

Abstract
Most of the proteome is considered undruggable, oftentimes hindering translational efforts for drug discovery. Identifying previously unknown druggable hotspots in proteins would enable strategies for pharmacologically interrogating these sites with small molecules. Activity-based protein profiling (ABPP) has arisen as a powerful chemoproteomic strategy that uses reactivity-based chemical probes to map reactive, functional, and ligandable hotspots in complex proteomes, which has enabled inhibitor discovery against various therapeutic protein targets. Here, we report an alkyne-functionalized N-hydroxysuccinimide-ester (NHS-ester) as a versatile reactivity-based probe for mapping the reactivity of a wide range of nucleophilic ligandable hotspots, including lysines, serines, threonines, and tyrosines, encompassing active sites, allosteric sites, post-translational modification sites, protein interaction sites, and previously uncharacterized potential binding sites. Surprisingly, we also show that fragment-based NHS-ester ligands can be made to confer selectivity for specific lysine hotspots on specific targets including Dpyd, Aldh2, and Gstt1. We thus put forth NHS-esters as promising reactivity-based probes and chemical scaffolds for covalent ligand discovery.
Graphical Abstract
Even with the discovery of thousands of proteins implicated in human disease, translating these potential therapeutic targets into drugs remains difficult, in part because most protein targets are considered “undruggable” or difficult to target with small molecules. Most of the proteome is devoid of pharmacological tools, hindering both basic and translational research efforts. Only ~2% of all predicted human gene products are currently targeted with small-molecule drugs, and 85–90% of the proteome is considered undruggable.1 A major limiting factor in defining the druggability of a target is the lack of apparent binding pockets that can be targeted with small molecules to modulate protein function; such pockets can be difficult to predict based on sequence or structures. Identifying ligandable binding pockets within the proteome would thus potentially enable us to identify sites that can be pharmacologically interrogated for drug discovery.
Activity-based protein profiling (ABPP) has arisen as a powerful chemoproteomic strategy to identify ligandable hotspots in complex proteomes, enabling inhibitor discovery.2–4 ABPP uses reactivity-based chemical probes to map reactive, functional, and ligandable hotspots in complex proteomes. When used in a competitive manner, covalently acting small molecules can be competed against the binding of reactivity-based probes to enable pharmacological interrogation of druggable hotspots.2,3,5 Previous studies have used ABPP platforms to identify irreversible small-molecule inhibitors against many different enzymes.3,6–8 Recently, covalent ligand discovery has also been coupled with isotopic tandem orthogonal proteolysis-enabled ABPP (isoTOP-ABPP) plat forms to map covalent ligands against many ligandable cysteine hotspots in complex proteomes.3,9,10
Cysteines within proteins have been particularly targeted for pharmacological tool and drug development due to well-established chemical scaffolds that exhibit high reactivity and specificity toward thiols, including haloacetamides, acrylamides, and maleamides.3,11–13 Thanks to these reactive scaffolds, there are many examples of cysteine-reactive small molecules targeting various druggable and undruggable protein targets.3,13–15 There have also been more recent efforts to develop reactivity-based probes and covalently acting small molecules for chemoproteomic studies targeting other nucleophilic residues, such as lysines, serines, threonines, and tyrosines. For example, Shannon et al. found that dichlorotriazines show lysine-selective reactivity and can be used for chemoproteomic applications.16 Identifying additional broadly reactive probes and scaffolds that can target these other nucleophilic hotspots beyond cysteines would drastically expand our ability to profile reactivity throughout the proteome and provide new handles for pharmacological interrogation of functional sites.
N-hydroxysuccinimide-esters (NHS-esters) are used extensively as functional groups in bioconjugation reactions, forming an amide bond with amines, such as lysines or N-termini. Other studies have also shown that NHS-esters can also react with other nucleophilic amino acids including serines, threonines, and tyrosines.17,18 We were thus intrigued as to whether NHS-esters could be used as reactivity-based probes to broadly map reactivity across these and other nucleophilic amino acid hotspots and whether, despite their high reactivity, small fragments designed containing an NHS-ester could target specific sites within the proteome.
To determine whether NHS-esters can be used as reactivity-based probes for chemoproteomic profiling, we used a commercially available alkyne-functionalized NHS-ester (propargyl-N-hydroxysuccinimidyl ester; NHS-ester-alkyne) to label mouse liver proteomes. We labeled proteomes with either a high (500 μM) or low (100 μM) concentration of NHS-ester-alkyne to map the relative reactivity of probe labeled sites using the isoTOP-ABPP platform (Figure 1; Table S1; Figure S1). We then interpreted the quantitative ratios of those probe-modified peptides that were identified in two out of four biological replicates.
Figure 1.
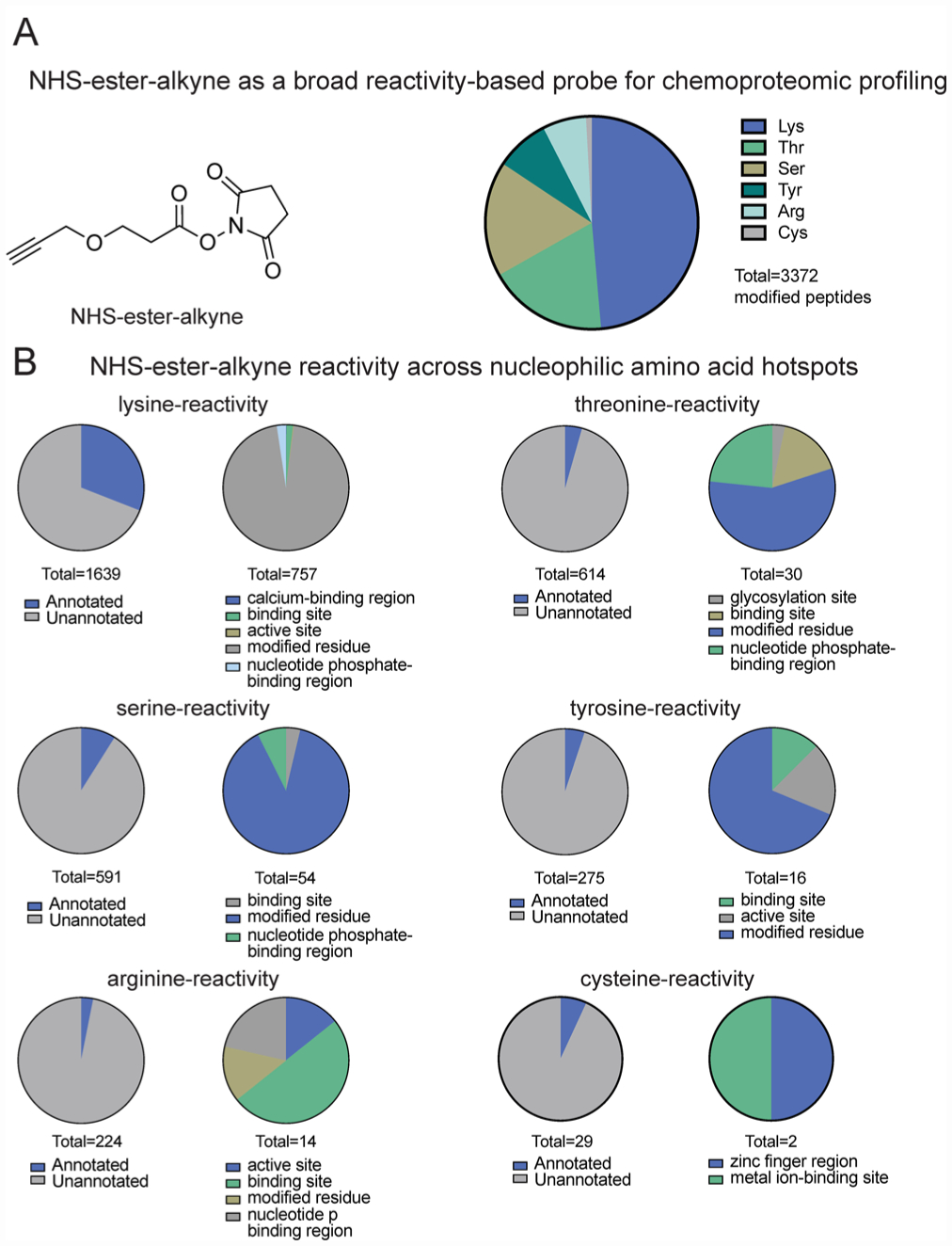
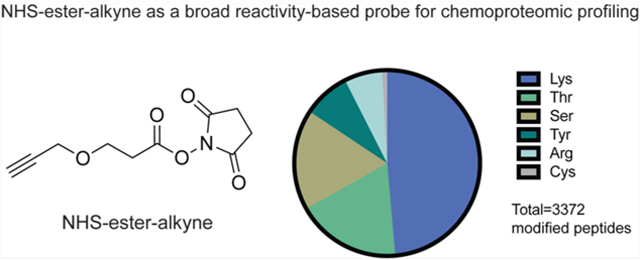
IsoTOP-ABPP of NHS-ester-alkyne reactivity in mouse liver proteome. (A) Structure of NHS-ester-alkyne (NHSyne) and distribution of probe-modified peptides in mouse liver proteome assessed by isoTOP-ABPP. Mouse liver proteomes were labeled with NHS-ester-alkyne (500 or 100 μM), followed by copper-catalyzed azide–alkyne cycloaddition (CuAAC) conjugation of a biotin-azide tag bearing an isotopically light (for 500 μM) or heavy (100 μM) mass tag to probe-labeled proteins. Probe-labeled proteins were subsequently avidin-enriched and digested with trypsin, and probe-modified tryptic peptides were isolated and eluted by TEV protease for subsequent LC-LC/MS/MS proteomic analysis. Raw data and ratiometric analysis of heavy to light peptides can be found in Figure S1 and Table S1. NHS-ester-alkyne predominantly reacted with lysines, showed significant reactivity with threonines and serines, and showed minor reactivity with tyrosines, arginines, and cysteines. (B) Breakdown of probe-labeled sites compared against annotated UniProt functional sites. While the majority of labeled residues are unannotated, the annotated sites were predominantly post-translationally modified or involved in ligand binding. Data shown in A and B are representative of probe-modified peptides found in two out of four biological replicates.
Through this effort, we identified over 3000 probe-modified peptides in mouse liver proteome. Nearly half of these sites (1639 sites) were lysines, consistent with the known preference of NHS-esters for amines. However, serines and threonines also represented ~17–18% each of total modified peptides. In addition, we also observed labeling of tyrosines and arginines, and to a lesser extent, cysteines (Figure 1A). Thus, we show that NHS-ester-alkyne is a versatile reactivity-based chemoproteomic probe for mapping the reactivity of proteome-wide accessible nucleophilic amino acids in complex proteomes. Most probe-modified peptides exhibited light (500 μM) to heavy (100 μM) ratios of 5 or greater (Figure S1; Table S1), while a subset of these peptides demonstrated ratios less than 5. Previous studies using isoTOP-ABPP to quantitatively map cysteine reactivity showed that those peptides with ratios less than 2 were considered hyper-reactive and enriched in functional residues.19 A subset of probe-labeled peptides also showed light to heavy ratios less than 2, suggesting that these sites exhibited saturable binding at lower concentrations of probes and may possibly represent sites of preferential reactivity, potentially due to conditions in their local protein microenvironment (Figure S1; Table S1). In contrast, we interpret those probe-modified peptides that show a light to heavy ratio much greater than 5 as essentially showing little to no labeling at 100 μM compared to 500 μM. We also performed a parallel study with the dichlorotriazine-alkyne lysine-reactive probe reported in Shannon et al. in mouse liver proteome and report 85 probe-modified lysines (Table S1), which is on par with the number of probe-labeled lysines reported previously.16 Overall, these data indicate that NHS-ester-alkyne is a promising reactivity-based probe that can be used in chemoproteomic experiments to map the reactivity of lysines and many other nucleophilic amino acids.
Further characterization of these NHS-ester-alkyne probe-modified peptides using the Uniprot database showed significant overlap with annotated functional sites (Figure 1B; Table S1). Among probe-modified lysines, 31% of these sites were annotated. Among these annotated residues, most were known sites of post-translational modifications such as lysine acetylation, succinylation, and malonylation. The remaining annotated lysine residues included active sites, binding sites, calcium binding regions, and nucleotide phosphate-binding regions (Figure 1B; Table S1). While most probe-modified amino acids that we identified across the other nucleophilic residues were unannotated, the annotated sites included active sites, binding sites, post-translational modification sites, and metabolite and metal binding regions (Figure 1B; Table S1).
We provide structural examples of both annotated sites as well as unannotated sites that may be functional, highlighting both the diversity of nucleophiles labeled as well as the variety of roles these nucleophiles possess. In the cases where solved crystal structures of mouse proteins were not available, the closest human homologue was used after confirming the labeled residue was conserved. Consistent with the role of lysines in enzyme catalysis, we observed several lysines in metabolic enzymes labeled with the NHS-ester-alkyne probe. First, in our isoTOP-ABPP data, we showed the active-site K230 labeled by our NHS-ester-alkyne probe in fructose bisphosphonate aldolase B (Aldob; shown in red), a glycolytic enzyme that catalyzes the reversible cleavage of fructose 1,6-bisphosphate to glyceraldehyde-3-phosphate and dihydroxyacetone phosphate.20 K230 forms a Schiff-base with dihydroxyacetonephosphate (Figure 2A, PDB: 1QO5).21 Consistent with the functional role of K230 in the catalytic mechanism of Aldob, we showed that the isotopic light to heavy ratio of this lysine is significantly less than 2, indicating this site is hyper-reactive (Table S1). Our data also revealed labeling of an active site tyrosine, Y55 in estradiol 17 beta-dehydrogenase 5 (Akr1c6), which acts as the proton donor during Akr1c6 catalyzed reduction of various androgens and estrogens (Figure 2B, PDB: 1J96).22 Beyond catalytic residues, we also demonstrated labeling of residues within enzyme active sites that are involved in substrate or cofactor binding, including K12 (shown in red) and R105 (shown in orange) labeled by our probe in nucleoside diphosphate kinase A (Nme1)—an enzyme that catalyzes the generation of nucleotide triphosphates.23 Both K12 and R105 interact with the α and β phosphates of the bound nucleoside diphosphate (Figure 2C, PDB: 2HVD). In addition to active-site lysines, we also observed labeling of post-translational modification sites within enzymes as well. K46 in glycine N-methyltransferase (Gnmt) was as another site of probe labeling (shown in red). Gnmt catalyzes the conversion of S-adenosyl-l-methionine to S-adenosyl-l-homocysteine and sarcosine.24 K46 is a known lysine succinylation site (Figure 2D, PDB: 1R8X).25
Figure 2.
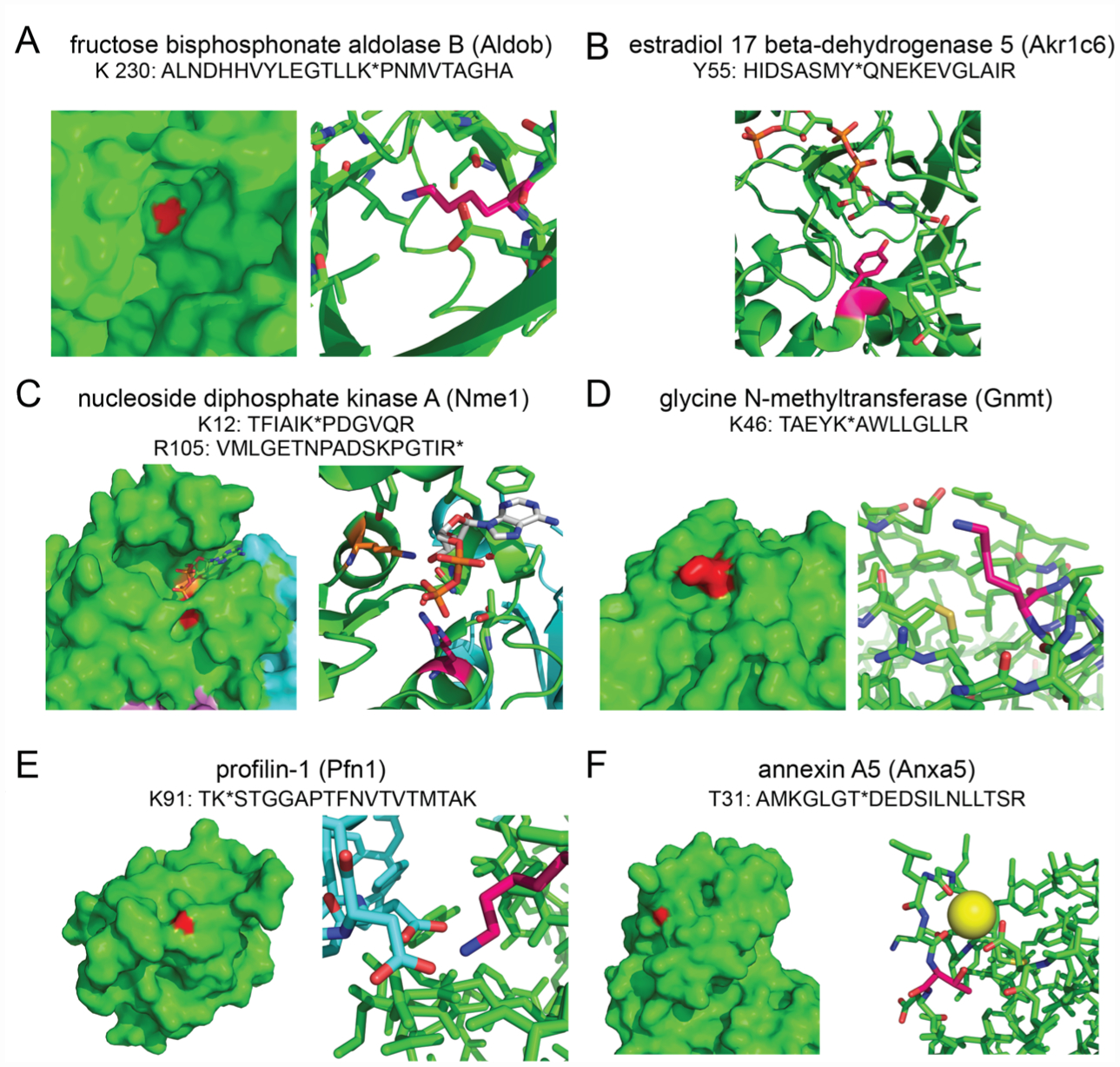
Examples of various types of nucleophilic residues targeted by NHS-ester-alkyne. (A) K230 on Aldob, a catalytic residue within the active site. (B) Y55 on Akr1c6, the catalytic proton donor within the active site. (C) K12 and R105 on Nme1, two positively charged residues involved in binding nucleoside diphosphates. (D) K46 on Gnmt, a surface lysine and known succinylation site. (E) K91 on Pfn1, a surface lysine which forms two salt bridges with aspartate residues at the actin–Pfn1 interface. (F) T31 on Anxa5, a surface threonine near the calcium binding site. PDB file designations are listed in the text. Specific structures are all from human counterparts.
Beyond metabolic enzymes, our isoTOP-ABPP data also uncovered labeling of lysines in other nonenzyme protein classes. Our isoTOP-ABPP data showed an unannotated K91 on profilin-1 (Pfn1) as a probe-labeled site (shown in red).26 Profilin is an actin-binding protein involved in restructuring the actin cytoskeleton and maintaining actin monomer homeostasis. Solved structures of human profilin-1 bound to actin show K91 forms salt bridges with D288 and D286 on actin, sitting ~3 Å from each Asp. Disruption of this interaction would likely destabilize that profilin-1–actin interface (Figure 2E, PDB: 1FIL, 2PBD). Another example of an unannotated site is T31 on annexin A5 that is labeled by our probe. Annexin A5 is a protein that binds to phosphatidylserine in a calcium-dependent manner.27 Structural analysis shows that T31 sits very closely (~4 Å) to the calcium binding site within annexin A5, suggesting that covalent modification of T31 could disrupt calcium binding and play a direct role in annexin A5 activity (Figure 2F, PDB: 1ANW).
Thus, by structurally examining sites of NHS-ester-alkyne probe labeling using isoTOP-ABPP, we demonstrate the versatility of NHS-ester-alkyne to provide a reactivity readout on a wide variety of functional and putatively ligandable sites, enabling both biological study and ligand discovery against these sites.
Following up on these data, we next sought to investigate whether NHS-esters could be used as scaffolds for covalent ligand discovery against specific nucleophilic hotspots targeted by the NHS-ester-alkyne probe. We synthesized two compounds CW 1–26 and CW 1–33, with varied structure around the NHS-ester reactive moiety (Figure 3). The starting carboxylic acid precursors for synthesizing CW 1–26 and CW 1–33 were chosen due to their commercial availability, price, and structural variety. While there were thousands of available carboxylic acids, the high reactivity of NHS esters led us to select relatively large structures that had a higher chance of possessing selectivity.
Figure 3.
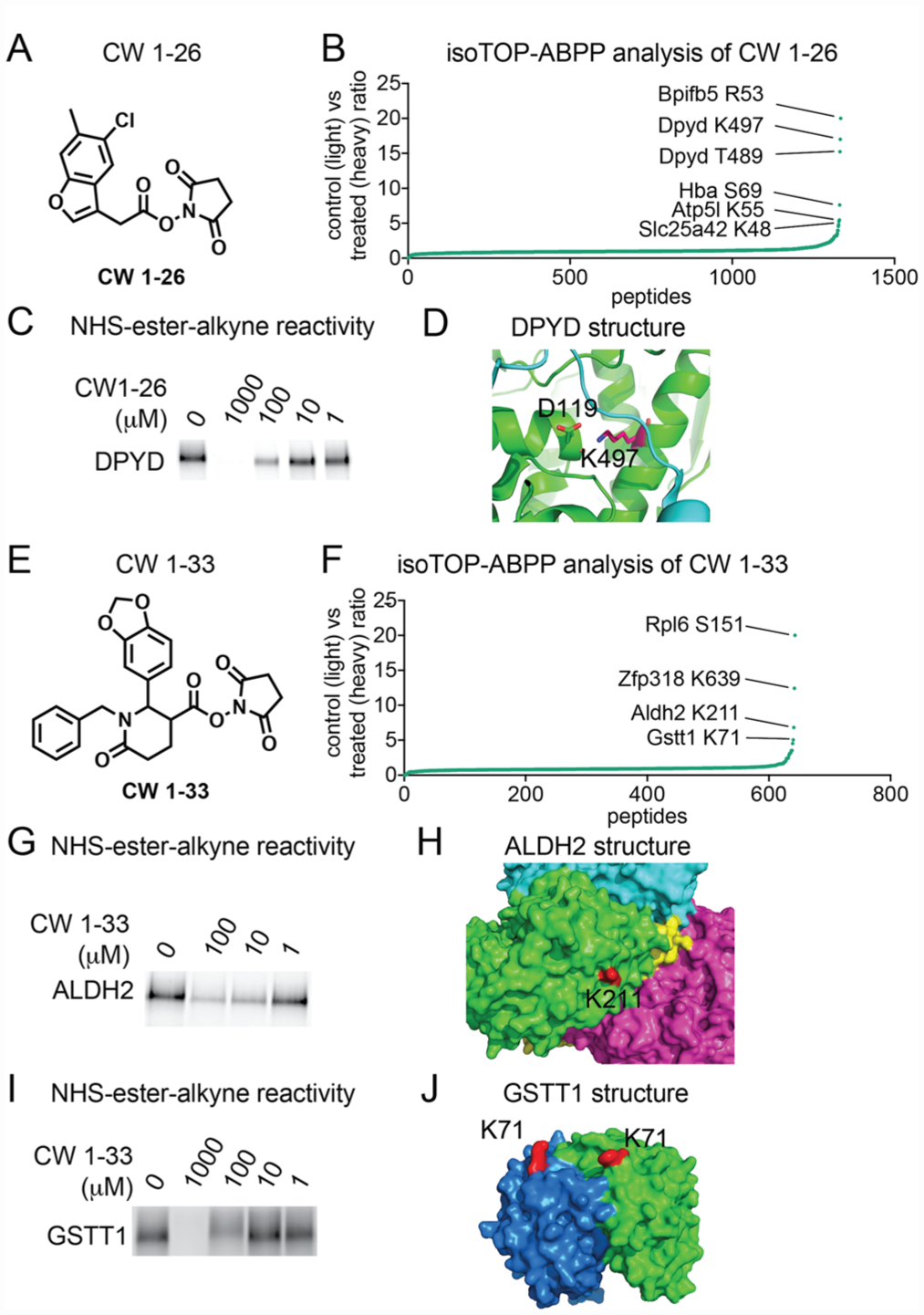
NHS-ester-based covalent ligands conferring selectivity for lysines on specific protein targets. (A) Structure of NHS-ester fragment CW 1–26. (B) Competitive isoTOP-ABPP analysis of CW 1–26 lysine reactivity in mouse liver proteomes. DMSO vehicle or CW 1–26 (100 μM) was preincubated with mouse liver proteomes for 30 min prior to labeling with NHS-ester-alkyne (500 μM) for 1 h. Probe-labeled samples were then taken through the isoTOP-ABPP procedure. Shown are individual isotopically light (vehicle-treated) to heavy (CW-1–26-treated) probe-labeled peptides, showing K497 as the primary target. (C) Competition of CW1–26 against NHS-ester alkyne labeling of recombinant pure human DPYD using gel-based in-gel fluorescence ABPP methods. (D) K497 is a surface lysine on DPYD which forms a salt bridge with D119 and resides at an interface between DPYD dimeric partners. (E) A second NHS-ester fragment CW1–33. (F) Competitive isoTOP-ABPP analysis of CW1–33 using the same approach described in B yielded three targets, K639 on Zfp318, K211 on Aldh2, and K71 Gstt1. (G–J) Validation of CW 1–33 competition against NHS-ester-alkyne labeling of pure human ALDH2 (G) and GSTT1 (I) protein and locations of lysines targeted by CW 1–33 on ALDH2 (H) and GSTT1 (J) structures. Data in B and D show average ratios for probe-labeled peptides identified in at least two out of three biological replicates. Gels shown in C, G, and I are representative gels from n = 3.
First, we demonstrated that neither of these compounds broadly impair NHS-ester-alkyne labeling of mouse liver proteomes, indicating already that these compounds are not reacting nonspecifically across the proteome (Figure S2). We next performed competitive isoTOP-ABPP studies to identify the specific targets of these NHS-ester-based covalent ligands in which we competed vehicle or CW 1–26 or CW 1–33 against the binding of NHS-ester-alkyne in mouse liver proteome followed by conjugation of isotopically light (for vehicle-treated) or heavy (for NHS-ester ligand-treated) biotin-enrichment handles bearing a TEV protease recognition sequence, followed by combining control and treated proteomes in a 1:1 ratio, avidin-enrichment and tryptic digestion of probe-modified proteins, and subsequent isolation and TEV protease release of probe-modified tryptic peptides for proteomic analysis (Figure 3). We then only interpreted the light to heavy isotopic ratios of probe-modified tryptic peptides for those peptides that were present in at least two out of three biological replicates. Among >600 probe-modified peptides profiled, we demonstrated that CW 1–26 selectively interacted with six targets, showing a light to heavy ratio of >5. These targets included R53 of Bpifb5, K497 and T489 of dihydropyrimidine dehydrogenase (Dpyd), S69 on Hba5, K55 on Atp5l, and K48 on Slc25a42, which possess an isotopically light to heavy probe-modified peptide ratio of >5 (Figure 3A,B; Table S1). We further validated one of these interactions showing competition of CW 1–26 against NHS-ester-alkyne labeling of pure human DPYD protein with a 50% inhibitory concentration (IC50) of 40 μM (Figure 3C). Dpyd is the initial and rate-limiting enzyme in uracil and thymidine catabolism. Examining a previously solved structure of human DPYD revealed that K497 is within 3 Å of D119 and is located where a strand from the other half of the DPYD homodimer crosses the protein surface. Disruption of this electrostatic interaction or addition by a bulky ligand could destabilize the DPYD dimer and affect enzyme function (Figure 3D).
The second ligand we tested, CW 1–33, also showed relatively selective interactions with four targets showing ratios of >5. These targets included S151 of Rpl6, K639 of Zfp318, K211 of aldehyde dehydrogenase 2 (Aldh2), and K71 of glutathione transferase t1 (Gstt1; Figure 3F; Table S1). We validated interaction of CW 1–33 with some of these targets and showed competition of CW 1–33 against NHS-ester alkyne labeling of human ALDH2 and GSTT1 and mapped the labeled sites in solved structures of these enzymes (Figure 3G–J). CW 1–33 showed more potent interactions with ALDH2 compared to GSTT1 with IC50 values of 4.1 and 100 μM, respectively (Figure 3G,I). Interestingly, both lysines targeted by CW 1–33 on ALDH2 and GSTT1 appear to be surface lysines with no obviously evident binding pocket (Figure 3H,J). These data may indicate that either there are subtle ligandable binding pockets, pockets that are formed by protein–protein interactions or conformational changes, or that these structures may dynamically form binding pockets upon interactions with CW 1–33 which may not be evident in these static structures. IC50 dose–response studies were performed by preincubating ligands with pure protein for 30 min followed by 1 h of labeling with NHS-ester-alkyne, and thus these reported IC50 values are relative potency values that can only be compared between the two probes under the same conditions. Most surprisingly, both CW 1–26 and CW 1–33 selectively interact with a only handful of targets out of hundreds of sites profiled, despite bearing their highly reactive NHS-ester core, as evidenced by most probe-modified peptides showing isotopic ratios of ~1 (Figure 3B,F). While these covalent ligands are clearly not selective enough for the reported targets, these data suggest that fragment-based NHS-ester covalent ligands can be potentially used to develop selective lead small-molecule modulators against specific nucleophilic hotspots targeted by the NHS-ester-alkyne probe. CW 1–33 does not inhibit ALDH2 activity based on a substrate activity assay in mouse liver proteome (data not shown). However, one could envision combining selective covalent ligands with small-molecule “degraders” that recruit E3 ubiquitin ligases for subsequent proteosomal degradation of specific protein targets28–30 as a means of leveraging this specificity for biological or therapeutic function.
Overall, our study demonstrates that the NHS-ester-alkyne probe is as a versatile reactivity-based probe for mapping the reactivity of a wide range of nucleophilic ligandable hotspots, including lysines, serines, threonines, and tyrosines encompassing active sites, allosteric sites, post-translational modification sites, protein interaction sites, and previously uncharacterized potential binding sites using chemoproteomic approaches. This commercially available probe can thus serve as a complementary tool to other previously developed reactivity-based probes that target lysines, such as with the dichlorotriazine-alkyne probe, as well as other nucleophilic residues.11,16,31,32 Surprisingly, we also demonstrate that selectivity for specific nucleophilic hotspots can be conferred with NHS-ester scaffolds, lending its future potential applicability for covalent ligand discovery against specific druggable hotspots targeted by reactivity-based probes.
METHODS
Materials.
2,5-Dioxopyrrolidin-1-yl-3-(prop-2-ynyloxy)propanoate (NHS-ester-alkyne) was obtained from Sigma-Aldrich. The dichlorotriazine-alkyne probe was synthesized according to the methods reported in Shannon et al.16 The synthesis of CW 1–26 and CW 1–33 is reported in the Supporting Information.
IsoTOP-ABPP.
IsoTOP-ABPP studies were done as previously reported.3,19 Proteome samples were derived from mouse livers, which were previously partitioned into three pieces, homogenized in PBS with a bead-blaster, and cleared of debris via high speed centrifugation. For isoTOP-ABPP studies, liver proteomes were diluted in PBS to 2 mg mL–1 and were labeled with NHS-ester-alkyne for 1 h at RT. CuAAC was used by sequential addition of tris(2-carboxyethyl)-phosphine (1 mM, Sigma), tris[(1-benzyl-1H-1,2,3-triazol-4-yl)-methyl]amine (34 μM, Sigma), copper(II) sulfate (1 mM, Sigma), and biotin-linker-azide, the linker functionalized with a TEV protease recognition sequence along with an isotopically light or heavy valine for treatment of control or treated proteome, respectively. Subsequent isoTOP-ABPP procedures and MS analysis were performed as previously reported.3,19,33,34 Cysteine residues were searched with a static modification for carboxyaminomethylation (+57.02146), and probe modification of peptides was searched with up to two differential modifications for methionine oxidation and either the light or heavy TEV tags (m/z + 550.22797 for light and +556.24178 for heavy for lysines, serines, threonines, arginines, and tyrosines and +493.20647 for light and +499.22028 for heavy for cysteine to account for the static modification). Peptides were required to have at least one tryptic end and to contain the TEV modification. ProLUCID data were filtered through DTASelect to achieve a peptide false-positive rate below 1%. For studies, with covalent ligands, proteomes were preincubated with DMSO or ligand for 30 min at RT prior to probe-labeling. The isoTOP-ABPP study with dichlorotriazine-alkyne was performed as described above, but the differential modifications for probe-modified lysines were m/z +550.22797 for light and +556.24178 for heavy. We note that the total numbers of NHS-ester-alkyne probe-modified peptides differ between the first runs compared with the competitive isoTOP-ABPP runs with ligands. This may be due to running four biological replicates, in Figure 1, compared to three biological replicates, in Figure 3; differences in mass spectrometry sensitivity at the time of running the samples; or instability of the probe with longer periods of storage.
Peptide Computational Analysis.
Quantified light/heavy ratios from each experiment that corresponded to the same peptide (including truncations) and labeling site were averaged, as long as the peptide had appeared in at least two experiments. These averages yielded a master list of labeled sites in the proteome for each residue of interest. The “feature” list from the uniprot records that each peptide mapped to was downloaded, and all features that overlapped with the labeled site in the protein and were shorter than 30 amino acids and were not in a list of ambigious/poorly annotated features (e.g., “strand,” “domain,” “site”) were kept as annotations for the peptide.
Gel-Based ABPP.
Gel-based ABPP methods were performed as previously described.35 Recombinant pure human proteins were either purified or purchased from Origene. Whole liver lysates were generated as previously described. Both samples were pretreated with DMSO or fragment in an incubation volume of 50 μL of PBS for 30 min at RT and were subsequently treated with NHS-ester-alkyne (10 μM final concentration) for 60 min at RT. CuAAC was performed to append rhodamine-azide onto probe-labeled proteins. The samples were separated by SDS/PAGE and scanned using a ChemiDoc MP (Bio-Rad Laboratories, Inc.).
Supplementary Material
ACKNOWLEDGMENTS
We thank the members of the Nomura Research Group for critical reading of the manuscript. This work was supported by grants from the National Institutes of Health (R01CA172667 and Chemical Biology Training Grant T32 GM066698), American Cancer Society Research Scholar Award (RSG14-242-01-TBE), and Department of Defense Breakthroughs Award (CDMRP W81XWH-15-1-0050).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.7b00125.
Supporting Methods, Table S1 legend, Figures S1 and S2 (PDF)
Table S1 (XLSX)
The authors declare no competing financial interest.
REFERENCES
- (1).Dixon SJ, and Stockwell BR (2009) Identifying druggable disease-modifying gene products. Curr. Opin. Chem. Biol 13, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Roberts AM, Ward CC, and Nomura DK (2017) Activity-based protein profiling for mapping and pharmacologically interrogating proteome-wide ligandable hotspots. Curr. Opin. Biotechnol 43, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Backus KM, Correia BE, Lum KM, Forli S, Horning BD, González-Páez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, Wolan DW, and Cravatt BF (2016) Proteome-wide covalent ligand discovery in native biological systems. Nature 534, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Evans MJ, and Cravatt BF (2006) Mechanism-based profiling of enzyme families. Chem. Rev 106, 3279–3301. [DOI] [PubMed] [Google Scholar]
- (5).Counihan JL, Ford B, and Nomura DK (2016) Mapping proteome-wide interactions of reactive chemicals using chemoproteomic platforms. Curr. Opin. Chem. Biol 30, 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Chang JW, Nomura DK, and Cravatt BF (2011) A potent and selective inhibitor of KIAA1363/AADACL1 that impairs prostate cancer pathogenesis. Chem. Biol 18, 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavón FJ, Serrano AM, Selley DE, Parsons LH, Lichtman AH, and Cravatt BF (2009) Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat. Chem. Biol 5, 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Chang JW, Zuhl AM, Speers AE, Niessen S, Brown SJ, Mulvihill MM, Fan YC, Spicer TP, Southern M, Scampavia L, Fernandez-Vega V, Dix MM, Cameron MD, Hodder PS, Rosen H, Nomura DK, Kwon O, Hsu K-L, and Cravatt BF (2015) Selective inhibitor of platelet-activating factor acetylhydrolases 1b2 and 1b3 that impairs cancer cell survival. ACS Chem. Biol 10, 925–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Roberts AM, Miyamoto DK, Huffman TR, Bateman LA, Ives AN, Heslin MJ, Akopian D, Contreras CM, Rape M, Skibola CF, and Nomura DK (2017) Chemoproteomic Screening of Covalent Ligands Reveals UBA5 as a Novel Pancreatic Cancer Target. ACS Chem. Biol 12, 899. [DOI] [PubMed] [Google Scholar]
- (10).Bateman LA, Nguyen TB, Roberts AM, Miyamoto DK, Ku W-M, Huffman TR, Petri Y, Heslin MJ, Contreras CM, Skibola CF, Olzmann JA, and Nomura DK (2017) Chemoproteomics-enabled covalent ligand screen reveals a cysteine hotspot in reticulon 4 that impairs ER morphology and cancer pathogenicity. Chem. Commun, DOI: 10.1039/C7CC01480E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Shannon DA, and Weerapana E (2015) Covalent protein modification: the current landscape of residue-specific electrophiles. Curr. Opin. Chem. Biol 24, 18–26. [DOI] [PubMed] [Google Scholar]
- (12).Pace NJ, and Weerapana E (2013) Diverse functional roles of reactive cysteines. ACS Chem. Biol 8, 283–296. [DOI] [PubMed] [Google Scholar]
- (13).Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, and Gray NS (2013) Developing irreversible inhibitors of the protein kinase cysteinome. Chem. Biol 20, 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ostrem JML, and Shokat KM (2016) Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat. Rev. Drug Discovery 15, 771–785. [DOI] [PubMed] [Google Scholar]
- (15).London N, Miller RM, Krishnan S, Uchida K, Irwin JJ, Eidam O, Gibold L, Cimermančič P, Bonnet R, Shoichet BK, and Taunton J (2014) Covalent docking of large libraries for the discovery of chemical probes. Nat. Chem. Biol 10, 1066–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Shannon DA, Banerjee R, Webster ER, Bak DW, Wang C, and Weerapana E (2014) Investigating the proteome reactivity and selectivity of aryl halides. J. Am. Chem. Soc 136, 3330–3333. [DOI] [PubMed] [Google Scholar]
- (17).Mädler S, Bich C, Touboul D, and Zenobi R (2009) Chemical cross-linking with NHS esters: a systematic study on amino acid reactivities. J. Mass Spectrom 44, 694–706. [DOI] [PubMed] [Google Scholar]
- (18).Miller BT, Collins TJ, Rogers ME, and Kurosky A (1997) Peptide Biotinylation with Amine-Reactive Esters: Differential Side Chain Reactivity. Peptides 18, 1585–1595. [DOI] [PubMed] [Google Scholar]
- (19).Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MBD, Bachovchin DA, Mowen K, Baker D, and Cravatt BF (2010) Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468, 790–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Cox TM (1994) Aldolase B and fructose intolerance. FASEB J 8, 62–71. [DOI] [PubMed] [Google Scholar]
- (21).Lorentzen E, Siebers B, Hensel R, and Pohl E (2005) Mechanism of the Schiff Base Forming Fructose-1,6-bisphosphate Aldolase: Structural Analysis of Reaction Intermediates. Biochemistry 44, 4222–4229. [DOI] [PubMed] [Google Scholar]
- (22).Barski OA, Tipparaju SM, and Bhatnagar A (2008) The Aldo-Keto Reductase Superfamily and its Role in Drug Metabolism and Detoxification. Drug Metab. Rev 40, 553–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Attwood PV, and Wieland T (2015) Nucleoside diphosphate kinase as protein histidine kinase. Naunyn-Schmiedeberg’s Arch. Pharmacol 388, 153–160. [DOI] [PubMed] [Google Scholar]
- (24).Luka Z, Mudd SH, and Wagner C (2009) Glycine N-methyltransferase and regulation of S-adenosylmethionine levels. J. Biol. Chem 284, 22507–22511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Park J, Chen Y, Tishkoff DX, Peng C, Tan M, Dai L, Xie Z, Zhang Y, Zwaans BMM, Skinner ME, Lombard DB, and Zhao Y (2013) SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 50, 919–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Alkam D, Feldman EZ, Singh A, and Kiaei M (2017) Profilin1 biology and its mutation, actin(g) in disease. Cell. Mol. Life Sci 74, 967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Bouter A, Carmeille R, Gounou C, Bouvet F, Degrelle SA, Evain-Brion D, and Brisson AR (2015) Review: Annexin-A5 and cell membrane repair. Placenta 36 (Suppl 1), S43–49. [DOI] [PubMed] [Google Scholar]
- (28).Neklesa TK, Winkler JD, and Crews CM (2017) Targeted protein degradation by PROTACs. Pharmacol. Ther, DOI: 10.1016/j.pharmthera.2017.02.027. [DOI] [PubMed] [Google Scholar]
- (29).Lai AC, Toure M, Hellerschmied D, Salami J, Jaime-Figueroa S, Ko E, Hines J, and Crews CM (2016) Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem., Int. Ed 55, 807–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, and Bradner JE (2015) DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Weerapana E, Simon GM, and Cravatt BF (2008) Disparate proteome reactivity profiles of carbon electrophiles. Nat. Chem. Biol 4, 405–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Nomura DK, Dix MM, and Cravatt BF (2010) Activity-based protein profiling for biochemical pathway discovery in cancer. Nat. Rev. Cancer 10, 630–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Ford B, Bateman LA, Gutierrez-Palominos L, Park R, and Nomura DK (2017) Mapping Proteome-wide Targets of Glyphosate in Mice. Cell Chem. Biol 24, 133. [DOI] [PubMed] [Google Scholar]
- (34).Counihan JL, Duckering M, Dalvie E, Ku W-M, Bateman LA, Fisher KJ, and Nomura DK (2017) Chemoproteomic Profiling of Acetanilide Herbicides Reveals Their Role in Inhibiting Fatty Acid Oxidation. ACS Chem. Biol 12, 635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Medina-Cleghorn D, Bateman LA, Ford B, Heslin A, Fisher KJ, Dalvie ED, and Nomura DK (2015) Mapping Proteome-Wide Targets of Environmental Chemicals Using Reactivity-Based Chemoproteomic Platforms. Chem. Biol 22, 1394–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.