

Abstract
Bone metastatic prostate cancer provokes extensive osteogenesis by driving the recruitment and osteoblastic differentiation of mesenchymal stromal cells (MSCs). The resulting lesions greatly contribute to patient morbidity and mortality, underscoring the need for defining how prostate metastases subvert the MSC–osteoblast differentiation program. To gain insights into this process we profiled the effects of co-culture of primary MSCs with validated bone metastatic prostate cancer cell line models. These analyses revealed a cast of shared differentially induced genes in MSC, including betaglycan, a co-receptor for TGFβ. Betaglycan has not been studied in the context of bone metastatic disease previously. Here we report that loss of betaglycan in MSC is sufficient to augment TGFβ signaling, proliferation and migration, and completely blocks the MSC–osteoblast differentiation program. Further, betaglycan was revealed as necessary for prostate cancer-induced osteogenesis in vivo. Mechanistically, gene expression analysis revealed betaglycan controls the expression of a large repertoire of genes in MSCs, and that betaglycan loss provokes >60-fold increase in the expression of Wnt5a that plays important roles in stemness. In accord with the increased Wnt5a levels, there was a marked induction of canonical Wnt signaling in betaglycan ablated MSCs, and the addition of recombinant Wnt5a to MSCs was sufficient to impair osteogenic differentiation. Finally, the addition of Wnt5a neutralizing antibody was sufficient to induce the expression of osteogenic genes in betaglycan-ablated MSCs. Collectively, these findings suggest a betaglycan–Wnt5a circuit represents an attractive vulnerability to ameliorate prostate cancer-induced osteogenesis.
Introduction
Prostate cancer metastasizes to bone more frequently than any other tissue [1]. In the bone, prostate cancer cells induce a vicious cycle of excessive osteolysis and osteogenesis, and indeed the latter is a hallmark of metastatic disease [1, 2]. Further, pathological bone formation induced by prostate cancer is of poor quality, causes severe pain and increased risk of fracture, and significantly compromises patient quality of life. Current therapies to treat this debilitating condition are mainly palliative, and there remains a need for understanding the mechanisms of how prostate cancer progresses in bone to inform new and effective treatments.
In the tumor–bone microenvironment, bone metastatic prostate cancer cells hijack and subvert the homeostatic processes of bone turnover, and this aberrant remodeling worsens during tumor progression [3, 4]. In particular, prostate cancer cells can promote osteoclast maturation that leads to bone resorption and the release of bone matrix-sequestered growth factors, including transforming growth factor beta (TGFβ), that support prostate cancer cell survival and growth [5, 6]. Further, we and others have shown that increased levels of TGFβ in bone metastatic prostate cancer promotes the recruitment of bone marrow-derived mesenchymal stromal cells (MSCs), which then undergo differentiation into bone-forming osteoblasts and contribute to the aberrant osteogenic features of the disease [7-9].
TGFβ and other members of the TGFβ superfamily, such as bone morphogenetic proteins (BMPs) play key roles in the development and turnover of the skeletal system, where they control the balance between bone resorption and bone formation [10, 11]. For example, loss of function studies have shown that ablation of TGFβ ligands or receptors compromises proper skeletal development [12, 13], and pharmacological agents that block TGFβ can promote increased bone formation under both normal and pathological conditions [14, 15]. TGFβ mediates its effects via binding to TGFβ receptor II (TβRII) and dimerization with TGFβ receptor I (TβRI), a serine/threonine receptor tyrosine kinase that leads to phosphorylation and nuclear translocation of SMAD transcription factors and to the transcription of TGFβ target genes. A third receptor, betaglycan (TβRIII), contains two separate TGFβ ligand-binding sites and a short cytoplasmic domain that has no kinase function. As such, betaglycan has been designated a TGFβ co-receptor [16]. Interestingly, betaglycan has been shown to both positively and negatively regulate TGFβ activity, where its opposing effects have been attributed to glycosaminoglycan modifications and/or proteolytic shedding [17].
Despite several studies regarding the roles of TGFβ in bone metastatic disease [5, 6, 18-22], the role of betaglycan in metastatic prostate cancer-associated bone disease has not been explored. Here, we report that prostate cancer cells promote the differential regulation of several TGFβ-related genes in MSCs, including a marked upregulation of betaglycan. Further, MSCs in prostate cancer patient bone biopsies express high levels of betaglycan, whereas there is little to no expression of betaglycan in adjacent prostate cancer cells. To assess the role of prostate cancer-induced betaglycan expression on MSC function and recruitment, we utilized CRISPR-mediated gene editing to ablate betaglycan expression in bone marrow-derived MSCs. These studies revealed key roles for betaglycan in controlling the MSC osteogenic program and mechanistically linked these effects to the control of Wnt5a expression. Collectively, the data presented herein support targeting the betaglycan-Wnt5a circuit to ameliorate aberrant osteogenesis in bone metastatic prostate cancer.
Results
Prostate cancer cells induce betaglycan expression in MSCs
To identify factors through which prostate cancer cells regulate MSC behavior, we incubated primary human MSCs with conditioned media derived from human prostate cancer cells lines having bone metastatic capabilities, C42B and PC3-2M. After 6 h of culture, MSC mRNA was isolated from biological replicates (n = 3/group) and used to perform gene microarray analysis. Of the 227 genes regulated by conditioned medium from both C42B and PC3-2M cells (Fig. 1a), gene ontology analyses (PANTHER [23]) revealed significant enrichment of MSC biological processes associated with development, cell receptor signaling, growth and differentiation (Fig. 1b). Several members of the TGFβ superfamily, including betaglycan, were up or down regulated compared to controls (Fig. 1c).
Fig. 1.

Prostate cancer cells induce betaglycan expression in bone marrow-derived MSCs. a Microarray analysis of differential gene expression induced in bone marrow MSCs in response to culture in C42B-conditioned and PC3-conditioned media (CM, 6h). b Gene ontology analysis of biological processes. c Expression of TGFβ pathway members in MSCs that are regulated by culture in CM from PC3-2M and C4-2B cells. d qRT-PCR analysis of betaglycan (BG) mRNA levels in MSCs after 6 h incubation in CM derived from C4-2B and PC3-2M, and from CM from the independent rodent prostate cancer cell line, PAIII. Data shows fold increase over basal BG expression. e Betaglycan (BG; red) localization in human bone metastatic prostate cancer specimens (n = 10). Representative images from three individual patient specimens are illustrated. CD90 (blue) was used to localize MSCs while pan-cytokeratin (green) was used to localize the prostate cancer metastases. DAPI (gray) was used as a nuclear counterstain
As betaglycan can positively or negatively regulate TGFβ signaling [17, 24-27] and has not been evaluated in the context of prostate cancer, we explored its potential functions in MSCs. Initially, we validated betaglycan induction in MSCs by prostate cancer-conditioned media via qRT-PCR, which demonstrated an 8–10-fold increase in betaglycan expression in response to C42B-conditioned and PC3-2M-conditioned media, and a four-fold increase in expression in response to conditioned media derived from the bone metastatic rodent prostate cancer cell line, PAIII (Fig. 1d). Notably, immunofluorescence analyses established the clinical relevance of these observations, where there were high levels of betaglycan expression in CD90+ MSCs surrounding the prostate cancer cells in the majority (70%) of bone metastatic prostate cancer samples analyzed (n = 10, Fig. 1e).
Ablation of betaglycan in MSCs enhances TGFβ signaling
To assess the role of betaglycan in MSCs, we ablated gene expression using CRISPR-Cas9 editing (SI Appendix, Table S1). Control (CTRL) and betaglycan knockdown (BGKD) clones were isolated, and two of five BGKD clones (4 and 5) exhibited complete loss of the full-length glycosylated protein (Fig. 2a). qRT-PCR analyses revealed that loss of betaglycan in MSCs was associated with a significant increase in TGFβ1 expression but with a significant reduction in TGFβ2 expression (Fig. 2b), the TGFβ isoform that has the highest affinity for betaglycan. Similarly, betaglycan loss had no effect on TβRI expression but did lead to increased levels of TβRII expression in MSCs (Fig. 2b).
Fig. 2.
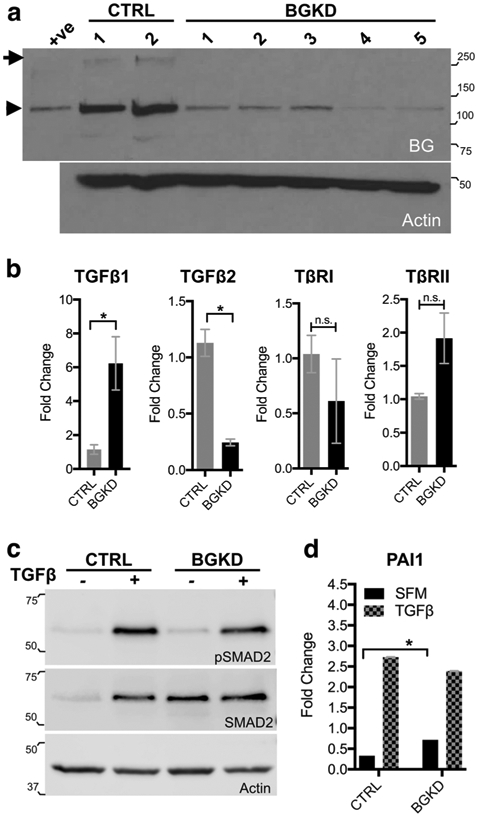
Betaglycan knockdown enhances TGFβ signaling in MSCs. a Analysis of Betaglycan protein levels in scrambled control (CTRL) and betaglycan CRISPR ablated (BGKD) clonal MSC populations. Arrow indicates full-length betaglycan protein while arrowhead indicates the core protein. Recombinant BG was used as a positive (+ve) control while the actin immunoblot was used as a loading control. b qRT-PCR analysis of TGFβ ligand isoforms (TGFβ1 and TGFβ2) and TGFβ receptor I and II (TβRI, TβRII) mRNA levels in betaglycan knockdown (BGKD) MSCs normalized to those in control (CTRL) MSCs. c Phosphorylated SMAD2 (pSMAD2) at basal levels (−) and in response to TGFβ treatment (+, 5 ng/ml for 30 min). Actin was used as a loading control. d PAI1 promoter-reporter outputs at basal levels in serum-free media (SFM) and in response to TGFβ treatment (5 ng/ml for 24 h). Molecular weights are in kDa
Canonical TGFβ signaling directs the phosphorylation and nuclear translocation of SMAD transcription factors [28]. Although the addition of exogenous TGFβ1 significantly elevated pSMAD2 and total SMAD2 in both CTRL and BGKD MSCs (Fig. 2c), we noted slightly elevated levels pSMAD2 in the BGKD MSCs under baseline serum-free conditions (Fig. 2c). Normalizing to actin, we also consistently observed higher baseline levels of total SMAD2 in BGKD MSCs (>5 fold) compared to CTRL. Plasminogen activator inhibitor 1 (PAI1) is a well-known TGFβ transcriptional target [29], and we confirmed the increased TGFβ signaling capacity in BGKD MSCs using a PAI1 promoter-reporter assay (Fig. 2d). We also validated these effects using an shRNA approach, where stable knockdown of betaglycan expression also augmented TGFβ signaling MSCs (SI Appendix, Fig. S1). Thus, betaglycan suppresses TGFβ signaling in MSCs.
Betaglycan promotes MSC differentiation
We and others have shown that TGFβ promotes MSC migration and proliferation [3, 8, 9, 30-33]. To test the effects of betaglycan on MSC motility, equal numbers of CTRL and BGKD MSCs were seeded into the upper wells of a modified Boyden chamber and allowed to freely migrate under serum-free conditions for 5 h. Analyses revealed that BGKD MSCs were three-fold more migratory than the CTRLs (Fig. 3a). Further, in long-term growth assays over 7 days, BGKD MSCs grew at faster rates compared to the CTRL MSCs (Fig. 3b). TGFβ is also a key mediator of cell stemness [34, 35], and given the inhibitory role for betaglycan in TGFβ signaling, we assessed the differentiation capacity of BGKD MSCs [27, 34]. Osteogenic assays using alizarin red as a readout for matrix mineralization demonstrated that BGKD MSCs were totally defective in MSC–osteoblast differentiation compared to CTRL MSCs (Fig. 3c). Again, similar effects of betaglycan on MSC migration, proliferation, and differentiation were independently validated using an shRNA approach (SI Appendix, Fig. S1). Finally, as measured by Oil Red O staining, BGKD MSCs were also defective in adipogenic differentiation compared to CTRL MSCs (Fig. 3d). Therefore, betaglycan is necessary for MSC differentiation into osteoblasts and adipocytes.
Fig. 3.

Betaglycan inhibits MSC migration and proliferation and is necessary for osteogenic and adipogenic MSC differentiation. a The number of migrating CTRL and BGKD MSCs in serum-free media was measured over a 5 h period using a Boyden chamber. Representative images from each group are shown. b CTRL and BGKD cell proliferation at 7 days was measured using cell counts. c Representative images of Alizarin Red staining for mineralization in CTRL and BGKD MSCs incubated with osteogenic media for 21 days. The amount of solubilized Alizarin Red was also measured in CTRL and BGKD MSCs. d The number of Oil Red O-positive cells as a function of total number of cells was measured in CTRL and BGKD MSCs incubated for 3 days in adipogenic media. Representative images from each group are shown. Asterisks denote statistical significance (**p < 0.01)
Betaglycan drives prostate cancer-induced osteogenesis
A hallmark of bone metastatic prostate cancer is aberrant osteogenesis, and this significantly contributes to patient morbidity. Given our in vitro data, we tested if MSC-derived betaglycan would impact prostate cancer-induced bone formation using the C4-2B model of osteogenic bone metastatic prostate cancer [30, 36]. Immunocompromised mice were intratibially co-injected with a 1:1 ratio of C4-2B:CTRL MSCs (n = 6) or C4-2B:BGKD MSCs (n = 7 per group). Mice were euthanized after 7 weeks of tumor progression, a time point at which the C4-2B cells have not yet breached the cortical bone [30, 37]. Tumor-bearing tibias were isolated and high-resolution μCT scan analysis revealed prostate cancer-induced osteogenesis was greatly reduced in the BGKD MSC group compared to the CTRL MSC-inoculated mice (Fig. 4a). We observed significantly reduced bone volume:total volume (BV/TV) ratios and decreased trabecular number and thickness in the C4-2B-BGKD MSC group vs. the C4-2B–CTRL MSC cohort (Fig. 4a). No effect on trabecular spacing was noted. Histomorphometry analysis for collagen using trichrome stain validated these μCT BV/TV results, demonstrating significantly lower BV/TV ratios in the C4-2B–BGKD MSC group compared to C4-2B–CTRL MSCs (Fig. 4b). Furthermore, tartrate-resistant acid phosphatase (TRAcP) staining of osteoclasts revealed increases in the numbers of multinucleated osteoclasts lining the tumor–bone interface in the C4-2B–BGKD MSC group compared to the C4-2B–CTRL MSC cohort, but this difference was not statistically significant (Fig. 4c). Finally, to validate these observations, these in vivo studies were repeated using the PAIII model of bone metastatic osteogenic prostate cancer. Although significant decreases in bone volume were observed via histomorphometry in the PAIII-BGKD MSC group compared to controls, the effects of betaglycan ablation were less robust than those observed with the C4-2B model (SI Appendix, Fig. S2). This may be due to the rapid nature of the PAIII model (2 weeks to endpoint) compared to that of C4-2B (7 weeks to endpoint). Overall, these data are consistent with the notion that MSC-derived betaglycan promotes prostate cancer-induced osteogenesis.
Fig. 4.

Betaglycan controls prostate cancer-induced osteogenesis. a Representative μCT images of tibias 7 weeks following co-inoculation with C4-2B prostate cancer cells and CTRL or BGKD MSCs. Graphs illustrate μCT values in each group for bone volume as function of total volume (BV/TV), trabecular thickness (TR. Thickness), trabecular number (TR. #), and trabecular spacing (TR. Spacing). b, c Representative images of trichrome b and TRAP c stained sections derived from CTRL/C4-2B and BGKD/C4-2B groups. BV/TV ratios b and osteoclast numbers/mm of tumor-bone interface (OCL#; red) were quantitated in non-sequential sections derived from each group. Asterisks denote statistical significance (*p < 0.05, ****p < 0.001) while n.s. indicates non-significance
Betaglycan controls expression of Wnt5a and Wnt signaling effectors in MSCs
To identify candidates through which betaglycan controls MSC differentiation, microarray analysis of CTRL and BGKD MSC cell lines cultured under normal serum-containing conditions was performed. These analyses revealed that ablation of betaglycan caused significant up-regulation and down regulation of genes associated with select biological processes, most prominently development and differentiation, as determined by heat maps and gene ontology analyses (Fig. 5a and SI Appendix, Fig. S3). Using a cutoff of greater than two-fold differential expression and positivity for at least two independent probe sets, we identified genes that were significantly up-regulated and down regulated in the BGKD MSCs compared to CTRL MSCs (Fig. 5b). Notably, Wnt5a expression was, on average, elevated to 63-fold in BGKD MSCs.
Fig. 5.

Betaglycan controls Wnt5a expression in MSCs. a Gene ontology analysis of microarray data obtained from CTRL and BGKD MSC grown under normal serum-containing culture conditions. b Microarray analysis of average gene expression identified the top-15 up-regulated and down-regulated genes in the BGKD MSCs compared to CTRL. c, d qRT-PCR c and immunoblot d analysis of Wnt5a expression in CTRL and BGKD MSCs. Arrow indicates Wnt5a. Recombinant Wnt5a was used as a positive control (+ve). Molecular weights shown are in kDa. e qPCR of Wnt5a mRNA levels following incubation of CTRL and BGKD MSCs with the TGFβ inhibitor 1D11 (10 μg/ml for 6 h). 1D11 effects are normalized relative to respective non-treatment controls. f–h qPCR analysis of β-catenin f, Wnt receptors g and Wnt signaling effectors h at baseline in CTRL and BGKD MSCs. Asterisks denote statistical significance (*p < 0.05, **p < 0.01, ***p < 0.005) while n.s. denotes non-significance
Given the interplay between TGFβ and the Wnt pathway in controlling stemness [35, 38-40], the relevance of the significant up-regulation of Wnt5a in BGKD MSCs was explored. A marked, 60-fold increase in Wnt5a levels in BGKD MSCs was verified by qRT-PCR (Fig. 5c). Further, analysis of conditioned media confirmed the presence of high levels of Wnt5a in the culture of MSC BGKD whereas no Wnt5a was detected in the MSC CTRLs (Fig. 5d). Immunoblot analyses revealed both the short and long isoforms of Wnt5a were selectively expressed at high levels in MSC BGKD cells [41].
To test if the increased Wnt5a expression in MSC BGKD cells was due to increases in TGFβ signaling, we incubated the MSC CTRL and MSC BGKD cells with a TGFβ inhibitor, 1D11, which blocks the binding of all TGFβ isoforms to TβRII [14, 15]. qRT-PCR analysis demonstrated that 1D11 treatment provoked a two-fold reduction in Wnt5a expression in MSC BGKD compared to MSC CTRL cells (Fig. 5e). Furthermore, consistent with increased Wnt5a signaling, β-catenin expression was also elevated in MSC BGKD cells vs. MSC CTRL cells (Fig. 5f). Interestingly, although there were no significant changes in the expression of Wnt receptors Ror2, Lrp5, or Lrp6, the expression of several Wnt signaling effectors was significantly upregulated in the MSC BGKD cells, including casein kinase 1 epsilon (CSK1ε), adenomatous polyposis coli (APC), and Axin (Fig. 5g, h and (SI Appendix, Table S2)). Thus, betaglycan-dependent differentiation of MSCs is associated with its control of genes that regulate stemness, such as Wnt5a and Wnt signaling effectors.
A betaglycan-Wnt5a circuit controls MSC osteoblast differentiation
Wnt signaling limits osteoblast differentiation by suppressing genes required for osteoblast maturation and the deposition of extracellular matrix [42]. qRT-PCR analysis revealed significantly reduced levels of runt-related transcription factor 2 (RUNX2), type-1-alpha collagen (COL1A1), and osterix (OSX) in MSC BGKD cells (Fig. 6a-c). Notably, the addition of a Wnt5a neutralizing antibody restored RUNX2 and COL1A1 levels in MSC BGKD to levels found in MSC CTRL cells (Fig. 6c). Furthermore, treatment of MSC CTRL cells with recombinant Wnt5a totally blocked their ability to mineralize the extracellular matrix cultured in osteogenic media, and in MSC BGKD cells (Fig. 6d), which are defective in osteogenesis (Fig. 3c, d). Thus a betaglycan-to-Wnt5a circuit controls the osteoblast differentiation program of MSC.
Fig. 6.

Betaglycan regulation of Wnt5a contributes to the MSC-osteoblast differentiation program. a–c qPCR analysis of RUNX2 (RUNX2) a, α-type I collagen (COL1A1; b), and osterix (OSX; c) in CTRL and BGKD MSCs treated in the presence or absence of a Wnt5a blocking antibody (αWnt5a; 1 μg/ml; 6 h). Fold changes are normalized to each respective non-treated control. d Representative images of Alizarin Red staining for mineralization in CTRL and BGKD MSCs incubated with osteogenic media (OM) for 21 days in the presence or absence of recombinant Wnt5a (rWnt5a; 0.5 μg/ml). Cells were also treated with non-OM media as a negative control (−ve). The amount of solubilized Alizarin Red was measured in the treated CTRL and BGKD MSCs. Asterisks denote statistical significance (**p < 0.01, ****p < 0.0001)
Discussion
A hallmark of bone metastatic prostate cancer is excessive bone formation, a phenomenon that is poorly understood relative to other tumor–bone microenvironmental processes such as osteolysis. Here we identified a mechanism of pathological bone formation by focusing on how prostate cancer cells regulate MSC function. We and others have previously found that the pleiotropic cytokine TGFβ recruits MSCs to sites of active bone remodeling [8, 9, 30]. Here we observed that bone metastatic prostate cancer cells induce the expression of the Type III TGFβ receptor, betaglycan, in MSC, and that betaglycan in turn promotes MSC osteoblast differentiation and the development of osteogenic lesions. Accordingly, CRISPR editing or shRNA knockdown studies revealed that betaglycan signaling is essential for osteogenic differentiation of MSCs. Finally, our expression analyses and functional studies revealed that a key mediator of betaglycan signaling in controlling MSC osteogenesis is Wnt5a, where betaglycan signaling represses the expression of Wnt5a and Wnt signaling effectors, and where blocking Wnt5a is sufficient to induce osteogenic gene expression in betaglycan-deficient MSCs, and treatment with Wnt5a blocks osteogenesis in control MSCs. Thus, a betaglycan–Wnt5a circuit is necessary for MSC osteogenesis.
Although not resolved here, it is plausible that TGFβ itself produced by prostate cancer cells is responsible for inducing the expression of betaglycan in MSC, particularly since binding sites for SMAD3, SMAD4, and SP1 are present in the betaglycan promoter region [43]. Regardless, betaglycan has been shown to play both activating and inhibitory roles in TGFβ signaling. This may in part be explained by post-translational modifications to this TGFβ receptor, where glycosaminoglycan chain modifications alter the ability of betaglycan to sequester TGFβ thereby regulating ligand interactions with TβRII. Germane to our work, recent studies have also shown that differences in the proteoglycan state of betaglycan can inhibit Wnt canonical signaling via the binding of Wnt ligands to heparin sulfate chains of the receptor. Therefore, ablating betaglycan in MSCs can lead to enhanced TGFβ and Wnt signaling and, as a consequence, promote cell stemness [24].
Here, our data show that betaglycan is a regulator of MSC stem-like properties where genetic ablation augments TGFβ signaling, cell motility, and proliferation, and totally blocks the ability of MSC to differentiate into osteoblasts. These findings establish roles for betaglycan in the biology of osteoblasts, where this receptor is highly expressed [44, 45]. In accord with our findings, betaglycan loss in mice provokes a lethal, diminutive embryonic phenotype that is associated with marked defects in skeletal ossification [46]. Interestingly, it is also notable that our studies revealed that betaglycan loss in MSCs leads to a significant reduction of TGFβ2. TGFβ2–null mice exhibit significant skeletal deformation as opposed to other TGFβ isoforms, supporting a role for the TGFβ2–betaglycan signaling axis in skeletal development [47]. Furthermore, TGFβ2 has the highest binding affinity for betaglycan and is known to induce betaglycan expression. This suggests that an interplay between TGFβ2 and betaglycan is potentially important in mediating bone development and the osteoblast differentiation program in MSCs [43]. In accord with this, a recent study demonstrated that betaglycan ablation prevented MSC commitment to osteoblast lineage in a mouse model of palatogenesis [48].
Notably the ablation of betaglycan in MSCs had profound effects on gene expression and leads to marked increases in the expression of Wnt5a, which is known to be induced by TGFβ signaling [39]. Consistent with these findings, we observed that inhibiting TGFβ signaling in the BGKD MSCs reduces Wnt5a expression [49, 50]. However, novel findings reported here are that inhibiting Wnt5a activity restores osteogenic gene expression and osteoblastic differentiation of BGKD MSCs, whereas treatment with recombinant Wnt5a blocks MSC osteogenesis. These findings are in accord with those showing that Wnt5a inhibits periodontal osteoblast differentiation and regulates the expression of bone-related genes [51]. Of note, however, Wnt5a has also been reported to promote osteoblast differentiation [52]. We suspect these differential effects may be due to the specific Wnt pathway node utilized and/or on the differentiation state of the MSC–osteoblast precursor at the time of exposure to Wnt5a. Further, the long and short isoforms of Wnt5a may also have differential effects on MSCs, and here it is notable that BGKD MSCs express both isoforms.
While our studies focused on Wnt5a given the interplay between TGFβ and Wnt pathways in development, it should be noted that betaglycan loss led to differential regulation of several other genes having roles in the MSC osteogenic program. For example, betaglycan loss was associated with a marked down-regulation (500-fold on average) in the levels of the ABI gene family member 3 (NESH)-binding protein (ABI3BP), a key mediator of MSC differentiation [53], as well as a 150-fold repression on average in pleiotrophin, also known as osteoblast-stimulating factor-1 (OSF-1). Thus, betaglycan signaling is necessary for a cast of genes that control MSC osteogenesis.
Importantly, our studies establish betaglycan as a key regulator of prostate cancer-induced osteogenesis, where betaglycan loss in MSCs significantly reduces aberrant osteogenesis in the intratibial C42B and PAIII models of bone metastatic prostate cancer. Our findings demonstrate that bone metastatic prostate cancer cells express little to no betaglycan but induce its expression in bone marrow MSCs which in turn leads to increased bone formation. Interestingly, other reports have shown that decreased betaglycan expression in cancer cells drives stemness, progression, and metastasis indicating differential roles for betaglycan in the cancer and host microenvironment [25, 54, 55]. Based our studies, we hypothesize that the induction of betaglycan in host MSCs by prostate cancer-derived factors also significantly suppresses the expression of Wnt5a. Clinically, high Wnt5a levels in prostate cancer correlate with favorable prognosis and out-comes [56], and causal roles for Wnt5a in limiting prostate cancer cell growth in bone have been described. For example, forced Wnt5a expression in PC3 cells significantly reduces cancer growth in bone [56, 57], and the induction of Wnt5a expression in osteoblasts has been reported to inhibit metastatic prostate cancer to growth [56-59]. Thus, the induction of betaglycan expression in MSCs may not only promote an osteogenic response but also suppress factors, such as Wnt5a that inhibit prostate cancer cell growth. Collectively, these data suggest that agents that mimic Wnt5a may be of therapeutic benefit administered alone or in combination with other targeted therapies. In this regard, the Wnt5a mimetic Foxy-05 reduces the invasion and metastasis of DU-145 prostate cancer cells in vivo [60] and is the subject of ongoing clinical trials (NCT02020291, NCT02655952). On the other hand, some studies have indicated Wnt5a contributes to prostate cancer disease progression, and Wnt5a expression in circulating tumor cells and in the bone environment correlates with development of castration-resistant disease [61, 62]. We speculate that the divergent roles for Wnt5a in prostate cancer reflects selective effects of Wnt5a isoforms. In particular, the short isoform (Wnt5a-S) augment cancer cell proliferation and is highly expressed in several cancers, whereas the long isoform (Wnt5a-L) suppresses cancer cell growth and is frequently suppressed in tumors [41].
Heretofore, how prostate cancers induce osteogenic responses is poorly understood. The findings presented herein explain this phenomenon, where we show that prostate cancer cells induce betaglycan signaling in MSCs and that a betaglycan-to-Wnt5a circuit is both essential for MSC osteoblastic differentiation and promotes prostate cancer-induced osteogenesis. Importantly, these findings support targeting this circuit for the treatment of bone metastatic osteogenic prostate cancer.
Materials and methods
Cell culture and patient specimens
Human MSCs were purchased from commercial vendors (Lonza, Cat# PT-2501) and kept at low passage. Mouse MSCs were previously isolated from bone marrow flushes of female FVB mice, as described [63] and characterized by flow (R&D Systems, #FMC003). MSCs stably transfected with CRISPR plasmids were cultured in low-glucose Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, platelet-derived growth factor (PDGF-BB;10 ng/ml), and puromycin (5 μg/ml). For differentiation, MSCs were cultured with lineage-specific supplements for 3–21 days, according to protocol (osteogenic, adipogenic, chondrogenic assays; R&D MSC Differentiation kits #CCM009, CCM011, CCM006). For analysis of effects of Wnt5a on differentiation, osteogenesis assays were supplemented with recombinant Wnt5a (0.5 μg/ml; 645-WN, R&D Biosystems) or anti-Wnt5a (1 μg/ml; #AF645, R&D Biosystems). Luciferase-expressing PAIII and C42B prostate cancer cell lines were cultured in complete Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and puromycin. All cell lines were periodically tested for mycoplasma (R&D, #CUL001B) and short tandem repeat (STR) verified at the Moffitt Molecular Genomics Core. De-identified tissue sections of bone metastatic prostate cancer were obtained from the University of Washington Rapid Autopsy Program (kind gift of Dr. Colm Morissey).
Microarray and gene expression analysis
MSC RNA was isolated from cells in serum-containing growth media using Trizol reagent, according to the manufacturer’s protocol, and genomic DNA removed using the RNeasy MinElute CleanUp kit (Qiagen). 100 ng of total RNA was amplified and labeled with biotin using the Ambion Message Amp Premier RNA Amplification Kit (ThermoFisher) [64]. Hybridization with the biotin-labeled RNA, staining, and scanning of the chips has been previously described [65]. The oligonucleotide probe arrays used were Affymetrix Human U133A 2.0 and Mouse 430 2.0 Arrays, which contain over >45,000 probe sets representing more than >39,000 transcripts. The array output files were visually inspected for hybridization artifacts and then analyzed using Affymetrix Expression Console v 1.4 using the MAS 5.0 algorithm, scaling probe sets to an average intensity of 500. Using the Affymetrix Transcriptome Analysis Console v3.0, a one-way unpaired ANOVA analysis was performed and differentially expressed probe sets with fold changes >2.0 and FDR adjusted p-values < 0.05 were retained. Gene ontology analyses were performed using PANTHER [23].
Intratibial prostate cancer studies
All animal experiments were performed with University of South Florida IACUC approval (R1762; CCL) and were conducted in accordance with the guidelines set forth in the Guidelines for the Care and Use of Laboratory Animals published by the National Institutes of Health. Six-week-old male C57BL/6 Rag-2 or SCID Beige mice were injected intratibially with luciferase-expressing PAIII or C4-2B cell lines (5 × 104 or 5 × 105, respectively, in 20 μl of saline) in a 1:1 ratio with either scrambled control (CTRL; n=6) and betaglycan CRISPR ablated (BGKD; n=7) clonal MSC populations. Contralateral limbs were injected with saline to serve as an injury control. Bioluminescence was measured (relative light units: RLU) longitudinally as a correlate of tumor growth (IVIS™ Perkin Elmer). Three days subsequent to inoculation, mice were randomized into groups based on obtained RLU values. Sample size was estimated based on our previous results with the C4-2B model [30]. The study endpoint was predetermined as 1 × 106 RLU. Mice were excluded in the event of histological evidence demonstrating tumor breach of cortical bone. At the study endpoint, mice were euthanized and tumor and saline-injected limbs were harvested and fixed in 10% neutral-buffered formalin for 24 h.
Statistical analysis
To determine statistical significance among groups, analysis of variance (ANOVA) was performed followed by Tukey’s multiple comparison test. Variance was further measured using the Brown–Forsythe test. Sample size was chosen based on a confidence level of 95% with a 5% margin of error. p-value<0.05 was considered as statistically significant. Error bars represent standard error from the mean (SEM). All statistical analyses were performed with Graph Pad Prism 6.0 (Graphpad Inc., LaJolla, CA, USA).
Supplementary Material
Acknowledgements
We thank Dr. Colm Morrissey, University of Washington for providing human bone metastatic prostate cancer specimens (P50-CA097186 16A1). LMC was supported by a fellowship from the American Cancer Society (PF-13-175-01-CSM). This work has been supported in part by the Molecular Genomics Core, the Analytic Microscopy Core, the Small Animal Imaging Laboratory Core and the Biostatistics and Bioinformatics Shared Resource at the H. Lee Moffitt Cancer Center & Research Institute, an NCI designated Comprehensive Cancer Center (P30-CA076292). Funding also provided by Miles for Moffitt (CCL), the Department of Defense PCRP program (W81XWH1810523; CCL), and by the Cortner-Couch Endowed Chair for Cancer Research from the University of South Florida (JLC).
Footnotes
Supplementary information The online version of this article (https://doi.org/10.1038/s41388-019-0913-4) contains supplementary material, which is available to authorized users.
Conflict of interest The authors declare that they have no conflict of interest.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Logothetis C, Morris MJ, Den R, Coleman RE. Current perspectives on bone metastases in castrate-resistant prostate cancer. Cancer Metastas-Rev. 2018;37:189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frieling JS, Basanta D, Lynch CC. Current and emerging therapies for bone metastatic castration-resistant prostate cancer. Cancer Control: J Moffitt Cancer Cent. 2015;22:109–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cook LM, Shay G, Aruajo A, Lynch CC. Integrating new discoveries into the “vicious cycle” paradigm of prostate to bone metastases. Cancer Metastas-Rev. 2014;33:511–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ren G, Esposito M, Kang Y. Bone metastasis and the metastatic niche. J Mol Med. 2015;93:1203–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Juarez P, Guise TA. TGF-beta in cancer and bone: implications for treatment of bone metastases. Bone. 2010;48:23–29. [DOI] [PubMed] [Google Scholar]
- 6.Fournier PG, Juarez P, Jiang G, Clines GA, Niewolna M, Kim HS, et al. The TGF-beta signaling regulator PMEPA1 suppresses prostate cancer metastases to bone. Cancer Cell. 2015;27:809–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Araujo A, Cook LM, Lynch CC, Basanta D. An integrated computational model of the bone microenvironment in bone-metastatic prostate cancer. Cancer Res. 2014;74:2391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crane JL, Cao X. Bone marrow mesenchymal stem cells and TGF-beta signaling in bone remodeling. J Clin Invest. 2014;124:466–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang Y, Wu X, Lei W, Pang L, Wan C, Shi Z, et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat Med. 2009;15:757–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen D, Zhao M, Mundy GR. Bone morphogenetic proteins 1. Growth Factors. 2004;22:233–41. [DOI] [PubMed] [Google Scholar]
- 11.Balooch G, Balooch M, Nalla RK, Schilling S, Filvaroff EH, Marshall GW, et al. TGF-beta regulates the mechanical properties and composition of bone matrix. Proc Natl Acad Sci USA. 2005;102:18813–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Janssens K, Ten Dijke P, Janssens S, Van Hul W. Transforming growth factor-beta1 to the bone. Endocr Rev. 2005;26:743–74. [DOI] [PubMed] [Google Scholar]
- 13.Erlebacher A, Derynck R. Increased expression of TGF-beta 2 in osteoblasts results in an osteoporosis-like phenotype. J Cell Biol. 1996;132:195–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edwards JR, Nyman JS, Lwin ST, Moore MM, Esparza J, O’Quinn EC, et al. Inhibition of TGF-beta signaling by 1D11 antibody treatment increases bone mass and quality in vivo. J Bone Min Res. 2010;25:2419–26. [DOI] [PubMed] [Google Scholar]
- 15.Biswas S, Nyman JS, Alvarez J, Chakrabarti A, Ayres A, Sterling J, et al. Anti-transforming growth factor β antibody treatment rescues bone loss and prevents breast cancer metastasis to bone. PLoS ONE. 2011;6:e27090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Todorovic-Rakovic N, Milovanovic J, Nikolic-Vukosavljevic D. TGF-beta and its coreceptors in cancerogenesis: an overview. Biomark Med. 2011;5:855–63. [DOI] [PubMed] [Google Scholar]
- 17.Bilandzic M, Stenvers KL. Betaglycan: a multifunctional accessory. Mol Cell Endocrinol. 2011;339:180–9. [DOI] [PubMed] [Google Scholar]
- 18.Mohammad KS, Javelaud D, Fournier PG, Niewolna M, McKenna CR, Peng XH, et al. TGF-beta-RI kinase inhibitor SD-208 reduces the development and progression of melanoma bone metastases. Cancer Res. 2011;71:175–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guise TA, Chirgwin JM. Transforming growth factor-beta in osteolytic breast cancer bone metastases 8. Clin Orthop Relat Res. 2003;415(Suppl):S32–38. [DOI] [PubMed] [Google Scholar]
- 20.Korpal M, Kang Y. Targeting the transforming growth factor-beta signalling pathway in metastatic cancer. Eur J Cancer. 2010;46:1232–40. [DOI] [PubMed] [Google Scholar]
- 21.Korpal M, Yan J, Lu X, Xu S, Lerit DA, Kang Y. Imaging transforming growth factor-beta signaling dynamics and therapeutic response in breast cancer bone metastasis. Nat Med. 2009;15:960–6. [DOI] [PubMed] [Google Scholar]
- 22.Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–49. [DOI] [PubMed] [Google Scholar]
- 23.Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, et al. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jenkins LM, Singh P, Varadaraj A, Lee NY, Shah S, Flores HV, et al. Altering the proteoglycan state of transforming growth factor beta Type III receptor (TbetaRIII)/betaglycan modulates canonical Wnt/beta-Catenin signaling. J Biol Chem. 2016;291:25716–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mythreye K, Blobe GC. The type III TGF-beta receptor regulates epithelial and cancer cell migration through beta-arrestin2-mediated activation of Cdc42. Proc Natl Acad Sci USA. 2009;106:8221–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turley RS, Finger EC, Hempel N, How T, Fields TA, Blobe GC. The type III transforming growth factor-beta receptor as a novel tumor suppressor gene in prostate cancer. Cancer Res. 2007;67:1090–8. [DOI] [PubMed] [Google Scholar]
- 27.Eickelberg O, Centrella M, Reiss M, Kashgarian M, Wells RG. Betaglycan inhibits TGF-beta signaling by preventing type I-type II receptor complex formation. Glycosaminoglycan modifications alter betaglycan function. J Biol Chem. 2002;277:823–9. [DOI] [PubMed] [Google Scholar]
- 28.Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6:506–20. [DOI] [PubMed] [Google Scholar]
- 29.Ohtsuki M, Massague J. Evidence for the involvement of protein kinase activity in transforming growth factor-beta signal transduction. Mol Cell Biol. 1992;12:261–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cook LM, Araujo A, Pow-Sang JM, Budzevich MM, Basanta D, Lynch CC. Predictive computational modeling to define effective treatment strategies for bone metastatic prostate cancer. Sci Rep. 2016;6:29384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Belotti D, Capelli C, Resovi A, Introna M, Taraboletti G. Thrombospondin-1 promotes mesenchymal stromal cell functions via TGFbeta and in cooperation with PDGF. Matrix Biol. 2016;55:106–16. [DOI] [PubMed] [Google Scholar]
- 32.Zhang SJ, Song XY, He M, Yu SB. Effect of TGF-beta1/SDF-1/CXCR4 signal on BM-MSCs homing in rat heart of ischemia/perfusion injury. Eur Rev Med Pharm Sci. 2016;20:899–905. [PubMed] [Google Scholar]
- 33.Shinojima N, Hossain A, Takezaki T, Fueyo J, Gumin J, Gao F, et al. TGF-beta mediates homing of bone marrow-derived human mesenchymal stem cells to glioma stem cells. Cancer Res. 2013;73:2333–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Watabe T, Miyazono K. Roles of TGF-beta family signaling in stem cell renewal and differentiation. Cell Res. 2009;19:103–15. [DOI] [PubMed] [Google Scholar]
- 35.Sakaki-Yumoto M, Katsuno Y, Derynck R. TGF-beta family signaling in stem cells. Biochim Biophys Acta. 2013;1830:2280–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu HC, Hsieh JT, Gleave ME, Brown NM, Pathak S, Chung LW. Derivation of androgen-independent human LNCaP prostatic cancer cell sublines: role of bone stromal cells. Int J Cancer. 1994;57:406–12. [DOI] [PubMed] [Google Scholar]
- 37.Dai J, Zhang H, Karatsinides A, Keller JM, Kozloff KM, Aftab DT, et al. Cabozantinib inhibits prostate cancer growth and prevents tumor-induced bone lesions. Clin Cancer Res. 2014;20:617–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luo K Signaling cross talk between TGF-beta/Smad and other signaling pathways. Cold Spring Harb Perspect Biol. 2017;9.pii: a022137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Serra R, Easter SL, Jiang W, Baxley SE. Wnt5a as an effector of TGFbeta in mammary development and cancer. J Mammary Gland Biol Neoplasia. 2011;16:157–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Katoh M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int J Oncol. 2017;51:1357–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bauer M, Benard J, Gaasterland T, Willert K, Cappellen D. WNT5A encodes two isoforms with distinct functions in cancers. PLoS ONE. 2013;8:e80526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krishnan V, Bryant HU, Macdougald OA. Regulation of bone mass by Wnt signaling. J Clin Invest. 2006;116:1202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lopez-Casillas F, Riquelme C, Perez-Kato Y, Ponce-Castaneda MV, Osses N, Esparza-Lopez J, et al. Betaglycan expression is transcriptionally up-regulated during skeletal muscle differentiation. Cloning of murine betaglycan gene promoter and its modulation by MyoD, retinoic acid, and transforming growth factor-beta. J Biol Chem. 2003;278:382–90. [DOI] [PubMed] [Google Scholar]
- 44.Bragado P, Estrada Y, Parikh F, Krause S, Capobianco C, Farina HG, et al. TGF-beta2 dictates disseminated tumour cell fate in target organs through TGF-beta-RIII and p38alpha/beta signalling. Nat Cell Biol. 2013;15:1351–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gebken J, Feydt A, Brinckmann J, Notbohm H, Muller PK, Batge B. Ligand-induced downregulation of receptors for TGF-beta in human osteoblast-like cells from adult donors. J Endocrinol. 1999;161:503–10. [DOI] [PubMed] [Google Scholar]
- 46.Stenvers KL, Tursky ML, Harder KW, Kountouri N, Amatayakul-Chantler S, Grail D, et al. Heart and liver defects and reduced transforming growth factor beta2 sensitivity in transforming growth factor beta type III receptor-deficient embryos. Mol Cell Biol. 2003;23:4371–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, et al. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hill CR, Jacobs BH, Brown CB, Barnett JV, Goudy SL. Type III transforming growth factor beta receptor regulates vascular and osteoblast development during palatogenesis. Dev Dyn. 2015;244:122–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roarty K, Serra R. Wnt5a is required for proper mammary gland development and TGF-beta-mediated inhibition of ductal growth. Development. 2007;134:3929–39. [DOI] [PubMed] [Google Scholar]
- 50.Kumawat K, Menzen MH, Bos IS, Baarsma HA, Borger P, Roth M, et al. Noncanonical WNT-5A signaling regulates TGF-beta-induced extracellular matrix production by airway smooth muscle cells. FASEB J. 2013;27:1631–43. [DOI] [PubMed] [Google Scholar]
- 51.Hasegawa D, Wada N, Yoshida S, Mitarai H, Arima M, Tomokiyo A, et al. Wnt5a suppresses osteoblastic differentiation of human periodontal ligament stem cell-like cells via Ror2/JNK signaling. J Cell Physiol. 2018;233:1752–62. [DOI] [PubMed] [Google Scholar]
- 52.Olivares-Navarrete R, Hyzy SL, Hutton DL, Dunn GR, Appert C, Boyan BD, et al. Role of non-canonical Wnt signaling in osteoblast maturation on microstructured titanium surfaces. Acta Biomater. 2011;7:2740–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hodgkinson CP, Naidoo V, Patti KG, Gomez JA, Schmeckpeper J, Zhang Z, et al. Abi3bp is a multifunctional autocrine/paracrine factor that regulates mesenchymal stem cell biology. Stem Cells. 2013;31:1669–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nishida J, Miyazono K, Ehata S. Decreased TGFBR3/betaglycan expression enhances the metastatic abilities of renal cell carcinoma cells through TGF-beta-dependent and -independent mechanisms. Oncogene. 2018;37:2197–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dong M, How T, Kirkbride KC, Gordon KJ, Lee JD, Hempel N, et al. The type III TGF-beta receptor suppresses breast cancer progression. J Clin Invest. 2007;117:206–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thiele S, Gobel A, Rachner TD, Fuessel S, Froehner M, Muders MH, et al. WNT5A has anti-prostate cancer effects in vitro and reduces tumor growth in the skeleton in vivo. J Bone Min Res. 2015;30:471–80. [DOI] [PubMed] [Google Scholar]
- 57.Thiele S, Zimmer A, Gobel A, Rachner TD, Rother S, Fuessel S, et al. Role of WNT5A receptors FZD5 and RYK in prostate cancer cells. Oncotarget. 2018;9:27293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thiele S, Rachner TD, Rauner M, Hofbauer LC. WNT5A and Its Receptors in the bone-cancer dialogue. J Bone Miner Res. 2016;31:1488–96. [DOI] [PubMed] [Google Scholar]
- 59.Ren D, Dai Y, Yang Q, Zhang X, Guo W, Ye L, et al. Wnt5a induces and maintains prostate cancer cells dormancy in bone. J Exp Med. 2018;216:428–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Canesin G, Evans-Axelsson S, Hellsten R, Krzyzanowska A, Prasad CP, Bjartell A, et al. Treatment with the WNT5A-mimicking peptide Foxy-5 effectively reduces the metastatic spread of WNT5A-low prostate cancer cells in an orthotopic mouse model. PLoS ONE. 2017;12:e0184418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miyamoto DT, Zheng Y, Wittner BS, Lee RJ, Zhu H, Broderick KT, et al. RNA-Seq of single prostate CTCs implicates non-canonical Wnt signaling in antiandrogen resistance. Science. 2015;349:1351–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee GT, Kwon SJ, Kim J, Kwon YS, Lee N, Hong JH, et al. WNT5A induces castration-resistant prostate cancer via CCL2 and tumour-infiltrating macrophages. Br J Cancer. 2018;118:670–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang S, Xu L, Sun Y, Wu T, Wang K, Li G. An improved protocol for isolation and culture of mesenchymal stem cells from mouse bone marrow. J Orthop Transl. 2015;3:26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Van Gelder RN, von Zastrow ME, Yool A, Dement WC, Barchas JD, Eberwine JH. Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proc Natl Acad Sci USA. 1990;87:1663–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Warrington JA, Nair A, Mahadevappa M, Tsyganskaya M. Comparison of human adult and fetal expression and identification of 535 housekeeping/maintenance genes. Physiol Genom. 2000;2:143–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.