

Abstract
OBJECTIVES:
Tenapanor is a first-in-class, minimally absorbed, small-molecule inhibitor of the gastrointestinal sodium/hydrogen exchanger isoform 3. This phase 3 trial assessed the efficacy and safety of tenapanor 50 mg b.i.d. for the treatment of patients with constipation-predominant irritable bowel syndrome (IBS-C).
METHODS:
In this phase 3, double-blind study (ClinicalTrials.gov identifier NCT02621892), patients with IBS-C were randomized to tenapanor 50 mg b.i.d. or placebo b.i.d. for 12 weeks followed by a 4-week randomized withdrawal period. The primary efficacy variable was the proportion of patients who reported a reduction in average weekly worst abdominal pain of ≥30.0% and an increase of ≥1 complete spontaneous bowel movement from baseline, both in the same week, for ≥6 weeks of the 12-week treatment period.
RESULTS:
Of the 629 randomized patients with IBS-C, 606 (96.3%) were included in the intention-to-treat analysis set (tenapanor: n = 307; placebo: n = 299) and 533 (84.7%) completed the 12-week treatment period. In the intention-to-treat analysis set (mean age 45 years, 81.4% women), a significantly greater proportion of patients treated with tenapanor met the primary endpoint than patients treated with placebo (27.0% vs 18.7%, P = 0.020). Abdominal symptoms and global symptoms of IBS also improved with tenapanor (P < 0.05 vs placebo). Diarrhea was the most commonly reported adverse event, resulting in study drug discontinuation in 6.5% and 0.7% of patients receiving tenapanor and placebo, respectively, during the 12-week treatment period.
DISCUSSION:
Tenapanor 50 mg b.i.d. improved IBS-C symptoms and was generally well tolerated, offering a potential new treatment option for patients with IBS-C.
INTRODUCTION
Irritable bowel syndrome (IBS) is a chronic symptom-based disorder characterized by abdominal pain and altered bowel movements (1). Symptoms of IBS can cause considerable morbidity, impair quality of life, and impact work productivity (1). The financial burden of IBS is comparable with other common chronic diseases of similar prevalence, including asthma, migraine, and congestive heart failure (2). The pathogenesis of IBS is heterogenous and may involve abnormalities in motility, visceral sensation, gut microbiota, intestinal permeability, and/or gut immune activation (3).
IBS affects approximately 11.2% of the global population, with constipation-predominant IBS (IBS-C) accounting for approximately one-third of IBS cases (4). Medications approved by the Food and Drug Administration for IBS-C act by increasing intestinal chloride secretion, albeit through different mechanisms. Lubiprostone stimulates chloride secretion through activation of type-2 chloride channels (5), whereas linaclotide (6) and plecanatide (7) both bind guanylate cyclase C receptors, stimulating chloride secretion indirectly by activating the cystic fibrosis transmembrane regulator. Given the heterogenous nature of IBS, there remains a need to develop compounds with novel mechanisms of action that can address the full range of symptoms experienced by affected patients (8).
Tenapanor is a first-in-class, minimally absorbed, small-molecule inhibitor of the gastrointestinal sodium/hydrogen exchanger isoform 3 (NHE3), a transporter that assists in maintaining salt and water balance through the electroneutral exchange of intestinal sodium ions for intracellular protons. NHE3 inhibition should therefore be expected to increase excretion of sodium and fluid in stool. Preclinical studies in rats and clinical studies have shown that tenapanor reduces sodium absorption, with minimal systemic drug exposure (9,10). To determine the efficacy of tenapanor in the treatment of constipation-related disorders, a phase 2b clinical study was conducted in 356 patients with IBS-C. In that trial, treatment with tenapanor 50 mg b.i.d. resulted in significant increases in complete spontaneous bowel movements (CSBMs) and reductions in abdominal pain relative to placebo (P < 0.05) and was generally well tolerated. The next-lowest dose that was assessed, tenapanor 20 mg b.i.d., did not significantly improve these outcomes (11).
Based on these results, tenapanor 50 mg b.i.d. was investigated in a phase 3, placebo-controlled clinical study in patients with IBS-C. Here, we report the efficacy and safety outcomes of this trial, which included a 12-week treatment period and a 4-week randomized withdrawal (RW) period.
METHODS
Study design
This multicenter, phase 3, randomized, double-blind, placebo-controlled study enrolled patients from 92 sites in the United States between November 2015 and March 2017 (last patient randomized on December 1, 2016). The sites conducting the study were gastroenterology or primary care practices (n = 22) or research practices specializing in internal medicine and/or gastroenterology (n = 70). After a 2-week screening period, eligible patients were randomly assigned to receive tenapanor hydrochloride (hereafter referred to as tenapanor) 50 mg b.i.d. or placebo b.i.d. for 12 weeks. Patients were instructed to take 1 tablet immediately before breakfast or the first meal of the day and another tablet immediately before dinner.
Patients who completed the 12-week treatment period entered a 4-week RW period. In the RW period, patients who had received 12 weeks of placebo were assigned to receive tenapanor for 4 weeks (hereafter referred to as “placebo/tenapanor”), and patients who had received 12 weeks of tenapanor were rerandomized to either receive placebo (hereafter “tenapanor/placebo”) or continue with tenapanor (hereafter “tenapanor/tenapanor”) for 4 weeks.
Efficacy variables were recorded daily by patients using a touch-tone telephone diary (interactive voice response system [IVRS]). Safety outcomes were assessed at patient visits, which occurred at weeks 1, 2, 4, 8, and 12 of the 12-week treatment period and weeks 2 and 4 of the RW period (study weeks 14 and 16). Adverse events (AEs) were recorded at all visits. Patient adherence to the study drug was monitored closely throughout the study.
This study (T3MPO-1; ClinicalTrials.gov identifier NCT02621892) was conducted in accordance with the Declaration of Helsinki. All participating sites obtained independent ethics committee/institutional review board approval, and all patients provided written informed consent before their participation in the trial.
Patients
Men and women aged 18–75 years who met the Rome III criteria for IBS-C (12) were eligible for study enrollment. To be included, patients were also required to have had a colonoscopy within the past 10 years if they were older than 50 years or had a colonoscopy at any age if they had experienced unexplained warning signs or symptoms, e.g., lower gastrointestinal bleeding, iron-deficiency anemia, clinically significant weight loss, and systemic signs of infection or colitis. Patients were also required to use appropriate methods of contraception or be surgically sterile or postmenopausal. In addition, patients needed to be able to communicate well with the investigator and to have daily access to a touch-tone telephone to record efficacy variables.
To be randomly assigned to study treatment, patients also had to not meet exclusion criteria and meet an additional set of study entry criteria during the 2-week screening period, which included completing their IVRS diary on ≥11 of 14 days to demonstrate diary adherence and the following patient-reported outcomes: an average weekly stool frequency of 5 or fewer spontaneous bowel movements (SBMs), defined as all nonaided bowel movements, and fewer than 3 CSBMs, defined as SBMs accompanied by a sensation of complete evacuation; an average weekly stool consistency score of less than 3 using the Bristol Stool Form Scale (BSFS) (13), an average weekly abdominal pain score of ≥3 on a scale of 0–10; no use of a prohibited medication, except rescue medication; and no liquid stools for any SBM or mushy stools for more than 1 SBM, in accordance with the BSFS. Full inclusion, exclusion, and study entry criteria have been published previously (11).
Rescue medication
Rescue medication use (bisacodyl 5-mg tablet or 10-mg suppository) was permitted for no more than 2 of the 14 screening period days. No rescue medication was allowed within 48 hours of being randomly assigned to the study drug. Throughout the 12-week treatment period and 4-week RW period, rescue medication was permitted to relieve severe constipation (defined as ≥72 hours without a bowel movement or when symptoms became intolerable). Bowel movements were not considered to be SBMs or CSBMs if they were reported within 24 hours after the use of rescue medication.
Treatment adherence
Patient adherence to study drug was closely monitored by clinical site staff and verified by the study monitor during site visits. Adherence with the IVRS was monitored by the site staff and through the IVRS itself. Accountability for the study drug at the site was the responsibility of the investigator or a pharmacist or designee appointed by the investigator. Each site kept accountability records that documented that patients were provided the doses as specified in the protocol and reconciled all the study drug received at the site. The study sponsor or a designee reviewed drug accountability on an ongoing basis during site visits.
Efficacy variables and assessments
The primary efficacy variable was the proportion of patients who had a combined response for ≥6 of 12 weeks of the treatment period (the “6/12-week” combined responder rate). A combined response was defined as a reduction in average weekly worst abdominal pain of ≥30.0% from baseline (“abdominal pain responder”) and an increase of ≥1 CSBM per week from baseline (“CSBM responder”), both in the same week. Baseline values for the treatment period were the average of values from weeks 1 and 2 of the screening period (study weeks −1 and −2). Key secondary variables were the 6/12-week CSBM responder rate and 6/12-week abdominal pain responder rate, and the 9/12-week combined, CSBM, and abdominal pain responder rates. Other secondary variables included “durable” combined, CSBM, and abdominal pain responder rates (i.e., 9/12-week responders who also met the relevant response criteria for ≥3 of the final 4 weeks of the treatment period); weekly proportion of patients with ≥3 CSBMs; average weekly numbers of CSBMs and SBMs, stool consistency, and straining scores; 6/12-week and 9/12-week responder rates for abdominal symptoms (discomfort, bloating, cramping, and fullness); weekly IBS severity and constipation severity scores; weekly adequate relief and degree of relief of IBS symptoms; and treatment satisfaction.
Recorded efficacy variables included the frequency and time of each bowel movement, sensation of complete bowel emptying (1 = yes and 2 = no), stool consistency (measured using the BSFS), abdominal symptom scores (pain, bloating, cramping, discomfort, and fullness; each on a scale of 0–10 [0 = absent and 10 = very severe]), straining score (on a scale of 1–5 [1 = not at all and 5 = an extreme amount]), and the use and time of rescue medication. Variables scored weekly included constipation severity, IBS severity (each on a scale of 1–5 [1 = none and 5 = very severe]), adequate relief of IBS (1 = yes and 2 = no), degree of relief from IBS symptoms (on a scale of 1–7 [1 = completely relieved and 7 = as bad as I can imagine]), and treatment satisfaction (on a scale of 1–5 [1 = not at all satisfied and 5 = very satisfied]).
Safety outcomes
Safety assessments were based on AEs, clinical laboratory tests, vital signs, electrocardiograms (ECGs), and physical examinations. All AEs that were spontaneously reported by the patient and/or in response to an open question from study personnel, or revealed by observation, physical examination, or other diagnostic procedures were recorded throughout the trial. Worsening of an IBS symptom, such as diarrhea, was deemed an AE only when its frequency and/or severity was beyond what the patient considered normal for their IBS.
Vital signs were assessed at each visit (systolic and diastolic blood pressure, heart rate, respiratory rate, body temperature, and body weight). Clinical laboratory tests (serum electrolytes, hematology, and urinalysis) were performed at weeks −2, 4, and 12 of the treatment period and at study week 16 and the end of the 4-week RW period. Physical examinations and 12-lead ECG monitoring were performed at weeks −2, 12, and 16.
Statistical methods
A computer-generated randomization scheme was made available to all clinical sites that met study requirements for participation, through an interactive web response system. The packaging and labeling of the study drug kits were based on a separate drug packaging randomization schedule.
The interactive web response system determined which drug package to administer to each patient, based on a randomization schedule in which each treatment was allocated once using a block size of 4 within each study site. This ensured that whole or partial block sizes were allocated, facilitating an even distribution of patients between the treatment groups.
Data collected through the IVRS throughout the study were automatically entered into a database; any abnormal values were flagged automatically, so that the relevant site could follow-up with the patient for clarification. Scores for weekly SBMs and CSBMs were standardized to 7-day frequencies, with missing days during the week being imputed with the average for the nonmissing days. A valid week required ≥4 nonmissing diary days and patients with less than 4 days' input were treated as nonresponders for that week.
All patients who met the study entry inclusion criteria and did not meet exclusion criteria who were randomized and who then received ≥1 dose of study drug were included in the intention-to-treat (ITT) analysis set. The ITT analysis set was used for the analysis of the primary, key secondary, and all other efficacy variables. All patients who received ≥1 dose of study drug were included in the safety analysis set.
A target sample size of 300 in each treatment group was selected, which would provide 95% power to detect a difference of 0.15 (15.0%) between the tenapanor and placebo groups for the 6/12-week combined responder rate (primary efficacy variable), assuming a responder rate of ≥45.0% for the tenapanor group.
All efficacy variables reported as proportions (e.g., responder rates) were analyzed using a Cochran–Mantel–Haenszel test with pooled investigator site as a stratification (adjustment) variable. Efficacy variables reported as changes from baseline were analyzed using an analysis of covariance model with terms for pooled investigator site and treatment and baseline values as the covariates. Degree of relief of IBS symptoms and treatment satisfaction were analyzed using an ANOVA model with terms for pooled investigator site and treatment as factors. Statistical analyses were performed at the 2-sided significance level of 0.050.
A sequential testing procedure was used to address the potential for multiplicity when testing numerous efficacy variables. The primary efficacy variable was tested at the 5% level of significance. If this test was significant, the first key secondary efficacy variable was to be tested at the 5% level. If this test was significant, then the next key secondary efficacy variable was tested at the 5% level. This procedure continued until one of the key secondary efficacy variables in the list (5 variables total) resulted in a P value greater than 0.05. Key secondary efficacy variables up to this point were declared statistically significant.
RESULTS
Twelve-week treatment period
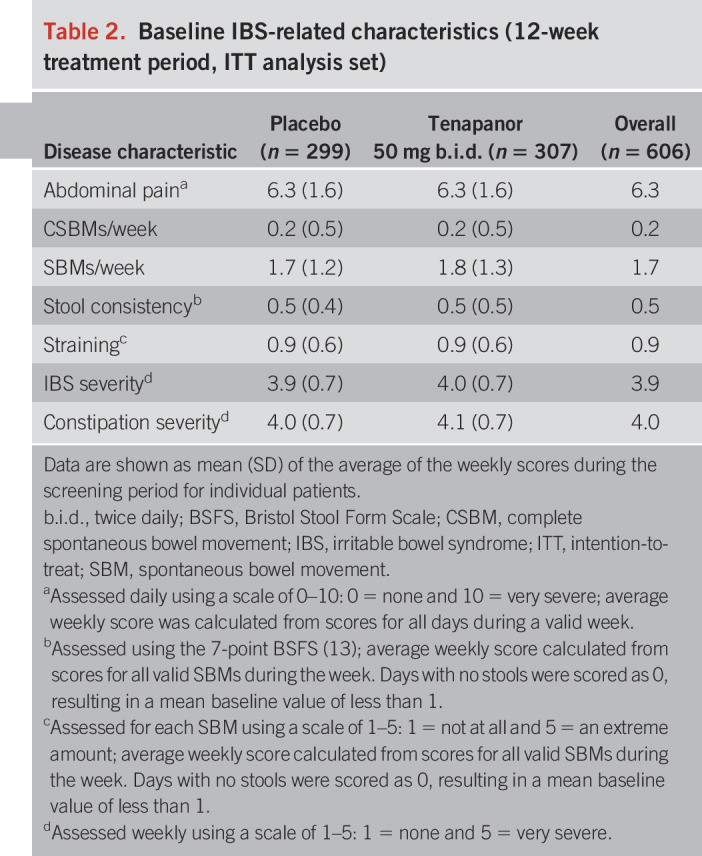
Of 1,599 patients who were screened, 629 (39.3%) were randomized (Figure 1). The treatment period was completed by 261 of the 319 patients (81.8%) who were randomized to receive tenapanor and 272 of the 310 patients (87.7%) who received placebo. A total of 610 patients were included in the safety analysis set, of whom 309 received tenapanor and 301 received placebo. Four patients in the safety analysis set were ineligible for inclusion in the ITT analysis set, leaving 307 and 299 subjects in the tenapanor and placebo groups, respectively. Patients in the ITT analysis set were well balanced across the tenapanor and placebo groups with respect to demographics and baseline IBS-related characteristics (Tables 1 and 2). The mean age of patients overall was 45 years; most were women (81.4%), and the majority were white (63.9%). Study drug adherence for the safety analysis set was similar between treatment groups (mean adherence of 96.4% for tenapanor and 98.0% for placebo).
Figure 1.

Overview of patient flow through the study. The safety analysis set includes all patients who received ≥1 dose of treatment. The ITT analysis set includes all patients who met the study entry inclusion/exclusion criteria, were randomized, and received ≥1 dose of the study drug. b.i.d., twice daily; ITT, intention-to-treat; PRO, patient-reported outcome.
Table 1.
Patient demographics and baseline characteristics (12-week treatment period, ITT analysis set)
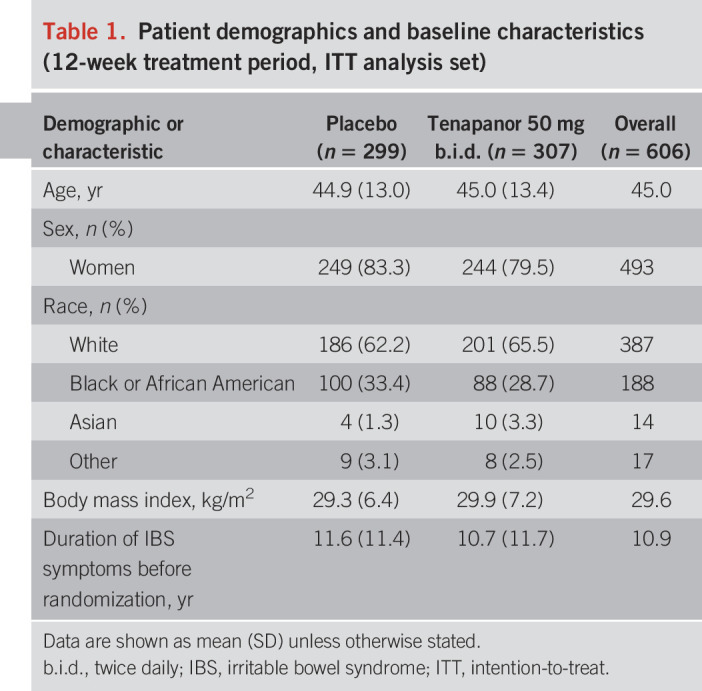
Table 2.
Baseline IBS-related characteristics (12-week treatment period, ITT analysis set)
In the ITT analysis set, a significantly greater proportion of patients receiving tenapanor met the primary endpoint (6/12-week combined responder rate) compared with those receiving placebo (27.0% vs 18.7%, P = 0.020; Figure 2a). Likewise, a significantly greater proportion of patients receiving tenapanor were 6/12-week abdominal pain responders compared with patients receiving placebo (44.0% vs 33.1%, P = 0.008; Figure 2b). The difference in the 6/12-week CSBM responder rate between the tenapanor and placebo groups was not statistically significant (33.9% vs 29.4%, P = 0.270; Figure 2c).
Figure 2.

Six- of 12-week responder rates (ITT analysis set): proportions of patients with (a) combined response for ≥6 of the 12 treatment weeks (primary efficacy variable), (b) abdominal pain response for ≥6 of 12 treatment weeks (key secondary efficacy variable), and (c) CSBM response for ≥6 of 12 treatment weeks (key secondary efficacy variable). aThe adjusted RR was based on the ratio of responder rates for tenapanor 50 mg b.i.d. vs placebo b.i.d., stratified by pooled investigator sites using the Mantel–Haenszel method. bThe CMH P value was based on a 1 degree of freedom test for association between treatment (tenapanor and placebo), stratified by pooled investigator sites. An abdominal pain response is defined as a decrease in average weekly worst abdominal pain of ≥30.0% from baseline. A CSBM response is defined as an increase of ≥1 CSBM/week from baseline. A combined response is defined as an abdominal pain response and CSBM response both occurring in the same week. b.i.d., twice daily; CI, confidence interval; CMH, Cochran–Mantel–Haenszel; CSBM, complete spontaneous bowel movement; ITT, intention-to-treat; RR, relative risk.
A significantly greater proportion of patients receiving tenapanor were 9/12-week combined responders compared with those receiving placebo (13.7% vs 3.3%, P < 0.001; Figure 3a). Similarly, a significantly greater proportion of patients receiving tenapanor were 9/12-week abdominal pain responders (30.3% vs 19.4%, P = 0.003; Figure 3b) and 9/12-week CSBM responders (16.9% vs 5.0%, P < 0.001; Figure 3c) compared with patients receiving placebo.
Figure 3.

Nine- of 12-week responder rates (ITT analysis set): proportions of patients with (a) combined response for ≥9 of 12 treatment weeks (key secondary efficacy variable), (b) abdominal pain response for ≥9 of 12 treatment weeks (key secondary efficacy variable), and (c) CSBM response for ≥9 of 12 treatment weeks (key secondary efficacy variable). aThe adjusted RR was based on the ratio of responder rates for tenapanor 50 mg b.i.d. vs placebo b.i.d., stratified by pooled investigator sites using the Mantel–Haenszel method. bThe CMH P value was based on a 1 degree of freedom test for association between treatment (tenapanor and placebo), stratified by pooled investigator sites. An abdominal pain response is defined as a decrease in average weekly worst abdominal pain of ≥30.0% from baseline. A CSBM response is defined as an increase of ≥1 CSBM/week from baseline. A combined response is defined as an abdominal pain response and CSBM response both occurring in the same week. b.i.d., twice daily; CI, confidence interval; CMH, Cochran–Mantel–Haenszel; CSBM, complete spontaneous bowel movement; ITT, intention-to-treat; RR, relative risk.
The durable combined responder rate was significantly higher with tenapanor treatment than with placebo (P < 0.001; Figure 4a). Treatment with tenapanor, compared with placebo, also resulted in significantly higher durable abdominal pain responder (P = 0.006; Figure 4b) and durable CSBM responder rates (P < 0.001; Figure 4c). Compared with placebo, patients receiving tenapanor had a significantly greater mean increase from baseline in average weekly number of CSBMs (P ≤ 0.001; Figure 5a) and greater mean decrease from baseline in average weekly abdominal pain score (P < 0.05; Figure 6a) during the treatment period. Patients treated with tenapanor had a significantly greater improvement in abdominal symptoms and global IBS treatment measures (Tables 3 and 4) compared with patients receiving placebo.
Figure 4.

Durable responder rates (ITT analysis set): proportions of patients with (a) durable combined response (secondary efficacy variable), (b) durable abdominal pain response (secondary efficacy variable), and (c) durable CSBM response (secondary efficacy variable). aThe adjusted RR was based on the ratio of responder rates for tenapanor 50 mg b.i.d. vs placebo b.i.d., stratified by pooled investigator sites using the Mantel–Haenszel method. bThe CMH P value was based on a 1 degree of freedom test for association between treatment (tenapanor and placebo), stratified by pooled investigator sites. A durable abdominal pain response is defined as a decrease in average weekly worst abdominal pain of ≥30.0% from baseline for ≥9 of 12 treatment weeks, including ≥3 of the final 4 weeks of the treatment period. A durable CSBM response is defined as an increase of ≥1 CSBM/week from baseline for ≥9 of 12 treatment weeks, including ≥3 of the final 4 weeks of the treatment period. A durable combined response is defined as an abdominal pain response and CSBM response, both occurring in the same week, for ≥9 of 12 treatment weeks, including ≥3 of the final 4 weeks of the treatment period. b.i.d., twice daily; CI, confidence interval; CMH, Cochran–Mantel–Haenszel; CSBM, complete spontaneous bowel movement; ITT, intention-to-treat; RR, relative risk.
Figure 5.

Mean change from baseline in average weekly number of CSBMs over time (ITT analysis set) during the (a) treatment period and (b) RW period. *P < 0.001, †P = 0.001 vs placebo. P values were based on an analysis of covariance model with treatment and pooled investigator site as factors and baseline value as a covariate. Baseline for the treatment period is defined as the average of the first and second weeks of the screening period. Baseline for the RW period is defined as the last valid week of the treatment period. A valid week required at least 4 days of SBM reporting. b.i.d., twice daily; CSBM, complete spontaneous bowel movement; ITT, intention-to-treat; RW, randomized withdrawal.
Figure 6.

Mean percentage change from baseline in average weekly abdominal pain score over time (ITT analysis set) during the (a) treatment period and (b) RW period. *P < 0.05, †P < 0.001 vs placebo. P values were based on an analysis of covariance model with treatment and pooled investigator site as factors and baseline value as a covariate. Baseline for the treatment period is defined as the average of the first and second weeks of the screening period. Baseline for the RW period is defined as the last valid week of the treatment period. A valid week required at least 4 days of abdominal pain reporting. b.i.d., twice daily; ITT, intention-to-treat; RW, randomized withdrawal.
Table 3.
Abdominal symptom responder rates (12-week treatment period, ITT analysis set)
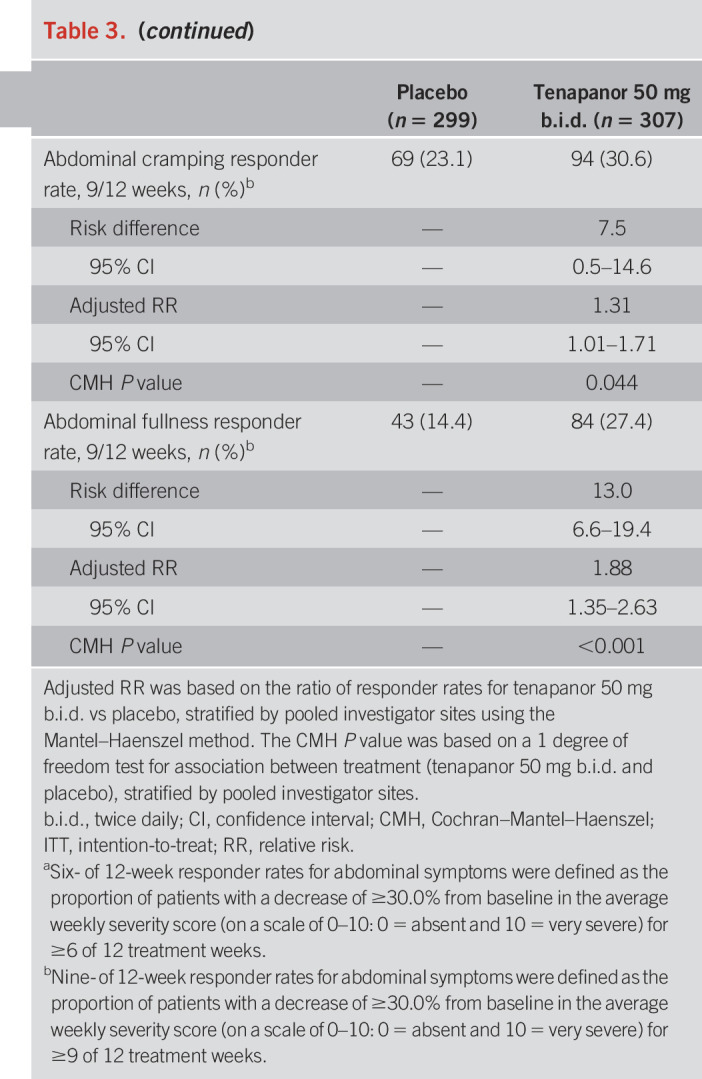
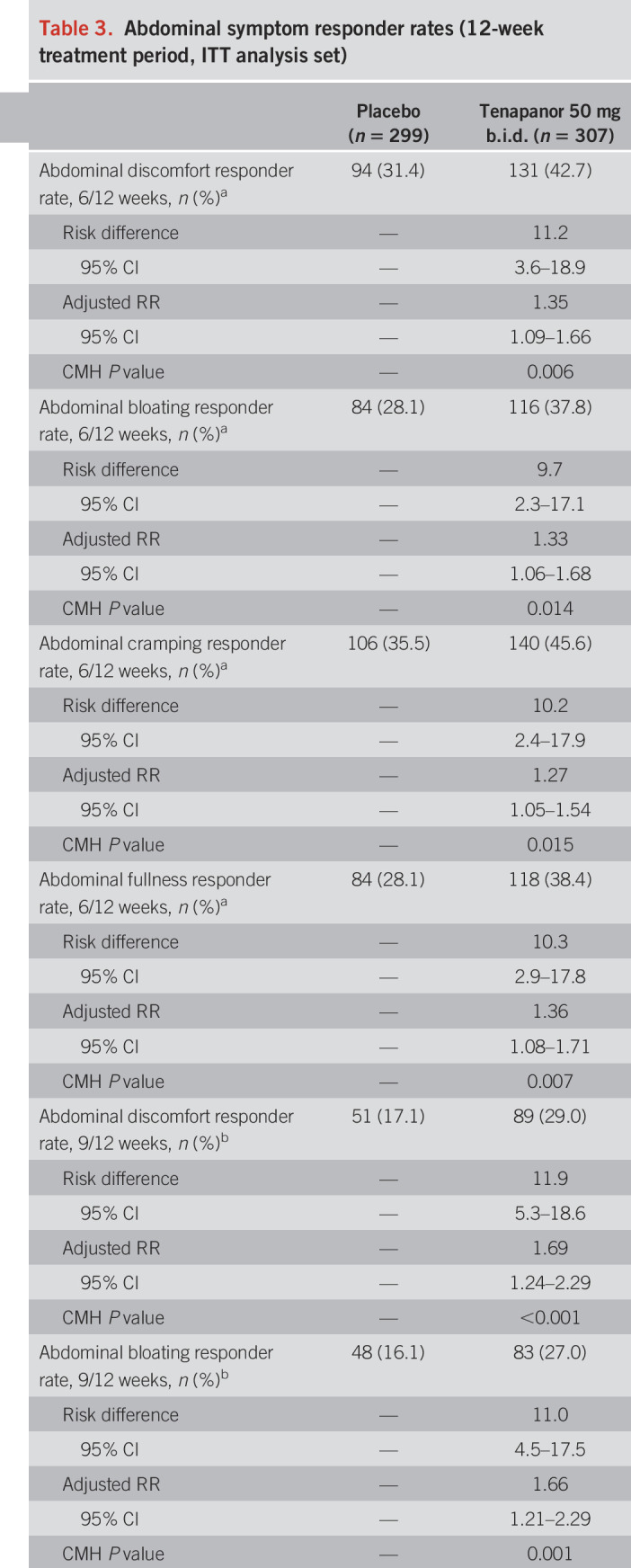
Table 4.
Other secondary variables (12-week treatment period, ITT analysis set)


Over the 12 weeks, 108 patients (35.2%) in the tenapanor group and 134 patients (44.8%) in the placebo group reported rescue medication use (P = 0.015).
Four-week RW period
Patients who completed 12 weeks of treatment with tenapanor (n = 261) were randomly assigned to receive placebo (tenapanor/placebo, n = 131) or tenapanor (tenapanor/tenapanor, n = 130) during the 4-week RW period (Figure 1). Patients who completed 12 weeks of treatment with placebo were assigned to receive tenapanor during the 4-week RW period (placebo/tenapanor, n = 272). The proportion of patients completing the 4-week RW period was in the range of 94.0%–97.0% for all 3 treatment groups. Demographic data were broadly similar for patients randomized into the 12-week treatment period and into the 4-week RW period.
At all points during the 4-week RW period, the placebo/tenapanor group had a significantly greater mean increase from baseline in average weekly number of CSBMs compared with the tenapanor/placebo group (P < 0.001; Figure 5b). The difference between the tenapanor/tenapanor group and the tenapanor/placebo group, in terms of change from baseline, was not statistically significant over the RW period.
From week 14 of the RW period, the placebo/tenapanor group had a significantly greater mean percentage decrease from baseline in average weekly abdominal pain score compared with the tenapanor/placebo group (P < 0.05; Figure 6b). With the exception of week 14, there was also a significantly greater mean percentage decrease from baseline for the tenapanor/tenapanor group compared with the tenapanor/placebo group (P < 0.05; Figure 6b).
Safety
Tables 5 and 6 provide an overview of the AEs that occurred during the 2 periods. Serious AEs (SAEs) were experienced by 4 patients receiving tenapanor during the 12-week treatment period and were not judged to be treatment-related by the site investigator. By preferred term, reported SAEs were panic attacks, osteoarthritis, migraine, and major depression. A further 2 SAEs (alcohol withdrawal syndrome and pineal neoplasm) occurred in the post-treatment period and were not judged to be treatment-related. In all instances, the patients recovered. No deaths occurred during the study.
Table 5.
Overview of AEs (12-week treatment period, safety analysis set)
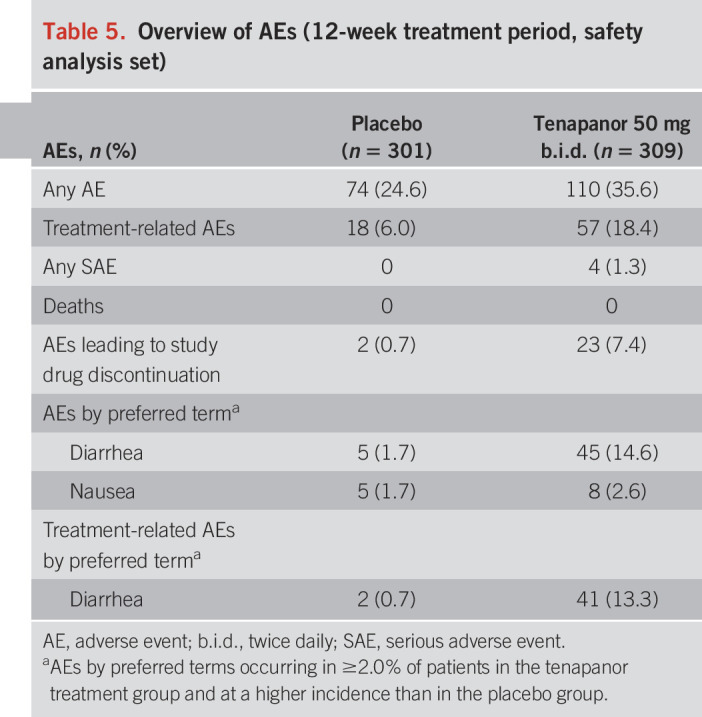
Table 6.
Overview of AEs (4-week RW period, safety analysis set)

During the 12-week treatment period, treatment-related AEs were reported for 57 patients (18.4%) receiving tenapanor and 18 patients (6.0%) receiving placebo. In the 4-week RW period, treatment-related AEs were reported by 1 patient (0.8%) in the tenapanor/placebo group, 7 patients (5.7%) in the tenapanor/tenapanor group, and 35 patients (13.3%) in the placebo/tenapanor group.
Most treatment-related AEs were gastrointestinal in nature. Diarrhea was the only AE by preferred term reported in ≥2.0% of patients receiving tenapanor and at a greater frequency than placebo, regardless of whether it was judged to be treatment-related. During the 12-week treatment period, diarrhea was reported by 45 patients (14.6%) receiving tenapanor and 5 patients (1.7%) receiving placebo. Diarrhea was judged to be treatment-related in 41 tenapanor-treated patients (13.3%) and led to study drug discontinuation in 20 patients (6.5%) and treatment interruption in 3 patients (1.0%). In most cases, diarrhea resolved on treatment interruption or withdrawal.
During the 4-week RW period, diarrhea was reported by 30 patients (11.4%) in the placebo/tenapanor group and was judged to be treatment-related in 27 patients (10.2%). Diarrhea led to study drug discontinuation in 9 patients (3.4%) in the placebo/tenapanor group. Diarrhea was reported by 2 patients (1.6%) in the tenapanor/tenapanor group, was judged to be treatment-related in both patients, and led to study drug discontinuation in 1 patient (0.8%). Patients in the tenapanor/placebo group did not report diarrhea during the 4-week RW period.
During the study, no notable changes from baseline were observed in laboratory parameters, vital signs, or physical examinations. During the 12-week treatment period, 1 patient in the placebo group had a clinically significant abnormal ECG finding of prolonged QT. The AE was considered nonserious, mild in severity, and judged to be most likely treatment-related. The QT interval for this patient was normal at screening and had returned to normal at week 16. During the 4-week RW period, 1 patient in the placebo/tenapanor group had a clinically significant abnormal 12-lead ECG finding of left precordial ST elevation (consistent with acute ischemia). The finding was considered nonserious, moderate in severity, judged to be possibly treatment-related, and resolved within 2 weeks without requiring a change to the tenapanor dose. The male patient aged 42 years was found to have abnormal, but not clinically significant, sinus bradycardia at screening and did not have a known history of cardiac events or associated risk factors.
DISCUSSION
In this phase 3 study, tenapanor significantly improved key IBS-C symptoms as demonstrated by the greater proportion of patients reporting a combined response—both an increase in the number of CSBMs and a reduction of abdominal pain in the same week, from baseline—for ≥6 weeks of the 12-week treatment period (the primary efficacy variable), compared with placebo. Similarly, a significantly greater proportion of patients receiving tenapanor experienced a combined response for ≥9 weeks of the 12-week treatment period compared with placebo. Improvements with tenapanor relative to placebo were also durable—a greater proportion of patients receiving tenapanor met both the abdominal pain and the CSBM responder criteria for ≥9 of 12 treatment weeks, including for ≥3 of the final 4 weeks of the treatment period.
Although the difference between tenapanor and placebo did not reach statistical significance in terms of the ≥6 of 12 treatment week CSBM responder rate, it is notable that statistical significance was achieved for the more stringent endpoints requiring ≥9 of 12 treatment weeks of CSBM response. In addition, across weeks 1–12, the tenapanor treatment group had significantly higher mean average weekly number of CSBMs than the placebo group, although both groups had an increase from baseline. At week 12, approximately one-third of patients treated with tenapanor reported ≥3 CSBMs/week. This bowel movement frequency falls within the healthy range for adults, based on the findings of 1 population-based study (14). For patients who continued with tenapanor in the 4-week RW period, the improvement in mean average weekly number of CSBMs from baseline was sustained to week 16. Patients who were switched from placebo to tenapanor for the 4-week RW period experienced an improvement in the mean average weekly number of CSBMs through to week 16, unlike patients who were switched from tenapanor to placebo, highlighting the efficacy of tenapanor in improving bowel movement frequency in patients with IBS-C. Constipation did not return to baseline levels after tenapanor withdrawal; during the RW period, the mean average weekly numbers of CSBMs for the tenapanor/placebo group exceeded those seen for the placebo group during the treatment period.
Tenapanor significantly improved a range of abdominal symptoms (discomfort, bloating, cramping, and fullness) for ≥9 of 12 weeks compared with placebo. The importance of improving the abdominal symptoms associated with IBS-C is highlighted by an online survey that found Japanese adults (n = 759) living with IBS-C felt greater anxiety in their daily lives—secondary to abdominal symptoms such as discomfort, pain, and bloating—compared to adults without IBS-C. Abdominal bloating was reported as the most bothersome symptom by more than one-quarter of respondents (15). Likewise, a US population-based internet survey of patients with IBS (identified by Rome II criteria) also found abdominal bloating to be a bothersome symptom, second only to abdominal cramping (16). Additional therapies are therefore needed to alleviate these abdominal symptoms.
Tenapanor is a first-in-class, minimally absorbed by design, NHE3 inhibitor that has been shown in preclinical and clinical studies to increase stool water content (9,10). This could account for the improvement in stool consistency reported by patients receiving tenapanor. An increase in intestinal fluid volume with tenapanor may also induce secondary peristalsis, leading to accelerated gastrointestinal transit time. In our study, these suggestions are supported by the observation that tenapanor treatment led to significant increases from baseline in the mean average weekly number of CSBMs and SBMs at week 12 compared with placebo.
The pathophysiology of abdominal pain in IBS may be attributed, in part, to visceral hypersensitivity (17), which may involve transient receptor potential vanilloid type 1 ion channels (18,19). Although the mechanism by which tenapanor alleviates abdominal pain in IBS-C has not been fully characterized, preliminary data from an animal model of IBS-like colonic hypersensitivity suggested that oral tenapanor treatment reduced visceral hypersensitivity and normalized colonic sensory neuronal excitability and transient receptor potential vanilloid type 1 currents (20). Further investigation is required to understand the antinociceptive action of tenapanor in IBS-C.
Tenapanor was generally well tolerated over the course of our study, with a safety profile consistent with that seen in the phase 2b clinical study (11). No deaths or treatment-related SAEs were reported in any of the study groups. Tenapanor was specifically designed to have minimal systemic availability (10). As such, the most common treatment-related AEs in this study involved the gastrointestinal system. In defining gastrointestinal AEs, patients were asked at each study visit to report occurrences of IBS symptoms that were not consistent with their “normal” experience of IBS, in terms of frequency and/or severity of symptoms. Diarrhea was typically mild to moderate in severity and resolved on treatment interruption or withdrawal in most cases. During the 4-week RW period, patients who continued with tenapanor (the tenapanor/tenapanor group) had a relatively low rate of diarrhea compared with the rate seen with patients treated with tenapanor during the 12-week treatment period. Long-term data are needed to understand the gastrointestinal AE profile of tenapanor over prolonged treatment periods.
There was 1 cardiac AE in each treatment group. During the treatment period, a patient in the placebo group developed transient QT interval prolongation. A patient in the placebo/tenapanor group developed transient ST elevation in the left precordial leads during the RW period. This event resolved without the need to discontinue or change the dose of tenapanor, suggesting that it was unrelated to tenapanor treatment. The negligible systemic exposure and lack of disparity in the number of cardiac AEs in patients included in the entire tenapanor development program, including patients with end-stage renal disease on maintenance dialysis who are at increased risk of ischemic heart disease, also support this conclusion (21,22).
Several potential limitations of the study should be acknowledged. Although the 12-week treatment period and 4-week RW period selected for T3MPO-1 exceed Food and Drug Administration recommendations for trial duration (23), additional studies will examine the effects of tenapanor for the management of IBS-C over longer periods (3). Most patients randomized were white women, which is the demographic group most likely to present with IBS-C in practice in western countries (24). Study drug adherence was almost double what may be expected in clinical practice (25), and further studies will be necessary to determine the efficacy of tenapanor in a real-world setting. Subanalyses of T3MPO-1 may also assist in determining the efficacy of tenapanor in other demographic groups.
In conclusion, tenapanor, with its unique mechanism of action, positive impact on key symptoms and satisfaction measures, and acceptable safety profile, could offer an exciting new treatment option for patients with IBS-C.
CONFLICTS OF INTEREST
Guarantor of the article: William D. Chey, MD, AGAF, FACG, FACP, RFF.
Specific author contributions: W.D.C. and A.J.L. contributed to the planning of the study, interpretation of the data, and critical revision of the manuscript for important intellectual content; D.P.R. contributed to the planning of the study, conduct of the study, interpretation of the data, and critical revision of the manuscript for important intellectual content; all authors approved the final version of the manuscript for submission.
Financial support: Medical writing support was provided by Svetha Sankar, BSc BVMS, and Steven Inglis, PhD, of Oxford PharmaGenesis and was funded by Ardelyx.
Potential competing interests: W.D.C. is a consultant for Allergan, Biomerica, IM Health, Ironwood, Outpost, Prometheus, QOL Medical, Ritter Pharmaceuticals, and Salix/Valeant and has received research funding from Ardelyx, Biomerica, Commonwealth Diagnostics International, Ironwood, Nestlé, QOL Medical, Salix/Valeant, Vibrant, and Zespri. A.J.L. is a consultant for Allergan, Ardelyx, Bioamerica, Ironwood, Prometheus, and Valeant and has received research funding from Prometheus, Bioamerica, Vibrant, and Ironwood. D.P.R. is an employee of, and has ownership interest in, Ardelyx, Inc.
Study Highlights.
WHAT IS KNOWN
✓ There are few treatment options for constipation-predominant IBS that effectively improve abdominal symptoms.
✓ In a phase 2b study, tenapanor improved CSBM frequency and abdominal pain rates in the same week (“combined response”) for ≥6 of 12 weeks, relative to placebo.
WHAT IS NEW HERE
✓ Consistent with the phase 2b study, tenapanor treatment improved the combined response rate for ≥6 of 12 weeks, relative to placebo.
✓ Tenapanor improved combined response rates for ≥9 of 12 weeks, including ≥3 of the final 4 treatment weeks, compared with placebo.
✓ Improvement from baseline in CSBMs per week was sustained to week 16 of tenapanor treatment.
✓ Tenapanor was generally well tolerated for up to 16 weeks; the most common AE was diarrhea.
ACKNOWLEDGMENTS
We thank the patients and their physicians who participated in the study. We also thank Yang Yang, Director of Biometrics at Ardelyx, for her assistance with revision of the manuscript and Andrew Yan, former employee of Ardelyx, for his assistance with statistical analyses and interpretation.
REFERENCES
- 1.Lacy BE, Chey WD, Lembo AJ. New and emerging treatment options for irritable bowel syndrome. Gastroenterol Hepatol (NY) 2015;11:1–19. [PMC free article] [PubMed] [Google Scholar]
- 2.Cash B, Sullivan S, Barghout V. Total costs of IBS: Employer and managed care perspective. Am J Manag Care 2005;11:S7–16. [PubMed] [Google Scholar]
- 3.Saha L. Irritable bowel syndrome: Pathogenesis, diagnosis, treatment, and evidence-based medicine. World J Gastroenterol 2014;20:6759–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Endo Y, Shoji T, Fukudo S. Epidemiology of irritable bowel syndrome. Ann Gastroenterol 2015;28:158–9. [PMC free article] [PubMed] [Google Scholar]
- 5.Drossman DA, Chey WD, Johanson JF, et al. Clinical trial: Lubiprostone in patients with constipation-associated irritable bowel syndrome—Results of two randomized, placebo-controlled studies. Aliment Pharmacol Ther 2009;29:329–41. [DOI] [PubMed] [Google Scholar]
- 6.Chey WD, Lembo AJ, Lavins BJ, et al. Linaclotide for irritable bowel syndrome with constipation: A 26-week, randomized, double-blind, placebo-controlled trial to evaluate efficacy and safety. Am J Gastroenterol 2012;107:1702–12. [DOI] [PubMed] [Google Scholar]
- 7.Brenner DM, Fogel R, Dorn SD, et al. Efficacy, safety, and tolerability of plecanatide in patients with irritable bowel syndrome with constipation: Results of two phase 3 randomized clinical trials. Am J Gastroenterol 2018;113:735–45. [DOI] [PubMed] [Google Scholar]
- 8.Schoenfeld PS. Advances in IBS 2016: A review of current and emerging data. Gastroenterol Hepatol (NY) 2016;12:1–11. [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenbaum DP, Yan A, Jacobs JW. Pharmacodynamics, safety, and tolerability of the NHE3 inhibitor tenapanor: Two trials in healthy volunteers. Clin Drug Investig 2018;38:341–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spencer AG, Labonte ED, Rosenbaum DP, et al. Intestinal inhibition of the Na+/H+ exchanger 3 prevents cardiorenal damage in rats and inhibits Na+ uptake in humans. Sci Transl Med 2014;6:227–36. [DOI] [PubMed] [Google Scholar]
- 11.Chey WD, Lembo AJ, Rosenbaum DP. Tenapanor treatment of patients with constipation-predominant irritable bowel syndrome: A phase 2, randomized, placebo-controlled efficacy and safety trial. Am J Gastroenterol 2017;112:763–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rome Foundation. Guidelines–rome III diagnostic criteria for functional gastrointestinal disorders. J Gastrointestin Liver Dis 2006;15:307–12. [PubMed] [Google Scholar]
- 13.Lewis SJ, Heaton KW. Stool form scale as a useful guide to intestinal transit time. Scand J Gastroenterol 1997;32:920–4. [DOI] [PubMed] [Google Scholar]
- 14.Mitsuhashi S, Ballou S, Jiang ZG, et al. Characterizing normal bowel frequency and consistency in a representative sample of adults in the United States (NHANES). Am J Gastroenterol 2018;113:115–23. [DOI] [PubMed] [Google Scholar]
- 15.Kanazawa M, Miwa H, Nakagawa A, et al. Abdominal bloating is the most bothersome symptom in irritable bowel syndrome with constipation (IBS-C): A large population-based internet survey in Japan. Biopsychosoc Med 2016;10:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ringel Y, Williams RE, Kalilani L, et al. Prevalence, characteristics, and impact of bloating symptoms in patients with irritable bowel syndrome. Clin Gastroenterol Hepatol 2009;7:68–72. [DOI] [PubMed] [Google Scholar]
- 17.Enck P, Aziz Q, Barbara G, et al. Irritable bowel syndrome. Nat Rev Dis Primers 2016;2:16014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kiyatkin M, Feng B, Schwartz E, et al. Combined genetic and pharmacological inhibition of TRPV1 and P2X3 attenuates colorectal hypersensitivity and afferent sensitization. Am J Physiol Gastrointest Liver Physiol 2013;305:G638–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wouters M, Balemans D, Wanrooy SV, et al. Histamine receptor H1-mediated sensitization of TRPV1 mediates visceral hypersensitivity and symptoms in patients with irritable bowel syndrome. Gastroenterology 2016;150:875–87. [DOI] [PubMed] [Google Scholar]
- 20.Li Q, King A, Liu L, et al. Tenapanor reduces IBS pain through inhibition of TRPV1-dependent neuronal hyperexcitability in vivo. Presented at the World Congress of Gastroenterology, October 13–18, 2017, Orlando, USA: American College of Gastroenterology.
- 21.Block GA, Rosenbaum DP, Leonsson-Zachrisson M, et al. Effect of tenapanor on serum phosphate in patients receiving hemodialysis. J Am Soc Nephrol 2017;28:1933–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aoki J, Ikari Y. Cardiovascular disease in patients with end-stage renal disease on hemodialysis. Ann Vasc Dis 2017;10:327–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Center for Drug Evaluation and Research (CDER). Guidance for industry: Irritable bowel syndrome—Clinical evaluation of drugs for treatment (https://www.fda.gov/regulatory-information/search-fda-guidance-documents/irritable-bowel-syndrome-clinical-evaluation-products-treatment2012). Accessed January 2, 2019.
- 24.Rey E, Talley NJ. Irritable bowel syndrome: Novel views on the epidemiology and potential risk factors. Dig Liver Dis 2009;41:772–80. [DOI] [PubMed] [Google Scholar]
- 25.Heitkemper M, Carter E, Ameen V, et al. Women with irritable bowel syndrome: Differences in patients' and physicians' perceptions. Gastroenterol Nurs 2002;25:192–200. [DOI] [PubMed] [Google Scholar]