

Abstract
Patients with pulmonary Langerhans cell histiocytosis (LCH) typically have a benign course but may have extensive cystic lung disease with rare life-threatening complications including multiple and recurrent pneumothoraces and respiratory failure. We report 7 severely affected pediatric patients treated with chemotherapy, aggressive chest tube management, and pleurodesis of whom 5 survived. Patients with extraordinary amounts of pulmonary cystic disease and multiple pneumothoraces due to LCH can have remarkable, curative outcomes with early recognition, optimal LCH-directed therapy, and supportive care.
Keywords: Langerhans cell histiocytosis, pneumothorax, cystic lung disease, pleurodesis, chemotherapy
Introduction
The overall incidence of Langerhans cell histiocytosis has been estimated at 3–5 per million children with pulmonary involvement occurring in 7–12% of pediatric patients and typically presenting during the first four years of life.(1–7) The most common sites of disease involvement in LCH are skin, bone, lungs, liver, spleen, and bone marrow. LCH lesions in the lungs most often represent a component of more extensive multisystem disease in children. Recent breakthroughs in the biologic understanding of this disease suggest that LCH originates from a myeloid dendritic cell with a neoplastic process driven by abnormal somatic cell-activating mutations in the MAPK pathway.(8) While the majority (50–65%) of LCH lesions have a single somatic BRAFV600E mutation in children, a series of pulmonary LCH lesions from adults demonstrated a large percentage of NRAS somatic mutations (40%) coincident with BRAFV600E mutations relative to non-pulmonary LCH lesions where no NRAS mutations were identified. (9;9)
Overall survival rates at 5 years for pediatric patients with LCH have been estimated at 90–95% with the highest risk for death occurring in patients with “risk organ” (liver, spleen, bone marrow) disease. Previous analyses of survival have demonstrated no significant differences between patients with only bone or bone with pulmonary involvement, resulting in the exclusion of lungs as a definition of risk organ involvement.(10) Cigarette smoking is clearly associated with increased risk for pulmonary LCH in teenagers and adults, and risk of death from respiratory failure is well known. (11)
Clinical symptoms at time of presentation include cough, dyspnea, and sudden onset of chest pain related to spontaneous pneumothoraces. Computed tomography (CT) often demonstrates nodules and cystic lung disease.(7;12) A majority of patients with pulmonary LCH have a modest number of cysts and nodules which are often asymptomatic and resolve quickly with treatment.
Although uncommon, some previously asymptomatic patients may present with severe respiratory distress and even respiratory failure from striking involvement of lung parenchyma with large cysts and lung collapse secondary to pneumothoraces.(13;14) Initial treatment of these patients may require admission to intensive care units, insertion of multiple chest tubes, and critical decisions regarding respiratory support, but many have remarkable improvement in lung function and imaging after a variety of treatments.
We report a series of 7 critically ill patients with severe respiratory failure due to pulmonary LCH seen at Texas Children’s Hospital (USA) and three hospitals in the United Kingdom (UK) over the past 25 years in whom aggressive management of pneumothoraces included multiple chest tubes, pleurodesis, and chemotherapy, with 5 of the 7 patients surviving. Most survivors eventually achieved near resolution of lung abnormalities on imaging and overall good functional outcome. There is little guidance in existing literature regarding potential for cure or management strategies in this group of patients. For all of these patients there was uncertainty about prognosis and difficult conversations regarding the best treatment course. In some cases, discussions included consideration of extracorporeal life support, lung transplantation and withdrawal of intensive care support.
Methods
All cases described here were reviewed in accordance with the relevant local research governance regulations. Medical records and imaging studies were reviewed and summarized under IRB approved protocols with informed consent waived. Clinical charts, chest radiographs and CT scans were reviewed for these children who had biopsy-proven LCH with response criteria based on clinical assessment (extent of respiratory support, oxygenation, and stabilization of pneumothoraces) and degree of improvement on imaging (CT scan, PET scan) where applicable.
Results
Seven patients with pulmonary LCH were reviewed ranging from 8 months to 16 years at diagnosis. All patients had multisystem disease with extra-pulmonary sites and one patient had high risk organ involvement (liver). Of the evaluable patients, 6 of 7 (85%) required pleurodesis, 4 of 6 (67%) required mechanical ventilation (median 12.5 days), 5 of 6 (83%) required critical care support (median 41.5 days), and 5 of 6 (83%) required supplemental oxygen (median 68 days). Overall survival was 71% with the two deaths occurring early on during treatment.
Case Descriptions: (Table 1 and Figure 1)
Table 1:
Clinical Characteristics of Patients with Pulmonary LCH
| Patient Number | Country | Age of onset | Other sites of LCH | Type of pleurodesis | Number of days | Chemotherapy | Outcome | ||
|---|---|---|---|---|---|---|---|---|---|
| Ventilated | ICU | oxygen | |||||||
| 1 | USA | Birth | Skin, skull bones, thyroid, thymus | Bilateral laser | 27 | 45 | 93 | Vbl/Pred | Death secondary to viral infection while on treatment |
| 2 | USA | 11 months | Skin, pituitary | Surgical | 4 | 113 | 144 | ARAC × 12 months | CR 2 years off treatment (Lansky 100%) |
| 3 | UK | 11 months | Skin, mastoid, temporal bone | Chemical (talc) – ineffective | 45 | 39 | 43 | 1) Pred/Vbl × 8 weeks with ARAC added at week 7 | Death after 8 weeks of therapy due to complications of air leaks (otorrhea and skin rash were resolving) |
| 4 | UK | 2 years | Vertebra plana | Chemical (talc) – ineffective | 21 | 44 | 215 | 1) Pred/Vbl x1 month, then ARAC added × 5 months 2) Vincristine and ARAC for 12 months (total treatment: 18 months) |
CR 9 months off treatment (Lansky 100%) |
| 5 | USA | 2.6 years | Skin, liver, lymph nodes | Surgical | --- | --- | --- | 1) Vbl/Pred/Mtx 2) ARAC/Clad × 1 month, then Clad × 6 months |
CR 13 years off treatment (Lansky 100%) |
| 6 | UK | 15 years | Scalp | None | 0 | 0 | 0 | 1) Vbl/Pred 2) ARAC |
Resolution of pneumothoraces; imaging showed smaller and fewer cysts but persistent fibrosis; currently still on chemotherapy (Lansky 100%) |
| 7 | USA | 16 years | Pituitary | Surgical with doxycycline | 0 | 7 | 36 | 1) ARAC × 12 months 2) Clofarabine × 6 months |
1 relapse/ CR 4 years off treatment (Karnofsky 80%) |
Abbreviations: Vbl: vinblastine, Pred: prednisone, Mtx: methotrexate, ARAC: cytarabine, Clad: cladribine, CR: complete response
Figure 1: Imaging series from patients.
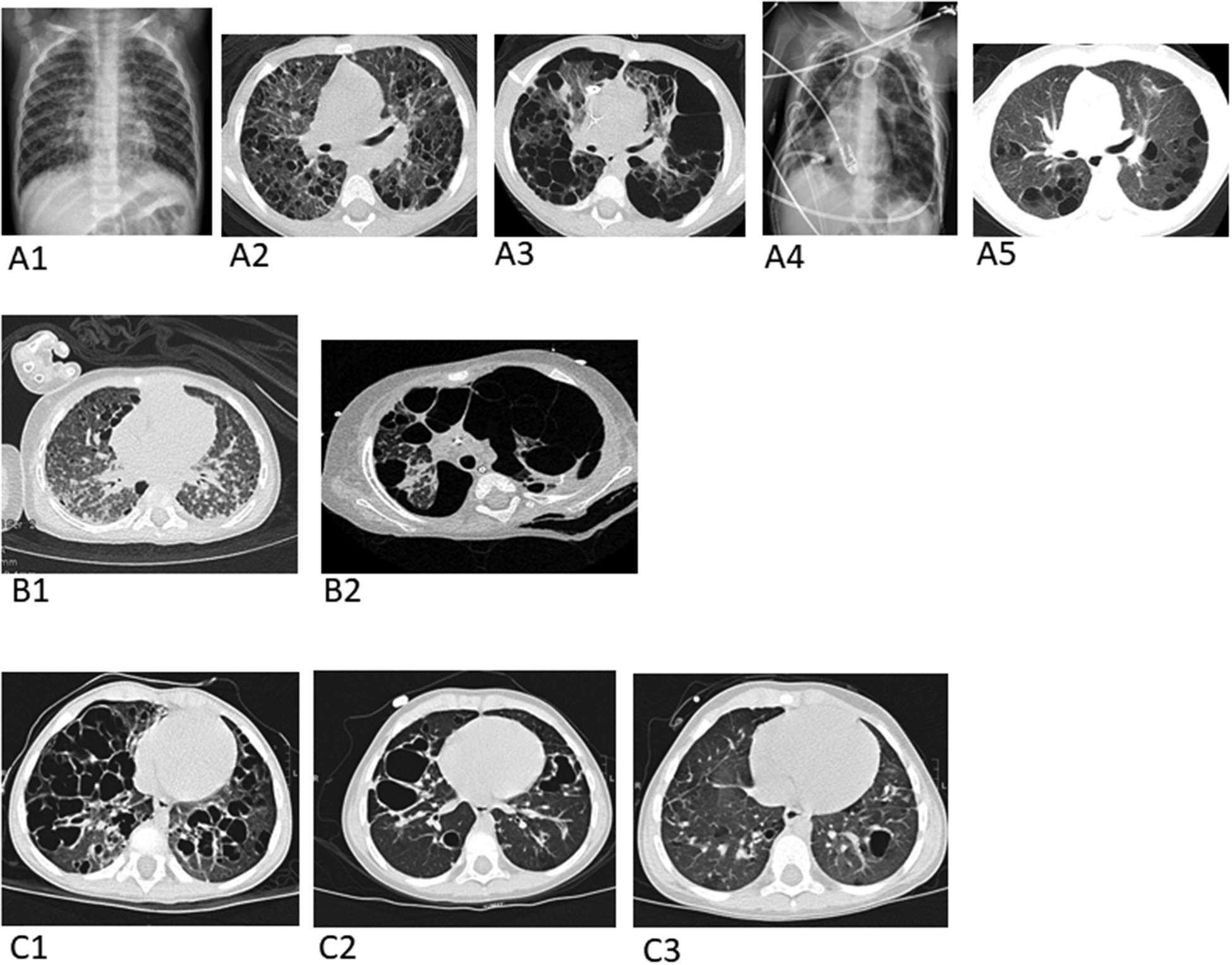
(A) Images from Patient 2: Baseline pre-treatment chest radiograph (A1) and axial chest CT image (A2), depicting multiple pulmonary cysts. An axial chest CT image (A3) one month later shows marked enlargement of many of the cysts with effacement of much of the normal lung parenchyma and bilateral pneumothoraces from cyst rupture. Chest radiograph (A4) three weeks later demonstrates three chest tubes and four pigtailed pleural-drainage catheters placed for refractory air leaks. Axial chest CT image (A5) 15 months later following chemotherapy with cytarabine shows a marked decrease in number and size of pulmonary cysts and reconstitution of normal-appearing intervening lung parenchyma.
(B) Images from Patient 3: Baseline pre-treatment axial chest CT (B1). Axial chest CT (B2) on the day the patient died due to complications of air leaks after 8 weeks of chemotherapy.
(C) Images from Patient 4: Baseline pre-treatment axial chest CT (C1). Axial chest CT(C2) 6 months after starting chemotherapy and axial CT chest (C3) 18 months after starting chemotherapy.
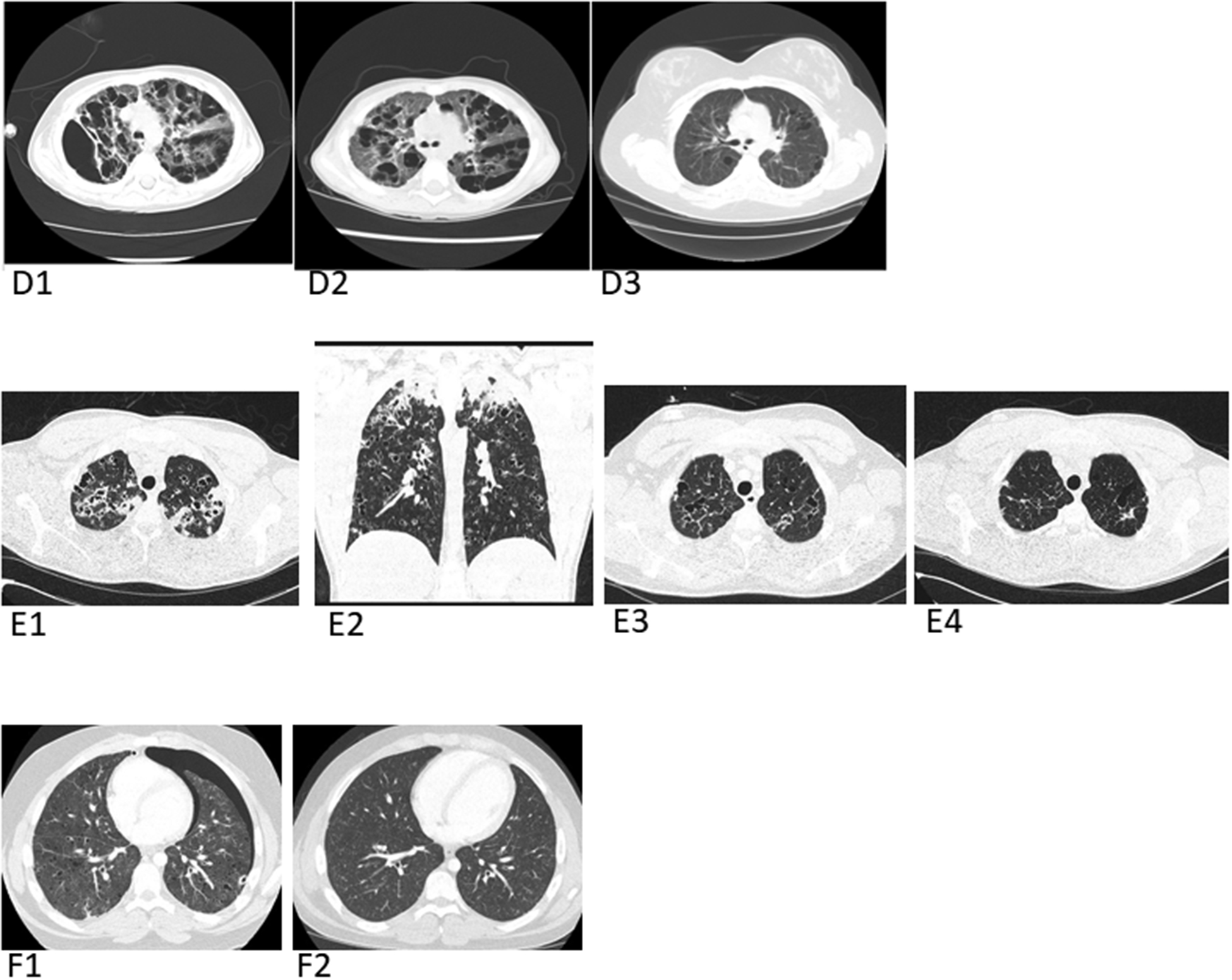
(D) Images from Patient 5: Baseline pre-treatment axial chest CT (D1) shows markedly enlarged pulmonary cysts and axial chest CT (D2) 2 months later shows suboptimal response to vinblastine, prednisone, and methotrexate. Patient was then treated with cytarabine/cladribine combination followed by cladribine alone for 6 months. Post-treatment axial chest CT (D3) several years later shows complete response (CR) of LCH and marked cyst involution with reconstitution of a large percentage of functional lung parenchyma.
(E) Images from Patient 6: Baseline pre-treatment axial chest CT (E1) and coronal chest CT (E2). Axial chest CT (E3) 3 months after starting chemotherapy; Axial chest CT (E4) 10 months after starting chemotherapy.
(F) Images from Patient 7: Baseline pre-treatment axial chest CT (F1) following bilateral chest tube placement for pneumothoraces, reveals a residual left pneumothorax and numerous cysts scattered throughout the lung parenchyma bilaterally. Axial chest CT image (F2) 3 years later off therapy including pleurodesis and chemotherapy, showing resolution of the pneumothoraces and lung cysts.
Patient 1 was an 8-month-old full term female infant who presented with history of rash since birth, recurrent respiratory distress, and intermittent stridor during the first few months of life. The patient was ultimately diagnosed with LCH of the skin, bones, thymus, thyroid and lungs. She developed multiple pneumothoraces, was treated with laser pleurodesis due to pneumothorax recurrence, and had initial response to vinblastine and prednisone resulting in stabilization of pulmonary LCH disease. However, despite significant improvement with supportive care measures and LCH-directed therapy, she developed recurrent respiratory distress secondary to RSV pneumonia and died at 11 months old (autopsy confirmed sequelae of RSV infection with no evidence of active pulmonary LCH).
Patient 2 was an 11-month-old girl whose initial presentation of LCH included dyspnea and hypoxia. She was diagnosed with skin, pituitary and lung LCH and had less than 20% normal lung parenchymal volume on baseline CT. Genetic analysis of tissue biopsy demonstrated no somatic BRAFV600E mutation. Recalcitrant pneumothoraces were managed with repeat surgical pleurodesis and up to 8 simultaneous chest tubes. Surgical pleurodesis of the right lung was first performed one month after start of treatment with cytarabine and was then performed a second time two months later due to persistent recurrence of pneumothorax in the left lung. After LCH-directed treatment with cytarabine for 12 months, she regained a significant volume of normal-appearing lung parenchyma on CT and is clinically well now, over 4 years off treatment with no evidence of recurrence.
Patient 3 was an 11-month-old boy who presented with a rash, otorrhea, fever and coughing. Imaging revealed bilateral multicystic lung disease, as well as temporal and mastoid bone disease. One week after the initiation of prednisolone and vinblastine, he developed a series of recurrent pneumothoraces requiring multiple bilateral chest drains and ventilation. Talc pleurodesis was ineffective. His course was complicated by ventilation-associated bacterial pneumonia, a progressive increase in bilateral intra-parenchymal bullae, and recurrent pneumothoraces. The LCH rash and otorrhea started to resolve but in the light of his increasing respiratory difficulties, cytarabine was added to his treatment. After 8 weeks of chemotherapy and 4 weeks of continued ventilation with active management of air leaks, oxygenation was no longer effective, and he succumbed to respiratory failure.
Patient 4 was a two-year-old boy who presented with cough and sudden respiratory deterioration due to a pneumothorax. Chest CT scan revealed bilateral diffuse cystic lung disease and T2 vertebra plana. While awaiting the result of a lung biopsy, he developed out-of-hospital cardiac arrest secondary to bilateral pneumothoraces and required cardiopulmonary resuscitation and emergency chest drains. Therapy with prednisone and vinblastine was initiated. He was treated with prolonged ventilation, multiple (26) chest drains, bilateral talc pleurodesis, airway pressure release ventilation (APRV), and inotropic support due to persistent pneumothoraces and pneumonia. Cytarabine was added to his treatment after 1 month, and 6 months later, therapy was changed to vincristine and cytarabine alone for an additional 12 months (total of 18 months of therapy). He gradually recovered and was weaned off supplemental oxygen after 215 days. Serial chest CT scans showed marked improvement. He remained asymptomatic and is in remission 9 months off treatment.
Patient 5 was a two-year-old girl who presented with skin, lung, lymph node and liver LCH. She initially required surgical pleurodesis for a pneumothorax at presentation. Due to the severity of her lung disease and suboptimal response to vinblastine, prednisone, and methotrexate, she was evaluated for possible lung transplant and LCH-directed therapy was then modified. After a single course of cytarabine/cladribine followed by 6 courses of cladribine, she achieved complete response (CR) with resolution of cysts and reconstitution of a large percentage of functional lung parenchyma. She has done well clinically and is now 14 years off treatment.
Patient 6 was a 15-year-old boy who presented with lethargy, weight loss, dyspnea, and a scalp rash. Chest CT scan revealed widespread lung cysts. Four days after starting vinblastine and prednisolone he presented acutely with a left-sided pneumothorax requiring a chest drain followed by a right-sided pneumothorax 11 days later. After 3 weeks, chemotherapy with vinblastine and prednisolone was switched to cytarabine. He had no further pneumothoraces, his symptoms resolved and there was marked improvement of the LCH chest CT manifestations. He completed 12 months of chemotherapy and remained in remission for 2 months after completing therapy. The pulmonary LCH has not recurred, but he is currently being treated for multisystem (skin, thyroid and pituitary) reactivation.
Patient 7 was a 16-year-old boy with pituitary and pulmonary LCH with prior medical history of acute lymphoblastic leukemia in remission from the age of 2 years. He had a significant cigarette smoking habit at the time of his LCH diagnosis and presented with recurrent pneumothoraces. The patient required supportive care with surgical and chemical pleurodesis and improved with LCH-directed treatment with cytarabine for 12 months. The patient had resolution of LCH with disappearance of most of the lung cysts on chest CT, but relapsed 4 months off therapy, possibly exacerbated or triggered by continued smoking. He was subsequently treated with salvage therapy with clofarabine for 12 months and is now clinically well in remission and more than 3 years off treatment.
Discussion
Typical clinical presentations for pulmonary LCH include chronic cough, dyspnea, tachypnea, wheezing, or chest pain, but some patients may have very mild or no symptoms.(7;12;14) In contrast to adults who usually have limited single system pulmonary LCH often precipitated by smoking, children most often develop pulmonary LCH as part of multisystem disease.(5;7;12) Imaging studies have revealed interstitial infiltrates, peribronchial thickening, nodules, and cysts in the lungs with a minority (11%) having a pneumothorax.(12;14–16) Sixteen cases of pulmonary LCH were summarized in a table from a report in 1964, including that of a 13-year-old girl with multiple pneumothoraces who required scarification of the parietal and visceral pleura with talc to stop recurrent air leaks.(17)
Patients with pulmonary LCH are usually curable and often have good outcomes with early diagnosis, treatment, and supportive care. For the unusual set of patients reported here, the combination of chemotherapy, intensive supportive care, multiple chest tube insertions, and pleurodesis in selected patients (6 of 7 patients received this therapy at least once) halted the recurrence of pneumothoraces and sustained 5 of 7 patients long enough to allow the chemotherapy to control the LCH. It is clear that successful responses to chemotherapy treatments led to patients gradually becoming less dependent on chest drains, ventilator support, and supplemental oxygen. A marked improvement in lung parenchyma appearance on chest CT was also observed in all surviving patients (Figure 1). It seems likely that these patients had restoration of some pulmonary parenchyma, although it is difficult to know how much, since resolution of the tension pneumothoraces would allow re-expansion of the atelectatic lung. Pulmonary function tests and exercise tolerance also improved following resolution of LCH in the five surviving patients at last clinical follow-up.
The range of chemotherapy agents used in the patients in this series does not allow for any conclusions regarding which may be the most effective in achieving rapid disease control, although previous retrospective reviews suggest cytarabine monotherapy may provide superior outcomes in upfront LCH therapy when compared to vinblastine/prednisone.(18) In a series of three cases reported by Yule and colleagues, treatment with prednisone and vinblastine was effective for one patient, but another required several pleurectomies due to 11 episodes of pneumothorax.(19) The third patient died from complications of the pneumothoraces and chest tubes despite treatment with vinblastine, prednisone, antibiotics, and oscillating ventilator support. Another patient with extensive cysts and recurrent pneumothoraces had a poor response to vinblastine, prednisone, and etoposide, but had improvement with cyclosporine, oral methotrexate and 6-mercaptopurine.(20) Two other children with very extensive cystic changes in the lungs and markedly decreased volume of normal pulmonary parenchyma had good outcomes after treatment with vinblastine, prednisone, 6-mercaptopurine and placement of multiple chest tubes.(14)
Identifying activating MAPK pathway mutations may be helpful diagnostically and therapeutically for patients with pulmonary LCH lesions. It is important to determine if a patient has the somatic BRAFV600E mutation as use of vemurafenib and dabrafenib can provide dramatic results in patients with refractory disease, but these agents are specifically effective for BRAF mutations that activate monomers (e.g. BRAF-V600E). In this series, somatic genetic testing was not available for the majority of patients due to their disease presentation occurring prior to the knowledge of the association of this pathway with LCH disease. MEK inhibitors have also been reported to be effective for non-pulmonary histiocytic lesions in the context of a variety of mutations.(9) In this series, an 18-year-old patient with pulmonary LCH and recurrent pneumothoraces with disease refractory to cladribine was successfully treated with trametinib after genotyping showed a somatic mutation in the MAP2K1 gene (MAP2K1, c.302–307del) (21). While MAPK pathway inhibitors appear to be quite promising in theory and in short term cases, they do not seem to be able to ultimately cure LCH. Additional chemotherapy or prolonged use of inhibitors may be needed.(22;23) A possible role for MAPK inhibitors alone or in combination with other therapies in the treatment of these patients merits further investigation.
Pneumothoraces occurred in this series as a presenting feature or soon after starting chemotherapy. Repeated insertion of chest tubes may be needed to control the recurrent pneumothoraces (23 chest drains required for Patient 5). Scarifying the lung surface by a chemical or surgical pleurodesis may provide the optimal management of air leaks. The decision to undertake pleurodesis is made with the input of surgical, interventional radiology, intensive care, pulmonary, and hematology/oncology consultants. Pleurodesis is not always effective and the more aggressive the pleural intervention, the less likely that the individual patient will be a suitable candidate for lung transplantation. A patient with recurrent pneumothoraces required chest tube placement and pleurodesis with talc before his air leaks could be controlled.(13) Other agents instilled in the pleural cavity include iodopovidone and bleomycin.(16;24) Three other patients with large tension pneumothoraces did not survive.(5;25;26) Some of the reported survivors required insertion of multiple chest drains and prolonged supportive care, as did patients 2, 3 and 5 in our series. The functional and radiologic outcomes in surviving patients are often better than would have been anticipated based on the initial presentation and course of the disease.
The goal of surgical management of air leaks in patients with severe pulmonary LCH is to maintain adequate lung function during the initial period of critical illness and minimize the long-term damage to the lung and pleural space. If LCH directed therapy and supportive care is successful, healing of lung parenchyma takes place and regeneration of healthy lung tissue is possible. Normal lung growth occurs via neo-alveolarization throughout childhood and into adolescence as demonstrated by hyperpolarized Helium3-MRI.(27;28) After pneumonectomy there is evidence of potential for lung growth over many months.(29) The primary surgical therapy is chest tube placement, which allows for drainage of large air collections and recruitment of collapsed lung. Given that it can be difficult to differentiate between a pneumothorax and a very large intra-parenchymal cyst, pleurodesis should be reserved for patients with persistent air leaks and evidence that the chest tubes are within the pleural space and not the lung parenchyma, often requiring CT imaging. Our experience suggests that a large air collection within the pleural cavity causing significant collapse of normal lung is better treated with an additional chest tube even if it already has an apparently functional chest tube within the pneumothorax, rather than with chemical or mechanical pleurodesis. We postulate that the volume of air leaking into that space exceeds the ability of a 10 French tube under 20 cm H2O negative pressure to efficiently drain the leak. Another possibility in these patients is that the pleural air collections can be loculated and separate tubes may be needed for all important loculations. For most patients, 10 French pigtail catheters provide adequate drainage of air and cause minimal damage to the lung when they are inadvertently placed into lung cysts. Pleurodesis is reserved for those patients who have a persistent air leak in spite of appropriately positioned pleural-drainage tubes.
Early recognition of LCH as a cause of pulmonary cystic disease provoking pneumothoraces and/or respiratory failure along with coordinated supportive care by a multidisciplinary team of subspecialists is essential for optimizing outcome. Our cases and the previously reported ones demonstrate that extensive involvement of the lung parenchyma and multiple pneumothoraces may be life threatening and necessitate intensive supportive care. A range of strategies were employed in different patients to deal with the risk and consequences of air leaks. These included efforts to avoid intubation and positive pressure ventilation where possible (e.g. insertion of chest tubes under sedation and with good pain relief in suitable patients), keeping inflation pressures as low as possible (if ventilation was required), and utilizing permissive hypercarbia to try to avoid progressive overexpansion of cystic lung spaces and additional barotrauma. Optimal nutrition support in the form of parenteral nutrition, enteric tube supplementation, and oral intake, when possible, was provided to promote healing. In the setting of chest tubes, pain control was particularly important to minimize splinting and allow for adequate lung expansion.
Mechanical ventilation in these patients is challenging as it adds infection risk and has potential to worsen disease with further barotrauma. The lung parenchyma is extraordinary fragile and prone to cyst rupture causing multiple pneumothoraces, with varying degrees of tensioning. The level of air leak means adequately ventilating alveoli becomes particularly challenging, as much of the ventilator driven gas flow is bypassed directly to the chest drains. Furthermore, the functional parenchyma has different time constants with different opening and closing pressures. In the case of patient 5, conventional ventilation failed, he deteriorated further on high frequency oscillation, but he stabilized with airway pressure release ventilation. Advanced pulmonary monitoring using electrical impedance tomography was helpful in providing precise knowledge of how to proceed.
Children with extensive pulmonary disease and life-threatening impairment in gas exchange present clinicians with the choice between continued chemotherapy and supportive care or consideration of extracorporeal membrane oxygenation (ECMO) and/or lung transplantation.(5) The reported successful use of ECMO as a bridge to lung transplantation and the excellent functional recovery of survivors in this series (without the need for transplantation) raise the question whether earlier initiation of ECMO can be used to allow sufficient time for chemotherapy to control the disease when other supportive measures fail.(30) Long ECMO runs are clearly undesirable.(31) The pace of recovery in the surviving patients who needed intensive care support in this series (median 12.5 days on ventilator (range 0–45); median 41.5 days in ICU; median 68 days on supplemental oxygen) suggest that an acceptable ECMO run duration may be sufficient. If this proves to be the case, the use of short term ECMO may be justified in the most severe cases, without the need to commit to lung transplantation. This requires further exploration.
Conclusion:
LCH patients with extensive cystic lung disease are at risk of life-threatening pneumothoraces. This series documents that patients with extensive pulmonary cystic LCH complicated by multiple pneumothoraces can be cured with chemotherapy, aggressive surgical management with chest tubes, and pleurodesis. Recovery from repeated air leaks and resolution of respiratory failure may take several months but can be achieved in some patients with good functional outcomes (potential for regeneration of new lung tissue and/or re-expansion of compressed lung parenchyma). When pediatric patients with extensive cystic pulmonary LCH present with respiratory failure, withdrawal of care and/or lung transplantation should be deferred until all efforts to treat have been exhausted, including standard or salvage chemotherapy, targeted MAPK inhibitors, sufficient respiratory support, and aggressive surgical interventions with multiple chest tubes and/or pleurodesis if needed.
Acknowledgments:
This work was performed with support from the HistioCure Foundation (Texas Children’s Cancer Center Histiocytosis Program), St. Baldrick’s Foundation which supports the North American Consortium for Histiocytosis (NACHO) (CEA, Consortium Award, Innovation Award), the Leukemia and Lymphoma Society (CEA, TRP Award), and the National Cancer Institute of the National Institutes of Health (OSE, K12CA090433; CEA, R01CA154489, R01CA154947, P50CA126752).
Abbreviations
- ARAC
Cytarabine
- Clad
Cladribine
- CT
Computed Tomography
- Pred
Prednisone
- LCH
Langerhans cell histiocytosis
- Vbl
Vinblastine
Footnotes
Conflict of Interest Disclosures: The authors have no significant financial interest in or other relationship with the manufacturer of any product or provider of any service mentioned in this article.
Prior Presentation of Study Data: This study was presented as a poster at the 32nd Annual Meeting of the Histiocyte Society in 2016 inclusive of five of the patients reported here. There are otherwise no prior publications or submissions with any overlapping information, including studies and patients.
Reference List
- (1).Salotti JA, Nanduri V, Pearce MS, Parker L, Lynn R, Windebank KP. Incidence and clinical features of Langerhans cell histiocytosis in the UK and Ireland. Arch Dis Child 2009. May;94(5):376–80. [DOI] [PubMed] [Google Scholar]
- (2).A multicentre retrospective survey of Langerhans’ cell histiocytosis: 348 cases observed between 1983 and 1993. The French Langerhans’ Cell Histiocytosis Study Group. Arch Dis Child 1996. July;75(1):17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Guyot-Goubin A, Donadieu J, Barkaoui M, Bellec S, Thomas C, Clavel J. Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000–2004. Pediatr Blood Cancer 2008. July;51(1):71–5. [DOI] [PubMed] [Google Scholar]
- (4).Alston RD, Tatevossian RG, McNally RJ, Kelsey A, Birch JM, Eden TO. Incidence and survival of childhood Langerhans cell histiocytosis in Northwest England from 1954 to 1998. Pediatr Blood Cancer 2007. May;48(5):555–60. [DOI] [PubMed] [Google Scholar]
- (5).Odame I, Li P, Lau L, Doda W, Noseworthy M, Babyn P, et al. Pulmonary Langerhans cell histiocytosis: a variable disease in childhood. Pediatr Blood Cancer 2006. December;47(7):889–93. [DOI] [PubMed] [Google Scholar]
- (6).Bernstrand C, Cederlund K, Sandstedt B, Ahstrom L, Lundell M, Dahlquist G, et al. Pulmonary abnormalities at long-term follow-up of patients with Langerhans cell histiocytosis. Med Pediatr Oncol 2001. April;36(4):459–68. [DOI] [PubMed] [Google Scholar]
- (7).Ha SY, Helms P, Fletcher M, Broadbent V, Pritchard J. Lung involvement in Langerhans’ cell histiocytosis: prevalence, clinical features, and outcome. Pediatrics 1992. March;89(3):466–9. [PubMed] [Google Scholar]
- (8).Allen CE, Merad M, McClain KL. Langerhans-Cell Histiocytosis. N Engl J Med 2018. August 30;379(9):856–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Mourah S, How-Kit A, Meignin V, Gossot D, Lorillon G, Bugnet E, et al. Recurrent NRAS mutations in pulmonary Langerhans cell histiocytosis. Eur Respir J 2016. June;47(6):1785–96. [DOI] [PubMed] [Google Scholar]
- (10).Ronceray L, Potschger U, Janka G, Gadner H, Minkov M. Pulmonary involvement in pediatric-onset multisystem Langerhans cell histiocytosis: effect on course and outcome. J Pediatr 2012. July;161(1):129–33. [DOI] [PubMed] [Google Scholar]
- (11).Vassallo R, Ryu JH, Colby TV, Hartman T, Limper AH. Pulmonary Langerhans’-cell histiocytosis. N Engl J Med 2000. June 29;342(26):1969–78. [DOI] [PubMed] [Google Scholar]
- (12).Braier J, Latella A, Balancini B, Castanos C, Rosso D, Chantada G, et al. Outcome in children with pulmonary Langerhans cell Histiocytosis. Pediatr Blood Cancer 2004. December;43(7):765–9. [DOI] [PubMed] [Google Scholar]
- (13).Chatkin JM, Bastos JC, Stein RT, Gaiger AM. Sole pulmonary involvement by Langerhans’ cell histiocytosis in a child. Eur Respir J 1993. September;6(8):1226–8. [PubMed] [Google Scholar]
- (14).Braier J, Latella A, Balancini B, Castanos C, Goldberg J. Isolated pulmonary Langerhans cell histiocytosis presenting with recurrent pneumothorax. Pediatr Blood Cancer 2007. February;48(2):241–4. [DOI] [PubMed] [Google Scholar]
- (15).Valliani L, Kanwar VS, Schwartz A. Isolated pulmonary Langerhans cell histiocytosis with recurrent bilateral pneumothoraces treated with chemotherapy and chemical pleurodesis. Pediatr Blood Cancer 2009. July;53(1):128–9. [DOI] [PubMed] [Google Scholar]
- (16).Verma S, Jondhale S, Bansal D, Radotra BD, Singhi SC. Iodopovidone pleurodesis for isolated pulmonary Langerhan’s cell histiocytosis in a two year old child. Indian J Pediatr 2014. July;81(7):715–8. [DOI] [PubMed] [Google Scholar]
- (17).ROLAND AS, MERDINGER WF, FROEB HF. RECURRENT SPONTANEOUS PNEUMOTHORAX; A CLUE TO THE DIAGNOSIS OF HISTIOCYTOSIS X. N Engl J Med 1964. January 9;270:73–7. [DOI] [PubMed] [Google Scholar]
- (18).Simko SJ, McClain KL, Allen CE. Up-front therapy for LCH: is it time to test an alternative to vinblastine/prednisone? Br J Haematol 2015. April;169(2):299–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Yule SM, Hamilton JR, Windebank KP. Recurrent pneumomediastinum and pneumothorax in Langerhans cell histiocytosis. Med Pediatr Oncol 1997. August;29(2):139–42. [DOI] [PubMed] [Google Scholar]
- (20).Zeller B, Storm-Mathisen I, Smevik B, Lie SO. Multisystem Langerhans-cell histiocytosis with life-threatening pulmonary involvement--good response to cyclosporine A. Med Pediatr Oncol 2000. October;35(4):438–42. [DOI] [PubMed] [Google Scholar]
- (21).Lorillon G, Jouenne F, Baroudjian B, de Margerie-Mellon C, Vercellino L, Meignin V, et al. Response to Trametinib of a Pulmonary Langerhans Cell Histiocytosis Harboring a MAP2K1 Deletion. Am J Respir Crit Care Med 2018. September 1;198(5):675–8. [DOI] [PubMed] [Google Scholar]
- (22).Eckstein OS, Visser J, Rodriguez-Galindo C, Allen CE. Clinical responses and persistent BRAF V600E(+) blood cells in children with LCH treated with MAPK pathway inhibition. Blood 2019. April 11;133(15):1691–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Donadieu J, Larabi IA, Tardieu M, Visser J, Hutter C, Sieni E, et al. Vemurafenib for Refractory Multisystem Langerhans Cell Histiocytosis in Children: An International Observational Study. J Clin Oncol 2019. September 12;37(31):2857–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Alavi S, Ashena Z, Paydar A, Hemmati N. Langerhans cell histiocytosis manifesting as recurrent simultaneous bilateral spontaneous pneumothorax in early infancy. Pediatr Int 2007. December;49(6):1020–2. [DOI] [PubMed] [Google Scholar]
- (25).Al-Trabolsi HA, Alshehri M, Al-Shomrani A, Shabanah M, Al-Barki AA. “Primary” pulmonary Langerhans cell histiocytosis in a two-year-old child: case report and literature review. J Pediatr Hematol Oncol 2006. February;28(2):79–81. [DOI] [PubMed] [Google Scholar]
- (26).Gunes T, Koklu E, Ozturk MA, Patiroglu T, Patiroglu T, Karakukcu M. A case of Langerhans cell histiocytosis presented with pneumothorax. J Pediatr Hematol Oncol 2007. January;29(1):60–2. [DOI] [PubMed] [Google Scholar]
- (27).Narayanan M, Owers-Bradley J, Beardsmore CS, Mada M, Ball I, Garipov R, et al. Alveolarization continues during childhood and adolescence: new evidence from helium-3 magnetic resonance. Am J Respir Crit Care Med 2012. January 15;185(2):186–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Burri PH. Structural aspects of postnatal lung development - alveolar formation and growth. Biol Neonate 2006;89(4):313–22. [DOI] [PubMed] [Google Scholar]
- (29).Shikuma K, Chen-Yoshikawa TF, Oguma T, Kubo T, Ohata K, Hamaji M, et al. Radiologic and Functional Analysis of Compensatory Lung Growth After Living-Donor Lobectomy. Ann Thorac Surg 2018. March;105(3):909–14. [DOI] [PubMed] [Google Scholar]
- (30).Sacco O, Moscatelli A, Conte M, Grasso C, Magnano GM, Sementa AR, et al. Long-Term Extracorporeal Membrane Oxygenation as Bridging Strategies to Lung Transplantation in Rapidly Devastating Isolated Langerhans Cell Histiocytosis. Pediatr Blood Cancer 2016. May;63(5):941–3. [DOI] [PubMed] [Google Scholar]
- (31).Biffi S, Di BS, Scaravilli V, Peri AM, Grasselli G, Alagna L, et al. Infections during extracorporeal membrane oxygenation: epidemiology, risk factors, pathogenesis and prevention. Int J Antimicrob Agents 2017. July;50(1):9–16. [DOI] [PubMed] [Google Scholar]