

Abstract
Erythropoiesis is tightly regulated by the growth factor erythropoietin (Epo). Signal activation begins when Epo engages its cognate receptor, Epo-R, triggering receptor homodimerization, and recruitment of signaling intermediates including Jak2 that phosphorylates both the receptor cytoplasmic tail and downstream effectors including the transcription factor, STAT5. Transcription factors subsequently activate transcription of prosurvival and prodifferentiation genes responsible for red blood cell production. The fidelity of Epo-R signaling is dependent upon residence within detergent insoluble membrane lipid raft fractions. Lipid rafts are membrane microdomains that serve as signaling scaffolds composed of densely packed sphingolipids and cholesterol where receptors and intermediate signaling proteins are recruited and interact to execute stimuli. Disruption of lipid rafts is detrimental to Epo signaling, a phenomenon that may be utilized to design novel therapeutics for conditions in which Epo signaling is deficient. Here, we review the Epo signaling cascade, particularly, as it relates to localization and dependence on lipid rafts, and discuss considerations for novel therapeutic design.
1. INTRODUCTION
Erythropoietin (Epo) is the primary cytokine responsible for erythropoiesis, stimulating both the proliferation and differentiation of erythroid progenitors into mature red blood cells (RBCs). Basal Epo levels are required to maintain steady state levels of RBCs in the blood, however, during times of blood loss or hypoxic stress, expression and secretion of the cytokine by renal interstitial fibroblasts is quickly upregulated in response to hypoxic sensors, thereby expanding the pool of erythroid progenitors and differentiation of mature erythrocytes. As such, tight regulation of the pathway is critical. Biological response to the cytokine is initiated upon engagement of Epo with its cognate receptor, Epo-R. This interaction induces conformational changes and receptor homodimerization that triggers downstream signaling events resulting in the activation of transcription factors that promote erythroid expansion. Recombinant human Epo (rhEpo) has widespread therapeutic use and is standard of care for a number of anemias including that arising from chronic kidney disease, chemotherapy, or infection, and a group of disorders known as the myelodysplastic syndromes (MDSs). In MDS, rhEpo treatment rescues diminished Epo signaling restoring erythropoiesis and correcting anemia resulting in transfusion independence (Hoefsloot, van Amelsvoort, Broeders, et al., 1997; Mantovani, Lentini, Hentschel, et al., 2000). Thorough investigation of Epo-R signaling is a key to understanding the pathobiology of disorders such as MDS, as well as identifying novel therapeutic strategies.
Lipid rafts are small (20–100 nm) plasma membrane microdomains composed of densely packed cholesterol and sphingolipids that are free to “float” laterally throughout the phospholipid bilayer of the plasma membrane. The role of lipid rafts in immune signaling has been well studied, particularly in B- and T-cell receptor (TCR) signal activation and in the formation of immunological synapses. These lipid microdomains act as docking stations whereby receptors and signal intermediates interact with one another. Protein recruitment and raft-associated spatial proximity facilitates downstream signal cascade activation (Kabouridis & Jury, 2008; McGraw, Fuhler, Johnson, et al., 2012). Here, we will explore the role of lipid rafts in erythroid precursors, and particularly, the relationship between lipid rafts and Epo-R signal fidelity. We will conclude that intact lipid raft microdomains are imperative to maximize Epo-R signal fidelity and that this phenomenon may be exploited to uncover novel therapeutic strategies to treat conditions with aberrant signaling.
2. EPO AND EPO RECEPTOR
Epo is the primary regulator of erythropoiesis and is critical for tissue oxygenation under both steady-state conditions and after acute blood loss or hypoxia. The Epo gene was first cloned in 1985 after purification by Miyake et al. who isolated the cytokine from the urine of patients with aplastic anemia. Identification of the amino acid sequence quickly led to identification of the nucleotide sequence followed by full characterization of the gene (Lacombe & Mayeux, 1998; Miyake, Kung, & Goldwasser, 1977). The Epo gene is located on chromosome 7q22, and transcriptional regulation is controlled by the transcription factors hypoxia-inducible factor (HIF)-1-alpha, signal transducer and activator of transcription 5 (STAT5), STAT3, and GATA-binding protein 1 (GATA-1) (Lacombe & Mayeux, 1998; Mulcahy, 2001; Zhao, Kitidis, Fleming, Lodish, & Ghaffari, 2006). These factors are critical to a cell’s ability to react to changes in physiologic needs for circulating erythrocytes. The Epo protein is a 34-kDa circulating glycoprotein that is produced predominately in the kidney and embryologically in the liver (Ebert & Bunn, 1999; Fisher, 2003; Lacombe & Mayeux, 1998). rhEpo protein has great clinical utility and is used to treat a number of conditions including anemia caused by chronic renal failure, chronic inflammatory disorders, chemotherapy, and MDS. Although rhEpo administration alleviates anemia by promoting the production of RBCs, rapid correction is associated with thromboembolism and chronic use with iron deficiency (Singbartl, 1994).
The Epo-R is the primary receptor that binds to and transmits the activation signal of the Epo cytokine. The Epo-R is a classical Type-I cytokine receptor with an extracellular domain that interacts with the microenvironment, a transmembrane region that spans the phospholipid bilayer, and a cytoplasmic tail that contains eight tyrosine phosphorylation sites that serve as docking locations for signaling adapters. Specific to the Type-I family, these cytokine receptors contain no intrinsic kinase activity and therefore rely upon the recruitment of kinases and adapter molecules to potentiate the receptor signal (D’Andrea & Zon, 1990; Lacombe & Mayeux, 1998). Epo-R maturation begins in the endoplasmic reticulum, after which it is transported to the Golgi apparatus. Only a small proportion of receptors will leave the Golgi, mature via heavy glycosylation, and be transported to and expressed on the plasma membrane. Expression of mature Epo-R on the plasma membrane is relatively low under stimulus-free conditions with less than 100 receptors predicted to be expressed at any given instance (D’Andrea & Zon, 1990; Lacombe & Mayeux, 1998). Importantly, transport of the receptor through its maturation process is dependent on its interaction with Janus Kinase 2 (Jak2) (Huang, Constantinescu, & Lodish, 2001). Jak2 is not only important for the surface expression of the receptor; it is also the primary effector kinase of the Epo/Epo-R signaling axis. Ligand engagement induces receptor conformational changes and homodimerization that results in the autophosphorylation of Jak2. Jak2, in turn, phosphorylates the receptor cytoplasmic tail to recruit a number of adaptor intermediates leading to transcriptional activation of genes involved in promoting the proliferation and differentiation of erythrocytes. The Epo-R signaling cascade and activation steps will be described in further detail in the next section. Epo-R signal attenuation occurs by negative-feedback inhibition via several mechanisms including suppressor of cytokine signaling (SOCS) proteins, negative-regulating phosphatases, receptor internalization and ubiquitination, and proteasomal degradation. The delicate balance between receptor maturation and receptor turnover is highly regulated and must occur rapidly as the body responds to external stimuli. As such, defects in this regulation, or in the receptor itself, will have damaging effects leading to disease pathogenesis and fluctuations in total red cell mass.
There are a number of primary bone marrow conditions that can be responsive to treatment with rhEpo such as MDS. MDS patients have diminished STAT5 transcription factor activity following Epo stimulation, indicating impairment in Epo-R signal response. A small proportion of patients do harbor single site Epo-R mutations; however, the functional consequence of these is unknown and does not appear to be detrimental to downstream signaling (Lewinski, Floru, Cohen, & Mittelmann, 1994). In myeloproliferative neoplasms (MPNs), such as polycythemia vera (PV), there is hypersensitivity to Epo despite lower levels of circulating cytokine, owing to somatic mutations involving JH2 pseudokinase autoinhibitory domain of JAK2, resulting in constitutive activation (McMullin & Percy, 1999). Similarly, in both erythroleukemia, a type of acute myeloid leukemia with pancytopenia, and Diamond Blackman Anemia, an inherited bone marrow failure syndrome with profound anemia, the Epo-R remains intact (McMullin & Percy, 1999). Alternatively, in Familial Erythrocytosis, a condition characterized by erythrocytosis, inherited mutations in the EpoR gene introduce a premature stop codon in the amino acid sequence resulting in halted translation. Interestingly, the truncated receptor is hypersensitive to circulating cytokine resulting in increased Jak2 and STAT5 activation (Watowich, Xie, Klingmuller, et al., 1999). This leads to sustained proliferation and increased RBC mass causing dizziness, excess bleeding, and increased risk for thrombus formation along with other symptoms in affected individuals. The lack of universal alterations in Epo-R or diminished circulating cytokine in these and other disorders, despite clear differences in Epo signaling, suggest that other factors involved in the signaling axis must be explored.
3. CONDITIONS WITH ABERRANT EPO SIGNALING
With the exception of Familial Erythrocytosis, aberrant Epo signaling found in a number of disorders cannot be attributed to dysfunctional Epo-R. Instead, aberrancies exist somewhere downstream of the receptor. MPNs are hematopoietic disorders characterized by increased production of particular cell lineages. The most common MPNs include PV characterized by increased red cell mass, platelets, and often leukocytes, primary myelofibrosis (PMF) characterized by a marked increase in bone marrow fibrosis, and essential thrombocytopenia (ET) characterized by sustained elevation in platelets. In PV patients, there are lower levels of circulating cytokine, and erythroid progenitor cells are able to proliferate independent of Epo. This is attributed to hypersensitivity to Epo, and other growth factors (McLornan, Percy, & McMullin, 2006). Just over a decade ago, a mutation in Jak2 that changes valine to phenylalanine at amino acid 617 (V617F) was identified. Since, this mutation has been identified in 95% of PV patients and 50% of PMF and ET patients. In those cases that do not harbor the V617F mutation, other mutations in either Jak2 or Jak2-regulating genes have been identified. These data demonstrate that aberrant Jak2 signaling downstream of the Epo-R underlies aberrant signaling. The V617F mutation causes loss of Jak2 autoinhibition resulting in constitutively active kinase activity signaling resulting in over transcribed erythroid promoting genes (Griner, McGraw, Johnson, List, & Reuther, 2013).
The MDSs are hematopoietic stem cell malignancies characterized by ineffective hematopoiesis. MDS patients display bone marrow failure, dysplastic bone marrow features, cytopenias, and an increased propensity for transformation to acute myeloid leukemia. MDS primarily affects older individuals (greater than 60 years). Survival depends on a number of factors and ranges from many years to only months. Prognostication is determined by bone barrow blast percentage, number and severity of cytopenias, cytogenetic abnormalities, and presence of somatic gene mutations. Anemia, thrombocytopenia, and neutropenia are common in MDS and the pathogenesis of the disease in unclear. Recent evidence suggests that dysregulation of innate immune signaling and chronic inflammation of the bone marrow microenvironment drive disease pathogenesis. Predisposing factors to MDS are also not clear; however, individuals with therapy-related toxicity such as that from chemotherapy or radiation are at increased risk for the MDS development. Clonal hematopoiesis also appears to contribute to MDS risk. Clonal hematopoiesis is the process of somatic gene mutation acquisition and clonal expansion. As a population ages, so does the frequency of acquiring somatic mutations in genes common to MDS. Clonal hematopoiesis is associated with increased risk for hematological malignancies including MDS (Genovese, Kahler, Handsaker, et al., 2014; Jaiswal, Fontanillas, Flannick, et al., 2014). Although our knowledge of the factors that contribute to MDS pathogenesis is increasing, we do not know what causes the deficient Epo-R signal. Therefore, it is imperative to interrogate the Epo-R signaling axis to determine its role in MDS pathogenesis.
4. EPO RECEPTOR SIGNALING
The Epo-R signaling cascade extends to a number of well-characterized enzymatic pathways that result in transcriptional activation of proliferation and antiapoptotic genes. These pathways are initiated when the Epo ligand engages the receptor, triggering conformational changes and receptor homodimerization. Dimerization, in turn, directs activation of Jak2 followed by signaling intermediates leading to increased activity of transcription factors including STAT3, STAT5, and GATA-1. As the Epo-R has no intrinsic kinase activity, it relies on these intermediates for downstream phosphorylation events. The most well-studied Epo-R signaling pathway is the Jak2/STAT5 axis. Epo-R homodimerization causes autophosphorylation of Jak2 kinase. Jak2, the key effecter of Epo signaling, is physically associated with Epo-R throughout receptor maturation and its transport to the plasma membrane. Jak2 phosphorylates tyrosine residues on the cytoplasmic tail of Epo-R that serve as docking sites for other signaling intermediates to bind to the receptor via their src-homology 2 (SH2) domains (Lacombe & Mayeux, 1998). In addition, Jak2 phosphorylates a number of key effectors. The most well-known substrate for Jak2 is STAT5. Jak2 phosphorylates STAT5, which is necessary for dimerization and translocation to the nucleus. In the nucleus, STAT5 binds to DNA and transcriptionally activates proliferation, antiapoptotic, and erythroid differentiation genes. The phosphatidyl-3 kinase (PI3K)/Akt pathway is also activated by Jak2 phosphorylation of Tyr479 on the Epo-R cytoplasmic tail as well as through interaction with adapter proteins IRS2 and Grb-2. The serine threonine kinase, Akt, downstream of PI3K, phosphorylates GATA-1 which promotes transcription of genes involved in erythroid differentiation and transcription of EpoR and GATA-1, thereby creating a positive-feedback loop (Zhao et al., 2006). The mitogen-activated protein kinase (MAPK) pathway is also activated upon Jak2 phosphorylation of Shc that interacts with Grb2. This activates Ras which in turn activates Raf-1, and then the MAPK protein family (Miura, Miura, Ihle, & Aoki, 1994). Another key kinase potentiating Epo-R signaling is Lyn kinase. Lyn kinase not only phosphorylates tyrosines on the cytoplasmic receptor tail but also phosphorylates STAT5 and regulates MAPK, further augmenting signal activity (Lacombe & Mayeux, 1998). In addition, Lyn kinase acts on negative-regulating phosphatases that dampen Jak2, STAT5, and MAPK activity, further increasing signal output (Slavova-Azmanova, Kucera, Louw, et al., 2014). Interestingly, Lyn kinase also negatively regulates the Epo-R signaling pathway via activation of the phosphatase SHP-1, which diminishes activity of Jak/STAT and MAPK (Slavova-Azmanova et al., 2014).
Negative regulation of Epo-R signaling occurs via several mechanisms. Primary regulation is accomplished via negative-feedback loops and SOCS family proteins. Eight proteins make up the SOCS family and each member contains an SH2 domain that can bind to phosphorylated tyrosines, and a SOCS box domain that directs target substrate proteins for degradation (Jegalian & Wu, 2002). These proteins suppress and extinguish cytokine signaling by several methods including inactivation of Jak2 and STAT5, displacing intermediates from receptor docking sites, and by regulating protein levels via ubiquitin tags targeting them for proteasomal degradation (Krebs & Hilton, 2001). Phosphatases also play a major role in negative regulation of cytokine signaling pathways including that of Epo/Epo-R. Hematopoietic cell phosphatase (HCP) binds to and dephosphorylates tyrosine residues on the Epo-R tail to inactivate signal effectors and kinases (Yi, Zhang, Miura, & Ihle, 1995). Additionally, CD45 phosphatase, and Src Homology-containing phosphatase-1, SHP-1, inactivate Jak2 by dephosphorylation. Last, Epo-R signaling is regulated by quick turnover of the receptor after ligand binding. Engagement with its cognate cytokine induces rapid internalization of the receptor via endocytosis in clathrin coated pits (Sulahian, Cleaver, & Huang, 2009). Once internalized, the receptor is ubiquitinated by E3 ligases including β-TRCP and ring finger protein 41 (RNF41) (Jing, Infante, Nachtman, & Jurecic, 2008; Meyer, Deau, Forejtnikova, et al., 2007). E3 ubiquitin ligases work by transferring a 76 amino acid peptide, ubiquitin, from the E2 subunit of the ligase to the protein substrate. The ubiquitin monomers form chains that target the protein for degradation by the proteasome. A summary of Epo-R signal activation and attenuation are provided in Fig. 1.
Fig. 1.
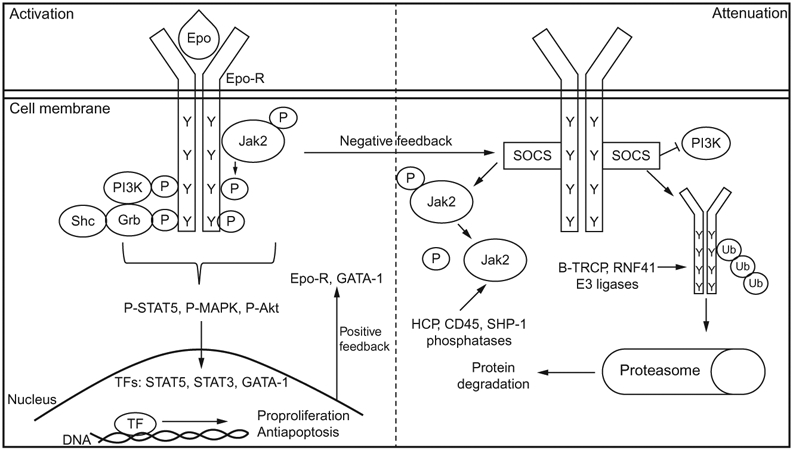
Epo-R signaling pathways. Epo-R signaling is a delicate balance between activation signals (left) and attenuation signals (right). Activation is initiated when Epo binds Epo-R which then dimerizes and causes the autophosphorylation of Jak2. Jak2 in turn phosphorylates tyrosine residues on the cytoplasmic tail of the receptor. This allows binding of other signal effectors via their SH2 domains such as PI3K, Shc, and Grb. Several pathways are collectively activated including the Jak/STAT, PI3K/Akt, and MAPK pathways. These result in the activation and nuclear translocation of transcription factors (TFs) including STAT5, STAT3, and GATA-1. These TFs induce the transcription of proproliferation and antiapoptotic signals. Furthermore, they provide positive feedback as levels of Epo-R and GATA-1 transcripts are also elevated. Alternatively, negative-feedback loops are also activated. Epo stimulation induces the expression of SOCS proteins which not only inactivate signal effectors but also bind to the receptor displacing adapter molecules. SOCS proteins also target the receptor for proteasomal degradation along with the E3 ubiquitin ligases. Addition of ubiquitin monomers by these proteins creates long polyubiquitin tails that direct the receptor to the proteasome for degradation. Phosphatases also play a major role in negative regulation of the Epo-R signaling. The proteins diminish signaling by removing activating phosphate groups.
5. LIPID RAFTS
Lipid or membrane rafts are integral components of the plasma membrane whose existence was confirmed just prior to the turn of the millennium. Lipid rafts are approximately 10–200 nm in size and constitute different proportions of the plasma membrane in different cell types. The plasma membrane consists of a bilayer of phospholipids with a hydrophilic head responsible for the interactions with the microenvironment or the cytoplasm, and a hydrophobic transmembrane tail responsible for interacting with the tail of the opposing phospholipid. This phospholipid bilayer creates a highly organized, although fluidic network that is the foundation of the cell’s structural integrity. Intricate compartmentalization of the plasma membrane is vital to regulate protein transport, to house receptors that detect environmental stimuli, and to facilitate engulfment of foreign matter such as bacteria or cellular debris for destruction. The plasma membrane also helps coordinate signal activation, signal inhibition, and protein turnover. The lipid raft microdomains “float” laterally throughout the plasma membrane and are indispensable for a variety of cellular processes. Lipid rafts contain high levels of very densely packed cholesterol and sphingolipids defining them as distinct entities within the plasma membrane. Sphingolipids contain amino alcohol backbones whose hydrophilic tails interact with the sterol, cholesterol, molecules forming tightly contained platforms by which they may coordinate cellular process. These sphingolipid, cholesterol-rich microdomains are relatively resistant to nonionic detergents at low temperatures that contributed to their isolation and identification. For this reason, they are sometimes referred to as detergent-resistant membranes. An example of lipid raft structure is provided in Fig. 2. It remains unclear how much of the cellular membrane is composed of lipid rafts, although we know that this differs from cell to cell. It is clear, however, that lipid rafts are involved in a number of cellular process including apoptosis, multidrug resistance, endocytosis, and perhaps most importantly cellular signaling. Lipid rafts are formed by trafficking subunits from the ER and Golgi via the actin cytoskeleton. The actin skeleton is an intricate network serving as the road structure for protein transport. The traffic signals that coordinate this complex highway are the GTPases, molecular switches that direct traffic via their fluent on/off activation state.
Fig. 2.

Lipid raft composition. Lipid rafts are composed of tightly packed sphingolipids and cholesterol-forming platforms that contain GPI anchored proteins, cytokine receptors, kinases, and GTPases that mediate cell signaling. From McGraw, K. (2013). Interrogation of EpoR fidelity in myelodysplastic syndrome hematopoiesis and stabilization by the immunomodulatory agent, lenalidomide (Doctoral Dissertation). Retrieved from Proquest Dissertations and Theses. (Accession Order No. 3588361).
Coalescence of rafts is dependent upon the contribution of several proteins including dedicator of cytokinesis-2 (DOCK2), a protein involved in hematopoietic cell actin cytoskeletal rearrangement, Arp 2/3, a protein complex that facilitates actin polymerization, Wiskott-Aldrich syndrome protein (WASp) that activates ARP2/3, and the family of proteins known as the Rac GTPases and their regulators. GTPases cycle between inactive GDP-bound states and active GTP-bound states. These proteins are required for the attachment of raft fractions to the membrane via isoprenylation. The most well-studied GTPases are the Rho-like family including Rho-A, Rho-B, and Rho-C; the Rac GTPases including Rac1, Rac2, and Rac3; and Cdc42-like GTPases. Each of these has known roles in regulating the cytoskeleton and is required for protein transport, raft coalescence and aggregation, and recruitment of signaling intermediates to receptors. The most well-studied pathway of raft aggregation is the Rho GTPases and the Rho kinase (ROCK). ROCKphosphorylates and activates LIMK which then deactivates the actin-depolymerizing protein coflin via an inhibitory phosphorylation. ROCK also inhibits the myosin light chain (MLC) phosphatase, activating MLC and stimulating actin polymerization. Furthermore, mDIA is a well-studied GTPase effector that promotes actin polymerization by attaching actin monomers to branched actin filaments creating the cytoskeletal network. A summary of these proteins and their roles in actin polymerization and raft aggregation is provided in Fig. 3.
Fig. 3.
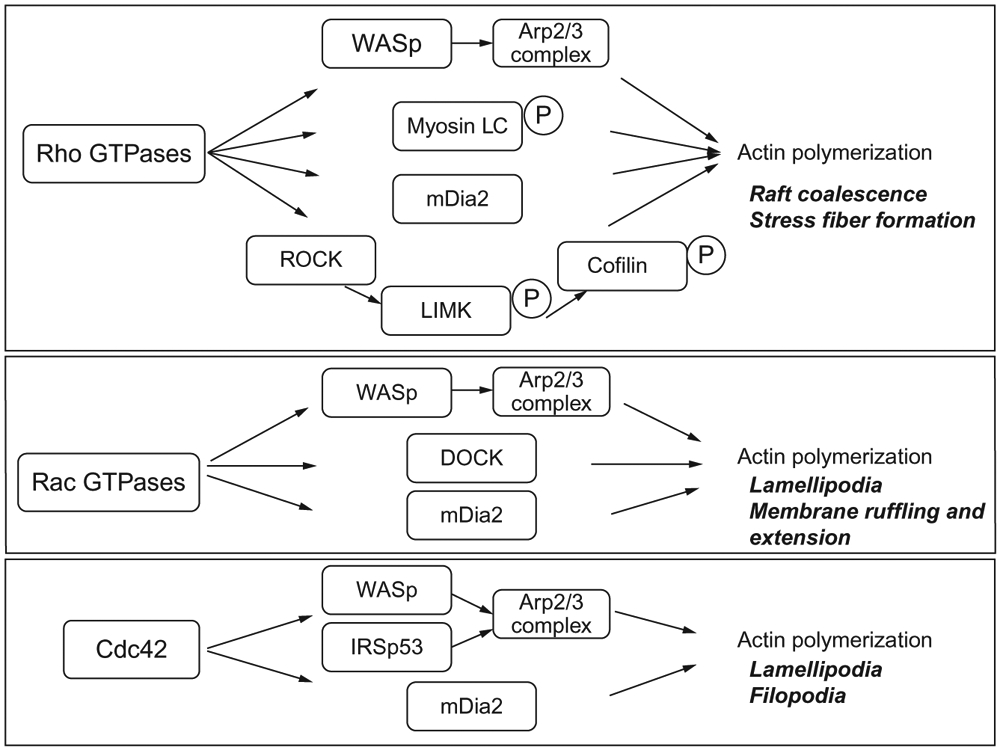
Rho GTPase activation of cytoskeletal reorganization. The Rho GTPases regulate actin cytoskeletal reorganization through a number of different pathways shown here. From McGraw, K. (2013). Interrogation of EpoR fidelity in myelodysplastic syndrome hematopoiesis and stabilization by the immunomodulatory agent, lenalidomide (Doctoral Dissertation). Retrieved from Proquest Dissertations and Theses. (Accession Order No. 3588361).
GTPases have long been known to have critical functions in hematopoiesis. They are involved in every step of hematopoietic differentiation and are critical for hematopoietic stem cell survival and proliferation. They are also required for erythropoiesis, as loss of Rac1 and Rac2 impairs erythropoiesis. These GTPases are critical for erythroid cytoskeletal organization owing to the cell’s structural integrity. The dedicator of cytokinesis 4 (DOCK4), a guanine exchange factor that regulates the activity of GTPases, is critical for erythrocyte structural integrity. The DOCK4 gene is expressed on chromosome 7, notably, a chromosomal region that is often deleted in MDS. Reduced DOCK4 expression in MDS patients with chromosome 7 deletions results in erythroid dysplasia due to disruption of the actin cytoskeleton (Sundaravel, Duggan, Bhagat, et al., 2015). This leads to erythroid frailty resulting in greater susceptibility to cell death and anemia in response to stress. These data highlight the critical role of not only the GTPases but also the actin cytoskeleton on erythropoiesis.
Residence of a variety of proteins found within lipid raft suggests their role in a number of cellular processes. Lipid rafts are highly involved in neuronal signal development, as well as signal transduction and axon directing. They are responsible for the regulation of the amyloid beta (Aβ) peptide. The Aβ peptides create plaques that affect neuronal communication and are prevalent in Alzheimer’s disease patients. The Fas/CD95 death receptor complex also resides in lipid rafts suggesting a role of these microdomains in apoptosis. Multidrug resistant proteins including MRP1 and PgP are localized to rafts and function in drug extrusion and drug resistance. Rafts are also the site of viral entry playing a major role in infection. Perhaps the most well-studied role of lipid rafts is in immune signaling. Both T- and B-cells utilize lipid rafts as the site of stimulus-triggered cell signaling. B-cells, key effectors in adaptive immunity, have receptors (BCR) that interact with CD20 and CD19/21. Engagement of these receptors leads to the activation of an extensive network of signaling intermediates, all of which are recruited to the receptor within the raft fractions. This results in activation of transcription factors responsible for carrying out the necessary signal whether it is cell differentiation, protein translation to coordinate immune response, or negative-feedback signals to halt activation of the receptor. T-cells, similarly key to adaptive immunity, include several subsets each with particular function. Some of these include cytotoxic T-cells (CD8+) which are responsible for killing infected cells, helper T-cells (CD4+) which facilitate a number of responses based on the different cytokines they secrete, and suppressor T-cells that function to regulate T-cell activation. Lipid rafts have perhaps been most extensively studied in T-cells. When stimulated, TCRs aggregate in coalesced lipid raft platforms. This localization initiates the recruitment of signaling intermediates Zap-70 and PLCγl. Of note, the negative regulatory phosphatase, CD45, is displaced from lipid raft fractions upon signal activation to further augment the signal. Lipid rafts are also largely aggregated at the immune synapse, the site of cell-to-cell contact between immune effectors such as T- and natural killer-cells. The well-studied role of lipid rafts in immune cells and receptor signaling, as well as the role of the cytoskeleton and GTPases on erythropoiesis, prompted the investigation of the role of lipid rafts in erythropoiesis, specifically as it relates to Epo signaling.
6. LIPID RAFTS AND EPO SIGNALING
Until recently the role of lipid rafts in Epo signaling had not been investigated. It was known, however, that rafts were abundant in RBC membranes (Mikhalyov & Samsonov, 2011; Murphy, Samuel, Harrison, et al., 2004). Most work on rafts in erythrocytes centered around infection by the malaria parasite, Plasmodium falciparum, which is facilitated by lipid rafts (Murphy, Fernandez-Pol, Chung, et al., 2007). There is also evidence to suggest that lipid rafts may have a role in erythrocyte enucleation; although reports are limited (Kalfa & Zheng, 2014). The role of lipid rafts in erythropoiesis, and Epo signaling was not investigated until recently by our laboratory. In order to determine what, if any, role of rafts existed in Epo/Epo-R signaling, we first investigated the EPO-responsive erythroleukemia cell line, UT-7. To confirm the presence of lipid rafts in these cells, we exploited the detergent-resistant properties of the cholesterol-rich fractions. Lipid rafts are undisturbed by traditional cell lysis with most nonionic detergents, and as such, one is able to remove these fractions from the cell lysates by gradient centrifugation. Purification of the rafts can be confirmed by detecting raft constituents, the most common of which is the ganglioside, GM-1, which can be detected in isolated raft fractions by immunoblotting. Indeed, GM-1 was demonstrable by immunoblotting after gradient centrifugation isolation in UT-7 cells in lower density fractions containing the lipid rafts. Direct visualization of lipid rafts can be performed by several methods including electron and fluorescence microcopy. In our study, we used fluorescent microscopy to visualize GM-1, the raft surrogate, in UT-7 cells. Unstimulated cells had a diffuse staining pattern and membrane rafts were present, although not prominently distinct on the membrane. However, after stimulation with rhEpo, there was a striking upregulation of both the number and size of rafts demonstrating rapid coalescence into clearly punctate, distinct fractions in the membrane. This was observed in both cultured UT-7 cells and erythroid progenitors isolated from healthy bone marrow. These findings are illustrated in Fig. 4. We next investigated whether the Epo-R was localized in the raft microdomains. To do so, we first utilized confocal microscopy and 3D rendering to demonstrate that upon Epo stimulation, there was colocalization of the receptor with raft fractions (Fig. 5). This was true in both cultured cell lines and primary erythroid progenitors. This was confirmed by raft fractionation by ultracentri-fugation and western blotting for Epo-R. Although there was basal colocalization of the receptor in the raft fractions, stimulation of the cells by rhEpo significantly increased receptor residence in raft fractions, similar to that observed in lymphocytes after TCR engagement. Importantly, upon Epo stimulation we observed not only recruitment of Epo-R to rafts but also recruitment of the downstream signaling intermediates Jak2, STAT5, MAPK, and Lyn kinase, shown in Fig. 6. Notably, the negative-regulating phosphatase, CD45, is repartitioned out of raft fractions upon Epo stimulation that further strengthens signal fidelity, analogous to that in T-cell signaling. As mentioned earlier, receptor turnover after ligand engagement is a rapid process and vital to assure extinction of the Epo-R signal. Epo-R is recruited to raft fractions within the first minute of stimulation and peaks at approximately 10 min. This time point coincides with the point at which the maximal activation of the Jak/STAT pathway occurs. After 30 min, however, the level of receptor residing in rafts is nearly undetectable, demonstrating a return to the steady state level (Fig. 6). We next investigated whether lipid rafts were integral to Epo-R signaling. In order to study this, we utilized two methods of raft disruption prior to Epo stimulation in the UT-7 cell line, and then studied downstream signal activation. Methyl, beta-cyclodextran (MβCD) is a raft disrupter that removes cholesterol from the membrane, thereby destroying integrity of the raft fraction. Nystatin, on the other hand, intercalates membrane cholesterol and is an alternative, albeit less effective raft disrupter. Importantly, upon disruption of lipid rafts by either method, Epo signaling is severely impaired as demonstrated by decreased phosphorylation of Jak2, STAT5, MAPK, and Akt. We next validated these findings in primary bone marrow erythroid progenitors isolated from healthy donors. Indeed, raft disruption in primary erythroid progenitors significantly diminished signaling initiated by Epo stimulation (McGraw et al., 2012). These results demonstrate that lipid rafts are critical for maximum signaling initiated by Epo-R engagement. But how does this occur? We know that aggregation of rafts and recruitment of Epo-R and signaling intermediates is dependent on ROCK and the Rho GTPases; however, the precise mechanisms by which this happens and the method by which rafts are aggregated remain unclear. Several potential mechanisms have been postulated to explain this. One proposal is that as rafts aggregate, there is increased cholesterol affinity, potentiating coalescence and recruitment of the receptors, and intermediates contributing to signal coordination. Alternatively, as receptors dimerize in response to ligand engagement, this may induce the aggregation of lipid raft fractions to support these growing protein complexes. Others have postulated that rafts themselves activate enzymes that drive downstream effectors and signal initiation (Brown & London, 1998). Further investigation is required to determine the precise mechanisms by which these processes occur.
Fig. 4.

Detection of lipid rafts in UT-7 and primary erythroid progenitors. (A) Dot blot detection of GM-1 in UT7 cell lysates in nonraft (fractions 5, 6) and raft fractions (fraction 2) in controls, and after Epo or MβCD (raft disrupter) treatment. (B) Immunofluorescence of UT7 cells showing an increase in raft (red) accumulation after Epo exposure. (C) Immunofluorescence of UT7 cells before and after Epo stimulation showing increased raft aggregates (red) in the plasma membrane. (D) Immunofluorescence of primary erythroid progenitors showing an increase in cellular membrane raft fluorescence intensity (red). McGraw, K. L., Fuhler, G. M., & Johnson, J. O., et al. (2012). Erythropoietin receptor signaling is membrane raft dependent. PloS One 7(4), e34477. doi:10.1371/journal.pone.0034477. Available under a Creative Commons Attribution (CC BY 4.0) license.
Fig. 5.

Epo-R colocalizes with lipid rafts. (A) Confocal immunofluorescence of UT-7 cells untreated or after Epo stimulation, lipid rafts: red, Epo-R: green, DAPI/Hoechst: blue. Right panel is a merged image showing lipid raft and Epo-R colocalization (yellow). (B) Confocal immunofluorescence in human primary erythroid progenitors. (C) Three-dimensional rendering of UT7 cells either untreated (left) or after Epo treatment (right). Top two rows display isosurfacing of the rafts (red), Epo-R (green), and nucleus (Dapi, blue). Dapi was removed from the middle row to further visualize association of the receptor with rafts in the second row of panels. The bottom row displays volume rendering of the same cells to illustrate membrane colocalization (yellow). McGraw, K. L., Fuhler, G. M., & Johnson, J. O., et al. (2012). Erythropoietin receptor signaling is membrane raft dependent. PloS One 7(4), e34477. doi:10.1371/journal.pone.0034477. Available under a Creative Commons Attribution (CC BY 4.0) license.
Fig. 6.
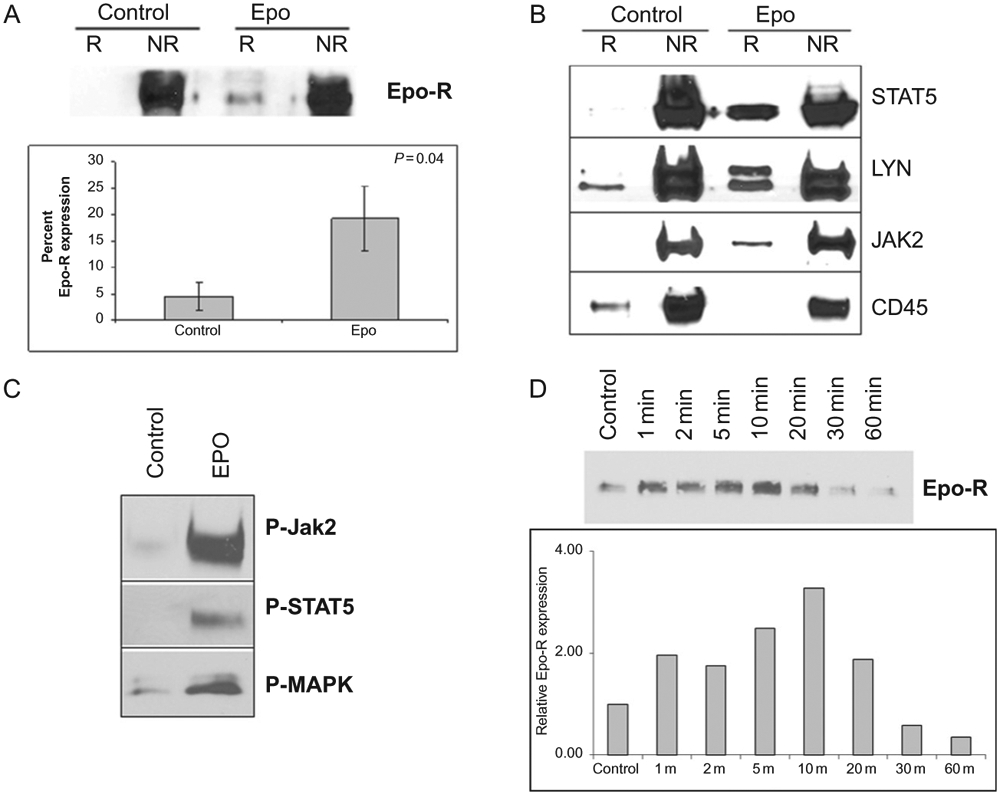
Epo stimulation recruits signal effectors into raft fractions. (A) Raft fractions (R) were separated from nonraft fractions (NR) by gradient centrifugation and immunoblotted for Epo-R to investigate receptor translocation into rafts after Epo stimulation. Corresponding quantitation represents the mean ± SE of receptor recruitment. (B) UT-7 cells were starved overnight then treated with Epo for 10 min. After fractionation, the nonraft (NR) fractions and raft (R) fractions were pooled and immunoblotted for the indicated proteins showing recruitment of these effector to the raft fractions after stimulation. (C) Activated (phosphorylated) forms of Jak2, STAT5, and MAPK were also increased in the raft fractions after Epo stimulation. (D) Raft fractions were isolated after stimulation with Epo at the indicated time points and immunoblotted for Epo-R. Results show that Epo-R is recruited into rafts within 1 min of Epo stimulation reaching maximum loading at 10 min, followed by gradual redistribution thereafter. Accompanying graphic quantitation of the representative experiment. McGraw, K. L., Fuhler, G. M., & Johnson, J. O., et al. (2012). Erythropoietin receptor signaling is membrane raft dependent. PloS One 7(4), e34477. doi:10.1371/journal.pone.0034477. Available under a Creative Commons Attribution (CC BY 4.0) license.
7. CLINICAL IMPLICATIONS
As discussed earlier, the MDSs include a spectrum of disorders characterized by ineffective hematopoiesis and diminished Epo-R signaling. Despite normal levels of circulating cytokine and comparable levels of cell surface receptor, Epo signaling is diminished as demonstrated by decreased STAT5 activation. Previous studies showed that neutrophils isolated from MDS patients had decreased density of lipid rafts, leading to diminished activation by granulocyte-macrophage colony-stimulating factor (GM-CSF) (Fuhler, Blom, Coffer, Drayer, & Vellenga, 2007). Based on this observation, and the requirements for intact lipid rafts for Epo signal fidelity, it reasons that lipid rafts may play a major role in the deficient signaling of MDS patients. And in fact, this is precisely the case. MDS erythroid progenitor cells have a marked reduction in lipid raft fractions compared to their normal counterparts (McGraw, Basiorka, Johnson, et al., 2014). In addition, the rafts are smaller in size, limiting recruitment of key signaling molecules and spatially impairing aggregation. As rafts are indispensable for Epo signal fidelity, lipid raft deficiency in MDS cells diminishes activation of STAT5 in response to Epo-R activation. Therapeutic strategies that promote accumulation of cholesterol-rich, lipid rafts, and recruitment of signaling intermediates may rescue anemia caused by deficient Epo signaling.
Lenalidomide is one of the most effective agents used to treat MDS patients. It is part of a family of compounds known as immunomodulatory (IMiDs) and a second generation analog of the known teratogen, thalidomide. Lenalidomide was approved for the treatment of transfusion-dependent patients with lower risk MDS and deletion in the long arm of chromosome 5 [del(5q)] in 2005, making it the first and only targeted therapy for these disorders. In del(5q) MDS, lenalidomide is cytotoxic to the clone, in part due to haploinsufficiency of the cereblon substrate casein kinase 1α, and another beta-catenin destruction complex member, protein phosphatase 2a, genes that are both located within or near the commonly deleted region on 5q (Kronke, Fink, Hollenbach, et al., 2015; Wei, Chen, Rocha, et al., 2009). Up to 75% of all del(5q) patients will respond to lenalidomide, with most experiencing sustained and prolonged transfusion independence and cytogenetic response. In non-del(5q) MDS patients, however, lenalidomide is able to restore erythropoiesis in approximately 25% of patients. The mechanism of how this occurs has been the focus of much research during the last decade. Our findings of the dependence of Epo-R signaling on lipid rafts led us to investigate whether lenalidomide had an effect on lipid rafts. Raft coalescence is dependent upon activation of GTPases, and it is well established that lenalidomide activates the Rho GTPases albeit through unknown mechanisms (Xu, Li, Ferguson, et al., 2009). In addition, lenalidomide augments Epo-induced signal activation evidenced by increased activation of signaling intermediates Jak2 and STAT5, and increased DNA binding of STAT5 to promote transcription of erythroid differentiation genes. Indeed, we found that treatment of erythroid cell lines and primary erythroid progenitors with lenalidomide-induced aggregation and accumulation of lipid raft fractions. In addition, lenalidomide fostered the recruitment of Jak2 and STAT5 into raft microdomains. And similar to Epo stimulation, lenalidomide treatment repartitioned CD45 outside of the raft fractions. Interestingly, the increase in raft accumulation in response to lenalidomide treatment was less in erythroid cells isolated from healthy donors compared to those from patients with lower risk non-del(5q) MDS. In the latter patient specimens, there was a striking and statistically significant upregulation of raft aggregation, correcting the deficiency to levels comparable to normal counterparts (McGraw et al., 2014). Furthermore, lenalidomide not only fosters lipid raft accumulation augmenting Epo signaling but also upregulates the protein expression of Epo-R. It does this via inhibition of the E3 ubiquitin ligase, RNF41 (Basiorka, McGraw, De Ceuninck, et al., 2016). RNF41 regulates expression of Jak-2 associated, Type-I receptors via ubiquitination and subsequent proteasomal degradation. Lenalidomide blocks RNF41 ubiquitinating function resulting in upregulation of Epo-R, another mechanism by which Epo signaling is augmenting by this IMiD agent. These data indicate novel mechanisms by which lenalidomide acts, as well as providing a rationale for the development and use of molecules targeting lipid rafts.
The role of lipid rafts in Epo signaling has led to the study of other disorders characterized by aberrant signaling. In contrast to diminished Epo-induced Jak2 and STAT5 activation in MDS, the MPNs have constitutive activation of these proteins. Although we know that Jak2 resides in the rafts, particularly after receptor stimulation, it has recently been shown that the Jak2 V617F mutant similarly is positioned within raft fractions. Importantly, raft disruption results in diminished Jak2 V617F constitutive signaling. Similar to the use of nystatin explained earlier in this chapter, other statins may also be utilized to disrupt rafts. Statins are highly prescribed to lower cholesterol in individuals with hyperlipidemia, and those with increased risk of coronary artery disease or stroke. The ability of statins to inhibit aberrant signaling caused by the constitutively active Jak2 V617F mutant, and growth inhibition of primary MPN cells, with no discernible effect on normal counterparts is an intriguing finding that may be exploitable in these disorders (Griner et al., 2013).
Recently, lipid rafts have been shown to play a major role in the pathogenesis paroxysmal nocturnal hemoglobinuria (PNH). PNH is a stem cell disorder in patients that harbor a mutation in PIG-A, which results in a decrease in expression of GPI-A anchored proteins on hematopoietic stem and progenitor cells (HSPCs), including CXCR4 and VLA4, adhesion receptors that facilitate stromal adherence of HSPCs in the bone marrow. It has recently been shown that CXCR4 and VLA-4 reside in lipid rafts that are diminished in PNH CD34+ bone marrow and peripheral blood cells. Raft deficiencies result in decreased stromal attachment via diminished CXCR4 ligand binding, increased mobility and clonal expansion. This results in differentiated cells including erythrocytes that are more susceptible to complement-mediated intravascular lysis resulting in the anemia observed in these patients (Ratajczak, Borkowska, Mierzejewska, et al., 2015). It reasons that compounds capable of manipulating rafts may offer benefit for this disorder.
8. CONCLUSION AND FUTURE DIRECTIONS
In conclusion, the findings discussed here demonstrate the importance of plasma membrane lipid rafts and the recruitment of receptors and signaling intermediates to these platforms for optimal Epo-R signal fidelity. These results explain, in part, differences in receptor signal potential in patients with MDS, where impaired Epo signaling is attributed in part to diminished raft fractions in the plasma membrane. Furthermore, the dependency of Epo signaling on rafts provides the possibility for new therapeutic strategies to treat these disorders, such as lenalidomide that, in addition to several other functions, induces lipid raft coalescence as well as the recruitment of signaling intermediates. Alternatively, in MPNs, disruption of rafts may be a key in abrogating the constitutive activation of Jak2 V617F that resides in raft fractions. Here, the use of statins has shown promising in vitro activity in decreasing growth of MPN cell models and primary cells. Together, as the roles of lipid rafts in disease pathogenesis continue to be discovered, we must look to future investigation to unravel the mechanisms by which lipid rafts assembly is controlled. Furthermore, we need to examine more closely the interaction between GTPases and lipid rafts to develop methods to exploit these interactions for future therapeutic benefit.
REFERENCES
- Basiorka A, McGraw K, De Ceuninck L, et al. (2016). Lenalidomide stabilizes the erythropoietin receptor by inhibiting the E3 ubiquitin ligase RNF41. Cancer Research, 76, 3531–3540. 10.1158/0008-5472. Published Online First April 6, 2016; CAN-15-1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, & London E (1998). Functions of lipid rafts in biological membranes. Annual Review of Cell and Developmental Biology, 14, 111–136. [DOI] [PubMed] [Google Scholar]
- D’Andrea AD, & Zon LI (1990). Erythropoietin receptor. Subunit structure and activation. The Journal of Clinical Investigation, 86(3), 681–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert BL, & Bunn HF (1999). Regulation of the erythropoietin gene. Blood, 94(6), 1864–1877. [PubMed] [Google Scholar]
- Fisher JW (2003). Erythropoietin: Physiology and pharmacology update. Experimental Biology and Medicine, 228(1), 1–14. [DOI] [PubMed] [Google Scholar]
- Fuhler GM, Blom NR, Coffer PJ, Drayer AL, & Vellenga E (2007). The reduced GM-CSF priming of ROS production in granulocytes from patients with myelodysplasia is associated with an impaired lipid raft formation. Journal of Leukocyte Biology, 81(2), 449–457. [DOI] [PubMed] [Google Scholar]
- Genovese G, Kahler AK, Handsaker RE, et al. (2014). Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. The New England Journal of Medicine, 371(26), 2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griner LN, McGraw KL, Johnson JO, List AF, & Reuther GW (2013). JAK2-V617F-mediated signalling is dependent on lipid rafts and statins inhibit JAK2-V617F-dependent cell growth. British Journal of Haematology, 160(2), 177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoefsloot LH, van Amelsvoort MP, Broeders LC, et al. (1997). Erythropoietin-induced activation of STAT5 is impaired in the myelodysplastic syndrome. Blood, 89(5), 1690–1700. [PubMed] [Google Scholar]
- Huang LJ, Constantinescu SN, & Lodish HF (2001). The N-terminal domain of Janus kinase 2 is required for Golgi processing and cell surface expression of erythropoietin receptor. Molecular Cell, 8(6), 1327–1338. [DOI] [PubMed] [Google Scholar]
- Jaiswal S, Fontanillas P, Flannick J, et al. (2014). Age-related clonal hematopoiesis associated with adverse outcomes. The New England Journal of Medicine, 371(26), 2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jegalian AG, & Wu H (2002). Differential roles of SOCS family members in EpoR signal transduction. Journal of Interferon & Cytokine Research, 22(8), 853–860. [DOI] [PubMed] [Google Scholar]
- Jing X, Infante J, Nachtman RG, & Jurecic R (2008). E3 ligase FLRF (Rnf41) regulates differentiation of hematopoietic progenitors by governing steady-state levels of cytokine and retinoic acid receptors. Experimental Hematology, 36(9), 1110–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabouridis PS, & Jury EC (2008). Lipid rafts and T-lymphocyte function: Implications for autoimmunity. FEBS Letters, 582(27), 3711–3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalfa TA, & Zheng Y (2014). Rho GTPases in erythroid maturation. Current Opinion in Hematology, 21(3), 165–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs DL, & Hilton DJ (2001). SOCS proteins: Negative regulators of cytokine signaling. Stem Cells, 19(5), 378–387. [DOI] [PubMed] [Google Scholar]
- Kronke J, Fink EC, Hollenbach PW, et al. (2015). Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature, 523(7559), 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacombe C, & Mayeux P (1998). Biology of erythropoietin. Haematologica, 83(8), 724–732. [PubMed] [Google Scholar]
- Lewinski UH, Floru S, Cohen AM, & Mittelmann M (1994). Recombinant human erythropoietin in the treatment of myelodysplastic syndromes—Response patterns. Leukemia & Lymphoma, 15(1–2), 149–152. [DOI] [PubMed] [Google Scholar]
- Mantovani L, Lentini G, Hentschel B, et al. (2000). Treatment of anaemia in myelodysplastic syndromes with prolonged administration of recombinant human granulocyte colony-stimulating factor and erythropoietin. British Journal of Haematology, 109(2), 367–375. [DOI] [PubMed] [Google Scholar]
- McGraw KL, Basiorka AA, Johnson JO, et al. (2014). Lenalidomide induces lipid raft assembly to enhance erythropoietin receptor signaling in myelodysplastic syndrome progenitors. PloS One, 9(12), e114249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGraw KL, Fuhler GM, Johnson JO, et al. (2012). Erythropoietin receptor signaling is membrane raft dependent. PloS One, 7(4), e34477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLornan D, Percy M, & McMullin MF (2006).JAK2 V617F: A single mutation in the myeloproliferative group of disorders. The Ulster Medical Journal, 75(2), 112–119. [PMC free article] [PubMed] [Google Scholar]
- McMullin MF, & Percy MJ (1999). Erythropoietin receptor and hematological disease. American Journal of Hematology, 60(1), 55–60. [DOI] [PubMed] [Google Scholar]
- Meyer L, Deau B, Forejtnikova H, et al. (2007). beta-Trcp mediates ubiquitination and degradation of the erythropoietin receptor and controls cell proliferation. Blood, 109(12), 5215–5222. [DOI] [PubMed] [Google Scholar]
- Mikhalyov I, & Samsonov A (2011). Lipid raft detecting in membranes of live erythrocytes. Biochimica et Biophysica Acta, 1808(7), 1930–1939. [DOI] [PubMed] [Google Scholar]
- Miura Y, Miura O, Ihle JN, & Aoki N (1994). Activation of the mitogen-activated protein kinase pathway by the erythropoietin receptor. The Journal of Biological Chemistry, 269(47), 29962–29969. [PubMed] [Google Scholar]
- Miyake T, Kung CK, & Goldwasser E (1977). Purification of human erythropoietin. The Journal of Biological Chemistry, 252(15), 5558–5564. [PubMed] [Google Scholar]
- Mulcahy L (2001). The erythropoietin receptor. Seminars in Oncology, 28(2 Suppl. 8), 19–23. [DOI] [PubMed] [Google Scholar]
- Murphy SC, Fernandez-Pol S, Chung PH, et al. (2007). Cytoplasmic remodeling of erythrocyte raft lipids during infection by the human malaria parasite Plasmodium falciparum. Blood, 110(6), 2132–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy SC, Samuel BU, Harrison T, et al. (2004). Erythrocyte detergent-resistant membrane proteins: Their characterization and selective uptake during malarial infection. Blood, 103(5), 1920–1928. [DOI] [PubMed] [Google Scholar]
- Ratajczak MZ, Borkowska S, Mierzejewska K, et al. (2015). Further evidence that paroxysmal nocturnal haemoglobinuria is a disorder of defective cell membrane lipid rafts. Journal of Cellular and Molecular Medicine, 19(9), 2193–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singbartl G (1994). Adverse events of erythropoietin in long-term and in acute/short-term treatment. The Clinical Investigator, 72(6 Suppl.), S36–S43. [PubMed] [Google Scholar]
- Slavova-Azmanova NS, Kucera N, Louw A, et al. (2014). Lyn kinase plays important roles in erythroid expansion, maturation and erythropoietin receptor signalling by regulating inhibitory signalling pathways that control survival. The Biochemical Journal, 459(3), 455–466. [DOI] [PubMed] [Google Scholar]
- Sulahian R, Cleaver O, & Huang LJ (2009). Ligand-induced EpoR internalization is mediated by JAK2 and p85 and is impaired by mutations responsible for primary familial and congenital polycythemia. Blood, 113(21), 5287–5297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaravel S, Duggan R, Bhagat T, et al. (2015). Reduced DOCK4 expression leads to erythroid dysplasia in myelodysplastic syndromes. Proceedings of the National Academy of Sciences of the United States of America, 112(46), E6359–E6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watowich SS, Xie X, Klingmuller U, et al. (1999). Erythropoietin receptor mutations associated with familial erythrocytosis cause hypersensitivity to erythropoietin in the heterozygous state. Blood, 94(7), 2530–2532. [PubMed] [Google Scholar]
- Wei S, Chen X, Rocha K, et al. (2009). A critical role for phosphatase haplodeficiency in the selective suppression of deletion 5q MDS by lenalidomide. Proceedings of the National Academy of Sciences of the United States of America, 106(31), 12974–12979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Li J, Ferguson GD, et al. (2009). Immunomodulatory drugs reorganize cytoskeleton by modulating Rho GTPases. Blood, 114(2), 338–345. [DOI] [PubMed] [Google Scholar]
- Yi T, Zhang J, Miura O, & Ihle JN (1995). Hematopoietic cell phosphatase associates with erythropoietin (Epo) receptor after Epo-induced receptor tyrosine phosphorylation: Identification of potential binding sites. Blood, 85(1), 87–95. [PubMed] [Google Scholar]
- Zhao W, Kitidis C, Fleming MD, Lodish HF, & Ghaffari S (2006). Erythropoietin stimulates phosphorylation and activation of GATA-1 via the PI3-kinase/AKT signaling pathway. Blood, 107(3), 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]