

Abstract
NF1 mutations predispose to neurofibromatosis type 1 (NF1) and women with NF1 have a moderately elevated risk for breast cancer, especially under age 50. Germline genomic analysis may better define the risk so screening and prevention can be applied to the individuals who benefit the most. Survey conducted in several neurofibromatosis clinics in the United States has demonstrated a 17.2% lifetime risk of breast cancer in women affected with NF1. Cumulated risk to age 50 is estimated to be 9.27%. For genomic profiling, fourteen women with NF1 and a history of breast cancer were recruited and underwent whole exome sequencing (WES), targeted genomic DNA based and RNA-based analysis of the NF1 gene. Deleterious NF1 pathogenic variants were identified in each woman. Frameshift mutations because of deletion/duplication/complex rearrangement were found in 50% (7/14) of the cases, nonsense mutations in 21% (3/14), in-frame splice mutations in 21% (3/14), and one case of missense mutation (7%, 1/14). No deleterious mutation was found in the following high/moderate-penetrance breast cancer genes: ATM, BRCA1, BRCA2, BARD1, BRIP1, CDH1, CHEK2, FANCC, MRE11A, NBN, PALB2, PTEN, RAD50, RAD51C, TP53, and STK11. Twenty-five rare or common variants in cancer related genes were discovered and may have contributed to the breast cancers in these individuals. Breast cancer predisposition modifiers in women with NF1 may involve a great variety of molecular and cellular functions.
1 |. INTRODUCTION
Neurofibromatosis type 1 (NF1) is a hereditary neoplasm predisposition syndrome. Besides multiple nonmalignant peripheral neurofibromas, individuals carrying a germline NF1 mutation have an increased risk for glioma and malignant peripheral nerve sheath tumor (MPNST). Increased risk is also seen for several other types of malignancies. Previous studies in Europe and the United States have shown a moderate predisposition for breast cancer, especially among women under age 50.1–6 Meanwhile, somatic NF1 mutations are found in 2–3% of the sporadic breast cancers as putative driver mutations (The Cancer Genome Atlas Network 2012; The Cancer Genome Atlas TCGA data bank, http://www.cbioportal.org/7; Catalogue of Somatic Mutations in Cancer COSMIC database, http://cancer.sanger.ac.uk/cancergenome/projects/cosmic8). These data suggest that neurofibromin, the protein encoded by the NF1 gene, plays a role in the development of breast cancer.
Despite the risk, only a few NF1 families have been reported where multiple affected also have breast cancer.1 From the perspective of targeted breast cancer screening and prevention, a cost-effective strategy may rely on genotype-phenotype correlations between the breast cancer and the types and locations of NF1 mutations. Besides environmental and life-style factors, additional germline variants may modify the neoplastic effect of NF1 mutations as well. Understanding the germline background of NF1 individuals who have developed breast cancer may shed light on the molecular mechanisms potentially leading to targeted treatment strategies.
Family studies have identified rare high-penetrance and moderate-penetrance risk variants for breast cancer in a number of genes, such as BRCA1, BRCA2, TP53, CDH1, ATM, CHEK2, PALB2, BRIP1, NBN, and STK11.9,10 Genome-wide association studies (GWAS) have also identified over 80 common low-penetrance loci associated with sporadic breast cancer.11 The levels of breast cancer risk appear to be a continuum with high-risk variants dominating in the high-risk group, medium-risk variants interacting with low-penetrance variants in the moderate-risk group and low-penetrance variants interplaying in the so called “sporadic cancer” group. Breast cancer in NF1 individuals could be a result of interaction between NF1 gene deleterious mutations and other germline variants.
To elucidate the relationship between NF1 and breast cancer, a multicenter effort was made to review medical records, collect specimens, and analyze germline NF1 mutations and whole exomes in NF1 women affected with breast cancer.
2 |. MATERIALS AND METHODS
2.1 |. Sample and data collection
To identify women affected with NF1 and breast cancer, medical history review was conducted in four Children’s Tumor Foundation (CTF) affiliated neurofibromatosis clinics, that is Henry Ford Health System (HFHS), University of Alabama at Birmingham (UAB), and Johns Hopkins University (JHU). Children’s National Medical Center (CNMC) in the District of Columbia also collected medical history of adult women with NF1 when they presented with their affected children in the clinic.1 This project was also publicized in the newsletters of CTF, NF MidWest advocacy organization’s annual conference and NF Michigan advocacy group’s annual fund raising events. With informed consent, adult women with NF1 and a previous diagnosis of breast cancer were enrolled. Their blood and breast tumor specimens were collected along with medical history, 3 generation family history, and pathology reports. The information collected includes age, ethnicity, age at menarche, typical physical features of NF1, and neoplasms. For breast cancer, the pathological type, stage, and age at diagnosis were collected.
2.2 |. Breast cancer lifetime risk assessment
For lifetime risk assessment, medical record review was conducted on consecutive cases in 4 participating NF clinics.1 This is not a population study. However, the cases represent NF1 women who seek neurofibromatosis specialty care in an outpatient setting in the following geographic areas of the United States: Washington DC, Baltimore in the State of Maryland, Detroit in the State of Michigan and Birmingham in the State of Alabama. Selection bias towards breast cancer is not suspected since these women came to the clinic for neurofibromatosis evaluation and treatment at the time when breast cancer risk in NF1 was not known to the patients.
The age-related cumulative breast cancer risks were assessed by the standard Kaplan-Meier methods using SAS software. A plot was generated using R software.
2.3 |. Germline NF1 gene analysis
Women with a breast cancer history who consented to participate in the germline genomic analysis received comprehensive genetic counseling provided by a certified genetic counselor. The counseling was targeted on NF1 gene testing and whole exome sequencing (WES). Subjects were given options to receive test results on NF1 gene and WES when the result may have impact on clinical care, that is, results that are “clinically actionable”. Fifty-seven genes were determined to be “actionable” based on the 2013 ACMG recommendations to report results when discovered as secondary (incidental) findings by WES.12
Blood specimens from 14 women with NF1 and history of breast cancer (NF+BrCa) were sent to the Medical Genomic Laboratory at the University of Alabama at Birmingham (UAB) for comprehensive NF1 gene analysis (including RNA-based analysis) as previously described.13 NF1 mutations were described following the recommendations of the Human Genome Variation Society using NM_000267.3 as the reference sequence. Exon numbering uses the historical numbering used by the NF1 consortium, followed by the NCBI numbering in square brackets.
2.4 |. Germline WES
Lymphocyte genomic DNA of this NF+BrCa cohort was also sent to the Applied Genomic Technology Center core laboratory in Wayne State University for WES. Libraries were built using the Illumina TruSeq DNA Library Prep Kit series. WES was carried out with 100 bp paired-end runs in rapid run flow cells by Illumina HiSeq 2500 Sequencer following Nextera Rapid Capture Exome protocol.
For the control group, 42 de-identified samples of genomic DNA extracted from lymphocytes were randomly selected from Henry Ford Hospital molecular diagnostic laboratory. These DNA samples belong to presumably healthy women who underwent prenatal hereditary genetic screening tests. WES for these samples was completed in WSU core laboratory and the data served as controls to analyze the data from NF+BrCa group.
WES data analysis was completed by Moffitt Cancer Center Cancer Informatics Core (CIC). Sequence reads were aligned to the reference human genome with the Burrows-Wheeler Aligner (BWA).14 Duplicate identification, insertion/deletion realignment, quality score recalibration, and variant identification was performed with PICARD (http://picard.sourceforge.net/) and the Genome Analysis ToolKit (GATK).15 Genotypes (reference and variant) at variant positions were determined using GATK on all samples simultaneously. Sequence variants were annotated to determine genic context (ie, nonsynonymous, missense, splicing) using ANNOVAR16 and summarized using spreadsheets and a genomic data visualization tool, VarSifter. Additional contextual information was incorporated, including allele frequency in other studies such as 1000 Genomes and the NHLBI Exome Sequence Project, in silico function impact predictions, as well as observed impacts from databases such as ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/),17 and cBioportal for Cancer Genomics.7 Sample stratification was assessed using multidimensional scaling via R and Plink. The samples were separated using genotypes from variants seen at minor allele frequency > 20%. The 1000 Genomes Phase 1 version 3 dataset was used as reference. Inherited variants were enriched for minor alleles by including only those observed at <30% in 1000 Genomes and an internal cohort. To ensure high quality, variants were only considered with GQ score > = 15 and VQSR Tranche level <= 99.0. Differences in mutation rates were assessed at the position and gene levels using the Fisher Exact test. Multiple testing was corrected using the Benjamini-Hochberg method.
For WES data interpretation, Omicia Genome Interpretation Software Opal™ platform (http://app.omicia.com) was also applied to the NF+BrCa group. Omicia generated predicted pathogenicity scores including SIFT, PolyPhen, CADD, MutationTaster, PhyloP, and the Omicia Variant Score. The Omicia score ranges from 0 to 1. A score of greater than 0.5 suggest that a variant is likely to be damaging or deleterious, with higher confidence at values closer to 1.
Omicia analysis was focused on cancer-related genes. Initial filter was set to include variants with an Omicia score of 0.7 or higher and MAF (minimal allele frequency) at 30% (0.3) or less in 1000 Genomes. Further examination was focused on variants fulfilling one or more of the following criteria: (1) MAF at 5% (0.05) or less; or (2) truncation mutations; or (3) Phylo value (conservation value) at 3.0 or higher; or (4) “Disease Causing” classification defined by MutationTaster, a disease-causing prediction software utilizing in silico tests on the DNA and protein levels.18
In this study, a rare variant is defined as MAF at 1% (0.01) or less in the general population, a common variant as MAF greater than 1% (0.01). To assess the significance of suspicious variants in cancer genes, a two-sided exact binomial test of a single proportion was performed comparing the rate for the sample to the population rate (European only). Then the P-values were adjusted for multiple testing using the Holm Stepdown approach—The Bonferroni-Holm correction.
This collaborative study was approved by the Institutional Review Board (IRB) of each participating site and Human Research Protection Organization (HRPO). This project is funded by the U.S. Army Medical Research.
3 |. RESULTS
3.1 |. Lifetime risk
Medical record survey of 423 NF1 women seen in 4 participating NF1 specialty centers was completed. Among them, 20 women had a history of breast cancer. The Kaplan-Meier estimate of breast cancer risk at age 70 is 17.20%, 95% CI [4.14% to 30.27%]. For age 50, the estimate is 9.27%, 95% CI [4.60% to 13.94%]. These estimates were generated using SAS software. The plot was generated using R software. For both of these analyses, estimates were generated using standard Kaplan-Meier methods (Figure 1).
FIGURE 1.
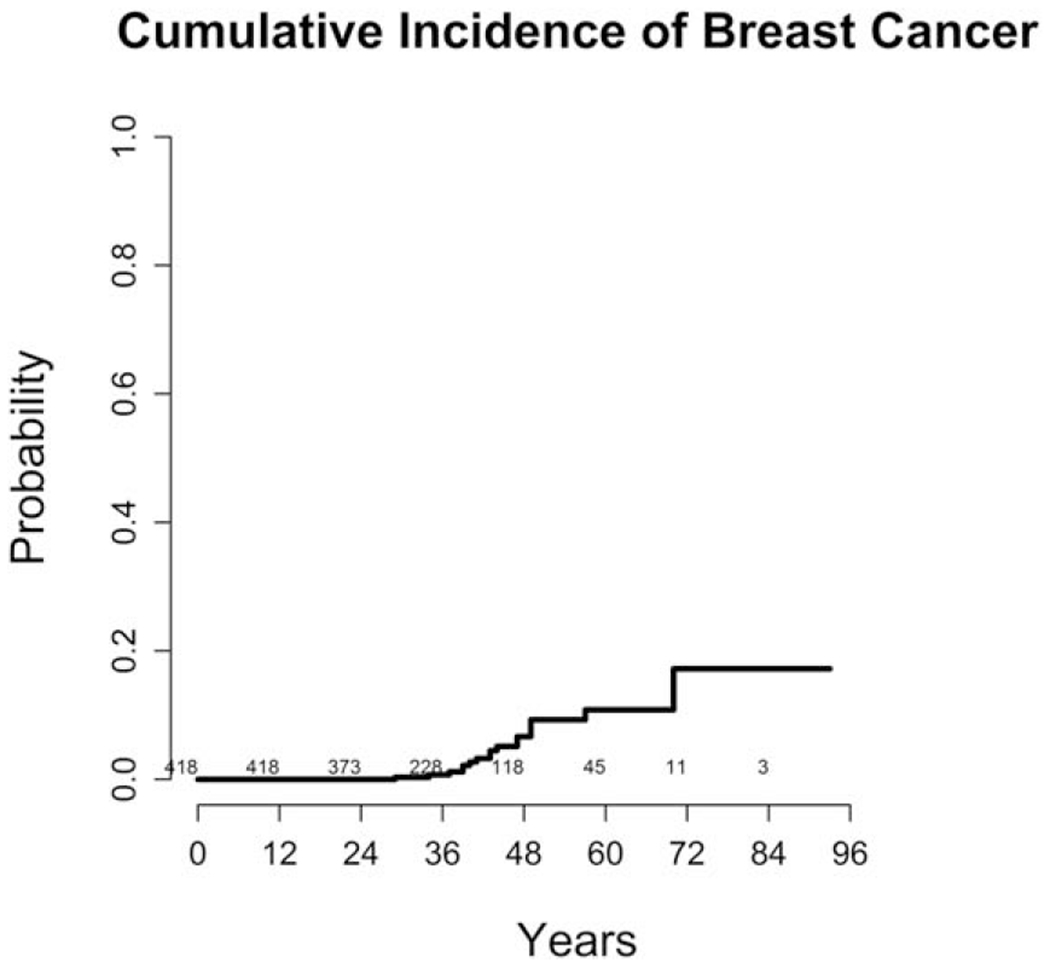
Breast cancer cumulative incidence in women with NF1
A total of 14 women were enrolled in the study and donated blood to undergo NF1 gene analysis and WES. Two women are biologically related. None of the women included in this study had a known history of brain tumor, optic glioma, or MPNST. Four cases reported here were also included in previous publications.1–3 The age at diagnosis, pathological type, personal and family history is described in Table 1.
TABLE 1.
Breast cancer pathology, clinical features and NF1 mutations
| Breast cancer |
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age |
Pathology |
Mutations (DNA level; RNA level; Protein level) |
||||||||||||||
| ID | Diagnosis | Menarche | Type | ER | PR | HER2 | Ki-67 | Grade | Other neoplasms | NF1 features | Family history cancer | Family history NF1 | Female relative withbreast ca and NF1 | Exon | Type | Description |
| 1 | 56 | 13 | IDC | NA | NA | NA | NA | III | − | LN, AF, CN, PN | − | De novo | − | 33 [42] | OOF skipping exon 33 [42] - PSC | c.6364G>A; r.6085_6364del; p.Val2029Lysfs*7 |
| 2 | 53 | NA | IDC DICS | + | + | − | NA | III | Carcinoid tumor; Pheochromocytoma | SS, LN, CAL, AF, SC, CN, PN | Breast; Ovary; Esophagus | Inherited | − | 10c [14] | frameshift - PSC | c,1541_1542delAG; r,1541_1542delag; Gln514Argfs*43 |
| 3 | 60 | 16 | DCIS | 98% | 22% | NA | NA | NA | − | LD, MC, LN, CAL, AF, SC, CN, PN | Breast; Ovary; Pancreas; gastrium | De novo | − | 37 [46] | IF skipping exon 37 [46] | c.6792C>G; r.6757_6858del; p.Ala2253_Lys2286del |
| 4 | 49 | NA | IDC DCIS | − | − | + | NA | II | Cervical cancer | LN, CAL, CN | Breast; Pheochromocytoma | Inherited | NA | 15 [20] | nonsense - PSC | c.2398G>T, r.2398g>u; p.Glu800* |
| 5 | 45 | 14 | NA | NA | NA | NA | NA | NA | − | SS, CAL, AF, CN | − | De novo | − | Intron 26 [34] | OOF splicing - PSC | c.4515-20_4515-18delAAG; r.4514_4515ins4515-14_4515-1; p.Argl505Serfs*53 |
| 6 | 47 | 11 | IDC DCIS LCIS | >90% | 70% | − | NA | II | − | SS, LN, CAL, AF, CN | Breast; Prostate | NA | − | 16 [21] | Truncation and low level OOF splicing - PSC | c.2621_2634dupAGGGTTCTATGATT; r.2621_2634dupaggguucuaugauu and r.2618_2850del; p.Ser879Argfs*4 and p.Lys874Phefs*4 |
| 7 | 39 | NA | IDC DCIS | 82% | 71% | NA | NA | III | − | SS, LN, CAL, AF, NF, PN | − | Inherited | − | 29–30 [38–39] | copy number variant - PSC | c.(5045_5337}_(5625_5796}del; r.5206_5749del; p.Glyl737Leufs*3 |
| 8 | 41 | 11 | IDC DCIS | + | − | Eq | NA | II | − | SS, CAL, AF, PN | Lung; Colon; Pheochromocytoma | Inherited | − | 12a [16] | Mis-sense | C.1733T>G; r,1733u>g; p.Leu578Arg |
| 9 | 49 | 14 | IDC DCIS | 30% | − | + | 20% | II | − | LN, CAL, AF | Breast | NA | NA | Intron 31 [40]) | OOF splicing - PSC | c.5943 + 1G>T; leads to out-of-frame splice r.5901_5943del; p.Met1967llefs*10 |
| 10 | 44 | NA | IDC DCIS | 80% | 100% | − | 15% | II | − | CAL, AF, SC, CN | Breast; Colon; Lung; Prostate | Inherited | NA | 16 [21] | frameshift - PSC | c.2728_2729delGG; r.2728_2729delgg; p.Gly910Thrfs*8 |
| 11 | 58 | NA | NA | NA | NA | NA | NA | NA | − | CAL, AF, BD, CN, PN | − | NA | NA | 28 [37] | frameshift - PSC | c.4910_4911delTT; r.4910_4911deluu; p.Phel637Serfs*3 |
| 12 | 52 | 13 | IDC | − | − | + | NA | III | − | LD, LN, AF, CN | Breast; Gastrium | De novo | − | 30 [39] | frameshift - PSC | c.5667dupT; r.5667dupu; p.llel890Tyrfs*2 |
| 13 | 47 | 13 | IDC DCIS | 24% | 2% | − | 59% | III | − | SS, MC, AF, CN | Breast; Ovary | Inherited | + | 9 [11] | deep intronic splice mutation - PSC | c.1260 + 1604A>G, r,1260_1261ins1260 + 1605_1260 +1646; p.Ser421_Val2818delinsLeuThrThr* |
| 14 | 42 | 13 | IDC DCIS | + | + | − | 10% | II | − | SS, CAL, AF, CN | Breast; Ovary | Inherited | + | 9 [11] | deep intronic splice mutation - PSC | c.1260+1604A>G, r,1260_1261ins1260 + 1605_1260 +1646; p.Ser421_Val2818delinsLeuThrThr* |
Abbreviations: IDC, invasive ductal carcinoma; DCIS, ductal carcinoma in situ; NA, information not available; ER, estrogen receptor status; PR, progesterone receptor status; HER2, human epidermal growth factor receptor 2 expression; Ki-67, Ki-67 proliferation marker; Eq, equivocal. Grade: I = Well differentiated, II = Moderately differentiated, III = Poorly differentiated. NF1 Features, that is physical features: SS (short stature); LD (learning disability); MC (macrocephaly); LN (Lisch nodules); CAL (>= 6 café au lait macules); AF (axillary freckling); BD (bony dysplasia); SC (scoliosis); CN (>= cutaneous neurofibromas); PN (plexiform neurofibroma). OOF, Out-of-frame; IF: In-frame; PSC: premature stop-codon.
3.2 |. Germline NF1 gene analysis
NF1 gene analysis discovered 3 cases (21%, 3/14) with mutations causing in-frame splicing error or exon skipping, 10 mutations (71%, 10/14) with truncation effect (3 of which were nonsense mutation and 7 of which were deletion/duplication/complex rearrangement), and one case (7%, 1/14) with missense variant (Table 1).
3.3 |. Germline WES
Variants were identified by the CIC bioinformatics pipeline and filtered to remove sequencing artifacts. The following types of variants were categorized and analyzed: nonsynonymous single nucleotide variant, nonframeshift insertion or deletion, splicing variant, and variant resulting in protein truncation (TrnV), such as nonsense single nucleotide variant, frameshift insertion or deletion, and stop codon gain. Stop codon loss is also placed in the category of “TrnV” for sorting purpose. Since the ancestries of majority of the cases were reported to be Europeans, the statistical analysis was focused on the individuals of European ancestry. Statistical analysis was not possible for the results from non-European individuals because of the small number. Out of 56 samples (14 cases and 42 controls), ancestry cluster analysis showed that 21 were non-Europeans (18 in control group and 3 in NF+BrCa group). The remaining 11 NF+BrCa cases and 24 controls were analyzed. The variant analysis was reported in 3 categories: (1) variants at the position level; (2) all variants collapsed to the gene level; and (3) truncating variants (TrnVs).
Variants filtered by CIC are described in Table 2. Even though numerous variants or genes in the NF+BrCa group reached P < 0.05, none of them satisfied the multiple tests with q < 0.05. TrnVs of NF1 gene have reached P = 4.91 × 10−5 and q = 0.079. Since the NF+BrCa cohort is selected based on NF1 germline mutations, the near significance of NF1 TrnV is expected.
TABLE 2.
Germline variants in NF+BrCa group detected by WES
| All V | All V p < 0.05 | Genes with V | Genes with V p < 0.05 | TrnV | TrnV p < 0.05 | |
|---|---|---|---|---|---|---|
| MAF =<5%) | 15,336 | 54 | 8,503 | 81 | 672 | 4 |
| 30% >= MAF >5% | 13,518 | 319 | 2,875 | 145 | 942 | 18 |
V, variant; MAF, minimal allele frequency in 1000 Genomes; TrnV, truncation variant.
Germline WES did not identify any pathogenic variant in the known or suspected breast cancer genes of moderate or high penetrance, such as ATM, BRCA1, BRCA2, BARD1, BRIP1, CDH1, CHEK2, FANCC, MRE11A, NBN, PALB2, PTEN, RAD50, RAD51C, TP53, or STK11. During medical record review, none of the NF+BrCa cases had a reported history of mutation in these genes either.
Omicia analysis of 499 cancer-related genes (Supporting Information Table 1) was applied to the 14 NF+BrCa datasets. The filtering strategy is described in the Materials and Methods. In addition, variants not meeting quality standard of CIC were eliminated. Variants of potential significance were curated. Twenty-five variants of potential significance were sorted based on the frequency encountered in the NF+BrCa group, MAF in the general population, the P-value comparing frequency with MAF in the population database, the level of conservation across 100 vertebrates selected by UCSC browser, and the observation as “confirmed somatic mutations” in databases such as COSMIC (Catalogue of Somatic Mutations in Cancer) or TCGA (The Cancer Genome Atlas) via cBioportal Cancer Genomic Browser. Rare variants are summarized in Table 3, common variants in Table 4.
TABLE 3.
Rare variants (aMAF =< 1%) in 499 cancer related genes in NF+BrCa samples
| Found in NF+BrCa samples | Nucleotide in UCSC browser | Somatic variant in COSMIC | Variants (described as Reference SNP Cluster ID or coordinates from Human GRCh37/hg19 Assembly) |
|---|---|---|---|
| N = 1 (found in one sample); bp is not significant | cHighly Conserved | – – –-> | APC rs150973053; ATG16L1 rs149927020; AXIN2 rs755990979;eEGFR rs121913418; ERCC2 rs149818919; ERCC6 rs142580756; FANCI chr15:89807202(T>C); MSH2 rs34136999; NELL1 rs144555978; PIGF rs367591899; PIK3C2A rs138982128; SOS1 rs56219475 |
| dLess Conserved | Observed | eAPC rs747362422;eMET rs56391007; | |
| N = 2 (found in two samples); bp=<0.02 | cHighly Conserved | – – –-> | MSH3 rs766742870; fUVRAG rs75431002 |
| dLess Conserved | – – –-> | fBUB1B rs56079734; PIK3C2A rs61755370; PLCG rs75472618; PLCG rs187956469; PMS2 rs63750668; fRECQL4 rs35842750; |
MAF: Minimal Allele Frequency in 1000 Genomes.
p-value of allele frequency (AF) in NF+BrCa compared with control group or with population AF in UCSC genomic browser.
Highly Conserved: PhyloP value is greater than 4.0.
Less Conserved: PhyloP value is less than 4.0.
Variants observed in COSMIC.
APC rs747362422 was observed in 3 samples of different type of cancers, all confirmed somatic.
EGFR rs121913418 was observed in 6 samples, 2 were confirmed somatic.
MET rs56391007 was observed in 23 samples, 8 were confirmed somatic.
Discordant population allele frequencies between ancestries.
RECQL4 rs35842750 and BUB1B rs56079734: Both alleles were observed in 2 non-European samples of NF+BrCa. MAF of non-Europeans in 1000 Genomes, such as Africans, is higher than 1%.
UVRAG rs75431002: This allele was observed in one European and one non-European NF+BrCa samples. Its population MAF is rare in Europeans, but is slightly higher in non-Europeans. MAF is 3% in Sierra Leon.
TABLE 4.
Common alleles in 499 cancer related genes and statistical status in NF+BrCa samples
| ID | NF1 Variant Description |
FANCI rs62020347 C>T Highly Conserved European AF = 0.068 Global AF = 0.03 NF+BrCa AF 6/22 = 0.273 Raw P = 0.0056 StepdownHolm P = 0.0560 |
ERCC4 rs1800067 G>A Highly Conserved European AF = 0.077 Global AF = 0.03 NF+BrCa AF 6/22 = 0.273 Raw P = 0.0102 StepdownHolm P = 0.0918 |
NOD2 rs2066847 G>GC Less Conserved European AF = 0.022 Global AF = 0.01 NF+BrCa AF 3/22 = 0.136 Raw P = 0.0245 StepdownHolm p = 0.1960 |
|---|---|---|---|---|
| 1 | c.6364G>A; r.6085_6364del; p.Val2029Lysfs*7 | T/T | A/G | G/G |
| 2 | c.1541_1542delAG; r.1541_1542delag; Gln514Argfs*43 | C/C | G/G | G/G |
| 3 | c.6792C>G; r.6757_6858del; p.Ala2253_Lys2286del | C/C | A/G | G/G |
| 4 | c.2398G>T, r.2398g>u; p.Glu800* | C/C | G/G | G/GC |
| 7 | c.(5045_5337)_(5625_5796)del; r.5206_5749del; p.Gly1737Leufs*3 | C/T | G/G | G/G |
| 8 | c.1733T>G; r.1733u>g; p.Leu578Arg | C/C | A/A | G/GC |
| 9 | c.5943 + 1G>T; leads to out-of-frame splice r.5901_5943del; p.Met1967Ilefs*10 | C/T | A/G | G/G |
| 11 | c.4910_4911delTT; r.4910_4911deluu; p.Phe1637Serfs*3 | C/T | A/G | G/G |
| 12 | c.5667dupT; r.5667dupu; p.Ile1890Tyrfs*2 | C/T | G/G | G/GC |
| 13 | c.1260 + 1604A>G, r.1260_1261ins1260 + 1605_1260 + 1646; p.Ser421_Val2818delinsLeuThrThr* | C/C | G/G | G/G |
| 14 | c.1260 + 1604A>G, r.1260_1261ins1260 + 1605_1260 + 1646; p.Ser421_Val2818delinsLeuThrThr* | C/C | G/G | G/G |
Highly Conserved, PhyloP value of nucleotide is greater than 4.0; Less Conserved, PhyloP value of nucleotide is less than 4.0; European AF, European population allele frequency in NHLBI Exome Sequencing Project; Global AF, 1000 Genomes global allele frequency; NF+BrCa AF, Allele rate in NF+BrCa samples; StepdownHolm: The Bonferroni-Holm correction (Holm S. A simple sequentially rejective Bonferroni test procedure. Scandinavian Journal of Statistics. 1979; 6: 65–70). Raw P-values are for the individual two-sided exact binomial test of a single proportion.
4 |. DISCUSSION
The lifetime breast cancer risk estimation of 17.2% is similar to the rate observed in Finland.19 A French NF1 cohort of n = 565 demonstrated 69.5% with a truncating mutation, 6.5% with an in-frame splicing mutation, and 7.5% with a missense mutation.20 The variant spectrum in the current NF+BrCa cohort is similar on the truncating (71%) and missense (7%) categories, but appears to be more in the in-frame splice category (21%, 3/14); however, the number is too small to draw a statistical conclusion.
Pathogenic mutations in NF1 gene only confer a moderate risk of breast cancer. However, these tumors appear to result in worsened survival in comparison to the sporadic counterparts.19 To define better the targetable individuals who may benefit the most from early screening or prevention, it is logical to search for additional cancer driving factors. In this cohort, 3 women have a female relative affected with both NF1 and breast cancer which suggest additional germline genetic factors.
Several articles have reported a total of 6 isolated cases of breast cancer carrying germline NF1 and BRCA1 gene mutations.4,21–23 A few cases of breast cancer in women with NF1 across the United States were reported to harbor a BRCA1 mutation as well (Wang, personal communication). However, WES of the current cohort did not find any deleterious pathogenic variants in the genes known to be associated with hereditary breast cancer risk (ATM, BRCA1, BRCA2, BARD1, BRIP1, CDH1, CHEK2, FANCC, MRE11A, NBN, PALB2, PTEN, RAD50, RAD51C, TP53, and STK11). It suggests that besides NF1 gene, low-penetrance alleles of various genes, rare or common, may have also contributed to the breast cancer collectively.
As expected, a significant amount of germline variants have been identified in individuals with breast cancer and NF1. We did not have the opportunity to recruit NF1 women without breast cancer as control group. The current control group was made of individuals representing general population. By comparing variants found in the study group and in the control group, effort has been made to identify variants potentially contributive to the breast cancer in NF1. The analysis would not distinguish any unique variants in NF1 women with breast cancer from the ones without breast cancer. Because of the small sample size, a definitive conclusion of association between the variants and NF1 breast cancer cannot be reached. Nevertheless, these findings offer clues and will contribute to the better understanding of germline genetic modifiers in the future. Multivariable statistical analysis cannot be used to determine if the rare variants contributed to the breast cancer because of small sample size. The following factors are considered for a rare variant (MAF =< 1%, 0.01 in the general population): (a) a rare variant appeared once or more times in the 14 case NF+BrCa cohort; (b) a rare variant occurs on a highly conserved nucleotide across 100 vertebrae species; (c) a rare variant has been observed as a somatic mutation in malignant neoplasms. These features may be additive to increase the suspicion of the association with breast cancer. On the other hand, for a common allele, defined as 1% (0.01) < MAF =< 30% (0.3) in the general population, the level of conservation across species and the frequency observed in the cancer somatic mutation databases are both contributory for possible association with breast cancer. In addition, it may be more suggestive of the association when the common variant rate in NF+BrCa cohort is significantly higher than the rate in the general population (Table 4). Variants of potential significance listed in Tables 3 and 4 belong to the genes of a wide spectrum of functions, such as signaling pathways, DNA repairs, autophagy, immune responses, growth factors and receptors, tyrosine kinase receptor, chromosome modulator, and cell cycle check-points. Some of these variants have had known functional significance. For example, NOD2 rs2066847 has a significant association with Crohn’s disease;24–26 nonsmall cell lung cancer (NSCLC) with EG FR rs121913418 (D761N variant) has sensitivity to certain EGFR inhibitor (AEE788)27; BUB1B rs56079734 was reported as a colon cancer pathogenicity allele and confers chromosomal instability.28 None of these variants appear to fit into a germline “pathogenic” or “likely pathogenic” category defined by ACMG variant classification guidelines.29 Some variants have never been reported before. Others have been reported and catalogued in ClinVar17 and are labeled as “benign” or “likely benign” or “variant of uncertain significance”.
Two individuals in the study group (NF+BrCa) are related to each other. However, none of the above variants exists in both individuals; thus this has not introduced bias into the P-value computation.
To summarize, the current study has not uncovered any high-penetrance variants for breast cancer in addition to those in the NF1 gene. For rare variants, low-penetrance variants or common variants, a much larger sample size is required to prove a significant cause-effect relationship with multiple analyses. In theory, NF1 families with multiple individuals affected with breast cancer may provide evidence of association between the cancer and genetic modifiers when NF1 pathogenic variant and other suspected risk gene variant are co-inherited in the family. However, the penetrance of each modifier may be too low, the number of modifiers may be too abundant for this method to be effective. Genomic modifiers in noncoding region can also be contributors and has not been studied so far for breast cancer in NF1.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank the personnel in Henry Ford Health System, Amy Decker, Carin Yates, Kristin Monaghan, and Helen Perez for their advice and assistance. The authors also wish to especially thank Dr. Howard McLeod, the chairman of the Department of Individualized Cancer Management in Moffitt Cancer Center, for his generous support to complete this project.
Funding information
This project was supported by the new investigator award from U.S. Army Medical Research. Medical Research and Materiel Command, Award number: W81XWH-11-1-0671
Footnotes
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the supporting information tab for this article.
REFERENCES
- [1].Wang X, Tousignant RN, Levin AM, et al. Indicator exploration for cancers in women with neurofibromatosis type 1—a multi-centre retrospective study. J Genet Syndr Gene Ther. 2016;7:292 10.4172/2157-7412.1000292 [DOI] [Google Scholar]
- [2].Seminog OO, Goldacre MJ. Age-specific risk of breast cancer in women with neurofibromatosis type 1. Br J Cancer 2015;112:1546–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wang X, Levin AM, Smolinski SE, Vigneau FD, Levin NK, Tainsky MA. Breast cancer and other neoplasms in women with neurofibromatosis type 1: a retrospective review of cases in the Detroit metropolitan area. Am J Med Genet A. 2012;158a:3061–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Madanikia SA, Bergner A, Ye X, Blakeley JO. Increased risk of breast cancer in women with NF1. Am J Med Genet A. 2012;158a:3056–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sharif S, Moran A, Huson SM, et al. Women with neurofibromatosis 1 are at a moderately increased risk of developing breast cancer and should be considered for early screening. J Med Genet. 2007; 44:481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Uusitalo E, Rantanen M, Kallionpaa RA, et al. Distinctive cancer associations in patients with neurofibromatosis type 1. J Clin Oncol. 2016; 34:1978–1986. [DOI] [PubMed] [Google Scholar]
- [7].Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Forbes SA, Beare D, Boutselakis H, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017; 45:D777–D783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Stratton MR, Rahman N. The emerging landscape of breast cancer susceptibility. Nat Genet. 2008; 40:17–22. [DOI] [PubMed] [Google Scholar]
- [10].Tung N, Domchek SM, Stadler Z, et al. Counselling framework for moderate-penetrance cancer-susceptibility mutations. Nat Rev Clin Oncol. 2016; 13:581–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Skol AD, Sasaki MM, Onel K. The genetics of breast cancer risk in the post-genome era: thoughts on study design to move past BRCA and towards clinical relevance. Breast Cancer Res. 2016; 18:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Green RC, Berg JS, Grody WW, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013; 15:565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Messiaen LM, Callens T, Mortier G, et al. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum Mutat. 2000; 15:541–555. [DOI] [PubMed] [Google Scholar]
- [14].Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011; 43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Landrum MJ, Lee JM, Benson M, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016; 44:D862–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014; 11:361–362. [DOI] [PubMed] [Google Scholar]
- [19].Uusitalo E, Kallionpaa RA, Kurki S, et al. Breast cancer in neurofibromatosis type 1: overrepresentation of unfavourable prognostic factors. Br J Cancer. 2017; 116:211–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sabbagh A, Pasmant E, Imbard A, et al. NF1 molecular characterization and neurofibromatosis type I genotype-phenotype correlation: the French experience. Hum Mutat. 2013; 34:1510–1518. [DOI] [PubMed] [Google Scholar]
- [21].Jeon YW, Kim RM, Lim ST, Choi HJ, Suh YJ. Early-onset breast cancer in a family with neurofibromatosis type 1 associated with a germline mutation in BRCA1. J Breast Cancer. 2015; 18:97–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ceccaroni M, Genuardi M, Legge F, et al. BRCA1-related malignancies in a family presenting with von Recklinghausen’s disease. Gynecol Oncol. 2002; 86:375–378. [DOI] [PubMed] [Google Scholar]
- [23].Campos B, Balmana J, Gardenyes J, et al. Germline mutations in NF1 and BRCA1 in a family with neurofibromatosis type 1 and early-onset breast cancer. Breast Cancer Res Treat. 2013; 139:597–602. [DOI] [PubMed] [Google Scholar]
- [24].Hugot JP, Chamaillard M, Zouali H, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001; 411:599–603. [DOI] [PubMed] [Google Scholar]
- [25].Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012; 491:119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Barrett JC, Hansoul S, Nicolae DL, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008; 40:955–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kancha RK, von Bubnoff N, Peschel C, Duyster J. Functional analysis of epidermal growth factor receptor (EGFR) mutations and potential implications for EGFR targeted therapy. Clin Cancer Res. 2009; 15:460–467. [DOI] [PubMed] [Google Scholar]
- [28].Cahill DP, Lengauer C, Yu J, et al. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998; 392:300–303. [DOI] [PubMed] [Google Scholar]
- [29].Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.