

Abstract
Background
Geranylgeranyltransferase I (GGTase I) catalyzes geranylgeranylation, a modification required for the function of many oncogenic RAS-related proteins. GGTI-2418 is a peptidomimetic small molecule inhibitor of GGTase I.
Objective
The aim of this study was to establish the maximum tolerated dose of GGTI-2418 in patients with advanced solid tumors.
Patients and Methods
This was a phase I, open-label, dose-escalation study conducted in two US centers (University of Pennsylvania and Indiana University) in adults with treatment-refractory advanced solid tumors. An accelerated dose-escalation schema was used across eight dose levels, from 120 to 2060 mg/m2, administered on days 1–5 of each 21-day cycle.
Results
Fourteen patients were enrolled in the dose-escalation cohort. No dose-limiting toxicities were observed, and 2060 mg/m2 was determined to be the maximum tolerated dose. The only potential drug-related grade 3 or 4 toxicities were elevated bilirubin and alkaline phosphatase in a single patient with concurrent malignant biliary obstruction. No objective responses were observed. Four of thirteen evaluable patients had stable disease for up to 6.7 months. The study was terminated prior to dose expansion based on a sponsor decision. Pharmacokinetic analysis demonstrated a mean terminal half-life of 1.1 h.
Conclusions
GGTI2418 was safe and tolerable at all tested dose levels with some evidence of disease stability. Due to rapid elimination, dosing of GGTI2418 in this study may have been inadequate to achieve optimal inhibition of its target, GGTase I.
1. Introduction
Geranylgeranyltransferase I (GGTase I) catalyzes geranylgeranylation, a lipid posttranslational modification required for the function of many RAS-related proteins such as the Ral, Rac and Rho family of small GTPases [1]. Geranylgeranylated RhoA and RhoC play important roles in the G1/S transition of the cell cycle and promote tumor growth, invasion, and metastasis [1-4]. GGTase I has been shown to be essential to tumorigenesis in RAS-driven tumor models but dispensable for normal tissue homeostasis, suggesting that GGTase I can be targeted therapeutically [5-9].
GGTI-2418 is a peptidomimetic small molecule with a design based on the tetrapeptide carboxyl termini of RhoA and RhoC (Fig. 1). It competitively inhibits GGTase I (half maximal inhibitory concentration [IC50] 9.5 μM), with selectivity over farnesyltransferase (IC50 53 μM) [10, 11]. GGTase I inhibitors (GGTIs) have been shown to induce G1 arrest at least in part by inducing the expression of cyclin-dependent kinase (CDK) inhibitors such as p27, inhibiting CDK2 and CDK4 and inducing hypophosphorylation of RB [12]. Further mechanistic studies have shown that GGTI-2417, the methylester pro-drug of GGTI-2418, requires p27 to induce breast tumor cell death and that it stabilizes p27 by inhibiting its CDK2-mediated phosphorylation at Thr187, inducing the accumulation of p27 in the nucleus [11]. In breast cancer xenografts grown in nude mice, daily or every 3-day peritoneal injections of GGTI-2418 resulted in 94% and 77% tumor growth inhibition, respectively, while continuous infusion via a subcutaneous osmotic mini pump over 14 days resulted in 60% tumor regression in ErbB2-mammary tumor transgenic mice and was associated with an increase in p27 and decreased P-Akt levels [11].
Fig. 1.
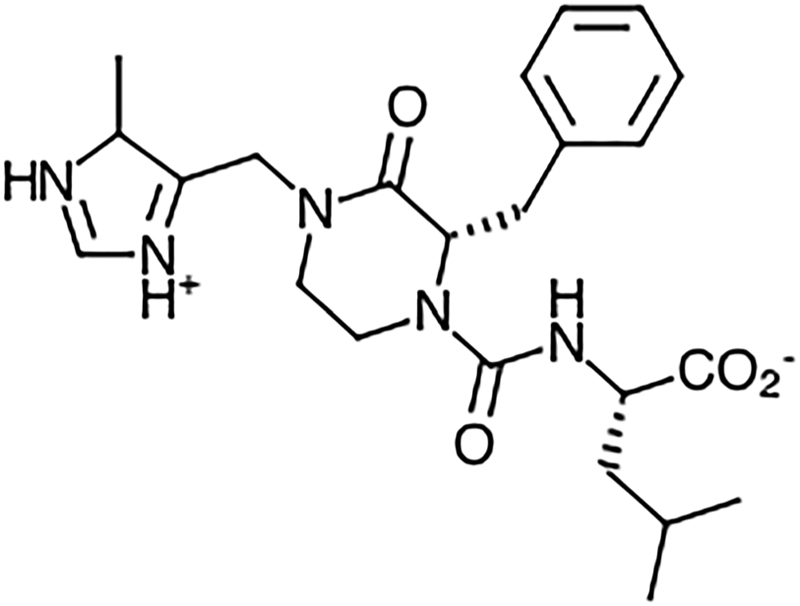
Structure of GGTI-2418
Preclinical dose-finding studies were performed in rats and dogs using two dosing strategies, daily and intermittent dosing [data on file]. Daily dosing was administered once daily for 7 days while intermittent dosing was administered once daily on Days 1–5 and Days 22–26 over a 6-week study period. Increases in maximum plasma concentration (Cmax) and area under the curve (AUC) were greater than dose proportional in both rats and dogs. The terminal half-life (t½) of GGTI-2418 ranged from 0.2 to 0.5 h in rats and from 0.4 to 1.2 h in dogs.
Hepatocyte metabolism studies in mice, rats, dogs, and humans indicate that GGTI-2418 undergoes transformation to five oxidized metabolites (M1–M5) that are detectable by liquid chromatography–mass spectrometry (LC/MS) but not high-performance liquid chromatography–ultraviolet (HPLC/UV) analysis. M1 was detectable in mice, rats, and dogs but not in humans.
Toxicology studies in rats established a maximum tolerated dose (MTD) of 200 mg/kg for single dosing and an MTD of 150 mg/kg/day for 7-day daily dosing. Infusion-related toxicities with red urine and rapid breathing were seen in both control and treated animals and attributed to the propylene glycol vehicle. Small foci of mononuclear inflammatory cells in the heart were seen in 4 of 20 rats at the 150-mg/kg/day dose and 2.9- to 3.7-fold increases in aspartate aminotransferase (AST) were seen at day 8 in rats dosed at 300 mg/kg/day.
In dogs, the MTD was 120 mg/kg/day using the intermittent schedule, and at least 80 mg/kg/day using the daily schedule. At the 120-mg/kg/day intermittent dose level, bronchiolar and alveolar hypertrophy with macrophage infiltration and fibrinous edema were observed in four of six dogs at day 27, but no clinical respiratory manifestations were observed, and no lung changes were observed at this dose in four dogs sacrificed at day 41. Infusion-related toxicity with shivering, vocalization, and soft stools was observed at multiple dose levels as well in controls. The highest non-severe toxic dose (HNSTD) was thus determined to be 60 mg/kg/day for 5 days, which approximates 1200 mg/m2/day in humans. Using a safety factor of 1/10, the initial starting dose of GGTI-2418 was determined to be 120 mg/m2 on days 1–5 of a 21-day cycle.
We conducted this first-in-human, open-label, phase I trial to establish the maximum tolerated dose, toxicity profile, pharmacokinetics, and preliminary antitumor efficacy of GGTI-2418 in patients with advanced solid tumors. The study was approved by the Institutional Review Board (IRB) of the University of Pennsylvania (IRB #5, protocol #809199) and all patients provided written informed consent prior to the initiation of any study procedures.
2. Methods
2.1. Patient Selection
Patients were eligible if they were at least 18 years of age, had a metastatic or unresectable solid tumor for which standard therapy did not exist or was ineffective, Eastern Cooperative Oncology Group Performance Status (ECOG PS) of 0–2, life expectancy of > 12 weeks, and normal bone marrow and organ function. Patients were excluded from the study if they were pregnant or had brain metastases, symptomatic pulmonary disease, uncontrolled intercurrent illness, HIV, or allergy to any component of the treatment. No therapy was permitted within 4 weeks of the initial dose of study drug.
2.2. Study Design
This was a phase I, open-label, dose-escalation study conducted in two US centers (University of Pennsylvania and Indiana University) in patients with treatment-refractory advanced solid tumors. The primary objectives were determination of dose-limiting toxicities (DLTs) and the MTD, and assessments of safety and tolerability. Secondary objectives were determination of overall response rate (ORR), characterization of pharmacokinetics, and exploration of biomarkers to predict response.
During the dose escalation, eight dose levels of GGTI-2418 were planned, from 120 to 2060 mg/m2, administered as a 30-min IV infusion on days 1–5 of each 21-day cycle. Doses of GGTI-2418 were supplied in 500 mM Tris aqueous buffer and diluted in dextrose or sterile water. An accelerated dose titration schema was used, with single patient cohorts until grade 2 or greater drug-related toxicity was observed during the 21-day DLT period, followed by a standard 3 + 3 dose escalation. At the MTD, an expansion cohort of an additional 10 patients with biomarker testing was planned. Patients were to continue on therapy until disease progression, unacceptable toxicity, or withdrawal of consent.
2.3. Evaluation of Patients
Physical examinations, hematological/biochemical laboratory evaluations, and electrocardiogram were performed at baseline and prior to the start of each 21-day cycle. Echocardiograms were performed at baseline and prior to cycle 3. Baseline radiologic tumor measurements were performed within 28 days prior to the start of the therapy, and subsequent radiographic assessments were performed every 2 cycles (6 weeks). Tumor response was evaluated according to Response Evaluation Criteria In Solid Tumors (RECIST) criteria. Toxicity was graded using Common Terminology Criteria for Adverse Events (CTCAE) v. 3.0.
2.4. Pharmacokinetic Analyses
Plasma was collected pre-dose and at 0.5 (end-of-infusion), 0.75, 1, 2, 4, 8, and 24 h from the start of the infusion on cycle 1, day 1 (C1D1) and cycle 2, day 4 (C2D4). High-performance liquid chromatography with tandem mass spectrometry (HPLC-MS/MS) was used to determine the plasma concentration of GGTI-2418. Plasma PK parameters were determined using non-compartmental analysis methods with WinNonlin Version 8.0. Actual (recorded) dosing and sampling times were used for the estimation of pharmacokinetic parameters.
3. Results
3.1. Patient Demographics
Fourteen patients were enrolled from April 2009 through April 2011. The baseline characteristics are listed in Table 1. The majority of patients had colorectal cancer (64%).
Table 1.
Demographics
| Total number of patients | 14 |
|---|---|
| Age | |
| Median | 60.9 |
| Range | 47–73 |
| Sex | |
| Male | 12 (86%) |
| Female | 2 (14%) |
| ECOG PS | |
| 0 | 4 (36%) |
| 1 | 7 (50%) |
| Unspecified | 2 (14%) |
| #Prior therapies | |
| 1 | 1 (7%) |
| 2 | 3 (21%) |
| 3 | 6 (43%) |
| 4 + | 3 (21%) |
| Unspecified | 1 (7%) |
| Tumor type | |
| Colorectal adenocarcinoma | 9 (64%) |
| Hepatocellular carcinoma | 1 (7%) |
| Duodenal adenocarcinoma | 1 (7%) |
| Colorectal neuroendocrine | 1 (7%) |
| Gallbladder | 1 (7%) |
| Unspecified | 1 (7%) |
ECOG PS Eastern Cooperative Oncology Group Performance Status
3.2. Dose Escalation and Maximum Tolerated Dose (MTD)
The dose escalation schema is shown in Table 2. No drug-related toxicities of grade 2 or higher occurred in the first 21-day cycle during dose escalation. At the 750 mg/m2 dose level, the first treated patient developed left facial droop and gait disturbance on cycle 1, day 1, and progressive ataxia on cycle 1, day 2. Brain magnetic resonance imaging demonstrated multiple metastases in the parietal lobe and cerebellum and he was discontinued from therapy due to disease progression and replaced. At each other dose level, a single patient was treated until the maximum planned dose of 2060 mg/m2 was reached. At this dose level, six patients were treated and no DLTs were observed.
Table 2.
Dose escalation
| Dose (mg/m2) | Increase from prior dose level |
Number of patients treated |
|---|---|---|
| 120 | – | 1 |
| 200 | 67% | 1 |
| 330 | 65% | 1 |
| 500 | 52% | 1 |
| 750 | 50% | 2a |
| 1050 | 40% | 1 |
| 1470 | 40% | 1 |
| 2060 | 40% | 6 |
One patient replaced due to disease progression on cycle 1 day 2 (new brain metastases)
3.3. Safety
All 14 patients received at least one dose of study treatment and were evaluable for toxicity (Table 3). The most common treatment-related toxicities were nausea (64%), diarrhea (28%), and fatigue (28%). A single instance of grade 3 alkaline phosphatase and grade 4 bilirubin elevation occurred in a patient with concurrent malignant biliary obstruction of the common hepatic duct from progressive disease. Treatment with GGTI-2418 was discontinued for disease progression and an external biliary drain was placed with resolution. All other toxicities attributed to GGTI-2418 were grades 1 or 2. One additional subject discontinued therapy for toxicity, with grade 2 bilirubin elevation and repeated elevations in alkaline phosphatase.
Table 3.
Toxicities attributed to GGTI-2418 occurring in at least two subjects or Grade ≥ 3
| Grade 1 | Grade 2 | Grade 3 | Grade 4 | |
|---|---|---|---|---|
| Nausea | 8 (57%) | 1 (7%) | ||
| Diarrhea | 2 (14%) | 2 (14%) | ||
| Fatigue | 4 (28%) | |||
| Pruritis | 1 (7%) | 2 (14%) | ||
| Neutropenia | 1 (7%) | 1 (7%) | ||
| Decreased appetite | 2 (14%) | |||
| Bilirubina | 1 (7%) | 1a (7%) | ||
| Elevated alkaline phosphatase | 1a (7%) | |||
| Elevated aspartate aminotransferase (AST) | 1 (7%) | 1a (7%) | ||
| Elevated alanine aminotransferase (ALT) | 2a (14%) |
Concurrent with malignant biliary obstruction due to disease progression
3.4. Efficacy
A total of 13 patients were evaluable for tumor response (Table 4). No objective responses were observed. Median number of cycles completed in the 13 evaluable patients was 2 (range 1–8). The best response was stable disease in 4 of 13 patients (31%), with duration of stable disease ranging from 2.6 to 6.7 months. The study discontinued enrollment prior to the planned expansion cohort due to a sponsor decision.
Table 4.
Best response and duration of therapy by dose and tumor type
| Dose (mg/m2) | No. of patients |
Primary tumor site (n) | Duration of therapy (mo) | Best response | Reason for discontinuation |
|---|---|---|---|---|---|
| 120 | 1 | CRC | 1.2 | PD | PD |
| 200 | 1 | CRC | 1.1 | PD | PD |
| 330 | 1 | HCC | 2.6 | SD | PD |
| 500 | 1 | CRC | 5.4 | SD | PD |
| 750 | 1 | Colon neuroendocrine | 4.3 | SD | Toxicity (LFTs) |
| 1050 | 1 | CRC | 0.8 | PD | PD |
| 1470 | 1 | CRC | 1.4 | PD | PD |
| 2060 | 6 | CRC (4) | Median: 1.3 | SD (1) | PD (5) |
| Gallbladder (1) | Range: 0.8–6.7 | PD (5) | Sponsor decision (1) | ||
| Unspecified (1) |
CRC colorectal cancer, HCC hepatocellular carcinoma, LFTs liver function tests, PD progressive disease, SD stable disease
3.5. Pharmacokinetics
Nine patients had pharmacokinetic analysis on C1D1, and six of these patients had subsequent analysis on C2D4 (Table 5). One subject at the 750-mg/m2 dose level and another at the 1050-mg/m2 dose had disease progression prior to cycle 2 pharmacokinetic analysis. Additionally, the sponsor only analyzed PK for C1D1 in a single subject at the 2060-mg/m2 dose level due to early study closure.
Table 5.
Pharmacokinetics of GGTI-2418
| Dose | AUC (μg × h/mL) |
Cmax (μg/mL) |
t½ (h) |
Vd (L) |
RAUC | ||||
|---|---|---|---|---|---|---|---|---|---|
| Cycle 1 Day 1 | Cycle 2 Day 4 | Cycle 1 Day 1 | Cycle 2 Day 4 | Cycle 1 Day 1 | Cycle 2 Day 4 | Cycle 1 Day 1 | Cycle 2 Day 4 | ||
| 120 | 2.6 | 2.5 | N/Aa | N/Aa | 1.2 | 1.0 | 157 | 133 | 1.0 |
| 200 | 15.2 | 21.9 | 18.5 | 26.8 | 1.1 | 1.4 | 40.4 | 36.1 | 1.4 |
| 330 | 16.0 | 16.2 | 17.8 | 19.0 | 1.2 | 1.3 | 75.8 | 56.0 | 1.0 |
| 500 | 36.3 | 23.0 | 56.1 | N/Aa | 1.0 | 1.1 | 42.8 | 70.6 | |
| 750 | 66.0 | 68.7 | 0.84 | 26.0 | |||||
| 750 | 126 | 186 | 93.7 | 117 | 1.1 | 1.1 | 18.3 | 12.2 | 1.5 |
| 1050 | 180 | 160 | 1.1 | 20.6 | |||||
| 1470 | 295 | 320 | 226 | 226 | 0.89 | 1.1 | 13.1 | 14.6 | 1.1 |
| 2060 | 462 | 209 | 0.99 | 12.8 | |||||
AUC area under the curve, Cmax maximal serum concentration, RAUC accumulation factor for area under the curve, t½ half-life, Vd volume of distribution
No end-of-infusion pharmacokinetic sample obtained; linear extrapolation applied and data excluded from summary analyses
The median elimination half-life was 1.1 h (range 0.84–1.2). Both Cmax and AUC increased linearly with dose (r2 for Cmax = 0.90, r2 for AUC = 0.97). Distribution volume (Vd) ranged from 13 to 150 L, suggesting some distribution outside the central compartment. There was only a minor degree of accumulation (R = 1.0–1.5) after repeat dosing over the evaluated dose range.
4. Discussion
This first-in-human trial of GGTI-2418 demonstrated tolerability across all dose levels tested. No dose-limiting toxicities were identified, and the only potential drug-related grade 3 or 4 toxicities were elevated bilirubin and alkaline phosphatase in a single patient with concurrent malignant biliary obstruction.
While efficacy was not the primary goal of this study, only limited evidence of disease stability was observed, with no objective responses. Stable disease was observed in four patients for up to 6.7 months; however, all four patients with disease stabilization eventually had progression of the disease and were discontinued. In light of limited efficacy and lack of toxicity, it is certainly possible that the drug did not adequately inhibit its target, GGTase I. Due to the early termination of the study, pharmacodynamic studies were not performed to test this hypothesis.
Pharmacokinetic testing demonstrated a t½ of only 1.1 hours, and it is possible that the rapid elimination of GGTI-2418 resulted in subtherapeutic drug concentrations for much of the 21-day treatment cycle with the 5-day infusion schedule. In preclinical murine models, intraperitoneal dosing every 3 days induced tumor stabilization despite the short half-life in plasma, suggesting either intratumoral accumulation or prolonged pharmacodynamics effects from downstream targets of GGTase I [11]. However, these murine models also showed that daily dosing produced greater disease stability compared with a higher dose every 3 days, and the use of a continuous subcutaneous pump induced tumor regression [11]. Thus, it is possible that more frequent dosing or an alternative formulation might be needed to achieve clinically relevant inhibition of GGTase I. An additional phase I study of GGTI-2418 with dose titration and pharmacodynamic correlation is planned in order to optimize dosing prior to phase II testing (NCT03900442).
In conclusion, GGTI2418 was safe and tolerable at all tested dose levels with some evidence of disease stability but no objective responses. Due to rapid elimination, dosing of GGTI2418 in this study may have been inadequate to achieve optimal inhibition of its target, GGTase I.
Key Points.
GGTI-2418 was safe and tolerable at all tested dose levels.
Dosing schedule may have been suboptimal due to the short half-life.
Some tumor stability was observed but there were no objective responses.
Acknowledgments
Funding This study was funded by Tigris Pharmaceuticals.
Footnotes
Conflict of interest Said Sebti is co-inventor of GGTI-2418 and chief scientific officer of Prescient Therapeutics, the manufacturer of GGTI-2418. Thomas Karasic declares that he has no conflict of interest. E. Gabriela Chiorean declares that she has no conflict of interest. Peter O’Dwyer declares that he has no conflict of interest.
Ethical Approval All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed Consent Informed consent was obtained from all individual participants included in the study.
References
- 1.Berndt N, Hamilton AD, Sebti SM. Targeting protein prenylation for cancer therapy. Nat Rev Cancer. 2011;11(11):775–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 2000;406:532–5. [DOI] [PubMed] [Google Scholar]
- 3.Hakem A, Sanchez-Sweatman O, You-Ten A, Duncan G, Wakeham A, Khokha R, Mak TW. RhoC is dispensable for embryogenesis and tumor initiation but essential for metastasis. Genes Dev. 2005;19:1974–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lambert JM, Lambert QT, Reuther GW, et al. Tiam1 mediates Ras activation of Rac by a PI(3)K-independent mechanism. Nat Cell Biol. 2002;4(8):621–5. [DOI] [PubMed] [Google Scholar]
- 5.Sjogren AK, Andersson KM, Liu M, et al. GGTase-I deficiency reduces tumor formation and improves survival in mice with K-RAS-induced lung cancer. J Clin Investig. 2007;117:1294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamad NM, Elconin JH, Karnoub A, et al. Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev. 2002;16:2045–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lim KH, Baines AT, Fiordalisi JJ, et al. Activation of RalA is critical for Ras-induced tumorigenesis of human cells. Cancer Cell. 2005;7:533–45. [DOI] [PubMed] [Google Scholar]
- 8.Malliri A, van der Kammen RA, Clark K, et al. Mice deficient in the Rac activator Tiam1 are resistant to Ras-induced skin tumours. Nature. 2002;417(6891):867–71. [DOI] [PubMed] [Google Scholar]
- 9.Khosravi-Far R, Solski PA, Clark GJ, et al. Activation of Rac1, RhoA, and mitogen-activated protein kinases is required for Ras transformation. Mol Cell Biol. 1995;15(11):6443–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peng H, Carrico D, Thai V, et al. Synthesis and evaluation of potent, highly-selective, 3-aryl-piperazinone inhibitors of protein geranylgeranyltransferase-I. Org Biomol Chem. 2006;4(9):1768–84. [DOI] [PubMed] [Google Scholar]
- 11.Kazi A, Carie A, Blaskovich MA, et al. Blockade of protein geranylgeranylation inhibits Cdk2-dependent p27Kip1 phosphorylation on Thr187 and accumulates p27Kip1 in the nucleus: implications for breast cancer therapy. Mol Cell Biol. 2009;29(8):2254–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun J, Qian Y, Chen Z, et al. The geranylgeranyltransferase I inhibitor GGTI-298 induces hypophosphorylation of retinoblastoma and partner switching of cyclin-dependent kinase inhibitors. A potential mechanism for GGTI-298 antitumor activity. J Biol Chem. 1999;274:6930–4. [DOI] [PubMed] [Google Scholar]