Allee” threshold, below which populations decline, and a is a parameter affecting the shape of the growth function. Because we are interested in the dynamics of tumor size at the phase of transition between large tumors (before initial treatment) and small tumors (which may or may not be eradicated following treatment), we define the per capita growth rate as a combination of these functions:
| (B) |
In the presence of chemotherapy, cancer cell mortality will increase depending on drug concentration, C(t), and the mean susceptibility of cancer cells to treatment, , such that:
| (C) |
Stochastic individual-based mode
The mean field model is an idealized representation of the population level tumor cell growth and decline. A comparable stochastic version of the deterministic population model can be created by assigning probabilities to the discrete events of birth and death (23). This framework can then be used to accurately simulate demographic stochasticity in continuous time (23). The stochastic analogue of the model considers the following 2 discrete events:
E1: Cell proliferation: division of a mother cell into two daughter cells
E2: Cell death
Where b(t) and d(t) are the per capita birth and death rates, respectively. By defining g(t) = b(t) + d(t) and relying on Eq. B, we obtain as can these follows:
| (D) |
To explicitly incorporate demographic stochasticity as well as evolutionary changes, we extend the traditional Gillespie algorithm by tracking each individual cell from the moment of birth to the moment of death. Hence, the two discrete events defined above are considered per individual cell. In practice, this implies that intrinsic rates are now considered on the individual level where probabilities of birth, λ, and death, μ, are a function of an individual cell’s trait value, x.
Cell variability
The tumor cell population is assumed to be phenotypically heterogeneous in terms of trait x. We assume that cancer cells with high x are less sensitive to the primary chemotherapy drug (see Fig. 2A) at the cost having lower proliferation rates (see Fig. 2B). This trade-off represents the cost of producing, maintaining and using the molecular machinery necessary for resistance (5, 24, 25). In all other aspects, cancer cells are assumed to be phenotypically identical. As a lower bound we assume that cancer cells with very low trait values of x (<0.02) have diminished survival. Figure 2A shows the dependency of a cell’s death rate from treatment, D, on its individual sensitivity level, xi, and the overall drug concentration at time t, C(t). We assume D(C(t), x) = max(0, β(C(t) − x)a). Figure 2B shows the relationship between a cell’s trait value (x) and the expected number of days for proliferation (1=λi). In the absence of drug pressure, we expect subpopulations with higher proliferation rates to grow faster, and in the large tumor regime where resources become limited and competition increases, natural selection will lead the fast replicating cells to dominate and outcompete the slower replicating cells. However, we assume that fast replicating cells are also more sensitive to treatment due to a tradeoff, such that in the presence of treatment, these fast replicating cells are at a disadvantage, and slow replicating cells have an opportunity to reappear and expand. In addition, we assume that during cell proliferation, two new daughter cells are born replacing the mother cell. Each daughter cell inherits the value of their maternal trait xmother with a slight mutation. These new x values are randomly sampled from a normal distribution with mean xmother and standard deviation of 0.02.
Figure 2.
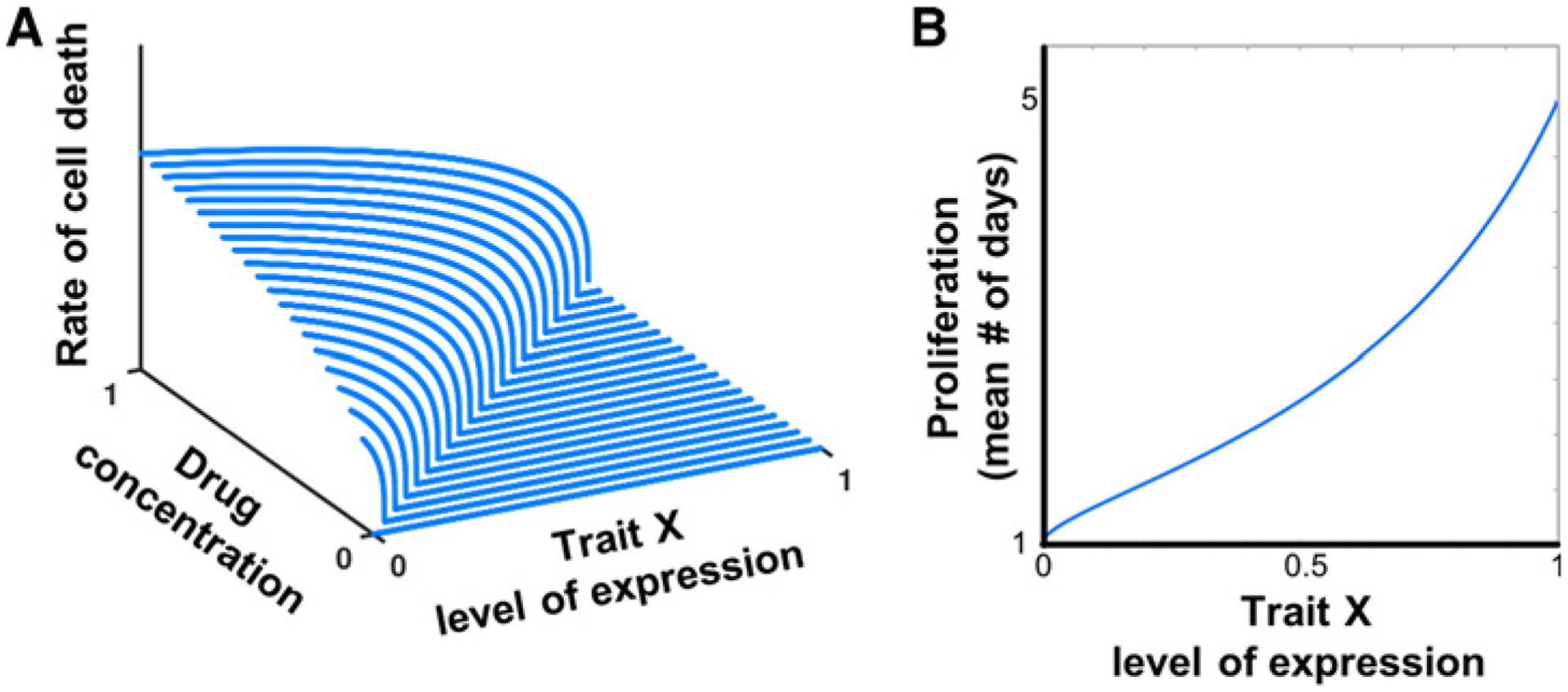
The tumor cell population is assumed to be genetically and phenotypically heterogeneous in the level of expression of trait x (see Materials and Methods). Depicted in A is the dependency of a cell’s death rate due to treatment on the overall level of drug pressure (C) and the degree of resistance of the cell to drug treatment (x). This is an “inverted” IC50 surface, defined as D(C, x) = max(0; β(C − x)α. For higher levels of resistance (higher values of trait x), death rate remains low even when drug concentrations increase. For intermediate levels of drug concentration, cancer cells with low resistance are cleared at a much faster rate, while those with high levels of resistance remain protected. High resistance comes with a cost, as shown in B, where λ = λmax − xs(λmax − λmin).
Conceptually, our modeling approach follows the concept of distributed evolutionary games as defined by Cohen (26, 27). Specifically, models structured with continuous traits that represent an abstract level of resistance have been used to study different aspects of population dynamics in cancer, such as the emergence of drug resistance (28), the effect of different categories of drugs (cytotoxic and cytostatic; ref. 29) on tumor populations, and the design of optimal drug combinations (30).
Implementation of the Gillespie algorithm
We implemented the Gillespie’s direct (23) method using the following steps:
Initialization of the system: define the full list of all possible demographic events and their respective rates (see E1 and E2, above); define the initial cell population size (N0) and the initial distribution of trait x in the tumor population (see next section); and define the end time of the simulation.
- Iteration of a two-step process:
- Determine when the next demographic even will occur. For a given point, this updates the rates of all possible events that can occur based on the current composition of the cancer population and the state of therapy. The procedure sums across all individual rates to obtain one total rate for the occurrence of the next event. We then sample a random number from a uniform distribution between 0 and 1 and use it to find the timing of the next event: Δt = −ln (Random)/Total Rate.
- Determine what demographic event will occur and for which individual cell. We turn rates into probabilities by dividing individual rates by the total sum of rates. We then sample another random number from a uniform distribution between 0 and 1 and use the list of individual rates to determine which of the events occurs next: Random < (Cumulative list individual rates)/(Total Rate). We then implement the event that took place, update the population count (either N → N + 1 or N → N − 1), and the new time (t → t + Δt).
Once the new time is larger or equal to the end time of the simulation, terminate and summarize the results.
Initial conditions
The initial distribution of trait x within the tumor population was randomly sampled from a predefined steady state distribution, that reflects a balance between natural birth-death-mutation processes in large population sizes limited by a carrying capacity, where natural selection favors faster proliferating cells. For a given set of parameters, we obtained the predefined x distribution as follows. We began with a small population of N0 = 200 cancer cells, with randomly sampled values of trait x drawn from a uniform distribution between x∈[0,1]. We ran the simulation without treatment, allowing the population of cells to grow. The population will approach and level off close to carrying capacity. Selection for the trait that maximizes proliferation rate and mutation will in time result in a dynamic-equilibrium distribution of trait values among the cancer cells. We ran the simulation and at time intervals of 10 days observed the cumulative probability distribution (CPD). The simulation for determining initial conditions was stopped once the CPD reached its steady state for the specific predefined parameter set. To ensure that the CPD was reliable, we repeated this process 100 times for three different initial population sizes (N = 5,000, 10,000 and 25,000) and found that for a given set of parameters, the resulting CPDs were identical. This distribution of x’s among cancer cells was then used as the tumor’s initial condition for the start of simulations with the various therapy strategies.
Our motivation for constructing the initial conditions in this way was 2-fold. First, we are interested in studying eco-evolutionary tumor dynamics as they transition from large tumor regimes dominated by selection for high proliferation under restricted carrying capacity, to small tumor regimes dominated by a selection pressure that is imposed through drug treatment. We therefore begin our simulations with initial conditions that reflect expected distributions of x in large tumors. Second, we are interested in clearly elucidating the effects of different treatment protocols on tumor dynamics in highly stochastic settings, which are likely characteristic of small tumors.
Parameters
Intrinsic death rate: ; proliferation: λmin = 0.2, λmax = 1, c = 0.25, K = 10, 000, A = −3, a = 100 (for results in Figs. 3–5) and A = 15, a = 0 (for the results in Fig. 6). First drug treatment and sensitivity: C = 0.4, α = 0.8, β = 0.2. Second drug treatment: one-time removal of 40% (for result in Fig. 4) and 20% (for the results in Figs. 5 and 6) of the cells, respectively.
Figure 3.
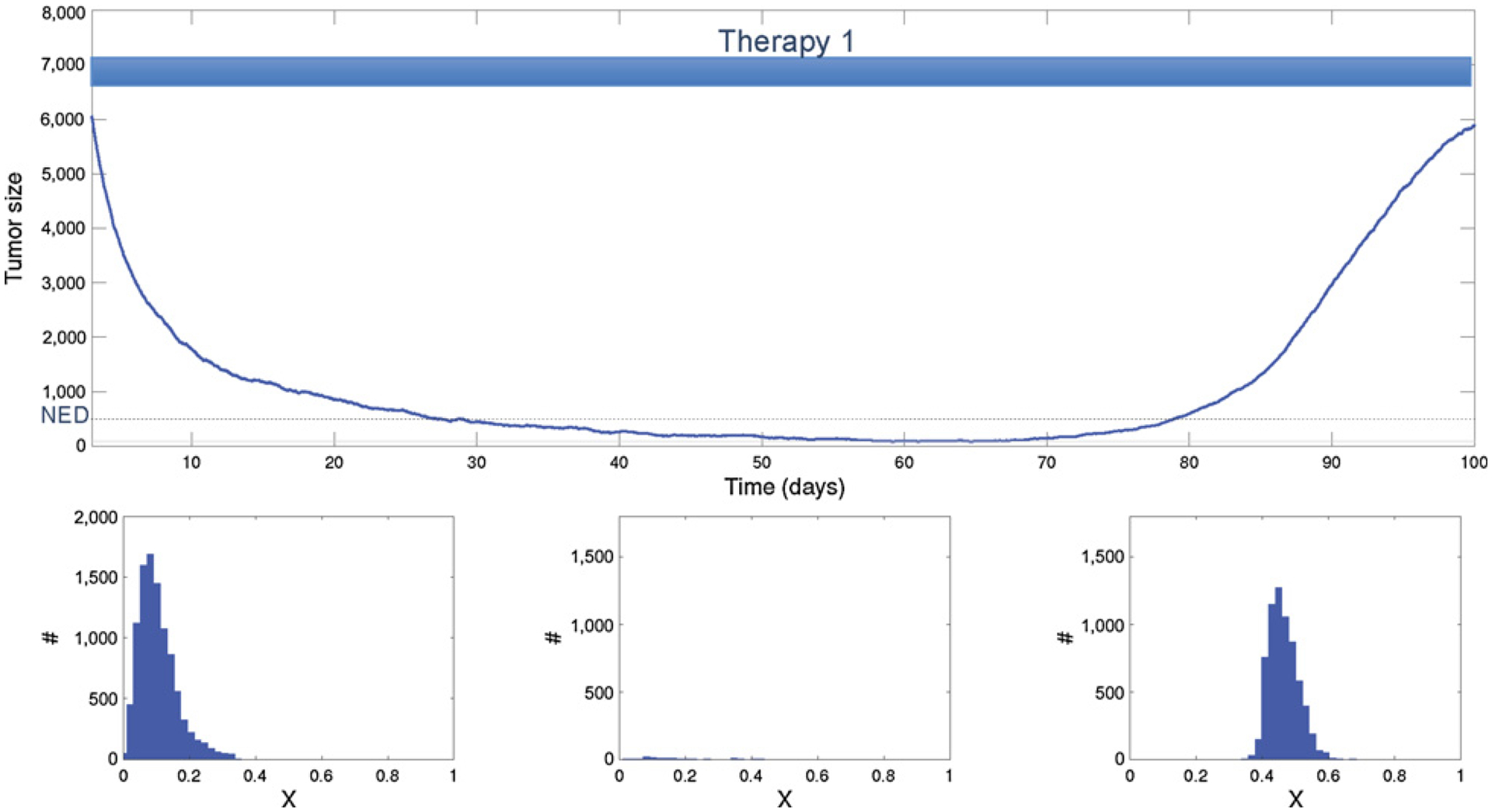
Effective initial treatment followed by maintenance therapy using the same drug. The histogram shows the phenotypic heterogeneity within the population before treatment (left), at no evidence of disease (NED; middle), and after recurrence (right). The initial therapy is highly effective, eliminating 90% of the cancer cells and rendering NED. Although the cancer cell population is small, the selection pressure during therapy eventually results in near universal resistance. Continued administration of the drug is ineffective because of an evolutionary rescue and the resistant cancer population recovers and then progresses.
Figure 5.
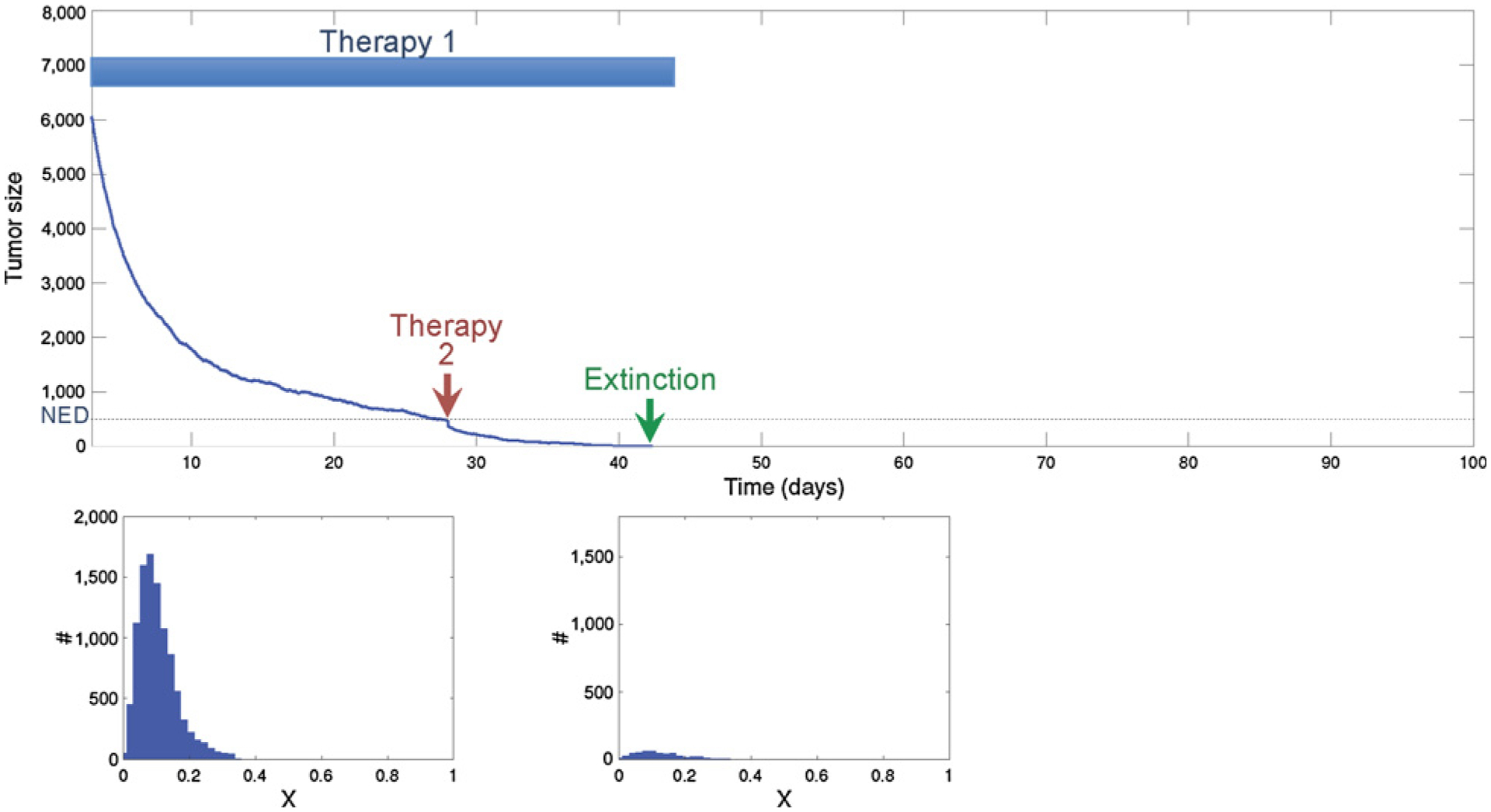
Using the proposed extinction strategy, the second treatment is applied when the cancer population reaches no evidence of disease (NED) following first-line treatment. Note that the tumor is NED and thus the efficacy of treatment is not measurable. However, because the population is small and homogeneous, the second drug, although only marginally effective with elimination of 40% of the population, is nonetheless sufficient to drive the small homogeneous tumor to extinction. The histogram shows the phenotypic heterogeneity within the population before treatment (left) and just before the second strike (right).
Figure 6.
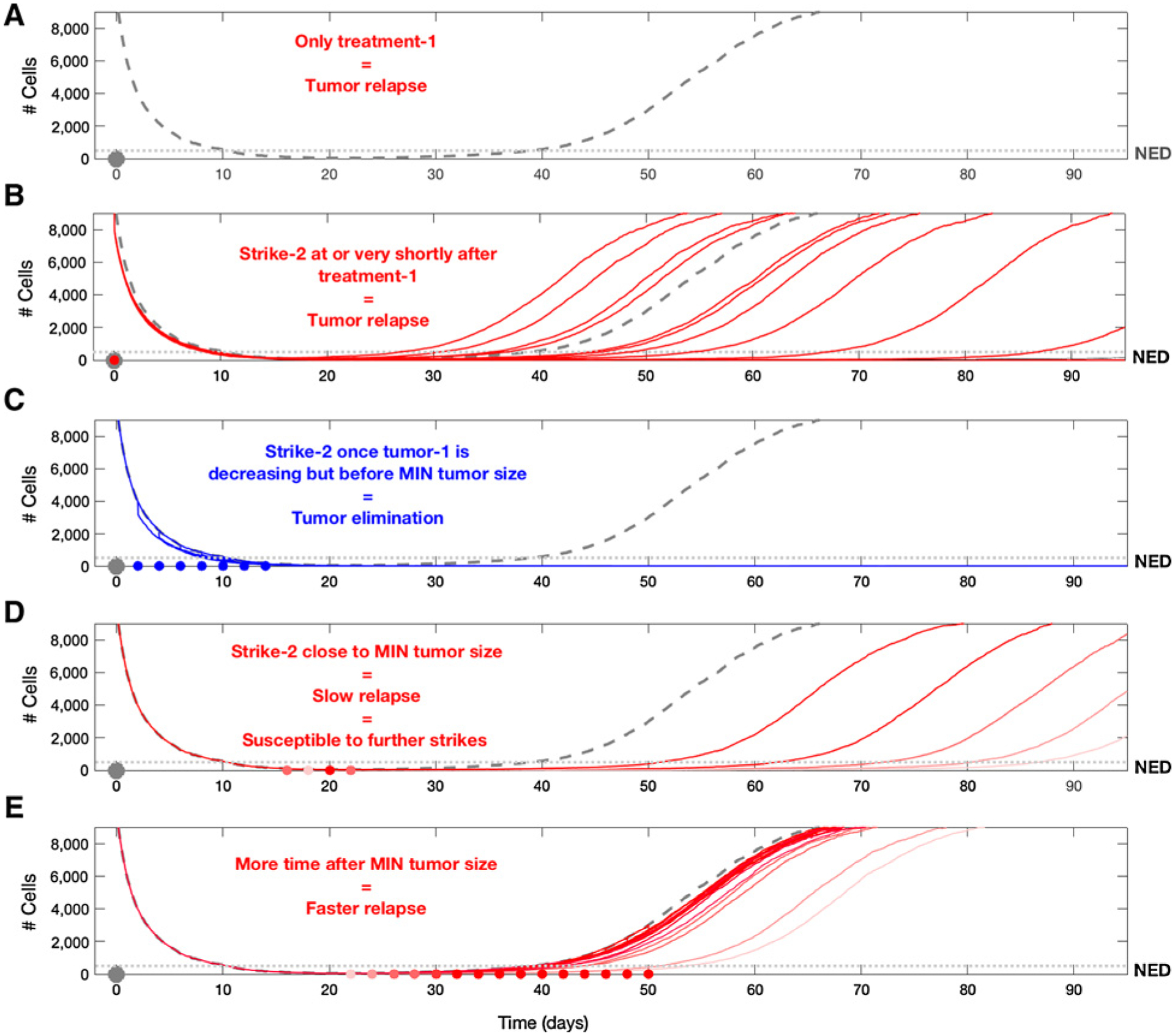
Here, we explore treatment outcomes when the second strike is applied at different time points. To easily compare between these outcomes, we display multiple overlapping simulations in the different subfigures. For each simulation, the second treatment was applied only once. Even though the second treatment reduced the tumor population by only 20%, it proved to be effective when applied in the appropriate timeframe (the times of treatment are marked as red/blue dots). A, The trajectory of relapse in the absence of a second strike. In B, we show 15 characteristic outcomes of what would happen if the first and second treatments were applied simultaneously. In 4 cases, relapse was earlier than it could have been in the absence of a second strike, in 9 cases, relapse was later than it would have been, and in 2 cases, the tumor was eliminated. As shown in C, applying treatment in the time window after the tumor starts to decline due to treatment, but before reaching the nadir, shows that all cases resulted in successful tumor elimination (blue dots indicate the time of application). D, Applying the second treatment in the time period where the tumor was near its nadir led to delayed relapses that could last several years. Despite the failure to eliminate the tumor in this window, there are opportunities to apply additional useful strikes (see Fig. 7). E, Demonstrates the ineffectiveness of a second treatment after the tumor is beyond its nadir and has begun growing again.
Figure 4.
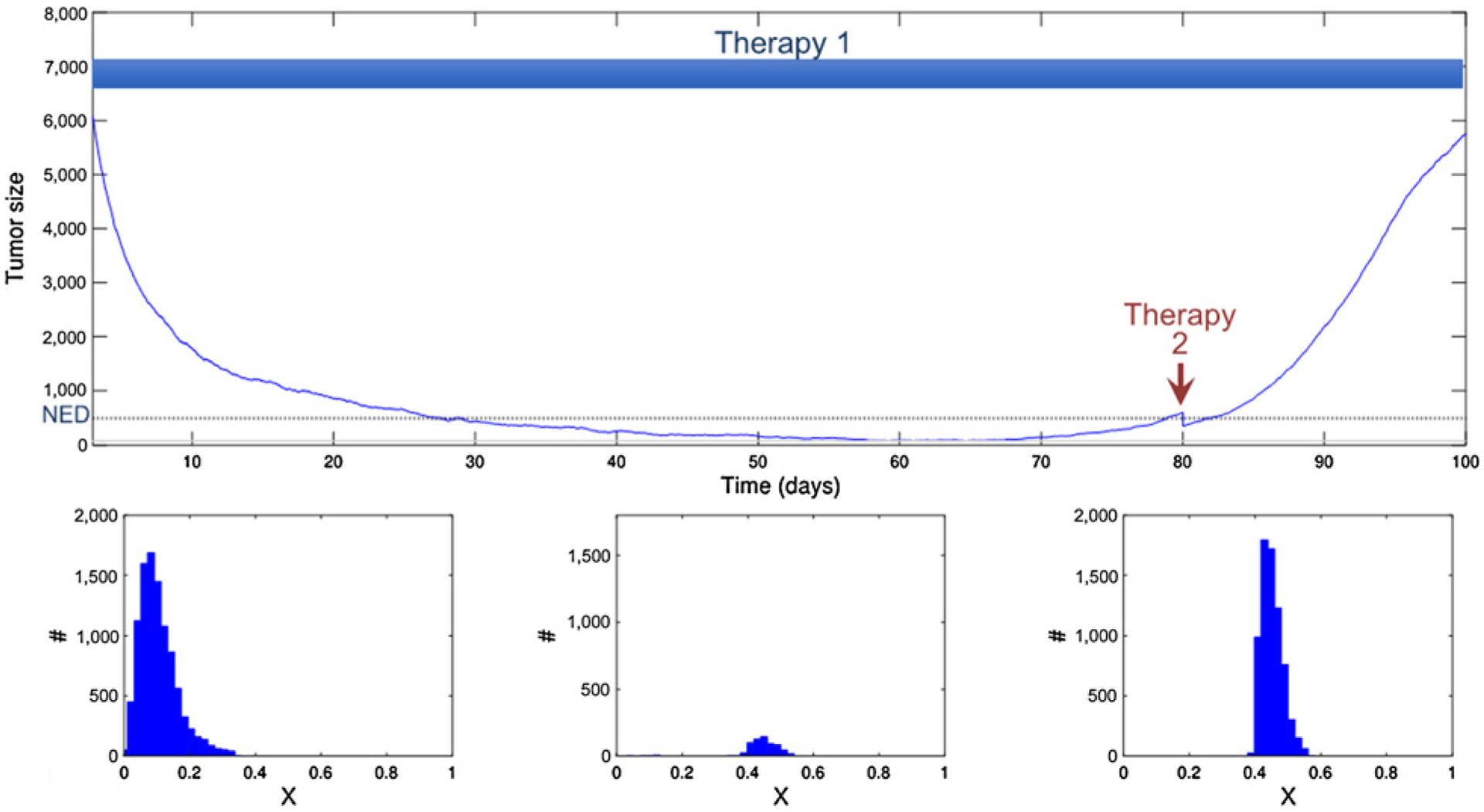
Initial therapy achieves a complete response and is continued at MTD until a measurable recurrence is observed. However, waiting for measurable tumor has allowed the tumor population to increase in size and diversity. The second drug is less effective than the first-line treatment and reduces the cancer population by 40%. This transiently reduces the population below NED, but resistance rapidly emerges leading to progression. The histogram shows the phenotypic heterogeneity within the population before treatment (left), just before the second strike (middle), and after recurrence (right).
The simulation code is available upon request.
Results
The mathematical model shows heuristically why and how the sequencing of two therapies can (i) preempt progression as a result of evolving resistance to the first strike; (ii) can succeed by switching therapies when tumor burden is declining and cancer cell populations are most damaged; and (iii) induce what otherwise might be modest additional mortality to drive the residual disease to below its MVP so that extinction is more probable.
In the initial set of simulations (Figs. 3–5), both the tumor burden and phenotypic heterogeneity are shown. In general, the depth and duration of response to treatment is dependent on the heterogeneity of the tumor populations (Figs. 3–5). There is, however, some degree of self-selection since curative treatment can only be applied after a successful initial treatment, which in turn requires some limit on heterogeneity in the pre-treatment population. Fortunately, an excellent initial response is seen in a wide range of tumors and treatments so that the proposed strategy can be applied in a variety of clinical settings.
In Fig. 3, the initial chemotherapy drug is administered at t = 0. The application of this drug favors higher trait values and does not affect trait values higher than 0.4. The phenotypic distribution of traits among cancer cells prior to therapy is shown in the lower left box of Fig. 3. Note that the mean trait value is about 0.15, which in the model represents fast replicating cells with a high level of drug sensitivity. The drug therapy kills about 90% of the initial population. This major collapse in population size can be viewed as a decrease to below the detection threshold (i.e., the patient is considered to have no evident disease, NED) creating a risk of extinction. However, the initial population size was sufficiently large and diverse to permit evolutionary rescue within a sufficiently fast time frame. As the population declines, the fast-replicating but drug-sensitive cells are removed, while the slower-replicating yet more drug-resistant cells are no longer limited by strong competition. This “competitive release” allows survivors with trait values closer to 0.4 to replicate more rapidly and evolutionarily explore higher values of x. Ultimately, population heterogeneity begins increasing again, which then becomes the basis for evolutionary rescue of the tumor population. In this simulation, the treatment protocol remains fixed and so a resistant population emerges and eventually undergoes unconstrained proliferation. This progression is inevitable due to the large and heterogeneous starting initial population.
In Fig. 4, we assume that the patient is treated according to the standard strategy of MTD until progression. Again, initial therapy reduces the population of cancer cells to NED. Although no tumor is visible, the simulation continues with MTD administration of the initial drug consistent with the standard oncology practice of “continuous application of drug at MTD until progression.” At the point of visible and measurable recurrence, a treatment containing one or more new agents is applied. This treatment is less effective than the initial therapy, with a killing capacity of only 40%, and a mortality effect that is independent of the cancer cell’s trait values. This second strike therapy was applied too late to prevent evolutionary rescue. This can also be seen in the middle histogram showing the distribution of trait x at the time of the second strike. Clearly, the population has shifted from being centered at low values of x, to values around x = 0.4, allowing resurgence of the tumor population.
In Fig. 5, we simulated the identical initial therapy, but this time, the second treatment occurred immediately when the tumor became NED. Although current oncology practice discourages treatment in which the effects of treatment cannot be measured, application of the second therapy when the tumor was not visible resulted in extinction. The histogram showing the distribution of trait x at the time of the second strike clearly demonstrates competitive release has not yet occurred, hence limiting the capacity for evolutionary rescue of the tumor population.
To further investigate the dynamics of extinction, in Fig. 6A–E we examined different time points along the tumor trajectory and minimum requirements for the efficacy of the second strike. Here we assumed that the second treatment only eliminates 20% of the surviving cancer population (i.e., even less effective than in the previous two examples). In Fig. 6B, we demonstrate a conventional strategy in which the second and first treatments are simply added together and administered simultaneously. Here, consistent with clinical observations, time to progression is generally (but not always) increased and, in a few cases, the tumor recovery is so slow that it does not become clinically apparent during the simulation. However, in Fig. 6 we demonstrate that even a marginally effective second strike (reducing the cancer population by only 20%) can drive the cancer population to extinction when administered within a “window of opportunity.” Although our original hypothesis assumed that the optimal time to apply the second strike for extinction was when the tumor size was at a nadir, we find that extinction can be achieved over a broader time period. In particular, Fig. 7A–C demonstrates that this window becomes broader as Allee effects are enhanced. In practice, such enhancements may be achieved, for example, by strategically applying additional treatments that promote angiogenesis, increase the immune response, or add systemic therapy that slightly increases tumor cell death rate or decreases birth rate. In other words, elimination of the tumor can be reached by targeting it in the period when it becomes NED or even earlier, during the initial period of decline following the first strike. Furthermore, when the cancer population is at its nadir in size, the extinction strategy may fail if, during the time it spent NED, the cells were able to adapt to therapy, thanks to the competitive release allowing an increase in the population’s heterogeneity.
Figure 7.
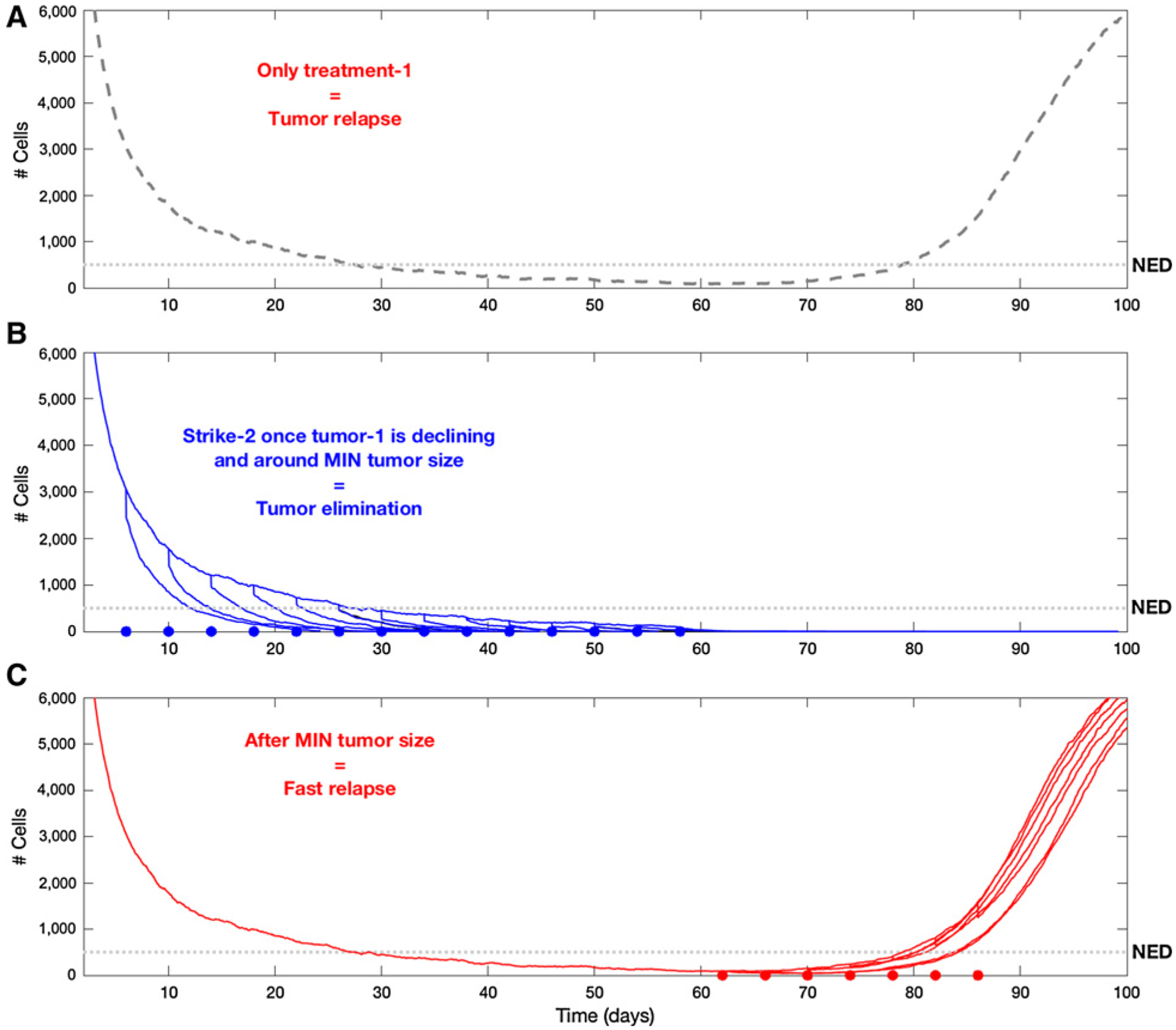
Here, we repeat the procedure carried out in Fig. 6, but increase the strength of the Allee effect on the population. Here, too, the figures show multiple overlapping simulations, and for each simulation, the second treatment was applied once. A, The trajectory of relapse in the absence of a second strike. B, The application of the second strike up to, and including, the time period of nadir, showing that all cases resulted in successful tumor elimination (blue dots indicate the time of application). In C, we see once again the ineffectiveness of a second strike after the tumor reached its minimum size and starts to grow again. The assumption of a stronger Allee effect can be seen as a proxy for what could be expected if “second strikes” were strategically designed for enhancing the Allee effects in small populations. This demonstrates that correct selection of second strike treatments can significantly widen the window of opportunity for successful tumor elimination.
In summary, we find the pathways to cancer extinction are governed by 3 interacting parameters: population size, population diversity, and the relative strength of the Allee effect. In general, consistent with the concept of the extinction vortex, a population in decline will remain in that trajectory often with relatively small applications of eco-evolutionary forces. Furthermore, our results emphasize that perturbations typically have disproportionately greater effects on small populations, a dynamic that can be exploited to driver cancers to extinction.
Discussion
Ideally, metastatic cancers will be eradicated by “magic bullet” agents that kill all cancer cells while sparing all normal ones. Currently available drugs fall short of this goal and, given the size and diversity of cancer populations, developing one is probably not achievable in the foreseeable future. Nevertheless, the evolutionary dynamics of Anthropocene extinctions suggest eradicating metastatic cancers may be possible through a strategic integration of several therapies, each of which, individually, cannot achieve a curative outcome and, in fact, may only be mildly effective.
This potential curative strategy requires two or more steps guided by eco-evolutionary principles. The first strike applies a therapy that is effective in reducing the population even though prior clinical experience has determined that it is rarely or never curative. The second strike, following immediately after the cancer cell population decline, exploits the unique properties of small populations. As generally seen in background extinctions, an identical perturbation may result in entirely different outcomes in small populations compared to large groups of the same species (17). This is due to the vulnerability of small populations to stochastic changes in birth and death rates (17), decreased cellular heterogeneity, and Allee effects that adversely affect small populations (19, 22).
Application of this strategy requires an initially effective first line treatment or sequence of treatments that can significantly reduce the cancer population ideally to NED, similar to standard first line therapy in current oncologic practice. Importantly, first-line treatment does not need to be a magic bullet that eradicates the entire cancer population. Rather, by significantly reducing the size and diversity of the cancer population, it renders it vulnerable to extinction. Effective first strikes are reasonably common. For example, androgen deprivation therapy for metastatic prostate cancer reduces PSA to normal or undetectable in >90% of patients. Similarly, initial chemotherapy for metastatic pediatric fusion-positive rhabdomyosarcoma (31, 32) and for small cell lung cancer renders most patients NED. However, clinical experience shows that curative outcomes are rare as small surviving resistant clones eventually repopulate the tumor.
Our proposed strategy differs from standard oncologic practice because it changes treatment even as the tumor is responding well to the first therapy. As demonstrated in Figs. 5 and 6, the standard practice of continued application of the same agent(s) at MTD therapy until tumor progression following an excellent initial response is ineffective because the surviving cancer cells are resistant. Furthermore, during tumor growth from NED to measurable disease, the cancer population increases in both size and diversity. Thus, application of second line therapy is too late to produce extinction.
Thus, we hypothesized curative outcomes may be achievable if, after effective initial therapy, new treatments are applied immediately after achieving NED. Figures 5 through 7 are consistent with the hypothesis but also find the opportunity for extinction is broader than expected. In fact, consistent with the concept of the extinction vortex, cancers that remain sufficiently large to be observable but are in decline are also vulnerable to extinction from a second strike. In contrast, as shown in Figs. 6 and 7, combining the first- and second-strike agents at the initiation of therapy is ineffective. However, if applied within an optimal window of opportunity when the cancer population is small and in decline, simulations found that addition of an even mildly effective second agent (killing only 20% of the surviving cells) consistently resulted in extinction of the cancer population.
Although extinction phase therapy is termed “Second Strike,” it is not meant to be a single therapy. Although a single second strategy was effective in eradicating the Galápagos goats, other Anthropocene extinction involving more numerous and geographically dispersed populations (e.g., the heath hen, passenger pigeon, and Rocky Mountain Locust) required multiple different perturbations. Second therapies can be theoretically divided into habitat perturbations, demographic perturbations, predator introduction and foraging restraints. Habitat disruption might be achievable with angiogenesis inhibitors. Demographic perturbations are simply treatments that will increase the death rate or decrease the birthrate of the surviving population. Similarly, introduction of a predator in the form of immunotherapy may be disproportionately effective in small populations. In addition, we point out that cancer cells must “forage” in the sense that they must acquire nutrients from their environment. It may be possible to disrupt cancer cell foraging when tumor cells remain in fragmented islands after the first strike by using, for example, disruption of amino acid transport or administration of racemic mixes of amino acids.
As an example of the Anthropocene extinction approach, consider metastatic pediatric fusion positive rhabdomyosarcoma. Typically, conventional chemotherapy produces NED in >90% of patients. However, in nearly all cases, the tumor recurs within 6 to 9 months and is typically resistant to the initial drugs. The most effective second line agent is vinorelbine, which typically produces a partial response. A planned trial will investigate introducing vinorelbine either in combination with the known effective first strike chemotherapy (augmented first strike) or as a second strike upon NED. Both interventions should improve upon traditional therapy. However, our model predicts the superiority of the sequential first-strike, second-strike strategy in which vinorelbine is applied when the tumor is small and in decline. Evolutionary rescue to vinorelbine is higher when all drugs are given initially and simultaneously in a large, diverse and growing tumor population.
Importantly, there is a precedent for an Anthropocene extinction approach in pediatric acute lymphoblastic leukemia. A highly successful, empirically derived therapy applies immediate and then intermediate “intensification” or “consolidation” therapy and then “maintenance treatment” with different agents following the initial “induction” therapy (33–35).
Finally, theoretical analysis suggests traditional criteria for cancer drug approval based on reduction of tumor size when the disease burden is high (i.e., as an initial therapy) may need to be altered. That is, some treatments (as shown in the simulations and in the Galápagos goat extinction analogy) that have limited efficacy in treating large tumor burdens may be quite effective in smaller and more homogeneous populations following the evolutionary bottleneck produced by the initial treatment. This invites a reevaluation and repurposing of therapeutic agents that otherwise have not been very effective in phase III trials. When applied strategically as a second-strike, they may prove decisive.
Significance:
Anthropocene extinctions suggest a strategy for eradicating metastatic cancers in which initial therapy, by reducing the size and diversity of the population, renders it vulnerable to extinction by rapidly applied additional perturbations.
Acknowledgments
We thank Drs. Katerina Stankova, Dawn Lamanne, Sandy (Alexander) Anderson, and Christopher Whelan for valuable discussion and feedback. This work was supported by the European Commission Horizon 2020 research and innovation program (grant agreement no. 690817), the James S. McDonnell Foundation grant, “Cancer therapy: Perturbing a complex adaptive system,” a V Foundation grant, NIH/NCI R01CA170595, Application of Evolutionary Principles to Maintain Cancer Control (PQ21), and NIH/NCI U54CA143970-05 [Physical Science Oncology Network (PSON)] “Cancer as a complex adaptive system,” R01CA187532-01 title: “Imaging Habitats in Sarcoma”, the National Pediatric Cancer Foundation (nationalpcf.org), and the Jacobson Foundation title: “Integrating evolution into cancer therapy.”
Footnotes
Disclosure of Potential Conflicts of Interest
D.R. Reed is on the advisory board at Loxo Oncology, Epizyme, Janssen, and Shire. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Walther V, Hiley CT, Shibata D, Swanton C, Turner PE, Maley CC. Can oncology recapitulate paleontology? Lessons from species extinctions. Nat Rev Clin Oncol 2015;12:273–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Korolev KS, Xavier JB, Gore J. Turning ecology and evolution against cancer. Nat Rev Cancer 2014;14:371–80. [DOI] [PubMed] [Google Scholar]
- 3.Gatenby RA. A change of strategy in the war on cancer. Nature 2009;459:508–9. [DOI] [PubMed] [Google Scholar]
- 4.Gatenby RA, Frieden BR. Inducing catastrophe in malignant growth. Math Med Biol 2008;25:267–83. [DOI] [PubMed] [Google Scholar]
- 5.Gatenby RA, Silva AS, Gillies RJ, Frieden BR. Adaptive therapy. Cancer Res 2009; 69:4894–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Enriquez-Navas PM, Kam Y, Das T, Hassan S, Silva A, Foroutan P, et al. Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Sci Transl Med 2016;8:327ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang J, Cunningham JJ, Brown JS, Gatenby RA. Integrating evolutionary dynamics into treatment of metastatic castrate-resistant prostate cancer. Nat Commun 2017;8:1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Norton L, Simon R. The Norton-Simon hypothesis revisited. Cancer Treat Rep 1986;70:163–9. [PubMed] [Google Scholar]
- 9.Murray GGR, Soares AER, Novak BJ, Schaefer NK, Cahill JA, Baker AJ, et al. Natural selection shaped the rise and fall of passenger pigeon genomic diversity. Science 2017;358:951–4. [DOI] [PubMed] [Google Scholar]
- 10.Pannell JH. The heath hen. Science 1943;98:174. [DOI] [PubMed] [Google Scholar]
- 11.Gomulkiewicz R, Holt RD. When does evolution by natural selection prevent extinction? Evolution 1995;49:201–7. [DOI] [PubMed] [Google Scholar]
- 12.Gonzalez A, Ronce O, Ferriere R, Hochberg ME. Evolutionary rescue: an emerging focus at the intersection between ecology and evolution. Philos Trans R Soc Lond B Biol Sci 2013;368:20120404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Foley P. Predicting extinction times from environmental stochasticity and carrying capacity. Conserv Biol 1994;8:124–37. [Google Scholar]
- 14.Chirakkal H, Gerber LR. Short- and long-term population response to changes in vital rates: implications for population viability analysis. Ecol Appl 2010;20:783–8. [DOI] [PubMed] [Google Scholar]
- 15.Beissinger SR, American Ornithologists’ Union. Modeling approaches in avian conservation and the role of field biologists. Washington, D.C.: American Ornithologists’ Union; 2006. p.56. [Google Scholar]
- 16.Beissinger SR, McCullough DR. Population viability analysis. Chicago, IL: University of Chicago Press; 2002. p.577. [Google Scholar]
- 17.Melbourne BA, Hastings A. Extinction risk depends strongly on factors contributing to stochasticity. Nature 2008;454:100–3. [DOI] [PubMed] [Google Scholar]
- 18.Boukal DS, Berec L. Single-species models of the Allee effect: extinction boundaries, sex ratios and mate encounters. J Theor Biol 2002;218:375–94. [DOI] [PubMed] [Google Scholar]
- 19.Johnson KE, Howard G, Mo W, Strasser MK, Lima E, Huang S, et al. Cancer cell population growth kinetics at low densities deviate from the exponential growth model and suggest an Allee effect. PLoS Biol 2019;17:e3000399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Konstorum A, Hillen T, Lowengrub J. Feedback regulation in a cancer stem cell model can cause an Allee effect. Bull Math Biol 2016;78:754–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neufeld Z, von Witt W, Lakatos D, Wang J, Hegedus B, Czirok A. The role of Allee effect in modelling post resection recurrence of glioblastoma. PLoS Comput Biol 2017;13:e1005818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown J, Cunningham JJ, Gatenby RA. Aggregation effects and population-based dynamics as a source of therapy resistance in cancer. IEEE Trans Biomed Eng 2017;64:512–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gillespie D. Exact stochastic simulation of coupled chemical reactions. J Phys Chem 1977;81:2340–61. [Google Scholar]
- 24.Broxterman HJ, Pinedo HM, Kuiper CM, Kaptein LC, Schuurhuis GJ, Lankelma J. Induction by verapamil of a rapid increase in ATP consumption in multidrug-resistant tumor cells. FASEB J 1988;2:2278–82. [DOI] [PubMed] [Google Scholar]
- 25.Broxterman HJ, Pinedo HM, Kuiper CM, Schuurhuis GJ, Lankelma J. Glycolysis in P-glycoprotein-overexpressing human tumor cell lines. Effects of resistance-modifying agents. Febs Lett 1989;247:405–10. [DOI] [PubMed] [Google Scholar]
- 26.Cohen Y. Distributed evolutionary games. Evol Ecol Res 2003;5:383–96. [Google Scholar]
- 27.Cohen Y. Evolutionary distribution. Evol Ecol Res 2009;11:611–4. [Google Scholar]
- 28.Clairambault J, Pouchol C. A survey of adaptive cell population dynamics models of emergence of drug resistance in cancer, and open questions about evolution and cancer. Biomath 2019;8 DOI: 10.11145/j.biomath.2019.05.147. [DOI] [Google Scholar]
- 29.Lorz A, Lorenzi T, Hochberg ME, Clairambault J, Perthame B. Populational adaptive evolution, chemotherapeutic resistance and multiple anti-cancer therapies. ESAIM 2013;47:377–99. [Google Scholar]
- 30.Pouchol C, Clairambault J, Lorz A, Trelat E. Asymptotic analysis and optimal control of an integro-differential system modelling healthy and cancer cells exposed to chemotherapy. J Math Pure Appl 2018;116:268–308. [Google Scholar]
- 31.Malempati S, Weigel BJ, Chi YY, Tian J, Anderson JR, Parham DM, et al. The addition of cixutumumab or temozolomide to intensive multiagent chemotherapy is feasible but does not improve outcome for patients with metastatic rhabdomyosarcoma: a report from the Children’s Oncology Group. Cancer 2019;125:290–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weigel BJ, Lyden E, Anderson JR, Meyer WH, Parham DM, Rodeberg DA, et al. Intensive multiagent therapy, including dose-compressed cycles of ifosfamide/etoposide and vincristine/doxorubicin/cyclophosphamide, irinotecan, and radiation, in patients with high-risk rhabdomyosarcoma: a report from the Children’s Oncology Group. J Clin Oncol 2016;34:117–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heikamp EB, Pui CH. Next-generation evaluation and treatment of pediatric acute lymphoblastic leukemia. J Pediatr 2018;203:14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Campana D, Pui CH. Minimal residual disease-guided therapy in childhood acute lymphoblastic leukemia. Blood 2017;129:1913–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pui CH, Yang JJ, Hunger SP, Pieters R, Schrappe M, Biondi A, et al. Childhood acute lymphoblastic leukemia: progress through collaboration. J Clin Oncol 2015;33:2938–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lloyd MC, Cunningham JJ, Bui MM, Gillies RJ, Brown JS, Gatenby RA. Darwinian dynamics of intratumoral heterogeneity: not solely random mutations but also variable environmental selection forces. Cancer Res 2016;76:3136–44. [DOI] [PMC free article] [PubMed] [Google Scholar]