

LETTER
Since 7 April 2020, our coronavirus disease 2019 (COVID-19) diagnostic laboratory (CLIAHUB) has received samples from multiple counties in California. Our reverse transcriptase PCR (RT-PCR) protocol (1) employs N-gene (NIID_2019-nCov_N_F2/R2ver3/P2 [Japan]) (2) and E-gene (E_Sarbeco_F/R/P1 [Germany]) (3) simplex assays. In July 2020, we identified 40+ samples from Madera County with poor N-gene assay performance relative to the E-gene assay.
Figure 1A shows the concordance of cycle threshold (CT) values for both assays in the 3,958 positive tests conducted during 27 May to 7 August 2020. For samples with positive E- and N-gene results (n = 3,629), the N- and E-gene CT value difference [ΔCT(N−E)] was 0.40 ± 1.18 (mean ± standard deviation).
FIG 1.
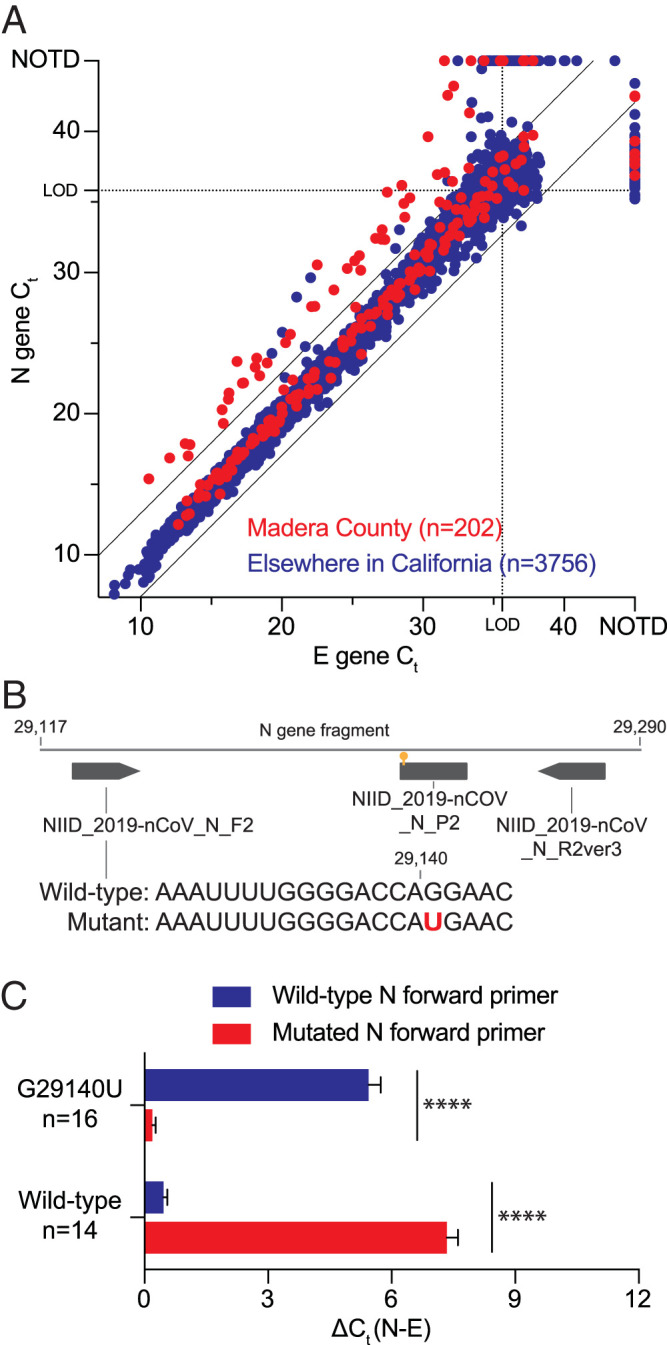

Single mutation in forward N-gene primer binding site, prevalent across the world, decreased SARS-CoV-2 RT-PCR sensitivity. (A) Potential SARS-CoV-2 mutants were identified by their increased ΔCT between the N- and E-gene assays (>2.5× standard deviation of average ΔCT; cutoff indicated by black lines). Dotted lines indicate the average CT value at the limit of detection (LOD) of each assay, above which more variation is expected. NOTD, not detected. (B) Diagram showing a fragment of the N gene, with the N-gene primers and probe originally developed by the National Institute of Infectious Diseases (NIID) in Tokyo, Japan (4), and the identified G29140U mutation indicated. (C) The increased ΔCT(N−E) of mutant lines using the conventional RT-PCR with the wild-type primer was reversed when a primer incorporating the mutation was used. The opposite was observed for wild-type samples that showed an increased ΔCT(N−E) when the mutated primer was used, further validating causality of the G29140U mutation for reduced N-gene RT-PCR performance. Error bars indicate standard errors of the means. ****, significant difference determined by a t test (P < 0.0001). (D) Phylogeny of SARS-CoV-2 isolates with N-gene mutation, including those with the G29140U mutation. Inferred mutation events on the tree are annotated with an asterisk that is colored depending on the allele. Both synonymous variants of the Q289H mutant are found, with the mutation estimated to have recurred 11 times on the tree, and only one of the mutant samples from GISAID was identical by descent to the Madera cluster. One of the wild-type Madera samples was closely related to the mutant cluster, with a common ancestor just before the mutation event. Sequence data are available in Table S1. All code used for analyses and figure generation is described in Text S1.
Sequencing of the detected N-gene fragments of 57 samples with a ΔCT(N−E) of ≥2.96 (2.5 standard deviations above the mean) identified 46 samples (45 from Madera) to have a G29140U mutation located in the forward primer binding site (16th of 20 nucleotides) of the N-gene assay (Fig. 1B). In 5 mutant samples, the N gene was undetectable by RT-PCR, but these cases were still recognized as positive for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) by the E-gene assay. The 11 wild-type samples with an increased ΔCT(N−E) are considered rare artifacts.
When the RT-PCR was repeated using a forward primer with full complementarity to the mutant sequence (Fig. 1C), the mean ΔCT(N−E) of 16 randomly selected mutant samples dropped from 5.44 with the canonical primer to 0.19 with the mutated primer. This trend was inverted for the 14 randomly selected wild-type samples where the ΔCT(N−E) increased from 0.46 to 7.34 with the canonical and mutated primer, respectively. These data validate causality of G29140U for the observed aberrant CT values of the N-gene assay, reducing its sensitivity by 67-fold.
G29140U encodes a Q289H amino acid mutation in the N gene that was also found in 27 other sequences available on GISAID (4), showing worldwide occurrence of these mutants. Q289H is located within the dimerization domain of the nucleocapsid protein but is not involved in any known dimer interface interactions, though tertiary-structure-level interactions could be impacted by mutations at this position (5).
Whole-genome sequencing of randomly selected mutant (n = 20) and wild-type (n = 11) samples from Madera showed little genetic diversity between our mutant samples and revealed that a GISAID sequence from San Diego was identical by descent (Fig. 1D). The remaining 26 mutants from GISAID fell on different clades of the tree, with 11 estimated recurrent mutation events at the locus.
Epidemiological data from Madera County indicated that the G29140U variant is replication competent, retains its virulence, and adequately transmits within and between different communities (see Text S1 in the supplemental material).
Our data show that even in areas of high SARS-CoV-2 community spread, replication-competent mutations that impair RT-PCR performance can emerge and spread, leading to reduced test sensitivity and potentially underdiagnosis if only one viral target is used. Since mutations have been described in primer/probe-binding regions of all published SARS-CoV-2 diagnostic assays (6), our findings strongly support continuous monitoring for mismatches and the routine use of at least two targets for SARS-CoV-2 detection by RT-PCR to avoid false-negative results.
Data availability.
All code and CT data are available in our Github repository: https://github.com/czbiohub/polymorphism_sarscov2_diagnostics. Sequence data are available via GISAID; see also Table S1 for a list of sequences used.
Supplementary Material
ACKNOWLEDGMENTS
We thank Peter Kim and Don Ganem for helpful discussions and editorial assistance on the manuscript.
This work was supported by the Chan Zuckerberg Biohub and the Chan Zuckerberg Initiative.
We have no conflicts of interest to declare.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Crawford ED, Acosta I, Ahyong V, Anderson EC, Arevalo S, Asarnow D, Axelrod S, Ayscue P, Azimi CS, Azumaya CM, Bachl S, Bachmutsky I, Bhaduri A, Bancroft Brown J, Batson J, Behnert A, Boileau RM, Bollam SR, Bonny AR, Booth D, Borja MJB, Brown D, Buie B, Burnett CE, Byrnes LE, Cabral KA, Cabrera JP, Caldera S, Canales G, Castañeda GR, Chan AP, Chang CR, Charles-Orszag AC, Cheung C, Chio U, Chow ED, Citron YR, Cohen A, Cohn LB, Chiu C, Cole MA, Conrad DN, Constantino A, Cote A, Crayton-Hall T, Darmanis S, Detweiler AM, Dial RL, Dong S, Duarte EM, et al.. 2020. Rapid deployment of SARS-CoV-2 testing: the CLIAHUB. PLoS Pathog 16:e1008966. doi: 10.1371/journal.ppat.1008966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shirato K, Nao N, Katano H, Takayama I, Saito S, Kato F, Katoh H, Sakata M, Nakatsu Y, Mori Y, Kageyama T, Matsuyama S, Takeda M. 2020. Development of genetic diagnostic methods for detection for novel coronavirus 2019(nCoV-2019) in Japan. Jpn J Infect Dis 73:304–307. doi: 10.7883/yoken.JJID.2020.061. [DOI] [PubMed] [Google Scholar]
- 3.Corman VM, Landt O, Kaiser M, Molenkamp R, Meijer A, Chu DKW, Bleicker T, Brünink S, Schneider J, Schmidt ML, Mulders DGJC, Haagmans BL, Van Der Veer B, Van Den Brink S, Wijsman L, Goderski G, Romette JL, Ellis J, Zambon M, Peiris M, Goossens H, Reusken C, Koopmans MPG, Drosten C. 2020. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 25:2000045. doi: 10.2807/1560-7917.ES.2020.25.3.2000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elbe S, Buckland-Merrett G. 2017. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob Chall 1:33–46. doi: 10.1002/gch2.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ye Q, West AMV, Silletti S, Corbett KD. 2020. Architecture and self-assembly of the SARS-CoV-2 nucleocapsid protein. Protein Sci 29:1890–1901. doi: 10.1002/pro.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang R, Hozumi Y, Yin C, Wei G-W. 2020. Mutations on COVID-19 diagnostic targets. Genomics 112:5204–5213. doi: 10.1016/j.ygeno.2020.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All code and CT data are available in our Github repository: https://github.com/czbiohub/polymorphism_sarscov2_diagnostics. Sequence data are available via GISAID; see also Table S1 for a list of sequences used.