

Abstract
Increasing evidence suggests key roles for members of the mammalian Sterile20-like (MST) family of kinases in many aspects of biology. MST3 is a member of the STRIPAK complex, the deregulation of which has recently been associated with cancer cell migration and metastasis. Targeting MST3 with small-molecule inhibitors may be beneficial for the treatment of certain cancers, but little information exists on the potential of kinase inhibitor scaffolds to engage with MST3. In this study we screened MST3 against a library of 277 kinase inhibitors using differential scanning fluorimetry and confirmed 14 previously unknown MST3 inhibitors by X-ray crystallography. These compounds, of which eight are in clinical trials or FDA approved, comprise nine distinct chemical scaffolds that inhibit MST3 enzymatic activity with IC50 values between 0.003 and 23 μm. The structure–activity relationships explain the differential inhibitory activity of these compounds against MST3 and the structural basis for high binding potential, the information of which may serve as a framework for the rational design of MST3-selective inhibitors as potential therapeutics and to interrogate the function of this enzyme in diseased cells.
Keywords: drug discovery, enzymes, inhibitors, protein structures, structure–activity relationships
Introduction
Members of the mammalian Sterile20-like (MST) family of kinases were named for their similarity to the yeast kinase Ste20, and increasing evidence suggests key roles for these proteins in many aspects of biology.[1] MST kinases have been broadly divided into two subgroups. MST1 (STK4) and MST2 (STK3) are key components of the Hippo signaling pathway and couple cellular context within tissues to growth control and regulation of migration. MST3 (STK24), MST4 (STK26), and YSK1 (STK25, also called SOK1), which have also been classified as germinal center subgroup III kinases (GckIII)[2] are involved in the regulation of the cytoskeleton and Golgi apparatus. They share ~90% sequence identity in the N-terminal kinase domain, but less than 20% in the C-terminal domain. MST3 can be cleaved by caspases during apoptosis, resulting in nuclear accumulation of the active kinase domain which can promote apoptosis.[3] A similar regulatory function of the C terminus has been reported for MST1 and MST2.[4] A common feature of MST kinases is their interaction with isoforms of the evolutionarily conserved MO25 scaffolding proteins,[5] which are known master regulators of the LKB1 tumor suppressor, through interaction with the Ste20-like pseudokinase STRAD.[6] MST3 is also activated by autophosphorylation of residue Thr178 of the activation loop, thereby dramatically increasing the kinase activity.[7]
Until recently, the involvement of MST3 in diseases was circumstantial. Altered MST3 levels have been reported in colorectal and non-small-cell lung cancer.[8] MST3 has been found to be up-regulated in the trophoblasts of human placenta as a result of oxidative stress, which has been linked with the spontaneous disruption of the fetal membrane tissue during natural childbirth, but also to the occurrence of spontaneous miscarriages.[9] MST3 (as well as MST4 and YSK1) are components of a large PP2A complex, termed the STRIPAK complex.[10] The STRIPAK complex has recently received attention for its involvement in cancer cell migration and metastasis.[11] Components of the STRIPAK complex, particularly FAM40A, negatively regulate MST3 and MST4 by dephosphorylation of the activation loop. Mutations of FAM40B, which is a competitive inhibitor of FAM40A, disrupt this regulation of MST3 and MST4, and together with high MST3 and MST4 levels promote malignancy by enabling enhanced migration.
As an orphan kinase, MST3 has received little attention for the development of small-molecule inhibitors. The only MST3–inhibitor co-crystal structures known were solved with staurosporine (PDB ID: 3CKX, no associated publication) and a set of recently disclosed pyrrolopyrimidine inhibitors of leucine-rich repeat kinase 2 (LRRK2), for which MST3 served as a surrogate.[12] In this work we screened MST3 against a library of diverse kinase inhibitors using differential scanning fluorimetry (DSF) and confirmed 14 compounds as MST3 inhibitors by X-ray crystallography. These inhibitors comprise nine distinct chemical scaffolds, and the provided structure–activity relationship (SAR) information may serve as a framework for the rational design of MST3-selective inhibitors as chemical probes and potential cancer therapeutics.
Results and Discussion
Discovery and confirmation of MST3 inhibitors
The kinase domain of MST3 (residues 1–303) was screened against the kinase inhibitor library from Selleck Chemicals using DSF. In the ligand-free state MST3 exhibited a melting temperature of 45 °C. Of the 277 compounds screened, 70 compounds showed ΔTm values between 1 and 5 °C and 17 compounds ΔTm values between 5 and 11 °C (Figure 1a; Supporting Information Table S1). Selected compounds along with three separately obtained kinase inhibitors, CDK1/2 inhibitor III, PKR inhibitor C16 and a hydroxyphenyltriazine fragment (TP fragment), which showed ΔTm values between 1.0 and 10.8 °C, were subjected to co-crystallization studies with MST3. Of these, 14 compounds were found to bind to the ATP site with full occupancy (Figure 2, Table S2). The inhibitory activity of thus confirmed ligands was determined in a 33P-radiolabeled kinase assay carried out by Reaction Biology (Figure 1b). Seven compounds were potent inhibitors, with IC50 values between 0.003 and 0.086 μm; five compounds were moderately active, with IC50 values between 0.16 and 1.3 μm; and three compounds were weak inhibitors, with IC50 values between 7.4 and 23 μm. Inhibitory activity and temperature shift were logarithmically proportional (Figure 1c), suggesting that DSF is a reliable method not only for the identification of small-molecule ligands of MST3 from compound libraries, but also to estimate the inhibitory activity of hit compounds and analogues thereof using a range of commercially available inhibitors as a standard. Under the conditions described in the Experimental Section, the data shown in Figure 1c follow Equation (1), thereby directly correlating differential melting temperature (y) with inhibitory activity [Eq. (1)]:
| (1) |
Figure 1.
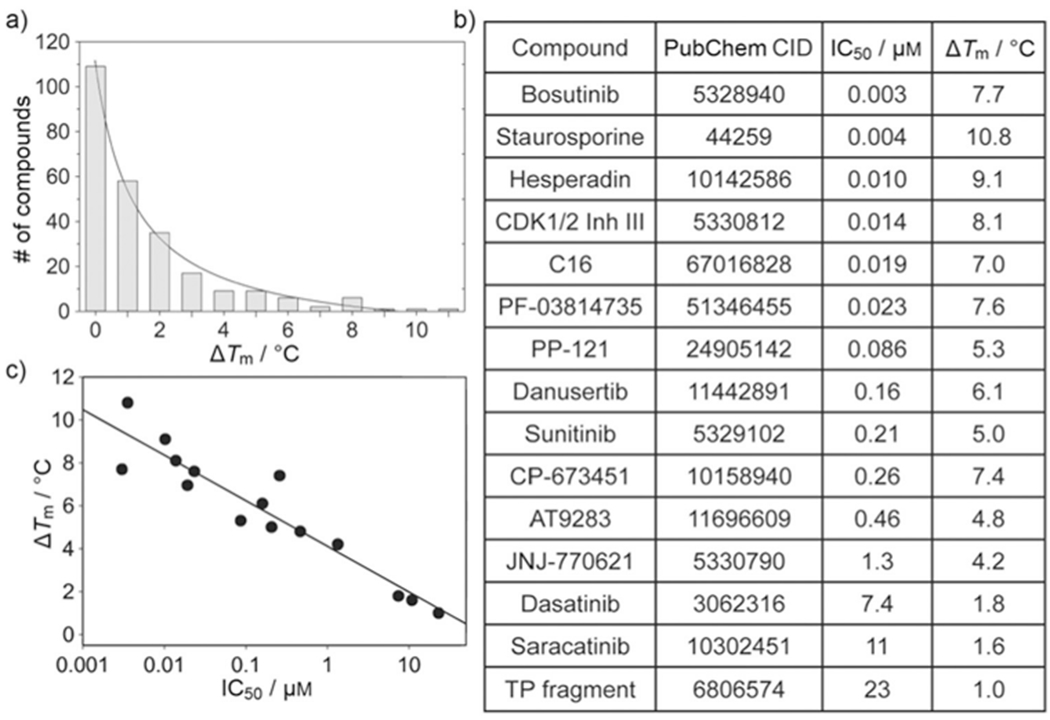
Discovery of small-molecule inhibitors of MST3 using DSF. a) Distribution of compounds as a function of ΔTm values. Of the 277 compounds screened, 23 compounds interfered with the assay due to intrinsic fluorescence or precipitation and were discarded from further evaluation; 109 compounds showed negative temperature shifts (−2.8 to −0.01 °C) and were denoted as zero. Each bar represents compounds with ΔTm ≤x-axis value, that is, 0: ΔTm≤0; 1: ΔTm≤1, etc. b) Identifiers and associated IC50 and ΔTm values of compounds confirmed as MST3 binders by X-ray crystallography. c) Logarithmic proportionality between ΔTm and IC50 values; data were fit to Equation (1), yielding y0 = 4.08±0.3 and a = −0.93±0.09.
Figure 2.
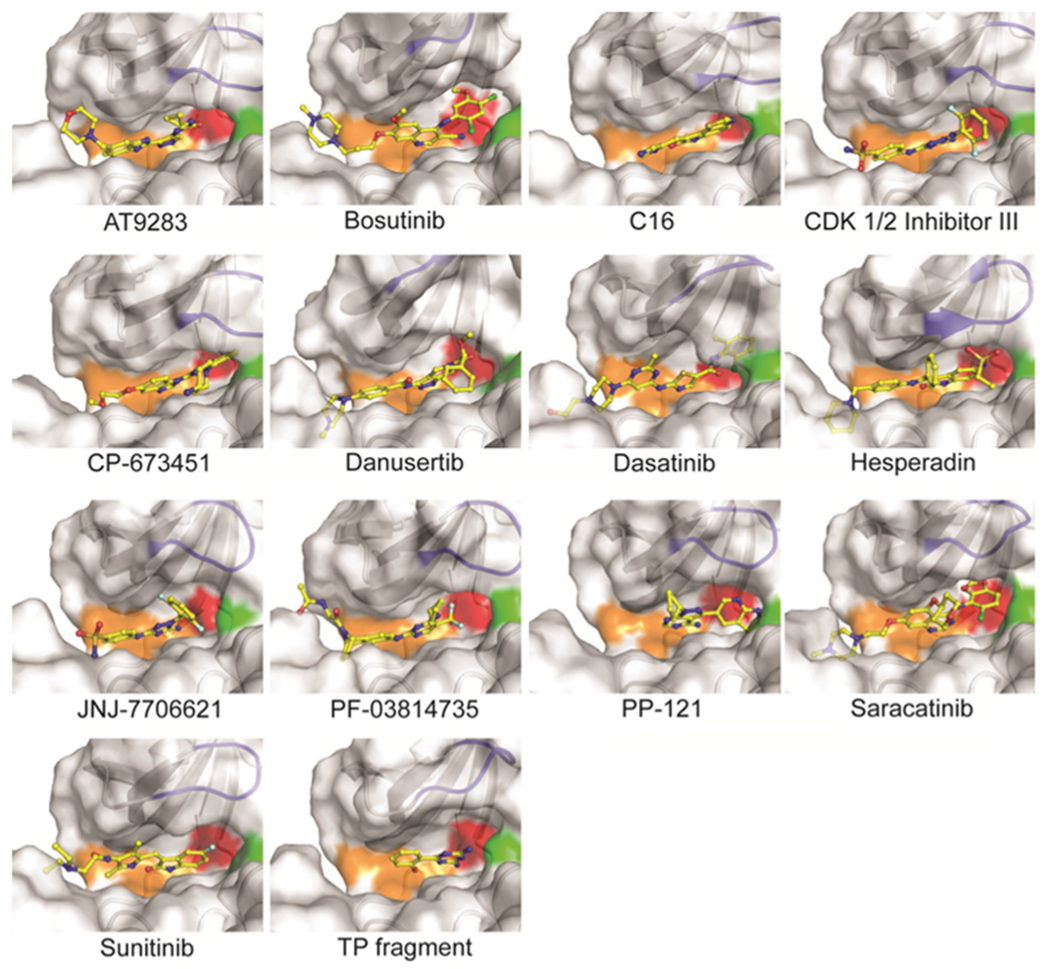
Co-crystal structures of MST3 with inhibitors identified by DSF. Shown are surface-cartoon presentations around the ATP site of MST3 with bound inhibitors (yellow). Indicated in orange, red, green, and blue are the hinge region, gatekeeper residue, DFG motif, and P-loop, respectively.
Compound-induced temperature shifts were similar for MST3 kinase domain (KD) and full-length protein (Table S3), suggesting that the ATP site is not obstructed by the C terminus, which is subject to cleavage by caspases.[3]
Ligand-induced structural changes in MST3
With the exception of staurosporine, the co-crystal structures determined herein provide novel information on the interaction of MST3 with diverse kinase inhibitors. All identified compounds are type I inhibitors that bind to the active (DFG-in) conformation of the ATP site through hydrogen bonding interactions with the hinge region (residues Glu100–Leu102; Figure 2, Figure S1). The activation loop is well defined in all co-crystal structures, stabilized by the autophosphorylated Thr178 residue, which establishes salt bridges with Arg143 and Arg176 (Figure 3a). No additional phosphorylation sites were identified in the MST3 KD. Superposition of all MST3–inhibitor complexes using the MST3–TP fragment complex as a reference allowed an assessment of global structural changes induced by ligand binding (Figure 3b–d). Apart from the flexible N terminus, the largest conformational changes occur in the P-loop including the flanking β strands (residues 31–37) with root-mean-square deviations (RMSD) of up to 6.5 Å and the C-helix (residues 63–76) with RMSD values of up to 3 Å. Other prominent regions defining the kinase domain, including the hinge region, the DFG motif, and the activation loop, are considerably less affected. It appears that structural changes do not correlate with inhibitory potency. For example, the complexes with bosutinib and staurosporine showed only small overall structural changes, whereas the equally potent hesperadin and CDK1/2 inhibitor III induced large structural changes in the P-loop and the C-helix, respectively.
Figure 3.
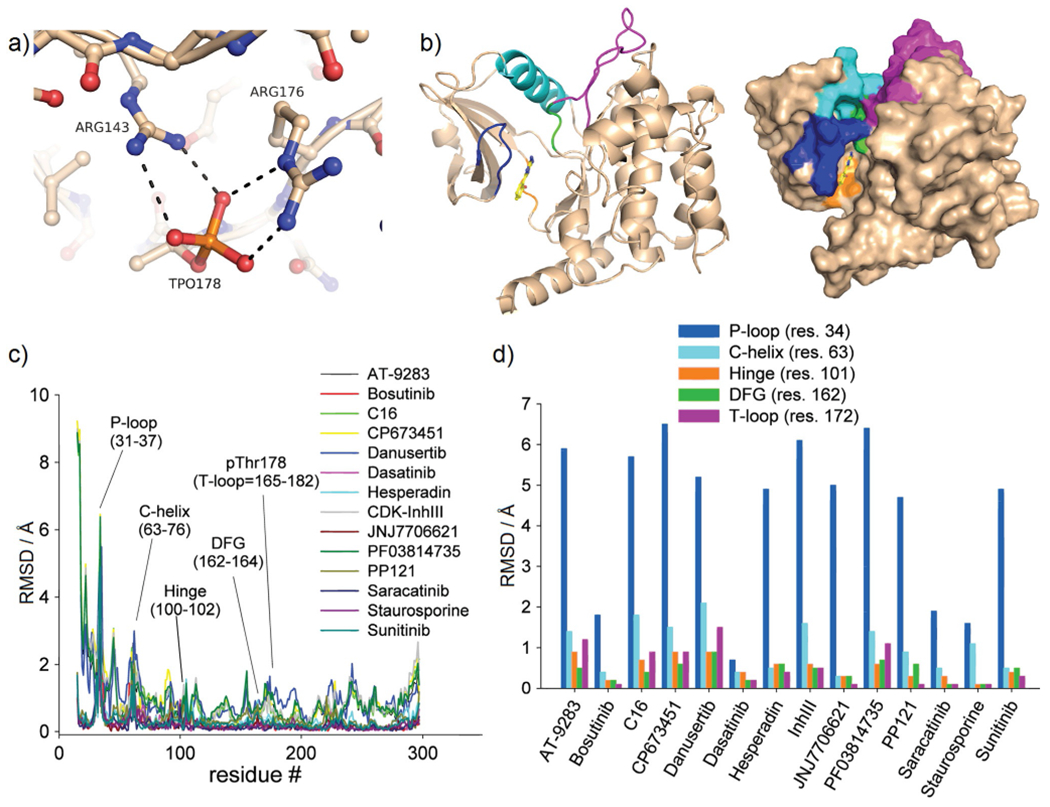
Inhibitor-induced conformational changes in MST3 occur primarily in the P-loop. a) The activation loop (T-loop) of MST3 (residues 165–182) is stabilized through salt bridges of the autophosphorylated Thr178 with Arg176 and Arg143. b) Overview of key structural elements of MST3 in cartoon presentation: P-loop (blue), C-helix (cyan), hinge (orange), DFG (green), and T-loop (magenta). c) RMSDs as a function of residues of liganded structures aligned through the MST3–TP fragment complex. d) Replot of panel c showing residues of each structural element with largest RMSD values.
SAR of MST3 inhibitors
The hinge binding moieties of the co-crystallized ligands can be grouped into nine distinct chemical scaffolds, four of which are shared among multiple inhibitors (Figure 4). Comparing the binding interactions of structurally related but differentially active compounds allows the identification of molecule–target properties that are critical for inhibitory potential.
Figure 4.
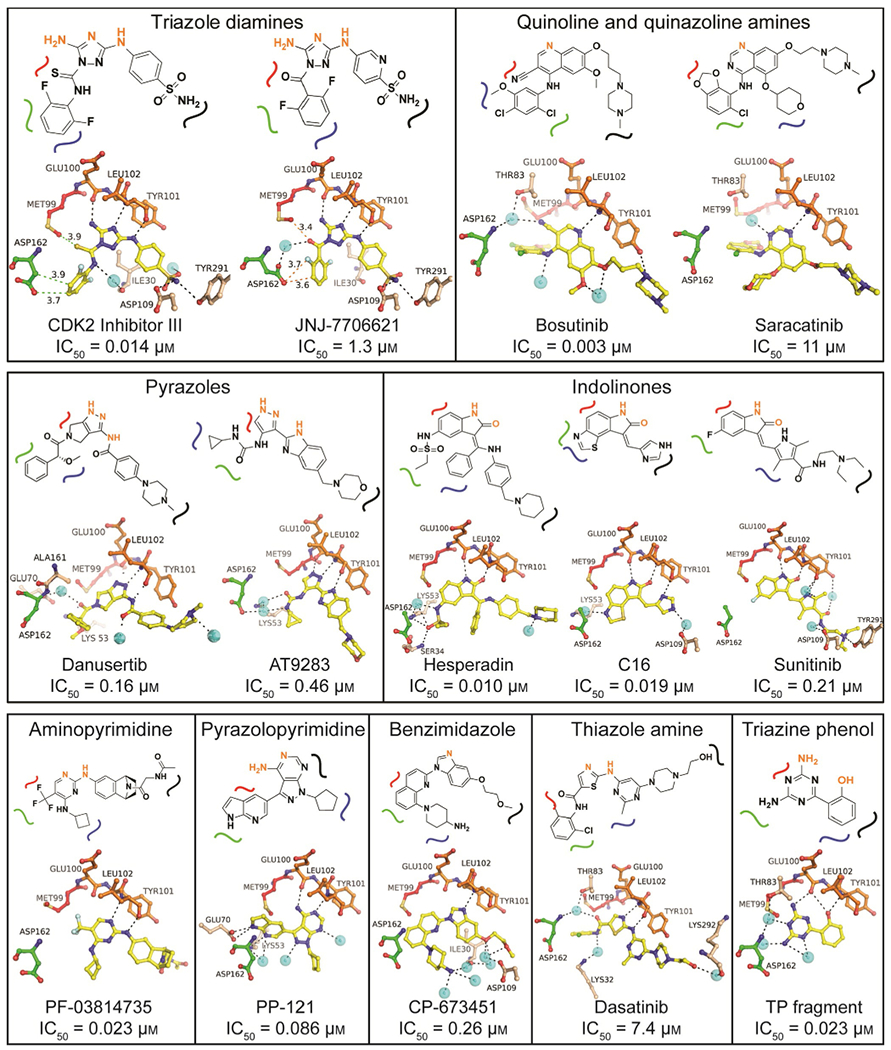
SAR of MST3 inhibitors. Compounds were classified according to their hinge binding core scaffolds. In the schematic representations, atoms highlighted in orange directly interact with the main chain of the hinge region (residues Glu100 and Tyr101). The curved lines indicate the position of the inhibitor relative to the gatekeeper residue Met99 (red), the DFG motif (residues 162–164; green), the P-loop (residues 31–37; blue), and the front specificity pocket toward solvent (black). The panels below the schematics show the crystallographic models with hydrogen bonding interactions as black dotted lines and other distances to neighboring residues indicated in green dotted lines. The complete hydrogen bonding and vdW interaction networks are shown in Figure S1 along with electron density maps of the bound ligand. Cyan spheres indicate water molecules.
The triazole diamine containing compounds CDK1/2 inhibitor III and JNJ-7706621 are highly similar in structure and binding mode to MST3, but differ 100-fold in inhibitory activity (IC50=0.014 and 1.3 μm, respectively). CDK1/2 inhibitor III was part of a series of 1-acyl-1H-[1,2,4]triazole-3,5-diamine analogues developed as CDK inhibitors with sub-nanomolar activity against CDK1 and CDK2, but also high activity against tyrosine kinases including VEGFR2.[13] JNJ-7706621 was developed as a cell-cycle regulatory kinase inhibitor primarily targeting CDKs and Aurora kinases which showed potent antiproliferative activity against a range of cancer cell lines.[13,14] In MST3, the number of hydrogen bonds, including water-mediated hydrogen bonds, is identical for both inhibitor complexes, and the differential inhibitory activity appears to be caused predominantly by differences in van der Waals (vdW) interactions along the gatekeeper–DFG interface of the ATP site. The thioamide group of the CDK inhibitor allows a more favorable vdW engagement with the gatekeeper residue Met99 than the polar carbonyl oxygen atom of JNJ-7706621, while the hydrophobic difluorophenyl ring is sandwiched between the side chain of Asp162 and residues of the P-loop (Gly31–Gly33). In contrast, the carbonyl group of JNJ-7706621 engages a water molecule attracting the side chain of Asp162, which positions the carboxyl group unfavorably close to the difluorophenyl ring.
The structurally related quinoline and quinazoline containing bosutinib and saracatinib bind similarly to MST3, but differ drastically in inhibitory potential (IC50=0.003 and 11 μm, respectively). Bosutinib (SKI-606) was originally developed as an SRC inhibitor[15] and is in clinical use for the treatment of chronic myelogenous leukemia under the tradename Bosulif. Saracatinib has been developed as a dual SRC/Bcr-Abl inhibitor[16] and is in several phase II and III trials for the treatment of solid tumors including ovarian, breast, lung and prostate cancer (www.clinicaltrials.gov). The differential inhibitory activity of bosutinib and saracatinib is presumably caused by the ability of the nitrile group of bosutinib to engage a tightly bound water molecule of a conserved water-mediated network recently reported for bosutinib-sensitive kinases.[17] Additionally, the substituents of the aniline ring of bosutinib interact with the sub-site confined by the DFG motif and the gatekeeper residue more favorably through multiple hydrophobic vdW interactions (Figure S1).
The pyrazole-containing inhibitors danusertib and AT9283 show similar potency against MST3, with IC50 values of 0.16 and 0.46 μm, respectively. Danusertib (PHA-739358) is a pan-Aurora kinase inhibitor that also inhibits the tyrosine kinases RET, NTRK1, FGFR1, and ABL1. It showed promising activity in rodent models of various cancers including colon, ovarian, breast and acute myelogenous leukemia,[18] and is currently in phase II trials for prostate cancer and leukemia. AT9283 is a broad-spectrum kinase inhibitor with high activity against JAK and Aurora kinases[19] and is in phase I and II trials for solid and hematological malignancies[20] (www.clinicaltrials.gov). Similar to the MST3–bosutinib complex, danusertib and AT9283 engage in water-mediated hydrogen bonds with Asp162 and neighboring residues of the DFG motif. Additionally, the N-methyl group of the piperazine ring of danusertib appears to establish a hydrogen bond with the carbonyl oxygen atom of Gly103 next to the hinge region which may explain the slightly higher inhibitory potential of danusertib over AT9283.
The indolinone-containing inhibitors hesperadin and C16 are highly potent against MST3, with IC50 values of 0.01 and 0.02 μm, respectively, while sunitinib is 20-fold less active. Hesperadin is a broad-spectrum kinase inhibitor with high activity against Aurora kinases.[21] C16 is a broad-spectrum kinase inhibitor with demonstrated activity against PKR in vitro and in vivo.[22] Sunitinib is a broad-spectrum tyrosine kinase inhibitor,[23] and is FDA approved for the treatment of renal cell carcinoma, gastrointestinal stromal tumor, and pancreatic neuroendocrine tumors (trade name Sutent). Hesperadin and C16 both engage a tightly bound water molecule bound to the side chain of Lys53 and positioned approximately midway between the gatekeeper residue and the DFG motif. The more hydrophobic properties of sunitinib in this region of the ATP site prevent a similar interaction potential. Although sunitinib shows increased hydrogen bonding potential toward residues of the front-specificity pocket of the ATP site, it appears that these interactions do not compensate for the lack of binding potential within the gatekeeper/DFG region.
Five inhibitors harbor unique hinge binding cores not shared with other inhibitors. PF-03814735 (aminopyrimidine) and PP-121 (pyrazolopyrimidine) are potent MST3 inhibitors, with IC50 values of 0.023 and 0.086 μm, respectively. PF-03814735, an Aurora kinase inhibitor, inhibited tumor growth in several xenograft models, including colon and breast cancer,[24] and a phase I trial for the treatment of advanced solid tumors was completed in 2012. PP-121 is a broad-spectrum kinase inhibitor active against several serine/threonine, tyrosine, and phosphoinositide kinases.[25] PF-03814735 establishes hydrogen bonds only with the hinge region, and its high activity against MST3 is presumably due to the optimal positioning of the trifluoromethyl moiety toward gatekeeper residue Met99 and DFG residue Asp162 for vdW interactions. By contrast, PP-121 appears to exert high inhibitory activity mostly through polar interactions. The benzimidazole CP-673451 was developed as a PDGFR inhibitor which significantly decreased tumor growth in xenograft models of glioblastoma, colon, and lung cancer,[26] With an IC50 value of 0.26 μm, it is moderately active against MST3 by establishing a single hydrogen bond with the hinge region and multiple water-mediated hydrogen bonds with residues of the front-specificity pocket and the P-loop. Dasatinib has been developed as a dual SRC/ABL inhibitor,[27] exhibits anti-proliferation and apoptotic activity against a wide range of cancers,[28] and is an FDA-approved antineoplastic agent (trade name Sprycel). With an IC50 value of 7.4 μm, dasatinib is a weak inhibitor of MST3 likely due to steric hindrance caused by the carboxamide linker protruding the chloromethylphenyl ring toward the gatekeeper residue, which moves away as a result. The hydroxyphenyltriazine (TP) fragment was previously identified as a weak CDK2 inhibitor in an HTS campaign (PDB ID: 3QQJ[29]) and served here to probe hinge binding potential in MST3.
Conclusions
MST3 can be considered an orphan kinase which, until recently, had no clear implications in disease. It has therefore received little attention as a potential target for the development of chemical probes and therapeutics. The discovery that alterations of the STRIPAK complex, of which MST3 is a component, impact cancer cell migration and metastasis[11] suggests that the targeting of MST3 by small-molecule inhibitors may be beneficial for the treatment of certain cancers. We identified, confirmed, and characterized 14 previously unknown inhibitors of MST3 representing nine distinct chemical scaffolds. Our findings indicate that MST3 is vulnerable to inhibition by various kinase inhibitors, some of which are commonly used as chemical probes and as cancer drugs in the clinic. It is conceivable that the most potent compounds synergize with MST3 and the primary target to achieve enhanced cancer cell killing. The lack of MST3-selective inhibitors and of suitable biomarkers makes it difficult to assess the consequences of chemical inhibition of MST3 in cell models of cancer and other diseases. The SAR information of this work provides the first solid basis for the rational design of MST3-selective inhibitors using diverse chemical scaffolds to interrogate the function of this enzyme in diseased cells.
Experimental Section
Reagents and compounds for biochemical and crystallographic experiments were purchased from Sigma–Aldrich, Selleck Chemicals, and Hampton Research unless otherwise indicated. The L1200 kinase inhibitor library was purchased from Selleck Chemicals. Protein concentration was determined by A280 molar absorbance using a Nanodrop ND-1000 spectrophotometer (Nanodrop Technologies).
Cloning and expression:
The gene coding for the kinase domain (KD) of human MST3 comprising residues 1–303 was amplified by PCR from MCF-7 cDNA (a kind gift from Dr. Lancaster, Moffitt Cancer Center) and subcloned into a modified pET28 plasmid to provide fusion with a cleavable His6 tag. The protein was overexpressed in E. coli BL21(DE3) CodonPlus® RIL cells at 16°C and 0.1 mm IPTG. The gene coding for the full-length (FL) human MST3 comprising residues 1–431 was amplified by PCR using DNASU clone (HsCD00042929 STK24 (FL) in pDONR221) as a template. The gene was then subcloned into a modified pET28 plasmid to provide fusion with a cleavable His6-tagged maltose binding protein (MBP). The protein was overexpressed in E. coli BL21(DE3) CodonPlus® RIL cells at 37 °C and 0.5 mm IPTG.
Protein purification:
The kinase domain was purified by FPLC using Ni2+-affinity chromatography (GE Healthcare) in 50 mM Tris (pH 8), 300 mM NaCl, and a gradient of 10–300 mm imidazole. The eluted fusion protein was digested overnight with PreScission protease at 4 °C, followed by a second Ni2+-affinity column. The flowthrough was then loaded onto a Superdex200 26/60 column (GE Healthcare) in 50 mm Tris (pH 8), 150 mm NaCl, 1 mm DTT. The resulting eluate yielded crystallization-grade monomeric enzyme. The full-length protein was purified using a similar approach with a few changes. The Ni2+-affinity columns were run using 50 mm Tris (pH 8), 150 mm NaCl, and a gradient of 10–250 mm imidazole. In addition, to remove PreScission from the solution, the protein was subjected to a glutathione column in 50 mm Tris (pH 8), 150 mm NaCl, 1 mm DTT, and a gradient of 0–10 mm reduced glutathione. The flow-through from this column was then loaded onto the Superdex200 26/60 in the same buffer as above. Purified MST3 KD was concentrated to 25 mgmL−1 for crystallization studies, and both KD and FL proteins were concentrated to 10 mgmL−1 for DSF studies and stored at −80°C.
Differential scanning fluorimetry (DSF):
The binding potential of compounds against MST3 was assessed by DSF using a StepOne-Plus real-time PCR system (Applied Biosystems, Grand Island, NY). Purified protein (4 μm final concentration; 50 mm HEPES (pH 7.5), 150 mm NaCI, 5 mm MgCl2, and 1 mm DTT) was assayed in a 96-well plate in quadruplicate. Inhibitors were added to a final concentration of 50 and 100 μm in 2% DMSO. Protein Thermal Shift Dye (Applied Biosystems, Grand Island, NY) was used as the fluorescent probe, and fluorescence was measured using the ROX Reporter channel (λ 620 nm). Protein stability was investigated by increasing temperature from 25 to 95 °C with a 1 % ramp speed. The inflection point of the transition curve/melting temperature (Tm) was calculated using the Boltzmann equation within the Protein Thermal Shift software package (ver. 1.1; Applied Biosystems, Grand Island, NY). The ΔTm was calculated by using DMSO control wells as a reference.
Determination of inhibitory activity:
Inhibitory activity of compounds was determined using a 33P-labeled assay performed by Reaction Biology Corp. The substrate was myelin basic protein at 10 μm, and reactions were carried out with 10 μm ATP. Compounds were assayed in 10-dose response with fourfold serial dilutions from 50 to 0.002 μm.
Crystallography:
Compounds were co-crystallized with MST3 using the hanging-drop vapor diffusion method. Robust crystallization conditions were established at 18 °C using 25 mgmL−1 protein in 50 mm Tris·HCl (pH 8), 150 mm NaCl, 1 mm DTT, 2mm compound mixed 1:1 with 200 mm MgCl2, 100 mm HEPES (pH 7.5), and 30% (v/v) PEG400. Final DMSO concentration in the droplet varied between 1 and 10% (v/v) depending on the aqueous solubility of compound. Crystals in mother liquor were mounted and flash frozen in a stream of nitrogen gas. X-ray diffraction data were recorded at −180°C at the beamlines 22-ID and 22-BM (SER-CAT, Advanced Photon Source, Argonne National Laboratories) and at the Moffitt Cancer Center Structural Biology Core using CuKα X-rays generated by a Rigaku Micro-Max 007-HF X-ray generator, focused by mirror optics and equipped with a Rigaku CCD Saturn 944 system. Data were reduced with HKL2000[30] or XDS;[31] PHENIX[32] was employed for phasing and refinement, and model building was performed using Coot.[33] Structures were solved by molecular replacement using the monomer of PDB ID: 3A7G[34] as search model. All crystal structures were validated by MolProbity[35] and phenix.model_vs_data[36] before deposition in the Protein Data Bank (PDB). Images were prepared using PyMOL (Schrödinger LLC). Data collection and refinement statistics along with the PDB entry codes are listed in Table S2.
Supplementary Material
Acknowledgements
We thank Christopher L. Watts and Donna J. Ingles (Moffitt Cancer Center) for assistance with data collection and protein purification, and the Southeast Regional Collaborative Access Team (SER-CAT, University of Georgia) for assistance with Synchrotron data collection at Argonne National Laboratory This work was supported in part by the Moffitt Chemical Biology Core Facility through grant P30-CA076292 and by the USF-Moffitt Anna Valentine Cancer Fund. The co-crystal structure coordinates and structure factors have been deposited in the PDB with accession codes 4QML, 4QMM, 4QMN, 4QMO, 4QMP, 4QMQ, 4QMR, 4QO9, 4QMT, 4QNA, 4QMU, 4QMV, 4QMW, 4QMX, 4QMY, and 4QMZ.
Footnotes
Supporting information for this article can be found under http://dx.doi.org/10.1002/cmdc.201600115.
References
- [1].Thompson BJ, Sahai E, J. Cell Biol 2015, 210, 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Sugden PH, McGuffin LJ, Clerk A, Biochem. J 2013, 454, 13–30. [DOI] [PubMed] [Google Scholar]
- [3].Huang CY, Wu YM, Hsu CY, Lee WS, Lai MD, Lu TJ, Huang CL, Leu TH, Shih HM, Fang HI, Robinson DR, Kung HJ, Yuan CJ, J. Biol. Chem 2002, 277, 34367–34374. [DOI] [PubMed] [Google Scholar]
- [4].a) Lee KK, Murakawa M, Nishida E, Tsubuki S, Kawashima S, Sakamaki K, Yonehara S, Oncogene 1998, 16, 3029–3037 [DOI] [PubMed] [Google Scholar]; b) Lee KK, Ohyama T, Yajima N, Tsubuki S, Yonehara S, J. Biol. Chem 2001, 276, 19276–19285. [DOI] [PubMed] [Google Scholar]
- [5].a) Filippi BM, de Los Heros P, Mehellou Y, Navratilova I, Gourlay R, Deak M, Plater L, Toth R, Zeqiraj E, Alessi DR, EMBO J. 2011, 30, 1730–1741 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hao Q, Feng M, Shi Z, Li C, Chen M, Wang W, Zhang M, Jiao S, Zhou Z, J. Struct. Biol 2014, 186, 224–233 [DOI] [PubMed] [Google Scholar]; c) Mehellou Y, Alessi DR, Macartney TJ, Szklarz M, Knapp S, Elkins JM, Biochem. Biophys. Res. Commun 2013, 431, 604–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Boudeau J, Baas AF, Deak M, Morrice NA, Kieloch A, Schutkowski M, Prescott AR, Clevers HC, Alessi DR, EMBO J. 2003, 22, 5102–5114 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Baas AF, Boudeau J, Sapkota GP, Smit L, Medema R, Morrice NA, Alessi DR, Clevers HC, EMBO J. 2003, 22, 3062–3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lu TJ, Lai WY, Huang CY, Hsieh WJ, Yu JS, Hsieh YJ, Chang WT, Leu TH, Chang WC, Chuang WJ, Tang MJ, Chen TY, Lu TL, Lai MD, J. Biol. Chem 2006, 281, 38405–38417. [DOI] [PubMed] [Google Scholar]
- [8].a) Hennig EE, Mikula M, Rubel T, Dadlez M, Ostrowski J, J. Mol. Med 2012, 90, 447–456 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wan YW, Sabbagh E, Raese R, Qian Y, Luo D, Denvir J, Vallyathan V, Castranova V, Guo NL, PLoS One 2010, 5, e12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Poston L, Raijmakers MT, Placenta 2004, 25, S72–S78 [DOI] [PubMed] [Google Scholar]; b) Tang C, Liang J, Qian J, Jin L, Du M, Li M, Li D, Int. J. Clin. Exp. Pathol 2014, 7, 959–968 [PMC free article] [PubMed] [Google Scholar]; c) Wu HY, Lin CY, Chen TC, Pan ST, Yuan CJ, Int. J. Biochem. Cell Biol 2011, 43, 742–750; [DOI] [PubMed] [Google Scholar]; d) Wu HY, Lin CY, Lin TY, Chen TC, Yuan CJ, Apoptosis 2008, 13, 283–294. [DOI] [PubMed] [Google Scholar]
- [10].a) Glatter T, Wepf A, Aebersold R, Gstaiger M, Mol. Syst. Biol 2009, 5, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kean MJ, Ceccarelli DF, Goudreault M, Sanches M, Tate S, Larsen B, Gibson LC, Derry WB, Scott IC Pelletier L, Baillie GS, Sicheri F, Gingras AC, J. Biol. Chem 2011,286, 25065–25075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Madsen CD, Hooper S, Tozluoglu M, Bruckbauer A, Fletcher G, Erler JT, Bates PA, Thompson B, Sahai E, Nat. Cell Biol 2015, 17, 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Henderson JL, Kormos BL, Hayward MM, Coffman KJ, Jasti J, Kurumbail RG, Wager TT, Verhoest PR, Noell GS, Chen Y, Needle E, Berger Z, Steyn SJ, Houle C, Hirst WD, Galatsis P, J. Med. Chem 2015, 58, 419–432. [DOI] [PubMed] [Google Scholar]
- [13].Lin R, Connolly PJ, Huang S, Wetter SK, Lu Y, Murray WV, Emanuel SL, Gruninger RH, Fuentes-Pesquera AR, Rugg CA, Middleton SA, Jolliffe LK, J. Med. Chem 2005, 48, 4208–4211. [DOI] [PubMed] [Google Scholar]
- [14].Emanuel S, Rugg CA, Gruninger RH, Lin R, Fuentes-Pesquera A, Connolly PJ, Wetter SK, Hollister B, Kruger WW, Napier C, Jolliffe L, Middleton SA, Cancer Res. 2005, 65, 9038–9046. [DOI] [PubMed] [Google Scholar]
- [15].Boschelli DH, Ye F, Wang YD, Dutia M, Johnson SL, Wu B, Miller K, Powell DW, Yaczko D, Young M, Tischler M, Arndt K, Discafani C, Etienne C, Gibbons J, Grod J, Lucas J, Weber JM, Boschelli F, J. Med. Chem 2001, 44, 3965–3977. [DOI] [PubMed] [Google Scholar]
- [16].Hennequin LF, Allen J, Breed J, Curwen J, Fennell M, Green TP, Lambert-van der Brempt C, Morgentin R, Norman RA, Olivier A, Otterbein L, Ple PA, Warin N, Costello G, J. Med. Chem 2006, 49, 6465–6488. [DOI] [PubMed] [Google Scholar]
- [17].Levinson NM, Boxer SG, Nat. Chem. Biol 2014, 10, 127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Carpinelli P, Ceruti R, Giorgini ML, Cappella P, Gianellini L, Croci V, Degrassi A, Texido G, Rocchetti M, Vianello P, Rusconi L, Storici P, Zugnoni P, Arrigoni C, Soncini C, Alli C, Patton V, Marsiglio A, Ballinari D, Pesenti E, Fancelli D, Moll J, Mol. Cancer Ther 2007, 6, 3158–3168. [DOI] [PubMed] [Google Scholar]
- [19].Howard S, Berdini V, Boulstridge JA, Carr MG, Cross DM, Curry J, Devine LA, Early TR, Fazal L, Gill AL, Heathcote M, Maman S, Matthews JE, McMenamin RL, Navarro EF, O’Brien MA, O’Reilly M, Rees DC, Reule M, Tisi D, Williams G, Vinkovic M, Wyatt PG, J. Med. Chem 2009, 52, 379–388. [DOI] [PubMed] [Google Scholar]
- [20].a) Hay AE, Murugesan A, DiPasquale AM, Kouroukis T, Sandhu I, Kukreti V, Bahlis NJ, Lategan J, Reece DE, Lyons JF, Sederias J, Xu H, Powers J, Seymour LK, Reiman T, Leuk. Lymphoma 2015, 1–4 [DOI] [PubMed] [Google Scholar]; b) Dent SF, Gelmon KA, Chi KN, Jonker DJ, Wainman N, Capier CA, Chen EX, Lyons JF, Seymour L, Invest. New Drugs 2013, 31, 1522–1529. [DOI] [PubMed] [Google Scholar]
- [21].Hauf S, Cole RW, LaTerra S, Zimmer C, Schnapp G, Walter R, Heckel A, van Meel J, Rieder CL, Peters JM, J. Cell Biol 2003, 767, 281–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].a) Jammi NV, Whitby LR, Beal PA, Biochem. Biophys. Res. Commun 2003, 308, 50–57 [DOI] [PubMed] [Google Scholar]; b) Ingrand S, Barrier L, Lafay-Chebassier C, Fauconneau B, Page G, Hugon J, FEBS Lett. 2007, 581, 4473–4478 [DOI] [PubMed] [Google Scholar]; c) Tronel C, Page G, Bodard S, Chalon S, Antier D, Neurochem. Int 2014, 64, 73–83. [DOI] [PubMed] [Google Scholar]
- [23].Chow LQ, Eckhardt SG, J. Clin. Oncol 2007, 25, 884–896. [DOI] [PubMed] [Google Scholar]
- [24].Jani JP, Arcari J, Bernardo V, Bhattacharya SK, Briere D, Cohen BD, Coleman K, Christensen JG, Emerson EO, Jakowski A, Hook K, Los G, Moyer JD, Pruimboom-Brees I, Pustilnik L, Rossi AM, Steyn SJ, Su C, Tsaparikos K, Wishka D, Yoon K, Jakubczak JL, Mol. Cancer Ther 2010, 9, 883–894. [DOI] [PubMed] [Google Scholar]
- [25].Apsel B, Blair JA, Gonzalez B, Nazif TM, Feldman ME, Aizenstein B, Hoffman R, Williams RL, Shokat KM, Knight ZA, Nat. Chem. Biol 2008, 4, 691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Roberts WG, Whalen PM, Soderstrom E, Moraski G, Lyssikatos JP, Wang HF, Cooper B, Baker DA, Savage D, Dalvie D, Atherton JA, Ralston S, Szewc R, Kath JC, Lin J, Soderstrom C, Tkalcevic G, Cohen BD, Pollack V, Barth W, Hungerford W, Ung E, Cancer Res. 2005, 65, 957–966. [PubMed] [Google Scholar]
- [27].Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K, Castaneda S, Cornelius LA, Das J, Doweyko AM, Fairchild C, Hunt JT, Inigo I, Johnston K, Kamath A, Kan D, Klei H, Marathe P, Pang S, Peterson R, Pitt S, Schieven GL, Schmidt RJ, Tokarski J, Wen ML, Wityak J, Borzilleri RM, J. Med. Chem 2004, 47, 6658–6661. [DOI] [PubMed] [Google Scholar]
- [28].Montero JC, Seoane S, Ocana A, Pandiella A, Clin. Cancer Res 2011, 17, 5546–5552. [DOI] [PubMed] [Google Scholar]
- [29].Schonbrunn E, Betzi S, Alam R, Martin MP, Becker A, Han H, Francis R, Chakrasali R, Jakkaraj S, Kazi A, Sebti SM, Cubitt CL, Gebhard AW, Hazlehurst LA, Tash JS, Georg GI, J. Med. Chem 2013, 56, 3768–3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Otwinowski Z, Minor W, Methods Enzymol. 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- [31].Kabsch W, Acta Crystallogr. Sect. D 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH, Acta Crystallogr. Sect. D 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Emsley P, Lohkamp B, Scott WG, Cowtan K, Acta Crystallogr. Sect. D 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ko TP, Jeng WY, Liu CI, Lai MD, Wu CL, Chang WJ, Shr HL, Lu TJ, Wang AH, Acta Crystallogr. Sect. D 2010, 66, 145–154. [DOI] [PubMed] [Google Scholar]
- [35].Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC, Acta Crystallogr. Sect. D 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Afonine PV, Grosse-Kunstleve RW, Chen VB, Headd JJ, Moriarty NW, Richardson JS, Richardson DC, Urzhumtsev A, Zwart PH, Adams PD, J. Appl. Crystallogr 2010, 43, 669–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.